

Abstract
Propionic academia (PA) occurs because of mutations in the PCCA or PCCB genes encoding the two subunits of propionyl-CoA carboxylase, a pivotal enzyme in the breakdown of certain amino acids and odd-chain fatty acids. There is no cure for PA, but dietary protein restriction and liver transplantation can attenuate its symptoms. We show here that a single intravenous injection of adeno-associated virus 2/8 (AAV8) or AAVrh10 expressing PCCA into PA hypomorphic mice decreased systemic propionylcarnitine and methyl citrate for up to 1.5 years. However, long-term phenotypic correction was always better in male mice. AAV-mediated PCCA expression was similar in most tissues in males and females at early time points and differed only in the liver. Over 1.5 years, luciferase and PCCA expression remained elevated in cardiac tissue for both sexes. In contrast, transgene expression in the liver and skeletal muscles of female, but not male, mice waned—suggesting that these tissues were major sinks for systemic phenotypic correction. These data indicate that single systemic intravenous therapy by AAV vectors can mediate long-term phenotype correction for PA. However, tissue-specific loss of expression in females reduces efficacy when compared with males. Whether similar sex-biased AAV effects occur in human gene therapy remains to be determined.
Introduction
Propionic acidemia (PA) is an inborn error of metabolism that results from mutations in the α or β subunit of propionyl-CoA carboxylase (PCC).1 There are early- and late-onset forms of disease, but symptoms generally manifest shortly following birth with early acute symptoms, including hyperammonemia, acidosis, and ketoacidosis, that can result in death if not treated. Late-onset cases are very heterogeneous in their presentation and can present any time from the first few months of life to adulthood. There are many chronic conditions observed in PA patients such as failure to thrive, seizures, immune deficiencies, and hypotonia; these are generally worsened by normal levels of protein consumption, infections, or periods of fasting, which can cause acute instances of metabolic decompensation.2,3 Repeated episodes of metabolic decompensation can result in neurological dysfunction, coma, and in some cases death.
While it is unclear exactly how all PA-related complications manifest, they can arise directly from high levels of circulating toxic compounds like propionic acid and ammonia or from secondary effects such as inhibition of energy-generating pathways like the Krebs' cycle or oxidative phosphorylation (OXPHOS) pathway by secondary metabolites. Propionic acid by itself can cause neurocognitive deficits like those observed in PA when this metabolite is injected alone into normal rats.4 Knowledge regarding the influence of local accumulation of potentially toxic metabolites in affected tissues is limited but growing.5 It is thought that elevations in propionyl-CoA and methyl citrate (MeCit) may be linked to certain pathological mechanisms of disease (Fig. 1). Propionyl-CoA is structurally similar to and competes with acetyl-CoA as a substrate for some biochemical processes; one example is the inhibition of pyruvate dehydrogenase,6 and another is formation of N-propionylglutamate by N-acytelglutamate synthase, a compound that is implicated in hyperammonemia.7,8 Propionyl-CoA also causes elevated levels of odd-number long-chain fatty acids (primarily C15:0, C17:0, and C17:1) as it acts as a primer for endogenous fatty acid synthesis.9 Finally, MeCit inhibits citrate synthase, aconitase, isocitrate dehydrogenase, and phosphofructokinase activity—enzymes that are involved in glycolysis and the citric acid cycle.10 Long-term pathology leading to organ failure in PA may be caused by OXPHOS inhibition, which has been demonstrated in multiple postmortem studies.5
FIG. 1.

Propionic academia (PA)-relevant metabolite pathways. Several relevant disease metabolites are discussed in this article and summarized here. The three main sources of propionyl-CoA are odd-chain fatty acids; the amino acids valine, isoleucine, methionine, and threonine; and production from gut bacteria. When deficiencies of the propionyl-CoA carboxylase (PCC) enzyme (1) exist, propionyl-CoA concentration in the mitochondria increases and is dealt with in two ways. Addition of carnitine by carnitine acyltransferase I (2) to form propionylcarnitine allows export across the mitochondrial membrane and eventually into the circulation. Alternatively, propionyl-CoA is combined with oxaloacetate by the action of citrate synthase to produce methyl citrate (3). Both propionyl-CoA and methyl citrate have been shown to cause inhibition of necessary energy-generating processes on the mitochondria.
In many countries, PA is detected during newborn screening by elevations in byproducts of disrupted PCC activity. In particular, elevations in propionylcarnitine (C3) and MeCit are diagnostic for PA.11 When PA is detected early, disease can be mediated by adopting a stringent protein-restricted diet sometimes coupled with carnitine supplementation at 100–200 mg/kg per day.2 Metronidazole is also given at 10–20 mg/kg per day either alone or alternating with other antibiotics to reduce production of propionyl-CoA from gut bacteria.2 While this has been successful at mitigating the worst symptoms of the disease in many patients and significantly increasing growth at young ages,12–14 no current treatments are capable of curing PA, and patients often continue to experience serious symptoms. Even with good dietary adherence, acute metabolic decompensation occurs in response to infections or other illnesses because of protein catabolism.15,16
More recently, elective liver transplantation has been performed in response to repeated episodes of metabolic decompensation. While this invasive intervention can blunt the worst metabolic issues in patients, PCC deficiency is corrected only in the liver, and patients still have high levels of propionic acid, propionyl-CoA, and MeCit.17–19 This likely occurs because liver transplantation does not account for the cell autonomous nature of PA where every cell in the body is deficient for PCC activity and could be producing these compounds. For example, orthotopic liver transplantation appears able to halt the progression of cardiomyopathy and neurocognitive function decline in some patients, but full recovery is not likely19,20; additionally, their long-term survival is 60–70% with transplantation.21 At this point it is unclear if these symptoms can be attributed to buildup of circulating compounds such as propionyl-CoA and MeCit, or if local production is the primary cause of pathology.
To explore other therapeutic options, we developed a hypomorphic model of PA in mice.22 These mice were generated on a PCCA null (Pcca−/−) mouse background that normally die within 36 hr of birth.23 They were rescued by transgenesis with a human cDNA encoding PCC with an A138T mutation identified in PA patients.24 These A138T mice survive to adulthood, have 2% of normal PCCA activity, and manifest many systemic and tissue-specific aspects of the disease that are observed in PA patients.22
We previously showed that single intravenous injection of first-generation adenovirus 5 or adeno-associated virus 2/8 (AAV8) vectors into the A138T mice at 5 or 10 weeks of age mediated dramatic reductions in propionylcarnitine and MeCit while improving weight gain on normal protein diets.22 However, phenotypic correction was lost as expected over 10 weeks after adenovirus therapy, but was stable after AAV8 therapy.
Given the efficacy of AAV8, we recently compared the ability of this liver-biased vector with muscle-biased AAV1 and more broadly tropic AAVrh10 vectors.25 A single intravenous injection resulted in significant corrections of circulating propionylcarnitine and MeCit by all vectors. When the muscle bias of AAV1 and the liver bias of AAV8 were made more specific by the use of tissue-specific promoters, liver-restricted AAV8-TTR-PCCAco mediated better correction than muscle-restricted AAV1-MCK6-PCCAco. This study helped elucidate two main points: (1) like liver transplantation, liver-restricted AAV8 therapy blunted, but did not ablate elevations in systemic PA metabolites, suggesting that liver-targeted gene therapy may be a viable and perhaps safer alternative to liver transplantation for liver-specific PA therapy. (2) The study also demonstrated that tissue-specific and tissue-biased gene therapy by AAV vectors could only blunt certain phenotypes when the disease is cell autonomous and not all cells are corrected. This finding suggests that treatment of nonhepatic tissues, particularly muscle, may provide a potential benefit in relieving some tissue-specific pathology in the disease.
In this work, we explored the ability of AAV vectors to express transgene in multiple tissues over time and to correct systemic PA symptoms over 1.5 years after single injection. We have documented the effects of host sex on the ability to mediate short- and long-term systemic metabolite reduction and maintenance of transgene expression in various tissues. These findings will have implication in the design of tissue-specific gene therapy for PA and possibly other metabolic diseases.
Materials and Methods
Animals
All mice were housed in animal facilities at Mayo Clinic and cared for by the department of comparative medicine according to Assessment and Accreditation of Laboratory Animal Care (AALAC) guidelines. All animal experiments were carried out according to the provisions of the PHS Animal Welfare Policy, Animal Welfare Act, and the principles of the NIH Guide for the Care and Use of Laboratory Animals. The Mayo Clinic Institutional Animal Care and Use Committee approved all procedures.
AAV vector production
All AAV vectors were produced by triple transfection of HEK 293 cells as published previously.22,26 Briefly, 293 cells were transfected with pHelper, pR/C(Rep/Cap), and PCCA or GFPLuc transgene plasmids. Cells were grown for 5 days in serum-free DMEM containing antibiotics, after which time NaCl was added to a final concentration of 0.5 M. After pelleting solid material the lysate was concentrated by tangential flow filtration, purified by ultracentrifugation through an Iodixanol gradient, and further concentrated using Amicon 100 kDa MWCO filter units (EMD Millipore). Vector titers were calculated by quantitative real-time PCR using SYBR green master mix and primers for the CMV or PCCA region of the vector DNA.
Vector administration
AAV vectors were diluted in PBS buffer to a concentration that was injectable in a 100 μl volume. From 5×1010 to 1×1012 viral genomes (vg) were injected intravenously via the tail vein.
Blood analyte assays
Blood was obtained from mice via submandibular puncture with a Goldenrod Lancet (MEDIpoint Inc.) and applied to Whatman 903 Protein Saver cards (GE Healthcare). Punches of blood-containing filter paper were then taken and assayed by tandem mass spectrometry as previously published.11,27
PCCA protein analysis
Tissues were removed from mice, rinsed thoroughly in PBS, and flash-frozen after euthanasia by exsanguination. Tissue pieces were then homogenized in T-PER buffer and quantitated using a BCA protein quantitation kit (Pierce). An amount of 50 μg of lysate was loaded into Mini-PROTEAN TGX gels (Bio-Rad), electrophoresed, and blotted onto PVDF membrane. The membrane was probed with custom-made anti-PCCA antibody (ProteinTech).
PCC enzyme activity assay
Mouse livers were homogenized in lysis buffer (50 mM Tris pH 8.0, 1 mM glutathione, 1 mM EDTA, protease inhibitor cocktail) and then spun at 15,000 rpm for 30 min. Protein concentration was determined using the Lowry method and 75 μg was used in the assay described previously for radiometric determination of PCC activity.28
Luciferase imaging
Mice were imaged at indicated time points after injection of 5×1011 vg of AAV8-GFPLuc. Before imaging, animals were anesthetized with a ketamine/xylezine injection, and then given an intraperitoneal injection of D Luciferin. Imaging was performed using a Kodak In Vivo F imaging system for a period of 10 min. Accumulation of total luminescence was then overlaid on top of a brightfield image.
Quantitative real-time PCR
Twenty-five days after injection with AAV8-PCCAco, DNA was extracted from mouse liver samples using a Maxwell 16 system and Maxwell 16 Mouse Tail DNA Purification kit (Promega). DNA was diluted and 10 ng per reaction was used with TaqMan qPCR primer probe sets (Life Technologies) specific for codon-optimized hPCCA and mouse Tfrc gene present in two copies. Reactions were run in triplicate and analyzed for ΔCt between the two genes.
Results
AAV treatment mediates sex-biased decreases in systemic PA metabolites
To remain consistent with previous studies, Pcca−/−(A138T) mice were treated with a single intravenous injection by tail vein at 5 weeks of age. Single-stranded AAV8-PCCAco vector expressing codon-optimized human PCCA was administered at 5 different doses from 5×1010 to 1×1012 vg into male or female mice. AAVrh10-PCCAco was also administered at the two highest doses of 5×1011 and 1×1012 vg per animal. Four weeks after injection, blood was collected and analyzed by tandem mass spectrometry for elevations in the two primary diagnostic metabolites for PA: propionylcarnitine (C3) and MeCit (Fig. 1). All doses of both AAV serotypes mediated significant decreases in both propionylcarnitine and MeCit when compared with untreated Pcca−/−(A138T) mice (Fig. 2A and B). However, there was a notable difference in the levels of PA metabolites in male and female mice. In most cases, female mice were outliers with the highest levels of propionylcarnitine and MeCit.
FIG. 2.

Dose-specific biomarker response to treatments. Five-week-old Pcca−/−(A138T) mice were administered indicated amounts of CMV-PCCAco expression vector pseudotyped with either the AAV8 or AAVrh10 capsid. Four weeks after IV administration of indicated vector, propionylcarnitine (A) and methyl citrate (B) levels were assayed in the blood. Mouse gender was also indicated by a closed circle for males or an open diamond for females. Asterisks indicate statistical significance of the entire treatment group compared with untreated Pcca−/−(A138T) mice. ****p<0.0001; **p<0.01. Error bars depict SEM.
AAV enhances PCCA expression in multiple tissues
Transplantation or gene therapy for PA has focused on correcting PCC deficiency in the liver. However, essentially every cell in the body is affected by PCC deficiency, and so the disease manifests in many tissues beyond the liver. To determine the degree to which AAV vectors can affect PCCA expression in different tissues, male and female mice were injected intravenously with 5×1011 vg of AAV1, AAV8, or AAVrh10 carrying PCCAco cDNA and were euthanized 4 weeks later. Western blots with PCCA antibody revealed increased PCCA protein in the liver, heart, and skeletal muscle by all AAV treatments, but little change in kidney, pancreas, or brain (Fig. 3 and Supplementary Fig. S1A; Supplementary Data are available online at www.liebertpub.com/hum). At this 4-week time point, PCCA levels differed between males and females only in the liver where quantitative real-time PCR revealed slightly lower, but statistically insignificant levels of AAV genome copies (Supplementary Fig. S1B). In contrast, cardiac and skeletal muscle PCCA expression was similar in male and female mice.
FIG. 3.

Tissue expression pattern of AAV vectors. Five-week-old mice of both genders were administered an IV dose of 5×1011 vg of AAV1-, AAV8-, or AAVrh10-PCCA. Twenty-five days after injection, mice were euthanized along with age-matched wild-type and untreated Pcca−/−(A138T) male controls. An amount of 50 μg of protein lysate for each indicated tissue was run on SDS-PAGE gel and blotted to PVDF membrane. Blots were then probed with anti-PCCA antibody and imaged.
Long-term biochemical effects of single AAV8-PCCAco therapy
Previous studies of AAV gene therapy for PA have revealed significant reductions in PA markers in mice at early time points.22,29,30 To determine if single intravenous AAV treatment could mediate long-lasting metabolite correction, propionylcarnitine and MeCit levels were monitored in male and female mice over 1.5 years after injection (Fig. 4). In untreated male or female Pcca−/−(A138T) mice, propionylcarnitine levels peaked at 13 weeks and then declined over the 1.5-year period (Fig. 4A dashed lines). Although AAV8-PCCAco therapy in males maintained low propionylcarnitine levels over time, waning levels in untreated males resulted in statistical significance only to 45 weeks. Liver PCC enzyme activity in 1.5-year-old untreated male and female mice revealed no differences (Supplementary Fig. S2), suggesting that PCC activity did not vary between the sexes over time. MeCit levels in untreated males also decreased over time (Fig. 4B dashed black line), but in this case the single AAV8-PCCAco treatment still resulted in significantly lower MeCit levels when compared with untreated PA males (p≤0.001 at 85 weeks by T-test). AAV8-PCCAco vector also reduced propionylcarnitine and MeCit levels in the female mice through 13 weeks, but the level of correction decreased by week 45 and was lost entirely by the 1.5-year time point (Fig. 4A and B).
FIG. 4.

Long-term PA metabolite monitoring. C3 (A) and MeCit (B) were monitored in Pcca−/−(A138T) mice for 1.5 years after injection of 5×1011 vg of AAV8-PCCAco. Age-matched untreated animals are denoted by dashed lines, while treated animals are denoted by solid lines. N=5 for each untreated and treated group. Asterisks (*p≤0.05; **p≤0.01 and ***p≤0.001) indicate statistical significance between sex-matched untreated and treated groups at indicated time points (Student's t-test). Error bars depict SEM.
Long-term expression of PCCA by AAV8 varies by tissue type and gender
At the 1.5-year posttreatment time point, the study was terminated and animals were euthanized. PCCA protein expression levels in the heart, liver, and gastrocnemius were measured by Western blot in three treated male and female mice (Fig. 5A). PCCA expression remained high in the hearts of both male and female animals. Expression in the liver and gastrocnemius muscle remained high in male mice. In contrast, expression in liver and skeletal muscle was barely detectable in treated female mice.
FIG. 5.

Long-term protein and enzyme activity response to AAV treatment. Pcca−/−(A138T) mice treated by IV injection of 5×1011 vg of AAV8-PCCAco along with age-matched wild type and untreated Pcca−/−(A138T) male controls were euthanized at 1.5 years after injection of the treatment animals. (A) An amount of 50 μg of protein lysate for each indicated tissue was run on SDS-PAGE gel and blotted to PVDF membrane. Blots were then probed with anti-PCCA antibody and/or neutravidin and imaged for protein expression. (B) Liver PCC activity was measured in male mice and compared with male wild type and untreated Pcca−/−(A138T) controls. *p<0.05 indicates statistical significance compared with untreated Pcca−/−(A138T) controls (one-way ANOVA). Error bars depict SEM.
AAV8-PCCA elicits long-term increases in liver PCC activity of male Pcca−/−(A138T) mice
PCC enzyme activity was also assayed in the AAV8-PCCAco-treated male mice 1.5 years after vector administration (insufficient numbers of females were available for these tests). Whereas untreated A138T mice had only 2% of wild-type PCC activity, a single treatment with AAV8-PCCAco increased PCC activity to 15% of wild-type levels even at this late time point (Fig. 5B).
In vivo luciferase expression mimics PCCA expression
To confirm the tropism effect observed in AAV8-PCCAco-treated mice, 5×1011 vg of AAV8 expressing a green fluorescent-luciferase fusion protein (AAV8-GFPLuc) were injected intravenously into male and female Pcca−/−(A138T) mice. Twenty-five days later, luciferase imaging revealed transgene expression in the skeletal muscle and heart of both genders, but strong activity was only observed in the livers of the male mice (Fig. 6A). This was consistent with differential expression of PCCA in the livers of male and female mice observed in Western blots (Fig. 3). Imaging 1.5 years after single AAV8-GFPLuc injection revealed lower, but persistent luciferase activity primarily in the heart and skeletal muscle of both male and female mice with little detectable activity in the livers (Fig. 6B).
FIG. 6.
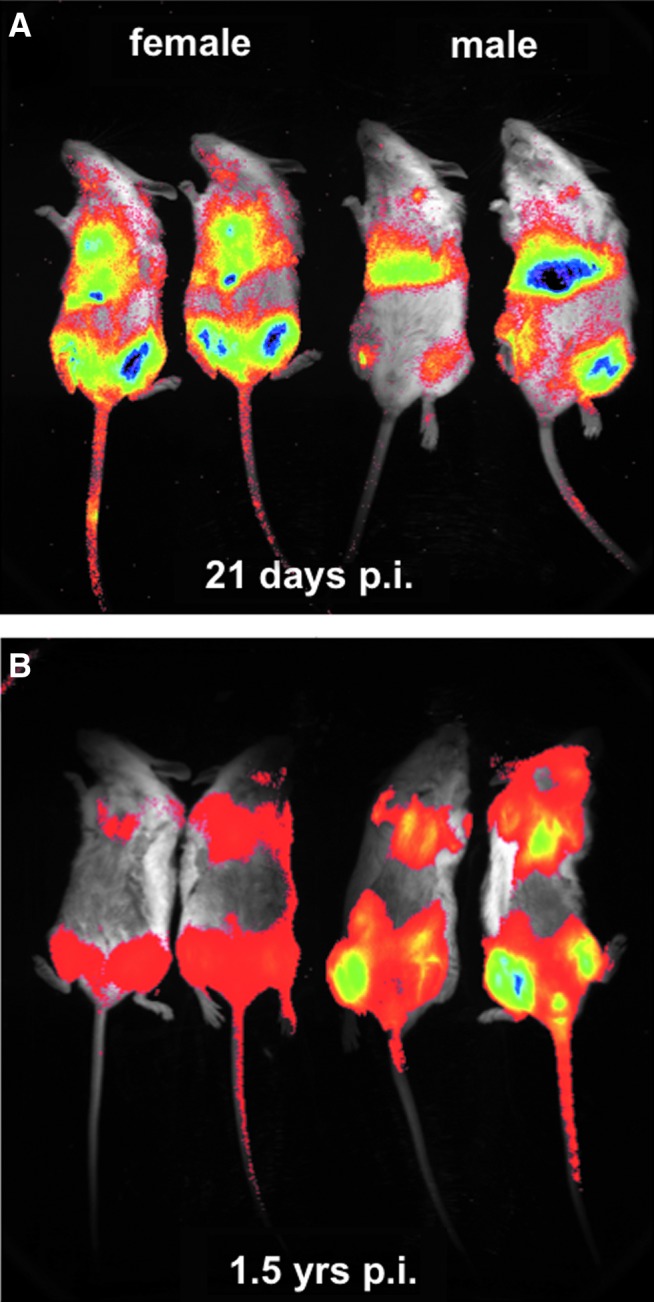
In vivo luciferase imaging of vector expression. Five-week-old Pcca−/−(A138T) mice of both gender were administered an IV dose of 5×1011 vg of AAV8-GFPLuc. The mice were injected with luciferin and imaged for 10 min at 21 days (A) and 1.5 years (B) after injection. Color images available online at www.liebertpub.com/hum
Discussion
This study demonstrates that AAV vectors are able to provide sustained long-term expression of the PCCA protein in Pcca−/−(A138T) mice over 1.5 years after single intravenous treatment. This expression resulted in levels of PCC activity that we believe are therapeutically relevant as determined by reductions in the levels of circulating propionylcarnitine and MeCit metabolites. Previous studies have provided evidence that AAV-mediated transfer of PCCA cDNA to Pcca-deficient mice mediates significant correction of circulating metabolite levels. However, these studies demonstrated efficacy only at early time points.22,29,30 This work supports the ability of single AAV treatment to mitigate some PA phenotypes over the lifespan of mice.
Previous studies demonstrated that AAV vectors mediate markedly different levels of liver transgene expression in male and female mice.31,32 Since transcription requires a double-stranded DNA template, proteins cannot be expressed directly from single-stranded AAV genomes. Therefore, incoming single-stranded genomes must be converted to double-stranded template by DNA repair mechanisms. Alternately, an expressible double-stranded template can be produced by hybridization of co-infected negative- and positive-strand AAV genomes. For this to occur, both must be delivered into the same cell and this is generally only likely to occur in tissues where many vector genomes are delivered.
Higher expression in males previously appeared to be mediated by higher levels of androgens in male mice.31 The observed higher expression in the livers of male mice was attributed to increased second-strand DNA synthesis of the AAV vector genome. In our studies, we observed no significant difference in the amount of AAV vector genomes in the livers of male and female mice by quantitative PCR (Supplementary Fig. S1B). We speculate that the female mice in our studies also have lower vector-mediated expression because of lack of androgens in female mice.
Previous studies comparing therapy in different sexes focused on relatively short-term expression of transgene primarily in the liver.31,33 Our observation of higher luciferase expression and higher therapeutic PCCA protein expression in the livers of male mice than in female mice agree with these earlier studies. This study in the PA mouse model extends these observations of differential expression between the sexes of mice by examining expression in muscle tissue and liver over substantially longer periods of time. While there was a large difference in luciferase and PCCA expression in the livers of male and female mice at 4 weeks, this effect was not observed to the same degree in muscle tissues at this early time point. This suggests that either we are providing a saturating dose of AAV to muscle tissue, or transgene expression in muscle of female mice is not attenuated in the same way the liver is.
The effect of sex on transgene expression was striking as far as 18 months after single treatment. Female mice had very little PCCA or luciferase expression in either the liver or skeletal muscle. In contrast, cardiac tissue was not affected by sex at early and late time points. There are several possible explanations for this cardiac effect: (1) there may be higher levels of co-transduction with both positive- and negative-stranded genomes in this tissue in both sexes, allowing for vector genome hybridization and expression; (2) cardiac tissues may better preserve single-stranded AAV vector genomes, allowing for better double-strand conversion; (3) the heart may have a higher capacity to convert single-stranded genomes to expressible double-stranded versions; and (4) cardiomyocytes may have a longer lifespan than hepatocytes,34 allowing more time and stability for AAV vector expression.
This study also builds on earlier work to explore how sex-biased transgene expression influences gene therapy. Four weeks after therapy, we document lower therapeutic gene expression in the liver of females, but not in other tissues. Higher propionylcarnitine and MeCit levels paralleled lower PCCA expression in the liver. This suggests that the liver is indeed an important source for or a metabolic sink for PA metabolites. This is consistent with our recent comparison of liver-specific versus muscle-specific AAV therapy in PA mice.25 In this case, liver-targeted therapy mediated markedly better control of metabolites in the blood than targeted expression in muscle. While liver PCCA expression was worse in females than in males after AAV therapy, the female mice still showed a therapeutic benefit from 1 to 45 weeks after treatment. This suggests that transgene expression beyond the liver also benefits the animals.
It is important to note that, regardless of the results in female mice, male Pcca−/−(A138T) mice retained significant PCCA expression in the heart, liver, and skeletal muscle throughout the entire 1.5-year duration of the experiment. This represents the majority of the life span of a mouse. Natural decreases in C3 and MeCit were observed in untreated PA animals, which could be explained by the normal waxing and waning of basal metabolic rate and food consumption of the mice as they become mature and then age over time. Another possibility is differences in the gut microbiome of male and female PA mice. Not only is gut bacteria a significant source of propionyl-CoA, but the microbiota has the ability to regulate androgens,35 which could partially account for differential AAV expression as well. In the study by Markle et al., the cecal contents of male nonobese diabetic mice were actually able to prevent diabetes onset in females.35 There has been little investigation into the effects of microbiome on organic acidemias, but future studies in this area may prove beneficial.
Both propionyl-CoA and MeCit have been directly linked to inhibition of several biochemical processes thought to cause pathology in PA patients.10,36 Increased PCC protein levels increased liver PCC enzyme activity and these corresponded with decreased levels of circulating propionylcarnitine and MeCit. The decreased levels of MeCit observed in male mice were particularly encouraging as this metabolite has been directly linked to metabolic perturbations.5,10
It is notable that AAV-mediated PCCA expression persisted in the heart of male and female mice even though it waned in other tissues. PA patients frequently suffer cardiac symptoms, including cardiomyopathy and arrhythmias,3 and there is growing evidence that these complications arise as a result of local OXPHOS complex inhibition in the cardiac tissue.36–38 Therefore, cardiac-specific treatment may be necessary to address the cardiac aspects of PA disease independently of liver therapy. In addition, certain hypomorphic PCC mutations do not have neonatal onset, but instead manifest as cardiac events in adults.39 For example, a founder A1606G mutation in Pccb is observed in Amish and Mennonite populations (mutation identified in Ref.40). PCC enzyme with Pccb A1606G retains significant enzyme activity, and so neonatal presentation is infrequent, but approximately 25% of patients with this mutation develop heart failure, usually secondary to respiratory infections.41
While AAV vectors have become favored for many in vivo gene therapy applications, there have been observations of tumor formation in AAV-treated mice.42,43 These tumors are thought to be because of preferential integration of transgene near transcriptionally active regions.44,45 More recently, liver cancers have been observed after neonatal AAV8 treatment of mouse models of methylmalonic acidemia (MMA).46 This is concerning as the MMA results from mutations in the enzyme methylmalonyl CoA mutase, the metabolic step immediately after PCC. In our studies, no increases in liver tumors were observed in any treated mice when compared with controls (data not shown). However, this was not a central experimental focus of these studies. This difference in tumor observation could be because of intrinsic differences in MMA and PA. Alternately, this difference is perhaps more likely because of differences in the age of AAV treatment in the MMA studies and ours in the PA mice. In the MMA studies, neonatal mice were injected intrahepatically with AAV8 early after birth at a time when their immune systems were barely functional. In contrast, we treated PA mice 5 weeks after birth by the intravenous route at a time when the animals' immune systems are better developed. We speculate that tumor formation in MMA mice may be amplified by reduced immune monitoring for neoplasia because of their age of treatment. Regardless, observations of this troubling side effect will require more stringent safety testing with AAV vectors prior to use in humans.
This study demonstrates that a single systemic injection with AAV vectors can mediate long-term phenotype correction in an animal model of PA. This supports translation of AAV vectors for systemic therapy of PA in humans. While robust, long-lasting efficacy was observed in male mice, tissue-specific loss of expression in females reduced the persistence and level of therapy in these mice. Whether AAV vectors mediate similar sex-biased effects in humans remains to be determined. The observation that AAV can mediate persistent PCCA expression in the hearts of both male and female mice suggests that this gene therapy may also be a viable option to prevent or treat cardiac phenotypes associated with early-onset or late-onset PA.
Supplementary Material
Acknowledgments
AAV constructs were obtained from the National Gene Vector Biorepository and the University of Pennsylvania Vector Core (Philadelphia, PA). We thank the Clinical Core of the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567), Optical Microscopy Core, the Gene Expression Core, and the Advanced Genomics Technology Core at Mayo Clinic for assistance with the work. This work was supported by funding to M.A.B. from the Propionic Acidemia Foundation and the Organic Acidemia Association. This work was also supported by the Mayo Clinic Department of Laboratory Medicine and Pathology, the Liver Regeneration Program in the Center for Regenerative Medicine, and the Department of Molecular Medicine. This gene therapy program was the Walter & Lucille Rubin Fund in Infectious Diseases Honoring Michael Camilleri, MD, at Mayo Clinic.
Author Disclosure Statement
The authors declare that no conflict of interest exists regarding the work in this publication.
References
- 1.Hsia YE, Scully KJ, Rosenberg LE. Inherited propionyl-Coa carboxylase deficiency in “ketotic hyperglycinemia.” J Clin Invest 1971;50:127–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baumgartner MR, Horster F, Dionisi-Vici C, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis 2014;9:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pena L, Franks J, Chapman KA, et al. Natural history of propionic acidemia. Mol Genet Metab 2012;105:5–9 [DOI] [PubMed] [Google Scholar]
- 4.Pettenuzzo LF, Schuck PF, Fontella F, et al. Ascorbic acid prevents cognitive deficits caused by chronic administration of propionic acid to rats in the water maze. Pharmacol Biochem Behav 2002;73:623–629 [DOI] [PubMed] [Google Scholar]
- 5.Kolker S, Burgard P, Sauer SW, et al. Current concepts in organic acidurias: understanding intra- and extracerebral disease manifestation. J Inherit Metab Dis 2013;36:635–644 [DOI] [PubMed] [Google Scholar]
- 6.Schwab MA, Sauer SW, Okun JG, et al. Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role for endogenous mitochondrial toxins. Biochem J 2006;398:107–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ah Mew N, Mccarter R, Daikhin Y, et al. N-carbamylglutamate augments ureagenesis and reduces ammonia and glutamine in propionic acidemia. Pediatrics 2010;126:e208–e214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tuchman M, Caldovic L, Daikhin Y, et al. N-carbamylglutamate markedly enhances ureagenesis in N-acetylglutamate deficiency and propionic acidemia as measured by isotopic incorporation and blood biomarkers. Pediatr Res 2008;64:213–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sperl W, Murr C, Skladal D, et al. Odd-numbered long-chain fatty acids in propionic acidaemia. Eur J Pediatr 2000;159:54–58 [DOI] [PubMed] [Google Scholar]
- 10.Cheema-Dhadli S, Leznoff CC, Halperin ML. Effect of 2-methylcitrate on citrate metabolism: implications for the management of patients with propionic acidemia and methylmalonic aciduria. Pediatr Res 1975;9:905–908 [DOI] [PubMed] [Google Scholar]
- 11.Turgeon CT, Magera MJ, Cuthbert CD, et al. Determination of total homocysteine, methylmalonic acid, and 2-methylcitric acid in dried blood spots by tandem mass spectrometry. Clin Chem 2010;56:1686–1695 [DOI] [PubMed] [Google Scholar]
- 12.Brandt IK, Hsia YE, Clement DH, et al. Propionicacidemia (ketotic hyperglycinemia): dietary treatment resulting in normal growth and development. Pediatrics 1974;53:391–395 [PubMed] [Google Scholar]
- 13.Touati G, Valayannopoulos V, Mention K, et al. Methylmalonic and propionic acidurias: management without or with a few supplements of specific amino acid mixture. J Inherit Metab Dis 2006;29:288–298 [DOI] [PubMed] [Google Scholar]
- 14.Yannicelli S, Acosta PB, Velazquez A, et al. Improved growth and nutrition status in children with methylmalonic or propionic acidemia fed an elemental medical food. Mol Genet Metab 2003;80:181–188 [DOI] [PubMed] [Google Scholar]
- 15.Sutton VR, Chapman KA, Gropman AL, et al. Chronic management and health supervision of individuals with propionic acidemia. Mol Genet Metab 2012;105:26–33 [DOI] [PubMed] [Google Scholar]
- 16.Thompson GN, Chalmers RA. Increased urinary metabolite excretion during fasting in disorders of propionate metabolism. Pediatr Res 1990;27:413–416 [DOI] [PubMed] [Google Scholar]
- 17.Rela M, Muiesan P, Andreani P, et al. Auxiliary liver transplantation for metabolic diseases. Transplant Proc 1997;29:444–445 [DOI] [PubMed] [Google Scholar]
- 18.Schlenzig JS, Poggi-Travert F, Laurent J, et al. Liver transplantation in two cases of propionic acidaemia. J Inherit Metab Dis 1995;18:448–461 [DOI] [PubMed] [Google Scholar]
- 19.Yorifuji T, Muroi J, Uematsu A, et al. Living-related liver transplantation for neonatal-onset propionic acidemia. J Pediatr 2000;37:572–574 [DOI] [PubMed] [Google Scholar]
- 20.Yorifuji T, Kawai M, Mamada M, et al. Living-donor liver transplantation for propionic acidaemia. J Inherit Metab Dis 2004;27:205–210 [DOI] [PubMed] [Google Scholar]
- 21.Barshes NR, Vanatta JM, Patel AJ, et al. Evaluation and management of patients with propionic acidemia undergoing liver transplantation: a comprehensive review. Pediatr Transplant 2006;10:773–781 [DOI] [PubMed] [Google Scholar]
- 22.Guenzel AJ, Hofherr SE, Hillestad M, et al. Generation of a hypomorphic model of propionic acidemia amenable to gene therapy testing. Mol Ther 2013;21:1316–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyazaki T, Ohura T, Kobayashi M, et al. Fatal propionic acidemia in mice lacking propionyl-CoA carboxylase and its rescue by postnatal, liver-specific supplementation via a transgene. J Biol Chem 2001;276:35995–35999 [DOI] [PubMed] [Google Scholar]
- 24.Campeau E, Desviat LR, Leclerc D, et al. Structure of the PCCA gene and distribution of mutations causing propionic acidemia. Mol Genet Metab 2001;74:238–247 [DOI] [PubMed] [Google Scholar]
- 25.Guenzel AJ, Kraus JP, Matern D, et al. Long-term sex-biased correction of circulating propionic acidemia disease markers by adeno-associated virus vectors. Hum Gene Ther 2014. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lock M, Alvira M, Vandenberghe LH, et al. Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum Gene Ther 2010;21:1259–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turgeon C, Magera MJ, Allard P, et al. Combined newborn screening for succinylacetone, amino acids, and acylcarnitines in dried blood spots. Clin Chem 2008:54:657–664 [DOI] [PubMed] [Google Scholar]
- 28.Jiang H, Rao KS, Yee VC, et al. Characterization of four variant forms of human propionyl-CoA carboxylase expressed in Escherichia coli. J Biol Chem 2005;280:27719–27727 [DOI] [PubMed] [Google Scholar]
- 29.Chandler RJ, Chandrasekaran S, Carrillo-Carrasco N, et al. Adeno-associated virus serotype 8 gene transfer rescues a neonatal lethal murine model of propionic acidemia. Hum Gene Ther 2011;22:477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hofherr SE, Senac JS, Chen CY, et al. Short-term rescue of neonatal lethality in a mouse model of propionic acidemia by gene therapy. Hum Gene Ther 2009;20:169–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davidoff AM, Ng CY, Zhou J, et al. Sex significantly influences transduction of murine liver by recombinant adeno-associated viral vectors through an androgen-dependent pathway. Blood 2003;102:480–488 [DOI] [PubMed] [Google Scholar]
- 32.Paneda A, Vanrell L, Mauleon I, et al. Effect of adeno-associated virus serotype and genomic structure on liver transduction and biodistribution in mice of both genders. Hum Gene Ther 2009;20:908–917 [DOI] [PubMed] [Google Scholar]
- 33.Dane AP, Cunningham SC, Graf NS, et al. Sexually dimorphic patterns of episomal rAAV genome persistence in the adult mouse liver and correlation with hepatocellular proliferation. Mol Ther 2009;17:1548–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bergmann O, Bhardwaj RD, Bernard S, et al. Evidence for cardiomyocyte renewal in humans. Science 2009;324:98–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Markle JG, Frank DN, Mortin-Toth S, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 2013;339:1084–1088 [DOI] [PubMed] [Google Scholar]
- 36.De Keyzer Y, Valayannopoulos V, Benoist JF, et al. Multiple OXPHOS deficiency in the liver, kidney, heart, and skeletal muscle of patients with methylmalonic aciduria and propionic aciduria. Pediatr Res 2009;66:91–95 [DOI] [PubMed] [Google Scholar]
- 37.Fragaki K, Cano A, Benoist JF, et al. Fatal heart failure associated with CoQ10 and multiple OXPHOS deficiency in a child with propionic acidemia. Mitochondrion 2011;11:533–536 [DOI] [PubMed] [Google Scholar]
- 38.Mardach R, Verity MA, Cederbaum SD. Clinical, pathological, and biochemical studies in a patient with propionic acidemia and fatal cardiomyopathy. Mol Genet Metab 2005;85:286–290 [DOI] [PubMed] [Google Scholar]
- 39.Bhan AK, Brody C. Propionic acidemia: a rare cause of cardiomyopathy. Congest Heart Fail 2001;7:218–219 [DOI] [PubMed] [Google Scholar]
- 40.Kraus JP, Spector E, Venezia S, et al. Mutation analysis in 54 propionic acidemia patients. J Inherit Metab Dis 2012;35:51–63 [DOI] [PubMed] [Google Scholar]
- 41.Morton DH, Donnelly P, Duffy A, et al. Cardiomyopathy is common in the propionic acidemia variant PCCB c.1606A>G and can be prevented & reversed by metabolic therapy. Mol Genet Metab 2014;111:262–263 [Google Scholar]
- 42.Donsante A, Miller DG, Li Y, et al. AAV vector integration sites in mouse hepatocellular carcinoma. Science 2007;317:477. [DOI] [PubMed] [Google Scholar]
- 43.Donsante A, Vogler C, Muzyczka N, et al. Observed incidence of tumorigenesis in long-term rodent studies of rAAV vectors. Gene Ther 2001;8:1343–1346 [DOI] [PubMed] [Google Scholar]
- 44.Miller DG, Trobridge GD, Petek LM, et al. Large-scale analysis of adeno-associated virus vector integration sites in normal human cells. J Virol 2005;79:11434–11442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakai H, Wu X, Fuess S, et al. Large-scale molecular characterization of adeno-associated virus vector integration in mouse liver. J Virol 2005;79:3606–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chandler RJ, LaFave MC, Varshney GK, et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J Clin Invest 2015. [Epub ahead of print] DOI: 10.1172/JCI79213 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.