

Abstract
STAT3 is a critical transcription factor activated downstream of cytokine signaling and is integral for the function of multiple immune cell types. Human mutations in STAT3 cause primary immunodeficiency resulting in impaired control of a variety of infections, including reactivation of latent viruses. In this study, we investigate how T-cell functions of STAT3 contribute to responses to viral infection by inducing chronic lymphocytic choriomeningitis virus (LCMV) infection in mice lacking STAT3 specifically in T cells. Although mice with conditional disruption of STAT3 in T cells were able to mount early responses to viral infection similar to control animals, including expansion of effector T cells, we found generation of T-follicular helper (Tfh) cells to be impaired. As a result, STAT3 T cell deficient mice produced attenuated germinal center reactions, and did not accumulate bone marrow virus specific IgG-secreting cells, resulting in failure to maintain levels of virus-specific IgG or mount neutralizing responses to LCMV in the serum. These effects were associated with reduced control of viral replication and prolonged infection. Our results demonstrate the importance of STAT3 in T cells for the generation of functional long-term humoral immunity to viral infections.
Keywords: Chronic infection, Humoral immunity, LCMV, STAT3, T follicular helper cells
Introduction
JAK-STAT signaling is a major cytokine effector pathway critical for the development and function of different immune cell types [1, 2]. In humans, mutations in STAT3 cause autosomal dominant hyper IgE syndrome (AD-HIES), a disorder with immunological manifestations including pneumonia, skin infections, and susceptibility to Staphylococcus aureus and mucocutaneous candidiasis [3, 4]. In addition to misregulation of IgE, AD-HIES syndrome patients also have impaired long-term IgG production following immunization [5–7].
Recent reports indicate that STAT3 mutations are further associated with reactivation of Epstein-Barr virus and Varicella zoster virus, suggesting that long term control of viruses may require STAT3 [8, 9]. However, a precise mechanistic understanding of viral responses in the absence of STAT3 is lacking.
STAT3 deficiency in humans is associated with reduced quantities of CD8+ memory T cells [8, 10]. Although memory CD8+ T-cell defects have also been observed in mice with CD8+ T-cell-deletion of STAT3 (GzB-cre+; Stat3fl/fl), effector function and ability to control primary acute lymphocytic choriomeningitis virus (LCMV) infection is preserved [11].
Antigen experienced CD4+ T follicular helper (Tfh) cells seed germinal center reactions during infection. Germinal center reactions promote differentiation of follicular B cells into antibody producing long-lived plasma cells and memory B cells [12, 13]. STAT3 is an important mediator of signaling for cytokines involved in the generation of Tfh cells including IL-6 and IL-21 [12, 13]. CD4-Cre conditional STAT3 knockout mice were recently examined following acute LCMV infection, revealing Tfh cell defects resulting in limited germinal center reactions [14]. However, these effects have not yet been examined in a chronic model of LCMV infection. Importantly, AD-HIES patients are also known to have reduced quantities of circulating Tfh cells [3, 15, 16].
IL-21 signaling via STAT3 in germinal centers is important for generation of plasma cells. LCMV infection of IL-21 deficient mice revealed a failure to maintain long lived virus specific IgG plasma cells in the bone marrow, effects which appear to be both T-cell and B-cell-dependent [17]. These data support a model whereby STAT3 is involved in both differentiation of Tfh cells to produce IL-21, and subsequent IL-21 signaling via STAT3 in B cells promotes differentiation into plasmablasts [3, 14, 18]. The direct role of T-cell STAT3 in maintenance of virus specific IgG-producing plasma cells during chronic infection has not yet been reported.
In order to better understand STAT3 function in T cells during viral infection, we chose to examine mice lacking T-cell STAT3 infected with chronic LCMV. Whereas initial control of viral infection including generation of virus specific T cells and production of virus specific antibodies was largely normal in the absence of STAT3, reduced quantities of Tfh cells were present. At later time points, STAT3 deficiency resulted greatly diminished accumulation of virus-specific IgG-producing cells in the bone marrow, and an inability to produce LCMV neutralizing antibodies or maintain serum levels of virus-specific IgG. These defects were associated with impaired long-term control of LCMV infection and reduced survival.
Results
STAT3 is dispensable for generation of virus-specific T cells but necessary for Tfh cells
STAT3 is critically required for development as homozygous Stat3 deficient mice arrest early during embryogenesis [19]. In an attempt to understand the role of STAT3 in T cells we investigated a model employing transgenic Lck-Cre to conditionally inactivate Stat3 across all T-cell lineages [20, 21]. Lck-Cre− Stat3fl/fl (referred to as WT in figures for simplicity) mice were indistinguishable from Lck-Cre+ Stat3fl/fl (also referred to as Stat3−/−) littermate controls in terms of appearance and long-term survival (Supporting Information Fig. 1A). We investigated basal levels of B cells, T cells and NK cells in the spleen of these mice, finding no major defects in cell population numbers associated with presence or absence of STAT3 (Fig.1A). Similarly, thymic T-cell populations were largely equivalent for Lck-Cre+ Stat3fl/fl and Lck-Cre− Stat3fl/fl mice indicating grossly normal T-cell development (Supporting Information Fig. 1B, C; also see general flow cytometry gating strategy in Supporting Information Fig. 2).
Figure 1.
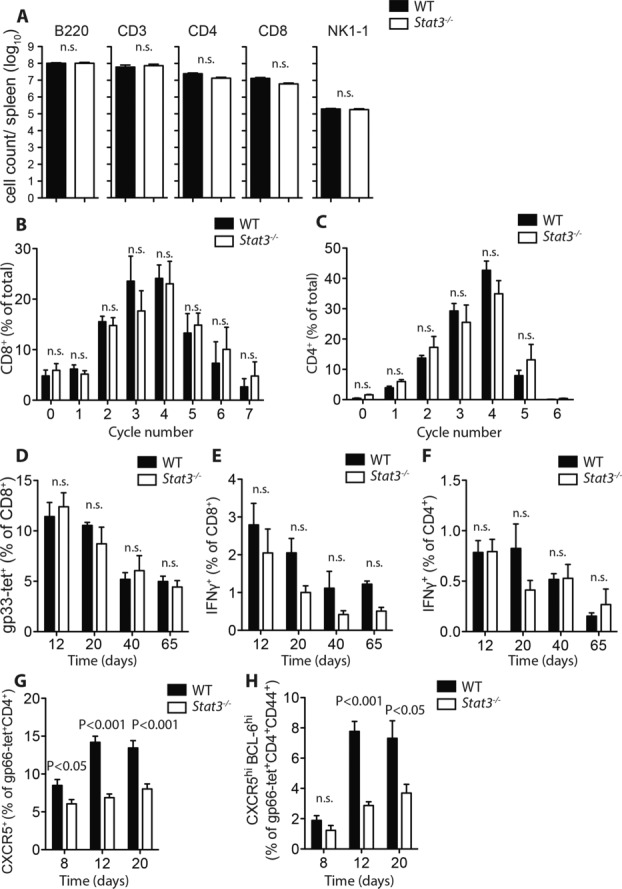
Generation of LCMV-specific Tfh cells requires STAT3. (A) Quantity of B220+, CD3+, CD4+, CD8+, and NK1-1+ cells were compared in naive spleens from Lck-Cre− Stat3fl/fl (WT) vs. Lck-Cre+ Stat3fl/fl (Stat3−/−) mice. (B–C) Quantification of CFSE dilution indicative of proliferation for Lck-Cre− Stat3fl/fl vs. Lck-Cre+ Stat3fl/fl (B) CD8+ T cells or (C) CD4+ T cells (both pregated on CD3+) in vitro. Percentage of cells in each division cycle for negatively selected, CFSE labeled Lck-Cre− Stat3fl/fl vs. Lck-Cre+ Stat3fl/fl T cells stimulated with 5 μg/mL anti-CD3 and 2 μg/mL anti-CD28 for 72 h as measured by flow cytometry. (D–H) Lck-Cre− Stat3fl/fl vs. Lck-Cre+ Stat3fl/fl mice were infected with 2 × 103 PFU LCMV-Docile. (D) Quantity of gp33 tetramer-specific CD8+ T cells in the spleen at days 12, 20, 40, and 65 after infection was measured (% of total CD8+ T cells). (E) Quantity of IFN-γ-producing CD8+ T cells present at days 12, 20, 40, and 65 after infection following in vitro restimulation with virus specific peptide gp33. (F) Quantity of IFN-γ-producing CD4+ T cells present at days 8, 12, 20, 40, and 65 after infection following in vitro restimulation with virus specific peptide gp61. (G) Quantity of virus-specific CXCR5+ cells in spleen at days 8, 12, and 20 postinfection was measured (% of gp66-tet+CD4+ T cells). (H) Quantification of CXCR5hiBCL-6hi virus-specific cells in spleen at days 12 and 20 postinfection (% of gp66-tet+CD44+CD4+ T cells). (A–H) Data are shown as mean ± SEM from (A) n = 5–6 mice/group, (B and C) n = 7 mice/group, (D–F) n = 4–7 mice/group, (G) n = 6 mice/group, (H) n = 3–4 mice/group. Data are pooled from two independent experiments (A–C), or pooled from one or two independent experiments per time point (D–H). Statistical significance between groups was determined by Student's t-test.
In order to better understand the function of STAT3 in T cells during antiviral immunity, we chose to infect mice with the LCMV-Docile strain which can result in a chronic course of infection [22, 23]. Control of LCMV infection relies on expansion of virus specific T cell populations. Therefore we first examined the ability of isolated T cells from Lck-Cre+ Stat3fl/fl and Lck-Cre− Stat3fl/fl mice to respond to non-specific proliferative signals. CD8+ and CD4+ T cells were isolated from spleens of uninfected Stat3fl/fl Lck-Cre+ Stat3fl/fl and Lck-Cre− Stat3fl/fl mice and cultured for 72 h in the presence of anti-CD3 and anti-CD28 activating antibodies. CFSE labeling revealed that both control and STAT3 deficient CD8+ T cells proliferated to the same extent (Fig.1B). Similar results were found when examining proliferative capacity of CD4+ T cells (Fig.1C). Next, we examined T-cell responses between 12 and 65 days following infection of mice with 2×103 PFU of LCMV-Docile. Following infection, comparably large numbers of virus specific CD8+ T cells recognizing the LCMV glycoprotein gp33 were evident in splenocytes isolated from both Lck-Cre+ Stat3fl/fl and Lck-Cre− Stat3fl/fl animals (Fig.1D). The ability of CD8+ and CD4+ T cells to be activated in response to LCMV peptides was tested by measuring interferon gamma (IFN-γ) production following re-stimulation with gp-33 and gp-61 peptides. Similar IFN-γ responses were detected following re-stimulation of both CD8+ and CD4+ T cells from infected Lck-Cre+ Stat3fl/fl and Lck-Cre− Stat3fl/fl mice (Fig.1E, F). As expected, we observed an overall decline in the quantity of virus specific cells and re-stimulated IFN-γ+ CD8+ and CD4+ T cells between 12 and 65 days post-infection in both the presence or absence of STAT3 (Fig.1D–F). These results indicate that in this setting, basal immune cell populations and the ability of the majority of T cells to be activated in response to chronic LCMV-Docile infection are not affected by the absence of STAT3. These results are also consistent with normal initial responses to acute infection with the LCMV-Armstrong strain (prior to the emergence of memory CD8+ defects) seen in CD8+ Stat3 conditional mice knockout mice (Granzyme B Cre+ Stat3fl/fl) [11]. However, similar to recent observations involving deletion of STAT3 in T cells [14], we observed reduced C-X-C chemokine receptor type 5 (CXCR5+) and BCL6+ virus specific CD4+ T cells in infected Lck-Cre+ Stat3fl/fl vs. Lck-Cre− Stat3fl/fl mice, indicating diminished Tfh populations at days 12 and 20 postinfection (Fig.1G and H).
STAT3 in T cells is necessary for sustained humoral immunity to LCMV
Since Tfh cells support germinal center reactions [13, 24, 25], we next examined the status of B-cell immunity to chronic LCMV in the presence or absence of STAT3. Total B-cell numbers in the spleen were similar across both types of animals (Fig.2A), as were proliferative responses of B cells to LPS or CD40-L (Supporting Information Fig. 3A, B), indicating that T cell specific deletion of STAT3 did not result in gross B-cell defects. Germinal center reactions were strongly induced in both genotypes, with similar levels of germinal center B cells present at day 12 following infection (Fig.2B). However, compared to controls, the quantity of germinal center B cells detectable in STAT3 deficient animals was markedly lower on days 20, 40, and 65 postinfection, indicating that normal germinal center reactions are not sustained in the absence of T cell STAT3 (Fig.2B–D).
Figure 2.
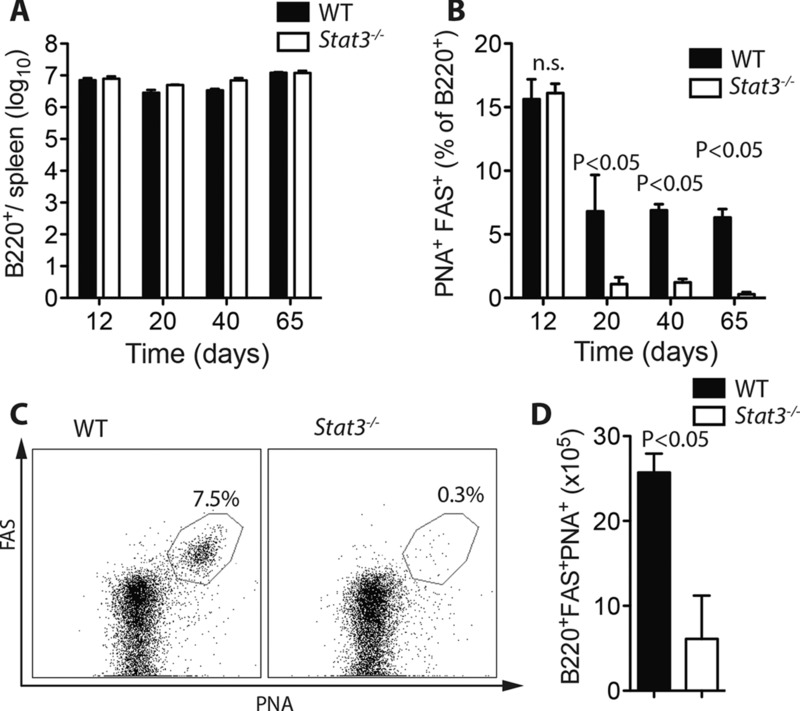
T-Cell STAT3 is required for the maintenance of germinal center B-cell reactions toward LCMV. (A–D) Lck-Cre− Stat3fl/fl vs. Lck-Cre+ Stat3fl/fl mice were infected with 2 × 103 PFU LCMV-Docile. (A) Total quantity of (B220+) B cells in the spleen on days 12, 20, 40, and 65 postinfection was measured by flow cytometry (mean ± SEM; n = 4–7 mice/group). (B–D) GC B cells (B220+PNA+FAS+) were measured on days 12, 20, 40, and 65 postinfection (n = 4–7 mice/group). (B) Quantity of B220+PNA+FAS+ cells as a percent of total B cells (B220+) at each time point (mean ± SEM), (C) representative dot plot from day 65 (gated on B220+ cells), and (D) numerical quantification of B220+PNA+FAS+ cells in each group at day 65 (mean ± SEM). Data are pooled from —one or two independent experiments per time point. Statistical significance between groups was determined by Student's t-test.
We next examined the kinetics of LCMV-GP specific IgG production in the serum by ELISA. Lck-Cre+ Stat3fl/fl and Lck-Cre− Stat3fl/fl mice had similar production of virus-specific IgG up until 20 days postinfection, however, while LCMV-GP-IgG titers continued to increase in control animals, titers progressively declined in the absence of STAT3, evident at time points up until 250 days postinfection (Fig.3A). STAT3 associated impairment in LCMV-specific antibody production was not evident for virus specific serum IgM (Fig.3B), suggesting that preclass switch B cell functions remained intact in T-cell STAT3 deficient animals. Serum antibodies with the capacity to neutralize LCMV appear with delayed kinetics relative to virus specific IgG in wild-type mice [26]. Consistently, we observed emergence of robust LCMV neutralization capacity in serum from infected control mice at day 65, which was maintained until at least day 250 (Fig.3C). In sharp contrast, neutralizing capacity of serum from STAT3 deficient animals was nearly undetectable across all time points (Fig.3C). Sustained IgG production is facilitated by long-lived IgG-producing plasma cells typically residing in the bone marrow [27]. Therefore, we measured the quantity of LCMV-specific IgG antibody-secreting cells (ASCs) present in both the spleen and bone marrow between zero and 65 days postinfection. Quantities of LCMV IgG ASCs appeared highest in the spleen at 12 days postinfection and were not significantly different between Lck-Cre+ Stat3fl/fl and Lck-Cre− Stat3fl/fl mice at any of the time points tested (Fig.3D). In contrast, STAT3 deficient mice possessed only minimal quantities of LCMV-specific IgG ASCs in the bone marrow compared to control mice (Fig.3E), consistent with the inability to sustain high serum titers of LCMV-specific IgG at later time points after infection. Taken together these data indicate that while early B-cell responses to chronic LCMV infection appear unaffected, absence of T-cell STAT3 is associated with reduced maintenance of germinal centers, failure to sustain serum virus-specific IgG levels or gain neutralization capacity and reduced accumulation of long lived virus specific IgG-producing cells in the bone marrow.
Figure 3.
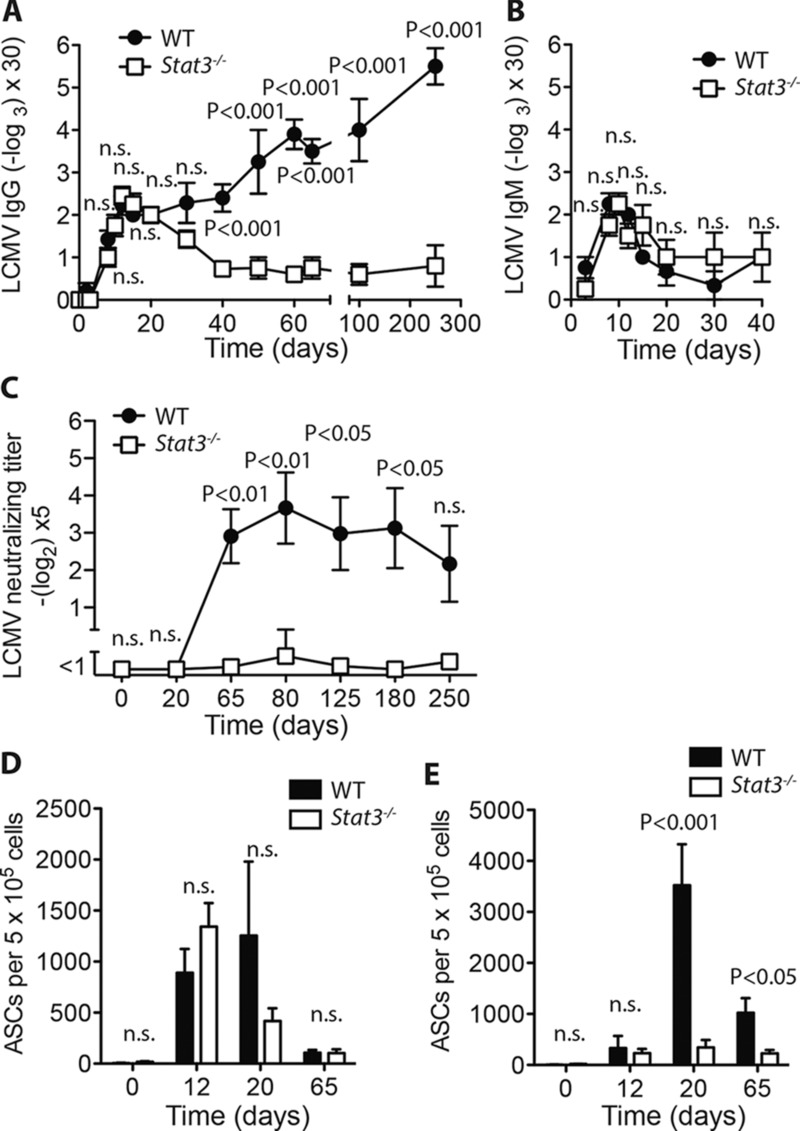
T-cell STAT3 is necessary for humoral immunity to chronic LCMV. (A–E) Lck-Cre− Stat3fl/fl vs. Lck-Cre+ Stat3fl/fl mice were infected with 2 × 103 pfu LCMV-Docile. (A) LCMV-specific IgG antibodies in serum were measured by serial sampling and ELISA on days 0, 2, 3, 8, 10, 12, 15, 20, 30, 40, 50, 60, 65, 100, and 250 postinfection (mean ± SEM; n = 4–15 mice/group (B) LCMV-specific IgM antibodies in serum were measured by serial sampling and ELISA at days 3, 8, 10, 12, 15, 20, 30, and 40 postinfection (mean ± SEM, n = 3–4 mice/group). (C) LCMV- neutralizing antibodies were measured in the serum after serial sampling on days 0, 20, 65, 80, 125, 180, 250 postinfection (mean ±SEM; n = 4–10 mice/group). (D–E) LCMV-specific ASC in (D) spleen or (E) bone marrow were counted on day 0, 12, 20, and 65 postinfection by ELISPOT (mean ± SEM; n = 4–7 mice/group). Data shown are pooled from (A) three independent experiments, (B and C) two independent experiments, or (D–E) —one or two experiments per time-point. Statistical significance between groups was determined by Student's t-test, significance between multiple time points, was determined by two-way ANOVA with Bonferroni posttest.
STAT3 T-cell deficiency results in impaired control of chronic LCMV
Cytotoxic T-cell responses toward LCMV are well described, yet viral persistence occurs in mice with impaired Tfh responses and impaired virus-specific antibody formation, indicating that humoral immunity is also important to control LCMV infection [14, 26, 28]. We therefore examined whether defects observed in Tfh cells and virus specific serum IgG production in the absence of STAT3 would be associated with reduced control of LCMV replication. Viral titers were measured in spleen, liver, lung, kidney, and brain of Lck-Cre+ Stat3fl/fl and Lck-Cre− Stat3fl/fl mice between 12 and 65 days postinfection. Virus was detected in all organs and was present in similar quantities in samples from control and STAT3 deficient mice at days 12 and 20 following infection (Fig.4A–E). However, by day 65 while many control mice had successfully cleared the infection, viral titers remained significantly elevated in organs from STAT3 deficient animals (Fig.4A–E). Particularly large differences between Lck-Cre+ Stat3fl/fl and Lck-Cre− Stat3fl/fl mice were evident in the liver (Fig.4B). Liver sections from these mice harvested at 65 days postinfection showed evidence of lymphocyte infiltration by hematoxylin and eosin staining in both settings, but strong LCMV-NP staining only in STAT3 deficient livers (Fig.4F). Taken together, viral replication appears to occur at normal levels in the initial phases following LCMV infection where CTL and early B-cell responses are also not significantly different between control and STAT3 deficient mice, however virus persists at later time points in STAT3 deficient animals coinciding with declining virus specific IgG levels.
Figure 4.
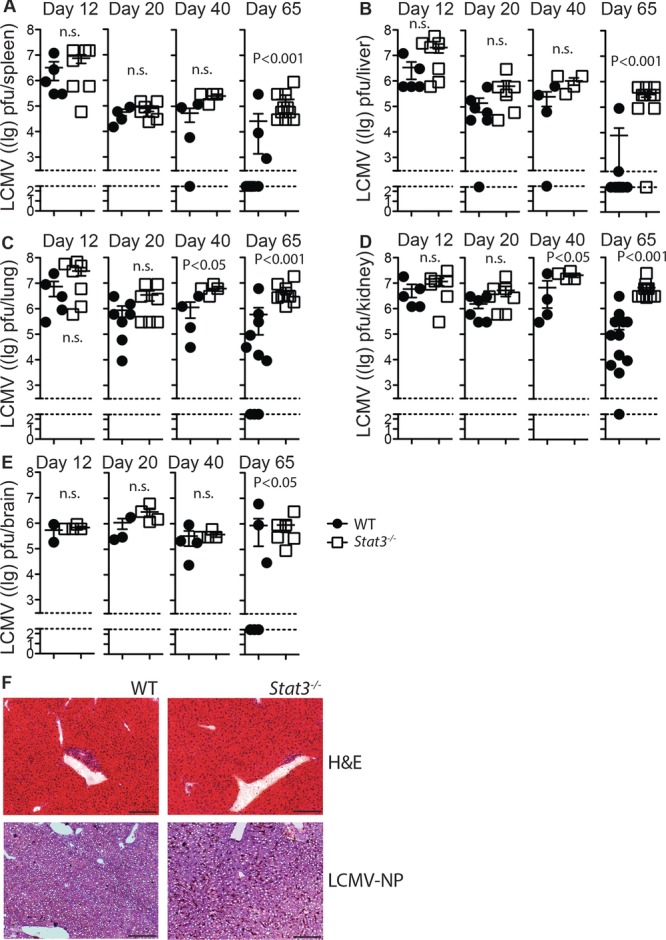
Long-term control of LCMV replication requires STAT3 in T cells. (A–F) Lck-Cre− Stat3fl/fl (closed circles) vs. Lck-Cre+ Stat3fl/fl (open squares) mice were infected with 2 × 103 pfu LCMV-Docile. On day 12, 20, 40, and 65 postinfection virus titer were determined in (A) spleen, (B) liver, (C) lung, (D) kidney, and (E) brain tissue (symbols represent individual mice, bars show mean ± SEM, n = 4–12 mice/group, data shown are pooled from one or two independent experiments per time point). (F) Snap-frozen liver sections from Lck-Cre− Stat3fl/fl and Lck-Cre+ Stat3fl/fl animals were stained with hematoxylin and eosin (upper panels), or immunostained for LCMV-NP with hematoxylin counterstaining (lower panels). One representative image from n = 4 mice/group from a single experiment is shown (10×, scale bar, 200 μm). Statistical significance between groups was determined by Student's t-test.
Prolonged infections of noncytolytic viruses can lead to toxic immunopathology [29–31]. When we tested long-term survival following LCMV infection we found that STAT3 deficiency was associated with significantly earlier demise (Fig.5). Thus impaired Tfh function, reduced humoral immunity, and diminished control of viral replication appears to reduce long-term survival of Lck-Cre+ Stat3fl/fl vs. Lck-Cre− Stat3fl/fl mice following chronic LCMV infection.
Figure 5.
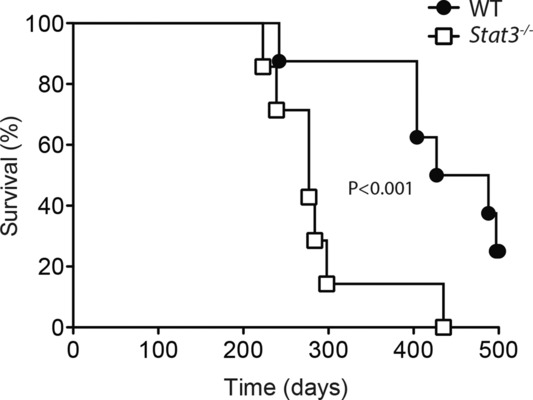
Reduced longevity of chronically infected STAT3 T-cell-deficient mice. Lck-Cre− Stat3fl/fl vs. Lck-Cre+ Stat3fl/fl mice were infected with 2 × 103 PFU LCMV-Docile. Survival of infected mice was monitored over time (n = 7–8 mice/group). Data shown are pooled from two independent experiments. Statistical significance was determined by Kaplan–Meier log rank test.
Discussion
In the setting examined here, chronic infection with LCMV revealed STAT3 to be dispensable for expansion and IFN–γ production by virus specific CD4+ and CD8+ T cells (Fig.1D–F). Mice lacking IL-21, a critical activator of STAT3, also generate normal levels of virus specific cytokine producing CD4+ T cells [17]. Furthermore, studies examining early time points after acute LCMV infection using mice with individual conditional deletion of STAT3 in either CD4+ [14] or CD8+ cells (Granzyme B-Cre) [11] corroborate that, although memory CD8+ T-cell responses are perturbed, virus specific CD4+ and CD8+ effector cells are generated in the absence of STAT3 signaling.
Despite similar overall quantities of T cells responding to virus in both genotypes, in the absence of STAT3 we observed a specific decrease in CXCR5+ Tfh cells during chronic LCMV infection, which are necessary to prime germinal center B cells (Fig.1G and H). These results are similar in magnitude to decreased Tfh cell populations recently observed during acute LCMV infection of CD4-Cre+ Stat3fl/fl mice [14]. Results are further consistent with previous work showing the importance of STAT3 in Tfh-cell function during acute LCMV infection in T cell transfer models [32], or following KLH antigen immunization [33]. Importantly, humans with STAT3 mutations also have reduced CD4+ CXCR5+ Tfh cells in vivo and defects in the ability of CD4+ T cells to help B cells in vitro [3, 15, 16].
Germinal center B cells were present at similar levels to control early after infection, however these declined more quickly in STAT3 deficiency (Fig.2B–D). Thus Tfh cells generated in response to LCMV in STAT3 deficient animals are able to seed, but not maintain germinal center reactions in the spleen. STAT3 deficiency was associated with reduced quantities of Tfh cells relative to controls, with differences more apparent at later stages after infection (Fig.1G). Interestingly, although the magnitude of the observed Tfh defect appeared similar between 12 and 20 days postinfection, the quantity of germinal center B cells dropped dramatically in STAT3 deficient mice during this same time frame (Figs. 1G–H and 2B–D). While Tfh cells in STAT3 deficient mice may not have reached sufficient numbers to trigger maintenance of germinal center B-cell reactions, this data raises an interesting alternative possibility for future investigation— that following infection, the functional capacity of Tfh cells may be impaired and progressively decline in the absence of STAT3. The kinetics of virus specific IgG production and germinal center involution in Lck-Cre+ Stat3fl/fl mice are also strikingly similar to those reported for mice lacking IL-21, suggesting that T cell STAT3 is a critical mediator of IL-21 in supporting germinal center reactions and long lived antiviral IgG production [17].
Mice with deletion of STAT3 in CD8+ T cells are able to control viral replication during primary LCMV infection [11]. Interestingly we find that mice lacking STAT3 throughout the T-cell compartment also have similar viral load to control animals at early time points postinfection, but fail to control viral replication at later stages (Fig.4A–E). Reduced ability to control chronic LCMV at later time points coincided with a decline in virus specific serum IgG from control levels at day 20, to dramatically lower levels by day 65 postinfection in STAT3 deficient animals (Figs.3A, and4A–E). These kinetics may be further explained by the emergence of serum antibodies with strong capacity to neutralize LCMV by day 65 postinfection in control mice, whereas neutralizing antibodies were nearly undetectable in STAT3 deficient mice for the duration of the observed period (Fig.3C).
Ray, et al. also recently observed decreased serum virus-specific IgG in the absence of STAT3 in T cells [14]. In addition to these findings, here we identify T-cell-specific expression of STAT3 to be important for the elimination of LCMV. Virus specific antibodies are known to be necessary to control persistent LCMV infection [26] and chronic infection can result in the demise of the host [30, 34]. Consistently, we observed reduced long term survival of infected Lck-Cre+ Stat3fl/fl mice compared to Lck-Cre− Stat3fl/fl controls (Fig.5).
Maintenance of long-term IgG responses to LCMV depend on accumulation of plasma cells in the bone marrow, independent of memory B cells [27]. We found significantly lower quantities of LCMV specific ASCs appearing in the bone marrow during STAT3 deficiency at later time points postinfection, coincident with declining serum anti-LCMV IgG (Fig.3E). Similar effects and kinetics have been observed in the absence of IL-21 [17]. Thus long-lived plasma cell maintenance and/or migration to the bone marrow appears to depend on both STAT3 production in T cells and IL-21 during LCMV infection.
Our observations support a model whereby limited IgG production in immunized AD-HIES patients reflects a failure to maintain long-lived plasma cells in the bone marrow [5–7]. In patients with inherited systemic mutations in STAT3, this could be a consequence of Tfh cell intrinsic defects, B-cell-intrinsic defects, or a combination of both. Nevertheless, our results demonstrate that absence of STAT3 in T cells is sufficient to prevent normal Tfh-cell development, block accumulation of long lived virus-specific antibody secreting cells in the bone marrow, and prevent maintenance of antiviral serum IgG.
Interestingly, in a report by Sheerin et al., primary antibody response to immunization in some AD-HIES patients was normal at early time points, but later declined below physiological levels [5], mimicking results seen in our mice. Similar defects to those observed in our mouse model may contribute to re-emergence of latent viral infections in humans with STAT3 mutations.
Materials and methods
Mice, viruses, virus titer
Mice used in this study were maintained on the C57BL/6-J genetic background. Stat3 fl/fl conditional knockout mice [20] and Lck-Cre transgenic mice [21] have been previously described. LCMV DOCILE (provided by R. M. Zinkernagel, University of Zurich) was propagated in L929 cells as previously described [35]. Virus titers were measured using a plaque forming assay also as previously described [36]. Mice were infected intravenously with 2 × 103 plaque forming units (PFU) of LCMV DOCILE. All experiments were performed in single ventilated cages. Animal experiments were carried out in accordance with the guidelines of the Ontario Cancer Institute and the German law for animal protection.
Purification of T cells and B cells
Total T-cell populations were purified from single cell suspended splenocytes by negative selection, and B220+-cell populations were purified from single cell suspended splenocytes by positive selection, both following the manufacturer's instructions (mouse Pan T cell Isolation Kit II, CD45R (B220) MicroBeads, mouse, Miltenyi, Bergisch Gladbach, Germany).
In vitro B-cell proliferation
Isolated B cells were labeled with 5-(and 6)-carboxyfluorescein diacetate succinimidyl ester (CFSE, Invitrogen, Carlsbad, CA, USA) for 10 min at 37°C. Labeled B cells were washed two times with RPMI 1640 culture medium (Biochrom, Berlin, Germany) with supplements (10% FCS, Biochrom, Berlin, Germany). B cells were cultured in 96-well flat-bottom tissue culture plates with either 100 ng/mL LPS or 10 ng/mL Il-4 (R&D Systems, Minneapolis, MN, USA) and 2 μg/mL CD40-L (2 × 105 cells/well). Cells were cultured at 37°C in humidified 5% CO2 incubator for 72 h prior to analysis by flow cytometry.
In vitro T-cell proliferation
Isolated CFSE-labeled T cells were cultured in 96 well flat-bottom tissue culture plates precoated with 5 μg/mL anti-CD3 (eBioscience, San Diego, CA, USA) and 2 μg/mL soluble anti-CD28 (BD Pharmingen, Franklin Lakes, New Jersey, USA) (2 × 105 cells/well). Cells were cultured at 37°C in humidified 5% CO2 incubator for 72 h prior to analysis by flow cytometry.
Flow cytometry analysis
gp33-H-2Db and np396-H-2Db tetramer production, surface, and intracellular staining, and analysis were performed as previously described [35]. Intracellular staining for IFN-γ was performed on single cell suspended splenocytes incubated with LCMV specific peptides (gp33 or gp61) for 1 h at 37°C followed by addition of Brefeldin A (eBioscience, San Diego, CA, USA) and subsequent incubation for 5 h at 37°C. After surface staining, cells were fixed with 2% formalin and permeabilized with 0.1% Saponin (Sigma-Aldrich, St. Louis, MO, USA) prior to staining with intracellular antibodies. Gp66-I-A(b) tetramer staining was performed for 30 min at 37°C prior to surface antibody staining at 4°C. Anti-CD3, anti-CD8, anti-IFN-γ, anti-CD4, anti-B220, anti-NK1.1, anti-CD25, anti-CD44, and anti-CD95 were obtained from eBioscience, San Diego, CA, USA. Anti-CXCR5 was obtained from BD Bioscience. PNA was procured from Vector Laboratories, Burlingame, CA, USA.
LCMV antibody Elisa
Analysis of LCMV glycoprotein specific antibodies was performed as previously described [37]. Briefly, plates were coated overnight with anti-human Fc (Jackson ImmunoResearch Laboratories, West Grove, PA, USA), blocked for 120 min with PBS + 2%BSA at RT and incubated with 100 μL/well Gp1-Fc supernatant overnight at 4°C. Sera were serial diluted on the plate in PBS + 2% BSA and incubated for 90 min at RT. Plates were washed with PBS + 0.1% Tween-20 (PBS-T) and diluted antimouse IgG (γ-chain specific)-HRP or anti-mouse IgM-HRP (Sigma-Aldrich, St. Louis, MO, USA) was added for 1 h at RT. Plates were washed and stained with 0.1M NaH2P04 (pH = 4) + ABST (10 102 946 001, Roche Diagnostics, Basel, Switzerland). Plates were read at 405 nm.
Histology
Immunohistochemistry was performed on snap-frozen samples. Tissue sections were stained with a monoclonal rat antibody recognizing LCMV-NP (clone: VL-4) and a biotinylated anti-rat secondary antibody(eBioscience, San Diego, CA, USA). Detection was performed by incubation with Streptavadin-peroxidase (Thermo Scientific, Waltham, MA, USA) and ImmPACT Nova Red substrate (Vector Laboratories, Burlingame, CA, USA). Immunohistochemistry sections were counterstained with hematoxylin (Vector Laboratories, Burlingame, CA, USA). Hematoxylin and Eosin (Sigma-Aldrich, St. Louis, MO, USA) staining was performed by standard methods on similarly processed tissue sections.
ASC (ELISPOT) LCMV assay
ELISPOT assay was performed as previously described [17]. Briefly, filter plates were coated overnight at RT with LCMV-infected BHK-cell lysates. Plates were washed with PBS-T and PBS and blocked with RPMI with supplements for 2 h at RT (10% FCS). Splenocytes and bone marrow cells were serial diluted onto filter plates and incubated for 8 h 37°C in humidified 5% CO2 incubator. After washing with PBS and PBS-T the plates were incubated with biotinylated anti-IgG (Vector Laboratories, Burlingame, CA, USA) overnight at 4°C. The plates were then incubated with HRP-conjugated avidin-D (Vector Laboratories, Burlingame, CA, USA) for 60 min at RT. Following washing with PBS-T and PBS plates were stained with AEC working solution and rinsed with dH2O.
LCMV neutralizing antibodies
Identification of serum antibodies capable of neutralizing LCMV was performed using an immunofocus reduction assay, as previously described [26]. Briefly, serum samples were diluted 1:5, then and serial diluted in α-MEM + 2% FCS (Biochrom, Berlin, Germany). Diluted serum samples were incubated with LCMV DOCILE for 90 min at 37°C followed by addition of MC57 cells (ATCC, Manassas, VA, USA) and 4 h incubation at 37°C to allow the virus to infect cells. Overlay-Media containing methylcellulose (Sigma-Aldrich, St. Louis, MO, USA) was added following the 4 h incubation and cells were fixed and stained after 48 h in a similar manner to the plaque forming assay indicated above.
Statistical analysis
Data are expressed as mean ± SEM. Statistical significance between two groups was analyzed using Student's t-test. For experiments involving analysis of multiple time points, two-way ANOVA with an additional Bonferroni post-test was used. Mantel-Cox test was used for analysis of survival curves. p-values < 0.05 were considered as statistically significant. Analysis performed using GraphPad Prism.
Acknowledgments
The authors are grateful for the technical assistance of Eugen Bäcker, and to Dr. Ata Ur Rasheed Mohammed at Emory University for protocol advice. This study was supported by the Alexander von Humboldt Foundation (SKA2008 and SKA2010) and the German Research Council (SFB974, LA2558/3-1, LA2558/5-1, TRR60, LA1419/5-1), Jürgen Manchot Foundation, Forschungskomission of the Heinrich Heine University Grant #9772555, a Canadian Institutes of Health Research fellowship 201210MFE-289576-150035 to D.R.M, and the NIH Tetramer Facility. G.P.N. is supported by grants NIH (U19 AI057229, U54CA149145, N01-HV-00242, 1U19AI100627, 5R01AI07372405, R01CA184968, 1 R33 CA183654, R33 CA183692, HHSF223201210194C, 41000411217, 7500108142), U.S. Department of Defense (OC110674, 11491122), and the European Commission (Health.2010.1.2-1).
Glossary
- AD-HIES
autosomal dominant hyper IgE syndrome
- ASC
antibody-secreting cell
- CXCR5
C-X-C chemokine receptor type 5
- LCMV
lymphocytic choriomeningitis virus
- Tfh
T-follicular helper
Conflict of interest
The authors declare no commercial or financial conflict of interest.
Additional supporting information may be found in the online version of this article at the publisher's web-site
References
- 1.Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 2003;3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 2.Kallal LE, Biron CA. Changing partners at the dance: variations in STAT concentrations for shaping cytokine function and immune responses to viral infections. JAK-STAT. 2013;2:e23504. doi: 10.4161/jkst.23504. DOI: 10.4161/jkst.23504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kane A, Deenick EK, Ma CS, Cook MC, Uzel G, Tangye SG. STAT3 is a central regulator of lymphocyte differentiation and function. Curr. Opin. Immunol. 2014;28C:49–57. doi: 10.1016/j.coi.2014.01.015. [DOI] [PubMed] [Google Scholar]
- 4.Mogensen TH. STAT3 and the Hyper-IgE syndrome: Clinical presentation, genetic origin, pathogenesis, novel findings and remaining uncertainties. JAK-STAT. 2013;2:e23435. doi: 10.4161/jkst.23435. DOI: 10.4161/jkst.23435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sheerin KA, Buckley RH. Antibody responses to protein, polysaccharide, and phi X174 antigens in the hyperimmunoglobulinemia E (hyper-IgE) syndrome. J. Allergy Clin. Immunol. 1991;87:803–811. doi: 10.1016/0091-6749(91)90126-9. [DOI] [PubMed] [Google Scholar]
- 6.Leung DY, Ambrosino DM, Arbeit RD, Newton JL, Geha RS. Impaired antibody responses in the hyperimmunoglobulin E syndrome. J. Allergy Clin. Immunol. 1988;81:1082–1087. doi: 10.1016/0091-6749(88)90873-1. [DOI] [PubMed] [Google Scholar]
- 7.Dreskin SC, Goldsmith PK, Gallin JI. Immunoglobulins in the hyperimmunoglobulin E and recurrent infection (Job's) syndrome. Deficiency of anti-Staphylococcus aureus immunoglobulin A. J. Clin. Invest. 1985;75:26–34. doi: 10.1172/JCI111683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siegel AM, Heimall J, Freeman AF, Hsu AP, Brittain E, Brenchley JM, Douek DC, et al. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity. 2011;35:806–818. doi: 10.1016/j.immuni.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chandesris M-O, Melki I. Natividad A, Puel A, Fieschi C, Yun L, Thumerelle C, et al. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: molecular, cellular, and clinical features from a French national survey. Medicine (Baltimore) 2012;91:e1–19. doi: 10.1097/MD.0b013e31825f95b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ives ML, Ma CS, Palendira U, Chan A, Bustamante J, Boisson-Dupuis S, Arkwright PD, et al. Signal transducer and activator of transcription 3 (STAT3) mutations underlying autosomal dominant hyper-IgE syndrome impair human CD8(+) T-cell memory formation and function. J. Allergy Clin. Immunol. 2013;132:400–11.e9. doi: 10.1016/j.jaci.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity. 2011;35:792–805. doi: 10.1016/j.immuni.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tangye SG, Ma CS, Brink R, Deenick EK. The good, the bad and the ugly - TFH cells in human health and disease. Nat. Rev. Immunol. 2013;13:412–426. doi: 10.1038/nri3447. [DOI] [PubMed] [Google Scholar]
- 13.Liu X, Nurieva RI, Dong C. Transcriptional regulation of follicular T-helper (Tfh) cells. Immunol. Rev. 2013;252:139–145. doi: 10.1111/imr.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ray JP, Marshall HD, Laidlaw BJ, Staron MM, Kaech SM, Craft J. Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity. 2014;40:367–377. doi: 10.1016/j.immuni.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma CS, Avery DT, Chan A, Batten M, Bustamante J, Boisson-Dupuis S, Arkwright PD, et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood. 2012;119:3997–4008. doi: 10.1182/blood-2011-11-392985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mazerolles F, Picard C, Kracker S, Fischer A, Durandy A. Blood CD4+CD45RO+CXCR5+ T cells are decreased but partially functional in signal transducer and activator of transcription 3 deficiency. J. Allergy Clin. Immunol. 2013;131:1146–56, 1156.e1–5. doi: 10.1016/j.jaci.2012.12.1519. [DOI] [PubMed] [Google Scholar]
- 17.Rasheed MAU, Latner DR, Aubert RD, Gourley T, Spolski R, Davis CW, Langley WA, et al. Interleukin-21 is a critical cytokine for the generation of virus-specific long-lived plasma cells. J. Virol. 2013;87:7737–7746. doi: 10.1128/JVI.00063-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berglund LJ, Avery DT, Ma CS, Moens L, Deenick EK, Bustamante J, Boisson-Dupuis S, et al. IL-21 signalling via STAT3 primes human naive B cells to respond to IL-2 to enhance their differentiation into plasmablasts. Blood. 2013;122:3940–3950. doi: 10.1182/blood-2013-06-506865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. U. S. A. 1997;94:3801–3804. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J. Immunol. 1998;161:4652–4660. [PubMed] [Google Scholar]
- 21.Orban PC, Chui D, Marth JD. Tissue- and site-specific DNA recombination in transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 1992;89:6861–6865. doi: 10.1073/pnas.89.15.6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 1993;362:758–761. doi: 10.1038/362758a0. [DOI] [PubMed] [Google Scholar]
- 23.Richter K, Perriard G, Oxenius A. Reversal of chronic to resolved infection by IL-10 blockade is LCMV strain dependent. Eur. J. Immunol. 2013;43:649–654. doi: 10.1002/eji.201242887. [DOI] [PubMed] [Google Scholar]
- 24.Chen M, Guo Z, Ju W, Ryffel B, He X, Zheng SG. The development and function of follicular helper T cells in immune responses. Cell. Mol. Immunol. 2012;9:375–379. doi: 10.1038/cmi.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crotty S. Follicular helper CD4 T cells (TFH) Annu. Rev. Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 26.Bergthaler A, Flatz L, Verschoor A, Hegazy AN, Holdener M, Fink K, Eschli B, et al. Impaired antibody response causes persistence of prototypic T cell-contained virus. PLoS Biol. 2009;7:e1000080. doi: 10.1371/journal.pbio.1000080. DOI: 10.1371/journal.pbio.1000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–372. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- 28.Fahey LM, Wilson EB, Elsaesser H, Fistonich CD, McGavern DB. Brooks DG. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J. Exp. Med. 2011;208:987–999. doi: 10.1084/jem.20101773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doherty PC, Zinkernagel RM. T-cell-mediated immunopathology in viral infections. Transplant. Rev. 1974;19:89–120. doi: 10.1111/j.1600-065x.1974.tb00129.x. [DOI] [PubMed] [Google Scholar]
- 30.Planz O, Ehl S, Furrer E, Horvath E, Bründler MA, Hengartner H, Zinkernagel RM. A critical role for neutralizing-antibody-producing B cells, CD4(+) T cells, and interferons in persistent and acute infections of mice with lymphocytic choriomeningitis virus: implications for adoptive immunotherapy of virus carriers. Proc. Natl. Acad. Sci. U. S. A. 1997;94:6874–6879. doi: 10.1073/pnas.94.13.6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou X, Ramachandran S, Mann M, Popkin DL. Role of lymphocytic choriomeningitis virus (LCMV) in understanding viral immunology: past, present and future. Viruses. 2012;4:2650–2669. doi: 10.3390/v4112650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi YS, Eto D, Yang JA, Lao C, Crotty S. Cutting edge: STAT1 is required for IL-6-mediated Bcl6 induction for early follicular helper cell differentiation. J. Immunol. 2013;190:3049–3053. doi: 10.4049/jimmunol.1203032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, Wang Y, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29:138–149. doi: 10.1016/j.immuni.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 35.Grusdat M, McIlwain DR, Xu HC, Pozdeev VI, Knievel J, Crome SQ, Robert-Tissot C, et al. IRF4 and BATF are critical for CD8(+) T-cell function following infection with LCMV. Cell Death Differ. 2014;21:1050–1060. doi: 10.1038/cdd.2014.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sedwick WD, Wiktor TJ. Reproducible plaquing system for rabies, lymphocytic choriomeningitis,k and other ribonucleic acid viruses in BHK-21-13S agarose suspensions. J. Virol. 1967;1:1224–1226. doi: 10.1128/jvi.1.6.1224-1226.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Recher M, Lang KS, Navarini A, Hunziker L, Lang PA, Fink K, Freigang S, et al. Extralymphatic virus sanctuaries as a consequence of potent T-cell activation. Nat. Med. 2007;13:1316–1323. doi: 10.1038/nm1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.