

Abstract
Caveolins are scaffolding proteins that play a pivotal role in numerous processes, including caveolae biogenesis, vesicular transport, cholesterol homeostasis and regulation of signal transduction. There are three different isoforms (Cav-1, -2 and -3) that form homo- and hetero-aggregates at the plasma membrane and modulate the activity of a number of intracellular binding proteins. Cav-1 and Cav-3, in particular, are respectively expressed in the reserve elements (e.g. satellite cells) and in mature myofibres of skeletal muscle and their expression interplay characterizes the switch from muscle precursors to differentiated elements. Recent findings have shown that caveolins are also expressed in rhabdomyosarcoma, a group of heterogeneous childhood soft-tissue sarcomas in which the cancer cells seem to derive from progenitors that resemble myogenic cells. In this review, we will focus on the role of caveolins in rhabdomyosarcomas and on their potential use as markers of the degree of differentiation in these paediatric tumours. Given that the function of Cav-1 as tumour conditional gene in cancer has been well-established, we will also discuss the relationship between Cav-1 and the progression of rhabdomyosarcoma.
Keywords: rhabdomyosarcoma, caveolins, skeletal muscle
Introduction
Caveolins (Cav-1, -2 and -3) are membrane scaffolding proteins that regulate numerous processes in a variety of tissues and cell types [1, 2]. They participate in the biogenesis of caveolae, flask-shaped invaginations of the plasma membrane where several molecules involved in the regulation of transduction pathways are specifically and highly enriched [3–5]. Caveolins are anchored at the inner leaflet of caveolae through a short hairpin hydrophobic domain and protrude towards the cytoplasm, where they can bind and influence the activity of several protein partners via a caveolin scaffolding domain (CSD) [6, 7]. In skeletal muscle, in particular, Cav-1 expression is restricted to satellite cells [8–10], whereas Cav-3 plays a pivotal role in the mature myofibres [11–13], as demonstrated by the fact that its deficiency is associated to a set of genetic muscular disorders, known as caveolinopathies [14–17]. Rhabdomyosarcoma (RMS) is the most frequent soft tissue sarcoma occurring in childhood and sharing features of myogenic cells [18–20]. Hence, cancerous RMS cells can be univocally identified by means of muscle markers [21]. Two different works have shown that caveolins are expressed in a cell stage–dependent manner in RMS [22, 23]. This review will summarize these findings and highlight the relevance of caveolins as diagnostic markers for the detection of RMS with a different grading. In addition, the potential contribution of caveolins to the progression of RMS is discussed, particularly for Cav-1.
Caveolae and caveolins
Caveolae are characteristic flask-shaped invaginations of the plasma membrane that configure as specialized microdomains involved in numerous functions, including cell signalling, lipid regulation and endocytosis [1–7]. Cav-1, Cav-2 and Cav-3 are the main structural proteins of caveolae and are codified by three different genes that have a high degree of homology [1, 2] (Table 1). In particular, caveolins are anchored to the inner leaflet of the plasma membrane via a short hydrophobic loop, which allow them to assume a unique hairpin structure, characterized by the presence of both N- and C-terminal portions facing towards the cytoplasm [1, 2]. Cav-1 and Cav-2 form hetero-oligomers and are ubiquitously co-expressed [24], whereas Cav-3 forms homo-oligomers that are predominantly expressed in skeletal and cardiac muscle [11, 12] (Table 1). Cav-1 and Cav-3 are unequivocally required for the biogenesis of caveolae [25], as corroborated by the lack of caveolae in Cav-1 null animals [26] and in muscle and cardiac tissues of Cav-3 null animals [27, 28], despite other key protein molecules, such as PTRF-Cavin [29, 30] and ARAF-1 [31], seem to have a role in the formation of these membranous structures. Although caveolins are predominantly recovered at the plasma membrane and in Golgi apparatus of cells [32–34], their subcellular localization may change upon post-translational modifications, as occurs for Cav-1, which may be targeted to cytoplasm or secretory vesicles upon phosphorylation on Tyr14 or Ser80 residues, respectively [35, 36].
Table 1.
The family of caveolins: genomic localization, cell- and tissue-specific expression and principal knockout mouse phenotypes
| Human gene | Chromosomal localization | Expression patterns | Knockout mouse phenotypes |
|---|---|---|---|
| Cav-1 | 7q31.1 | Adipocytes | Diabetes [40, 41] |
| Cardiac fibroblasts | Lung diseases [26, 42, 43] | ||
| Endothelia | Heart diseases [42, 44] | ||
| Macrophages | Cerebral ischaemia [45–47] | ||
| Neural cells | Predisposition to skin and breast cancer [54, 75–79] | ||
| Pneumocytes | Protection from prostate cancer [80] | ||
| Smooth muscle cells | |||
| Striated muscle cells | |||
| Cav-2 | 7q31.1 | Same as Cav-1 | Impaired pulmonary functionality [48] |
| Abnormalities in skeletal muscle [49] | |||
| Cav-3 | 3p25 | Striated muscle cells | Mild myopathic changes [27, 28] |
| Smooth muscle cells | Cardiomyopathy [50] | ||
| Cardiac myocytes | Insulin resistance and increased adiposity [51] |
Caveolins as scaffolding proteins
The most important feature of caveolins relies on their ability to bind and modulate the biological activity of a multitude of intracellular protein partners through the so-called CSD, which recognises motifs enriched of aromatic residues [6, 37]. Although the CSD sequence results highly conserved in both Cav-1 and Cav-3, it is slightly different for Cav-2, which in fact seems to be clearly less prone to bind proteins. To date, more than 90 proteins are known to be bound and regulated by caveolins, including G-protein–coupled receptors and G-proteins, different membrane receptorial (insulin receptor, PDGFR, TGF-β receptors, etc.) and non-receptorial proteins (Src, Fyn, PKA, etc.), enzymes (adenylyl cyclase, e- and nNOS, phospholipases, etc.), GTPases (H-RAS, RhoA, etc.), protein adaptors (Shc), nuclear proteins (estrogen and androgen receptors) as well as a miscellaneous of other proteins involved in disparate processes (E-cadherin, β- and γ-catenin, calsequestrin, calreticulin, etc.) [1–7]. Cav-1, furthermore, is a cholesterol-binding protein and contributes to regulate its homeostasis [38].
Impact of the lack of caveolins in the whole body physiology
Mice deficient in Cav-1, Cav-2 or Cav-3 are viable and fertile but display several alterations in the whole body physiology [15, 39] (Table 1). In particular, Cav-1 null mice develop a complex spectrum of diseases, such as diabetes [40, 41], impaired lung [26, 42, 43] and heart [42, 44] functionality and cerebral ischaemia [45–47]. Moreover, Cav-1 null mice are predisposed to certain tumours whereas are protected from others [15, 39] (for a more detailed discussion please refer to section ‘Relevance of Cav-1 as tumour conditional gene’). Cav-2 null mice show exercise intolerance associated to impaired pulmonary functionality [48] and peculiar abnormalities in skeletal muscle, such as tubular aggregate formation, mitochondrial proliferation/aggregation and increased number of satellite cells [49]. On the other hand, because of the restricted tissue-specificity of Cav-3 expression within mature myofibres and cardiac myocytes, Cav-3 null mice exhibit mild myopathic changes, such as mononuclear cell infiltration, variable fibre size and presence of necrosis [27, 28], as well as a progressive cardiomyopathy due to extracellular regulated kinases (ERK) pathway hyperactivation [50]. In addition, Cav-3 null mice develop insulin resistance and increased adiposity [51], suggesting an involvement of Cav-3 in the regulation of the whole body glucose metabolism.
Relevance of Cav-1 as tumour conditional gene
The human Cav-1 gene is localized on a suspected tumour suppressor locus of chromosome 7q31.1 [52]. Targeted down-regulation of Cav-1, indeed, promotes cell transformation of NIH3T3 fibroblasts, anchorage-independent growth in vitro [53] and tumour growth in vivo [54, 55]. In addition, Cav-1 overexpression blocks mouse embryonic fibroblasts in the G0/G1 phase of the cell cycle [56] and abrogates the transformed cell phenotype [57, 58], suggesting that Cav-1 acts as a tumour-suppressor in non-neoplastic tissues by mainly limiting the ERK signalling pathway [53, 59]. Accordingly, Cav-1 is down-regulated in different tumours, such as ovarian [60], lung [61] and breast carcinomas [57, 62–64], mesenchymal sarcomas [65] as well as in cell lines derived from human tumours or transformed by oncogenes [66]. Paradoxically, Cav-1 is up-regulated in many other malignancies, such as colon adenocarcinoma [67], bladder carcinoma [68], oesophageal squamous cell carcinoma [69] and prostate cancer [70], suggesting that Cav-1 plays a dual role in cancer progression depending on the different type and stage of cancer [71–74]. The complex relationship between Cav-1 and cancer has been particularly outlined by the employment of Cav-1 null mice (Table 1). In particular, although ablation of Cav-1 seems to be not sufficient to induce spontaneous tumour formation, it significantly predisposes mice to skin and breast tumours by stimulating cellular hyperplasia [54, 75–79]. In striking contrast, genetic loss of Cav-1 in the TRAMP model (transgenic adenocarcinoma of mouse prostate) decreases incidence of prostate tumours and metastasis [80]. Collectively, a growing body of evidence derived from different models suggests that loss of Cav-1 cooperates to cell transformation in the early phases of tumour growth, whereas a later Cav-1 re-expression favours tumour metastases and multi-drug resistance [81–84]. A possible explanation to this ambiguous behaviour could be referred to the presence of multiple alterations in the cellular environment, such as the expression of different subsets of caveolin partners during tumour progression or the occurrence of inactivating mutations [85, 86] or post-translational modifications of Cav-1 [87, 88], which may promote a shift of Cav-1 activity from tumour-suppressor to proto-oncogene. In this sense, Cav-1 should be considered a tumour-conditional gene [71–74].
Role of caveolins in skeletal muscle
In skeletal muscle, Cav-1 and Cav-3 reside in satellite cells [8–10] and myofibres [11–13], respectively. Interposed among the fibres, satellite cells represent a pool of reserve elements which are recruited in different processes, such as the repair of damaged fibres [89], stretch [90] and fibre hypertrophy [91]. In particular, Cav-1 has been shown to play an important role in maintaining the quiescence of satellite cells by antagonizing the RAS/ERK signalling [10]. On muscle injury, Cav-1 is transcriptionally down-regulated through an HGF/cMET axis pathway, allowing the satellite cells to escape quiescence, migrate and repair the injured site [10]. On the other side, in mature myofibres Cav-3 regulates the activity of different signalling proteins [14–16], associates with the T-tubules structures [27] and stabilizes the dystrophin–glycoprotein complex through a WW-like binding domain [92, 93]. In this regard, a deficit of Cav-3 expression due to inherited Cav-3 gene mutations is responsible of a set of distinct neuromuscular and cardiac disorders [14–17]. For instance, the missense Cav-3 (P104L) substitution, which has been predominantly associated to the Limb Girdle Muscular Dystrophy 1-C (LGMD1-C) [94], affects the integrity of skeletal muscle in vivo [95, 96] and the myogenic program in vitro [97]. Remarkably, the homeostasis of skeletal muscle is even compromised by an excess of Cav-3, as observed in muscles derived from patients affected by Duchenne’s muscular dystrophy [98, 99] and confirmed by studies of Cav-3 overexpression [97, 100]. Collectively, these observations suggest that the switch from Cav-1 to Cav-3 expression represents a pivotal step for the overall myogenic program, leading the muscle precursors from quiescence towards differentiation.
Histopathological, genetical and molecular signatures of RMS
Soft tissue sarcomas arise from primitive mesenchymal cells located throughout the body and make up to approximately 7% of all cancer cases in patients under the age of 20 [101, 102]. These tumours can be subdivided into two major groups: RMS and non-RMS soft tissue sarcomas, the latter including a miscellaneous of tumours, such as the synovial sarcoma, malignant fibrous histiocytoma, malignant peripheral nerve sheath tumour and fibrosarcoma [101]. The immunohistochemical or molecular detection of myogenic regulatory factors, such as MyoD and myogenin [103–106], allows an RMS diagnosis, whereas the detection of myosin and other contractile proteins identifies more mature RMS phenotypes [107]. The current classification of RMS into two major histological variants, termed embryonal (ERMS) and alveolar (ARMS), is supported by histopathological criteria and genetic signatures (Fig. 1).
Fig 1.

Molecular alterations, animal models and human syndromes associated to RMS. MIM ID numbers linked to each syndrome can be used to retrieve further informations at the following site: http://www.ncbi.nlm.nih.gov/omim.
Embryonal RMS
ERMS accounts for up to 80% of RMS in children of less than 10 years of age and is the most common and more treatable subtype. ERMS can occur at any site, including nasopharynx and biliary tract, but they are most commonly observed in the head and neck or genitourinary region [108]. On histologic examination, ERMS are highly heterogeneous, ranging from poorly differentiated lesions with immature tumour cells to highly differentiated lesions containing rhabdomyoblasts with large eosinophilic cytoplasm. ERMS also comprise different histological subtypes formed by botryoid and spindle cells. A severe genomic instability generally characterizes ERMS subsets (Fig. 1): loss of heterozygosis (LOH) and the loss of imprinting (LOI) on chromosome region 11p15.5 are the most frequent signatures that retrieve the inactive allele and cause the loss of the active one [109–113]. This genetical signature impairs the expression of different putative tumour suppressor genes on chromosome 11, including H19 [114], CDKN1C (p57/KIP2) [115] and SLC22AIL (BWR1A) [116]; in contrast, the gene encoding for IGF-2, imprinted in the opposite direction, is overexpressed [110, 117]. In accordance, the tumourigenesis of the RMS RD cell line is suppressed by transferring a normal human chromosome 11 [118]. Interestingly, inherited alterations of the 11p15.5 locus are retrieved in patients affected by the Beckwith–Wiedemann syndrome, an overgrowth syndrome associated to an increased risk of developing Wilms tumour, hepatoblastoma, adrenocortical carcinoma, neuroblastoma and also RMS [119]. LOH is also frequently observed on chromosome 9q22 (Fig. 1), causing deficiency in Patched (PTCH) gene [120] and predisposing to high incidence of medulloblastoma and ERMS [121]. In addition, even activating mutations in RAS gene, which is intriguingly localized within the 11p15.5 locus, are associated to ERMS [122–124].
Alveolar RMS
ARMS mainly affect adolescents and adults and are characterized by a poorer prognosis. ARMS cells resemble lung alveoli, with clusters of eosinophilic tumour cells arranged loosely and disposed in an alveolar pattern. ARMS typically occur in the trunk and body extremities [108] and frequently harbour non-random chromosomal translocations [125] (Fig. 1). In particular, translocations t(2;13)(q35;q14) and t(1;13)(p36;q14) account respectively for about 70% and 10% of ARMS and give rise to chimeric proteins that are formed by the fusion of the paired and homeo-DNA binding domain of PAX3 or PAX7 factors with the transactivation domain of FKHR (FOXO1) [126–128]. The so-called PAX3-FKHR and PAX7-FKHR transcription factors enable an aberrant transcriptional program and significantly contribute to RMS progression through multiple mechanisms. In particular, PAX3-FKHR and PAX7-FKHR are frequently overexpressed in ARMS [129], display incremented accessibility to chromatin due to localization exclusively nuclear [130–132] and reach a 10- to 100-fold increase in the transcriptional activation of downstream target genes in comparison to wild-type PAX3 and PAX7 proteins [133, 134]. Ectopic expression of PAX3-FKHR triggers a transformed phenotype in chicken embryo [134] and NIH3T3 fibroblasts [135] and increases the tumourigenicity of two ERMS cell lines [136]. Moreover, PAX3-FKHR prevents apoptosis [137, 138] and even abrogates myoblast terminal differentiation [139, 140]. Transgenic mice carrying PAX3-FKHR develop defects in hindlimb skeletal muscle formation and neural crest migration [140], but do not undergo tumour formation, supporting the idea that PAX3-FKHR expression is required but is per se not sufficient to cause oncogenic transformation.
Principal pathways and targets deregulated in RMS
The network of pathways deregulated in RMS is rather complex (Fig. 1). The loss of p53 seems to be central for RMS development [141–145], in addition to the deregulation of components for receptorial signalling pathways, including HGF [146, 147], IGF1-R [148–150], EGFR/HER-1, HER-2 and HER-3 [151, 152], PDGFR [150, 153], VEGF [154–156], FGFR4 [157] and IL-4R [158, 159]. The overactivation of the RAS pathway occurs rather frequently [122–124]. In addition to the peculiar PAX3/7-FKHR expression [126–128], there are other molecular signatures that are commonly retrieved in RMS, such as the overexpression of cMET/HGF receptor [160], IGF-2 [110, 117], IGF-2–binding protein (IGFBP5) [161], N- and C-MYC [162–164], MDM2 [144, 165] and some mutations in the cyclin-dependent kinases (CDKs) genes [166]. Moreover, as previously mentioned (section ‘Embryonal RMS’), loss of expression of H19 [114], CDKN1C [115], SLC22AIL [116] and PTCH [120, 121] may occur. It is worth remembering that RMS cells are committed to myogenic lineage and therefore express muscle markers. In this regard, a paradoxical feature of RMS cells is that the expression of MyoD does not overlap with its full functionality because of multiple altered mechanisms [167], such as the inactivation of the mitogen-activated protein kinase (MAPK) p38 [168], whose function is required to enable MyoD transcriptional activity [169], or the presence of E-proteins complexes that compete for the generation of active full-length E-protein/MyoD heterodimers [170]. Moreover, PAX3-FKHR factor has been shown to increment the transcriptional levels of MyoD, but then abrogates its activity through protein phosphorylation [171, 172].
Animal models and human syndromes associated to RMS
A growing body of evidence derived from different animal models suggests that the development of RMS frequently occurs upon suppression of p53 pathway in conjunction with deregulated activities of receptorial systems along the RAS axis and/or the expression of PAX3/7-FKHR gene products (Fig. 1). In particular, RMS development occurs in p53-mutant mice [173, 174] and tumour incidence increases in conjunction with loss of FOS [175], activation of HER-2/neu [176], RAS [177] or the expression of PAX3/7-FKHR proteins [178]. In addition, mice with ablated PTCH gene [179] or with aberrant HGF signalling [146, 147] and non-transgenic dystrophin-deficient mdx mice [180] are prone to develop RMS. In zebrafish, transgenic expression of oncogenic K-RAS in activated satellite cells predisposes to RMS and simultaneous p53 inactivation accelerates tumour formation [181]. Finally, important clues on the risk factors predisposing to RMS came also from the evidence of a significant RMS incidence in certain familial cancer syndromes [145], including the Li–Fraumeni syndrome [141], Beckwith–Wiedemann syndrome [119], neurofibromatosis-1 [182], Costello syndrome [183], Gorlin syndrome [184], retinoblastoma [185], mosaic variegated aneuploidy syndrome [186] and mismatch repair deficiency syndrome [187] (Fig. 1).
Origins of RMS
Despite great efforts have been made to understand the molecular signatures of RMS, the identity of the tumour-initiating cell remains to be clarified. Satellite cells and multipotent mesenchymal stem cells (MSCs) are thought to be the most credited source for ERMS and ARMS, respectively [188–190]. In particular, inactivation of p53 cooperates with oncogenic RAS in muscle precursors and satellite cells to induce ERMS in mice [191] and zebrafish models [181]. On the other side, despite the delivery of PAX3-FKHR or PAX7-FKHR in MSCs derived from mouse bone marrow is effective to induce expression of MyoD and myogenin, ARMS tumours form only in conjunction with the expression of a dominant-negative p53, SV40 early region or a constitutively activated H-RAS [192, 193]. These findings strongly support the notion that PAX3/7-FKHR factors may confer the myogenic identity to MSCs elements [188], but then secondary deregulated mechanisms are required to induce tumour formation.
Expression of caveolins in RMS tumours and cell lines
Validation of caveolins expression in RMS has drawn the attention only recently. In a large screening of different tumours, including RMS, malignant mullerian tumour and a spectrum of different neoplasms, Cav-3 was indicated as specific marker for the detection of mature RMS, being particularly expressed in those cell elements with abundant eosinophilic cytoplasm and striation [22]. In a recent study, we have confirmed this evidence by means of immunohistochemical analyses [23]. In particular, Cav-3 and Cav-1 were predominantly associated to mature or immature RMS tumours, respectively (Fig. 2). Given the heterogeneity in degree of maturation present in the RMS cell components, the expression of Cav-1 or Cav-3 cannot be univocally associated to a certain RMS histotype. Nevertheless, a simplified model would indicate such a relationship between the expression of Cav-1 or Cav-3 and a status of poor or advanced cell differentiation, respectively, as analogously observed in skeletal muscle (Fig. 2) [10]. To further substantiate the in vivo findings, the expression of caveolins has been analysed in vitro [23]. In particular, Cav-1 expression was retrieved in the majority of the human ERMS cell lines analysed (Fig. 3A), except for TE671 cells (personal unpublished results), which intriguingly displayed low expression of MyoD. This evidence should deserve attention as might suggest an unappreciated relationship between MyoD levels and the expression of Cav-1 in immature muscle precursors, as much as a relationship between myogenin and the levels of Cav-3 has been already shown in differentiating myoblasts [194, 195]. The analysis of Cav-1 behaviour was then particularly characterized by employing RD cells, an RMS model line in which RAS mutations are long known to counteract the myogenic differentiation via hyperactivation of the ERK pathway [196]. In RD cells, Cav-1 was found to be properly localized at the plasma membrane (Fig. 3B), thus excluding the presence of inactivating gene mutations leading to protein mislocalization, as occurs in some cancer types [62, 85, 86]. In particular, high levels of Cav-1 were associated to proliferation of RD cells, whereas pharmacological inhibition of the ERK pathway, eliciting block of cell growth and subsequent differentiation, lead to Cav-1 down-regulation and increase of Cav-3, myogenin and MHC (Fig. 3C). Although it remains to be established if the decrease in Cav-1 might be related to growth arrest alone or to both withdrawal from cell cycle and subsequent myogenic differentiation, Cav-1 and Cav-3 seem to be associated to an immature and mature RMS cell phenotype, respectively, thereby confirming the previous in vivo observations. Keeping in mind that activating RAS mutations are frequently detectable in ERMS cells lines [122–124] and are critically involved in RMS tumour formation [177, 181, 183, 190–193], the relationship between Cav-1 expression and the RAS/ERK pathway deserves current attention. In fact, although Cav-1 is a marker of quiescence in muscle satellite cells [10], it configures as a marker of proliferation in RMS cells, suggesting that deregulated mechanisms in RMS cells might impair the ability of Cav-1 to overcome the RAS/ERK pathway. In this perspective, it is crucial to assess how targeted silencing of Cav-1 might influence RMS cell growth. Importantly, expression of Cav-2 has been also observed in RMS (personal unpublished data). Cav-2, indeed, is almost always co-expressed with Cav-1 and its phosphorylation can regulate the formation of caveolae and mitosis [197, 198]. Thus, the presumed impaired ability of Cav-1 to control cell proliferation in RMS cells needs to be ascertained also in relation to Cav-2 functionality.
Fig 2.
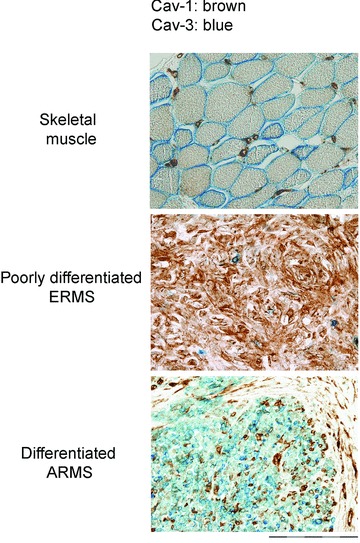
Expression of caveolins in RMS tumours. Double immunostain showing that in skeletal muscle Cav-1 and Cav-3 mark satellite cells and the plasmalemma of myofibres, respectively. In RMS, Cav-1 and Cav-3 are predominantly associated to immature and mature tumours, respectively. Bars = 50 μm.
Fig 3.
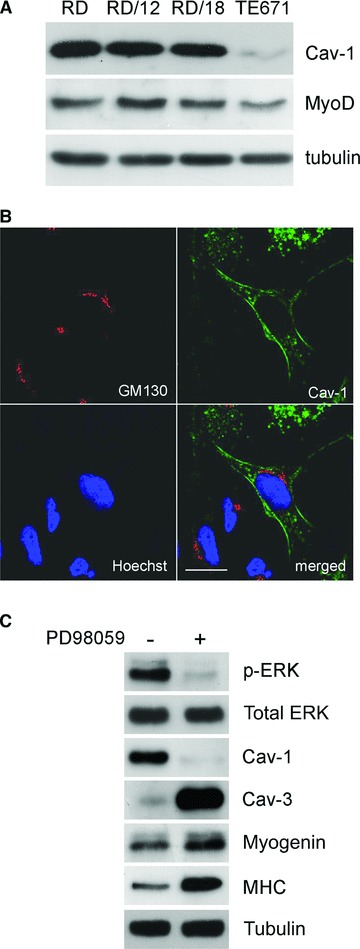
Expression of caveolins in RMS cell lines. (A) Western blot analyses showing the expression of Cav-1 in several MyoD-positive ERMS cell lines. Tubulin was used as loading control. (B) As shown by confocal microscopy analysis, Cav-1 localizes at the plasma membrane or in intracellular vesicles of embryonal RD cells. GM130 marker was employed to stain the Golgi apparatus. Bars = 100 μm. (C) Ten microliters of PD98059 administration attenuates the ERK phosphorylation in RD cells and allows the transition from proliferation to differentiation, leading to Cav-1 down-regulation and increase of myogenin, MHC and Cav-3. Tubulin was used as loading control.
Future perspectives: assessing the role of caveolins in RMS tumour progression
At the plasma membrane caveolins control the activity of several proteins involved in pathways transduction, thereby representing a checkpoint of cellular signalling. As such, in the following paragraphs the potential relationship between caveolins and different pathways which are central to RMS development will be discussed (Table 2).
Table 2.
The table summarizes the physiological role exerted by Cav-1 on different pathways which are involved in RMS
| Pathway | Physiological role of Cav-1 | Alterations linked to RMS |
|---|---|---|
| p53 | Cav-1 may positively regulate p53 tumour suppressor function by sequestering MDM2 [202]. | Germline p53 mutations predispose to different tumours (Li–Fraumeni syndrome), including RMS [141]. |
| Overexpression of MDM2, a p53 binding protein, is associated to ARMS tumours and cell lines [144, 165]. | ||
| IGF1-R | Cav-1 is a positive regulator of IGF-IR signalling pathway [204–207]. | Alterations in IGF1-R signalling are involved in RMS [148–150]. |
| EGFR | Cav-1 inhibits the autophosphorylation of the EGF-R kinase in vitro [208]. | HER1/EGFR is mainly expressed in ERMS, HER-2/EGFR in ARMS [151, 152]. |
| PDGFR | Cav-1 inhibits the autophosphorylation of PDGF receptors in a dose-dependent manner [209]. | Both ERMS and ARMS overexpress PDGFR-A and its ligands PDGF-C and PDGF-A [150, 153]. |
| VEGFR | Cav-1 acts as a negative regulator of VEGFR-2 activity in endothelial caveolae [210]. | Autocrine VEGF secretion stimulates RMS cell growth [154–156]. |
| RAS/ERK | Cav-1 limits the RAS/ERK pathway activation in several cell types [53, 59, 211], including muscle satellite cells [10]. | Activating RAS mutations are detected in ERMS cell lines [122–124, 196] and favour ERMS tumours [177, 181, 183, 190–193]. |
| TGF-β/myostatin | Cav-1 interacts with TGF-β type I receptors (ALK receptors) to limit the activation of TGF-β pathways [213]. | TGF-β superfamily members, such as TGF-β and myostatin, may impair RMS differentiation [214–218]. |
| HGF/cMET | Cav-1 is a downstream target of the HGF/cMET signalling axis in muscle satellite cells [10]. | Transgenic mice overexpressing HGF develop cancer, including RMS [146, 147]. |
| cMET is frequently overexpressed in RMS [160]. | ||
| RAGE | Cav-1 is a downstream target of RAGE-mediated Src activation in Schwann cells [227] and endothelial smooth muscle cells [228]. | Low RAGE expression in myoblasts and RMS cells correlates with increased proliferation, migration, invasiveness and tumour growth [229, 230]. |
Cav-1 and p53 signalling
Inactivation of p53 pathway, as occurs for spontaneous p53 germline mutations [141] or overexpression of MDM2 [144, 165], plays a critical role for RMS development. As effect of p53 loss of function, cancerous cells elude senescence and divide indefinitely. In this context, cellular senescence may represent a tumour-suppressor mechanism. In fibroblasts subjected to oxidative stress, Cav-1 has emerged as promoter of cell senescence [199–201] in virtue of its ability to sequester MDM2 [202], a negative regulator of p53 activity, or inhibit the activity of anti-oxidant enzymes, such as thioredoxin reductase 1 [203]. These findings suggest an investigation on the potential role of Cav-1 in controlling the p53 pathway in RMS cells.
Cav-1 and multiple control of receptorial systems for growth factors
Different receptorial systems for growth factors have been shown to signal aberrantly in RMS cells, including those for IGF-1R [148–150], EGFR [151, 152], PDGFR [150, 153] and VEGFR [154–156].
Remarkably, Cav-1 exerts a multiple physiological control on several components of these pathways through multiple mechanisms, such as physical interaction, regulation of receptor phosphorylation and internalization. In particular, Cav-1 configures as a positive regulator of IGF-1R pathways [204–207], although it is a negative regulator of EGFR [208], PDGFR [209] and VEGFR-2 activities [210]. Therefore, the ability of these receptorial systems to generate downstream signalling may be influenced, at least in part, by deregulated expression and/or localization of Cav-1, leading to a significant change in the biological outcome promoted by each pathway.
Cav-1 and RAS/ERK signalling
Overactivation of RAS/ERK signalling is one of the factors predisposing to RMS [122–124, 177, 181, 106]. Historically, Cav-1 configures as a strong inhibitor of this pathway in several cell types and tissues [10, 53, 59, 211]. Actually, the elevated Cav-1 expression detected in human RMS embryonal RD cells has been correlated to hyperactivation of the ERK pathway (Fig. 3C) [23]. These findings indicate that, at least in RD cells, Cav-1 seems to be aberrantly converted to a downstream target of the ERK pathway, giving rise to an ambiguous situation, in which cell proliferation is associated to persistent Cav-1 expression, suggesting that Cav-1 has lost the ability to antagonize the RAS/ERK signalling. The main hypotheses explaining this behaviour could be referred to an impaired activity of additional proteins which might cooperate with Cav-1 in the inhibition of the RAS/ERK pathway; alternatively, the antagonistic role of Cav-1 on the ERK pathway might occur upstream RAS, thus rendering ineffective its inhibition in the presence of activating RAS mutations. In this regard, it is known that hyperactivation of the ERK pathway cooperates to shift the function of some proteins, as occurs for Sprouty-1, which changes its function from antagonist to agonist of the RAS/ERK pathway in ERMS tumours harbouring activating RAS mutations [212]. Thus, it is important to establish how the association between high Cav-1 expression and elevated RAS/ERK pathway might influence RMS cell behaviour.
Caveolins and TGFβ/myostatin signalling
Caveolins are long known to limit the TGF-β pathway [213], which is supposed to play a not less important role in RMS [214–218].
Transforming growth factor-β (TGF-β) signalling pathways regulate numerous physiological and pathological processes [219–221] and proceed from the cell membrane to the nucleus through the cooperation of the types I and II serine/threonine kinase receptors (TβR-I and -II) and their downstream SMAD effectors [222]. Different TGF-β ligands, such as TGF-β[214] and myostatin [223], have been implicated in poor differentiation of RD cells [214–218]. Remarkably, either Cav-1 or Cav-3 interacts with and inhibits the activity of TβR-I receptors at the membrane [97, 213, 224], thereby limiting the downstream signalling. In view of these findings, the expression of caveolins may influence the cell behaviour of RMS cells in response to TGF-β/myostatin ligands.
Cav-1 and HGF/cMET signalling
Elevated HGF/cMET signalling has been implicated in RMS [146, 147, 160] as well as in different tumours [225]. In particular, cMET overexpression plays a significant role in RMS maintenance, as its silencing reduces cell invasiveness and tumour growth in a model of RMS xenograft [160]. In muscle satellite cells, HGF/cMET signalling axis plays a major role to elicit the transcriptional suppression of Cav-1 during muscle regeneration, a mechanism which is supposed to be central for the repair of the injured site [10]. Hence, these findings suggest that the aberrant HGF/cMET signalling observed in RMS cells might possibly reflect on the expression levels of Cav-1.
Caveolins and RAGE signalling
Receptor for advanced glycation end-products (RAGE) is a multi-ligand receptor of the immunoglobulin superfamily with a positive or negative role in cancer progression and metastasis depending on the tumour type [226]. A role of RAGE in regulating Cav-1 and Cav-3 expression/phosphorylation has been reported in several cell types. Indeed, (i) RAGE-mediated Src activation induces Cav-1 phosphorylation in Schwann cells [227] and endothelial smooth muscle cells [228]; and (ii) RAGE engagement induces Cav-3 expression in a myoblast cell line, whereas functional inactivation of RAGE in normal myoblasts results in the acquisition of a tumour behaviour with concomitant reduced expression of Cav-3 [229]. In RMS cells, RAGE activity is predictive of reduced proliferation, invasiveness and increased differentiation [230]. These findings suggest that RAGE signalling might have a role in regulating caveolins expression in RMS cells.
Conclusions
This review outlines the great amount of data indicating that caveolins play an important role in tumour development and progression. In RMS, the most frequent childhood soft-tissue sarcomas sharing myogenic features, Cav-1 and Cav-3 configure as markers of immature or mature tumours, respectively. Hence, immunohistochemical detection of caveolins in conjunction with strengthened myogenic markers may provide a useful diagnostic tool for establishing more accurately the grading in tumour samples. In this regard, the availability of novel molecular signatures may be critical to discriminate particular RMS variants and implement the current classification. Two major criteria support the demand to establish the role of caveolins in RMS tumour progression. First, caveolins are scaffolding proteins regulating several targets and pathways which are central to RMS development, and therefore loss or gain of caveolins function may generate multiple effects on the tumour behaviour. Finally, Cav-1 represents a tumour conditional gene in cancer and, thereby, recognizing its precise role in RMS progression will be crucial to elaborate targeted therapies. In summary, this review open up new interesting perspectives for investigating the role of caveolins in a large variety of pathological mechanisms predisposing to RMS.
Acknowledgments
The authors thank Raffaella Vescovi, Fiorella Faggi, Francesca Longhena and Maria Luisa Giudici for providing a critical revision of the paper. This work was supported by Associazione Amici per il Cuore-ONLUS, Chiari (Brescia), Italy, to A.F. and E.M., by Fondazione Cariplo grant to E.M. and P.L.P. and by University of Brescia research fund (ex 60%) to A.F.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002;54:431–67. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- 2.Williams TM, Lisanti MP. The caveolin proteins. Genome Biol. 2004;5:214. doi: 10.1186/gb-2004-5-3-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kurzchalia TV, Parton RG. Membrane microdomains and caveolae. Curr Opin Cell Biol. 1999;11:424–31. doi: 10.1016/s0955-0674(99)80061-1. [DOI] [PubMed] [Google Scholar]
- 4.Parton RG. Caveolae—from ultrastructure to molecular mechanisms. Nat Rev Mol Cell Biol. 2003;4:162–7. doi: 10.1038/nrm1017. [DOI] [PubMed] [Google Scholar]
- 5.Van Deurs B, Roepstorff K, et al. Caveolae: anchored, multifunctional platforms in the lipid ocean. Trends Cell Biol. 2003;13:92–100. doi: 10.1016/s0962-8924(02)00039-9. [DOI] [PubMed] [Google Scholar]
- 6.Couet J, Li S, Okamoto T, et al. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J Biol Chem. 1997;272:6525–33. doi: 10.1074/jbc.272.10.6525. [DOI] [PubMed] [Google Scholar]
- 7.Patel HH, Murray F, Insel PA. Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu Rev Pharmacol Toxicol. 2008;48:359–91. doi: 10.1146/annurev.pharmtox.48.121506.124841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;9:493–5. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Grand F, Rudnicki MA. Skeletal muscle satellite cells and adult myogenesis. Curr Opin Cell Biol. 2007;19:628–33. doi: 10.1016/j.ceb.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Volonte D, Liu Y, Galbiati F. The modulation of caveolin-1 expression controls satellite cell activation during muscle repair. FASEB J. 2005;19:237–9. doi: 10.1096/fj.04-2215fje. [DOI] [PubMed] [Google Scholar]
- 11.Way M, Parton RG. M-caveolin, a muscle-specific caveolin-related protein. FEBS Lett. 1996;378:108–12. doi: 10.1016/0014-5793(96)82884-5. [DOI] [PubMed] [Google Scholar]
- 12.Tang Z, Scherer PE, Okamoto T, et al. Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J Biol Chem. 1996;271:2255–61. doi: 10.1074/jbc.271.4.2255. [DOI] [PubMed] [Google Scholar]
- 13.Fanzani A, Musarò A, Stoppani E, et al. Hypertrophy and atrophy inversely regulate Caveolin-3 expression in myoblasts. Biochem Biophys Res Commun. 2007;357:314–8. doi: 10.1016/j.bbrc.2007.03.148. [DOI] [PubMed] [Google Scholar]
- 14.Hnasko R, Lisanti MP. The biology of caveolae: lessons from caveolin knockout mice and implications for human disease. Mol. Interv. 2003;3:445–64. doi: 10.1124/mi.3.8.445. [DOI] [PubMed] [Google Scholar]
- 15.Cohen AW, Hnasko R, Schubert W, et al. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84:1341–79. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- 16.Woodman SE, Sotgia F, Galbiati F, et al. Caveolinopathies: mutations in caveolin-3 cause four distinct autosomal dominant muscle diseases. Neurology. 2004;62:538–43. doi: 10.1212/wnl.62.4.538. [DOI] [PubMed] [Google Scholar]
- 17.Gazzerro E, Sotgia F, Bruno C, et al. Caveolinopathies: from the biology of caveolin-3 to human diseases. Eur J Hum Genet. 2010;18:137–45. doi: 10.1038/ejhg.2009.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barr FG. The role of chimeric paired box transcription factors in the pathogenesis of pediatric rhabdomysarcoma. Cancer Res. 1999;59:1711s–5s. [PubMed] [Google Scholar]
- 19.Merlino G, Helman LJ. Rhabdomyosarcoma-working out the pathways. Oncogene. 1999;18:5340–8. doi: 10.1038/sj.onc.1203038. [DOI] [PubMed] [Google Scholar]
- 20.De Giovanni C, Landuzzi L, Nicoletti G, et al. Molecular and cellular biology of rhabdomyosarcoma. Future Oncol. 2009;5:1449–75. doi: 10.2217/fon.09.97. [DOI] [PubMed] [Google Scholar]
- 21.Tonin PN, Scrable H, Shimada H, et al. Muscle-specific gene expression in rhabdomyosarcomas and stages of human fetal skeletal muscle development. Cancer Res. 1991;51:5100–6. [PubMed] [Google Scholar]
- 22.Fine SW, Lisanti MP, Argani P, et al. Caveolin-3 is a sensitive and specific marker for rhabdomyosarcoma. Appl Immunohistochem Mol Morphol. 2005;13:231–6. doi: 10.1097/00129039-200509000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Rossi S, Poliani PL, Cominelli M, et al. Caveolin 1 is a marker of poor differentiation in Rhabdomyosarcoma. Eur J Cancer. 2011;47:561–72. doi: 10.1016/j.ejca.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 24.Scherer PE, Lewis RY, Volonte D, et al. Cell-type and tissue-specific expression of caveolin-2. Caveolins 1 and 2 co-localize and form a stable hetero-oligomeric complex in vivo. J Biol Chem. 1997;272:29337–46. doi: 10.1074/jbc.272.46.29337. [DOI] [PubMed] [Google Scholar]
- 25.Fra AM, Williamson E, Simons K, et al. De novo formation of caveolae in lymphocytes by expression of VIP21-caveolin. Proc Natl Acad Sci USA. 1995;92:8655–9. doi: 10.1073/pnas.92.19.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drab M, Verkade P, Elger M, et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–52. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- 27.Galbiati F, Engelman JA, Volonte D, et al. Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J Biol Chem. 2001;276:21425–33. doi: 10.1074/jbc.M100828200. [DOI] [PubMed] [Google Scholar]
- 28.Hagiwara Y, Sasaoka T, Araishi K, et al. Caveolin-3 deficiency causes muscle degeneration in mice. Hum Mol Genet. 2000;9:3047–54. doi: 10.1093/hmg/9.20.3047. [DOI] [PubMed] [Google Scholar]
- 29.Hill MM, Bastiani M, Luetterforst R, et al. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell. 2008;132:113–24. doi: 10.1016/j.cell.2007.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu L, Pilch PF. A critical role of cavin (polymerase I and transcript release factor) in caveolae formation and organization. J Biol Chem. 2008;283:4314–22. doi: 10.1074/jbc.M707890200. [DOI] [PubMed] [Google Scholar]
- 31.Pelkmans L, Bürli T, Zerial M, et al. Caveolin-stabilized membrane domains as multifunctional transport and sorting devices in endocytic membrane traffic. Cell. 2004;118:767–80. doi: 10.1016/j.cell.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 32.Kurzchalia TV, Dupree P, Parton RG, et al. VIP21, a 21-kDa membrane protein is an integral component of trans-Golgi-network-derived transport vesicles. J Cell Biol. 1992;118:1003–14. doi: 10.1083/jcb.118.5.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glenney JR, Jr, Soppet D. Sequence and expression of caveolin, a protein component of caveolae plasma membrane domains phosphorylated on tyrosine in Rous sarcoma virus-transformed fibroblasts. Proc Natl Acad Sci USA. 1992;89:10517–21. doi: 10.1073/pnas.89.21.10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schlegel A, Lisanti MP. A molecular dissection of caveolin-1 membrane attachment and oligomerization. Two separate regions of the caveolin-1 carboxy-terminal domain mediate membrane binding and oligomer/oligomer interactions in vivo. J Biol Chem. 2000;275:21605–17. doi: 10.1074/jbc.M002558200. [DOI] [PubMed] [Google Scholar]
- 35.Liu P, Rudick M, Anderson RG. Multiple functions of caveolin-1. J Biol Chem. 2002;277:41295–8. doi: 10.1074/jbc.R200020200. [DOI] [PubMed] [Google Scholar]
- 36.Li WP, Liu P, Pilcher BK, et al. Cell-specific targeting of caveolin-1 to caveolae, secretory vesicles, cytoplasm or mitochondria. J Cell Sci. 2001;114:1397–408. doi: 10.1242/jcs.114.7.1397. [DOI] [PubMed] [Google Scholar]
- 37.Okamoto T, Schlegel A, Scherer PE, et al. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J Biol Chem. 1998;273:5419–22. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- 38.Murata M, Peränen J, Schreiner R, et al. VIP21/caveolin is a cholesterol-binding protein. Proc Natl Acad Sci USA. 1995;92:10339–43. doi: 10.1073/pnas.92.22.10339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mercier I, Jasmin JF, Pavlides S, et al. Clinical and translational implications of the caveolin gene family: lessons from mouse models and human genetic disorders. Lab Invest. 2009;89:614–23. doi: 10.1038/labinvest.2009.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Razani B, Combs TP, Wang XB, et al. Caveolin-1-deficient mice are lean, resistant to diet-induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. J Biol Chem. 2002;277:8635–47. doi: 10.1074/jbc.M110970200. [DOI] [PubMed] [Google Scholar]
- 41.Cohen AW, Razani B, Wang XB, et al. Caveolin-1-deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue. Am J Physiol Cell Physiol. 2003;285:222–35. doi: 10.1152/ajpcell.00006.2003. [DOI] [PubMed] [Google Scholar]
- 42.Zhao YY, Liu Y, Stan RV, et al. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci USA. 2002;99:11375–80. doi: 10.1073/pnas.172360799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jasmin JF, Mercier I, Hnasko R, et al. Lung remodeling and pulmonary hypertension after myocardial infarction: pathogenic role of reduced caveolin expression. Cardiovasc Res. 2004;63:747–55. doi: 10.1016/j.cardiores.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 44.Cohen AW, Park DS, Woodman SE, et al. Caveolin-1 null mice develop cardiac hypertrophy with hyperactivation of p42/44 MAP kinase in cardiac fibroblasts. Am J Physiol Cell Physiol. 2003;284:457–74. doi: 10.1152/ajpcell.00380.2002. [DOI] [PubMed] [Google Scholar]
- 45.Sonveaux P, Martinive P, DeWever J, et al. Caveolin-1 expression is critical for vascular endothelial growth factor-induced ischemic hindlimb collateralization and nitric oxide-mediated angiogenesis. Circ Res. 2004;95:154–61. doi: 10.1161/01.RES.0000136344.27825.72. [DOI] [PubMed] [Google Scholar]
- 46.Jasmin JF, Malhotra S, Singh Dhallu M, et al. Caveolin-1 deficiency increases cerebral ischemic injury. Circ Res. 2007;100:721–9. doi: 10.1161/01.RES.0000260180.42709.29. [DOI] [PubMed] [Google Scholar]
- 47.Patel HH, Tsutsumi YM, Head BP, et al. Mechanisms of cardiac protection from ischemia/reperfusion injury: a role for caveolae and caveolin-1. FASEB J. 2007;21:1565–74. doi: 10.1096/fj.06-7719com. [DOI] [PubMed] [Google Scholar]
- 48.Razani B, Wang XB, Engelman JA, et al. Caveolin-2-deficient mice show evidence of severe pulmonary dysfunction without disruption of caveolae. Mol Cell Biol. 2002;22:2329–44. doi: 10.1128/MCB.22.7.2329-2344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schubert W, Sotgia F, Cohen AW, et al. Caveolin-1(−/−)- and caveolin-2(−/−)-deficient mice both display numerous skeletal muscle abnormalities, with tubular aggregate formation. Am J Pathol. 2007;170:316–33. doi: 10.2353/ajpath.2007.060687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Woodman SE, Park DS, Cohen AW, et al. Caveolin-3 knock-out mice develop a progressive cardiomyopathy and show hyperactivation of the p42/44 MAPK cascade. J Biol Chem. 2002;277:38988–97. doi: 10.1074/jbc.M205511200. [DOI] [PubMed] [Google Scholar]
- 51.Capozza F, Combs TP, Cohen AW, et al. Caveolin-3 knockout mice show increased adiposity and whole body insulin resistance, with ligand-induced insulin receptor instability in skeletal muscle. Am J Physiol Cell Physiol. 2005;288:1317–31. doi: 10.1152/ajpcell.00489.2004. [DOI] [PubMed] [Google Scholar]
- 52.Engelman JA, Zhang XL, Lisanti MP. Genes encoding human caveolin-1 and -2 are co-localized to the D7S522 locus (7q31.1), a known fragile site (FRA7G) that is frequently deleted in human cancers. FEBS Lett. 1998;436:403–10. doi: 10.1016/s0014-5793(98)01134-x. [DOI] [PubMed] [Google Scholar]
- 53.Galbiati F, Volonte D, Engelman JA, et al. Targeted downregulation of caveolin-1 is sufficient to drive cell transformation and hyperactivate the p42/44 MAP kinase cascade. EMBO J. 1998;17:6633–48. doi: 10.1093/emboj/17.22.6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Capozza F, Williams TM, Schubert W, et al. Absence of caveolin-1 sensitizes mouse skin to carcinogen-induced epidermal hyperplasia and tumour formation. Am J Pathol. 2003;162:2029–39. doi: 10.1016/S0002-9440(10)64335-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Razani B, Engelman JA, Wang XB, et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- 56.Galbiati F, Volonte D, Liu J, et al. Caveolin-1 expression negatively regulates cell cycle progression by inducing G(0)/G(1) arrest via a p53/p21(WAF1/Cip1)-dependent mechanism. Mol Biol Cell. 2001;12:2229–44. doi: 10.1091/mbc.12.8.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Engelman JA, Wycoff CC, Yasuhara S, et al. Recombinant expression of caveolin-1 in oncogenically transformed cells abrogates anchorage-independent growth. J Biol Chem. 1997;272:16374–81. doi: 10.1074/jbc.272.26.16374. [DOI] [PubMed] [Google Scholar]
- 58.Lee SW, Reimer CL, Oh P, et al. Tumour cell growth inhibition by caveolin re-expression in human breast cancer cells. Oncogene. 1998;16:1391–7. doi: 10.1038/sj.onc.1201661. [DOI] [PubMed] [Google Scholar]
- 59.Engelman JA, Chu C, Lin A, et al. Caveolin-mediated regulation of signaling along the p42/44 MAP kinase cascade in vivo. A role for the caveolin-scaffolding domain. FEBS Lett. 1998;428:205–11. doi: 10.1016/s0014-5793(98)00470-0. [DOI] [PubMed] [Google Scholar]
- 60.Wiechen K, Diatchenko L, Agoulnik A, et al. Caveolin-1 is down-regulated in human ovarian carcinoma and acts as a candidate tumour suppressor gene. Am J Pathol. 2001;159:1635–43. doi: 10.1016/S0002-9440(10)63010-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Racine C, Belanger M, Hirabayashi H, et al. Reduction of caveolin 1 gene expression in lung carcinoma cell lines. Biochem Biophys Res Commun. 1999;255:580–6. doi: 10.1006/bbrc.1999.0236. [DOI] [PubMed] [Google Scholar]
- 62.Bonuccelli G, Casimiro MC, Sotgia F, et al. Caveolin-1 (P132L), a common breast cancer mutation, confers mammary cell invasiveness and defines a novel stem cell/metastasis-associated gene signature. Am J Pathol. 2009;174:1650–62. doi: 10.2353/ajpath.2009.080648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Engelman JA, Lee RJ, Karnezis A, et al. Reciprocal regulation of neu tyrosine kinase activity and caveolin-1 protein expression in vitro and in vivo. Implications for mammary tumorigenesis. J Biol Chem. 1998;273:20448–55. doi: 10.1074/jbc.273.32.20448. [DOI] [PubMed] [Google Scholar]
- 64.Sloan EK, Stanley KL, Anderson RL. Caveolin-1 inhibits breast cancer growth and metastasis. Oncogene. 2004;23:7893–7. doi: 10.1038/sj.onc.1208062. [DOI] [PubMed] [Google Scholar]
- 65.Wiechen K, Sers C, Agoulnik A, et al. Down-regulation of caveolin-1, a candidate tumour suppressor gene, in sarcomas. Am J Pathol. 2001;158:833–9. doi: 10.1016/S0002-9440(10)64031-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koleske AJ, Baltimore D, Lisanti MP. Reduction of caveolin and caveolae in oncogenically transformed cells. Proc Natl Acad Sci USA. 1995;92:1381–5. doi: 10.1073/pnas.92.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Patlolla JM, Swamy MV, Raju J, et al. Overespression of caveolin-1 in experimental colon adenocarcinoma and human colon cancer cell lines. Oncol Rep. 2004;11:957–63. [PubMed] [Google Scholar]
- 68.Rajjayabun PH, Garg S, Durkan GC, et al. Caveolin-1 expression is associated with high-grade bladder cancer. Urology. 2001;58:811–4. doi: 10.1016/s0090-4295(01)01337-1. [DOI] [PubMed] [Google Scholar]
- 69.Ando T, Ishiguro H, Kimura M, et al. The overexpression of caveolin-1 and caveolin-2 correlates with poor prognosis and tumour progression in esophageal squamous cell carcinoma. Oncol Rep. 2007;18:601–9. [PubMed] [Google Scholar]
- 70.Yang G, Truong LD, Wheeler TM, et al. Caveolin-1 expression in clinically confined human prostate cancer: a novel prognostic marker. Cancer Res. 1999;59:5719–23. [PubMed] [Google Scholar]
- 71.Williams TM, Lisanti MP. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am J Physiol Cell Physiol. 2005;288:494–506. doi: 10.1152/ajpcell.00458.2004. [DOI] [PubMed] [Google Scholar]
- 72.Burgermeister E, Liscovitch M, Röcken C, et al. Caveats of caveolin-1 in cancer progression. Cancer Lett. 2008;268:187–201. doi: 10.1016/j.canlet.2008.03.055. [DOI] [PubMed] [Google Scholar]
- 73.Goetz JG, Lajoie P, Wiseman SM, et al. Caveolin-1 in tumour progression: the good, the bad and the ugly. Cancer Metastasis Rev. 2008;27:715–35. doi: 10.1007/s10555-008-9160-9. [DOI] [PubMed] [Google Scholar]
- 74.Quest AF, Gutierrez-Pajares JL, Torres VA. Caveolin-1: an ambiguous partner in cell signalling and cancer. J Cell Mol Med. 2008;12:1130–50. doi: 10.1111/j.1582-4934.2008.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee H, Park DS, Razani B, et al. Caveolin-1 mutations (P132L and null) and the pathogenesis of breast cancer: caveolin-1 (P132L) behaves in a dominant-negative manner and caveolin-1(−/−) null mice show mammary epithelial cell hyperplasia. Am J Pathol. 2002;161:1357–69. doi: 10.1016/S0002-9440(10)64412-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Park DS, Lee H, Frank PG, et al. Caveolin-1-deficient mice show accelerated mammary gland development during pregnancy, premature lactation and hyperactivation of the Jak-2/STAT5a signaling cascade. Mol Biol Cell. 2002;13:3416–30. doi: 10.1091/mbc.02-05-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Williams TM, Cheung MW, Park DS, et al. Loss of caveolin-1 gene expression accelerates the development of dysplastic mammary lesions in tumour-prone transgenic mice. Mol Biol Cell. 2003;14:1027–42. doi: 10.1091/mbc.E02-08-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Williams TM, Medina F, Badano I, et al. Caveolin-1 gene disruption promotes mammary tumorigenesis and dramatically enhances lung metastasis in vivo role of Cav-1 in cell invasiveness and matrix metalloproteinase (MMP-2/9) secretion. J Biol Chem. 2004;279:51630–46. doi: 10.1074/jbc.M409214200. [DOI] [PubMed] [Google Scholar]
- 79.Williams TM, Lee H, Cheung MW, et al. Combined loss of INK4a and caveolin-1 synergistically enhances cell proliferation and oncogene induced tumorigenesis: role of INK4a/CAV-1 in mammary epithelial cell hyperplasia. J Biol Chem. 2004;279:24745–56. doi: 10.1074/jbc.M402064200. [DOI] [PubMed] [Google Scholar]
- 80.Williams TM, Hassan GS, Li J, et al. Caveolin-1 promotes tumour progression in an autochthonous mouse model of prostate cancer: genetic ablation of Cav-1 delays advanced prostate tumour development in tramp mice. J Biol Chem. 2005;280:25134–45. doi: 10.1074/jbc.M501186200. [DOI] [PubMed] [Google Scholar]
- 81.Nestl A, Von Stein OD, Zatloukal K, et al. Gene expression patterns associated with the metastatic phenotype in rodent and human tumours. Cancer Res. 2001;61:1569–77. [PubMed] [Google Scholar]
- 82.Ravid D, Maor S, Werner H, et al. Caveolin-1 inhibits cell detachment-induced p53 activation and anoikis by upregulation of insulin-like growth factor-I receptors and signaling. Oncogene. 2005;24:1338–47. doi: 10.1038/sj.onc.1208337. [DOI] [PubMed] [Google Scholar]
- 83.Belanger MM, Roussel E, Couet J. Up-regulation of caveolin expression by cytotoxic agents in drug-sensitive cancer cells. Anticancer Drugs. 2003;14:281–7. doi: 10.1097/00001813-200304000-00005. [DOI] [PubMed] [Google Scholar]
- 84.Ravid D, Maor S, Werner H, et al. Caveolin-1 inhibits anoikis and promotes survival signaling in cancer cells. Advances in Enzyme Regulation. 2006;46:163–75. doi: 10.1016/j.advenzreg.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 85.Hayashi K, Matsuda S, Machida K, et al. Invasion activating caveolin-1 mutation in human scirrhous breast cancers. Cancer Res. 2001;61:2361–4. [PubMed] [Google Scholar]
- 86.Han SE, Park KH, Lee G, et al. Mutation and aberrant expression of Caveolin-1 in human oral squamous cell carcinomas and oral cancer cell lines. Int J Oncol. 2004;24:435–40. [PubMed] [Google Scholar]
- 87.Lee H, Volonte D, Galbiati F, et al. Constitutive and growth factor-regulated phosphorylation of caveolin-1 occurs at the same site (Tyr-14) in vivo: identification of a c-Src/Cav-1/Grb7 signaling cassette. Mol Endocrinol. 2000;14:1750–75. doi: 10.1210/mend.14.11.0553. [DOI] [PubMed] [Google Scholar]
- 88.Schlegel A, Arvan P, Lisanti MP. Caveolin-1 binding to endoplasmic reticulum membranes and entry into the regulated secretory pathway are regulated by serine phosphorylation. Protein sorting at the level of the endoplasmic reticulum. J Biol Chem. 2001;276:4398–408. doi: 10.1074/jbc.M005448200. [DOI] [PubMed] [Google Scholar]
- 89.Cooper RN, Tajbakhsh S, Mouly V, et al. In vivo satellite cell activation via Myf5 and MyoD in regenerating mouse skeletal muscle. J Cell Sci. 1999;112:2895–901. doi: 10.1242/jcs.112.17.2895. [DOI] [PubMed] [Google Scholar]
- 90.Winchester PK, Davis ME, Alway SE, et al. Satellite cell activation in the stretch-enlarged anterior latissimus dorsi muscle of the adult quail. Am J Physiol. 1991;260:206–12. doi: 10.1152/ajpcell.1991.260.2.C206. [DOI] [PubMed] [Google Scholar]
- 91.Snow MH. Satellite cell response in rat soleus muscle undergoing hypertrophy due to surgical ablation of synergists. Anat Rec. 1990;227:437–46. doi: 10.1002/ar.1092270407. [DOI] [PubMed] [Google Scholar]
- 92.Song KS, Scherer PE, Tang Z, et al. Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J Biol Chem. 1996;271:15160–5. doi: 10.1074/jbc.271.25.15160. [DOI] [PubMed] [Google Scholar]
- 93.Sotgia F, Lee JK, Das K, et al. Caveolin-3 directly interacts with the carboxy-terminal tail of beta-dystroglycan. Identification of a central WW-like domain within caveolin family members. J Biol Chem. 2000;275:38048–58. doi: 10.1074/jbc.M005321200. [DOI] [PubMed] [Google Scholar]
- 94.Minetti C, Sotgia F, Bruno C, et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet. 1998;18:365–8. doi: 10.1038/ng0498-365. [DOI] [PubMed] [Google Scholar]
- 95.Sunada Y, Ohi H, Hase A, et al. Transgenic mice expressing mutant caveolin-3 show severe myopathy associated with increased nNOS activity. Hum Mol Genet. 2001;10:173–8. doi: 10.1093/hmg/10.3.173. [DOI] [PubMed] [Google Scholar]
- 96.Nixon SJ, Wegner J, Ferguson C, et al. Zebrafish as a model for caveolin-associated muscle disease; caveolin-3 is required for myofibril organization and muscle cell patterning. Hum Mol Genet. 2005;14:1727–43. doi: 10.1093/hmg/ddi179. [DOI] [PubMed] [Google Scholar]
- 97.Stoppani E, Rossi S, Meacci E, et al. Point mutated caveolin-3 form (P104L) impairs myoblast differentiation via Akt and p38 signalling reduction, leading to an immature cell signature. Biochim Biophys Acta. 2011;1812:468–79. doi: 10.1016/j.bbadis.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 98.Vaghy PL, Fang J, Wu W, et al. Increased caveolin-3 levels in mdx mouse muscles. FEBS Lett. 1998;431:125–7. doi: 10.1016/s0014-5793(98)00738-8. [DOI] [PubMed] [Google Scholar]
- 99.Repetto S, Bado M, Broda P, et al. Increased number of caveolae and caveolin-3 overexpression in Duchenne muscular dystrophy. Biochem Biophys Res Commun. 1999;261:547–50. doi: 10.1006/bbrc.1999.1055. [DOI] [PubMed] [Google Scholar]
- 100.Galbiati F, Volonte D, Chu JB, et al. Transgenic overexpression of caveolin-3 in skeletal muscle fibers induces a Duchenne-like muscular dystrophy phenotype. Proc Natl Acad Sci USA. 2000;97:9689–94. doi: 10.1073/pnas.160249097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Meyer WH, Spunt SL. Soft tissue sarcomas of childhood. Cancer Treat Rev. 2004;30:269–80. doi: 10.1016/j.ctrv.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 102.Ognjanovic S, Linabery AM, Charbonneau B, et al. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975–2005. Cancer. 2009;115:4218–26. doi: 10.1002/cncr.24465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Berkes CA, Tapscott SJ. MyoD and the transcriptional control of myogenesis. Semin Cell Dev Biol. 2005;16:585–95. doi: 10.1016/j.semcdb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 104.Buckingham M, Bajard L, Chang T, et al. The formation of skeletal muscle: from somite to limb. J Anat. 2003;202:59–68. doi: 10.1046/j.1469-7580.2003.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Buckingham M, Montarras D. Skeletal muscle stem cells. Curr Opin Genet Dev. 2008;18:330–6. doi: 10.1016/j.gde.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 106.Bergstrom DA, Tapscott SJ. Molecular distinction between specification and differentiation in the myogenic basic helix-loop-helix transcription factor family. Mol Cell Biol. 2001;21:2404–12. doi: 10.1128/MCB.21.7.2404-2412.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Morotti RA, Nicol KK, Parham DM, et al. An immunohistochemical algorithm to facilitate diagnosis and subtyping of rhabdomyosarcoma: the Children’s Oncology Group experience. Am J Surg Pathol. 2006;30:962–8. doi: 10.1097/00000478-200608000-00005. [DOI] [PubMed] [Google Scholar]
- 108.Tsokos M, Webber BL, Parham DM, et al. Rhabdomyosarcoma. A new classification scheme related to prognosis. Arch Pathol Lab Med. 1992;116:847–55. [PubMed] [Google Scholar]
- 109.Koufos A, Hansen MF, Copeland NG, et al. Loss of heterozygosity in three embryonal tumours suggests a common pathogenetic mechanism. Nature. 1985;316:330–4. doi: 10.1038/316330a0. [DOI] [PubMed] [Google Scholar]
- 110.Scrable HJ, Cavenee W, Ghavimi F, et al. A model for embryonal rhabdomyosarcoma tumorigenesis that involves genome imprinting. Proc Natl Acad Sci USA. 1989;86:7480–4. doi: 10.1073/pnas.86.19.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Scrable HJ, Witte DP, Lampkin BC, et al. Chromosomal localization of the human rhabdomyosarcoma locus by mitotic recombination mapping. Nature. 1987;329:645–7. doi: 10.1038/329645a0. [DOI] [PubMed] [Google Scholar]
- 112.Loh WE, Jr, Scrable HJ, Livanos E, et al. Human chromosome 11 contains two different growth suppressor genes for embryonal rhabdomyosarcoma. Proc Natl Acad Sci USA. 1992;89:1755–9. doi: 10.1073/pnas.89.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Anderson J, Gordon A, McManus A, et al. Disruption of imprinted genes at chromosome region 11p15.5 in paediatric rhabdomyosarcoma. Neoplasia. 1999;1:340–8. doi: 10.1038/sj.neo.7900052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hao Y, Crenshaw T, Moulton T, et al. Tumour-suppressor activity of H19 RNA. Nature. 1993;365:764–7. doi: 10.1038/365764a0. [DOI] [PubMed] [Google Scholar]
- 115.Matsuoka S, Thompson JS, Edwards MC, et al. Imprinting of the gene encoding a human cyclin-dependent kinase inhibitor, p57KIP2, on chromosome 11p15. Proc Natl Acad Sci USA. 1996;93:3026–30. doi: 10.1073/pnas.93.7.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schwienbacher C, Sabbioni S, Campi M, et al. Transcriptional map of 170-kb region at chromosome 11p15.5: identification and mutational analysis of the BWR1A gene reveals the presence of mutations in tumour samples. Proc Natl Acad Sci USA. 1998;95:3873–8. doi: 10.1073/pnas.95.7.3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhan S, Shapiro DN, Helman LJ. Activation of an imprinted allele of the insulin-like growth factor II gene implicated in rhabdomyosarcoma. J Clin Invest. 1994;94:445–8. doi: 10.1172/JCI117344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Koi M, Johnson LA, Kalikin LM, et al. Tumour cell growth arrest caused by subchromosomal transferable DNA fragments from chromosome 11. Science. 1993;260:361–4. doi: 10.1126/science.8469989. [DOI] [PubMed] [Google Scholar]
- 119.Hoovers JM, Kalikin LM, Johnson LA, et al. Multiple genetic loci within 11p15 defined by Beckwith-Wiedemann syndrome rearrangement breakpoints and subchromosomal transferable fragments. Proc Natl Acad Sci USA. 1995;92:12456–60. doi: 10.1073/pnas.92.26.12456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bridge JA, Liu J, Weibolt V, et al. Novel genomic imbalances in embryonal rhabdomyosarcoma revealed by comparative genomic hybridization and fluorescence in situ hybridization: an intergroup rhabdomyosarcoma study. Genes Chromosomes Cancer. 2000;27:337–44. doi: 10.1002/(sici)1098-2264(200004)27:4<337::aid-gcc1>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 121.Hahn H, Wojnowski L, Specht K, et al. Patched target Igf2 is indispensable for the formation of medulloblastoma and rhabdomyosarcoma. J Biol Chem. 2000;275:28341–4. doi: 10.1074/jbc.C000352200. [DOI] [PubMed] [Google Scholar]
- 122.Stratton MR, Fisher C, Gusterson BA, et al. Detection of point mutations in N-ras and K-ras genes of human embryonal rhabdomyosarcomas using oligonucleotide probes and the polymerase chain reaction. Cancer Res. 1989;49:6324–7. [PubMed] [Google Scholar]
- 123.Wilke W, Maillet M, Robinson R. H-ras-1 point mutations in soft tissue sarcomas. Mod Pathol. 1993;6:129–32. [PubMed] [Google Scholar]
- 124.Yoo J, Robinson RA. H-ras and K-ras mutations in soft tissue sarcoma: comparative studies of sarcomas from Korean and American patients. Cancer. 1999;86:58–63. [PubMed] [Google Scholar]
- 125.Barr FG. Molecular genetics and pathogenesis of rhabdomyosarcoma. J Pediatr Hematol Oncol. 1997;19:483–91. doi: 10.1097/00043426-199711000-00001. [DOI] [PubMed] [Google Scholar]
- 126.Barr FG, Galili N, Holick J, et al. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993;3:113–7. doi: 10.1038/ng0293-113. [DOI] [PubMed] [Google Scholar]
- 127.Galili N, Davis RJ, Fredericks WJ, et al. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993;5:230–5. doi: 10.1038/ng1193-230. [DOI] [PubMed] [Google Scholar]
- 128.Davis RJ, D’Cruz CM, Lovell MA, et al. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. 1994;54:2869–72. [PubMed] [Google Scholar]
- 129.Davis RJ, Barr FG. Fusion genes resulting from alternative chromosomal translocations are overexpressed by gene-specific mechanisms in alveolar rhabdomyosarcoma. Proc Natl Acad Sci USA. 1997;94:8047–51. doi: 10.1073/pnas.94.15.8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Del Peso L, Gonzalez VM, Hernandez R, et al. Regulation of the forkhead transcription factor FKHR, but not the PAX3-FKHR fusion protein, by the serine/threonine kinase Akt. Oncogene. 1999;18:7328–33. doi: 10.1038/sj.onc.1203159. [DOI] [PubMed] [Google Scholar]
- 131.Fredericks WJ, Galili N, Mukhopadhyay S, et al. The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol Cell Biol. 1995;15:1522–35. doi: 10.1128/mcb.15.3.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bennicelli JL, Edwards RH, Barr FG. Mechanism for transcriptional gain of function resulting from chromosomal translocation in alveolar rhabdomyosarcoma. Proc Natl Acad Sci USA. 1996;93:5455–9. doi: 10.1073/pnas.93.11.5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bennicelli JL, Advani S, Schafer BW, et al. PAX3 and PAX7 exhibit conserved cis-acting transcription repression domains and utilize a common gain of function mechanism in alveolar rhabdomyosarcoma. Oncogene. 1999;18:4348–56. doi: 10.1038/sj.onc.1202812. [DOI] [PubMed] [Google Scholar]
- 134.Scheidler S, Fredericks WJ, Rauscher FJ, 3rd, et al. The hybrid PAX3-FKHR fusion protein of alveolar rhabdomyosarcoma transforms fibroblasts in culture. Proc Natl Acad Sci USA. 1996;93:9805–9. doi: 10.1073/pnas.93.18.9805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lam PY, Sublett JE, Hollenbach AD, et al. The oncogenic potential of the Pax3-FKHR fusion protein requires the Pax3 homeodomain recognition helix but not the Pax3 paired-box DNA binding domain. Mol Cell Biol. 1999;19:594–601. doi: 10.1128/mcb.19.1.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Anderson J, Ramsay A, Gould S, et al. Pax3-fkhr induces morphological change and enhances cellular proliferation and invasion in rhabdomyosarcoma. Am J Pathol. 2001;159:1089–96. doi: 10.1016/S0002-9440(10)61784-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bernasconi M, Remppis A, Fredericks WJ, et al. Induction of apoptosis in rhabdomyosarcoma cells through down-regulation of PAX proteins. Proc Natl Acad Sci USA. 1996;93:13164–9. doi: 10.1073/pnas.93.23.13164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Margue CM, Bernasconi M, Barr FG, et al. Transcriptional modulation of the anti-apoptotic protein BCL-XL by the paired box transcription factors PAX3 and PAX3/FKHR. Oncogene. 2000;19:2921–9. doi: 10.1038/sj.onc.1203607. [DOI] [PubMed] [Google Scholar]
- 139.Epstein JA, Lam P, Jepeal L, et al. Pax3 inhibits myogenic differentiation of cultured myoblast cells. J Biol Chem. 1995;270:11719–22. doi: 10.1074/jbc.270.20.11719. [DOI] [PubMed] [Google Scholar]
- 140.Anderson MJ, Shelton GD, Cavenee WK, et al. Embryonic expression of the tumour associated PAX3-FKHR fusion protein interferes with the developmental functions of Pax3. Proc Natl Acad Sci USA. 2001;98:1589–94. doi: 10.1073/pnas.98.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–8. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 142.Felix CA, Kappel CC, Mitsudomi T, et al. Frequency and diversity of p53 mutations in childhood rhabdomyosarcoma. Cancer Res. 1992;52:2243–7. [PubMed] [Google Scholar]
- 143.Diller L, Sexsmith E, Gottlieb A, et al. Germline p53 mutations are frequently detected in young children with rhabdomyosarcoma. J Clin Invest. 1995;95:1606–11. doi: 10.1172/JCI117834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Taylor AC, Shu L, Danks MK, et al. P53 mutation and MDM2 amplification frequency in pediatric rhabdomyosarcoma tumours and cell lines. Med Pediatr Oncol. 2000;35:96–103. doi: 10.1002/1096-911x(200008)35:2<96::aid-mpo2>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 145.Lindor NM, McMaster ML, Lindor CJ, et al. Concise handbook of familial cancer susceptibility syndromes-second edition. J Natl Cancer Inst Monogr. 2008;38:1–93. doi: 10.1093/jncimonographs/lgn001. [DOI] [PubMed] [Google Scholar]
- 146.Takayama H, LaRochelle WJ, Sharp R, et al. Diverse tumorigenesis associated with aberrant development in mice overexpressing hepatocyte growth factor/scatter factor. Proc Natl Acad Sci USA. 1997;94:701–6. doi: 10.1073/pnas.94.2.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sharp R, Recio JA, Jhappan C, et al. Synergism between INK4a/ARF inactivation and aberrant HGF/SF signaling in rhabdomyosarcomagenesis. Nat Med. 2002;8:1276–80. doi: 10.1038/nm787. [DOI] [PubMed] [Google Scholar]
- 148.Kim SY, Toretsky JA, Scher D, et al. The role of IGF-1R in pediatric malignancies. Oncologist. 2009;14:83–91. doi: 10.1634/theoncologist.2008-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ayalon D, Glaser T, Werner H. Transcriptional regulation of IGF-I receptor gene expression by the PAX3-FKHR oncoprotein. Growth Horm IGF Res. 2001;11:289–97. doi: 10.1054/ghir.2001.0244. [DOI] [PubMed] [Google Scholar]
- 150.Blandford MC, Barr FG, Lynch JC, et al. Rhabdomyosarcomas utilize developmental, myogenic growth factors for disease advantage: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2006;46:329–38. doi: 10.1002/pbc.20466. [DOI] [PubMed] [Google Scholar]
- 151.De Giovanni C, Landuzzi L, Frabetti F, et al. Antisense epidermal growth factor receptor transfection impairs the proliferative ability of human rhabdomyosarcoma cells. Cancer Res. 1996;56:3898–901. [PubMed] [Google Scholar]
- 152.Ganti R, Skapek SX, Zhang J, et al. Expression and genomic status of EGFR and ErbB-2 in alveolar and embryonal rhabdomyosarcoma. Mod Pathol. 2006;19:1213–20. doi: 10.1038/modpathol.3800636. [DOI] [PubMed] [Google Scholar]
- 153.Taniguchi E, Nishijo K, McCleish AT, et al. PDGFR-A is a therapeutic target in alveolar rhabdomyosarcoma. Oncogene. 2008;27:6550–60. doi: 10.1038/onc.2008.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Onisto M, Slongo ML, Gregnanin L, et al. Expression and activity of vascular endothelial growth factor and metalloproteinases in alveolar and embryonal rhabdomyosarcoma cell lines. Int J Oncol. 2005;27:791–8. [PubMed] [Google Scholar]
- 155.Gee MF, Tsuchida R, Eichler-Jonsson C, et al. Vascular endothelial growth factor acts in an autocrine manner in rhabdomyosarcoma cell lines and can be inhibited with all-trans-retinoic acid. Oncogene. 2005;24:8025–37. doi: 10.1038/sj.onc.1208939. [DOI] [PubMed] [Google Scholar]
- 156.Wang W, Slevin M, Kumar S, et al. The cooperative transforming effects of PAX3-FKHR and IGF-II on mouse myoblasts. Int J Oncol. 2005;27:1087–96. doi: 10.3892/ijo.27.4.1087. [DOI] [PubMed] [Google Scholar]
- 157.Taylor JG, 6th, Cheuk AT, Tsang PS, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest. 2009;119:3395–407. doi: 10.1172/JCI39703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ebauer M, Wachtel M, Niggli FK, et al. Comparative expression profiling identifies an in vivo target gene signature with TFAP2B as a mediator of the survival function of PAX3/FKHR. Oncogene. 2007;26:7267–81. doi: 10.1038/sj.onc.1210525. [DOI] [PubMed] [Google Scholar]
- 159.Nanni P, Nicoletti G, Palladini A, et al. Opposing control of rhabdomyosarcoma growth and differentiation by myogenin and interleukin 4. Mol Cancer Ther. 2009;8:754–61. doi: 10.1158/1535-7163.MCT-08-0678. [DOI] [PubMed] [Google Scholar]
- 160.Taulli R, Scuoppo C, Bersani F, et al. Validation of met as a therapeutic target in alveolar and embryonal rhabdomyosarcoma. Cancer Res. 2006;66:4742–9. doi: 10.1158/0008-5472.CAN-05-4292. [DOI] [PubMed] [Google Scholar]
- 161.Khan J, Bittner ML, Saal LH, et al. cDNA microarrays detect activation of a myogenic transcription program by the PAX3-FKHR fusion oncogene. Proc Natl Acad Sci USA. 1999;96:13264–9. doi: 10.1073/pnas.96.23.13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dias P, Kumar P, Marsden HB, et al. N-myc gene is amplified in alveolar rhabdomyosarcomas (RMS) but not in embryonal RMS. Int J Cancer. 1990;45:593–6. doi: 10.1002/ijc.2910450403. [DOI] [PubMed] [Google Scholar]
- 163.Driman D, Thorner PS, Greenberg ML, et al. MYCN gene amplification in rhabdomyosarcoma. Cancer. 1994;73:2231–7. doi: 10.1002/1097-0142(19940415)73:8<2231::aid-cncr2820730832>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 164.Marampon F, Ciccarelli C, Zani BM. Down-regulation of c-Myc following MEK/ERK inhibition halts the expression of malignant phenotype in rhabdomyosarcoma and in non muscle-derived human tumours. Mol Cancer. 2006;5:31. doi: 10.1186/1476-4598-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Keleti J, Quezado MM, Abaza MM, et al. The MDM2 oncoprotein is overexpressed in rhabdomyosarcoma cell lines and stabilizes wild-type p53 protein. Am J Pathol. 1996;149:143–51. [PMC free article] [PubMed] [Google Scholar]
- 166.Iolascon A, Faienza MF, Coppola B, et al. Analysis of cyclin-dependent kinase inhibitor genes (CDKN2A, CDKN2B and CDKN2C) in childhood rhabdomyosarcoma. Genes Chromosomes Cancer. 1996;15:217–22. doi: 10.1002/(SICI)1098-2264(199604)15:4<217::AID-GCC3>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 167.Tapscott SJ, Thayer MJ, Weintraub H. Deficiency in rhabdomyosarcomas of a factor required for MyoD activity and myogenesis. Science. 1993;259:1450–3. doi: 10.1126/science.8383879. [DOI] [PubMed] [Google Scholar]
- 168.Puri PL, Wu Z, Zhang P, et al. Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes Dev. 2000;14:574–84. [PMC free article] [PubMed] [Google Scholar]
- 169.Puri PL, Sartorelli V. Regulation of muscle regulatory factors by DNA-binding, interacting proteins, and post-transcriptional modifications. J Cell Physiol. 2000;185:155–73. doi: 10.1002/1097-4652(200011)185:2<155::AID-JCP1>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 170.Yang Z, MacQuarrie KL, Analau E, et al. MyoD and E-protein heterodimers switch rhabdomyosarcoma cells from an arrested myoblast phase to a differentiated state. Genes Dev. 2009;23:694–707. doi: 10.1101/gad.1765109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Davicioni E, Finckenstein FG, Shahbazian V, et al. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res. 2006;66:6936–46. doi: 10.1158/0008-5472.CAN-05-4578. [DOI] [PubMed] [Google Scholar]
- 172.Graf Finckenstein F, Shahbazian V, Davicioni E, et al. PAX-FKHR function as pangenes by simultaneously inducing and inhibiting myogenesis. Oncogene. 2008;27:2004–14. doi: 10.1038/sj.onc.1210835. [DOI] [PubMed] [Google Scholar]
- 173.Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 174.Jacks T, Remington L, Williams BO, et al. Tumour spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 175.Fleischmann A, Jochum W, Eferl R, et al. Rhabdomyosarcoma development in mice lacking Trp53 and Fos: tumour suppression by the Fos protooncogene. Cancer Cell. 2003;4:477–82. doi: 10.1016/s1535-6108(03)00280-0. [DOI] [PubMed] [Google Scholar]
- 176.Nanni P, Nicoletti G, De Giovanni C, et al. Development of rhabdomyosarcoma in HER-2/neu transgenic p53 mutant mice. Cancer Res. 2003;63:2728–32. [PubMed] [Google Scholar]
- 177.Tsumura H, Yoshida T, Saito H, et al. Cooperation of oncogenic K-ras and p53 deficiency in pleomorphic rhabdomyosarcoma development in adult mice. Oncogene. 2006;25:7673–9. doi: 10.1038/sj.onc.1209749. [DOI] [PubMed] [Google Scholar]
- 178.Keller C, Arenkiel BR, Coffin CM, et al. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 2004;18:2614–26. doi: 10.1101/gad.1244004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hahn H, Wojnowski L, Zimmer AM, et al. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat Med. 1998;4:619–22. doi: 10.1038/nm0598-619. [DOI] [PubMed] [Google Scholar]
- 180.Chamberlain JS, Metzger J, Reyes M, et al. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 2007;21:2195–204. doi: 10.1096/fj.06-7353com. [DOI] [PubMed] [Google Scholar]
- 181.Langenau DM, Keefe MD, Storer NY, et al. Effects of RAS on the genesis of embryonal rhabdomyosarcoma. Genes Dev. 2007;21:1382–95. doi: 10.1101/gad.1545007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ferrari A, Bisogno G, Macaluso A, et al. Soft-tissue sarcomas in children and adolescents with neurofibromatosis type 1. Cancer. 2007;109:1406–12. doi: 10.1002/cncr.22533. [DOI] [PubMed] [Google Scholar]
- 183.Kratz CP, Steinemann D, Niemeyer CM, et al. Uniparental disomy at chromosome 11p15.5 followed by HRAS mutations in embryonal rhabdomyosarcoma: lessons from Costello syndrome. Hum Mol Genet. 2007;16:374–9. doi: 10.1093/hmg/ddl458. [DOI] [PubMed] [Google Scholar]
- 184.Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med. 2004;6:530–9. doi: 10.1097/01.gim.0000144188.15902.c4. [DOI] [PubMed] [Google Scholar]
- 185.Kleinerman RA, Tucker MA, Abramson DH, et al. Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst. 2007;99:24–31. doi: 10.1093/jnci/djk002. [DOI] [PubMed] [Google Scholar]
- 186.Hanks S, Coleman K, Summersgill B, et al. Comparative genomic hybridization and BUB1B mutation analyses in childhood cancers associated with mosaic variegated aneuploidy syndrome. Cancer Lett. 2006;239:234–8. doi: 10.1016/j.canlet.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 187.Kratz CP, Holter S, Etzler J, et al. Rhabdomyosarcoma in patients with constitutional mismatch-repair-deficiency syndrome. J Med Genet. 2009;46:418–20. doi: 10.1136/jmg.2008.064212. [DOI] [PubMed] [Google Scholar]
- 188.Charytonowicz E, Cordon-Cardo C, Matushansky I, et al. Alveolar rhabdomyosarcoma: is the cell of origin a mesenchymal stem cell. Cancer Lett. 2009;279:126–36. doi: 10.1016/j.canlet.2008.09.039. [DOI] [PubMed] [Google Scholar]
- 189.Mercado GE, Barr FG. Fusions involving PAX and FOX genes in the molecular pathogenesis of alveolar rhabdomyosarcoma: recent advances. Curr Mol Med. 2007;7:47–61. doi: 10.2174/156652407779940440. [DOI] [PubMed] [Google Scholar]
- 190.Merlino G, Khanna C. Fishing for the origins of cancer. Genes Dev. 2007;21:1275–9. doi: 10.1101/gad.1563707. [DOI] [PubMed] [Google Scholar]
- 191.Linardic CM, Downie DL, Qualman S, et al. Genetic modeling of human rhabdomyosarcoma. Cancer Res. 2005;65:4490–5. doi: 10.1158/0008-5472.CAN-04-3194. [DOI] [PubMed] [Google Scholar]
- 192.Ren YX, Finckenstein FG, Abdueva DA, et al. Mouse mesenchymal stem cells expressing PAX-FKHR form alveolar rhabdomyosarcomas by cooperating with secondary mutations. Cancer Res. 2008;68:6587–97. doi: 10.1158/0008-5472.CAN-08-0859. [DOI] [PubMed] [Google Scholar]
- 193.Keller C, Capecchi MR. New genetic tactics to model alveolar rhabdomyosarcoma in the mouse. Cancer Res. 2005;65:7530–2. doi: 10.1158/0008-5472.CAN-05-0477. [DOI] [PubMed] [Google Scholar]
- 194.Biederer CH, Ries SJ, Moser M, et al. The basic helix-loop-helix transcription factors myogenin and Id2 mediate specific induction of caveolin-3 gene expression during embryonic development. J Biol Chem. 2000;275:26245–51. doi: 10.1074/jbc.M001430200. [DOI] [PubMed] [Google Scholar]
- 195.Martìnez-Moreno M, Martìnez-Ruiz A, Alvarez-Barrientos A, et al. Nitric oxide down-regulates caveolin-3 levels through the interaction with myogenin, its transcription factor. J Biol Chem. 2007;282:23044–54. doi: 10.1074/jbc.M610751200. [DOI] [PubMed] [Google Scholar]
- 196.Chardin P, Yeramian P, Madaule P, et al. N-ras gene activation in the RD human rhabdomyosarcoma cell line. Int J Cancer. 1985;35:647–52. doi: 10.1002/ijc.2910350513. [DOI] [PubMed] [Google Scholar]
- 197.Sowa G, Pypaert M, Fulton D, et al. The phosphorylation of caveolin-2 on serines 23 and 36 modulates caveolin-1-dependent caveolae formation. Proc Natl Acad Sci USA. 2003;100:6511–6. doi: 10.1073/pnas.1031672100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sowa G, Xie L, Xu L, et al. Serine 23 and 36 phosphorylation of caveolin-2 is differentially regulated by targeting to lipid raft/caveolae and in mitotic endothelial cells. Biochemistry. 2008;47:101–11. doi: 10.1021/bi701709s. [DOI] [PubMed] [Google Scholar]
- 199.Volonte D, Zhang K, Lisanti MP, et al. Expression of caveolin-1 induces premature cellular senescence in primary cultures of murine fibroblasts. Mol Biol Cell. 2002;13:2502–17. doi: 10.1091/mbc.01-11-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Dasari A, Bartholomew JN, Volonte D, et al. Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Cancer Res. 2006;66:10805–14. doi: 10.1158/0008-5472.CAN-06-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bartholomew JN, Galbiati F. Mapping of oxidative stress response elements of the caveolin-1 promoter. Methods Mol Biol. 2010;594:409–23. doi: 10.1007/978-1-60761-411-1_29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Bartholomew JN, Volonte D, Galbiati F. Caveolin-1 regulates the antagonistic pleiotropic properties of cellular senescence through a novel Mdm2/p53-mediated pathway. Cancer Res. 2009;69:2878–86. doi: 10.1158/0008-5472.CAN-08-2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Volonte D, Galbiati F. Inhibition of thioredoxin reductase 1 by caveolin 1 promotes stress-induced premature senescence. EMBO Rep. 2009;10:1334–40. doi: 10.1038/embor.2009.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Panetta D, Biedi C, Repetto S, et al. IGF-I regulates caveolin 1 and IRS1 interaction in caveolae. Biochem Biophys Res Commun. 2004;316:240–3. doi: 10.1016/j.bbrc.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 205.Salani B, Briatore L, Garibaldi S, et al. Caveolin-1 down regulation inhibits IGF-IR signal transduction in H9C2 rat cardio myoblasts. Endocrinology. 2008;149:461–5. doi: 10.1210/en.2007-0312. [DOI] [PubMed] [Google Scholar]
- 206.Chen J, Capozza F, Wu A, et al. Regulation of insulin receptor substrate-1 expression levels by caveolin-1. J Cell Physiol. 2008;217:281–9. doi: 10.1002/jcp.21498. [DOI] [PubMed] [Google Scholar]
- 207.Salani B, Passalacqua M, Maffioli S, et al. IGF-IR internalizes with Caveolin-1 and PTRF/Cavin in Hacat cells. PLoS One. 2010;5:14157. doi: 10.1371/journal.pone.0014157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Couet J, Sargiacomo M, Lisanti MP. Interaction of a receptor tyrosine kinase, EGF-R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J Biol Chem. 1997;272:30429–38. doi: 10.1074/jbc.272.48.30429. [DOI] [PubMed] [Google Scholar]
- 209.Yamamoto M, Toya Y, Jensen RA, et al. Caveolin is an inhibitor of platelet-derived growth factor receptor signaling. Exp Cell Res. 1999;247:380–8. doi: 10.1006/excr.1998.4379. [DOI] [PubMed] [Google Scholar]
- 210.Labrecque L, Royal I, Surprenant DS, et al. Regulation of vascular endothelial growth factor receptor-2 activity by caveolin-1 and plasma membrane cholesterol. Mol Biol Cell. 2003;14:334–47. doi: 10.1091/mbc.E02-07-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Li S, Couet J, Lisanti MP. Src tyrosine kinases, Galpha subunits, and H-Ras share a common membrane-anchored scaffolding protein, caveolin. Caveolin binding negatively regulates the auto-activation of Src tyrosine kinases. J Biol Chem. 1996;271:29182–90. doi: 10.1074/jbc.271.46.29182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Schaaf G, Hamdi M, Zwijnenburg D, et al. Silencing of SPRY1 triggers complete regression of rhabdomyosarcoma tumours carrying a mutated RAS gene. Cancer Res. 2010;70:762–71. doi: 10.1158/0008-5472.CAN-09-2532. [DOI] [PubMed] [Google Scholar]
- 213.Razani B, Zhang XL, Bitzer M, et al. Caveolin-1 regulates transforming growth factor (TGF)-beta/SMAD signaling through an interaction with the TGF-beta type I receptor. J Biol Chem. 2001;276:6727–38. doi: 10.1074/jbc.M008340200. [DOI] [PubMed] [Google Scholar]
- 214.Bouché M, Canipari R, Melchionna R, et al. TGF-beta autocrine loop regulates cell growth and myogenic differentiation in human rhabdomyosarcoma cells. FASEB J. 2000;14:1147–58. doi: 10.1096/fasebj.14.9.1147. [DOI] [PubMed] [Google Scholar]
- 215.Ricaud S, Vernus B, Duclos M, et al. Inhibition of autocrine secretion of myostatin enhances terminal differentiation in human rhabdomyosarcoma cells. Oncogene. 2003;22:8221–32. doi: 10.1038/sj.onc.1207177. [DOI] [PubMed] [Google Scholar]
- 216.Langley B, Thomas M, McFarlane C, et al. Myostatin inhibits rhabdomyosarcoma cell proliferation through an Rb-independent pathway. Oncogene. 2004;23:524–34. doi: 10.1038/sj.onc.1207144. [DOI] [PubMed] [Google Scholar]
- 217.Yang Z, Zhang J, Cong H, et al. A retrovirus-based system to stably silence GDF-8 expression and enhance myogenic differentiation in human rhabdomyosarcoma cells. J Gene Med. 2008;10:825–33. doi: 10.1002/jgm.1216. [DOI] [PubMed] [Google Scholar]
- 218.Rossi S, Stoppani E, Puri PL, et al. Differentiation of human rhabdomyosarcoma RD cells is regulated by reciprocal, functional interactions between myostatin, p38 and extracellular regulated kinase signalling pathways. Eur J Cancer. 2011;47:1095–105. doi: 10.1016/j.ejca.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 219.Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 220.Massagué J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–78. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 221.Gordon KJ, Blobe GC. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim Biophys Acta. 2008;1782:197–228. doi: 10.1016/j.bbadis.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 222.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 223.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 224.Ohsawa Y, Hagiwara H, Nakatani M, et al. Muscular atrophy of caveolin-3-deficient mice is rescued by myostatin inhibition. J Clin Invest. 2006;116:2924–34. doi: 10.1172/JCI28520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Danilkovitch-Miagkova A, Zbar B. Dysregulation of Met receptor tyrosine kinase activity in invasive tumours. J Clin Invest. 2002;109:863–7. doi: 10.1172/JCI15418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Sparvero LJ, Asafu-Adjei D, Kang R, et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med. 2009;7:17. doi: 10.1186/1479-5876-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Perrone L, Peluso G, Melone MA. RAGE recycles at the plasma membrane in S100B secretory vesicles and promotes Schwann cells morphological changes. J Cell Physiol. 2008;217:60–71. doi: 10.1002/jcp.21474. [DOI] [PubMed] [Google Scholar]
- 228.Reddy MA, Li SL, Sahar S, et al. Key role of Src kinase in S100B-induced activation of the receptor for advanced glycation end products in vascular smooth muscle cells. J Biol Chem. 2006;281:13685–93. doi: 10.1074/jbc.M511425200. [DOI] [PubMed] [Google Scholar]
- 229.Riuzzi F, Sorci G, Donato R. The amphoterin (HMGB1)/receptor for advanced glycation end products (RAGE) pair modulates myoblast proliferation, apoptosis, adhesiveness, migration, and invasiveness. Functional inactivation of RAGE in L6 myoblasts results in tumour formation in vivo. J Biol Chem. 2006;281:8242–53. doi: 10.1074/jbc.M509436200. [DOI] [PubMed] [Google Scholar]
- 230.Riuzzi F, Sorci G, Donato R. RAGE expression in rhabdomyosarcoma cells results in myogenic differentiation and reduced proliferation, migration, invasiveness, and tumour growth. Am J Pathol. 2007;171:947–61. doi: 10.2353/ajpath.2007.070049. [DOI] [PMC free article] [PubMed] [Google Scholar]