

Abstract
Calpastatin is an endogenous specific inhibitor of calpain, a calcium-dependent cysteine protease. Here we show that loss-of-function mutations in calpastatin (CAST) are the genetic causes of an autosomal-recessive condition characterized by generalized peeling skin, leukonychia, acral punctate keratoses, cheilitis, and knuckle pads, which we propose to be given the acronym PLACK syndrome. In affected individuals with PLACK syndrome from three families of different ethnicities, we identified homozygous mutations (c.607dup, c.424A>T, and c.1750delG) in CAST, all of which were predicted to encode truncated proteins (p.Ile203Asnfs∗8, p.Lys142∗, and p.Val584Trpfs∗37). Immunohistochemistry shows that staining of calpastatin is reduced in skin from affected individuals. Transmission electron microscopy revealed widening of intercellular spaces with chromatin condensation and margination in the upper stratum spinosum in lesional skin, suggesting impaired intercellular adhesion as well as keratinocyte apoptosis. A significant increase of apoptotic keratinocytes was also observed in TUNEL assays. In vitro studies utilizing siRNA-mediated CAST knockdown revealed a role for calpastatin in keratinocyte adhesion. In summary, we describe PLACK syndrome, as a clinical entity of defective epidermal adhesion, caused by loss-of-function mutations in CAST.
Main Text
Peeling skin syndrome (PSS) is characterized by continuous shedding of the stratum corneum of the epidermis with onset from birth or infancy and lasting throughout life.1 Skin peeling can be accompanied by erythema, vesicular lesions, or other ectodermal features including fragile hair and nail abnormalities.2 PSS can be divided into two clinical forms: acral PSS (APSS [MIM 609796]) and generalized PSS (GPSS [MIM 270300]). APSS involves the palmar, plantar, and dorsal surfaces of hands and feet and can be associated with mutations in transglutaminase 5 (TGM5 [MIM 603805]).3 In addition, mutations in the gene encoding for the cysteine protease inhibitor cystatin A (CSTA [MIM 184600]) have recently been associated with autosomal-recessive exfoliative ichthyosis (MIM 607936) and also APSS.4,5 Individuals with inflammatory GPSS associated with mutations in corneodesmosin (CDSN [MIM 602593]) can also present with severe pruritus, food allergies, and repeated episodes of angioedema, urticaria, and asthma.6 A similar entity termed SAM syndrome (severe dermatitis, multiple allergies, and metabolic wasting [MIM 615508]), which is caused by desmoglein 1 (DSG1 [MIM 125670]) deficiency, shares clinical features with inflammatory GPSS and atopy.7 A homozygous missense mutation was identified within CHST8 (MIM 610190), encoding N-acetylgalactosamine-4-O-sulfotransferase, in a large consanguineous family with non-inflammatory GPSS.8 However, the genetic basis of a number of PSS cases is still unresolved.9,10
Here we show that homozygous loss-of-function (LOF) mutations in CAST (MIM 114090) underlie autosomal-recessive generalized PSS with leukonychia, acral punctate keratoses, cheilitis, and knuckle pads. We propose this clinical entity to be given the acronym PLACK syndrome. Via exome sequencing and Sanger sequencing we demonstrate that distinct homozygous LOF CAST mutations segregate with the disorder in all three families with PLACK syndrome. CAST encodes calpastatin, an endogenous protease inhibitor. Our findings emphasize the important role of the protease-inhibitor balance in epidermal homeostasis.
We ascertained a 28-year-old Chinese female (individual 1, V-3 in Figure 1A) affected with PLACK syndrome who was born to second-cousin parents (Figure 1A). She was found to have trauma-induced recurrent blistering prominently on the extremities since infancy, which was worse in summer. In winter, asymptomatic skin peeling was more prominent, either spontaneously or after the remission of blistering, leaving underlying erythema. The blistering improved and was confined to distal extremities after puberty, and skin peeling progressed to involve nearly the entire body. She had mild pruritus and no history of atopic eczema, food allergy, allergic rhinitis, or asthma. Physical examination revealed generalized dry, scaly skin with superficial exfoliation and underlying erythema (Figure 1B). Cheilitis with dry, shedding scales was observed (Figure 1B). Several blisters were noted on her wrists and soles. Punctate palmoplantar keratoderma was seen, which coalesced into focal keratoderma predominately on the weight-bearing areas (Figure 1B). Knuckle pads with multiple hyperkeratotic micropapules were also found involving all the interphalangeal joints. Leukonychia affected the proximal half of the nails with mild distal onycholysis (Figure 1B). No other systemic abnormalities were identified. Serum total IgE level and blood eosinophil count were within normal range. Histological examination of a biopsy from the scaly skin of her leg showed hyperkeratosis, acanthosis, and intraepidermal clefting with irregular acantholysis (Figure 1C).
Figure 1.
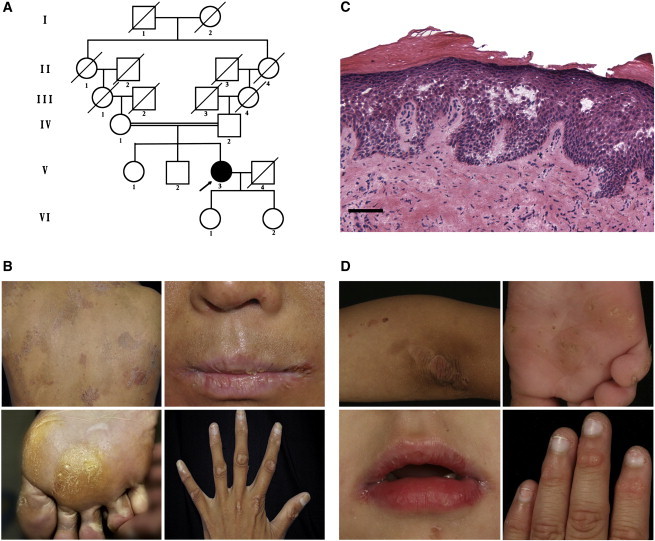
Clinical Findings of PLACK Syndrome
(A) Family pedigree of individual 1. The arrow indicates individual 1 (V-3).
(B) Individual 1 showed clinical features of generalized skin peeling, cheilitis, plantar keratoses on weight-bearing areas, blistering, leukonychia, and knuckle pads with multiple hyperkeratotic micropapules.
(C) Histopathology of skin biopsy from lower extremities of individual 1 demonstrated intraepidermal clefting with irregular acantholysis (hematoxylin-eosin staining; scale bar represents 50 μm).
(D) Similar clinical features were found in individual 2, including skin peeling, cheilitis, punctate keratoses of the soles, leukonychia, and knuckle pads with hyperkeratotic micropapules.
We also investigated an affected Nepalese female (individual 2) from non-consanguineous parents who presented to dermatologists at age 3 with a 2-year history of painful lesions on the palms and soles. Clinical examination revealed punctate keratoses on the palms, extending onto the flexor aspect of the wrists and soles, plaques with hyperkeratotic micropapules over the interphalangeal joints, cheilitis of the upper lip, angular cheilitis, subtle telangiectasia on the cheeks, follicular hyperkeratosis on the extensor surface of the knees, and leukonychia of 70%–100% of the nails (Figures 1D and S1). At review at age 4, peeling areas had developed on the distal limbs including the extensor surface of the knees and elbows (Figure 1D). No history of atopic eczema, hay fever, or asthma was recorded and serum total IgE and eosinophil count were normal.
An additional family with known consanguinity, previously reported as having a recessive form of pachyonychia congenita, was also enrolled in this study.11 In brief, both affected European male siblings (individuals 3 and 4, now ages 54 and 58 years old) give a history of blistering and peeling of skin from the age of about 3 months on the hands, feet, knuckles, elbows, and knees. Skin fragility is induced by minor trauma and heat and continues to be the greatest problem for these individuals. Both individuals also have leukonychia, leukokeratosis, angular cheilitis, papules on the extensor surface of the fingers and toes, and punctate palmar keratoses and a plantar keratoderma, described in more detail by Haber and Rose.11
We collected blood and saliva samples from the three families after obtaining informed consent. This project was approved by the Clinical Research Ethics Committee of the Peking University First Hospital, East London and City Health Authority Research Ethics Committee, and the Western Institutional Review Board, which all comply with all principles of the Helsinki Accord. After exclusion of pathogenic mutations in TGM5, CSTA, CDSN, and CHST8 by Sanger sequencing, we performed exome sequencing in individual 1 using 3 μg of genomic DNA. Exome capture was performed with the NimbleGen SeqCap EZ Library (Roche) for enrichment and then sequenced on an Illumina HiSeq2000 according to the manufacturer’s protocols. Variants were filtered against dbSNP137, the 1000 Genomes Project, HapMap8, and BGI internal databases, as described in our previous study. Because her parents were consanguineous, we focused on homozygous variants to identify the underlying defective gene. Among all the 148 variants fulfilling these criteria, only 15 variants lay in coding exons or splicing boundaries and were predicted to be “not tolerated” by SIFT.12 These included a one-nucleotide duplication in CAST that was predicted to lead to a frameshift and truncation of the protein. By Sanger sequencing (primers are listed in Table S1), we confirmed that this mutation, c.607dup (p.Ile203Asnfs∗8; RefSeq accession numbers NM_001042440.3, NP_001035905.1), was homozygous in individual 1 and heterozygous in her unaffected parents, siblings, and daughters (Figures 1A and 2A). This mutation was not found in 200 ethnically matched controls.
Figure 2.
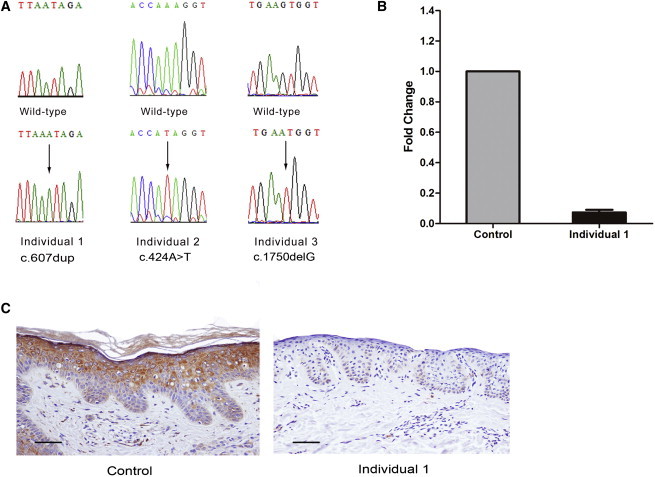
Homozygous Mutations in CAST and Its Expression and Localization in the Skin
(A) Sequence chromatograms showing homozygous mutations c.607dup, c.424A>T, and c.1750delG in the affected individuals. Arrows indicate the position of the mutation.
(B) Homozygous frameshift mutation c.607dup-mediated mRNA decay of calpastatin in individual 1 is demonstrated by qRT-PCR compared to the normal control. The error bar represents the SEM.
(C) Immunohistochemistry shows absent calpastatin staining in individual 1 (right) compared to normal staining throughout the epidermis in the control (left). The scale bars represent 50 μm.
Exome sequencing was performed on genomic DNA from individual 2 using the NimbleGen SeqCap EZ Library SR (Roche) and sequenced on an Illumina HiSeq2000 according to the manufacturer’s protocols. Further details of analysis have been described previously.12 Analysis of novel variants revealed a homozygous nonsense mutation (c.424A>T [p.Lys142∗]) in CAST. Sanger sequencing showed co-segregation with the disorder in this family (Figures 2A and S1A). Subsequent exome sequencing and Sanger sequencing also showed a homozygous frameshift mutation, c.1750delG (p.Val584Trpfs∗37), in CAST in individuals 3 and 4, which co-segregated with the disorder in the family (primers are listed in Table S1). All three identified mutations (including c.607dup) in CAST are absent in dbSNP137, HapMap8, the 1000 Genomes Project database, the National Heart, Lung, and Blood Institute Exome Sequencing Project (ESP) Exome Variant Server, and the BGI internal database, highly suggestive of the pathogenicity of these mutations based on their extreme rarity.
All three mutations identified in CAST are predicted to encode truncated proteins and lead to a loss of function. To investigate the consequences of homozygosity for the c.607dup mutation in vivo, we examined the mRNA expression of CAST in the skin from individual 1 by using quantitative real-time PCR (qRT-PCR) (primers are listed in Table S2). The level of CAST transcription was determined on the basis of the comparative cycle threshold (2−ΔΔCt) method using cDNA templates generated from a corresponding area of the skin from a gender- and age-matched healthy control as the calibrator. In contrast to the control, we could detect only trace expression of CAST mRNA in individual 1, probably due to mechanisms of nonsense-mediated mRNA decay (Figure 2B). Immunohistochemical staining was subsequently performed with a calpastatin antibody (catalog number: sc-20779, Santa Cruz) in paraffin-embedded sections from the pretibial skin lesion of individual 1. A similar analysis was performed on paraffin-embedded sections taken from xerotic left thigh skin of individual 2, but using immunofluorescence. Consistent with the predicted effect of the two mutations and the results of the qRT-PCR analysis of the c.607dup mutation, calpastatin staining was absent or reduced in the skin from both individuals, whereas in the normal control skin, calpastatin is localized throughout the epidermis and has a cytoplasmic localization (Figures 2C and S2). No skin biopsy material was available from individuals 3 or 4.
Calpastatin is a specific endogenous protease inhibitor of calpains (μ-calpain and m-calpain). Calpains are intracellular cysteine proteases that require calcium or epidermal growth factor for their catalytic activity.13 In skin, calpains have been reported as being involved in a range of cellular processes, including keratinocyte growth, migration, and cell cycle regulation.14 These processes are particularly important in epidermal terminal differentiation, characterized by expression of specific structural proteins. Immunohistochemical staining of lesional skin sections from individual 1 revealed a slight upregulation of loricrin (antibody catalog number ab24722, Abcam), keratin 1 (antibody catalog number ab24643, Abcam), and keratin 10 (antibody catalog number ab76318, Abcam) compared to control (Figure S3). The increased labeling of these proteins might be a compensatory mechanism for epidermal barrier disruption.15 In contrast, the staining of filaggrin in the epidermis of individual 1 (antibody catalog number ab81468, Abcam) and in the epidermis of individual 2 (antibody catalog number NCL-Filaggrin, Leica Biosystems), as well as the staining of the tight junction component claudin-116 (antibody catalog number 13255, Cell Signaling Technology) in the epidermis of individual 1 was indistinguishable from control (data not shown).
Because increased activity of calpains can induce apoptosis,17 in situ apoptotic examination of keratinocytes in the lesional skin biopsy from individual 1 was performed by a TUNEL assay (In Situ Cell Death Detection Kit, Roche). Skin sections from an unrelated healthy subject were used as a control. Images above the stratum basale were taken randomly with a fluorescence microscope (IX71, Olympus), with the same settings for both control and individual skin sections. Total cells and apoptotic cells (with fluorescent nuclei) in five random high-power fields (400×) were counted. The number of apoptotic cells in the imaged fields was significantly increased in lesional skin compared to normal control (Figures 3A and 3B). Ultrastructural analysis of lesional skin sections from individual 1 was performed by transmission electron microscopy (TEM), which showed expanded intercellular spaces (Figure 3D), apoptotic chromatin condensation, and margination (Figure 3E), supporting the results of the TUNEL assay.
Figure 3.
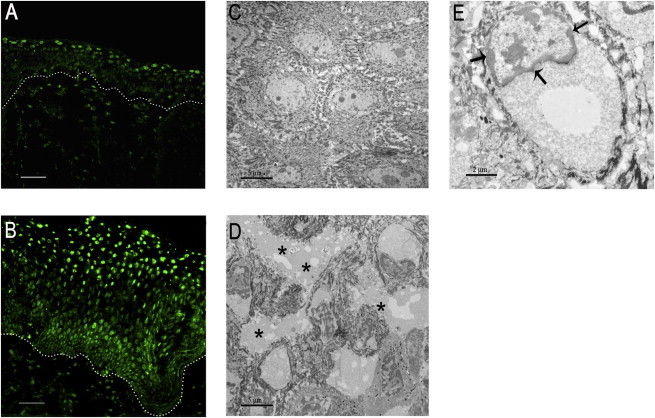
Increased Keratinocyte Apoptosis in Lesional Skin
(A and B) TUNEL assay demonstrated increased apoptotic level of keratinocytes in lesional skin from individual 1 (B) compared to the control (A). The scale bars represent 50 μm.
(C and D) TEM of lesional skin from individual 1 showed expanded intercellular spaces (D, asterisks) compared to the control (C). The scale bars represent 5 μm.
(E) At higher magnification, chromatin condensation and margination (arrows) were apparent. The scale bar represents 2 μm.
To determine the functional consequences of CAST LOF mutations in vitro, we performed siRNA-mediated knockdown (KD) of CAST by using a specific siRNA pool (ON-TARGETplus Human CAST siRNA-SMARTpool, GE Healthcare Dharmacon) in the immortalized keratinocyte cell line, HaCaT. Non-targeting pool siRNA (ON-TARGETplus Non-targeting Pool, GE Healthcare Dharmacon) was used as a control. Immunocytochemistry of HaCaT cell monolayers treated with CAST siRNA and analysis of total protein lysates by immunoblotting showed robust CAST knockdown (Figures 4A–4C).
Figure 4.
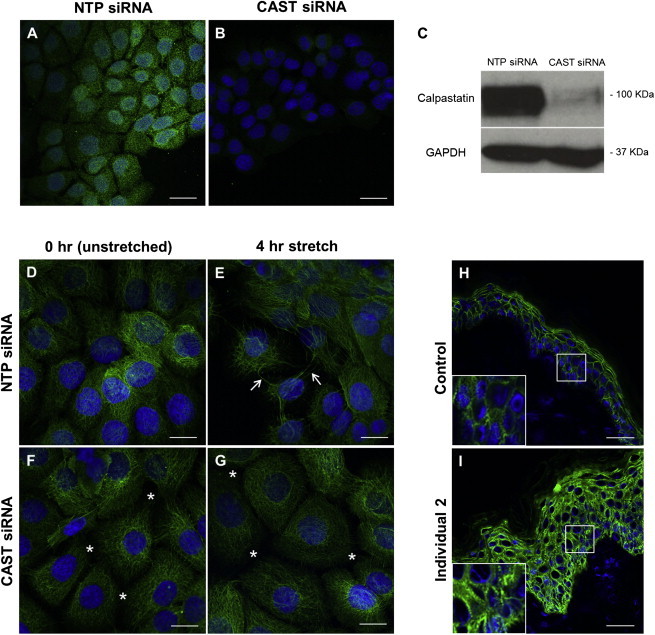
CAST Knockdown Decreases Intercellular Adhesion and Increases Desmoplakin Abundance
(A–C) HaCaT cells transfected with a pool of NTP siRNA (A) or CAST siRNA (B) were stained with an anti-calpastatin (green) antibody and DAPI (blue) as a nuclear marker. Total protein from HaCaT cell lysates after transfection with NTP siRNA or CAST siRNA were incubated with an anti-calpastatin antibody (C). GAPDH was used as loading control. A significant reduction in calpastatin protein abundance can be observed in the CAST KD cells compared to NTP cells, both by immunocytochemistry and immunoblotting.
(D–G) CAST KD (F and G) and NTP (D and E) cells were subjected to cyclic stretching for 0 hr (unstretched; D, F) and 4 hr (E, G) followed by staining with an anti-keratin 14 antibody. Stretching of the keratin filaments can be observed after 4 hr mechanical stress in the NTP cells (arrows) compared to unstretched monolayers. In the CAST KD cells, breakage of these filaments together with widening of the intercellular spaces was observed independent of mechanical stress (asterisks).
(H and I) Immunohistochemistry with an anti-desmoplakin antibody on skin sections from individual 2 revealed an upregulation and change in localization of this protein from a membranous appearance (H) to a both membranous and cytoplasmic appearance (I) in comparison to control.
The scale bars in each image represent 20 μm.
We then used an in vitro mechanical-induced stress assay to investigate the role of calpastatin in keratinocyte adhesion in CAST siRNA-treated cells (CAST KD cells) and NTP siRNA-treated cells (NTP cells). For this we have used the Flexcell FX-4000 Tension System (Flexcell), which uses vacuum pressure to apply cyclic or static strain to cells cultured on flexible-bottomed culture plates. CAST KD cells (mimicking the homozygous LOF mutations) and NTP cells were subjected to mechanical stretch at a frequency of 5 Hz (five cycles of stretch and relaxation per second) and an elongation of amplitude ranging from 10% to 14% (increase in diameter across the silicone deformable membrane from 10% to 14%). Cells were stretched for 0 hr (non-stretched) and 4 hr. Immunocytochemistry with an in-house LL001 monoclonal keratin 14 antibody18 performed on siRNA-treated cells before and after mechanical stress revealed breakage of the intercellular connections in CAST KD cell monolayers, independent of whether they had been subjected to mechanical stress. In contrast, NTP cells presented with stretched keratin filaments after stretching but no disruption in intercellular adhesion prior to mechanical stress (Figures 4D–4G).
A recent study by Nassar et al. looking at CAST overexpression in a mouse model reported significant changes in the wound-healing process compared to normal mice.19 CAST-overexpression mice showed a striking delay in wound healing with reduced proliferation and re-epithelialization, particularly in the early steps of the wound-healing process.19 The possible effects of CAST LOF mutations on keratinocyte migration were assessed by performing a scratch assay on CAST KD cell monolayers compared to NTP cells. Because CAST KD scratch wounds appeared to close at the same rate as NTP scratch wounds, we concluded that cell migration in monolayers was not altered by CAST knockdown in three independent experiments (Figure S4).
After observations in CAST LOF skin and our in vitro studies indicating a key role for calpastatin in keratinocyte adhesion, we examined desmosomal protein abundance. Immunofluorescence with an antibody targeting desmoplakin (DSP; 11-5F mouse monoclonal, a gift from David Garrod),20 the major protein of the desmosome, was performed on frozen skin sections from individuals 1 and 2. This showed an apparent increase in DSP abundance, with both a plasma membrane and cytoplasmic localization pattern compared to a specific membranous localization pattern in control skin (Figures 4H, 4I, S3G, and S3H). Furthermore, our in vitro studies displayed a general trend of DSP upregulation in CAST KD cells independent of mechanical stress when compared to NTP cells (data not shown).
Calpain and its endogenous specific inhibitor calpastatin constitute an intracellular non-lysosomal proteolytic system ubiquitously distributed in mammal cells and many other organisms. By catalyzing the controlled proteolysis of target proteins, calpains play an important role in various cell functions, including cell proliferation, differentiation, mobility, and cell cycle progression, as well as cell-type-specific functions like cell fusions in myoblasts.13,21 Also, activation of calpains has been suggested to trigger apoptosis by cleaving either pro-apoptotic or anti-apoptotic proteins.22 It has been demonstrated in vitro that increased activity of m-calpain results in apoptosis of HaCaT cells.23 LOF mutations in CAST lead to disinhibition of calpains, thus enhancing apoptosis of keratinocytes, as showed in our TUNEL and TEM results. Elevated apoptotic levels of keratinocytes can result in skin hyperkeratosis,24,25 leading to the clinical phenotypes of acral punctate keratoses, knuckle pads with hyperkeratotic micropapules, and leukonychia.
Previous studies have shown that calpain could promote focal adhesion disassembly by proteolysis of talin and focal adhesion kinase.26,27 Our CAST KD experiments indicate that the calpain/calpastatin system might also be critical for intercellular adhesion. Increased calpain activity due to LOF mutations in CAST might lead to excessive proteolysis of certain epidermal desmosomal components, which though not extensively described in the present study, result in impaired desmosome function characterized by decreased resistance of the epidermis to mechanical stretch (blistering, skin peeling, and acantholysis). This speculation was further strengthened by the remarkable phenotypic overlap between PLACK syndrome and other inherited disorders with desmosome deficiency, including inflammatory GPSS,6 SAM syndrome,7 and Netherton syndrome (MIM 256500), the last of which was caused by LOF mutations in SPINK5 (MIM 605010) encoding protease inhibitor LEKTI,28 leading to over-degradation of CDSN and DSG1, two major components of the desmosome.29 The increased abundance of DSP in keratinocytes observed in the two affected individuals might be a compensatory mechanism in response to desmosomal damage. Although calpastatin was suggested to play diverse physiological roles in neurological, musculoskeletal, and ocular systems,30 no significant related symptoms were observed in our affected individuals. Further studies are required to elucidate whether there is redundancy of the calpain proteolytic system in these organs, explaining the lack of abnormalities. Notably, Cast-knockout mice, which showed increased activity of calpains, exhibit no defect under normal conditions31 and only slight behavioral changes in a stressful environment.32 These phenotypic differences indicate different physiological functions of the calpain/calpastatin system between humans and mice.
In summary, we describe the clinical features of an autosomal-recessive entity termed PLACK syndrome with generalized skin peeling, leukonychia, acral punctate keratoses, cheilitis, and knuckle pads, distinct from epidermolysis bullosa, pachyonychia congenita, and Bart-Pumphrey syndrome (MIM 149200), a syndrome associated with sensorineural deafness, leukonychia, and knuckle pads. In three families with PLACK syndrome, homozygous LOF mutations in CAST were identified, leading to reduced abundance of calpastatin, the only known inhibitor of calpains. Mutations of protease inhibitors can disrupt the skin barrier, impair keratinocyte adhesion, affect cell signaling, and cause various genetic skin conditions,4,33 such as Netherton syndrome caused by mutations in SPINK528 and Nagashima-type palmoplantar keratosis (MIM 615598) caused by mutations in SERPINB7 (MIM 603357).34,35 Our findings expand the spectrum of these conditions and explore new avenues for proteolytic pathways in skin.
Acknowledgments
We are grateful to the patients and their family members for participation in this study. We gratefully acknowledge Dr. Deirdre Buckley (Bath, UK) who referred individual 2 to us. This work was supported in part by China National Funds for Distinguished Young Scientists (81425020 to Y.Y.), National Natural Science Foundation of China (81271744 to Y.Y. and 81201220 to Z.L.), Beijing Higher Education Young Elite Teacher Project (YETP0069 for Z.L.), and Shenzhen Key Laboratory of Cognitive Genomics (CXB201108250094A). F.J.D.S. and N.J.W. are supported by grants from the Pachyonychia Congenita Project. The Centre for Dermatology and Genetic Medicine at the University of Dundee is supported by a Wellcome Trust Strategic Award (098439/Z/12/Z to W.H.I.M.). This study is also funded, in part, by Barts and the London Charity (D.P.K.).
Contributor Information
David P. Kelsell, Email: d.p.kelsell@qmul.ac.uk.
Yong Yang, Email: dryongyang@bjmu.edu.cn.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
Pachyonychia Congenita Project, http://www.pachyonychia.org/
References
- 1.Hacham-Zadeh S., Holubar K. Skin peeling syndrome in a Kurdish family. Arch. Dermatol. 1985;121:545–546. [PubMed] [Google Scholar]
- 2.Cassidy A.J., van Steensel M.A., Steijlen P.M., van Geel M., van der Velden J., Morley S.M., Terrinoni A., Melino G., Candi E., McLean W.H. A homozygous missense mutation in TGM5 abolishes epidermal transglutaminase 5 activity and causes acral peeling skin syndrome. Am. J. Hum. Genet. 2005;77:909–917. doi: 10.1086/497707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kharfi M., El Fekih N., Ammar D., Jaafoura H., Schwonbeck S., van Steensel M.A., Fazaa B., Kamoun M.R., Fischer J. A missense mutation in TGM5 causes acral peeling skin syndrome in a Tunisian family. J. Invest. Dermatol. 2009;129:2512–2515. doi: 10.1038/jid.2009.118. [DOI] [PubMed] [Google Scholar]
- 4.Blaydon D.C., Nitoiu D., Eckl K.M., Cabral R.M., Bland P., Hausser I., van Heel D.A., Rajpopat S., Fischer J., Oji V. Mutations in CSTA, encoding Cystatin A, underlie exfoliative ichthyosis and reveal a role for this protease inhibitor in cell-cell adhesion. Am. J. Hum. Genet. 2011;89:564–571. doi: 10.1016/j.ajhg.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krunic A.L., Stone K.L., Simpson M.A., McGrath J.A. Acral peeling skin syndrome resulting from a homozygous nonsense mutation in the CSTA gene encoding cystatin A. Pediatr. Dermatol. 2013;30:e87–e88. doi: 10.1111/pde.12092. [DOI] [PubMed] [Google Scholar]
- 6.Oji V., Eckl K.M., Aufenvenne K., Nätebus M., Tarinski T., Ackermann K., Seller N., Metze D., Nürnberg G., Fölster-Holst R. Loss of corneodesmosin leads to severe skin barrier defect, pruritus, and atopy: unraveling the peeling skin disease. Am. J. Hum. Genet. 2010;87:274–281. doi: 10.1016/j.ajhg.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Samuelov L., Sarig O., Harmon R.M., Rapaport D., Ishida-Yamamoto A., Isakov O., Koetsier J.L., Gat A., Goldberg I., Bergman R. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat. Genet. 2013;45:1244–1248. doi: 10.1038/ng.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cabral R.M., Kurban M., Wajid M., Shimomura Y., Petukhova L., Christiano A.M. Whole-exome sequencing in a single proband reveals a mutation in the CHST8 gene in autosomal recessive peeling skin syndrome. Genomics. 2012;99:202–208. doi: 10.1016/j.ygeno.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pavlovic S., Krunic A.L., Bulj T.K., Medenica M.M., Fong K., Arita K., McGrath J.A. Acral peeling skin syndrome: a clinically and genetically heterogeneous disorder. Pediatr. Dermatol. 2012;29:258–263. doi: 10.1111/j.1525-1470.2011.01563.x. [DOI] [PubMed] [Google Scholar]
- 10.Chang Y.Y., van der Velden J., van der Wier G., Kramer D., Diercks G.F., van Geel M., Coenraads P.J., Zeeuwen P.L., Jonkman M.F. Keratolysis exfoliativa (dyshidrosis lamellosa sicca): a distinct peeling entity. Br. J. Dermatol. 2012;167:1076–1084. doi: 10.1111/j.1365-2133.2012.11175.x. [DOI] [PubMed] [Google Scholar]
- 11.Haber R.M., Rose T.H. Autosomal recessive pachyonychia congenita. Arch. Dermatol. 1986;122:919–923. [PubMed] [Google Scholar]
- 12.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 13.Goll D.E., Thompson V.F., Li H., Wei W., Cong J. The calpain system. Physiol. Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 14.Carragher N.O., Frame M.C. Focal adhesion and actin dynamics: a place where kinases and proteases meet to promote invasion. Trends Cell Biol. 2004;14:241–249. doi: 10.1016/j.tcb.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 15.Pigors M., Kiritsi D., Cobzaru C., Schwieger-Briel A., Suárez J., Faletra F., Aho H., Mäkelä L., Kern J.S., Bruckner-Tuderman L., Has C. TGM5 mutations impact epidermal differentiation in acral peeling skin syndrome. J. Invest. Dermatol. 2012;132:2422–2429. doi: 10.1038/jid.2012.166. [DOI] [PubMed] [Google Scholar]
- 16.Furuse M., Hata M., Furuse K., Yoshida Y., Haratake A., Sugitani Y., Noda T., Kubo A., Tsukita S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J. Cell Biol. 2002;156:1099–1111. doi: 10.1083/jcb.200110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith M.A., Schnellmann R.G. Calpains, mitochondria, and apoptosis. Cardiovasc. Res. 2012;96:32–37. doi: 10.1093/cvr/cvs163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Purkis P.E., Steel J.B., Mackenzie I.C., Nathrath W.B., Leigh I.M., Lane E.B. Antibody markers of basal cells in complex epithelia. J. Cell Sci. 1990;97:39–50. doi: 10.1242/jcs.97.1.39. [DOI] [PubMed] [Google Scholar]
- 19.Nassar D., Letavernier E., Baud L., Aractingi S., Khosrotehrani K. Calpain activity is essential in skin wound healing and contributes to scar formation. PLoS ONE. 2012;7:e37084. doi: 10.1371/journal.pone.0037084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parrish E.P., Steart P.V., Garrod D.R., Weller R.O. Antidesmosomal monoclonal antibody in the diagnosis of intracranial tumours. J. Pathol. 1987;153:265–273. doi: 10.1002/path.1711530311. [DOI] [PubMed] [Google Scholar]
- 21.Barnoy S., Maki M., Kosower N.S. Overexpression of calpastatin inhibits L8 myoblast fusion. Biochem. Biophys. Res. Commun. 2005;332:697–701. doi: 10.1016/j.bbrc.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 22.Tan Y., Dourdin N., Wu C., De Veyra T., Elce J.S., Greer P.A. Ubiquitous calpains promote caspase-12 and JNK activation during endoplasmic reticulum stress-induced apoptosis. J. Biol. Chem. 2006;281:16016–16024. doi: 10.1074/jbc.M601299200. [DOI] [PubMed] [Google Scholar]
- 23.Inoue A., Yamazaki M., Ishidoh K., Ogawa H. Epidermal growth factor activates m-calpain, resulting in apoptosis of HaCaT keratinocytes. J. Dermatol. Sci. 2004;36:60–62. doi: 10.1016/j.jdermsci.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 24.Lin Z., Chen Q., Lee M., Cao X., Zhang J., Ma D., Chen L., Hu X., Wang H., Wang X. Exome sequencing reveals mutations in TRPV3 as a cause of Olmsted syndrome. Am. J. Hum. Genet. 2012;90:558–564. doi: 10.1016/j.ajhg.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H., Cao X., Lin Z., Lee M., Jia X., Ren Y., Dai L., Guan L., Zhang J., Lin X. Exome sequencing reveals mutation in GJA1 as a cause of keratoderma-hypotrichosis-leukonychia totalis syndrome. Hum. Mol. Genet. 2015;24:243–250. doi: 10.1093/hmg/ddu442. [DOI] [PubMed] [Google Scholar]
- 26.Franco S.J., Rodgers M.A., Perrin B.J., Han J., Bennin D.A., Critchley D.R., Huttenlocher A. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat. Cell Biol. 2004;6:977–983. doi: 10.1038/ncb1175. [DOI] [PubMed] [Google Scholar]
- 27.Chan K.T., Bennin D.A., Huttenlocher A. Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK) J. Biol. Chem. 2010;285:11418–11426. doi: 10.1074/jbc.M109.090746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chavanas S., Bodemer C., Rochat A., Hamel-Teillac D., Ali M., Irvine A.D., Bonafé J.L., Wilkinson J., Taïeb A., Barrandon Y. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat. Genet. 2000;25:141–142. doi: 10.1038/75977. [DOI] [PubMed] [Google Scholar]
- 29.Samuelov L., Sprecher E. Peeling off the genetics of atopic dermatitis-like congenital disorders. J. Allergy Clin. Immunol. 2014;134:808–815. doi: 10.1016/j.jaci.2014.07.061. [DOI] [PubMed] [Google Scholar]
- 30.Carragher N.O. Calpain inhibition: a therapeutic strategy targeting multiple disease states. Curr. Pharm. Des. 2006;12:615–638. doi: 10.2174/138161206775474314. [DOI] [PubMed] [Google Scholar]
- 31.Takano J., Tomioka M., Tsubuki S., Higuchi M., Iwata N., Itohara S., Maki M., Saido T.C. Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains: evidence from calpastatin mutant mice. J. Biol. Chem. 2005;280:16175–16184. doi: 10.1074/jbc.M414552200. [DOI] [PubMed] [Google Scholar]
- 32.Nakajima R., Takao K., Huang S.M., Takano J., Iwata N., Miyakawa T., Saido T.C. Comprehensive behavioral phenotyping of calpastatin-knockout mice. Mol. Brain. 2008;1:7. doi: 10.1186/1756-6606-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Veer S.J., Furio L., Harris J.M., Hovnanian A. Proteases: common culprits in human skin disorders. Trends Mol. Med. 2014;20:166–178. doi: 10.1016/j.molmed.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 34.Kubo A., Shiohama A., Sasaki T., Nakabayashi K., Kawasaki H., Atsugi T., Sato S., Shimizu A., Mikami S., Tanizaki H. Mutations in SERPINB7, encoding a member of the serine protease inhibitor superfamily, cause Nagashima-type palmoplantar keratosis. Am. J. Hum. Genet. 2013;93:945–956. doi: 10.1016/j.ajhg.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin J., Xu G., Wang H., Zhao J., Duo L., Cao X., Tang Z., Lin Z., Yang Y. New and recurrent SERPINB7 mutations in seven Chinese patients with Nagashima-type palmoplantar keratosis. J. Invest. Dermatol. 2014;134:2269–2272. doi: 10.1038/jid.2014.80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.