

Abstract
Serotyping forms the basis of national and international surveillance networks for Salmonella, one of the most prevalent foodborne pathogens worldwide (1–3). Public health microbiology is currently being transformed by whole-genome sequencing (WGS), which opens the door to serotype determination using WGS data. SeqSero (www.denglab.info/SeqSero) is a novel Web-based tool for determining Salmonella serotypes using high-throughput genome sequencing data. SeqSero is based on curated databases of Salmonella serotype determinants (rfb gene cluster, fliC and fljB alleles) and is predicted to determine serotype rapidly and accurately for nearly the full spectrum of Salmonella serotypes (more than 2,300 serotypes), from both raw sequencing reads and genome assemblies. The performance of SeqSero was evaluated by testing (i) raw reads from genomes of 308 Salmonella isolates of known serotype; (ii) raw reads from genomes of 3,306 Salmonella isolates sequenced and made publicly available by GenomeTrakr, a U.S. national monitoring network operated by the Food and Drug Administration; and (iii) 354 other publicly available draft or complete Salmonella genomes. We also demonstrated Salmonella serotype determination from raw sequencing reads of fecal metagenomes from mice orally infected with this pathogen. SeqSero can help to maintain the well-established utility of Salmonella serotyping when integrated into a platform of WGS-based pathogen subtyping and characterization.
INTRODUCTION
Salmonella is the most prevalent foodborne pathogen in the United States, causing 1.2 million cases of illness annually and the largest health burden among all bacterial pathogens (4). The U.S. National Salmonella Surveillance System has been built upon serotyping in public health laboratories, a subtyping method traditionally performed through the agglutination of Salmonella cells with specific antisera that detect lipopolysaccharide O antigen and flagellar H antigens. Specific combinations of O and H antigenic types represent serotypes (or serovars). More than 2,500 Salmonella serotypes have been described in the White-Kauffmann-Le Minor scheme (5, 6). The phenotypic determination of serotypes is labor-intensive and time-consuming (taking at least 2 days), which has led to the development of genetic methods for serotype determination (7, 8). These methods generally use two categories of targets for serotype determination: (i) indirect targets, requiring the use of random surrogate genomic markers associated with particular serotypes, and (ii) direct targets, requiring the use of genetic determinants of serotypes, including the rfb gene cluster responsible for somatic (O) group synthesis (9, 10) and the fliC (11) and fljB (12) genes encoding the two flagellar antigens present in Salmonella. The latter approach has the advantage of determining serotypes using the same markers as the phenotypic method, providing continuity between the serotypes determined by phenotypic and genetic markers (13, 14). While this approach may result in distinct genetic lineages being assigned the same serotype due to horizontal gene transfer of the serotype determinants, phylogenetic reconstruction is beyond the scope of serotyping and can be better performed by other subtyping methods. Also, through the identification of individual serotype determinants, methods based on serotype determinants have the potential to predict a wide range of Salmonella serotypes. In contrast, methods based on random surrogate genomic markers rely on the presumed correspondence between the markers and particular serotypes and therefore need to be validated for each new serotype tested.
Routine and real-time implementation of whole-genome sequencing (WGS) (15, 16) is poised to transform public health microbiology. Efforts have been made to enable a variety of pathogen subtyping and characterization analyses through the use of WGS data, such as multilocus sequence typing (17, 18), antimicrobial resistance identification (19), and virulence characterization (16). Beyond WGS of pure cultures, recent application of metagenome sequencing in diagnosis and outbreak investigation of infectious diseases (20, 21) has demonstrated the potential for culture-independent detection of pathogens from complex clinical samples.
Here we present a novel application of whole-genome and metagenome sequence data for Salmonella serotype determination. Curated databases for major serotype determinants were constructed that included the rfb gene clusters responsible for somatic O-group antigen synthesis (22); the wzx O-antigen flipase gene and the wzy O-antigen polymerase gene, which are typically found in the rfb cluster and are highly specific for the majority of O groups (23); additional genes from the rfb cluster useful for characterization of specific O groups; and the fliC and fljB genes that encode Salmonella flagellar antigens. Based on mapping raw sequencing reads to these databases for the identification of individual antigen types, our bioinformatics approach allows robust and comprehensive prediction of Salmonella serotype without genome assembly. A Web application of our serotyping tool (named “SeqSero”) is publicly available at www.denglab.info/SeqSero.
MATERIALS AND METHODS
Whole-genome sequences.
A total of 229 Salmonella enterica isolates of various relatively uncommon serotypes (see Table S1 in the supplemental material) were sequenced on an Illumina HiSeq 2000 platform (100-bp, paired-end reads) per the manufacturer's instruction by the 100K Food-borne Pathogen Genome Project at University of California, Davis (http://100kgenome.vetmed.ucdavis.edu/). An additional 79 Salmonella genomes representing common serotypes from the WGS collection of CDC (NCBI BioProject PRJNA186441) were included, for a total of 308 genomes in the CDC strain set. The serotypes of these isolates were confirmed using traditional (24) and genetic (13, 14) serotyping assays. For the GenomeTrakr strain set, Salmonella genomes sequenced by the Illumina platform and uploaded to the GenomeTrakr depository (NCBI BioProject 183844) as of 1 June 2014 were reviewed for suitability for inclusion in a validation data set. Genomes were excluded for the following reasons: (i) no serotype or two or more serotypes indicated for a specific genome (n = 766); (ii) rough, nonmotile strains (n = 39); (iii) monophasic variants (n = 76); and (iv) less than 10× sequencing coverage (n = 11). A total of 354 assembled genomes with a N50 contig size of >150,000 bases were downloaded from GenBank for validation analysis.
Mouse infections, feces sample preparation, and metagenome sequencing.
Mouse infections, feces sample preparation, and DNA extraction were performed as previously described (25). S. enterica serotype Typhimurium strain 14028s was used to orally challenge female, age-matched (6-to-8-week-old) 129SvJ mice (25). Fecal samples from control mice had not been sequenced and were not available for the current study. For deep metagenomic sequencing, extracted DNAs were assigned bar codes, multiplexed, and sequenced using the Illumina V3 chemistry on the HiSeq 2000 platform. We implemented automation for the construction of up to 96 fragment or paired-end libraries at one time. Paired-end libraries were constructed using the Illumina TruSeq protocol. Approximately 1 Gb of shotgun sequence data per sample was generated.
Databases for Salmonella serotype determinants.
For O-group determination, two databases were built: (i) sequences from the entire rfb cluster were used for O-group determination from genome assemblies and (ii) wzx (O-antigen flippase), wzy (O-antigen polymerase), and other genes or markers from the rfb cluster useful for O-group determination (see Table S4 in the supplemental material) were used when the input data were raw sequencing reads. Two O-antigen groups, those that possess O9 (O9,O2, O9,46, and O9,46,27) and those that possess O3 (O3,10 and O1,3,19), require additional markers for differentiation, including the rfb sequence specific to serotype O3,10 and a frameshift mutation in tyv (see Table S4). The combined use of the six markers allowed the differentiation of 273 (O3,10) and 72 (O1,3,19) strains (data not shown). In the two O-group databases, each of the 46 O antigens was represented by a single rfb cluster (26) or a single allele of the wzx or wzy gene (27).
For H antigen determination, a single database that contained both fliC and fljB alleles was built; the sequences were primarily from reference 28 and were supplemented with fliC and fljB gene sequences extracted from Salmonella genomes (closed and draft assemblies) available at GenBank. Multiple, distinct alleles for the same flagellar antigenic type were allowed to accommodate the multiphyletic nature of some H antigens (28).
For the multiple rounds of reads mapping for H antigen determination, three additional data sets were developed. (i) fliC and fljB alleles were grouped into clusters based on sequence similarity (see Table S5 in the supplemental material). This grouping was used to identify the mostly likely H antigen group after the first two rounds of reads mapping (see details below). (ii) A representative allele for each H antigen type was selected and used to extract sequencing reads relevant to H antigens in the third round of reads mapping. This allele was near the midpoint between the root and the tip of longest branch of the phylogenetic tree that contained all the alleles for an antigen. (iii) For H antigen clusters that had multiple antigen types (see Table S5) and therefore required a BLAST analysis for final H antigen determination, a database of the middle, variable sequences of the alleles for every antigen in the cluster was used for the BLAST alignment (see details below). All the databases and additional data sets are available at www.denglab.info/SeqSero. They are regularly curated and updated when new sequences become available. Text S1 in the supplemental material provides a discussion of considerations for Salmonella serotype determination using the conventions of the White-Kauffmann-Le Minor scheme.
Serotype prediction from raw sequencing reads.
A reads mapping-based strategy was developed for prediction of O and H antigenic types. In general, raw sequencing reads without any quality filtering or trimming were mapped to individual antigen sequence databases using Burrows-Wheeler Aligner (BWA) with the default parameter setting of the sampe/samse algorithm (29). The allele to which the highest number of reads mapped was chosen as the allele potentially present in the genome tested.
Some fliC and fljB alleles share high levels of sequence similarity (28), creating challenges for the determination of antigenic types based on DNA sequence. This issue was aggravated in our pipeline because multiple, closely related alleles were present in the database. When the test genome contains a gene for an antigen type that is represented by a single allele in the database, most reads map to that one allele and only a few to other alleles in the database, producing a pronounced difference. When the database contains multiple closely related alleles, reads can map to multiple alleles, diminishing or even eliminating the otherwise pronounced excess in the number of reads mapped to the allele expected for the genome being tested (see Fig. S1 in the supplemental material). To minimize these problems, we implemented a stepwise identification approach using two rounds of reads mapping for all analyses and incorporating an additional round of mapping plus a subsequent BLAST analysis in cases where multiple antigenic types are present in a predefined H antigen cluster (see Table S5).
An example workflow of fliC identification is depicted in Fig. 1; a similar workflow is used for fljB determination. (i) In round 1 mapping, the raw sequencing reads of a serotype Typhimurium genome (NCBI SRA accession no. SRX528051) were mapped to the entire H antigen database. The fliC alleles were then ranked according to the number of reads mapped to each allele, from the largest to the smallest. Up to three antigen clusters (see Table S5 in the supplemental material) that contained the highest-ranking alleles were selected. In this example, clusters fliC_eh (including antigenic type “e,h”), fliC_ir (including antigenic types “i,” “r,” and “r,i”), and fliC_z35 (including antigenic type “z35”) were selected. (ii) In round 2 mapping, one allele in each cluster that had the most mapped reads was selected and reads were mapped to just those alleles. The alleles were again ranked as described above. In this example, the order of the top ranking clusters changed to fliC_ir, fliC_eh, and fliC_z35, suggesting that an error caused by the “dilution effect” (see Fig. S1 in the supplemental material) between clusters fliC_ir and fliC_eh had been corrected, and the antigen of the test genome was determined to belong to cluster fliC_ir. (iii) In round 3 mapping, the representative alleles for the antigenic types in cluster fliC_ir were used in another round of reads mapping to extract relevant reads with homology to the fliC locus. (iv) In BLAST analysis, the extracted reads were aligned using BLAST (30) to a collection of variable regions of the alleles in cluster fliC_ir and a BLAST score was assigned to each read/allele alignment. BLAST scores of all alignments associated with the same allele were summed, and the highest score pointed to the most likely allele and its corresponding antigen for the test genome, in this example, flagellar antigen “i.”
FIG 1.

An example workflow of fliC H antigen prediction. A detailed description can be found in Materials and Methods. fliC_eh>fliC_ir>fliC_z35a, predefined antigen clusters are summarized in Table S5 in the supplemental material.
Serotype prediction from genome assembly.
For O-antigen group determination, the galF and gnd genes that flank the rfb cluster were located by aligning the two genes against a Salmonella genome assembly (30). When both genes resided in the same contig, the rfb gene cluster between the two loci was extracted. When the two genes fell into two separate contigs, the corresponding contigs were split at galF or gnd in order to separate the sequence with homology to the rfb cluster from flanking sequences, producing four contig fragments. The rfb cluster or the set of 4 contig fragments that might or might not contain a partial rfb cluster was then aligned against the rfb database using BLAST. The resulting hits were ranked by BLAST scores, with the highest-ranking rfb hit determining the O-antigen group of the genome. For H antigen determination, fliC and fljB alleles were obtained from a genome assembly by in silico PCR (http://hgwdev.cse.ucsc.edu/cgi-bin/hgPcr). Primers used for in silico PCR are summarized in Table S7 in the supplemental material. Since the sequences flanking fljB may vary, multiple sets of primers were used to maximize the possibility of obtaining fljB amplicons. In silico amplicons of fliC and fljB were aligned against the H antigen database using BLAST, and the antigen types were identified using a method similar to that used for the determination of O antigen as described above.
Statistical analysis.
We assessed how well we could identify fliC and fljB antigens using the GenomeTrakr data set by calculating the difference between the numbers of reads (x and y) aligned to the top two best-mapped alleles for each of the two genes in the H antigen database. We used logistic regression to estimate the probability of making an incorrect identification given the size of the mapped reads difference (x − y). The outcome of the model was a binary indicator of whether the correct H antigen was identified. The covariate was the logarithmically scaled reads difference. We used scaling to account for the fact that the larger number of sequencing reads (z) tends to yield a bigger reads difference. The scaled reads difference (α) was calculated as follows: α = [(x − y)/z] × 106.
Nucleotide sequence accession numbers.
The sequences determined in this study have been deposited in the NCBI Sequence Read Archive under accession numbers SAMN03264859 to SAMN03264906, SAMN03264909 to SAMN03265006, and SAMN03265010 to SAMN03265087.
RESULTS
SeqSero pipeline.
The major components and workflows of the SeqSero system are outlined in Fig. 2 and detailed in Materials and Methods.
FIG 2.

Major components and workflows of SeqSero. Two workflows are represented, including serotype determination from (i) genome assembly and (ii) raw sequencing reads.
Databases of antigen determinants.
A total of 473 alleles representing 56 antigenic types for fliC and a total of 190 alleles representing 18 antigenic types for fljB were included in a combined H antigen database. A second database consisting of the 46 described rfb clusters was used for O-group determination from genome assemblies. A third database containing wzx, wzy, and other targets (see Table S4 in the supplemental material) was used for O-group determinations from raw sequencing reads (see Materials and Methods for details). The alleles represented in the databases theoretically identify 2,389 of the 2,577 serotypes described in the White-Kauffmann-Le Minor scheme.
Serotype prediction from whole-genome sequencing.
The results of the predictions are summarized in Table 1. For raw sequencing reads, two sets of isolates were tested: (i) 308 isolates that were serotyped at CDC and represented 72 serotypes (see Table S1 in the supplemental material) and (ii) 3,306 isolates of 228 serotypes sequenced as of June 2014 by GenomeTrakr of the Food and Drug Administration, a network of state and federal public health laboratories for the monitoring of foodborne pathogens isolated from food; the serotype of the strain was extracted from the metadata deposited with the sequence (see Table S2). For genome assemblies, 354 draft or finished genomes of 44 serotypes were tested, including all the assemblies deposited in GenBank as of April 2014 with serotype information available in the associated metadata and an N50 contig size (31) of more than 150,000 bases (see Table S3 in the supplemental material). This empirical N50 cutoff was used to exclude poorly assembled genomes.
TABLE 1.
Accuracy of serotype predictions
| Result | No. of genomes (% of total) |
||
|---|---|---|---|
| Reads mapping, CDC strains | Reads mapping, GenomeTrakr strains | Assembled genomes | |
| Expected serotypea | 304 (98.7) | 3,061 (92.6) | 324 (91.5) |
| Unexpected serotype | 2 (0.65) | 205 (6.2)b | 11 (3.1)b |
| Partial or no serotypec | 2 (0.65) | 40 (1.2) | 19 (5.4) |
| Total tested | 308 | 3306 | 354 |
The identification of the predicted serotype was considered correct when the serotype antigens detected corresponded to the antigens detected by conventional methods. See Text S1 in the supplemental material for a discussion of interpretation of serotype results. For GenomeTrakr and genome assembly datasets, serotype prediction in consensus with annotated serotype was considered correct.
Numbers represent serotype predictions inconsistent with the annotated serotype; the accuracy of the annotated serotype is unknown.
Some or all of the expected serotype determinants were not detected.
Of the 308 isolates with a confirmed serotype, 304 (98.7%) were correctly identified, 2 produced partial serotype information, and 2 produced an unexpected serotype result (Table 1). The accuracies of serotype predictions based on annotated serotypes were 92.6% and 91.5% for the GenomeTrakr and assembled genome data sets, respectively.
We analyzed the four whole-genome sequences from the confirmed serotype data set that produced a partial or unexpected serotype result in order to determine whether the result pointed to a problem in the SeqSero pipeline. Two of the six serotype Hvittingfoss (antigenic formula I 16:b:e,n,x) genomes tested failed to generate O-antigen calls, resulting in a partial serotype. Those genomes lacked sequencing reads that mapped to any rfb cluster that included the wzx gene and the wzy gene. One of the three serotype London (antigenic formula I 3,10:l,v:1,6) genomes produced a fljB determination of “e,n,x” instead of the expected “1,6”; reads that could be assembled into both “1,6” and “e,n,x” alleles were found. One of the five serotype Weltevreden (antigenic formula I 3,10:r:z6) genomes produced a fliC determination “i” instead of the expected “r” allele. Again, reads corresponding to both “r” and “i” were found to be present in the WGS.
Together, 200 serotypes were successfully predicted from the three data sets (see Table S6 in the supplemental material), including 85 of the top 100 Salmonella serotypes from human infections most commonly reported to the U.S. national Salmonella surveillance system (http://www.cdc.gov/nationalsurveillance/salmonella-surveillance.html).
Robustness of H antigen identification by reads mapping.
The phenotypic nature of serotyping and the diversity of Salmonella flagellar antigens make it difficult to map specific antigen types to individual genotypes or sequence variations (e.g., point mutations, insertions, and deletions). Closely related H antigens such as the G complex and 1 complex (11) constitute a particular challenge for robust identification of an antigenic type based on sequence comparison. We defined and calculated median scaled reads difference values (see Materials and Methods for details) to evaluate how well we can use reads mapping to identify H antigens of the genomes in the GenomeTrakr data set. The median scaled reads difference values were 3.59 for fliC and 1.82 for fljB, corresponding to predicted probabilities of an incorrect antigen call of 2.7% and 5.6%, respectively (Fig. 3). These results suggested that our method of H antigen identification based on reads mapping was robust. It should be noted that the statistical modeling was based on the results obtained after only the first round of reads mapping (Fig. 1); therefore, it included errors that might later have been corrected by the subsequent mapping and BLAST analyses.
FIG 3.

Predicted incorrect H antigen identification using reads mapping with 95% confidence limits. (A) Prediction for fliC identification. (B) Prediction for fljB identification. Logistic regression was used to estimate the probability of making an incorrect identification given the size of the mapped reads difference scaled by total number of reads sequenced from a genome. The GenomeTrakr data set selected for SeqSero validation was used for this analysis. Observed correct and incorrect antigens calls were based on the first round of reads mapping.
Serotype prediction from metagenome sequencing.
Serotype Typhimurium was detected in metagenomes of mouse stool samples 1 day before and 3, 6, and 14 days after the oral infection of S. enterica serotype Typhimurium strain 14028s (Table 2). A small number of reads were mapped to the serotype determinants on day −1, far fewer than were seen with later samples (Table 2). The strain detected on day −1 appeared to be phylogenetically distinct from the strain used for infection and serotyped on days 3, 6, and 14 (Fig. 4).
TABLE 2.
Serotype determination from stool metagenomes of mice orally infected with Salmonella
| Sampling time | Sample accession no.a | No. of reads mapped to individual antigen allelesc |
||
|---|---|---|---|---|
| wzx/wzy (O4)b | fliC (i) | fljB (1,2) | ||
| Day −1 | SRR916930 | 273 | 2 | 2 |
| Day 3 | SRR916932 | 521 | 10 | 11 |
| Day 6 | SRR916933 | 519 | 12 | 10 |
| Day 14 | SRR916931 | 1,572 | 21 | 21 |
NCBI SRA accession number of the metagenome sequence.
Predicted antigen type.
The number of reads aligned to the best-mapped antigen allele after the first round of reads mapping.
FIG 4.
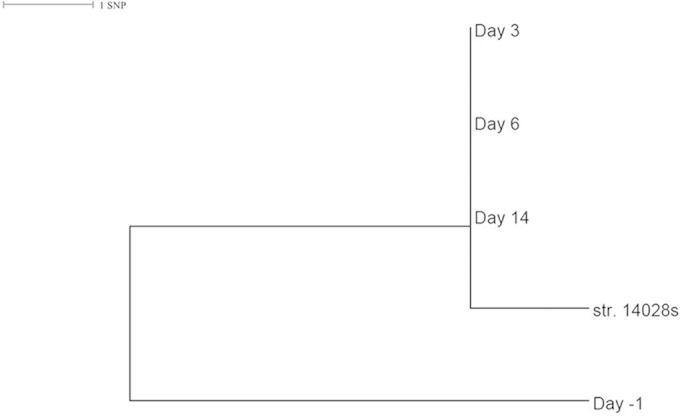
Phylogenetic relationship among detected Salmonella enterica serotype Typhimurium strains from fecal metagenomes of mice. A maximum likelihood tree shows the phylogenetic distance among the Salmonella strains serotyped from stool metagenomes of mice before and after oral infection. Raw reads from each metagenome were mapped to the genome (GenBank accession number CP001363) of the infection strain (str. 14028s), high-quality single nucleotide polymorphisms (SNPs) were identified, and a core genome SNP maximum likelihood tree was built using methods similar to those previously described in reference 36.
We also tested metagenome sequencing reads from a study performed to detect Salmonella spp. in a tomato phyllosphere microbiome (32); we did not find any Salmonella serotype markers, likely due to the low abundance of Salmonella in those samples and consistent with the fact that no Salmonella sp. was detected in that study using real-time PCR or culture methods.
To test whether Escherichia coli DNA might produce a false-positive signal in metagenomic samples, we tested metagenome sequences from 45 fecal specimens from patients involved in the 2011 outbreak of Shiga-toxigenic E. coli (STEC) O104:H4 in Germany (21). No reads from any of the metagenomes mapped to any allele in the Salmonella antigen databases.
DISCUSSION
The bioinformatics pipeline reported here determined Salmonella serotypes directly from raw sequencing reads or assembled genomes. The O group is determined primarily by analysis of wzx and wzy sequences for raw reads and by analysis of the rfb cluster for assembled genomes. Both H phases are determined through analysis of fliC and fljB sequences combined in the same H antigen database. Serotype determination from raw reads is recommended for high-throughput sequencing technologies that generate short reads, such as Illumina. Using raw sequencing reads reduces analysis time and allows serotype determination from raw data without the need for high-quality genome assembly and subsequent extraction of serotype determinants. With a computing capacity of 4 central processing unit (CPU) cores and 4 GB of random access memory (RAM), the serotype predictions of most isolates from raw WGS reads (an average of 2.17 million reads per genome) were finished within 10 min.
SeqSero proved accurate in determining serotypes by the use of genomes from strains in the CDC collection, which represented most of the 100 serotypes most commonly identified in the United States (Table 1). An O group was not identified for two isolates because no reads with homology to the entirety or the vast majority (the first 11,325 bases of the 12,901 bases of O16 rfb) of the rfb cluster were present in the WGS. Since an O group was detected in these strains using conventional methods, the rfb cluster is presumably present in those strains; we are currently investigating why no sequence reads were generated. Two additional isolates were not identified as the expected serotype due to the identification of a flagellar antigenic type that was not detected by conventional methods; for those genomes, reads corresponding to all three antigenic types (two expected for the confirmed serotype and a third detected by SeqSero) were identified, suggesting that these strains may have a third flagellin allele. This phenomenon has been described before (33). The accuracy of the GenomeTrakr and assembled genomes data set was somewhat lower; we were unable to confirm the accuracy of the annotated serotype for those strains. Since the serotypes of those strains had likely been determined in a variety of laboratories and reported to GenomeTrakr, it is possible that at least some of these misidentifications were serotyping errors and not errors of our application. Since the isolates of these sequences were not available to us, we could not confirm whether the results of the original serotype determination were correct. Also, they represented a somewhat more diverse set of serotypes; partial serotype determination may be due allelic diversity in previously uncharacterized serotypes.
The option to input genome assemblies for analysis was designed to support high-quality assemblies, especially those made possible by long-read sequencing platforms, such as PacBio. However, since O-group prediction from assembled genomes is based on the entire rfb cluster and Salmonella spp. and E. coli share some rfb clusters (26), the presence of an E. coli genome may produce a false-positive Salmonella O-group call (data not shown). The raw sequencing reads approach uses the more discriminatory targets wzx and wzy for O-group identification and is less likely to produce false-positive calls. Also, the genome assemblies in our validation data set produced a higher proportion of partial serotypes than did raw reads (Table 1), likely due to the failure in extracting serotype determinants from draft assemblies.
To improve differentiation of closely related H antigens, the assembly-free approach used a combination of reads mapping for efficiency and BLAST alignment for resolution. The first two of three rounds of mapping were used to identify a group of related H antigens (see Table S5 in the supplemental material). The third round extracted reads that could be aligned to fliC and fljB loci, followed by a BLAST alignment to determine specific fliC and fljB antigenic types. This strategy has the potential to detect Salmonella serotypes from voluminous and noise-rich metagenome sequences of complex microbial communities such as the fecal samples used for culture-independent diagnosis.
Rough, nonmotile, and monophasic variants were excluded from the initial validation of the tool because they may possess nonexpressed serotype determinants and may serotype differently by phenotypic and genetic methods. fljB may be deleted in some monophasic strains, in which case they type the same by phenotypic and genetic methods. In other instances, some or all of fljB remains or the monophasic nature arises from mutation in the phase inversion mechanism; for those strains, flagellar antigen determinants not detected by phenotypic method may be detected by genetic methods. Although they were excluded here, the ability to more fully characterize these strains is an added benefit of serotyping by genetic markers.
We were able to detect serotype Typhimurium from mouse fecal samples at four sampling times, including 1 day before oral infection. The strain on day −1 appeared to be present in a small amount and phylogenetically distinct from the challenge strain; its origin is unknown. Metagenomic samples known to contain E. coli O104:H4 did not produce any signal, suggesting that no false-positive serotyping had been generated by pathogenic or commensal Enterobacteriaceae spp. other than Salmonella spp. in the fecal samples. Due the limited data available for the evaluation of serotype determinations from metagenomic data sets, further investigation is needed to test the sensitivity and specificity of our tool when applied to metagenome sequencing data, especially when multiple strains of Salmonella with different serotypes are present in the same sample.
While serotype determination from the WGS workflow consists of multiple steps and relies on various databases for reads mapping and BLAST alignment, a self-explanatory and easy-to-use Web user interface is provided for public access to the tool. The Web application runs on a cloud server and is compatible with all major Internet browsers and mobile devices; it requires no empirical or arbitrary parameters to be set for analysis and is thus user friendly for novice users.
Since serotype antigens are subject to horizontal transfer, serotypes do not always correlate with phylogenetic relationships among Salmonella strains; i.e., strains from distinct genetic lineages may have the same complement of serotype antigens. It has been suggested that Salmonella serotyping should be replaced by a genetic subtyping scheme, such as multilocus sequence typing (MLST) (34). However, serotyping continues to serve a key role as a first-line subtyping method for Salmonella, with decades of epidemiological data based on serotype identification. Our tool provides a simple and fast means for determining serotypes from a WGS using the determinants responsible for serotypes. MLST and other genetic subtyping methods play an important role in public health surveillance and can provide a phylogenetic context within a serotype when needed. The ongoing transition into advanced technologies such as WGS (35) will enable the integration of the multiple identification, subtyping, and characterization workflows typically employed in public health laboratories into a single comprehensive and highly efficient platform, featuring in silico identification and prediction of various genotypic and phenotypic features (e.g., https://cge.cbs.dtu.dk/services/). Multiple methods can then be selected depending on the nature and scale of a particular investigation. Toward this prospect, the serotyping tool we present here maintains the well-established utility of Salmonella serotyping by bridging the gap between this historically important subtyping method and the cutting-edge application of whole-genome and metagenome sequencing in clinical and public health practices.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the members of the FDA GenomeTrakr network for making large volumes of Salmonella whole-genome sequences publicly available. We thank the members of the 100K Food-borne Pathogen Genome Project for sequencing CDC isolates used in this study.
This work was supported in part by contributions from the Board of Advisors, Center for Food Safety, University of Georgia, and by University of Georgia startup funds to X.D.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.00323-15.
REFERENCES
- 1.Herikstad H, Motarjemi Y, Tauxe RV. 2002. Salmonella surveillance: a global survey of public health serotyping. Epidemiol Infect 129:1–8. doi: 10.1017/S0950268802006842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weinberger M, Keller N. 2005. Recent trends in the epidemiology of non-typhoid Salmonella and antimicrobial resistance: the Israeli experience and worldwide review. Curr Opin Infect Dis 18:513–521. doi: 10.1097/01.qco.0000186851.33844.b2. [DOI] [PubMed] [Google Scholar]
- 3.Ran L, Wu S, Gao Y, Zhang X, Feng Z, Wang Z, Kan B, Klena JD, Lo Fo Wong DM, Angulo FJ, Varma JK. 2011. Laboratory-based surveillance of nontyphoidal Salmonella infections in China. Foodborne Pathog Dis 8:921–927. doi: 10.1089/fpd.2010.0827. [DOI] [PubMed] [Google Scholar]
- 4.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, Jones JL, Griffin PM. 2011. Foodborne illness acquired in the United States—major pathogens. Emerg Infect Dis 17:7–15. doi: 10.3201/eid1701.09-1101p1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grimont P, Weill F. 2007. Antigenic formulae of the Salmonella serovars, 9th ed WHO Collaborating Centre for Reference and Research on Salmonella. WHO, Geneva, Switzerland. [Google Scholar]
- 6.Guibourdenche M, Roggentin P, Mikoleit M, Fields PI, Bockemuhl J, Grimont PA, Weill FX. 2010. Supplement 2003-2007 (no. 47) to the White-Kauffmann-Le Minor scheme. Res Microbiol 161:26–29. doi: 10.1016/j.resmic.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Wattiau P, Boland C, Bertrand S. 2011. Methodologies for Salmonella enterica subsp. enterica subtyping: gold standards and alternatives. Appl Environ Microbiol 77:7877–7885. doi: 10.1128/AEM.05527-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi C, Singh P, Ranieri ML, Wiedmann M, Moreno Switt AI. 14 November 2013, posting date Molecular methods for serovar determination of Salmonella. Crit Rev Microbiol doi: 10.3109/1040841X.2013.837862. [DOI] [PubMed] [Google Scholar]
- 9.Samuel G, Reeves P. 2003. Biosynthesis of O-antigens: genes and pathways involved in nucleotide sugar precursor synthesis and O-antigen assembly. Carbohydr Res 338:2503–2519. doi: 10.1016/j.carres.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 10.Jiang XM, Neal B, Santiago F, Lee SJ, Romana LK, Reeves PR. 1991. Structure and sequence of the rfb (O antigen) gene cluster of Salmonella serovar typhimurium (strain LT2). Mol Microbiol 5:695–713. doi: 10.1111/j.1365-2958.1991.tb00741.x. [DOI] [PubMed] [Google Scholar]
- 11.Smith NH, Selander RK. 1990. Sequence invariance of the antigen-coding central region of the phase 1 flagellar filament gene (fliC) among strains of Salmonella typhimurium. J Bacteriol 172:603–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vanegas RA, Joys TM. 1995. Molecular analyses of the phase-2 antigen complex 1,2,.. of Salmonella spp. J Bacteriol 177:3863–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fitzgerald C, Collins M, van Duyne S, Mikoleit M, Brown T, Fields P. 2007. Multiplex, bead-based suspension array for molecular determination of common Salmonella serogroups. J Clin Microbiol 45:3323–3334. doi: 10.1128/JCM.00025-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McQuiston JR, Waters RJ, Dinsmore BA, Mikoleit ML, Fields PI. 2011. Molecular determination of H antigens of Salmonella by use of a microsphere-based liquid array. J Clin Microbiol 49:565–573. doi: 10.1128/JCM.01323-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Köser CU, Ellington MJ, Cartwright EJ, Gillespie SH, Brown NM, Farrington M, Holden MT, Dougan G, Bentley SD, Parkhill J, Peacock SJ. 2012. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog 8:e1002824. doi: 10.1371/journal.ppat.1002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joensen KG, Scheutz F, Lund O, Hasman H, Kaas RS, Nielsen EM, Aarestrup FM. 2014. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J Clin Microbiol 52:1501–1510. doi: 10.1128/JCM.03617-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, Jelsbak L, Sicheritz-Ponten T, Ussery DW, Aarestrup FM, Lund O. 2012. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol 50:1355–1361. doi: 10.1128/JCM.06094-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inouye M, Conway TC, Zobel J, Holt KE. 2012. Short read sequence typing (SRST): multi-locus sequence types from short reads. BMC Genomics 13:338. doi: 10.1186/1471-2164-13-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. 2012. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67:2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu G, Greninger AL, Isa P, Phan TG, Martinez MA, de la Luz Sanchez M, Contreras JF, Santos-Preciado JI, Parsonnet J, Miller S, DeRisi JL, Delwart E, Arias CF, Chiu CY. 2012. Discovery of a novel polyomavirus in acute diarrheal samples from children. PLoS One 7:e49449. doi: 10.1371/journal.pone.0049449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loman NJ, Constantinidou C, Christner M, Rohde H, Chan JZ, Quick J, Weir JC, Quince C, Smith GP, Betley JR, Aepfelbacher M, Pallen MJ. 2013. A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli O104:H4. JAMA 309:1502–1510. doi: 10.1001/jama.2013.3231. [DOI] [PubMed] [Google Scholar]
- 22.Eisenstein TK. 1975. Evidence for O antigens as the antigenic determinants in “ribosomal” vaccines prepared from Salmonella. Infect Immun 12:364–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong Y, Cunneen MM, Reeves PR. 2012. The Wzx translocases for Salmonella enterica O-antigen processing have unexpected serotype specificity. Mol Microbiol 84:620–630. doi: 10.1111/j.1365-2958.2012.08048.x. [DOI] [PubMed] [Google Scholar]
- 24.Brenner FW, McWhorter-Murlin AC. 1998. Identification and serotyping of Salmonella. Centers for Diseases Control and Prevention, Atlanta, GA. [Google Scholar]
- 25.Deatherage Kaiser BL, Li J, Sanford JA, Kim YM, Kronewitter SR, Jones MB, Peterson CT, Peterson SN, Frank BC, Purvine SO, Brown JN, Metz TO, Smith RD, Heffron F, Adkins JN. 2013. A multi-omic view of host-pathogen-commensal interplay in Salmonella-mediated intestinal infection. PLoS One 8:e67155. doi: 10.1371/journal.pone.0067155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu B, Knirel YA, Feng L, Perepelov AV, Senchenkova SN, Reeves PR, Wang L. 2014. Structural diversity in Salmonella O antigens and its genetic basis. FEMS Microbiol Rev 38:56–89. doi: 10.1111/1574-6976.12034. [DOI] [PubMed] [Google Scholar]
- 27.Fitzgerald C, Sherwood R, Gheesling LL, Brenner FW, Fields PI. 2003. Molecular analysis of the rfb O antigen gene cluster of Salmonella enterica serogroup O:6,14 and development of a serogroup-specific PCR assay. Appl Environ Microbiol 69:6099–6105. doi: 10.1128/AEM.69.10.6099-6105.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McQuiston JR, Parrenas R, Ortiz-Rivera M, Gheesling L, Brenner F, Fields PI. 2004. Sequencing and comparative analysis of flagellin genes fliC, fljB, and flpA from Salmonella. J Clin Microbiol 42:1923–1932. doi: 10.1128/JCM.42.5.1923-1932.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller JR, Koren S, Sutton G. 2010. Assembly algorithms for next-generation sequencing data. Genomics 95:315–327. doi: 10.1016/j.ygeno.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ottesen AR, Gonzalez A, Bell R, Arce C, Rideout S, Allard M, Evans P, Strain E, Musser S, Knight R, Brown E, Pettengill JB. 2013. Co-enriching microflora associated with culture based methods to detect Salmonella from tomato phyllosphere. PLoS One 8:e73079. doi: 10.1371/journal.pone.0073079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith NH, Selander RK. 1991. Molecular genetic basis for complex flagellar antigen expression in a triphasic serovar of Salmonella. Proc Natl Acad Sci U S A 88:956–960. doi: 10.1073/pnas.88.3.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Achtman M, Wain J, Weill FX, Nair S, Zhou Z, Sangal V, Krauland MG, Hale JL, Harbottle H, Uesbeck A, Dougan G, Harrison LH, Brisse S, S. Enterica MLST Study Group. 2012. Multilocus sequence typing as a replacement for serotyping in Salmonella enterica. PLoS Pathog 8:e1002776. doi: 10.1371/journal.ppat.1002776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kupferschmidt K. 2011. Epidemiology. Outbreak detectives embrace the genome era. Science 333:1818–1819. doi: 10.1126/science.333.6051.1818. [DOI] [PubMed] [Google Scholar]
- 36.Deng X, Desai PT, den Bakker HC, Mikoleit M, Tolar B, Trees E, Hendriksen RS, Frye JG, Porwollik S, Weimer BC, Wiedmann M, Weinstock GM, Fields PI, McClelland M. 2014. Genomic epidemiology of Salmonella enterica serotype Enteritidis based on population structure of prevalent lineages. Emerg Infect Dis 20:1481–1489. doi: 10.3201/eid2009.131095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.