

Abstract
Background and Purpose
Staphylococcal enterotoxin B (SEB) is a potent activator of Vβ8+T-cells resulting in the clonal expansion of ∼30% of the T-cell pool. Consequently, this leads to the release of inflammatory cytokines, toxic shock, and eventually death. In the current study, we investigated if Δ9tetrahydrocannabinol (THC), a cannabinoid known for its anti-inflammatory properties, could prevent SEB-induced mortality and alleviate symptoms of toxic shock.
Experimental Approach
We investigated the efficacy of THC against the dual administration (intranasal and i.p.) of SEB into C3H/HeJ mice based on the measurement of SEB-mediated clinical parameters, including cytokine production, cellular infiltration, vascular leak, and airway resistance. In addition, the molecular mechanism of action was elucidated in vitro by the activation of splenocytes with SEB.
Key Results
Exposure to SEB resulted in acute mortality, while THC treatment led to 100% survival of mice. SEB induced the miRNA-17-92 cluster, specifically miRNA-18a, which targeted Pten (phosphatase and tensin homologue), an inhibitor of the PI3K/Akt signalling pathway, thereby suppressing T-regulatory cells. In contrast, THC treatment inhibited the individual miRNAs in the cluster, reversing the effects of SEB.
Conclusions and Implications
We report, for the first time a role for the miRNA 17–92 cluster in SEB-mediated inflammation. Furthermore, our results suggest that THC is a potent anti-inflammatory compound that may serve as a novel therapeutic to suppress SEB-induced pulmonary inflammation by modulating critical miRNA involved in SEB-induced toxicity and death.
Tables of Links
| TARGETS | |
|---|---|
| GPCRsa | Enzymesd |
| CB1 receptor | Akt (PKB) |
| CB2 receptor | CASP2 (caspase 2) |
| Nuclear hormone receptorsb | CASP8 (caspase 8) |
| RORG (RAR-related orphan receptor-γ) | ERK |
| Catalytic receptorsc | JAK |
| Death receptor | MAPK |
| IL1RL1 | PI3K |
| IL6ST | PIK3R1 |
| TGFBR2 | PTEN |
| TGFBR3 | |
| TNFRSF1B |
| LIGANDS | |
|---|---|
| Cannabidiol (CBD) | MCP-1 (CCL2) |
| Dexamethasone | Rapamycin |
| IFN-γ | TGF-β |
| IL-2 | THC |
| IL-6 | TNF-α |
| IL-10 | TNFSF11 |
| IL-12 | VEGF-A |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c,dAlexander et al., 2013a,b,c,d).
Introduction
Staphylococcal enterotoxin B (SEB) is a potent activator of the immune system resulting in the clonal expansion of 5–30% of the T-cell pool and massive release of cytokines (Choi et al., 1989; Faulkner et al., 2005). As a consequence, it is associated with a number of diseases ranging from food poisoning, multi-organ failure and lethal toxic shock (Dinges et al., 2000; Alouf and Muller-Alouf, 2003; Larkin et al., 2009). When SEB is inhaled, the combination of cellular infiltration and cytokine production, results in vascular leak, pulmonary oedema, tissue damage and eventually acute inflammatory lung injury (Saeed et al., 2012; Rao et al., 2014). Due to its ability to be easily aerosolized and for its possible role as a biological weapon, SEB is considered a Center for Disease Control and Prevention – Category B select agent (Ulrich et al., 2001). While it is known that the interaction of SEB with the T-cell receptor (TCR) results in the activation of inflammatory pathways such as the PI3K, MAPK and NFκB (Krakauer, 2013), a recent study from our laboratory has suggested that microRNA (miRNA) may play an important role in SEB-mediated inflammation (Rao et al., 2014).
miRNA are ∼22 nucleotide, small, non-coding RNA that target mRNA, leading to its degradation and/or translational repression (Guo et al., 2010). Consequently, they control the development and differentiation of various immune cells thereby leading to the regulation of immune responses (Lindsay, 2008). Upon receiving inflammatory signals, active changes occur within the transcriptional repertoire accompanied with altered expression of a number of miRNA resulting in the up- or down-regulation of several important genes (O'Connell et al., 2012). One such modulator of gene expression is the miR-17-92 cluster. Originally recognized as an oncogenic miRNA, this cluster that comprises of six miRNA (miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1 and miR-92a-1) is known to be an important regulator of B and T-cell responses (Olive et al., 2010). For example, miR-17-92 transgenic mice that express the cluster in both B and T-cells develop lymphoproliferative disease (Xiao et al., 2008). In addition, miR-19b and miR-17 within the cluster are reported to regulate CD4 + T-cells and enhance Th1 responses (Liu et al., 2014), thereby demonstrating an important role for the cluster during inflammation.
Although current therapeutic strategies against SEB include the use of non-specific human immunoglobulins (intravenous immunoglobulins or IVIg) as well as monoclonal antibodies against SEB-induced cytokines (Miethke et al., 1992; Matthys et al., 1995; Larkin et al., 2009, 2010) its efficacy remains inefficient (Darenberg et al., 2004). Additionally, corticosteroids have been shown to attenuate SEB-induced toxic shock and acute lung injury (Krakauer and Buckley, 2006; Huzella et al., 2009), but the immunosuppressive property of corticosteroids is either accompanied by a number of side effects or have remained ineffective clinically (Bernard et al., 1987; Meduri et al., 1998; Wajanaponsan et al., 2007). As a result, there is a need for alternative agents that mitigate SEB-triggered inflammation with the potential to modulate SEB-induced inflammatory miRNA.
Δ9-Tetrahydrocannabinol (THC) is a marijuana plant-derived cannabinoid known for its robust anti-inflammatory and immunosuppressive properties (Klein, 2005; Nagarkatti et al., 2009, 2010). It mediates its action by binding to two main cannabinoid receptors, CB1 and CB2, found primarily in the brain and on immune cells respectively (Felder and Glass, 1998). Previously, we have demonstrated that THC induces apoptosis in Jurkat leukemia T-cells and dendritic cells (Do et al., 2004). THC suppresses the production of Th1 cytokines IFN-γ and TNF-α (Klein et al., 1995; Srivastava et al., 1998; Klein, 2005; Sun et al., 2008), while increasing Th2 cytokines, IL-10 and TGF-β (Sun et al., 2008). Additionally, while we have earlier reported that THC induces the production of myeloid derived suppressor cells (MDSCs; Hegde et al., 2010),we have for the first time demonstrated that THC-mediated miRNA control the development of these MDSCs (Hegde et al., 2013). Taken together, THC, by virtue of its anti-inflammatory and immunosuppressive properties and its recently discovered ability to regulate miRNA expression could serve as an effective therapeutic agent in the attenuation of SEB-mediated lung injury.
Thus, in the current study, we tested the hypothesis that THC treatment ameliorates SEB-induced toxicity through regulation of miRNA. Our data demonstrate that THC treatment down-regulates the members of the miR-17-92 cluster and ameliorates inflammatory symptoms associated with SEB exposure in the lungs.
Methods
Mice
Female C3H/HeJ mice (6–8 weeks) were purchased from The Jackson laboratory. All mice were housed at the Animal Resource Facility (ARF), University of South Carolina, under specific pathogen free conditions with a maximum of five animals per cage. Animals were kept under 12 light/12 dark cycle at a temperature of ∼18–23 °C and 40–60% humidity. Food and water were available ad libitum. All experiments involving the use of vertebrate animals were conducted under protocols approved by the Institutional Animal Care and Use Committee (IACUC) at USC and the US National Research Council's ‘Guide for the Care and Use of Laboratory Animals’. A total of 80 animals were used in the experiments described here. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
SEB administration and THC treatment schedule
THC was provided by National Institute on Drug Abuse (Bethesda, MD, USA) and SEB was procured from Toxin Technologies (Sarasota, FL, USA). The treatment schedule comprised of injecting vehicle or THC i.p. at a concentration of 20 mg·kg−1 body weight. The rationale behind administering the current dose of THC is based on body surface area normalization (Reagan-Shaw et al., 2008), whereby 20 mg·kg−1 THC dose converts to 60 mg·m−2, which is within the range of synthetic THC used in the clinic (90 mg·m−2·day−1). THC or vehicle was first administered in a 100 μL volume dissolved in ethanol (day 1). The following day (day 2), THC (20 mg·kg−1) was once again administered via the i.p. route. Thirty minutes later, SEB was delivered as a ‘Dual Dose’ as described previously (Huzella et al., 2009). Briefly, SEB dissolved in sterile PBS (2 mg·mL−1) was administered first by the intranasal (i.n.) route at a concentration of 5 μg per mouse in a volume of 25 μL. Two hours later, a second dose of SEB was delivered i.p. at a concentration of 2 μg per mouse in a 100 μL volume. On day 3, mice were treated i.p. with THC (20 mg·kg−1). Survival of mice was monitored up to 5 days after SEB exposure and any moribund mice were immediately killed.
Assessment of lung damage and function
Vascular leak in the lungs was determined as described previously (Rieder et al., 2011, 2012; Saeed et al., 2012). Briefly, mice were injected with 1% Evans blue in sterile PBS i.v., 72 h after the second dose of SEB. Two hours later, mice were killed and lungs were perfused with heparin-containing PBS. Lungs were incubated in formamide at 37°C for 24 h to extract the dye. The OD of the supernatant was measured spectrophotometrically at 620 nm and % increase in vascular leak was calculated using the following formula – (ODsample − ODcontrol/ODcontrol) × 100. Airway resistance was measured using whole-body plethysmography (Buxco, Troy, NY, USA). Each mouse was restrained in a two-chamber plethysmographic tube and was first allowed to acclimatize, followed by exposure to saline for 2 min. This was followed by a 2 min exposure to increasing doses of methacholine. The specific airway resistance (sRaw) measurement at each methacholine dose was calculated and plotted as % airway resistance. To examine lung morphology and histology, lungs were fixed in 10% formalin, paraffin embedded and stained with haematoxylin and eosin. The slides were observed under a light microscope at 20× magnification.
Cell preparation and flow cytometry
Vehicle, SEB + vehicle and SEB + THC mice (five mice per group) were killed 72 h after dual exposure to SEB. The lungs were perfused with heparin-containing PBS, harvested and homogenized using Stomacher® 80 Biomaster blender from Seward (Davie, FL, USA) in 10 mL of sterile PBS. After being washed with sterile PBS, cells were layered carefully onto Ficoll-Histopaque ®-1077 from Sigma-Aldrich (St Louis, MO, USA) at a 1:1 ratio. Mononuclear cells were separated by density gradient centrifugation as a distinct layer as described previously (Rieder et al., 2012; Rao et al., 2014) and enumerated by Trypan blue exclusion. To determine the phenotypical characteristics of the infiltrating cells, mononuclear cells were stained with the following fluorescent conjugated antibodies – FITC-conjugated anti-CD8 (clone: 53-6.7), anti-CD3 (clone: 145.2 C11). Phycoerthyrin (PE)-conjugated anti-CD4 (clone: GK 1.5), anti NK1.1 (clone: PK136) from Biolegend (San Diego, CA, USA). FITC-conjugated anti-Vβ8 (clone: K516) from Ebioscience (San Diego, CA, USA). Intracellular staining of Foxp3 was carried out using Biolegend's Foxp3 Fix/Perm buffer set following manufacturer's instructions and using anti-foxp3 alexa flour 488 (clone MF-14) from Biolegend.
Cytokine analysis
To assess serum cytokines, mice were bled 3 h after dual exposure to SEB. Cytokines from the broncheoalveolar lavage fluid (BALF) were obtained at the time by binding the trachea with a suture and excising the lung along with the trachea, as described previously (Rieder et al., 2012; Rao et al., 2014). Sterile, ice-cold PBS was injected through the trachea to aspirate the fluid. The samples were centrifuged to obtain the supernatants. All cytokine levels were measured using Biolegend elisa MAX™ standard kits.
miRNA target predictions and transfections
miRNA target candidate Pten was predicted using Ingenuity Pathway Analysis (IPA) software from Ingenuity Systems® (Mountain View, CA, USA). Briefly, highly predicted and experimentally observed targets of the individual miRNA in the miR-17-92 cluster were selected. A core analysis was carried out and significant (Fisher's exact test) biological functions associated with the data set were generated. Additionally, a bar graph highlighting key canonical pathways associated with the data set was also generated. miRSVR score and alignment of miR-18a with Pten was obtained from www.microRNA.org, target prediction website. To validate Pten as a target of miR-18a, splenocytes from naïve C3H/HeJ mice were harvested and cultured in complete (10% FBS, 10 mM L-glutamine, 10 mM HEPES, 50 μM β-mercaptoethanol and 100 μg·mL−1 penicillin) RPMI 1640 medium (Gibco Laboratories, Grand Island, NY, USA). Cells were seeded at 2 × 105 cells per well in a 24-well plate and transfected for 24 h with 40 nM synthetic mmu-miR-18a (MSY0000528) or mock transfected with HiperFect transfection reagent from Qiagen (Valencia, CA, USA). For inhibition of miR-18a, SEB-activated cells were similarly transfected for 24 h with 100 nM synthetic mmu-miR-18a (MIN0000528) or mock transfected.
Total RNA extraction and qRT-PCR
Total RNA (including small RNA) was isolated from lung-infiltrating mononuclear cells or in vitro from splenocytes using miRNeasy kit from Qiagen following the manufacturer's instructions. The purity and concentration of the RNA was confirmed spectrophotometrically using Nanodrop 2000c from Thermo Scientific (Wilmington, DE, USA). For miRNA validation and quantification, we used SYBR Green PCR kit (Qiagen) and for mRNA validation, SSO Advanced™ SYBR green PCR kit from Biorad (Hercules, CA, USA). Fold change of miRNA was determined by normalization to Snord96_an internal control, whereas mRNA levels were normalized to β-actin. The following qRT-PCR primers were used: β-actin (F) 5'GGCTGTATTCCCCTCCAT G-3′ and (R) 5′-CCAGTT GGTAACAATGCCATGT-3′; Foxp3 (F) 5′ AGCAGTCCACTTCACCAAGG 3′ and (R) 5′ GGATAACGCCAGAGGAGCTG 3′; Pten (F) 5′ TGGATTCGACTTAGACTTGACCT 3′ and (R) 5′ GCGGTGTCATAATGTCTCTCAG 3′.
In vitro cell culture assays
Splenocytes from naïve C3H/HeJ mice were harvested and cultured in complete RPMI. Cells were seeded at 1 × 106 cells per well of a 96-well plate and either left unstimulated or stimulated with SEB (1 μg·mL−1). Cell were either treated with THC or with an allosteric Akt 1/2 kinase inhibitor (A6730), that is pleckstrin homology (PH) domain dependent and does not have an inhibitory effect against PH domain lacking Akts, or related kinases (Sigma-Aldrich) at the doses indicated. Twenty-four hours later, cells were harvested and centrifuged. The cell supernatants were collected for assessment of IFN-γ levels by elisa and the cell pellets were used for total RNA extraction and qRT-PCR. To determine the effect of other immunosuppressive compounds on the miR-17-92 cluster, SEB-activated splenocytes were treated with cannabidiol (CBD) obtained from the National Institute on Drug Abuse (Bethesda, MD, USA), dexamethasone (Dexa) (#D4902) and rapamycin (Rapa) (#R8781) from Sigma. Cell proliferation was determined by incubating the cells as described above for 48 h. [3H]-thymidine (2μCi) was added to the cell cultures in the last 12 h of incubation. Cultures were collected using a cell harvester and thymidine incorporation was measured using a scintillation counter (Perkin Elmer, Waltham, MA, USA).
Western blots
SEB-activated splenocytes were treated with THC (20 μM) for 18 h and protein extracts (∼15 μg) were separated on a 10% SDS-PAGE by electrophoresis (60V for ∼2 h). Separated protein was transferred onto a nitrocellulose membrane. The membrane was probed with antibodies against pan-Akt (#4685), phosphor-Akt-Ser473 ($9271S), β-actin 13E5 (#4970) from Cell Signaling Technology® (Danvers, MA, USA) and phosphatase and tensin homologue (PTEN) (#SC 6817-R) from Santa Cruz Biotechnology®, Inc (Dallas, TX, USA).
Statistical analysis
All statistical analyses were carried out using GraphPad Prism Software (San Diego, CA, USA). In all experiments, the number of mice used was 4–5 per group, unless otherwise specified. Results are expressed as means ± SEM. Student's t-test was used to compare the two groups, whereas multiple comparisons were made using one-way anova, followed by post hoc analysis using Tukey's method. A P-value of <0.5 was considered statistically significant. Individual experiments were performed in triplicate and each experiment was performed independently at least three times to test reproducibility of results. Survival analysis was carried out using a log-rank test.
Results
THC strongly attenuates SEB-mediated inflammation and prevents acute mortality
Dual SEB exposure has been previously used to study acute lung injury leading to 100% death in C3H/HeJ mice (Huzella et al., 2009). In the current study, we found that 100% of the mice exposed to SEB died between 96 and 120 h. Remarkably, in the THC-treated groups, all mice survived (Figure 1A). SEB-exposed mice displayed signs of lethargy, hunching, ruffled fur and respiratory stress, whereas THC-treated mice were as active as vehicle-treated mice and did not display hunched posture, ruffling of fur or signs of respiratory distress. To gauge the extent of pulmonary damage, we measured airway resistance using whole-body plethysmography and found that SEB exposure resulted in a significant percent increase in airway resistance, while THC-treated mice recorded sRAW values similar to vehicle alone (Figure 1B). Further, we measured the percent increase in vascular leak by administration of Evans blue dye. Evans blue binds to serum albumin and is a measure of vascular permeability, as shown previously (Rieder et al., 2012). Our results demonstrated that while SEB exposure had a profound increase in vascular leak when compared with vehicle only, THC treatment caused a significant decrease in vascular leak (Figure 1C). Acute inflammatory lung injury is characterized by massive immune cell infiltration into the lung. Accordingly, we found an increase in the total number of mononuclear cells after SEB exposure and a subsequent decrease with THC treatment (Figure 1D). This was confirmed by histopathological examination of the lungs, whereby SEB-exposed mice displayed an increase in infiltrating immune cells around the bronchioles and air vessels while the THC-treated mice, yielded significantly fewer layers (Figure 1E). To identify the immune subsets among the infiltrating mononuclear cells, we stained the cells with various fluorescein-conjugated anti-mouse antibodies. We found that exposure to SEB resulted in increased CD3+ (T-cells), CD4+ (T-helper cells), CD8+ (cytotoxic T-cells), Vβ8+ NK+ (NK cells) and NK1.1 + CD3+ (NK T-cells), THC treatment caused an overall decrease in the absolute cell numbers (Figure 1F).
Figure 1.

THC prevents mortality and alleviates SEB-induced inflammation in the lung. (A) Survival curve of mice receiving SEB+ vehicle when compared with mice treated with SEB+THC. (B) Measurement of airway hyperreactivity (sRAW) using whole-body plethysmography. (C) Assessment of vascular leak in the lungs; % vascular leak was calculated by measuring absorbance at 620 nm. (D) Histopathological examination of lungs as determined by haemotoxylin and eosin staining. Total layers of infiltrating cells were counted around 10 different capillaries and enumerated in the bar graph. (E) Total number of infiltrating mononuclear cells obtained from the lung was enumerated by trypan blue exclusion method. (F) Flow cytometric analysis to identify immune subsets was carried out. Mononuclear cells were stained with antibodies against T-cells (CD3), T-helper cells (CD4), cytotoxic T-cells (CD8), Vβ8-region of the T-cell receptor (Vβ8), NK cells and natural killer T-cells (NKT). Absolute cell numbers were calculated using the formula: total number of cells isolated from the lungs × % of specific cells/100, and plotted as a bar graph. Bar graphs summarize the means ±SEM from 3–5 independent experiments. All experiments above were carried out 72 h after exposure to SEB. Statistical significance is indicated as follows: *P < 0.005; **P < 0.01.
A hallmark of SEB-mediated inflammation is the abundant release of cytokines. To determine if THC was able to blunt cytokine secretion, we first analysed the concentration of early cytokines IL-2 and MCP-1 in the serum. Mice were bled at 3 h, 6 h and 24 h after SEB exposure. While IL-2 and MCP-1 peaked at 3 h (data not shown), we found that the THC-treated group showed diminished secretion of both IL-2 and MCP-1 as early as 3 h after SEB exposure (Figure 2A), supporting the potent anti-inflammatory role of THC in this model. Moreover, an examination of cytokines in the BALF revealed that THC treatment led to the substantial decrease in IFN-γ, IL-6, IL-12 and IL-10 (Figure 2B). Overall, these data suggest that THC attenuates SEB-induced immune cell infiltration, decreases early and late cytokine secretion, and prevents mortality of the mice.
Figure 2.

THC decreases SEB-induced cytokine secretion. (A) Measurement of early cytokines, IL-2 and MCP-1 in serum 3 h after SEB exposure. (B) Measurement of IFN-γ, IL-12, IL-10 and IL-6 in the BALF. All cytokine concentrations were determined using elisa. Bar graphs summarize the means ±SEM from 3–5 independent experiments. For cytokines from the serum, unpaired, two-tailed t-test was used to determine significance from SEB. For cytokines from the BALF, one-way anova, followed by post hoc analysis using Tukey's method was used.
THC modulates the expression of the miR-17-92 cluster
Antigenic stimulation and the activation of the TCR is known to result in the induction of miR-17-92 cluster (Wu et al., 2012). Consequently, we reasoned that SEB exposure would lead to the expression of this prominent miRNA cluster. Seventy-two hours after exposure to SEB, we measured miRNA expression in mononuclear cells isolated from the lung by qRT-PCR. Interestingly, we observed high levels (up to 600-fold) of the miRNA cluster, although individual miRNA were induced to different levels. More interestingly, THC treatment was able to down-regulate the individual members of the cluster significantly (Figure 3), strongly suggesting that THC may exhibit its powerful anti-inflammatory activity through the modulation of inflammatory miRNA. In order to establish that the ability to modulate the cluster was unique to THC alone, we also compared the effect of other known immunomodulatory compounds dexamethasone, rapamycin and CBD in vitro on the cluster. We observed that while CBD only significantly down-regulated miR-19a and miR-20a, dexamethasone only affected miR-19a and miR-19b-1 and rapamycin, decreased levels of miR-19b-1, miR-92a-1 and miR-17 suggesting that while these compounds may affect a few miRNA derived from the cluster via a mechanism distinct from THC, they are unable to modulate the entire cluster (Supporting information Figure S1).
Figure 3.
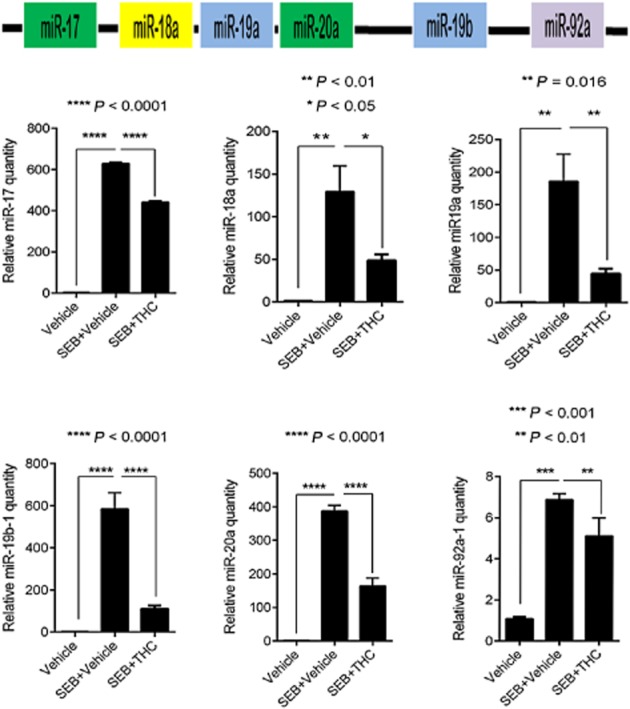
THC significantly down-regulates SEB-induced expression of the miR-17-92 cluster. Real-time (RT) PCR validation of the individual miRNA (miR-17, miR-18a, miR-19a, miR-19b-1, miR20a and miR-92a-1) of the miR-17-92 cluster obtained from lung-infiltrating mononuclear cells. Data are normalized to internal control Snord_96a. Statistical significance was assessed using anova, Tukey's multiple comparison test.
miR-17-92 cluster is linked to the activation of the PI3K/Akt/pathway
Because SEB exposure resulted in the strong induction of the miR-17-92 cluster, we sought to explore the significance of this particular cluster in our study. Therefore, we carried out computational analysis on the highly predicted and experimentally observed targets of the cluster (miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1 and miR-92a-1) using IPA. IPA results suggested that the members of the cluster were involved in a number of biological functions relevant to the expansion of T-helper lymphocytes, the development of regulatory T-cells (T-regs), the proliferation of cells and apoptotic processes (Figure 4A). In addition, analysis of the canonical pathways associated with the cluster indicated the involvement of the MAPK, NFκB and PI3K/Akt signalling pathways, (processes that are all reported to be SEB-triggered). Interestingly, while the PTEN signalling and mTOR pathways were highlighted by IPA as significant (Figure 4B), pathway analysis also indicated that the miRNA in the cluster are highly predicted or experimentally observed to converge on Pten (Figure 4C). As a result, we reasoned that the SEB-induced miRNA cluster is involved in the activation of the PI3K/Akt signalling pathway while THC-mediated down-regulation of this cluster inhibits the aforementioned activation. To test if the cluster was indeed involved in the activation of the PI3K/Akt pathway, we activated splenocytes with SEB and treated them with an Akt1/2 inhibitor. Among the individual members of the cluster that were up-regulated by SEB activation, miR-18a in particular was significantly down-regulated upon Akt1/2 inhibition, while the other members of the cluster either remained unaltered, were insignificantly down-regulated or found to be further up-regulated after Akt1/2 inhibition (Figure 5A). This indicated the direct involvement of miR-18a in SEB-mediated activation of the PI3K/Akt pathway.
Figure 4.

The involvement of the miR-17-92 cluster in key biological pathways. (A) Graphical representation of the biological functions associated with significantly up-regulated SEB-induced miRNA as determined by IPA. (B) Canonical pathways associated with miRNA target genes. IPA was used to filter highly predicted and experimentally observed targets of only the significantly up-regulated miRNA in response to SEB. A graphical representation of the significant (Fisher's exact test) pathways of these particular target genes was generated. (C) IPA pathway demonstrating the convergence of the members of the miR-17-92 cluster on Pten.
Figure 5.
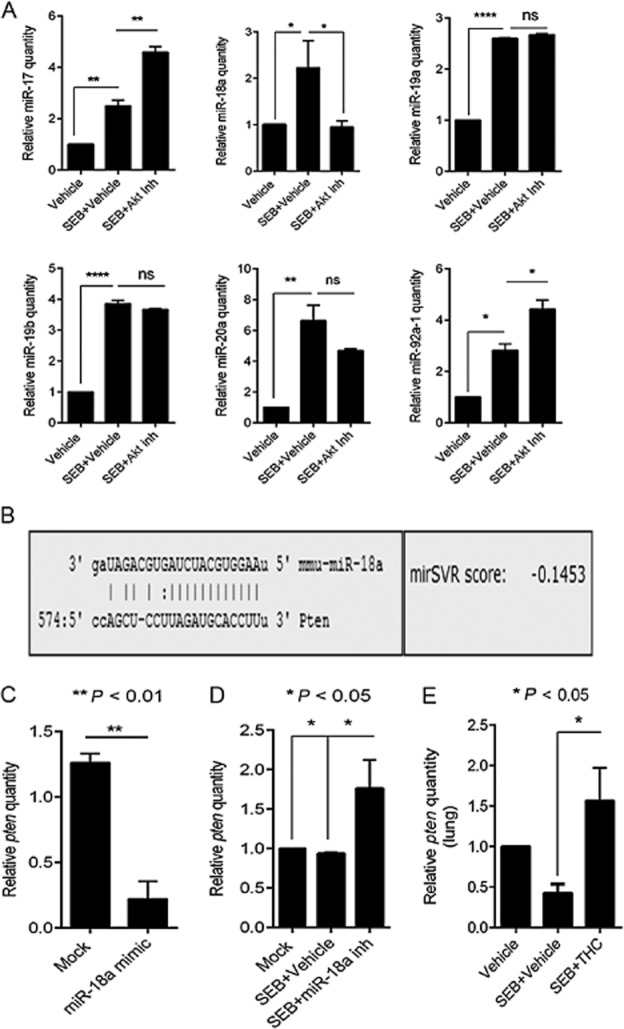
miR-18a targets Pten, an inhibitor of the PI3K/Akt pathway. (A) RT-PCR of members of the miR-17-92 cluster in splenocytes that were activated with SEB and treated with AKT inhibitor (20 μM). (B) Target prediction of miR-18a using miRanda (www.microrna.org), showing the alignment of the mature miRNA to the 3′ UTR of Pten mRNA. The miRSVR score represents the probability of mRNA target down-regulation and the cut-off for a good score was set at ≤ −0.01. (C) RT-PCR quantification of miR-18a and its target Pten. Splenocytes were transfected with 40 nmol of a miR-18a mimic for 24 h. (D) RT-PCR quantification of miR-18a and Pten after transfection with a synthetic miR-18a inhibitor. Splenocytes that were activated with SEB (1 μg·mL−1) were also transfected with miR-18a inhibitor for 24 h. (E) Real-time PCR quantification of Pten levels in lung-infiltrating mononuclear cells. All miRNA levels were measured relative to internal control Snord_96a and mRNA levels were normalized to β-actin. Statistical significance was assessed using anova, Tukey's multiple comparison test.
miR-18a targets PTEN, a negative regulator of the PI3K/Akt pathway
Previous reports have established that a principal target of the miR-17-92 cluster is PTEN, an antagonist of the PI3K/Akt pathway (Xiao et al., 2008; Liu et al., 2014). Using the www.miRNA.org alignment tool, we found that miR18a is predicted to target the 3′ UTR of Pten with a good miRSVR score of −0.1453 (Figure 5B). To assess if miR-18a indeed targets Pten, we first transfected splenocytes with synthetic miR-18a mimic. Interestingly, we found that miR-18a mimic repressed Pten (Figure 5C). Further, the inhibition of SEB-activated cells with a miR-18a synthetic inhibitor led to the derepression of Pten (Figure 5D), suggesting that miR-18a, belonging to the cluster, plays a prominent role in the repression of Pten and consequently results in the activation of SEB-triggered PI3K/Akt pathway. Earlier, we observed that THC was able to dramatically decrease the expression of the miRNA cluster (Figure 3). Therefore, we wondered if THC treatment would be able to subsequently restore the SEB-induced suppression of Pten. Upon assessing Pten expression in lung-infiltrating mononuclear cells by qRT-PCR, we observed that while SEB exposure indeed resulted in the repression of Pten, THC treatment led to its increase (Figures 5E and 6A). Taken together, these data indicated that THC, via down-regulation of miR-18a, leads to the release of Pten and in doing so, may act as an Akt inhibitor.
Figure 6.

THC is an inhibitor of the Akt pathway and leads to the induction of CD4+Foxp3+ T-regulatory cells. (A) IFN-γ levels in supernatants of splenocytes that were activated with SEB and treated with varying doses of THC (5 μM,10 μM and 20 μM) or Akt inhibitor (5 μM,10 μM or 20 μM) as indicated. (B) Thymidine incorporation to measure proliferation of splenocytes activated with SEB or treated with the indicated doses of THC (5 μM, 10 μM and 20 μM) or Akt inhibitor (5 μM,10 μM or 20 μM). (C) RT-PCR of Foxp3 levels in splenocytes that were treated either with THC (20 μM )or Akt inhibitor (20 μM). mRNA levels measured relative to β-actin. (D) Flow cytometric analysis of CD4+Foxp3+ T-regulatory cells in lung-infiltrating mononuclear cells for the groups indicated. The bar graph represents the RT-PCR expression of Foxp3 in lung-infiltrating mononuclear cells. In all in vitro experiments, splenocytes were activated with 1 μg·mL−1 SEB. Statistical significance was assessed using anova, Tukey's multiple comparison test.
THC functions as an Akt inhibitor
The activation of the PI3K/Akt pathway leads to cellular proliferation, the release of IFN-γ and inhibition of T-regulatory cells. Our results suggested that by antagonizing the PI3K/Akt axis via the down-regulation of miR-18a and the subsequent release of Pten, THC may mimic the properties of an Akt inhibitor. To confirm this notion, we activated splenocytes for 18 h with SEB or treated cells with THC and conducted a Western blot. We found a reduction in phosphorylated Akt in THC-treated cells when compared with SEB activation (Figure 6A). Next, we compared the properties of THC with an Akt inhibitor on IFN-γ production in vitro. Splenocytes were activated with SEB and treated with THC or Akt inhibitor. The resulting concentration of IFN-γ was measured by elisa. As expected, Akt inhibition led to the dose-dependent decrease of IFN-γ (Figure 6B). Interestingly, THC treatment also demonstrated a trend in the dose-dependent decrease of IFN-γ, with a significant blunting of this cytokine at the highest dose (Figure 6C). Further, when cellular proliferation of cells was assayed by thymidine incorporation, we observed a similar dose-dependent decrease in cellular proliferation (Figure 6D) with THC and the Akt inhibitor. Moreover, the interruption of Akt signalling results in the induction of CD4+Foxp3+ T-regulatory cells (Sauer et al., 2008; Merkenschlager and von Boehmer, 2010). Upon activating splenocytes with SEB in vitro and treating with either the Akt inhibitor or THC, we found significant induction of Foxp3 (Figure 6E) compared with SEB alone. The induction was further confirmed by flow cytometric analysis and qRT-PCR of lung-infiltrating mononuclear cells (Figure 6E).Taken together, these data indicate that THC inhibits Akt signalling by modulating miR-18a and allowing for the release of Pten. The resulting decrease in cellular proliferation, IFN-γ production and induction T-regulatory cells, prevent SEB-mediated acute inflammatory lung injury and death.
Discussion
In the current study, we investigated the role of THC in preventing SEB-induced inflammatory lung injury and subsequent mortality via the modulation of miRNA. Through the use of qRT-PCR and computational tools, we found that the SEB-induced miRNA 17–92 cluster, which was overexpressed in the lungs, is down-regulated with THC treatment. Specifically, by performing gain-and loss-of function analysis using synthetic mimic and inhibitor, we confirmed the predominant role of miR-18a in the SEB-mediated activation of the PI3K/Akt pathway. Our studies also suggested that THC may function as an antagonist of the Akt pathway, at least in part, by its ability to significantly decrease the expression of miR-18a. Our results highlight the role of miRNA in facilitating severe inflammation and the ability of cannabinoids to suppress its expression. Importantly, we show that the previously reported potent anti-inflammatory role of THC can be explained, at least in part, by its ability to act upon SEB-mediated miRNA.
SEB specifically expands a large number of T-cells, by virtue of engaging the Vβ8 region of the TCR following binding to the MHC II on antigen presenting cells (Choi et al., 1989; Hurley et al., 1995). Consequently, SEB exposure leads to the massive release of inflammatory cytokines, proliferation of T-cells, tissue damage and SEB-mediated shock (Miethke et al., 1992; DeVries et al., 2011; Kissner et al., 2011). Most models developed to study the effects of SEB exposure in mice, have employed the use of transgenic mice or external agents such as LPS or D-galactosamine to potentiate SEB-mediated inflammatory response (DaSilva et al., 2002; Savransky et al., 2003). In the dual administration of SEB as used in this study, microgram quantities of SEB were sufficient to cause inflammatory symptoms and toxicity reminiscent of SEB exposure in humans (Huzella et al., 2009). In this model, mice succumb to SEB-mediated shock and severe respiratory damage (Huzella et al., 2009). Consistent with these reports, we observed an overall increase in mononuclear cells in the lung, particularly T-cells. We also found SEB-mediated release of early monocyte and T-cell recruiting cytokines IL-2 and MCP-1 in the serum. Additionally, we noted the abundant release of late cytokines, especially IFN-γ in the BALF of the lungs resulting in compromised vascular permeability and the ultimate death of mice.
Earlier studies have reported that the molecular mechanism of action behind SEB-mediated inflammation and toxicity begins soon after the activation of the TCR (Goldbach-Mansky et al., 1992; Linsley and Ledbetter, 1993). Following the increase in co-stimulatory molecules, a number of signalling pathways such as the MAPK, ERK, JNK and PI3K/Akt are simultaneously activated (Krakauer, 2013). These pathways culminate in the stimulation of various transcription factors such as NFκB and NFAT (Trede et al., 1995; Tsytsykova and Goldfeld, 2000). However, with the recent discovery of miRNA, our understanding of the molecular mechanisms that govern gene regulation has been revolutionized. It is now evident that these small single-stranded RNA molecules are capable of targeting the 3′ UTR of mRNA thereby regulating biological processes such as cellular proliferation, differentiation and development (Davidson-Moncada et al., 2010). miRNA are also induced upon a number of inflammatory cues and subsequently influence immune responses and immune cell development (Dai and Ahmed, 2011).
The miR-17-92 cluster found on mouse chromosome 14 is among the many miRNA that are found to be overexpressed under inflammatory conditions (Sonkoly and Pivarcsi, 2009). It is transcribed as a single polycistronic unit and comprises of six miRNA – miR-17, miR-18a, miR-19a, miR-19b-1, miR-20a and miR-92-1 (Wu et al., 2012). Previously, it has been shown that naïve CD4+ T-cells polarized under Th1 inducing conditions show a significant increase in the miR-17-92 cluster. (Sasaki et al., 2010) Further, a recent report demonstrated the critical role of the cluster in the expansion of T-cells upon antigenic stimulation, although they observed that individual miRNA within the cluster have differential roles in promoting Th1 responses. (Liu et al., 2014). Prompted by these reports, we first investigated the role of miRNA in facilitating SEB-mediated lethality in mice and found that SEB exposure led to the overexpression of the miR-17-92 cluster in lung-infiltrating mononuclear cells. Similar to previous studies, we also found that the members of the cluster, were expressed to different levels, suggesting that one or more miRNA within the cluster may play a more prominent role.
It was reported that the overexpression of miR-17-92 cluster causes lymphoproliferation, autoimmunity and premature death of mice by targeting Pten, a well-established antagonist of the PI3K/Akt pathway (Xiao et al., 2008). In the present study, we demonstrate that Pten is suppressed after SEB exposure. Whereas previous studies have shown a primary role for miR-19b in the targeting of Pten (Olive et al., 2009; Liu et al., 2014), we attribute the SEB-mediated suppression of Pten to the predominant role of miR-18a. These data are surprising because while we demonstrate a clear reversal in the suppression of Pten using a miR-18a inhibitor, a few reports have demonstrated its role in inhibiting cellular proliferation (Tsang and Kwok, 2009). It is possible that the functional targets of miRNA may differ based on the type of antigenic stimulation. Alternatively, the critical timing of SEB dual administration could contribute to the specific regulation of Pten by miR-18a. Moreover, it is also possible that because the cluster comprises of six miRNA with several predicted target genes, Pten is simply one of many genes simultaneously targeted during the course of inflammation. Nevertheless, our results show a clear induction of the miR-17-92 cluster and specifically the suppression of Pten by miR-18a.
The anti-inflammatory and immunosuppressive effects of THC are diverse and function effectively to abrogate a number of inflammatory processes. For example, THC has previously been reported to prevent the development of a murine model of multiple sclerosis and colitis (Lyman et al., 1989; Jamontt et al., 2010). In a mouse model of Con-A induced hepatitis, we have demonstrated its ability to act on acute inflammation, where it not only decreased the production of inflammatory cytokines, but also reduced cellular proliferation (Hegde et al., 2008). Consistent with these reports, our data demonstrate a significant reduction in infiltrating immune cells in the lung, blunting of cytokines as early as 3 h after SEB exposure and more astonishingly, the 100% survival of mice.
We have previously demonstrated that THC's anti-inflammatory properties can be credited, in part, to the induction of immunosuppressive cells such as T-regs and MDSC (Hegde et al., 2008, 2010). Recently, we have identified a novel role for THC in modulating miRNA involved in the development of MDSCs (Hegde et al., 2013), a finding that could shed mechanistic light on THC's mode of action. Thus, we rationalized that THC could potentially exert its strong anti-inflammatory activities by modulating SEB-induced miRNA. Our present findings validate our hypothesis and we observed a potent down-regulation of the miR-17-92 cluster. Interestingly, CBD, a cannabinoid also derived from the marijuana plant did not decrease all members of the cluster. Similarly immunosuppressive agents dexamethasone and rapamycin, previously shown to rescue C3H/HeJ mice from SEB induced toxicity (Krakauer and Buckley, 2006, 2012), also failed to effectively target the cluster, highlighting the unique property of THC to act specifically on the miR-17-92 cluster. Our results demonstrate the derepression of Pten suggesting that THC, via the down-regulation of the cluster might be an inhibitor of the PI3K/Akt pathway. Interestingly, earlier studies in cancer models have demonstrated that THC disrupts the PI3K/Akt signalling pathway (Greenhough et al., 2007; Leelawat et al., 2010). However, in light of our current observation that it modulates key miRNA, its anti-inflammatory properties via its role as a PI3K/Akt inhibitor are made more evident.
The disruption of the PI3K/Akt/mTOR axis using Akt inhibitors has been previously reported to decrease cellular proliferation, induce apoptosis and decrease IFN-γ production (Shin et al., 2002; Mandal et al., 2005). Further, rapamycin, an inhibitor of mTORC1, specifically diminishes SEB-induced IL-2, IFN-γ and T-cell proliferation (Krakauer et al., 2010). In line with these reports, our results also demonstrated decreased cellular proliferation and IFN-γ production with THC treatment. Additionally, the PI3K/Akt activation of PTEN-deficient T-cells led to the suppression of CD4+Foxp3+ T-cells, which was reversed with PI3K/Akt inhibition (Sauer et al., 2008). Similarly, we observed the induction of T-regs in the lung upon THC treatment, further confirming that THC is an inhibitor of the PI3K/Akt signalling pathway, and that part of its mechanism involves down-regulation of the miR-17-92 cluster, particularly miR-18a.
Taken together, our data demonstrate that THC is a strong anti-inflammatory agent capable of rescuing mice from SEB-mediated toxicity and death. By affecting the SEB-induced miR-17-92 cluster, it restores Pten, and enables the proper regulation of the PI3K/Akt signalling pathway. Consequently, it reduces cellular proliferation, diminishes the production of pro-inflammatory cytokine and induces T-regs (Figure 7).
Figure 7.
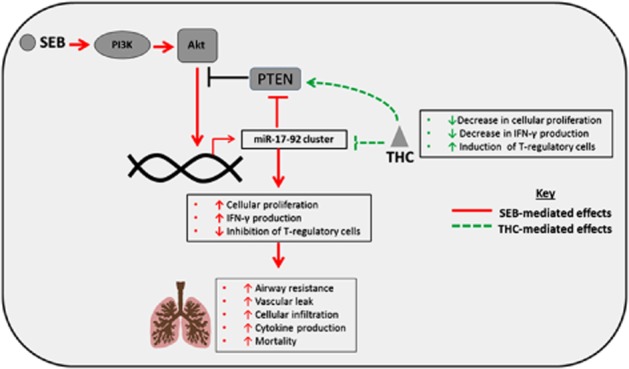
Schematic of the proposed working model. SEB administration leads to activation of the PI3K/Akt pathway via the induction of the miR-17-92 cluster and the subsequent down-regulation of Pten. As a result, SEB exposure leads to increased cellular proliferation, cytokine production, pulmonary damage and acute mortality. The down-regulation of the cluster by THC restores Pten levels and allows for the inhibition of the PI3K/Akt axis. This leads to attenuation of acute inflammatory lung injury and induction of T-regulatory cells, which together prevent SEB-induced mortality.
Acknowledgments
This work was supported by NIH grants P01AT003961, R01AT006888, R01ES019313, R01MH094755, P20RR032684 and VA Merit Award BX001357
Glossary
- miRNA
microRNA
- PTEN
phosphatase and tensin homologue
- SEB
Staphylococcal enterotoxin B
- T-regs
regulatory T-cells
- THC
Δ9tetrahydrocannabinol
Authorship contribution
Statement of author contributions: R. R., M. N., P. S. N. contributed to the design of the experiments. R. R. performed the experiments. R. R., M. N. and P. S. N. analysed the data. R. R., M. N. and P. S. N. contributed to the writing of the manuscript. M. N. and P. S. N. directed the research.
Conflict of interest
The authors have no conflicts to report.
Supporting Information
Figure S1 Regulation of miR-17-92 cluster by THC, dexamethasone (Dexa), rapamycin (Rapa) and cannabidiol (CBD) in vitro. Splenocytes were activated with SEB and treated with THC (20 μM), CBD (20 μM ), dexamethasone (200 nM) and rapamycin (100 nM). Total RNA was extracted and RT-PCR was conducted to measure miRNA levels relative to Snord_96a internal control. Statistical significance was assessed using anova, Tukey's multiple comparison test.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Nuclear hormone receptors. Br J Pharmacol. 2013b;170:1652–1675. doi: 10.1111/bph.12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic receptors. Br J Pharmacol. 2013c;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013d;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alouf JE, Muller-Alouf H. Staphylococcal and streptococcal superantigens: molecular, biological and clinical aspects. Int J Med Microbiol. 2003;292:429–440. doi: 10.1078/1438-4221-00232. [DOI] [PubMed] [Google Scholar]
- Bernard GR, Luce JM, Sprung CL, Rinaldo JE, Tate RM, Sibbald WJ, et al. High-dose corticosteroids in patients with the adult respiratory distress syndrome. N Engl J Med. 1987;317:1565–1570. doi: 10.1056/NEJM198712173172504. [DOI] [PubMed] [Google Scholar]
- Choi YW, Kotzin B, Herron L, Callahan J, Marrack P, Kappler J. Interaction of Staphylococcus aureus toxin ‘superantigens’ with human T cells. Proc Natl Acad Sci U S A. 1989;86:8941–8945. doi: 10.1073/pnas.86.22.8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai R, Ahmed SA. MicroRNA, a new paradigm for understanding immunoregulation, inflammation, and autoimmune diseases. Transl Res. 2011;157:163–179. doi: 10.1016/j.trsl.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darenberg J, Soderquist B, Normark BH, Norrby-Teglund A. Differences in potency of intravenous polyspecific immunoglobulin G against streptococcal and staphylococcal superantigens: implications for therapy of toxic shock syndrome. Clin Infect Dis. 2004;38:836–842. doi: 10.1086/381979. [DOI] [PubMed] [Google Scholar]
- DaSilva L, Welcher BC, Ulrich RG, Aman MJ, David CS, Bavari S. Humanlike immune response of human leukocyte antigen-DR3 transgenic mice to staphylococcal enterotoxins: a novel model for superantigen vaccines. J Infect Dis. 2002;185:1754–1760. doi: 10.1086/340828. [DOI] [PubMed] [Google Scholar]
- Davidson-Moncada J, Papavasiliou FN, Tam W. MicroRNAs of the immune system: roles in inflammation and cancer. Ann N Y Acad Sci. 2010;1183:183–194. doi: 10.1111/j.1749-6632.2009.05121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries AS, Lesher L, Schlievert PM, Rogers T, Villaume LG, Danila R, et al. Staphylococcal toxic shock syndrome 2000–2006: epidemiology, clinical features, and molecular characteristics. PLoS ONE. 2011;6:e22997. doi: 10.1371/journal.pone.0022997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinges MM, Orwin PM, Schlievert PM. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev. 2000;13:16–34. doi: 10.1128/cmr.13.1.16-34.2000. , table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do Y, McKallip RJ, Nagarkatti M, Nagarkatti PS. Activation through cannabinoid receptors 1 and 2 on dendritic cells triggers NF-kappaB-dependent apoptosis: novel role for endogenous and exogenous cannabinoids in immunoregulation. J Immunol. 2004;173:2373–2382. doi: 10.4049/jimmunol.173.4.2373. [DOI] [PubMed] [Google Scholar]
- Faulkner L, Cooper A, Fantino C, Altmann DM, Sriskandan S. The mechanism of superantigen-mediated toxic shock: not a simple Th1 cytokine storm. J Immunol. 2005;175:6870–6877. doi: 10.4049/jimmunol.175.10.6870. [DOI] [PubMed] [Google Scholar]
- Felder CC, Glass M. Cannabinoid receptors and their endogenous agonists. Annu Rev Pharmacol Toxicol. 1998;38:179–200. doi: 10.1146/annurev.pharmtox.38.1.179. [DOI] [PubMed] [Google Scholar]
- Goldbach-Mansky R, King PD, Taylor AP, Dupont B. A co-stimulatory role for CD28 in the activation of CD4+ T lymphocytes by staphylococcal enterotoxin B. Int Immunol. 1992;4:1351–1360. doi: 10.1093/intimm/4.12.1351. [DOI] [PubMed] [Google Scholar]
- Greenhough A, Patsos HA, Williams AC, Paraskeva C. The cannabinoid delta(9)-tetrahydrocannabinol inhibits RAS-MAPK and PI3K-AKT survival signalling and induces BAD-mediated apoptosis in colorectal cancer cells. Int J Cancer. 2007;121:2172–2180. doi: 10.1002/ijc.22917. [DOI] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde VL, Hegde S, Cravatt BF, Hofseth LJ, Nagarkatti M, Nagarkatti PS. Attenuation of experimental autoimmune hepatitis by exogenous and endogenous cannabinoids: involvement of regulatory T cells. Mol Pharmacol. 2008;74:20–33. doi: 10.1124/mol.108.047035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde VL, Nagarkatti M, Nagarkatti PS. Cannabinoid receptor activation leads to massive mobilization of myeloid-derived suppressor cells with potent immunosuppressive properties. Eur J Immunol. 2010;40:3358–3371. doi: 10.1002/eji.201040667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde VL, Tomar S, Jackson A, Rao R, Yang X, Singh UP, et al. Distinct MicroRNA expression profile and targeted biological pathways in functional myeloid-derived suppressor cells induced by delta9-tetrahydrocannabinol in vivo: regulation of CCAAT/enhancer-binding protein alpha by microRNA-690. J Biol Chem. 2013;288:36810–36826. doi: 10.1074/jbc.M113.503037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JM, Shimonkevitz R, Hanagan A, Enney K, Boen E, Malmstrom S, et al. Identification of class II major histocompatibility complex and T cell receptor binding sites in the superantigen toxic shock syndrome toxin 1. J Exp Med. 1995;181:2229–2235. doi: 10.1084/jem.181.6.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huzella LM, Buckley MJ, Alves DA, Stiles BG, Krakauer T. Central roles for IL-2 and MCP-1 following intranasal exposure to SEB: a new mouse model. Res Vet Sci. 2009;86:241–247. doi: 10.1016/j.rvsc.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Jamontt JM, Molleman A, Pertwee RG, Parsons ME. The effects of Delta-tetrahydrocannabinol and cannabidiol alone and in combination on damage, inflammation and in vitro motility disturbances in rat colitis. Br J Pharmacol. 2010;160:712–723. doi: 10.1111/j.1476-5381.2010.00791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissner TL, Ruthel G, Alam S, Ulrich RG, Fernandez S, Saikh KU. Activation of MyD88 signaling upon staphylococcal enterotoxin binding to MHC class II molecules. PLoS ONE. 2011;6:e15985. doi: 10.1371/journal.pone.0015985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein TW. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol. 2005;5:400–411. doi: 10.1038/nri1602. [DOI] [PubMed] [Google Scholar]
- Klein TW, Newton C, Zhu W, Daaka Y, Friedman H. delta 9-Tetrahydrocannabinol, cytokines, and immunity to Legionella pneumophila. Proc Soc Exp Biol Med. 1995;209:205–212. doi: 10.3181/00379727-209-43897b. [DOI] [PubMed] [Google Scholar]
- Krakauer T. Update on staphylococcal superantigen-induced signaling pathways and therapeutic interventions. Toxins (Basel) 2013;5:1629–1654. doi: 10.3390/toxins5091629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakauer T, Buckley M. Dexamethasone attenuates staphylococcal enterotoxin B-induced hypothermic response and protects mice from superantigen-induced toxic shock. Antimicrob Agents Chemother. 2006;50:391–395. doi: 10.1128/AAC.50.1.391-395.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakauer T, Buckley M. Intranasal rapamycin rescues mice from staphylococcal enterotoxin B-induced shock. Toxins (Basel) 2012;4:718–728. doi: 10.3390/toxins4090718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakauer T, Buckley M, Issaq HJ, Fox SD. Rapamycin protects mice from staphylococcal enterotoxin B-induced toxic shock and blocks cytokine release in vitro and in vivo. Antimicrob Agents Chemother. 2010;54:1125–1131. doi: 10.1128/AAC.01015-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin EA, Carman RJ, Krakauer T, Stiles BG. Staphylococcus aureus: the toxic presence of a pathogen extraordinaire. Curr Med Chem. 2009;16:4003–4019. doi: 10.2174/092986709789352321. [DOI] [PubMed] [Google Scholar]
- Larkin EA, Stiles BG, Ulrich RG. Inhibition of toxic shock by human monoclonal antibodies against staphylococcal enterotoxin B. PLoS ONE. 2010;5:e13253. doi: 10.1371/journal.pone.0013253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leelawat S, Leelawat K, Narong S, Matangkasombut O. The dual effects of delta(9)-tetrahydrocannabinol on cholangiocarcinoma cells: anti-invasion activity at low concentration and apoptosis induction at high concentration. Cancer Invest. 2010;28:357–363. doi: 10.3109/07357900903405934. [DOI] [PubMed] [Google Scholar]
- Lindsay MA. microRNAs and the immune response. Trends Immunol. 2008;29:343–351. doi: 10.1016/j.it.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Linsley PS, Ledbetter JA. The role of the CD28 receptor during T cell responses to antigen. Annu Rev Immunol. 1993;11:191–212. doi: 10.1146/annurev.iy.11.040193.001203. [DOI] [PubMed] [Google Scholar]
- Liu SQ, Jiang S, Li C, Zhang B, Li QJ. miR-17-92 cluster targets phosphatase and tensin homology and ikaros family zinc finger 4 to promote TH17-mediated inflammation. J Biol Chem. 2014;289:12446–12456. doi: 10.1074/jbc.M114.550723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman WD, Sonett JR, Brosnan CF, Elkin R, Bornstein MB. Delta 9-tetrahydrocannabinol: a novel treatment for experimental autoimmune encephalomyelitis. J Neuroimmunol. 1989;23:73–81. doi: 10.1016/0165-5728(89)90075-1. [DOI] [PubMed] [Google Scholar]
- Mandal M, Kim S, Younes MN, Jasser SA, El-Naggar AK, Mills GB, et al. The Akt inhibitor KP372-1 suppresses Akt activity and cell proliferation and induces apoptosis in thyroid cancer cells. Br J Cancer. 2005;92:1899–1905. doi: 10.1038/sj.bjc.6602595. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Matthys P, Mitera T, Heremans H, Van Damme J, Billiau A. Anti-gamma interferon and anti-interleukin-6 antibodies affect staphylococcal enterotoxin B-induced weight loss, hypoglycemia, and cytokine release in D-galactosamine-sensitized and unsensitized mice. Infect Immun. 1995;63:1158–1164. doi: 10.1128/iai.63.4.1158-1164.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meduri GU, Headley AS, Golden E, Carson SJ, Umberger RA, Kelso T, et al. Effect of prolonged methylprednisolone therapy in unresolving acute respiratory distress syndrome: a randomized controlled trial. JAMA. 1998;280:159–165. doi: 10.1001/jama.280.2.159. [DOI] [PubMed] [Google Scholar]
- Merkenschlager M, von Boehmer H. PI3 kinase signalling blocks Foxp3 expression by sequestering Foxo factors. J Exp Med. 2010;207:1347–1350. doi: 10.1084/jem.20101156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miethke T, Wahl C, Heeg K, Echtenacher B, Krammer PH, Wagner H. T cell-mediated lethal shock triggered in mice by the superantigen staphylococcal enterotoxin B: critical role of tumor necrosis factor. J Exp Med. 1992;175:91–98. doi: 10.1084/jem.175.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarkatti M, Rieder SA, Hegde VL, Kanada S, Nagarkatti P. Do cannabinoids have a therapeutic role in transplantation? Trends Pharmacol Sci. 2010;31:345–350. doi: 10.1016/j.tips.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarkatti P, Pandey R, Rieder SA, Hegde VL, Nagarkatti M. Cannabinoids as novel anti-inflammatory drugs. Future Med Chem. 2009;1:1333–1349. doi: 10.4155/fmc.09.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell RM, Rao DS, Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30:295–312. doi: 10.1146/annurev-immunol-020711-075013. [DOI] [PubMed] [Google Scholar]
- Olive V, Bennett MJ, Walker JC, Ma C, Jiang I, Cordon-Cardo C, et al. miR-19 is a key oncogenic component of mir-17-92. Genes Dev. 2009;23:2839–2849. doi: 10.1101/gad.1861409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive V, Jiang I, He L. mir-17-92, a cluster of miRNAs in the midst of the cancer network. Int J Biochem Cell Biol. 2010;42:1348–1354. doi: 10.1016/j.biocel.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42:D1098–1106. doi: 10.1093/nar/gkt1143. (Database Issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R, Nagarkatti P, Nagarkatti M. Staphylococcal enterotoxin B (SEB) -induced microRNA-155 targets suppressor of cytokine signaling-1 (SOCS1) to promote acute inflammatory lung injury. Infect Immun. 2014;82:2971–2979. doi: 10.1128/IAI.01666-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- Rieder SA, Nagarkatti P, Nagarkatti M. CD1d-independent activation of invariant natural killer T cells by staphylococcal enterotoxin B through major histocompatibility complex class II/T cell receptor interaction results in acute lung injury. Infect Immun. 2011;79:3141–3148. doi: 10.1128/IAI.00177-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder SA, Nagarkatti P, Nagarkatti M. Multiple anti-inflammatory pathways triggered by resveratrol lead to amelioration of staphylococcal enterotoxin B-induced lung injury. Br J Pharmacol. 2012;167:1244–1258. doi: 10.1111/j.1476-5381.2012.02063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed AI, Rieder SA, Price RL, Barker J, Nagarkatti P, Nagarkatti M. Acute lung injury induced by Staphylococcal enterotoxin B: disruption of terminal vessels as a mechanism of induction of vascular leak. Microsc Microanal. 2012;18:445–452. doi: 10.1017/S1431927612000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Kohanbash G, Hoji A, Ueda R, McDonald HA, Reinhart TA, et al. miR-17-92 expression in differentiated T cells – implications for cancer immunotherapy. J Transl Med. 2010;8:17. doi: 10.1186/1479-5876-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A. 2008;105:7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savransky V, Rostapshov V, Pinelis D, Polotsky Y, Korolev S, Komisar J, et al. Murine lethal toxic shock caused by intranasal administration of staphylococcal enterotoxin B. Toxicol Pathol. 2003;31:373–378. doi: 10.1080/01926230390201093. [DOI] [PubMed] [Google Scholar]
- Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8:1145–1152. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- Sonkoly E, Pivarcsi A. Advances in microRNAs: implications for immunity and inflammatory diseases. J Cell Mol Med. 2009;13:24–38. doi: 10.1111/j.1582-4934.2008.00534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava MD, Srivastava BI, Brouhard B. Delta9 tetrahydrocannabinol and cannabidiol alter cytokine production by human immune cells. Immunopharmacology. 1998;40:179–185. doi: 10.1016/s0162-3109(98)00041-1. [DOI] [PubMed] [Google Scholar]
- Sun F, Fu H, Liu Q, Tie Y, Zhu J, Xing R, et al. Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett. 2008;582:1564–1568. doi: 10.1016/j.febslet.2008.03.057. [DOI] [PubMed] [Google Scholar]
- Trede NS, Tsytsykova AV, Chatila T, Goldfeld AE, Geha RS. Transcriptional activation of the human TNF-alpha promoter by superantigen in human monocytic cells: role of NF-kappa B. J Immunol. 1995;155:902–908. [PubMed] [Google Scholar]
- Tsang WP, Kwok TT. The miR-18a* microRNA functions as a potential tumor suppressor by targeting on K-Ras. Carcinogenesis. 2009;30:953–959. doi: 10.1093/carcin/bgp094. [DOI] [PubMed] [Google Scholar]
- Tsytsykova AV, Goldfeld AE. Nuclear factor of activated T cells transcription factor NFATp controls superantigen-induced lethal shock. J Exp Med. 2000;192:581–586. doi: 10.1084/jem.192.4.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich RG, Sidell S, Taylor TJ, Wilhelmsen CL, Franz DR. 2001. Textbook of military medicine: medical aspects of chemical and biological warfare. Office of The Surgeon General, Department of the Army, United States of America.
- Wajanaponsan N, Reade MC, Milbrandt EB. Steroids in late ARDS? Crit Care. 2007;11:310. doi: 10.1186/cc5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Wieland A, Araki K, Davis CW, Ye L, Hale JS, et al. Temporal expression of microRNA cluster miR-17-92 regulates effector and memory CD8+ T-cell differentiation. Proc Natl Acad Sci U S A. 2012;109:9965–9970. doi: 10.1073/pnas.1207327109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Regulation of miR-17-92 cluster by THC, dexamethasone (Dexa), rapamycin (Rapa) and cannabidiol (CBD) in vitro. Splenocytes were activated with SEB and treated with THC (20 μM), CBD (20 μM ), dexamethasone (200 nM) and rapamycin (100 nM). Total RNA was extracted and RT-PCR was conducted to measure miRNA levels relative to Snord_96a internal control. Statistical significance was assessed using anova, Tukey's multiple comparison test.