

Abstract
One third of all drugs in clinical use owe their pharmacological activity to the functional inhibition of enzymes, highlighting the importance of enzymatic targets for drug development. Because of the close relationship between inhibition and catalysis, understanding the recognition and turnover of enzymatic substrates is essential for rational drug design. Although the Staphylococcus aureus enoyl-acyl carrier protein reductase (saFabI) involved in bacterial fatty acid biosynthesis constitutes a very promising target for the development of novel, urgently needed anti-staphylococcal agents, the substrate binding mode and catalytic mechanism remained unclear for this enzyme. Using a combined crystallographic, kinetic and computational approach, we have explored the chemical properties of the saFabI binding cavity, obtaining a consistent mechanistic model for substrate binding and turnover. We identified a water-molecule network linking the active site with a water basin inside the homo-tetrameric protein, which seems to be crucial for the closure of the flexible substrate binding loop as well as for an effective hydride and proton transfer during catalysis. Based on our results, we also derive a new model for the FabI-ACP complex that reveals how the ACP-bound acyl-substrate is injected into the FabI binding crevice. These findings support the future development of novel FabI inhibitors that target the FabI-ACP interface leading to the disruption of the interaction between these two proteins.
As one third of all currently used drugs are directed against enzyme targets, understanding the functionality of enzymes is of particular importance for rational drug design 1. Enzymes are tailored to stabilize the transition state (TS) of the catalyzed reaction, which generally requires perfect shape complementarity with the corresponding binding pocket 2. This complementarity, however, is often achieved via induced-fit effects upon substrate binding and turnover – effects that are still insufficiently characterized at the structural level 3.
Many enzymes harbor flexible loops that close upon substrate binding and are most likely required for precise substrate orientation, avoidance of the release of reaction intermediates, and exclusion of water from the binding-site 4. Members of the short-chain dehydrogenase/reductase (SDR) superfamily contain such a flexible loop, termed the substrate binding loop (SBL) 5. Two distinct SDR proteins are involved in the bacterial fatty acid biosynthesis (FAS-II) pathway, namely the β-ketoacyl-ACP reductase (FabG) and the enoyl-ACP reductase (FabI, EC 1.3.1.9/10) 6. Since the FAS-II acyl substrates are attached to the small acidic acyl carrier protein (ACP) via a 4’-phosphopantetheine linker (PPant), protein-protein interactions are additionally involved in substrate recognition and turnover. However, little is known about the interaction of ACP with FAS-II enzymes.
Enoyl-ACP reductases catalyze the last step of the FAS-II elongation pathway - the reduction of the trans-2-enoyl-ACP double bond 6. Most FabI enzymes catalyze substrate reduction through an ordered bi-bi mechanism, in which binding of the cofactor NAD(P)H precedes substrate binding 6. Subsequently, the reaction proceeds via the transfer of the pseudoaxially oriented pro-4S hydride of NAD(P)H 7 from the 3-si face to the C3 carbon of the C2-C3 enoyl-ACP double bond followed by the stereospecific protonation to the 2-re face of the resultant enolate intermediate (Figure 1A) 8-10. To enable effective hydride transfer, the catalytic triad residue Lys164 likely plays a crucial role in the correct positioning of the cofactor’s reactive nicotinamide ring (residues are numbered according to the Staphylococcus aureus homologue) 8. NAD(P)H is known to bind at the bottom of an elongated cavity with two channels leading to the solvent - a minor portal adjacent to the nicotinamide ring and a major portal next to the adenine ring (Figure 1B) 8. However, it is currently unclear whether the enoyl-substrate enters via the minor or major portal 8, 11. A structure of the substrate analog trans-2-hexadecenoyl-(N-acetylcysteamine)-thioester (C16-NAC) bound to the FabI homologue of Mycobacterium tuberculosis (InhA) indicated that ACP should be located at the major portal 8. However, in an Escherichia coli FabI (ecFabI) - NAD+ - trans-2-dodecenoyl-ACP (DD-ACP) structure, ACP was oriented for substrate delivery to proceed via the minor portal. Based on this structure, which lacked the ACP amino acid side chains, the enoyl-substrate, and the PPant linker due to missing electron density in a presumably very flexible complex, a model of the FabI-ACP complex was generated via molecular dynamics (MD) simulations 11. According to this model the thioester carbonyl oxygen would interact with the catalytic residue Tyr147, and the partial C1-C2 double bond has to be in s-trans conformation due to the defined stereochemical outcome of the reaction (Figure 1C) 9-11. However, if the ACP is located at the major portal, the substrate would be in the energetically more favorable s-cis conformation, and the thioester carbonyl would be hydrogen bonded to Tyr157, complementing the catalytic triad (Figure 1D) 8, 9, 11.
Figure 1.
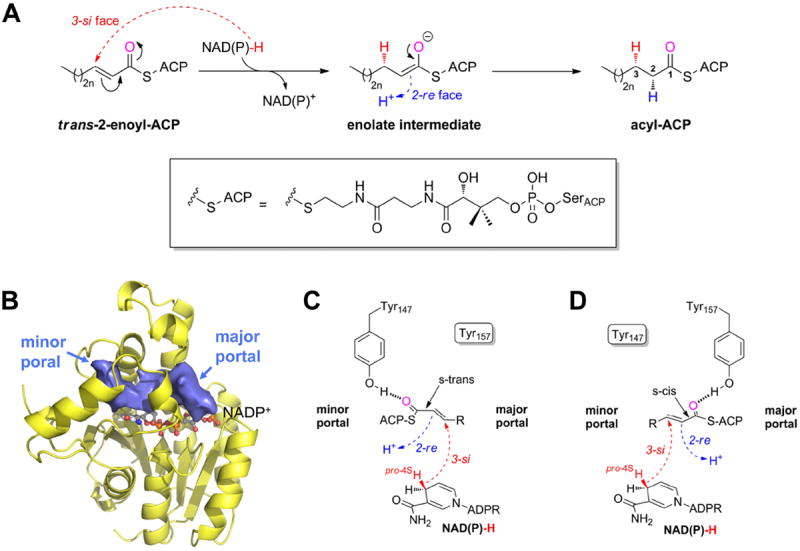
The FabI-catalyzed reaction 9-11. (A) Syn-addition of H2 by a two-step mechanism (n = 0-8). Dashed arrows indicate an attack from below the paper plane. The pro-4S hydride of NAD(P)H (red) is transferred from the 3-si face. In a second step, the enolate intermediate is protonated stereospecifically from the re face at C2 as observed for ecFabI and InhA (proton shown in blue) 9, 10. The thioester oxygen, which is hydrogen bonded to either Tyr147 (panel C) or Tyr157 (panel D), is indicated in magenta. During the reaction, the substrate is attached to ACP via a phosphopantetheine moiety (depicted in the box). (B) Binding-pocket topology including portals towards the solvent. Povme 47 was used to calculate the binding-pocket volume indicated as blue surface (PT400-bound saFabI structure). NADP+ is depicted as ball-and-stick model. (C) Substrate delivery via the minor portal. With the cofactor located below the substrate in the chosen view (see panel B), the trans-2-enoyl-ACP has to be in the s-trans conformation to enable the correct stereochemical outcome in a scenario where ACP binds close to the minor portal, and the thioester carbonyl is, thus, bound to Tyr147. Dashed arrows indicate attack vectors from below. (D) Substrate delivery via the major portal. If ACP is located at the major portal, the thioester carbonyl will be hydrogen bonded to Tyr157 and, thus, the substrate has to be in the s-cis conformation for consistency with the observed stereochemistry.
During our efforts to develop inhibitors against the target S. aureus FabI (saFabI), for which three drug candidates have reached clinical trials 12, we identified a close relationship between catalysis and inhibition 13, 14. Hence, small-molecule inhibitors can be used as chemical probes to explore substrate recognition and catalysis of this enzyme. Using a combined crystallographic, kinetic, and computational approach, we analyze here the properties of the saFabI binding cavity, the substrate binding mode, and the protonation source to arrive at a consistent mechanistic and dynamic model for the enzymatic reaction of this important member of the enoyl-ACP-reductase antibacterial target family.
Experimental Procedures
Crystallization, Data Collection and Structure Determination
SaFabI purification, crystallization and structure determination followed our recently reported protocol 13, 15. SaFabI protein samples (15 mg/ml in 20 mM citrate pH 5.6, 280 mM NaCl, 280 mM potassium glutamate and 1 mM EDTA; 19 mg/ml in 20 mM citrate pH 5.6, 250 mM NaCl, 250 mM potassium glutamate and 1 mM EDTA for PT449) were incubated for 2 hours at 4 °C with a 10-fold molar excess of NADP+ (dissolved in water) and a 15- to 20-fold molar excess of inhibitor (dissolved in DMSO). Using the vapor diffusion method, crystals were obtained in 0.1 M Na/K-phosphate pH 6.5 and 39 - 47% 2-methyl-2,4-pentanediol (MPD) (Table S1). For diffraction experiments, crystals were directly flash-frozen without the addition of a cryoprotectant. Similarly, ecFabI (purified as described previously 16, 13 mg/ml in 20 mM PIPES pH 7.5, 200 mM NaCl and 1 mM EDTA) was incubated with a 10-fold molar excess of NAD+ (dissolved in water) and a 20-fold molar excess of PT449 (dissolved in DMSO). Crystals grew in a precipitant solution composed of 0.1 M CAPS pH 10.5, 0.08 M NH4Ac and 20% PEG 3350 and were cryo-protected using the mother liquid supplemented by 20% ethylene glycol.
Diffraction data were collected at beamline 14.1 of the BESSY II synchrotron (λ = 0.918 Å, T = 100 K) using a MarMosaic 225 (PT12, PT400, PT404, MUT37307) or Pilatus 6M detector (PT449), integrated with Imosflm 17 or XDSAPP 18, respectively, and scaled with Scala19. After molecular replacement with Phaser 20 (search models: PDB-entries 4ALK and 1QSG for saFabI and ecFabI, respectively), alternate cycles of model building in Coot 21 and refinement using Refmac 5 22 led to the final models. In order to keep model bias to a minimum, we copied the Rfree flags from the starting model (4ALK). Since the electron density for the ecFabI amino acid region 193-210 was only weakly defined, this part of the protein could not be fully modeled. Data collection and refinement statistics are given in Table S1. All structural figures were prepared using PyMOL 23. Distances between individual atoms within a particular structure were measured for all eight subunits and are given as mean values ± standard deviations.
Site-directed mutagenesis, expression and purification of saFabI variants
Site-directed mutagenesis was performed using the QuikChange Mutagenesis Kit from Stratagene to construct the D249A, S189A and Y147F variants from a pET-16b plasmid containing the wild-type fabI gene. The primers are listed in Table S2. Expression and purification of the variants was performed as described for the wild-type enzyme 15.
Steady-state kinetics
Kinetic experiments were performed as described previously 15. Briefly, reaction velocities were measured by monitoring the oxidation of NAD(P)H to NAD(P)+ at 340 nm (ε = 6220 M-1 cm-1). Kinetic parameters for saFabI D249A, S189A and Y147F were determined by measuring initial velocities at varying concentrations of trans-2-octenoyl CoA and a constant concentration of NADPH (350 μM).
Inhibition kinetics
Progress curves at a single inhibitor concentration were used to determine the kinetics and thermodynamics of inhibition by the compounds in Table 1, as described previously 13.
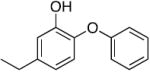
Table 1.
Kinetic parameters for saFabI inhibition by diphenyl ether analogues a
| Name | Structure | kon (x 1010 M-1 hr-1) | koff (hr-1) | Ki (nM) |
|---|---|---|---|---|
| PT01 b |
|
1.01 | 0.94 | 0.09 |
| PT04 b |
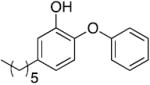
|
1.30 | 0.13 ± 0.04 c | 0.01 d |
| PT12 |
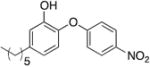
|
1.69 | 0.86 | 0.05 |
| PT18 |
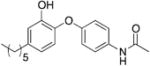
|
0.67 | 9.98 | 1.50 |
| PT55 b |
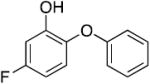
|
0.68 | 9.67 | 1.42 |
| PT104 b |
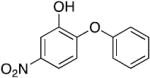
|
0.61 | 9.03 | 1.49 |
| PT400 |
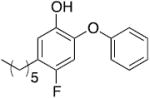
|
1.30 | 0.13 e | 0.01 e |
| PT404 |
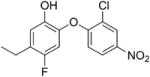
|
0.04 | 0.31 | 0.75 |
| PT411 |
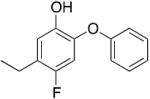
|
0.61 | 0.55 | 0.09 |
| PT449 |
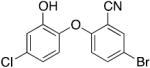
|
0.25 | 0.43 | 0.17 |
| MUT37307 |
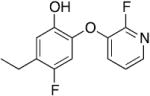
|
0.17 | 0.60 | 0.35 |
| MUT056399 |
|
0.34 | 0.27 | 0.08 |
Determined via single point progress curve analysis, as described in 13 unless otherwise specified.
Data obtained from 13
Calculated by fitting the 32P-NAD-based dissociation curve as described in 13
Determined using the koff value obtained from the 32P-NAD dissociation assay as described in 13
Based on identical behavior to PT04 in forward progress curve analysis.
Thermal shift assay
ThermoFluor experiments were carried out in 96-well plates (Concord) using the CFX96 Real-Time PCR Detection System and C1000 Thermal Cycler (Bio-Rad), as described previously13.
Molecular Dynamics Simulations
In total, eight MD simulations were conducted based on the saFabI-NADP+-PT400 structure. The full homo-tetramer (subunits E-H, referred to as monomers 1-4) was simulated for the wild-type protein as well as the Y147F, S189A and D249A variants with bound NADP+ and PT400. Moreover, simulations were initiated using the tetrameric complex structure without PT400 or NADP+ and PT400, respectively. Since saFabI can exist as a dimer in the apo form 15, we also simulated the dimeric protein lacking both ligands (subunits F-G). As a reference, we additionally performed a simulation for the monomeric saFabI-NADP+-PT400 complex (subunit G).
The AMBER suite of programs (University of California, San Francisco, 2011/2012) was used for the setup, performance and analysis of all MD simulations. For the setup of the MD simulations only those water molecules of the binding pocket and water-chain were included from the original structure which displayed clear electron density (56 waters for the tetrameric protein). The binding site of the protein was defined by residues 93-99, 102, 121, 146-147, 154-157, 160, 164, 190-193, 195, 197-204, 207 and NADP+. Guided by visual inspection, the most likely asparagine, glutamine and histidine conformations and protonation states were assigned (in particular, His247 was assumed to be doubly protonated due to its interactions with Glu151 and Glu244’).
Using the tleap module of AMBER11 24, hydrogens were added and ff99SB force field parameters assigned to the protein atoms. Atomic charges for NADP+ and PT400 were determined with Gaussian 03 25 using the RESP methodology 26 at the HF/6-31G* level. The AMBER11 parmchk module 27 was used to assign the missing force field parameters according to the GAFF force field 28. The correct GAFF atom and bond types were determined using antechamber 27. A short energy minimization over 200 steps using a generalized Born implicit solvent model was performed with the SANDER module of AMBER11 24. Using tleap, the minimized systems were then solvated in a box of TIP3P water molecules and neutralized using sodium counter ions. The wild-type and mutant simulations were performed using the SANDER module of AMBER11 24 or the PMEMD module of AMBER12 29, respectively. First, the solvent molecules were disordered in a 25 ps constant-volume simulation, keeping the protein atoms fixed using the ibelly option. This was achieved by heating the systems from 100 to 300 K over 20 ps and then cooling back down to 100 K within 5 ps, applying the Berendsen weak coupling algorithm with a 0.5 ps time constant. In a second step, the entire system was allowed to move freely and was heated gradually to 300 K over 25 ps. To enable a time step of 2 fs during the following MD run, all bonds involving hydrogen atoms were constrained by the SHAKE algorithm 30. Moreover, the constant-volume periodic boundary conditions were switched to constant-pressure by applying isotropic position scaling with a reference pressure of 1 bar and a relaxation time of 1 ps. In addition, the temperature was fixed by coupling to a heat bath with a time constant of 1 ps. An 8 Å cutoff was used for van der Waals interactions, and long-range electrostatic interactions were treated using the particle mesh Ewald method 31. Coordinates were saved at intervals of 1 ps and MD trajectory data were collected over a period of 16 ns for each system. Processing and analyses of the trajectories were performed using the ptraj module of AMBER11, carnal of AMBER7 and VMD 32. Hierarchical clustering analysis was conducted using SYBYL-X (Tripos, St. Louis, 2009), version 1.0 33.
Computational Docking
Putative substrate binding poses were generated using our validated approach described previously for the docking of pyridone inhibitors to saFabI 34. Briefly, binding poses were produced using FlexX (BioSolveIT, Sankt Augustin, 2009), version 3.1.4 35, and rescored with DrugScoreX (G. Neudert and G. Klebe, University of Marburg, 2008) 36. The most likely binding mode was selected based on the DrugScoreX score and the selection criteria specified in the results section. Re-docking of PT400 yielded rmsd values below 0.77 Å for the 10 best-ranked poses (0.60 Å for the top-ranked pose) and thus confirms the reliability of our approach. During the docking procedure, the hydroxyl dihedral angles of Tyr147 and Tyr157 were set to 0° or 120° and 0°, respectively.
An octanoyl-NAC enolate intermediate (4) was docked into the saFabI-NADP+-PT400 structure (omitting PT400) whereas the S-trans-4-methyl-2-hexenoyl-NAC (anteiso, 1), trans-5-methyl-2-hexenoyl-NAC (iso, 2), trans-2-octenoyl-NAC (3) and trans-2-octenoyl-PPantMe substrate analogs were docked into the same receptor containing the reduced cofactor NADPH (derived from a superimposition with an NADPH-bound saFabI structure; PDB-entry 4CV1, subunit C). For the five compounds mentioned above, the docking poses with rank 1, 18, 5, 2 and 1 (with respect to the DrugScoreX score) were chosen for further analysis, respectively (for pose selection criteria see results section).
Results
Inhibition of SaFabI
We have investigated the inhibition of saFabI by the diphenyl ethers PT12, PT400, PT404, MUT37307, and PT449 using enzyme kinetics (Table 1) and X-ray crystallography (Figure 2). During the development of the drug candidate MUT056399 by the company Mutabilis, MUT37307 was found to be one of the most promising FabI inhibitors amongst a large series of diphenyl ether inhibitors 37. Both compounds contain more hydrophilic B-rings leading to lowered clogD values while maintaining their ability to inhibit saFabI. To understand enzyme-inhibitor recognition, we solved the structure of saFabI in complex with NADP+ and MUT37307 revealing the binding mode of this promising compound. Interestingly, the fluoropyridine B-ring does not engage in direct interactions with the protein but is well tolerated at this position (Figure 2B, Table 1). MUT056399, MUT37307, PT404 and PT411 share the same phenolic A-ring comprising a 4-fluoro and 5-ethyl group. The former substituent, which is also present in PT400 (Figure 2A), was suggested to act as a metabolic regulator 38 and did not affect the affinity significantly (Table 1 - PT01 and PT04 compared to PT411 and PT400, respectively). Moreover, there was no change in the pKa of the 2’-hydroxyl group between PT01 and PT411 (data not shown). Since fluorine has only a slightly increased atomic radius compared to hydrogen, it is well tolerated in a small side pocket created by Phe204 and Ala198 without affecting the closed state of the substrate binding loop (Figures 2A-C). For instance, the PT404 fluoro-substituent contacts one of the Phe204 Cε-H atoms at a C-F distance of 3.20 ± 0.12 Å (Figure 2C). Moreover, a direct weak hydrogen bonding interaction of the F with the Cα-H of Ala198 was observed at a C-F distance of 3.38 ± 0.05 Å.
Figure 2.
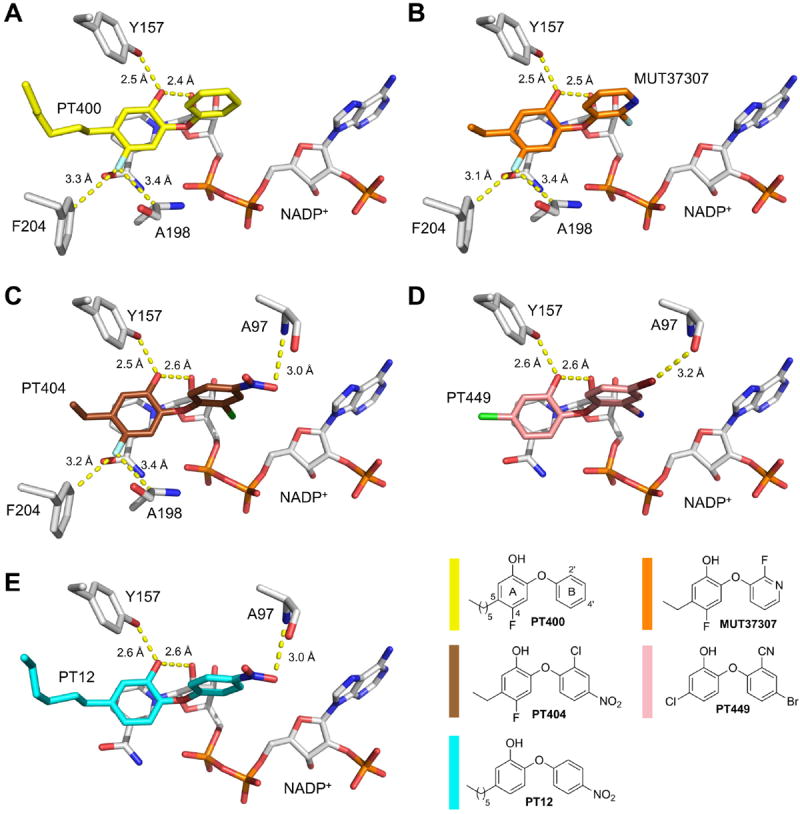
Inhibitor binding modes. Direct interactions are highlighted by yellow dashed lines. The color code used for the different inhibitors is depicted in the lower, right corner. Selected protein residues and the cofactor are shown as gray sticks. (A) SaFabI inhibition by PT400 (yellow). The PT400 hydroxyl interacts with the Tyr157 hydroxyl group and the 2’-OH of the nicotinamide ribose at distances of 2.54 ± 0.09 Å and 2.44 ± 0.07 Å, respectively. Furthermore, the 4-F substituent forms weak hydrogen bonds with one of the two Phe204 Cε hydrogens as well as with the Ala198 Cα hydrogen (distances of 3.31 ± 0.07 Å and 3.35 ± 0.04 Å, respectively; for more details regarding such interactions see also reference 39). (B) SaFabI inhibition by MUT37307 (orange). As PT400, MUT37307 is hydrogen bonded to the Tyr157 and nicotinamide ribose hydroxyl groups at distances of 2.51 ± 0.04 Å and 2.48 ± 0.04 Å, respectively. The distances of the Phe204 Cε and Ala198 Cα atoms to the 4-F substituent of MUT37307 are 3.13 ± 0.04 Å and 3.41 ± 0.02 Å, respectively. (C) SaFabI inhibition by PT404 (brown). The PT404 Aring hydroxyl group is hydrogen bonded to the Tyr157 hydroxyl group and the 2’-OH of the nicotinamide ribose at distances of 2.50 ± 0.05 Å and 2.57 ± 0.03 Å, respectively. The 4’-nitro group interacts with the amide nitrogen of Ala97 at a distance of 2.99 ± 0.10 Å. As for PT400 and MUT37307, the 4-F substituent contacts one of the two Phe204 Cε hydrogens as well as the Ala198 Cα hydrogen at distances of 3.20 ± 0.12 Å and 3.38 ± 0.05 Å, respectively. (D) SaFabI inhibition by PT449 (pink). The OH group of PT449 is bound to Tyr157 and the 2’-OH of the NADP+ nicotinamide ribose at distances of 2.59 ± 0.14 Å and 2.64 ± 0.10 Å, respectively. Furthermore, a linear halogen bond is formed between the Ala97 carbonyl oxygen and the 4’-Br group of PT449 at a distance of 3.16 ± 0.11 Å 39. (E) SaFabI inhibition by PT12 (blue). As for the other inhibitors, the central interaction is formed between the PT12 A-ring hydroxyl group and the Tyr157 hydroxyl group as well as with the 2’-OH of the nicotinamide ribose at distances of 2.60 ± 0.08 Å and 2.59 ± 0.09 Å, respectively. In addition, the 4’-nitro group contacts the amide nitrogen of Ala97 at a distance of 3.00 ± 0.12 Å.
In order to strengthen the linear halogen bond observed between the 4’-Cl substituent of the antimicrobial agent triclosan and the Ala97 carbonyl oxygen 15, 39, we introduced a 4’-Br substituent combined with the potent 2’-CN group leading to PT449. However, compared to the corresponding analogue lacking the bromine substituent (PT447) 13, PT449 displayed a 3-fold increased Ki value although a halogen bond was observed with for saFabI (d = 3.16 ± 0.11 Å, Figure 2D) and ecFabI (d = 3.26 Å). Based on the kinetic results, the reduced affinity towards saFabI is mostly attributed to a slower rate of inhibitor association (kon) compared to PT447. Similarly, among PT404, PT411 and MUT37307, the combination of 2’- and 4’-substituents (PT404 carrying a 2’-Cl and 4’-nitro group) results in the slowest inhibitor association rate despite relatively similar rates of inhibitor dissociation. These observations may be structurally correlated with the barrier to placing bulky substituents into the saFabI binding crevice (Figures 2C and 2D). Although PT12 harbors the same 4’-nitro substituent as PT404 which interacts with Ala97 (Figure 2E), this ligand has a comparable rate of association as PT04 lacking the nitro group. Hence, kon seems to be decreased especially for diphenyl ethers harboring both, bulky 2’- and 4’-substituents.
To investigate whether and how the saFabI binding-site residues are influenced by the accommodation of different inhibitors, we compared the five saFabI structures by means of a per-residue rmsd analysis (Figure S1A). With a maximum of 0.79 Å for Ile207, these values do not indicate drastic structural changes, in accordance with the conserved binding mode of diphenyl ether inhibitors. Overall, the structural variations mostly follow the trend expected from a B-factor analysis (Figure S1B), with the largest values obtained for the inherently flexible SBL. Visual inspection of the superimposed structures revealed that only Ile207 shows a clear directed side-chain movement away from those inhibitors that carry the long 5-hexyl chain instead of an ethyl or chloro group (Figure S1C). We observed this movement of Ile207 previously and assume that this residue is important for the recognition of longer substrates 13. Importantly, our analysis reveals low mobility for the three catalytic triad residues Tyr147, Tyr157 and Lys164 (Figure S1B). For Tyr147, the comparison of the five structures shows the largest positional variability for the phenolic oxygen (rmsd = 0.41 Å), whereas the hydroxyl group of Tyr157 occupies very similar positions in all 5 structures (rmsd = 0.13 Å), despite some variation in its main chain atoms (rmsd of the Cα atom = 0.21 Å; see also Figure S1C). This is a clear indication that the hydroxyl group of Tyr157 might be of particular importance for inhibitor and substrate recognition.
Analysis of the SaFabI Binding Pocket Characteristics
Small-molecule inhibitors are valuable tools to investigate the mechanism of enzymatic reactions 3, 14. In the case of saFabI, we used diphenyl ether-based inhibitors 13 to probe substrate binding and turnover for this important drug target. These TS-analogues trigger the full closure of the SBL 15 and establish an elongated binding pocket in contact with the solvent via the minor and major portals (Figure 3A). Given the conserved binding mode of the diphenyl ethers 13, substituents in the 5-position of the A-ring can be used to explore the chemical properties of the left part of the pocket (“left” subpocket) (Figure 3B, surrounded by gray residues), whereas substituted B-rings are ideal probes for the right part of the pocket (“right” subpocket) (Figure 3B, surrounded by green residues). Thus, we kinetically investigated a series of diphenyl ethers (Table 1) including the inhibitors bound in the five saFabI ternary complex structures described above (PT12, PT400, PT404, MUT37307 37, and PT449). Due to the very hydrophobic environment of the left subpocket, lipophilic alkyl chains are optimal substituents at the 5-position (Figures 3A-B, Table 1), whereas the more hydrophilic 5-nitro and 5-fluoro substituents lead to a decrease in binding affinity by one order of magnitude compared to more hydrophobic similarly sized 5-substituents 13. A comparable trend was observed for ecFabI 37 indicating that the hydrophobicity of the left subpocket is conserved across species and that this pocket is ideally suited to bind the hydrophobic enoyl chain of the substrate.
Figure 3.
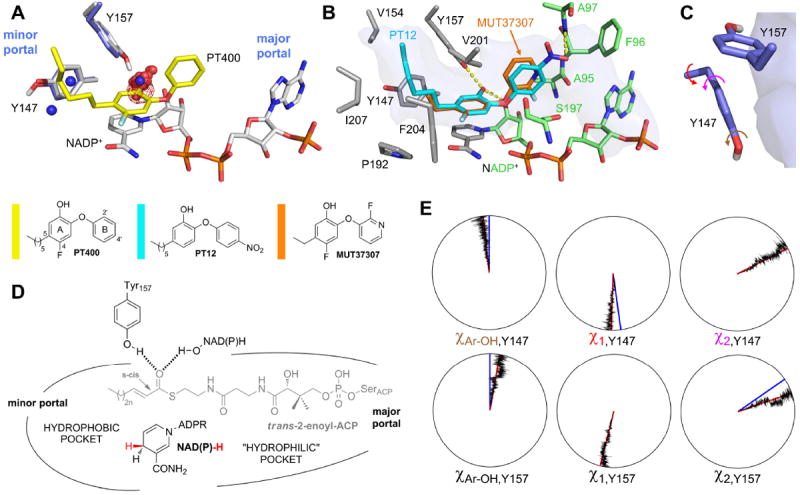
SaFabI binding pocket characteristics. (A) Hydrogen bonding hotspot. PT400 (yellow) binds to the saFabI cavity and is hydrogen bonded to Tyr157 (gray; saFabI-NADP+-PT400 structure). The protonated Tyr147 and Tyr157 residues are shown for the representative structure of the largest cluster from a hierarchical clustering analysis of the saFabI4-NADP+-PT400 MD simulation (Figure S4). Red (blue) spheres are shown for all oxygen (nitrogen) FabI inhibitor atoms within a radius of 3.5 Å around Tyr147 or Tyr157, respectively (for this purpose, all structurally known inhibitor-bound FabI complexes were superimposed on the saFabI-NADP+-PT400 structure). The water occupancy map for one monomer of the saFabI4-NADP+ MD simulation is depicted as red mesh at the 50% level. (B) Probing the pocket characteristics with diphenyl ether inhibitors. The lipophilic, left subpocket (defined by the residues shown in gray) is occupied by hydrophobic alkyl chains. In contrast, the 4’-nitro substituent of PT12 as well as the fluoropyridine moiety of MUT37307 highlight the less hydrophobic character of the right subpocket (residues defining this part of the pocket are indicated in green; saFabI-NADP+-PT12 structure). The binding pocket topology is highlighted in transparent blue surface representation (PT400-bound structure). (C) The catalytic tyrosines Tyr157 and Tyr147 and their orientation with respect to the binding pocket (blue surface). (D) Summary of the saFabI binding pocket properties. The expected substrate binding mode based on these observations is indicated in gray. (E) Dials plots for Tyr147 and Tyr157. The dihedral angles Cε-Cζ-O-H (Ar-OH), χ1 and χ2 (indicated by the arrows in panel C) are exemplarily plotted for one monomer of the saFabI4-NADP+-PT400 trajectory. Red lines indicate the simulation averages (χAr-OH(Y147/Y157) = -7 ± 11°/11 ± 11°, χ1 (Y147/Y157) = -178 ± 8°/-167 ± 7°, χ2 (Y147/Y157) = 63 ± 10°/71 ± 10°) whereas blue lines depict the corresponding values for the crystal structure (for χAr-OH the reference values were set to 0° since no hydrogen atoms were observed during the crystallographic experiments, χ1 (Y147/Y157) = 172°/-166°, χ2 (Y147/Y157) = 63°/56°).
In contrast, the right subpocket is less hydrophobic as highlighted by the tolerance for the hydrophilic B-rings of PT12 and MUT37307 (Figure 3B, Table 1). Our crystal structures show that the 4’-nitro groups of PT12 and PT404 are hydrogen bonded to the amide nitrogen of Ala97 at a distance of 3.0 ± 0.1 Å (Figures 2E and C). Importantly, the three saFabI inhibitors, which are currently in clinical trials, were also suggested to interact with the backbone of Ala97 34, thus defining an important hydrophilic anchor in the right subpocket and a possible contribution to PPant-binding. In agreement with this suggestion, the clinical candidate MUT056399, which carries a hydrophilic 4’-amide group, displays high affinity towards saFabI (Table 1).
Based on the observed binding pocket characteristics of saFabI, we speculated that the substrate enters the protein via the major portal because only in this orientation the lipophilic acyl chain can be delivered to the hydrophobic left subpocket while the less lipophilic PPant moiety remains in the more hydrophilic right subpocket (Figures 1D and 3D). To investigate whether such a model is in agreement with the dynamic properties of saFabI, we performed five different 16 ns MD simulations using our saFabI-NADP+-PT400 structure. Since saFabI is a tetramer in its ligand-bound state but can exist as a dimer in the apo-form 15, MD simulations were conducted for different oligomeric states of the protein (indicated by a subscript) including or excluding cofactor and/or inhibitor. We refer to the simulations as saFabI4-NADP+-PT400, saFabI1-NADP+-PT400, saFabI4-NADP+, saFabI4 and saFabI2 (cf. Table S3 and Figure S2 for trajectory rmsd analyses). The orientation and dynamic behavior of the Tyr147 and Tyr157 hydroxyl groups are of particular importance for inhibitor and substrate recognition since the substrate thioester carbonyl oxygen has to be held in a fixed position by one of these two residues to ensure the precise orientation of the enoyl-ACP double bond with respect to the NAD(P)H pro-4S hydride. As usually observed for tyrosine OH-groups 40, the analysis of the five MD trajectories revealed that the hydroxyl hydrogens of both active-site tyrosines are exclusively located almost within the phenyl-ring plane (in both cases, the Cε-Cζ-O-H dihedral angle approximates 0°, as seen in Figure 3E; only in some simulations it was rarely found at ~180° for Tyr147). In addition, the average χ1 and χ2 torsion angles are close to the crystallographic values and are very stable along the MD trajectories, leading to the precise orientation of the OH hydrogens. Preciseness in the orientation of catalytic residues seems to be a crucial component of enzymatic activity 4. In agreement, the saFabI catalytic triad residues were found to be amongst the least flexible binding-site residues in the simulations (exemplarily shown for saFabI4-NADP+-PT400 in Figures S3 and S4; this trend is confirmed by the B-factor analysis of the experimentally determined structures depicted in Figure S1B). Importantly, only the Tyr157 but not the Tyr147 hydroxyl group points towards the saFabI binding pocket and, thus, displays an ideal geometry to bind the substrate thioester carbonyl group (Figure 3C). Accordingly, all investigated diphenyl ether inhibitors bind with their hydroxyl groups to the oxyanion hole formed by Tyr157 and the 2’-OH of the nicotinamide ribose, which likely mimics the interaction with the enolate intermediate (Figures 2 and 3). The resulting hydrogen bonds were conserved during the saFabI4-NADP+-PT400 MD simulation to 79% and 67%, respectively, leading to low rmsd values for the PT400 A-ring (1.30 ± 0.55 Å; all-atom fit) and its 5-hexyl substituent (1.47 ± 0.40 Å) compared to the B-ring (2.11 ± 1.11 Å). If PT400 was omitted during the MD simulation (saFabI4-NADP+), a water molecule binds to the oxyanion hole instead (Figure 3A). An analysis of all structurally characterized inhibitor-bound FabI-complexes revealed that all inhibitors from 8 different scaffold classes bind with an oxygen or nitrogen atom (rarely water-mediated) to this oxyanion hole, whereas only two polar atoms are found in the vicinity of Tyr147 (spheres in Figure 3A). These findings are thus highly indicative of substrate delivery via the major portal (Figures 1D and 3D), which also explains why Tyr147 is not conserved in the M. tuberculosis homologue InhA, where this residue is a phenylalanine 8.
Putative Substrate Binding Modes
Since the proceeding chemical reaction hampers the structural characterization of intact enzyme-substrate complexes, we decided to use our validated computational docking approach to generate meaningful substrate binding modes 34. An octanoyl-NAC enolate intermediate (4, Figure 4) was docked into the saFabI-NADP+-PT400 structure (omitting PT400) whereas different substrate analogs (S-trans-4-methyl-2-hexenoyl-NAC = 1, trans-5-methyl-2-hexenoyl-NAC = 2, trans-2-octenoyl-NAC = 3) were docked into the same receptor containing NADPH (details are given in the Experimental Procedures section). Putative binding poses were selected based on the DrugScoreX score and the following three criteria: (1) the thioester carbonyl group has to bind to Tyr157 or Tyr147; (2) to fulfill the stereochemical requirements 9, 10, the C1-C2 bond of the substrate has to be in s-cis (Figure 1D) or s-trans (Figure 1C) conformation, respectively; and (3) the C3 atom of the substrate has to be located adjacent to the reactive pro-4S NADPH hydride. Consistently, all selected binding modes exhibit an interaction between the thioester carbonyl and the oxyanion hole formed by Tyr157 and the nicotinamide ribose 2’-OH (Figures 4A-B, interaction distances are given for 3 throughout the manuscript: 3.2 Å and 2.7 Å, respectively). Remarkably, even when forcing the hydroxyl hydrogen of Tyr147 to point towards the binding pocket with an artificial, energetically unfavorable Tyr147-OH torsion angle of 120° 40, the docking program generated no poses where the thioester carbonyl binds to this residue. Similar to the PT12 and PT404 4’-nitro substituents, the amide group of the substrate analogs interacts with the Ala97 backbone nitrogen (d = 2.9 Å) and is additionally bound to Ser197 (Figures 4A-B). A comparison with the InhA-NAD+-C16-NAC structure revealed consistency with the experimental substrate binding mode (Figure 4C) 8.
Figure 4.
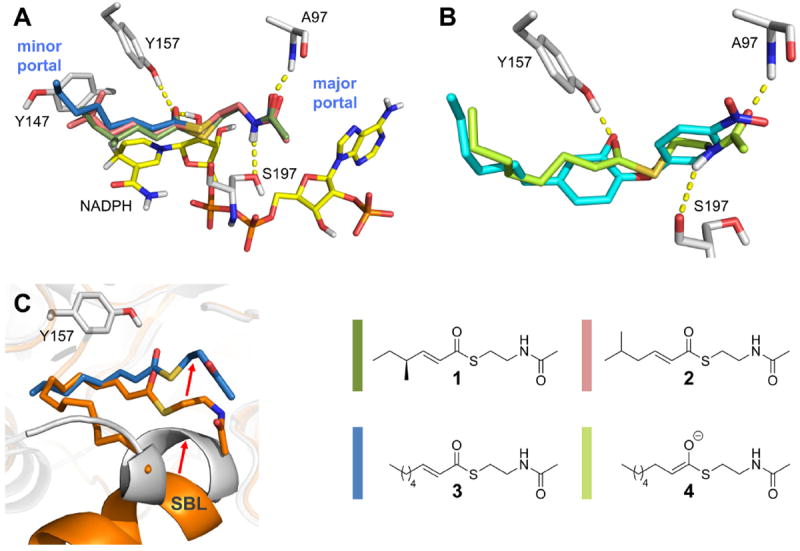
Putative substrate binding mode. (A) Enoyl-substrate analogs bound to saFabI-NADPH (gray). The selected docking poses for S-trans-4-methyl-2-hexenoyl-NAC (1), trans-5-methyl-2-hexenoyl-NAC (2) and trans-2-octenoyl-NAC (3) are depicted (color code according to the legend). (B) Model of the octanoyl-NAC enolate intermediate (4) bound to saFabI-NADP+ (gray). The inhibitor PT12 is shown as reference (cyan). (C) Comparison with the InhA-NAD+- C16-NAC complex (orange). The putative saFabI-NADPH-3 complex is shown in gray and blue, respectively. Red arrows indicate shifts in the substrate binding mode and SBL conformations.
Hydride Transfer and Proton Delivery
The putative saFabI-NADPH-3 complex is consistent with the transfer of the pro-4S hydride to the C3 atom of the substrate in the s-cis conformation (d = 3.9 Å), whereas the thioester carbonyl should be protected from reduction (d = 5.0 Å, Figures 1D and 5A). However, the question remains how the enolate-like TS is stabilized by the action of FabI. Clearly, the oxyanion hole is ideally suited to stabilize the evolving negative charge of the enolate. The 2’-OH of the nicotinamide ribose is involved in a hydrogen-bonding network consisting of Lys164 and two structurally and temporally conserved water molecules presumably leading to a delocalization of the evolving negative charge of the enolate (Figure 5A). Furthermore, the FabI enzyme contains a SBL, and the closure of a similar loop in triosephosphate isomerase (TIM) was proposed to be responsible for the stabilization of the two transition states 4, 41. In this regard, the desolvation of the binding site, which can be effectively achieved by the gradual closure of a flexible loop, was suggested to transfer the substrate from the solvent into a protein cavity characterized by a much lower dielectric constant, resulting in a strengthening of electrostatic interactions 2, 41. The negative charge of the enolate generated during the FabI-catalyzed reaction might therefore be most effectively stabilized upon full closure of the SBL. Importantly, a strong electrostatic interaction develops between the enolate and the evolving positive charge on the NAD(P)δ+-H nicotinamide ring (Figure 5B). Also several other enzymes take advantage of positively charged moieties to stabilize negatively charged intermediates including enolates 41, 42.
Figure 5.
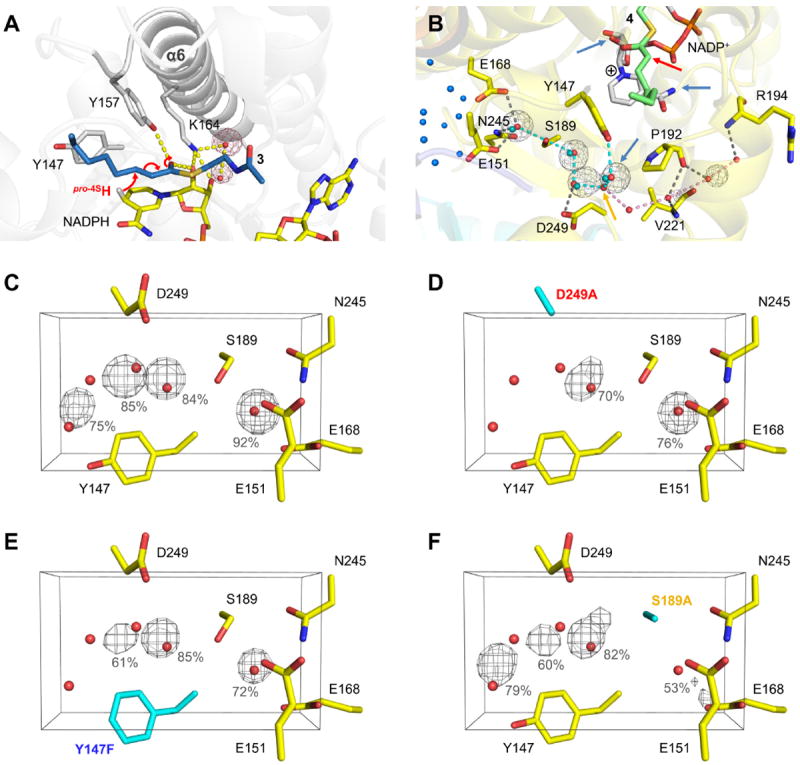
Hydride and proton transfer. (A) Hydride transfer. The pro-4S hydride of NADPH is transferred to the C3-atom of the substrate (indicated by red arrows; the putative saFabI-NADPH-3 complex is shown, see also Figure 4). Two highly conserved water molecules bound to Lys164 are depicted as red spheres. The corresponding water occupancy maps for one monomer of the saFabI4-NADP+-PT400 MD trajectory are depicted as red meshes at the 50% level. (B) Water-wire originating from Tyr147. In the saFabI-NADP+-PT400 structure, a chain of water molecules (red spheres) connects Tyr147 with a water basin inside the tetramer (blue spheres; cyan dashes) and with the bulk solvent (pink dashes), respectively. The branch between these two water-chains is indicated by the orange arrow. Whereas the water-chain towards the interior of the tetramer is highly conserved during the saFabI4-NADP+-PT400 MD simulation (occupancy maps are shown as gray meshes at 50%), the water-wire towards the exterior seems not to be established permanently. Blue arrows indicate possible proton sources and the red arrow the C2 atom of the enolate intermediate (4 shown in green, see Figure 4), which is protonated during the reaction. Cyan spheres represent water molecules of the ecFabI-NAD+-PT449 structure towards the inside of the tetramer. (C-F) Water occupancies for the wild-type and mutant (D249A, Y147F, S189A; highlighted in cyan) saFabI4-NADP+-PT400 MD simulations. The corresponding water occupancy maps (averaged for all monomers) are shown as gray meshes at the 50% level. Occupancies higher than 50% are given explicitly.
Since the saFabI enzyme has evolved to stabilize an enolate-like transition state, dissociation of the tightly bound enolate intermediate may limit the rate of catalysis. Thus, an alternate route to dissociation via protonation of the enolate intermediate is very important. To effectively protonate this intermediate in the desolvated saFabI binding pocket, a defined water-mediated connection to bulk solvent can be envisioned. Indeed, our saFabI structures reveal a conserved water-chain originating from Tyr147 and extending towards the inside of the homo-tetramer and the exterior of the protein (Figure 5B). We therefore propose the following model: After the transfer of the hydride, the natural fluctuations along the reaction coordinate might cause a slight opening of the SBL, which could be further triggered by the re-hydration of the binding site via this water chain. The water molecule directly bound to Tyr147 will be the first to enter the active site and due to steric reasons will only be able to intercalate between the enolate and nicotinamide, thus enabling the stereospecific protonation from the 2-re face (Figures 1D and 5B) 9, 10. In principle, the NADP+ amide group could be alternatively responsible for the protonation step, but its hydrogen atoms are involved in a tight hydrogen bonding network. In addition, an enol formed by protonation of the enolate oxygen via the oxyanion hole cannot be the final product dissociating from the enzyme due to the defined stereochemical outcome of the reaction 9. In fact, it has been proposed that water networks linking substrate binding sites with bulk solvent might be a general feature of all enzymes to enable protonation steps as well as dehydration and rehydration of the active site upon substrate binding and product release, respectively 43.
To improve our understanding of such networks, we investigated the FabI water-wire using a combined experimental and computational approach. Interestingly, our saFabI4-NADP+-PT400 MD simulation clearly indicates that only the water-chain towards the water chamber inside the tetrameric protein is conserved over time (Figure 5B, cyan dashes). However, the water molecule of the saFabI-NADP+-PT400 structure defining the branch of the water-wire towards the exterior of the protein (Figure 5B, orange arrow) is not observed in other related structures (such as the ecFabI-NAD+-PT449 solved in this study; cf. Figure 5B, cyan spheres) and is also quickly released during the saFabI4-NADP+-PT400 MD simulation, leading to the same network composed of four water molecules and Ser189 (Figure S5) as in the mentioned ecFabI structure. Residues Tyr147, Glu168 (rarely an aspartate), Ser189 and Asp249 involved in the formation of the water-chain are strictly conserved within the FabI-familiy (mycobacterial enoyl-reductases are an exception and have an alternative water-network 8). Thus, we explored the kinetic and dynamic behavior of the saFabI Y147F, S189A and D249A variants. A comparison of wild-type and variant saFabI4-NADP+-PT400 MD simulations (Table S4) clearly indicates that the water occupancies and, thus, the stability of the directed water-chain towards the interior of the tetramer are reduced in all three variants, most drastically in D249A (Figures 5C-F). Accordingly, the catalytic efficiency decreased from 8 to 10,000-fold for the water-channel variants (Table 2). Both thermal shift assays and CD-spectra suggest that each variant assumes a folded and stable structure (Table 3 and Figure S6). Pyridone- and diphenyl ether-based TS analogues 34 were used to probe the ability of the mutant saFabI enzymes to stabilize the TS of the hydride transfer. Thermal shifts of the S189A and D249A ternary complexes are significantly diminished, suggesting increased TS energies (Table 3). Moreover, the increased Km, oct-CoA suggests decreased binding affinity for the acyl-CoA substrate. Given that these residues are located outside of the active site and the substrate binding pocket, these findings suggest that the water chain may be crucial for SBL closure and, consequently, hydride transfer.
Table 2.
Kinetic characterization of saFabI water-channel variants
| Km,oct-CoA (μM) a,b | kcat (min-1) a | kcat/Km,oct-CoA (min-1 μM-1) | |
|---|---|---|---|
| wt saFabI c | 20.7 ± 2.7 | 2645.4 ± 107.3 | 127.8 ± 17.5 |
| saFabI D249A | > 100 | 0.012 ± 0.001 d | |
| saFabI S189A | > 100 | 1.50 ± 0.04 d | |
| saFabI Y147F | 44.2 ± 14.5 | 723.9 ± 73.3 | 16.4 ± 5.6 |
Km,oct-CoA and kcat values were determined at a fixed NADPH concentration (350 μM)
Oct-CoA = trans-2-octenoyl-CoA
Data obtained from 15.
Estimated based on linear slope of the Michaelis-Menten plot at low oct-CoA concentrations
Table 3.
| Wild-type saFabI | saFabI D249A | saFabI S189A | saFabI Y147F | |
|---|---|---|---|---|
| E | 62.6 | 63.4 | 61.2 | 63.2 |
| E-NADPH-CG400549 | 71.2 | 64.6 | 64.2 | 71.2 |
| E-NADP+-PT119 | 70.4 | 63.6 | 61.8 | 72.2 |
Although one could argue that these mutations might cause slight structural perturbations that may be transferred to the active site, the remote location and significant effects of the mutations suggest that more global processes are compromised. The particularly dramatic effect of the D249A mutation (>10,000-fold reduced catalytic efficiency) is consistent with our prediction that Asp249 has the greatest impact on water occupancy (Figure 5). We propose that water molecules, which are trapped in between the entering substrate and the left subpocket, cannot properly leave the binding site via the disrupted water-channel of the D249A and S189A variants, thereby hindering substrate binding and the closure of the SBL. Similar long-distance effects have been observed for other enzymes, such as dihydrofolate reductase, where mutations up to 20 Å away from the active site were suggested to impair catalytic activity by changing the mobility of loop regions that are important for catalysis 2.
Interestingly, the Y147F saFabI variant maintains a thermal shift similar to the wild-type enzyme upon ternary complex formation with a TS analogue (Table 3). This suggests comparable TS energies for hydride transfer in both mutant and wild-type enzymes. Given similar enzyme-substrate complex energies (reflected by Km, oct-CoA) and an unchanged TS energy, the energetic barrier to hydride transfer is expected to be very similar for the two enzymes. The experimental observation of a 4-fold smaller kcat for the Y147F variant may, therefore, be indicative of a new rate limiting step. One possibility is that we successfully raised the barrier to enolate protonation, thereby making it the new bottleneck step in saFabI catalysis.
Discussion
In the course of these studies we analyzed the inhibition, chemical binding-pocket properties and conformational dynamics of the saFabI enzyme. In each case the diphenyl ether inhibitors bind so that their phenolic oxygen atoms interact with the hydroxyl group of Tyr157 (Figure 2). We previously proposed that such inhibitors closely resemble the transition state for the reaction (TS-analogues)13 and, thus, anticipate that the substrate would also hydrogen bond to Tyr157. To support this hypothesis, we performed docking experiments and indeed found that the substrate thioester carbonyl oxygen binds to Tyr157 (Figure 4), and clearly not to the second catalytic tyrosine (Tyr147). In fact, our MD-simulations revealed that only Tyr157 is perfectly situated to donate a hydrogen-bond to the incoming substrate (Figure 3C). Together with the 2’-hydroxyl group of the cofactor, this residue can form an oxyanion hole, which seems to be crucial for the recognition of most known FabI-inhibitors (Figure 3A) and, by analogy, for the binding and activation of the substrate.
In contrast to Tyr157, the hydroxyl group of Tyr147 points away from the binding pocket towards a conserved water-chain (Figures 5B and S5). Importantly, we found that catalytic efficiency of the saFabI enzyme is compromised when mutating residues that are involved in the stabilization of this water wire. In particular, the water network is in contact with a water chamber inside the homo-tetrameric enzyme generating a path for efficient water and proton transport. Upon occlusion of the binding site by the presumably concerted movement of the flexible SBL and injection of the substrate, this path likely ensures the continuing contact to bulk solvent and is used for the desolvation of the binding pocket which is essential for catalysis. Furthermore, this path likely defines the proton source required for the protonation of the enolate intermediate and subsequent product release.
Based on the defined stereochemical outcome of the FabI-catalyzed reaction (Figure 1) and the finding that Tyr157, and not Tyr147, binds the thioester oxygen, the ACP substrate must be delivered via the major portal. Using small-molecule inhibitors as tools, we further probed this hypothesis and found that more hydrophilic moieties are tolerated in the right subpocket towards the major portal of the protein, whereas the left part of the binding cavity is very hydrophobic and prefers to accommodate lipophilic substituents (Figure 3). According to these polarity trends, the more hydrophilic PPant moiety of the substrate is expected to bind to the right subpocket whereas the lipophilic acyl chain is accommodated in the left part of the cavity. This is fully consistent with our substrate docking poses, which highlight the close analogy between recognition of the diphenyl ether inhibitors and of the FabI substrate: Figure 4B highlights the similarities between substrate and inhibitor recognition regarding the hydrophobic chain, the hydrogen bond with Tyr157 and the interaction with Ala97. Although our results clearly indicate that the substrate should be delivered via the major portal, the ecFabI-NAD+-DD-ACP structure suggested that the ACP residue Ser36 carrying the substrate is located adjacent to the minor portal 11. To resolve this issue, we first had a closer look at the intrinsic protein fluctuations since protein-protein interactions usually require structural adaptations 44. Our apo MD simulations revealed an anti-correlated opening movement of the SBL and helix α2, facilitating cofactor and substrate uptake and providing an ideal platform for the interaction with ACP adjacent to the major portal (cf. Figures S7 and S8, Movie S1 and Supplemental Results).
We therefore revisited the ecFabI-NAD+-DD-ACP structure (PDB-entry 2FHS) and found that ACP not only contacts the FabI molecule in the asymmetric unit, which was interpreted to be the biologically relevant interaction partner (Figure 6A-B, yellow), but also interacts with a symmetry-related FabI molecule (Figure 6A-B, gray). Since the complex with the symmetry-related monomer, in contrast to the original interpretation 11, enables the simultaneous binding of four ACP molecules per homo-tetramer without any clashes (Figures 6D-E), this could likely be the biologically relevant assembly assuming a 1:1 stoichiometry. We superimposed a decanoylecACP structure (PDB-entry 2FAE) onto the partial ACP model of the 2FHS-structure to create a full model for the FabI-ACP complex (Figure 6B), which displays a much larger interaction surface for the assembly with the symmetry-related monomer compared to the original FabI-ACP complex (344 vs. 93 Å2 according to the PISA-server 45, Figure 6C). In this new complex the acidic ACP is located at the major portal and can favorably interact with the positively charged FabI surface patches surrounding the SBL and helix α2 as proposed by our MD simulations (Figure S9; further supporting arguments for the new interpretation of the FabI-ACP complex structure can be found in the Supplemental Discussion section). Moreover, we note that for the closely related FabG protein, stereochemistry alone can reveal the location of ACP because the positions of the pro-4S hydride donor and β-keto acceptor groups are fixed (as depicted in Figure S10). Indeed, the R-β-hydroxyacyl-ACP is formed during the FabG-catalyzed reaction 46, providing evidence that ACP is located at the major portal for FabG as we propose for FabI.
Figure 6.
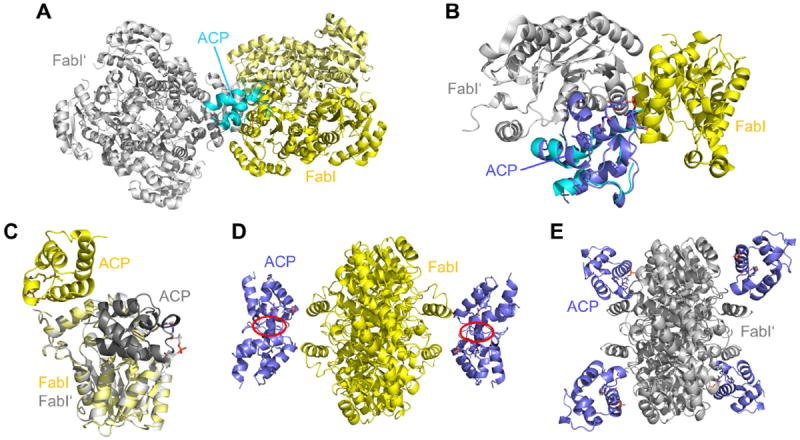
Interpretation of the ecFabI-ACP complex structure. (A) Symmetry-related ecFabI homo-tetramer (gray) within the ecFabI-NAD+-DD-ACP structure (yellow, PDB-code: 2FHS; two FabI monomers and one ACP in the asymmetric unit). The partial ACP model is depicted in cyan. (B) ACP environment in the 2FHS-structure. To derive a full ACP model, decanoyl-ecACP (lilac, PDB-code: 2FAE) was superimposed onto the partial ACP model of the 2FHS-structure considering only main chain atoms. (C) Superposition of the two crystallographically feasible FabI-ACP interaction models. The originally proposed model is depicted in yellow, the model derived from the interaction with a symmetry related FabI monomer is colored in gray. (D, E) FabI-ACP clash-test. The simultaneous interaction of the FabI homo-tetramer with four ACP substrates will lead to severe clashes for the original model (highlighted in red; panel D) 11, whereas no clashes are observed for the new model (panel E). These figures were created by superimposing both models (panel C) on each subunit of the full FabI homo-tetramer.
In summary, our results indicate that Tyr157, and not Tyr147, binds the thioester oxygen of the ACP substrate and thereby correctly positions the enoyl double bond relative to the cofactor for an effective reduction. This leads to a new FabI-ACP model fully consistent with our findings and supporting the notion that the substrate should enter via the major portal. This is also consistent with the structure of the C16-NAC substrate analog bound to the FabI protein from Mycobacterium tuberculosis 8. While Tyr157 is important for substrate recognition, Tyr147 seems to be involved in the protonation of the enolate intermediate by establishing a water-mediated connection between bulk solvent and the catalytic site. This new insight into substrate binding and turnover will now facilitate medicinal chemistry efforts aimed at mimicking important substrate moieties within the adaptable FabI binding pocket, such as the interaction of Ala97 with the terminal PPant amide, or at blocking the essential interaction of this enzyme with ACP.
Supplementary Material
Acknowledgments
We thank the staff at the BESSY II beamline 14.1 (Berlin) for technical support.
Funding Sources
This work was supported in part by the NIH grant GM102864 to P.J.T., and through the Deutsche Forschungsgemeinschaft to C.K. (SFB630 and Forschungszentrum FZ82) and C.A.S. (SFB630). A.C. was supported by the Chemical Biology Training Program (NIH grant T32GM092714) and by the Medical Scientist Training Program (NIH Grant T32GM008444). J.S. was supported by a grant of the German Excellence Initiative to the Graduate School of Life Sciences, University of Würzburg.
Abbreviations
- ACP
acyl carrier protein
- CAPS
3-(Cyclohexylamino)-1-propanesulfonic acid
- CD
circular dichroism
- CoA
coenzyme A
- DD
dodecenoyl
- ecFabI
Escherichia coli enoyl-ACP reductase
- DMSO
dimethyl sulfoxide
- EDTA
ethylenediaminetetraacetic acid
- FabI
trans-2-enoyl-ACP reductase
- FabG
β-ketoacyl-ACP reductase
- FAS-II
type 2 fatty acid biosynthesis
- GAFF
general AMBER force field
- InhA
Mycobacterium tuberculosis enoyl-ACP reductase
- MD
molecular dynamics
- MPD
2-methyl-2,4-pentanediol
- MRSA
methicillin-resistant Staphylococcus aureus
- NAC
N-acetylcysteamine
- NAD+
nicotinamide adenine dinucleotide
- NADH
nicotinamide adenine dinucleotide, reduced form
- NADP+
nicotinamide adenine dinucleotide phosphate
- NADPH
nicotinamide adenine dinucleotide phosphate, reduced form
- PDB
protein data bank
- PEG
polyethylene glycol
- PIPES
1,4-Piperazinediethanesulfonic acid
- PPant
phosphopantetheine
- RESP
restrained electrostatic potential
- saFabI
Staphylococcus aureus enoyl-ACP reductase
- SBL
substrate binding loop
- SDR
short-chain dehydrogenase/reductase superfamiliy
- TIM
triosephosphate isomerase
- TS
transition state
Footnotes
Supporting Information
Supporting Information Available. The supplement contains four tables, ten figures and a movie as well as supplemental experimental procedures, results and discussion sections. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Accession Codes
The coordinates and structure factors of saFabI in complex with NADP+ and PT12, PT400, PT404, PT449, or MUT37307 as well as ecFabI in complex with NAD+ and PT449 have been deposited in the PDB with the codes 4D41, 4D42, 4D43, 4D45, 4D44 and 4D46, respectively.
References
- 1.Schramm VL. Enzymatic transition states, transition-state analogs, dynamics, thermodynamics, and lifetimes. Annu Rev Biochem. 2011;80:703–732. doi: 10.1146/annurev-biochem-061809-100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- 3.Fieulaine S, Boularot A, Artaud I, Desmadril M, Dardel F, Meinnel T, Giglione C. Trapping conformational states along ligand-binding dynamics of peptide deformylase: the impact of induced fit on enzyme catalysis. PLoS Biol. 2011;9:e1001066. doi: 10.1371/journal.pbio.1001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knowles JR. Enzyme catalysis: not different, just better. Nature. 1991;350:121–124. doi: 10.1038/350121a0. [DOI] [PubMed] [Google Scholar]
- 5.Grimm C, Maser E, Mobus E, Klebe G, Reuter K, Ficner R. The crystal structure of 3alpha -hydroxysteroid dehydrogenase/carbonyl reductase from Comamonas testosteroni shows a novel oligomerization pattern within the short chain dehydrogenase/reductase family. J Biol Chem. 2000;275:41333–41339. doi: 10.1074/jbc.M007559200. [DOI] [PubMed] [Google Scholar]
- 6.White SW, Zheng J, Zhang YM, Rock The structural biology of type II fatty acid biosynthesis. Annu Rev Biochem. 2005;74:791–831. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- 7.Bell AF, Stratton CF, Zhang X, Novichenok P, Jaye AA, Nair PA, Parikh S, Rawat R, Tonge PJ. Evidence from Raman spectroscopy that InhA, the mycobacterial enoyl reductase, modulates the conformation of the NADH cofactor to promote catalysis. J Am Chem Soc. 2007;129:6425–6431. doi: 10.1021/ja068219m. [DOI] [PubMed] [Google Scholar]
- 8.Rozwarski DA, Vilcheze C, Sugantino M, Bittman R, Sacchettini JC. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J Biol Chem. 1999;274:15582–15589. doi: 10.1074/jbc.274.22.15582. [DOI] [PubMed] [Google Scholar]
- 9.Fillgrove KL, Anderson VE. Orientation of coenzyme A substrates, nicotinamide and active site functional groups in (Di)enoyl-coenzyme A reductases. Biochemistry. 2000;39:7001–7011. doi: 10.1021/bi0000566. [DOI] [PubMed] [Google Scholar]
- 10.Saito K, Kawaguchi A, Seyama Y, Yamakawa T, Okuda S. Steric course of reaction catalyzed by the enoyl acyl-carrier-protein reductase of Escherichia coli. Eur J Biochem. 1981;116:581–586. doi: 10.1111/j.1432-1033.1981.tb05375.x. [DOI] [PubMed] [Google Scholar]
- 11.Rafi S, Novichenok P, Kolappan S, Zhang X, Stratton CF, Rawat R, Kisker C, Simmerling C, Tonge PJ. Structure of acyl carrier protein bound to FabI, the FASII enoyl reductase from Escherichia coli. J Biol Chem. 2006;281:39285–39293. doi: 10.1074/jbc.M608758200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerusz V. Recent Advances in the Inhibition of Bacterial Fatty Acid Biosynthesis. In: John EM, editor. Annual Reports in Medicinal Chemistry. Academic Press; 2010. pp. 295–311. [Google Scholar]
- 13.Chang A, Schiebel J, Yu W, Bommineni GR, Pan P, Baxter MV, Khanna A, Sotriffer CA, Kisker C, Tonge PJ. Rational optimization of drug-target residence time: insights from inhibitor binding to the Staphylococcus aureus FabI enzyme-product complex. Biochemistry. 2013;52:4217–4228. doi: 10.1021/bi400413c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hiratake J. Enzyme inhibitors as chemical tools to study enzyme catalysis: rational design, synthesis, and applications. Chem Rec. 2005;5:209–228. doi: 10.1002/tcr.20045. [DOI] [PubMed] [Google Scholar]
- 15.Schiebel J, Chang A, Lu H, Baxter MV, Tonge PJ, Kisker C. Staphylococcus aureus FabI: inhibition, substrate recognition, and potential implications for in vivo essentiality. Structure. 2012;20:802–813. doi: 10.1016/j.str.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sivaraman S, Zwahlen J, Bell AF, Hedstrom L, Tonge PJ. Structure-activity studies of the inhibition of FabI, the enoyl reductase from Escherichia coli, by triclosan: kinetic analysis of mutant FabIs. Biochemistry. 2003;42:4406–4413. doi: 10.1021/bi0300229. [DOI] [PubMed] [Google Scholar]
- 17.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography. 1992;26 [Google Scholar]
- 18.Krug M, Weiss MS, Heinemann U, Mueller U. XDSAPP: a graphical user interface for the convenient processing of diffraction data using XDS. Journal of Applied Crystallography. 2012;45:568–572. [Google Scholar]
- 19.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 20.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 22.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 23.The PyMOL Molecular Graphics System, Version 1.3. Schrödinger, LLC; [Google Scholar]
- 24.Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Wang B, Hayik S, Roitberg A, Seabra G, Kolossvai I, Wong KF, Paesani F, Vanicek J, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. AMBER 11. University of California; San Francisco: 2010. [Google Scholar]
- 25.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, T V, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03. Wallingford CT; 2004. [Google Scholar]
- 26.Bayly CI, Cieplak P, Cornell W, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. The Journal of Physical Chemistry. 1993;97:10269–10280. [Google Scholar]
- 27.Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 29.Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Hayik S, Roitberg A, Seabra G, Swails J, Goetz AW, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. AMBER 12. University of California; San Francisco: 2012. [Google Scholar]
- 30.Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. Journal of Computational Physics. 1977;23:327–341. [Google Scholar]
- 31.Darden T, York D, Pedersen L. Particle mesh Ewald: An N [center-dot] log(N) method for Ewald sums in large systems. The Journal of Chemical Physics. 1993;98:10089–10092. [Google Scholar]
- 32.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 33.Tripos. SYBYL-X 1.0. 1699 South Hanley Rd.; St. Louis, Missouri, 63144, USA: 2009. [Google Scholar]
- 34.Schiebel J, Chang A, Shah S, Lu Y, Liu L, Pan P, Hirschbeck MW, Tareilus M, Eltschkner S, Yu W, Cummings JE, Knudson SE, Bommineni GR, Walker SG, Slayden RA, Sotriffer CA, Tonge PJ, Kisker C. Rational Design of Broad Spectrum Antibacterial Activity Based on a Clinically Relevant Enoyl-Acyl Carrier Protein (ACP) Reductase Inhibitor. J Biol Chem. 2014;289:15987–16005. doi: 10.1074/jbc.M113.532804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 36.Velec HF, Gohlke H, Klebe G. DrugScore(CSD)-knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction. J Med Chem. 2005;48:6296–6303. doi: 10.1021/jm050436v. [DOI] [PubMed] [Google Scholar]
- 37.Gerusz V, Denis A, Faivre F, Bonvin Y, Oxoby M, Briet S, Lefralliec G, Oliveira C, Desroy N, Raymond C, Peltier L, Moreau F, Escaich S, Vongsouthi V, Floquet S, Drocourt E, Walton A, Prouvensier L, Saccomani M, Durant L, Genevard JM, Sam-Sambo V, Soulama-Mouze C. From triclosan toward the clinic: discovery of nonbiocidal, potent FabI inhibitors for the treatment of resistant bacteria. J Med Chem. 2012;55:9914–9928. doi: 10.1021/jm301113w. [DOI] [PubMed] [Google Scholar]
- 38.Moreau F, Denis A, Faivre F, Bonvin Y, Gerusz V, Briet S, Oxoby M, Desroy N, Soulama-Mouze C, Floquet S, Genevard JM, Oliveira C, Escaich S. Intersci Conf Antimicrob Agents Chemother. American Society for Microbiology; Washington, DC: 2008. Design, Synthesis and Antibacterial properties of new potent Aryloxy-phenol FabI Inhibitors. [Google Scholar]
- 39.Bissantz C, Kuhn B, Stahl M. A medicinal chemist’s guide to molecular interactions. J Med Chem. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kossiakoff AA, Shpungin J, Sintchak MD. Hydroxyl hydrogen conformations in trypsin determined by the neutron diffraction solvent difference map method: relative importance of steric and electrostatic factors in defining hydrogen-bonding geometries. Proc Natl Acad Sci U S A. 1990;87:4468–4472. doi: 10.1073/pnas.87.12.4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malabanan MM, Amyes TL, Richard JP. A role for flexible loops in enzyme catalysis. Curr Opin Struct Biol. 2010;20:702–710. doi: 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jez JM, Ferrer JL, Bowman ME, Dixon RA, Noel JP. Dissection of malonyl-coenzyme A decarboxylation from polyketide formation in the reaction mechanism of a plant polyketide synthase. Biochemistry. 2000;39:890–902. doi: 10.1021/bi991489f. [DOI] [PubMed] [Google Scholar]
- 43.Meyer E. Internal water molecules and H-bonding in biological macromolecules: a review of structural features with functional implications. Protein Sci. 1992;1:1543–1562. doi: 10.1002/pro.5560011203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tobi D, Bahar I. Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc Natl Acad Sci U S A. 2005;102:18908–18913. doi: 10.1073/pnas.0507603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krissinel E. Crystal contacts as nature’s docking solutions. J Comput Chem. 2010;31:133–143. doi: 10.1002/jcc.21303. [DOI] [PubMed] [Google Scholar]
- 46.Glasfeld A, Leanz GF, Benner SA. The stereospecificities of seven dehydrogenases from Acholeplasma laidlawii. The simplest historical model that explains dehydrogenase stereospecificity. J Biol Chem. 1990;265:11692–11699. [PubMed] [Google Scholar]
- 47.Durrant JD, de Oliveira CA, McCammon JA. POVME: an algorithm for measuring binding-pocket volumes. J Mol Graph Model. 2011;29:773–776. doi: 10.1016/j.jmgm.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.