

Abstract
Excessive manganese (Mn) in the brain promotes a variety of abnormal behaviors, including memory deficits, decreased motor skills and psychotic behavior resembling Parkinson’s disease. Hereditary hemochromatosis (HH) is a prevalent genetic iron overload disorder worldwide. Dysfunction in HFE gene is the major cause of HH. Our previous study has demonstrated that olfactory Mn uptake is altered by HFE deficiency, suggesting that loss of HFE function could alter manganese-associated neurotoxicity. To test this hypothesis, Hfe-knockout (Hfe−/−) and wild-type (Hfe+/+) mice mice were intranasally-instilled with manganese chloride (MnCl2 5 mg/kg) or water daily for 3 weeks and examined for memory function. Olfactory Mn diminished both short-term recognition and spatial memory in Hfe+/+ mice, as examined by novel object recognition task and Barnes maze test, respectively. Interestingly, Hfe−/− mice did not show impaired recognition memory caused by Mn exposure, suggesting a potential protective effect of Hfe deficiency against Mn-induced memory deficits. Since many of the neurotoxic effects of manganese are thought to result from increased oxidative stress, we quantified activities of anti-oxidant enzymes in the prefrontal cortex (PFC). Mn instillation decreased superoxide dismutase 1 (SOD1) activity in Hfe+/+ mice, but not in Hfe−/− mice. In addition, Hfe deficiency up-regulated SOD1 and glutathione peroxidase activities. These results suggest a beneficial role of Hfe deficiency in attenuating Mn-induced oxidative stress in the PFC. Furthermore, Mn exposure reduced nicotinic acetylcholine receptor levels in the PFC, indicating that blunted acetylcholine signaling could contribute to impaired memory associated with intranasal manganese. Together, our model suggests that disrupted cholinergic system in the brain is involved in airborne Mn-induced memory deficits and loss of HFE function could in part prevent memory loss via a potential up-regulation of anti-oxidant enzymes in the PFC.
Keywords: Acetylcholine receptor, Glutathione peroxidase, Hemochromatosis, Iron overload, Novel object recognition, Superoxide dismutase
INTRODUCTION
While manganese (Mn) is an indispensable metal for proper brain function, Mn in excess is neurotoxic. Increased attention has been paid to manganese intoxication by inhalation because this route of exposure significantly affects the health of people in occupational settings (1), such as workers employed in mining and Mn ore processing and agricultural workers exposed to Mn-containing pesticide (2). Compared with oral exposure, the olfactory route provides a greater extent of Mn transport to the systemic circulation due to lack of hepatic first-pass elimination mediated by biliary excretion (3). Moreover, olfactory transport circumvents the brain-blood barrier (4) and thus enables direct entry of Mn into the brain. Excessive Mn in the brain results in a variety of abnormal behaviors, including memory deficits, decreased motor skills and psychotic behavior resembling Parkinson’s disease (1).
Hereditary hemochromatosis is one of the most common genetic diseases in the North American Caucasian population (5), characterized by uncontrollable absorption of dietary iron and accumulation of the metal in several parenchymal tissues, such as liver and heart, which results in organ failure. Mutations in the HFE (High iron or Fe) gene are the primary cause of this disease, and the two most prevalent HFE missense variants (C282Y and H63D) are found worldwide: North American (36.9%), European (34.9%), African/Middle Eastern (16.7%) and Asian (2.8%) populations (6). Although a large body of evidence has indicated that altered iron metabolism can influence manganese transport and toxicity (7-11), it is unclear whether loss of HFE function found in hemochromatosis modifies olfactory transport and behavioral toxicity of inhaled manganese. In the present study, we conducted memory-related behavioral tests after intranasal instillation of Mn into Hfedeficient mice, a mouse model that recapitulates iron overload hemochromatosis in humans (12). Our data demonstrate that olfactory exposure to manganese impairs memory function, which could be restored in part by Hfe deficiency. Our model further suggests that loss of Hfe function could provide a potential resistance mechanism against Mninduced cognitive impairment by up-regulating brain redox status.
MATERIALS AND METHODS
Ethics statement. This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Northeastern University Animal Care and Use Committee.
Animals and manganese exposure. Hfe-deficient (Hfe−/−) mice (12) and their wild-type control (Hfe+/+) mice were kindly provided by Dr. Nancy Andrews (Duke University, NC, USA). All mice used for these studies were on the 129S6/SvEvTac background (12). One-month old male mice were fed facility chow (Prolab Isopro RMH 3000, LabDiet; 96 mg manganese and 380 mg iron per kg diet) and given water ad libitum. Neither manganese nor iron was detected in drinking water. Male mice were chosen because estrogen affects iron metabolism (13,14). For olfactory exposure to manganese, mice were intranasally-instilled with manganese chloride (5 mg MnCl2/kg body weight; 0.08 mL/kg) or double distilled water as vehicle control daily for 22 days. During this period, all animals were subject to novel object recognition task (exposure days 13~14; postnatal days 43~44) and Barnes maze (exposure days 17~22; postnatal days 47~52).
Novel object recognition task. Mice were acclimated to an open field (Med Associates, VT, USA; 27 L × 27 W × 20 H cm) for 6 min per day over 3 days prior to the novel object recognition test. For the short-term memory test, in the first trial two identical objects were placed symmetrically in the open field. The mouse was placed in the center of the open field, allowed to explore freely for 5 min and returned to home cage. To avoid olfactory cues, the open field was cleaned with Quatricide TB (Pharmacal Research Laboratories Inc., Waterbury, CT, USA) thoroughly between the trials. After 30 min, the mouse was placed again for another 5 min in the open field, in which one of the familiar objects was replaced by a novel object with different shape and color. After this session, the mouse returned to home cage. A long-term memory test was carried out 24 hr later in the presence of another novel object. The time spent with both novel and familiar objects and total distance traveled during the session were recorded and analyzed by ANY-maze software (Stoelting Co., Wood Dale, IL, USA). Exploration with the objects was defined as the mouse nose was within 2 cm toward the object (15). The percentage of time spent with the novel object to the total exploration time (time spent with both novel and familiar objects) was defined as recognition index.
Barnes maze test. Barnes maze (Stoelting Co.) consisted of a round table (91 cm in diameter, 90 cm in height) with 20 holes (5 cm in diameter) evenly distributed around the edge. An escape box was placed underneath one of the holes, 4 cm in depth. Visual cues with different colors and shapes were placed above 4 quadrants of the maze. The location of the escape box remained the same during the training session for 4 days and was removed on the test day (day 5). Four floodlights (4 × 150 watts) were placed above the maze and used as a stimulus. The advantage of the Barnes maze is that it does not require extensive physical activity, like swimming in the water maze, which may affect the performance of memory capacity. Barnes maze test comprised 3 sessions: acclimation, training and test. In the acclimation period, each mouse was placed by the edge of the escape box. The mouse was gently put into the box if it had not entered the escape box within 1 min. During the training period, the mouse was placed in the center of the maze and covered by an opaque plastic box. After 15 s, the floodlights were turned on and the box was then removed. The mouse was allowed to walk around the maze for 3 min to find and enter the escape box. If the mouse failed to enter the escape box within 3 min, it was gently guided to the box. Once the mouse entered the box, the floodlights were turned off and it was allowed to stay in the box for 1 min. Three trials were performed per training day for consecutive 4 days. The latency to enter into the escape box was recorded and analyzed by ANY-maze. On day 5 (test), the escape box was removed and the mouse was allowed to walk around the maze for 90 s with floodlights on. Time spent in the target hole where the escape box used to locate, latency to reach the target hole and total distance traveled were recorded and analyzed by ANY-maze. The maze was wiped thoroughly with Quatricide TB to avoid olfactory cues.
Tissue collection. After the last behavioral test, mice (postnatal day 53) were euthanized by isofluane overdose, followed by exsanguination and brain collection. The brain was further microdissected to harvest the prefrontal cortex and hippocampus (16). All tissues were flash-frozen in liquid nitrogen and stored at −80℃ until analysis.
Western blot analysis. Prefrontal cortex and hippocampus were homogenized, electrophoresed on 7.5% SDS-polyacrylamide gels (30 μg proteins) and transferred to polyvinylidene difluoride (prefrontal cortex) or nitrocellulose (hippocampus) membranes. After blocking with 5% non-fat milk, the membrane was incubated in rabbit antibody against α7 nicotinic acetylcholine receptor (1:800; Abcam, Cambridge, MA, USA) (17). As a loading control, the immunoblot was incubated with mouse anti-actin (1:5,000, MP Biomedicals, Solon, OH, USA). The blots were incubated with donkey anti-rabbit antibody conjugated with HRP (1:1,000, GE Healthcare, Piscataway, NJ, USA) or sheep anti-mouse antibody (1:5,000, GE Healthcare, USA). Immunoreactivity was detected using ECL West Dura substrate (Thermo Scientific, Tewksbury, MA, USA). Protein bands were visualized by ChemiDoc XRS (Bio-Rad, Hercules, CA, USA) and intensities of protein bands were quantified using Image Lab (version 4.1, Bio-Rad).
Analysis of anti-oxidant enzyme activity. Prefrontal cortex and hippocampus were homogenized using Tris buffer (100 mM, pH 7.4; 10-time dilution) and centrifuged at 10,000 g for 15 min. Activities of superoxide dismutase 1 (SOD1), catalase and glutathione peroxidase (GPx) were determined using assay reagents (Cayman Chemical, Ann Arbor, MI, USA) according to manufacturer’s instructions. Protein concentration was determined by Bradford assay. SOD and catalase activities were presented as unit per mg protein and GPx activity was shown as unit/min per mg protein.
Statistical analysis. Values reported were expressed as means ± SEM. A two-way ANOVA was performed using SigmaPlot (version 12.3; Systat Software Inc., San Jose, CA, USA) to determine individual main effects and an interaction effect between Hfe deficiency and olfactory manganese exposure. Post-hoc comparisons were performed by the Holm-Sidak method. Differences were considered significant at p < 0.05.
RESULTS
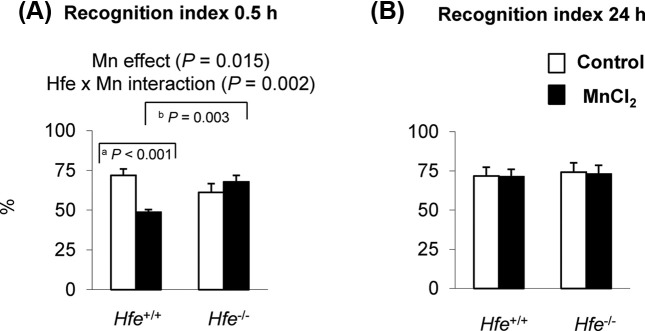
Intranasal manganese impairs a short-term recognition memory in wild-type mice, but not in Hfe−/−mice. To evaluate if Mn exposure decreases recognition memory, mice were tested for the novel object recognition task. The percent time spent with a novel object was significantly decreased in Hfe+/+ mice after intranasal Mn (Fig. 1A) (p < 0.001), indicating an impaired short-term (0.5 hr) memory upon Mn exposure in the presence of Hfe. Notably, Hfe−/− mice did not show Mn-induced memory deficit (Fig. 1A), representing a significant interaction effect between Hfe deficiency and Mn exposure (p = 0.002). In addition, Mninstilled Hfe−/− mice significantly increased 0.5-h recognition index than Mn-instilled Hfe+/+ mice (p = 0.003). In contrast, a long-term recognition memory, defined as memory 24 hr after initial recognition, was not different, regardless of Mn exposure or Hfe deficiency (Fig. 1B). Total distance traveled or total time interacted with both objects was not altered upon Mn exposure or Hfe deficiency (data not shown). Combined, our results indicate that olfactory Mn reduces a short-term recognition memory, which is restored by Hfe deficiency.
Fig. 1. Effect of Hfe deficiency on recognition memory in manganese-instilled mice. Hfe+/+ and Hfe−/− mice exposed to olfactory manganese were tested for the novel recognition memory task in an open field. Following 30-min of acclimation with two identical objects, one object was replaced with a different object and mouse behavior was recorded for 5 min. Time of interaction with each object was recorded to quantify a shortterm recognition index (A). Mice were tested 24 hr later with another novel object in order to determine 24-hr recognition index (B). Empty and closed bars represent water-instilled and MnCl2-instilled mice, respectively. Data were presented as means ± SEM (n = 7~11 per group) and were analyzed using two-way ANOVA followed by post-hoc comparisons. a p < 0.05 for manganese effect in Hfe+/+ mice; b p < 0.05 for Hfe effect in MnCl2-instilled mice.
Spatial memory capacity is reduced by olfactory manganese. We further tested if intranasal Mn and/or Hfe deficiency alters spatial memory capacity using the Barnes maze, a “dry-land” version of the Morris water maze. During the training sessions for four days, neither Mn exposure nor Hfe deficiency demonstrated altered learning capacity (Fig. 2A). On day 5, however, Mn-instilled mice spent less time near the target hole, indicating a reduced spatial memory (Fig. 2B) (p = 0.048), whereas both Hfe+/+ and Hfe−/− mice showed a similar spatial memory performance. The overall activity, as assessed by total distance traveled, was unchanged among four groups of treatment (Fig. 2C), suggesting that the reduced memory capacity caused by Mn exposure was not due to lack of explorative activity or motor function. These results demonstrate that intranasal manganese negatively influences spatial memory, which is not altered by Hfe deficiency.
Fig. 2. Effect of olfactory manganese exposure on spatial memory in Hfe-deficient mice. Mice instilled with MnCl2 or water were trained on the Barnes maze for 4 days with an escape box (A). One day after last training, mice were tested in the maze without the escape box to quantify time in target hole area (B) and total distance traveled (C). Empty and closed bars represent water-instilled and MnCl2-instilled mice, respectively. Data were presented as means ± SEM (n = 10~13 per group) and were analyzed using two-way ANOVA.

Superoxide dismutase 1 activity decreases after intranasal manganese and increases upon Hfe deficiency in prefrontal cortex. Our findings that Hfe−/− mice were resistant to Mn-induced impaired recognition memory led us to further explore potential molecular mechanisms of these different behavioral responses. Since Mn neurotoxicity is associated with elevated oxidative stress (18,19), we set out to determine and compare anti-oxidant enzymes in the prefrontal cortex and hippocampus. Superoxide dismutase 1 (SOD1) activity, also known as cytosolic SOD, was reduced in Hfe+/+ mice upon Mn instillation in the prefrontal cortex (Fig. 3A). In addition, we found that the SOD1 activity was significantly higher in the prefrontal cortex of Hfe−/− mice compared with Hfe+/+ mice (Fig. 3A) (p = 0.020, Hfe effect). Furthermore, there was an Hfe x Mn interaction effect on SOD1 activity (p = 0.010); Mn-instilled Hfe+/+ mice decreased SOD1 activity compared with waterinstilled Hfe+/+ mice (Fig. 3A) (p = 0.046), whereas Hfe−/− mice did not show Mn effect on SOD1 activity. Notably, Hfe deficiency increased glutathione peroxidase activity (Fig. 3B) (p = 0.029, Hfe effect). However, activities of SOD1 and Gpx did not change in the hippocampus either by Mn exposure or by Hfe deficiency. Activities of catalase (Fig. 3C) were not significantly different among four groups in either brain region. Collectively, loss of Hfe function results in elevated anti-oxidant enzyme activities in the brain, which could compensate for Mn-induced oxidative stress.
Fig. 3. Effect of loss of Hfe function and olfactory manganese exposure on anti-oxidant enzymes in the prefrontal cortex and hippocampus of mice. After behavioral tests, mice were euthanized and prefrontal cortex and hippocampus were homogenized to determine the activities of anti-oxidant enzymes, including superoxide dismutase (A), glutathione peroxidase (B) and catalase (C). Empty and closed bars represent water-instilled and MnCl2 -instilled mice, respectively. Data were presented as means ± SEM (n = 4~8 per group) and were analyzed using twoway ANOVA followed by post-hoc comparisons. a p < 0.05 for manganese effect in Hfe+/+ mice; b p < 0.05 for Hfe effect in MnCl2-instilled mice.

Intranasal manganese down-regulates acetylcholine receptors in prefrontal cortex. We also examined if olfactory Mn exposure alters acetylcholine receptor expression in the two brain regions associated with memory process, the prefrontal cortex and hippocampus. The levels of α7 nicotinic acetylcholine receptors (nAChR) were reduced in the prefrontal cortex of Mn-treated mice (Fig. 4A) (p = 0.031, Mn effect), but not in the hippocampus (Fig. 4B). These results indicate that olfactory Mn exposure attenuates the expression levels of acetylcholine receptors, which is associated with impaired spatial memory examined by the Barnes maze test.
Fig. 4. Effect of manganese instillation and Hfe deficiency on the expression of nicotinic acetylcholine receptor in prefrontal cortex and hippocampus. Prefrontal cortex (A) and hippocampus (B) collected from mice intranasally instilled with MnCl2 were homogenized to determine the expression levels of α7-nicotinic acetylcholine receptor (nAChR). Relative intensities of protein bands normalized to actin were determined using Image Lab (version 4.1). Empty and closed bars represent water-instilled and MnCl2-instilled mice, respectively. Data were presented as means ± SEM (n = 4 per group) and were analyzed using two-way ANOVA.

DISCUSSION
A large body of evidence indicates a close relationship between Mn exposure and memory problems (20,21). Many of the neurotoxic effects of Mn are thought to result from oxidative toxicity (18,19), which could affect anti-oxidant enzymes in the brain. Here we found decreased SOD1 activity in the PFC of wild-type mice after Mn instillation, while Hfe-deficient mice showed increased activities of SOD1 and GPx upon Mn exposure. It should be noted that HFE mutation is associated with increased oxidative stress and promotes the progression of several neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis (ALS) (22). Furthermore, Ali-Rahmani et al. (23) demonstrated that HFE mutant mice exhibit decreased memory capacity compared with wild-type control mice at the age of 14~18 months old. In contrast, our Hfe-deficient mice did not show any abnormal cognitive/memory performance or oxidative stress. There are at least two explanations. First, neurodegenerative diseases may result from gain of mutant function rather than loss of function (24). Age is another possibility. For example, while the neurodegenerative process would require prolonged exposure to oxidative stress (23), mice in our study were 2-month-old and thus may not present the pathological symptom. Collectively, our results offer a novel idea that loss of Hfe function corrects attenuated SOD1 activity resulting from Mn exposure and likely prevents loss of recognition memory induced by olfactory manganese.
While acetylcholine plays a well-established role in cognitive/memory function, recent studies have shown that Mn perturbs the cholinergic system (25,26). Yet, it is largely unknown whether acetylcholine receptor is modified upon Mn exposure in vivo. We found decreased acetylcholine receptor expression in both genotypes after manganese exposure. Since an elevated cholinergic activity in the prefrontal regions could contribute to attentional and cognitive functions (17,25,27), our data suggest that there is a close association between spatial memory deficits and down-regulation of acetylcholine effect through the receptor by Mn exposure. Our findings agree with previous results from a cell culture system in which Mn inhibits the synthesis of acetylcholine receptor (28). We also found that Mn-mediated receptor down-regulation was not significant in the hippocampus. While other neurotransmitters may be involved in impaired memory after intranasal instillation of manganese, our results demonstrate that olfactory Mn could lessen acetylcholine response by receptor down-regulation, likely resulting in reduced spatial memory capacity.
To our knowledge, the current study is the first to characterize the effect of olfactory manganese with loss of Hfe function on recognition and spatial memory. Our results demonstrate that olfactory manganese exposure decreases memory function by modulating oxidative stress and cholinergic neurotransmission, particularly in the prefrontal cortex. In addition, we show that Hfe-deficient mice do not display manganese-related problems in short-term recognition memory. Our data suggest that loss of Hfe function protects from manganese-induced neurotoxicity. KwikUribe and Smith demonstrated that low to moderate exposures to manganese result in impaired cellular response similar to iron deficiency and disrupt cellular iron metabolism (29). Since iron homeostasis is adequately regulated in physiologic conditions by an iron-dependent manner, which is disrupted by Hfe deficiency (30), it is conceivable that Hfe−/− mice fail to elicit manganese-induced neurotoxic responses that are perhaps mediated by iron metabolism. Also, altered nAChR expression and SOD1 activity in the prefrontal cortex could be mixed consequences in the downstream pathways triggered by combinatorial effects of both neurotoxicity (mediated by manganese inhalation) and neuroprotection (associated with Hfe deficiency). Our model suggests that individuals with hemochromatosis by loss of HFE function may be less susceptible to neurological problems resulting from manganese inhalation and further provides a therapeutic basis for memory-related neurodegenerative diseases.
Acknowledgments
The authors are grateful to Dr. Abitha Sukumaran, Ms. JuOae Chang and Ms. Murui Han for help during animal experiments and Dr. Timothy Maher at MCPHS University (Boston, MA, USA) for helpful comments on neurobehavioral tests.
References
- 1.Cotzias G.C. Manganese in health and disease. Physiol. Rev. (1958);38:503–532. doi: 10.1152/physrev.1958.38.3.503. [DOI] [PubMed] [Google Scholar]
- 2.Zielhuis R.L., del Castilho P., Herber R.F., Wibowo A.A. Levels of lead and other metals in human blood: suggestive relationships, determining factors. Environ. Health Perspect. (1978);25:103–109. doi: 10.1289/ehp.7825103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bertinchamps A.J., Miller S.T., Cotzias G.C. Interdependence of routes excreting manganese. Am. J. Physiol. (1966);211:217–224. doi: 10.1152/ajplegacy.1966.211.1.217. [DOI] [PubMed] [Google Scholar]
- 4.Brenneman K.A., Wong B.A., Buccellato M.A., Costa E.R., Gross E.A., Dorman D.C. Direct olfactory transport of inhaled manganese ((54)MnCl(2)) to the rat brain: toxicokinetic investigations in a unilateral nasal occlusion model. Toxicol. Appl. Pharmacol. (2000);169:238–248. doi: 10.1006/taap.2000.9073. [DOI] [PubMed] [Google Scholar]
- 5.Merryweather-Clarke A.T., Pointon J.J., Jouanolle A.M., Rochette J., Robson K.J. Geography of HFE C282Y and H63D mutations. Genet. Test. (2000);4:183–198. doi: 10.1089/10906570050114902. [DOI] [PubMed] [Google Scholar]
- 6.Hanson E.H., Imperatore G., Burke W. HFE gene and hereditary hemochromatosis: a HuGE review. Human Genome Epidemiology. Am. J. Epidemiol. (2001);154:193–206. doi: 10.1093/aje/154.3.193. [DOI] [PubMed] [Google Scholar]
- 7.Kim J., Li Y., Buckett P.D., Böhlke M., Thompson K.J., Takahashi M., Maher T.J., Wessling-Resnick M. Iron-responsive olfactory uptake of manganese improves motor function deficits associated with iron deficiency. PLoS One. (2012);7:e33533. doi: 10.1371/journal.pone.0033533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim J., Buckett P.D., Wessling-Resnick M. Absorption of manganese and iron in a mouse model of hemochromatosis. PLoS One. (2013);8:e64944. doi: 10.1371/journal.pone.0064944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia S.J., Gellein K., Syversen T., Aschner M. Iron deficient and manganese supplemented diets alter metals and transporters in the developing rat brain. Toxicol. Sci. (2007);95:205–214. doi: 10.1093/toxsci/kfl139. [DOI] [PubMed] [Google Scholar]
- 10.Phattanarudee S., Han M., Kim J. Effect of olfactory manganese dose on motor coordination in iron-deficient rats. MO. J. Toxicol. (2014);1:1–7. [Google Scholar]
- 11.Claus Henn B., Kim J., Wessling-Resnick M., Téllez-Rojo M.M., Jayawardene I., Ettinger A.S., Hernández-Avila M., Schwartz J., Christiani D.C., Hu H., Wright R.O. Associations of iron metabolism genes with blood manganese levels: a population-based study with validation data from animal models. Environ. Health. (2011);10:97. doi: 10.1186/1476-069X-10-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levy J.E., Montross L.K., Cohen D.E., Fleming M.D., Andrews N.C. The C282Y mutation causing hereditary hemochromatosis does not produce a null allele. Blood. (1999);94:9–11. [PubMed] [Google Scholar]
- 13.Hou Y., Zhang S., Wang L., Li J., Qu G., He J., Rong H., Ji H., Liu S. Estrogen regulates iron homeostasis through governing hepatic hepcidin expression via an estrogen response element. Gene. (2012);511:398–403. doi: 10.1016/j.gene.2012.09.060. [DOI] [PubMed] [Google Scholar]
- 14.Kim J., Wessling-Resnick M. Iron and mechanisms of emotional behavior. J. Nutr. Biochem. (2014);25:1101–1107. doi: 10.1016/j.jnutbio.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dobarro M., Orejana L., Aguirre N., Ramírez M.J. Propranolol restores cognitive deficits and improves amyloid and Tau pathologies in a senescence-accelerated mouse model. Neuropharmacology. (2013);64:137–144. doi: 10.1016/j.neuropharm.2012.06.047. [DOI] [PubMed] [Google Scholar]
- 16.Chang J., Kueon C., Kim J. Influence of lead on repetitive behavior and dopamine metabolism in a mouse model of iron overload. Toxicol. Res. (2014);30:267–276. doi: 10.5487/TR.2014.30.4.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han M., Kim J. Effect of dietary iron loading on recognition memory in growing rats. PLoS One. (2015);10:e0120609. doi: 10.1371/journal.pone.0120609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.HaMai D., Bondy S.C. Oxidative basis of manganese neurotoxicity. Ann. N. Y. Acad. Sci. (2004);1012:129–141. doi: 10.1196/annals.1306.010. [DOI] [PubMed] [Google Scholar]
- 19.Donaldson J., LaBella F.S., Gesser D. Enhanced autoxidation of dopamine as a possible basis of manganese neurotoxicity. Neurotoxicology. (1981);2:53–64. [PubMed] [Google Scholar]
- 20.Guilarte T.R., Chen M.K. Manganese inhibits NMDA receptor channel function: implications to psychiatric and cognitive effects. Neurotoxicology. (2007);28:1147–1152. doi: 10.1016/j.neuro.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blecharz-Klin K., Piechal A., Joniec-Maciejak I., Pyrzanowska J., Widy-Tyszkiewicz E. Effect of intranasal manganese administration on neurotransmission and spatial learning in rats. Toxicol. Appl. Pharmacol. (2012);265:1–9. doi: 10.1016/j.taap.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 22.Zecca L., Youdim M.B., Riederer P., Connor J.R., Crichton R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. (2004);5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- 23.Ali-Rahmani F., Grigson P.S., Lee S., Neely E., Connor J.R., Schengrund C.L. H63D mutation in hemochromatosis alters cholesterol metabolism and induces memory impairment. Neurobiol. Aging. (2014);35:e1–12. doi: 10.1016/j.neurobiolaging.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 24.Nandar W., Connor J.R. HFE gene variants affect iron in the brain. J. Nutr. (2011);141:729S–739S. doi: 10.3945/jn.110.130351. [DOI] [PubMed] [Google Scholar]
- 25.Finkelstein Y., Milatovic D., Aschner M. Modulation of cholinergic systems by manganese. Neurotoxicology. (2007);28:1003–1014. doi: 10.1016/j.neuro.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Chtourou Y., Fetoui H., Garoui el M., Boudawara T., Zeghal N. Improvement of cerebellum redox states and cholinergic functions contribute to the beneficial effects of silymarin against manganese-induced neurotoxicity. Neurochem. Res. (2012);37:469–479. doi: 10.1007/s11064-011-0632-x. [DOI] [PubMed] [Google Scholar]
- 27.Sarter M., Gehring W.J., Kozak R. More attention must be paid: the neurobiology of attentional effort. Brain Res. Rev. (2006);51:145–160. doi: 10.1016/j.brainresrev.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 28.Lorković H., Feyrer A. Manganese ions inhibit acetylcholine receptor synthesis in cultured mouse soleus muscles. Neurosci. Lett. (1984);51:331–335. doi: 10.1016/0304-3940(84)90398-7. [DOI] [PubMed] [Google Scholar]
- 29.Kwik-Uribe C., Smith D.R. Temporal responses in the disruption of iron regulation by manganese. J. Neurosci. Res. (2006);83:1601–1610. doi: 10.1002/jnr.20836. [DOI] [PubMed] [Google Scholar]
- 30.Feder J.N., Penny D.M., Irrinki A., Lee V.K., Lebrón J.A., Watson N., Tsuchihashi Z., Sigal E., Bjorkman P.J., Schatzman R.C. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc. Natl. Acad. Sci U.S.A. (1998);95:1472–1477. doi: 10.1073/pnas.95.4.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]