

Abstract
Lateral gene transfer (LGT) has been crucial in the evolution of the cholera pathogen, Vibrio cholerae. The two major virulence factors are present on two different mobile genetic elements, a bacteriophage containing the cholera toxin genes and a genomic island (GI) containing the intestinal adhesin genes. Non-toxigenic V. cholerae in the aquatic environment are a major source of novel DNA that allows the pathogen to morph via LGT. In this study, we report a novel GI from a non-toxigenic V. cholerae strain containing multiple genes involved in DNA repair including the recombination repair gene recA that is 23% divergent from the indigenous recA and genes involved in the translesion synthesis pathway. This is the first report of a GI containing the critical gene recA and the first report of a GI that targets insertion into a specific site within recA. We show that possession of the island in Escherichia coli is protective against DNA damage induced by UV-irradiation and DNA targeting antibiotics. This study highlights the importance of genetic elements such as GIs in the evolution of V. cholerae and emphasizes the importance of environmental strains as a source of novel DNA that can influence the pathogenicity of toxigenic strains.
Introduction
Vibrio cholerae is a common inhabitant of marine and estuarine waters and is the causative agent of the diarrheal disease cholera. Although there are over 200 O-antigen serogroups among V. cholerae strains, only two, O1 and O139, are known to cause pandemics of cholera disease (Kaper et al., 1995). Lateral gene transfer (LGT) has largely contributed to the emergence of new pandemic strains of cholera (Faruque and Mekalanos, 2003; Keymer and Boehm, 2011). The appearance of the O139 serogroup and the so-called hybrid strains in the early 1990s are prime examples (Ramamurthy et al., 2003; Safa et al., 2009). Mobile genetic elements (MGEs) have been pivotal in the evolution of V. cholerae including diverse elements, such as the genomic islands (GIs) VPI-1, VPI-2, VSP-1, VSP-2, an integrative conjugative element, and the bacteriophage CTX (Faruque and Mekalanos, 2003; Grim et al., 2010). GIs are defined as large chromosomal regions that have features suggestive of recent LGT (Boyd et al., 2008). They have the capacity to excise and form circular intermediates and often target tRNA loci for their integration. In V. cholerae, GIs have been implicated in causing human disease and in environmental survival. For example, the replacement of the O1 classical biotype by the O1 El Tor biotype in the 1960s is suggested to be due to the acquisition of VSP-1 and VSP-2 that have probably enhanced epidemic spread (Faruque and Mekalanos, 2003). VPI-2 is a 57.3 kb island integrated at tRNA-Ser and encodes a neuraminidase important for converting higher-order sialogangliosides to GM1 gangliosides, the receptor for cholera toxin (Galen et al., 1992). Moreover, VPI-1 encodes for the toxin-coregulated pilus (TCP), an essential intestinal colonization factor, as well as the accessory colonization factor (ACF), and virulence regulators ToxT and TcpPH (Everiss et al., 1994; Murphy and Boyd, 2008). Non-O1/O139 V. cholerae strains are considered to be the major source of laterally acquired DNA for O1/O139 strains (Meibom et al., 2005) thus, a better understanding of the diverse genetic elements present in the V. cholerae species is important for predicting and mitigating the emergence of new pandemic strains.
In bacteria, errors in DNA can occur as part of normal DNA replication or can be induced by external stimuli (e.g. UV irradiation) (Janion, 2008). There are several genetic systems involved in error-free DNA repair including base excision repair (BER), nucleotide excision repair (NER), recombinational DNA repair and mismatch repair (MMR) (Rattray and Strathern, 2003; Janion, 2008; Polosina and Cupples, 2010; Lenhart et al., 2012). However, if DNA damage is extensive the mutagenic phase of the SOS response is triggered (Goodman, 2002). This response is mediated by DNA polymerases that replicate past template lesions in a process called translesion DNA synthesis (TLS) that is inherently error-prone (Goodman, 2002). For example, DNA polymerase V, encoded by the umuDC operon (Patel et al., 2010). The SOS induction of error-prone polymerases is considered a final response where although induced mutation(s) may be deleterious to the host cell, this is balanced against the need for rapid DNA repair (Goodman, 2002). An alternative view for the function of error-prone polymerases is that they act to generate genetic diversity that may have a role in environments where the host is maladapted by providing a bank of pre-existing genetic diversity within that population, some of which may confer a positive selective advantage. To support this second view, transcription of error-prone polymerases has been observed in the absence of SOS inducing DNA damage (Yeiser et al., 2002). Furthermore, error-prone polymerase mutants are less competitive than the parent cells during starvation (McKenzie et al., 2000; Yeiser et al., 2002; Tark et al., 2005), and some antibiotics (e.g. quinolones) induce the SOS mutagenic response increasing the frequency of resistant mutants (Piddock and Wise, 1987; Ysern et al., 1990).
In this study we report a novel GI inserted into recA of V. cholerae non-O1/O139 strain S24 isolated from an estuarine river in Sydney, Australia. This strain lacks the major virulence factors: cholera toxin and the toxin-coregulated pilus, thus is not capable of causing cholera. The GI carries (i) a recA gene phylogenetically distant from the disrupted host recA, designated recARME; (ii) a umuDC operon, designated umuDCRME, encoding DNA polymerase V; and (iii) genes encoding hypothetical proteins, proteins with DNA processing domains including a MutL domain involved in MMR, and proteins involved in site-specific recombination. The GI can excise as a closed circle and preferentially inserts into a specific site within recA. We also show that recARME is functional and provides protection from UV irradiation, a common source of DNA damage encountered in the shallow waters of marine and estuarine environments. Furthermore, the GI provides protection from the antibiotics bleomycin and ciprofloxacin. Acquisition of this GI by O1/O139 toxigenic V. cholerae would not only enhance survival of this pathogen in the natural environment but may also provide enhanced protection from DNA targeting antibiotics such as ciprofloxacin.
Results and discussion
Identification of a novel genomic island in V. cholerae S24 containing recA
S24 is an environmental, non-O1/O139 V. cholerae strain isolated from Georges River in Sydney, Australia, as described in a previous study (Islam et al., 2013). It was noted during multilocus sequence analysis of housekeeping genes adk, gyrB, mdh and recA using primers designed to amplify the V. cholerae S24 recA, (designated here recAS24), that a product of ∼ 1.5 kb was identified instead of the expected ∼ 850 bp. When the V. cholerae S24 draft genome sequence (to be released at a later date) was interrogated, it was noted that recAS24 was present on two separate contigs. PCR, using primers designed to sequence within these contigs, was used to close this region of the genome (described in Experimental procedures) resulting in a final contig of 262,869 bp. Within this contig and disrupting recAS24 at 494 bp into the 1065 bp gene was a GI of 32,787 bp we have designated recA mobile element (RME). Consistent with RME being a mobile genetic element, the GC content is 41.3% compared with the genome average of 47.2%, it encodes mobility functions (see below) and is bordered by 9 bp inverted repeats, designated IRR (for recA end) and IRi (for integrase end) (Fig. 1). Bioinformatic analysis of the GI identified 23 coding sequences (CDSs) (Fig. 1) including a complete copy of recA, designated recARME at the IRR end and a phage integrase at the IRi end. To our knowledge, this is the first mobile genetic element associated with the lateral movement of the critical gene, recA.
Fig 1.

Genetic structure and gene content of the recA genomic island. The RME contains 9 bp inverted repeats at each end (IRR and IRi) and 23 ORFs inclusive of the transposase genes from the ISVuv4 elements (striped boxes). The ISVuv4 elements are abutted by 7 bp direct repeats (DR) indicating insertion by transposition. RME contains multiple genes in DNA repair including a full copy of recA (RME001), the umuDC operon (RME017 and RME018) encoding the two subunits of DNA polymerase V and a gene encoding a protein with a MutL mismatch repair domain (RME004).
A number of other genes similar to those known to be involved in DNA processing are also found in RME including umuDC encoding the error-prone DNA polymerase V and a gene encoding a protein with a partial domain found in MutL (COG0323; Fig. 1), a component of the MMR pathway (Polosina and Cupples, 2010). A number other genes on RME, homologous to those involved in DNA processes include those encoding a ParB-like nuclease (91% identity to Vibrio alginolyticus 12G01; WP_005381205.1), a redox sensitive transcriptional activator with a SoxR-domain (96% identity to Vibrio sp. 712i1; WP_017634100.1), a type II restriction enzyme containing a methylase subunit (77% identity to Vibrio splendidus; WP_017082665.1) and a helicase (90% identity to Vibrio brasiliensis LMG 20546; WP_006880978.1).
Two insertion sequence (ISVvu4) elements were identified at positions 12,877 – 14,083 and 15,897 – 17,103 (striped boxes in Fig. 1) of RME. In both instances, 7 bp direct repeats (DR) were evident bordering the ISVvu4 elements indicating insertion by transposition. The DR for each ISVvu4 element is different, indicating independent insertion events. In silico removal of the ISVvu4 elements from the sequence did not restore any CDSs indicating that their insertion had not led to gene disruption. As expected, the promoter regions of both recARME and the umuDCRME operon have the characteristic LexA binding sequence of CTGT-(AT)4-ACAG indicating control by the SOS response (Wertman and Mount, 1985; Sanchez-Alberola et al., 2012). Present on RME are also genes putatively involved in mobilization/integration such as a phage integrase (RME022) and a site-specific recombinase XerS (RME019) (Fig. 1).
Phylogenetic analysis of recA sequences from the Vibrionaceae determined that recAS24 is characteristic of recA genes found within the V. cholerae clade, whereas recARME is not. It does, however, group with other more distantly related recA genes found in other members of the Vibrio genus (Fig. 2). This is consistent with recARME having been acquired by LGT. recA is an excellent phylogenetic marker for resolving relationships within the Vibrionaceae family (Stine et al., 2000; Thompson et al., 2004). Although the acquisition of a divergent recA in V. cholerae S24 is easily evident, this data reminds us that LGT of critical housekeeping genes like recA can and does occur. Less evident would be LGT of recA between closely related strains within the V. cholerae species confounding phylogenetic trees using a single marker (Bapteste et al., 2004; Creevey et al., 2004).
Fig 2.
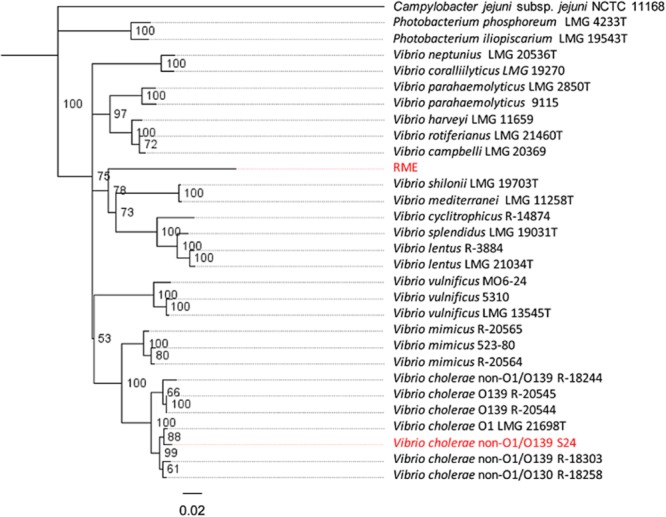
Phylogenetic analysis of recAS24 and recARME (RME highlighted in red) recAS24 (also highlighted in red) groups with V. cholerae strains whereas, recARME groups with recA from other Vibrio species indicating that recARME was mobilized from another member of the Vibrio genus.
The recA mobile element excises as a closed circle and targets a specific site in recA
Many GIs are known to excise from their location in the chromosome (Boyd et al., 2008). Analysis of RME suggested that it integrated into recAS24 using site-specific recombination. In site-specific recombination, a DNA recombinase recognizes specific sequences (usually inverted sequences) allowing for DNA breakage and joining reactions that result in integration or excision of the element (Hallet and Sherratt, 1997). Exact excision of RME at IRR and IRi was predicted to leave behind a 4 bp scar introducing a frame shift in recAS24 (Fig. 3A). To determine whether RME excision would leave behind an excision scar, an inverse PCR was conducted using primers reading out from the IRR and IRi ends (primers RME-F/RME-R in Table 1). A product of ∼ 560 bp was amplified (see gel image in Fig. 3B) and sequenced. Analysis of the sequence showed that excision occurred in one of two possible ways (Fig. 3B): (1) Excision occurred at 2 bp on either side of the IRR and IRi ends (black arrows in Fig. 3A) and/or (2) precise excision occurred at the end of IRi and at 4 bp before the end of IRR (grey arrows in Fig. 3A). Either way, excision was predicted to restore an uninterrupted and therefore functional copy of recAS24 in the chromosome, consistent with site-specific recombination. This was confirmed by amplification of an intact ‘empty’ insertion site using primers S24-cinA-F/S24-recX-R and excising the predicted ∼ 1.6 kb fragment (marked with a diamond in Fig. 3C). A nested PCR was then performed on the purified excised fragment using primers EcoRI-recA-F/EcoRI-recA-R (see gel image in Fig. 3C) and the product sequenced (Fig. 3C).
Fig 3.

A. Sequence abutting insertion of the recA genomic island in V. cholerae S24. The black and grey arrows demarcate the possible excision points for the RME.B. Sequence and gel image of the product derived from inverse PCR of the excised RME. The sequence shows that excision does not precisely occur at IRR and IRi and either occurs by two possible methods shown in Fig. 3A (see text for more details).C. Sequence of the ‘empty’ recAS24 insertion site and translated peptide sequence shows excision restores an uninterrupted recAS24. The asterisk marks the point of RME insertion. Amplification of the ‘empty’ recAS24 site gave a faint product (marked by diamond in left gel image). This was excised, purified and a nested PCR (right gel image) was conducted to generate sufficient product for sequencing.
Table 1.
Primers used in this study
| Primer | Sequence (5′-3′) | Target | Source |
|---|---|---|---|
| RME-R | GACGAGTCCAGCTCATGACA | integrase end of recA genomic island | This study |
| RME-F | GCTGCTAACGCTTTCTGCTT | recA end of recA genomic island | This study |
| S24-ctg675-F | CGGTTAGGAGGGGCTTTTAG | 3′ end of contig 675 | This study |
| S24-ctg708-R | TATCGGCTGTGGTTGTTTGA | 5′ end of contig 675 | This study |
| S24-ctg367-F | TAGCTAGAGCATTTGTCATAAGAAAAAGTAAG | 3′ end of contog 675 | This study |
| S24-ctg367-R | ACTGGCAGCAGAAGAAGCAT | 5′ end contig 708 | This study |
| S24-cinA-F | CAAGGTTGGCTCAAAGTG | cinA in V. cholerae S24 | This study |
| S24-recX-R | GGCATCACTCAAATACCCTA | recX in V. cholerae S24 | This study |
| S24-recA-F | CTGGAAATTTGTGATGCATT | recA in V. cholerae S24 | This study |
| EcoRI-recA-Fa | TTTTGAATTCTGGACGAGAATAAACAGAAGG | recA in V. cholerae S22 & S24 | This study |
| EcoRI-recA-Ra | TTTTGAATTCAAACTCTTCTGGCACCGC | recA in V. cholerae S22 & S24 | This study |
| EcoRI-Ori700-Ra | TTTTGAATTCCGCGCTATCGCTTGTCG | oripB1067 of pOriVn700 | This study |
| EcoRI-OriR6K-Fa | TTTTGAATTCGTGTTCCTGTGTCACTCAAAATTG | ori6k | This study |
| Ori700-F | CCCTATTCCTCTTTAGTCCTGC | oripB1067 of pOriVn700 | This study |
| Ori6K-R | TAACGCACTGAGAAGCCC | ori6k | This study |
| S24-phage-Int-F | GCCAAGATATGGCAGGAAAA | Integrase in recA genomic island | This study |
| S24-phage-Int-R | GGACGCTACCCAGTGAATGT | Integrase in recA genomic island | This study |
| recA-F | TGGACGAGAATAAACAGAAGGC | recA | (Boucher et al., 2011) |
| recA-R | CCGTTATAGCTGTACCAAGCGCCC | recA | (Boucher et al., 2011) |
| pCC2FOS-FP | GTACAACGACACCTAGAC | pCC2FOS sequencing primers (F) | Epicentre Biotechnologies |
| pCC2FOS-RP | CAGGAAACAGCCTAGGAA | pCC2FOS sequencing primers (R) | Epicentre Biotechnologies |
| recA-Tn5-F | CGCTCATAAGTCAGTAATGCTTCA | recA on genomic island. Used to screen for Tn5 insertion. | This study |
| umuC-Tn5-F | GATGTATGGCTGAATCGACCA | umuC on genomic island. Used to screen for Tn5 insertion. | This study |
| KAN-2 FP-1 | ACCTACAACAAAGCTCTCATCAACC | Forward primer inside Tn5 used to screen for Tn5 insertion. | Epicentre Biotechnologies |
| KAN-2 RP-1 | GCAATGTAACATCAGAGATTTTGAG | Reverse primer inside Tn5 used to screen for Tn5 insertion. | Epicentre Biotechnologies |
Bold and underlined sequence shows the EcoRI restriction site.
To determine whether the RME was capable of translocating from the genome of V. cholerae S24 into a new location, a vector (pOriVn700-recAS22; see Fig. 4A) containing the recA gene from a closely related strain of V. cholerae S24, strain S22 (Islam et al., 2013), was introduced into V. cholerae S24 by conjugation. Here, the RME is expected to excise from the genome of V. cholerae S24 and insert into recAS22 present on pOriVn700-recAS22. A control vector substituting recAS22 with gfp (pOriVn700-Placgfp; see Fig. 4A) was also introduced into V. cholerae S24 by conjugation as a control. Primers (ori6k-R and ori700-F; see Fig. 4A) targeting the vector backbone and the ends of the RME were used in a PCR reaction to determine whether the RME had mobilized into either pOriVn700-recAS22 or pOriVn700-Placgfp. In four independent experiments where pOriVn700-Placgfp was successfully introduced into V. cholerae S24 by conjugation, a product was never detected in the transconjugates (see representative gel in Fig. 4B). However, when pOriVn700-recAS22 was successfully introduced into V. cholerae S24, products were amplified (see representative gel in Fig. 4B) demonstrating insertion of the RME in the equivalent DNA site of recAS22 and in both orientations, with respect to recAS22. Sequence of the PCR products are shown in Fig. 4C, demonstrating successful insertion of RME into the equivalent recAS24 insertion site into recAS22 in pOriVn700-recAS22.
Fig 4.

Translocation of the recA genomic island from the genome of V. cholerae S24 into a replicating vector containing recAS22.A. Genetic structure of the replicating vectors pOriVn700-recAS22 and pOriVn700-Placgfp and the placement of primers ori6k-R and ori700-F used for amplifying the boundaries of the translocated genomic island are shown.B. Representative gel showing amplification using vector specific and RME specific primers from colonies derived from conjugation of pOriVn700-recAS22 (lanes 1 and 2) and pOriVn700-Placgfp (lanes 3 and 4) into V. cholerae S24. Lane 5 shows negative dH2O control.C. Sequence of products derived using vector-specific and RME-specific primers from PCR of V. cholerae S24 transconjugates from four independent conjugations. Each transconjugant is denoted by i, ii, iii and iv. In most instances (iib, iiib and ivb), the same transconjugate showed insertion of RME in both orientations relative to recAS22. The sequences indicate specific insertion of RME into the same site of recAS22 (the equivalent insertion site in recAS24).
It should be noted that homologous recombination between recAS22 in pOriVn700-recAS22 and recAS24 in the V. cholerae S24 genome could result in merodiploids that generate the same amplicons as those for RME inserted in the orientation (relative to recAS24) found in V. cholerae S24 (see Supporting Information Fig. S1 on expected merodiploids). However, in the immediate 2 bp of the IRR end for three of the transconjugates (Fig. 4C; ia, iiia and iva), there is a G to T substitution and at the immediate 3 bp of the IRi end, two transconjugants (Fig. 4C; iiia and iva) showed a T to G substitution. Since the recAS22 sequence is identical to recAS24 around the insertion point, homologous recombination should result in identical sequences immediately surrounding the RME. Furthermore, the RME was also found in both orientations, with respect to recAS22 (Fig. 4C). Consequently, homologous recombination is unable to explain these results.
These data show that RME is capable of mobilization and preferentially targets a specific site within recA. By carrying its own functional copy of recA, the GI does not affect any of the vital cell pathways associated with disruption of this gene during integration. Furthermore, specific targeting of recA may be necessary to ensure successful maintenance and dissemination of the GI. Since RecA does not function as a monomer but polymerizes to form a filament structure (Yu et al., 2004), disruption of the indigenous recA prevents a situation where two divergent RecA proteins might negatively interact resulting in reduced cell fitness.
The recA mobile element provides E. coli protection from UV irradiation
The presence of multiple genes involved in DNA repair prompted us to look at whether the GI could protect against a common DNA-damaging process faced by V. cholerae – UV irradiation. To investigate if recARME has a role in protecting the cell from DNA damage, the RME was cloned into a fosmid and used to transform recA- E. coli strain EPI300. The resultant transformant was subjected to UV-C irradiation. Fig. 5A shows that the presence of the RME element conferred enhanced bacterial cell survival when exposed to 0.8 mJ cm−2 of UV-C. From Fig. 5A and B it can be seen that EPI300 and EPI300 transformed by vector only controls are completely killed by exposure to 20 s 0.8 mJ cm−2 of UV-C. However, EPI300 transformants containing the RME survive for up to 60 s of UV-C exposure and show a 100-fold increase in survival at 10 s and up to 10,000,000-fold higher survival after 20 s UV-C exposure. Fig. 5A shows that when recARME is insertionally inactivated, the level of cell survivability decreases to a level comparable with the vector-only control (Fig. 5B). This demonstrates that recARME is functional and is the gene mainly responsible for the protection provided by the presence of the RME. An interesting future question would be whether recARME is more efficient in DNA repair than the host recA (i.e. recAS24). There is precedent for such an idea, in a strain of Clostridium difficile, a 4.2 kb insert disrupts a gene encoding a thymidylate synthetase (involved in DNA synthesis and repair) but contains a more functionally active version of the disrupted gene (Knetsch et al., 2011).
Fig 5.
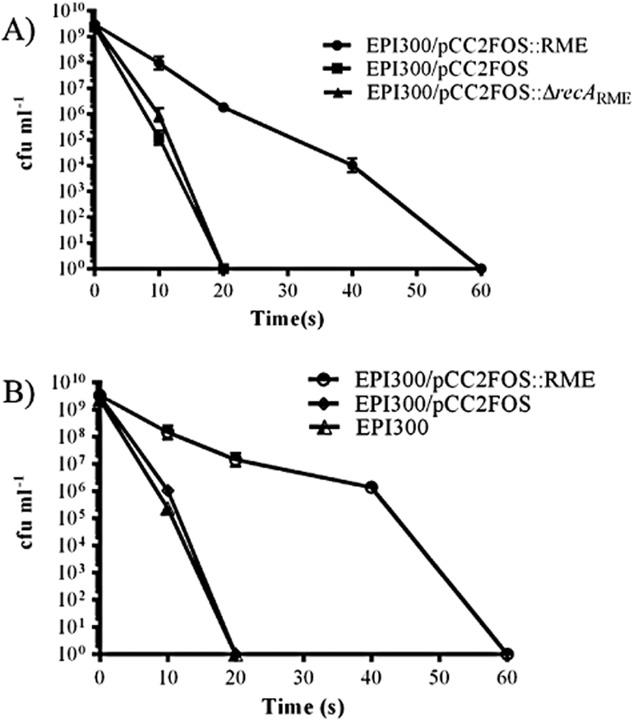
Survival of E. coli carrying the recA genomic island and control strains when exposed to UV-C stress. Time points are given at 0, 10, 20, 40 and 60 s. UV-C exposure was set to 0.8 mJ cm−2.
The recA mobile element provides E. coli with increased protection against antibiotics
Since RME has multiple genes involved in DNA repair, we tested whether RME provided enhanced protection against three DNA-targeting antibiotics: nalidixic acid, ciprofloxacin and bleomycin. Minimum inhibitory concentrations (MICs) were determined using nalidixic acid, ciprofloxacin and bleomycin (Table 2). Interestingly, the MIC of the first-generation quinolone, nalidixic acid, did not vary between any of the tested strains, including the RME (data not shown). However, when the strains given in Table 2 were tested using ciprofloxacin, a second-generation quinolone, a fourfold increase in MIC was observed for strains containing the RME. In the case of ciprofloxacin, it is apparent that recARME is responsible for the increased resistance. When recARME is insertionaslly inactivated from the genomic island, the MIC drops to a level equivalent to that seen for the E. coli strain EPI300. The importance of recA in protection against ciprofloxacin and other antibiotics has previously been documented [e.g. in Acinetobacter baumannii (Aranda et al., 2011)]. RME also provided protection from bleomycin in E. coli EPI300. However, in contrast to ciprofloxacin, when recARME and umuC from the RME (designated umuCRME) are both inactivated independently, the MIC against bleomycin is the same as E. coli EPI300, indicating that RecARME activation of the DNA polymerase V subunit UmuD to UmuD’ encoded on the GI is responsible for protections against bleomycin (Patel et al., 2010). Since umuDCRME was able to provide protection from bleomycin in a genetic background that already contains umuDC, it is hypothesized that in the V. cholerae species, where umuDC is only sporadically found, this element may provide increased protection from DNA damage compared with what was observed here in E. coli EPI300.
Table 2.
Minimal inhibitory concentration (MICsa)
| Strain | Ciprofloxacin | Bleomycin |
|---|---|---|
| EPI300 | 0.015625 | 8 |
| EPI300/pCC2FOS | 0.015625 | 8 |
| EPI300/pCC2FOS::RME | 0.0625 | 16 |
| EPI300/pCC2FOS::RMEΔumuCRME | 0.0625 | 8 |
| EPI300/pCC2FOS::RMEΔ recARME | 0.015625 | 8 |
MIC given as μg ml−1.
Apart from DNA repair, recA and DNA polymerase V (encoded by umuDC) are known to increase spontaneous mutation frequencies resulting in the emergence of antibiotic resistance mutants (Thi et al., 2011). Spontaneous mutation in V. cholerae is well documented to cause resistance to a variety of antibiotics (Goss et al., 1965; Gellert et al., 1977; Sugino et al., 1977; Allen et al., 1979; Kitaoka et al., 2011). Here we chose to examine the mutation frequency of two antibiotics, rifampicin which acts on protein synthesis and nalidixic acid which targets DNA replication by inhibiting the A subunit of DNA gyrase. Mutation frequencies after 24 and 48 h showed no differences between E. coli containing RME and the controls on 100 μg ml−1 rifampicin (data not shown). This may be because of rifampicin acting on protein synthesis and therefore not inducing the SOS response which induces transcription of umuDC. However, when the experiments were repeated with 50 μg ml−1 nalidixic acid, E. coli EPI300 and the vector-only control consistently did not produce any spontaneous mutants, while E. coli EPI300/pCC2FOS::RME showed an increased mutation frequency after both 24 and 48 h incubation in the presence of nalidixic acid (Table 3). E. coli EPI300/pCC2FOS::RMEΔumuCRME generally had the same mutation frequency as the complete RME. However, experiment 3 (Table 3) showed that this strain produced no mutants after 24 and 48 h. It can be concluded from these experiments that E. coli EPI300 with the complete RME provides an adaptive advantage by increasing the mutation rate resulting in subsequent resistance to nalidixic acid, but this effect could not be wholly attributed to the activity of umuCRME. One possible explanation is the activation of the indigenous E. coli UmuD by RecA provided by the GI. However, it cannot be excluded that other genes on the RME are elevating the spontaneous mutation rate. Nevertheless, umuDC-like operons are commonly associated with mobile genetic elements (Permina et al., 2002; Tark et al., 2005; Hare et al., 2012) and do provide a general adaptive advantage to hosts that house them (Yeiser et al., 2002; Tark et al., 2005). Although we failed in our attempts to transfer RME into seven non-O1/O139 V. cholerae strains from Sydney using chitin transformation, this element is likely to do the same in a V. cholerae genetic background.
Table 3.
Nalidixic acida mutation frequencies
| Experiment 1 | ||
|---|---|---|
| Strain | Mutation frequency 24 h | Mutation frequency 48 h |
| EPI300 | < 1.1 × 10-11 | < 1.1 × 10-11 |
| EPI300/pCC2FOS | < 1.7 × 10-11 | < 1.7 × 10-11 |
| EPI300/pCC2FOS::RME | 1.4 × 10-8 (4) | 1.5 × 10-8 (4) |
| EPI300/pCC2FOS::RMEΔumuCRME | 1.6 × 10-9 (3) | 2.1 × 10-9 (4) |
| Experiment 2 | ||
|---|---|---|
| Strain | Mutation frequency 24 h | Mutation frequency 48 h |
| EPI300 | < 1.1 × 10-11 | < 1.1 × 10-11 |
| EPI300/pCC2FOS | < 1.7 × 10-11 | < 1.7 × 10-11 |
| EPI300/pCC2FOS::RME | 8.3 × 10-10 (1) | 1.4 × 10-9 (3) |
| EPI300/pCC2FOS::RMEΔumuCRME | 2.8 × 10-9 (6) | 4.4 × 10-9 (7) |
| Experiment 3 | ||
|---|---|---|
| Strain | Mutation frequency 24 h | Mutation frequency 48 h |
| EPI300 | < 1.1 × 10-11 | < 1.1 × 10-11 |
| EPI300/pCC2FOS | < 1.3 × 10-11 | < 1.3 × 10-11 |
| EPI300/pCC2FOS::RME | 4.8 × 10-10 (1) | 1.7 × 10-9 (4) |
| EPI300/pCC2FOS::RMEΔumuCRME | < 9.1 × 10-12 | < 9.1 × 10-12 |
Concentration of nalidixic acid = 50 μg ml−1.
‘<’ indicates that zero colonies appeared in all 10 replicates (see Experimental procedures).
Numbers in brackets indicates the number of replicates in which one or more colonies appeared.
To conclude, this study reports a novel GI in V. cholerae that contains genes involved in multiple DNA repair pathways, including the critical housekeeping gene recA and genes encoding DNA polymerase V which in this study, we show to be functional. The presence of other DNA processing genes may provide V. cholerae with alternative DNA repair pathways. Since this element can excise from its chromosomal location, it has the potential to mobilize into other strains, such as cholera toxin-producing O1/O139 pandemic strains. Such mobilization could have implications for increased environmental survival or resistance to certain antibiotics.
Experimental procedures
Bacterial strains, plasmids and growth conditions
All strains and plasmids used are shown in Table 4. V. cholerae strain S24 was collected from Georges River in the greater Sydney (Australia) urban area as previously described (Islam et al., 2013). All E. coli and V. cholerae strains were routinely grown on Luria–Bertani (LB) broth at 37°C under aerobic conditions. For E. coli WM3064, diaminopimelic acid (DAP) was added to a final concentration of 0.3 mM. Spectinomycin was used for E. coli and V. cholerae at 50 μg ml−1 and 125 μg ml−1 respectively. Chloramphenicol was used at 12.5 μg ml−1.
Table 4.
List of strains and plasmids
| Strain or plasmid | Relevant genotypea | Reference or source |
|---|---|---|
| V. cholerae | ||
| S24 | Wild-type (non-O1/O139) | This study |
| S22 | Wild-type (non-O1/O139) | (Islam et al., 2013) |
| E. coli | ||
| DH5αλpir | endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG Φ80dlacZΔM15 Δ(lacZYA-argF)U169 hsdR17 λpir | (Demarre et al., 2005) |
| WM3064 | Donor strain for conjugation: thrB1004 pro thi rpsL hsdS lacZΔM15 RP4-1360 Δ(araBAD)567 ΔdapA1341::[erm pir], SmR | (Saltikov and Newman, 2003) |
| EPI300™-T1R | [F- mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ- rpsL nupG trfA tonA dhfr], SmR, TpR | Epicentre Biotechnologies |
| Plasmids/fosmids | ||
| pCC2FOS | Cloning vector, CmR | |
| pCC2FOS-RME | pCC2FOS vector containing 32 kb insert from V. cholerae S24. The insert contains the recA GI and surrounding sequence, CmR | This study |
| pCC2FOS::RMEΔrecARME | pCC2FOS vector containing 32 kb insert from V. cholerae S24. The insert contains the recA GI and surrounding sequence and has recA on the GI insertionally inactivated by Tn5, KmR, CmR | This study |
| pCC2FOS::RMEΔumuCRME | pCC2FOS vector containing 32 kb insert from V. cholerae S24. The insert contains the recA GI and surrounding sequence and has umuC present on the GI insertionally inactivated by Tn5, KmR, CmR | This study |
| pOriVn700 | Low copy mobilizable vector containing oripB1067 (vibrio specific) and ori6K, SpR | (Le Roux et al., 2011) |
| pOriVn700-recAS22 | pOriVn700 with recA from V. cholerae S22 in between oripB1067 and ori6K. The recA gene is reading toward oripB1067, SpR | This study |
| pOriVn700-Placgfp | pOriVn700 with Placgfp cloned in between oripB1067 and ori6K, SpR | (Le Roux et al., 2011) |
TcR, tetracycline resistance; SmR, streptomycin resistance; SpR, spectinomycin resistance; CmR, chloramphenicol resistance, KmR, kanamycin resistance.
Whole genome sequencing, PCR, DNA extraction and sequencing methods
DNA was extracted using the Wizard genomic DNA purification kit (Promega). Plasmid and PCR/gel extractions were done using PureYield Plasmid Miniprep and Wizard SV Gel and PCR clean-up systems respectively (Promega). Purified DNA from V. cholerae S24 was sequenced at the Wellcome Trust Sanger Institute using Illumina-based technology.
All primers used in this study are shown in Table 1. Standard PCR was performed using the PCR master mix (Promega) containing 25 units ml−1 of Taq DNA polymerase, 800 μM dNTPs and 1.5 mM MgCl2. Primers were used at a final concentration of 0.5 μM each. All PCRs were performed with 30 cycles of denaturation at 94°C for 30 s, the appropriate annealing temperature for 30 s and an extension of 72°C (1 min kb−1) and sequencing performed at Macrogen. From whole genome sequencing (Wellcome Trust Sanger Institute) it became evident that the host recA had been disrupted and was present on two separate contigs. The two contigs (contigs 675 and 708) were pieced together by PCR and joined to an intervening third contig to produce a contig of 262,869 bp (contig 367) using primers described in Table 1. The accession number for RME is KJ123688.
Cloning of RME and transposon mutagenesis of recA genomic island
To clone the recA genomic island (RME) from V. cholerae strain S24, genomic DNA was digested with NaeI and a library constructed using the CopyControl Fosmid Library Production Kit (Epicentre). NaeI digestion of V. cholerae strain S24 genomic DNA creates a fragment of 38, 913 bp containing the entire 32, 787 bp RME. The library was screened for a fosmid clone containing the 38, 913-bp NaeI fragment using primers targeting the phage integrase in the RME (Table 1). A positive clone designated pCC2FOS-RME was confirmed by sequencing the ends of the cloned insert using the pCC2FOS vector primers FP and RP (Table 1). To create the pCC2FOS no insert control, linearized and dephosphorylated pCC2FOS (Epicentre) was treated with T4 polynucleotide kinase and circularized by ligation. A mutant library of pCC2FOS-RME was constructed using the EZ-Tn5 Kan-2 Insertion Kit (Epicentre Biotechnologies) according to manufacturer instructions. Mutants containing knockouts of individual genes present on the genomic island were screened by PCR using primers reading out from EZ-Tn5 Kan-2 and a primer targeting the gene of interest (Table 1).
Phylogenetic analysis
Phylogenetic analysis of recAS24 and recARME was done using bioinformatics program Geneious version 6.1.6 and FigTree version 1.4.0. Phylogenetic tree parameters were taken from (Thompson et al., 2004). Distance estimations were obtained using the Jukes and Cantor model and tree built using the neighbour-joining method. Bootstrap percentages were calculated after 100 simulations. The Campylobacter jejuni subsp. jejuni NCTC 11168 recA sequence was used as an outgroup.
recA targeting experiments
Vector pOriVn700 and recA from a strain of V. cholerae S22 that is closely related to V. cholerae S24 were amplified using primer pairs EcoRI-Ori700-R/EcoRI-Ori6K-F and EcoRI-recA-F/EcoRI-recA-R respectively (Table 1). Since the primers contained engineered EcoRI sites, the resulting amplicons of recAS22 and pOriVn700 were purified, digested with EcoR1 and then ligated together using T4 DNA ligase (Fermentas). The ligation mix was then transformed into E. coli DH5αλpir to produce pOriVn700-recAS22. The construct was then extracted and transformed into the conjugation donor strain E. coli WM3064.
Conjugations using pOriVn700-recAS22 and pOriVn700-Placgfp were performed by combining equal volumes of overnight cultures in LB from both donor and recipient strains. These were then centrifuged at 3000 × g and re-suspended in 50 μL of LB and spotted onto a 0.2 μM filter (Millipore) that had been placed on an LB agar plate containing 0.3 mM DAP. Donor and recipient cells were left to incubate for 4 h at 37°C and cells were then removed from the filter by vortexing. The re-suspended cells were then plated on LB + 125 μg ml−1 spectinomycin and incubated at 37°C overnight. One colony per mating was picked and appropriate junction PCR was conducted using primers in plasmid backbone (Table 1; Ori700-F/Ori6K-R) and primers reading out from RME (Table 1; RME-F/RME-R).
UV stress experiments
UV stress experiments were adapted from Lin and Wang (2001). Strains were grown for 16–20 h at 37°C with shaking at 230 r.p.m. in 5 ml LB broth supplemented with appropriate antibiotic. Cells were centrifuged at 4000 × g, corrected for differences in optical density (OD) at 600 nm and re-suspended in equal volumes of M9 salts (Sambrook et al., 1989) supplemented with MgSO4.7H2O to a final concentration of 0.002 M. The entire cell suspension was placed in a clear bottom 10 cm plastic petri dish and subjected to 0.8 mJ cm−2 UV-C for 0, 10, 20, 40 and 60 s using an Amersham Life Science Ultraviolet Crosslinker. After each time interval, 150 ul aliquot was removed and placed in a 1.5 ml Eppendorf tube in the dark. The remaining liquid culture was thoroughly re-suspended using a pipette to avoid clumping of cells. After the final UV-C exposure time point, cells were diluted in M9 salts+ MgSO4.7H2O to 10-6 and enumerated by the drop plate method on LB agar. Plates were incubated in the dark to prevent photoreactivation at 37°C overnight and colony-forming units (CFUs) were calculated the following day.
Minimum inhibitory concentration experiments and antibiotic mutation frequency experiments
MICs of nalidixic acid, ciprofloxacin and bleomycin were determined by broth microdilution using standard methods (Clinical and Laboratory Standards Institute, 2003) except that LB broth was used as the growth medium instead of Mueller Hinton. Each MIC was performed in triplicate. The mutation frequency experiment was designed using the guidelines described in (Pope et al., 2008). Specifically, mutation frequencies were determined using LB supplemented with 50 μg ml−1 nalidixic acid and 100 μg ml−1 rifampicin. Ten replicate overnight cultures for each strain were grown in 5 ml LB (chloramphenicol was added for those strains carrying pCC2FOS and derivatives). Each overnight culture was then diluted to ∼ 104 CFU ml−1 with fresh LB5 (no chloramphenicol added) and 5 ml for each replicate was transferred into a 15 ml tube and incubated for 16–20 h at 37°C with shaking at 230 r.p.m. The following day, 200 μl from each tube was spread plated onto LB5 agar supplemented with the appropriate antibiotic (rifampicin or nalidixic acid) and incubated for 24 h and then 48 h at 37°C when colonies were counted. This was repeated in triplicate. In order to calculate total colony counts, cells were enumerated on LB5 agar with no antibiotic. Note that these experiments were performed in a Class II Biosafety Hood to avoid any contamination. Mutation frequencies were calculated as number of antibiotic-resistant CFUs/total number of CFUs after 24 and 48 h.
Acknowledgments
We would like to acknowledge Associate Professor Aaron Darling for his help with the phylogenetic analysis. This work was supported by the ithree institute, UTS. Rita Rapa is a recipient of a UTS postgraduate scholarship. The sequencing of V. cholerae S24 was funded under Wellcome Trust grant 098051.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Fig. S1. Possible insertion and homologous recombination events of the recA genomic island with pOriVn700-recAS22. Production of merodiploids because of homologous recombination between recAS22 and recAS24 could result in two genetic structures (crossover 1 and crossover 2). In both instances, PCR products could be generated using the vector-specific and RME-specific primers used to detect insertion of the RME into recAS22 (broken lines) in the orientation found in the V. cholerae S24 genome (insertion 1). Insertion of the RME in the inverse orientation (insertion 2) would generate products using inverse primer pairs. The inverse insertion cannot be explained by homologous recombination and indicates an integration event.
Appendix S1. Genbank file of the recA mobile element (RME); Accession Number KJ123688.
References
- Allen JG, Atherton FR, Hall MJ, Hassall CH, Holmes SW, Lambert RW, et al. Phosphonopeptides as antibacterial agents: alaphosphin and related phosphonopeptides. Antimicrob Agents Chemother. 1979;15:684–695. doi: 10.1128/aac.15.5.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda J, Bardina C, Beceiro A, Rumbo S, Cabral MP, Barbe J, Bou G. Acinetobacter baumannii RecA protein in repair of DNA damage, antimicrobial resistance, general stress response, and virulence. J Bacteriol. 2011;193:3740–3747. doi: 10.1128/JB.00389-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapteste E, Boucher Y, Leigh J, Doolittle WF. Phylogenetic reconstruction and lateral gene transfer. Trends Microbiol. 2004;12:406–411. doi: 10.1016/j.tim.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Boucher Y, Cordero OX, Takemura A, Hunt DE, Schliep K, Bapteste E, et al. Local mobile gene pools rapidly cross species boundaries to create endemicity within global Vibrio cholerae populations. MBio. 2011;2:e00335-10. doi: 10.1128/mBio.00335-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd EF, Almagro-Moreno S, Parent MA. Genomic islands are dynamic, ancient integrative elements in bacterial evolution. Trends Microbiol. 2008;17:47–53. doi: 10.1016/j.tim.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Clinical and Laboratory Standards Institute. 2003. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically.
- Creevey CJ, Fitzpatrick DA, Philip GK, Kinsella RJ, O'Connell MJ, Pentony MM, et al. Does a tree-like phylogeny only exist at the tips in the prokaryotes? Proc Biol Sci. 2004;271:2551–2558. doi: 10.1098/rspb.2004.2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarre G, Guérout A, Matsumoto-Mashimo C, Rowe-Magnus DA, Marliére P, Mazel D. A new family of mobilizable suicide plasmids based on broad host range R388 plasmid (IncW) and RP4 plasmid (IncPα) conjugative machineries and their cognate Escherichia coli host strains. Res Microbiol. 2005;156:245–255. doi: 10.1016/j.resmic.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Everiss KD, Hughes KJ, Peterson KM. The accessory colonization factor and toxin-coregulated pilus gene clusters are physically linked on the Vibrio cholerae 0395 chromosome. DNA Seq. 1994;5:51–55. doi: 10.3109/10425179409039704. [DOI] [PubMed] [Google Scholar]
- Faruque SM, Mekalanos JJ. Pathogenicity islands and phages in Vibrio cholerae evolution. Trends Microbiol. 2003;11:505–510. doi: 10.1016/j.tim.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Galen JE, Ketley JM, Fasano A, Richardson SH, Wasserman SS, Kaper JB. Role of Vibrio cholerae neuraminidase in the function of cholera toxin. Infect Immun. 1992;60:406–415. doi: 10.1128/iai.60.2.406-415.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellert M, Mizuuchi K, O'Dea MH, Itoh T, Tomizawa JI. Nalidixic acid resistance: a second genetic character involved in DNA gyrase activity. Proc Natl Acad Sci USA. 1977;74:4772–4776. doi: 10.1073/pnas.74.11.4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu Rev Biochem. 2002;71:17–50. doi: 10.1146/annurev.biochem.71.083101.124707. [DOI] [PubMed] [Google Scholar]
- Goss WA, Deitz WH, Cook TM. Mechanism of action of nalidixic acid on Escherichia coli Inhibition of deoxyribonucleic acid synthesis. J Bacteriol. 1965;89:1068–1074. doi: 10.1128/jb.89.4.1068-1074.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grim CJ, Hasan NA, Taviani E, Haley B, Chun J, Brettin TS, et al. Genome sequence of hybrid Vibrio cholerae O1 MJ-1236, B-33, and CIRS101 and comparative genomics with V. cholerae. J Bacteriol. 2010;192:3524–3533. doi: 10.1128/JB.00040-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallet B, Sherratt DJ. Transposition and site-specific recombination: adapting DNA cut-and-paste mechanisms to a variety of genetic rearrangements. FEMS Microbiol Rev. 1997;21:157–178. doi: 10.1111/j.1574-6976.1997.tb00349.x. [DOI] [PubMed] [Google Scholar]
- Hare JM, Bradley JA, Lin C, Elam TJ. Diverse responses to UV light exposure in Acinetobacter include the capacity for DNA damage-induced mutagenesis in the opportunistic pathogens Acinetobacter baumannii and Acinetobacter ursingii. Microbiology. 2012;158:601–611. doi: 10.1099/mic.0.054668-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam A, Labbate M, Djordjevic SP, Alam M, Darling A, Melvold J, et al. Indigenous Vibrio cholerae strains from a non-endemic region are pathogenic. Open Biol. 2013;3:120181. doi: 10.1098/rsob.120181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janion C. Inducible SOS response system of DNA repair and mutagenesis in Escherichia coli. Int J Biol Sci. 2008;4:338–344. doi: 10.7150/ijbs.4.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaper J, Morris J, Jr, Levine M. Cholera. Clin Microbiol Rev. 1995;8:48–86. doi: 10.1128/cmr.8.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keymer DP, Boehm AB. Recombination shapes the structure of an environmental Vibrio cholerae population. Appl Environ Microbiol. 2011;77:537–544. doi: 10.1128/AEM.02062-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaoka M, Miyata ST, Unterweger D, Pukatzki S. Antibiotic resistance mechanisms of Vibrio cholerae. J Med Microbiol. 2011;60:397–407. doi: 10.1099/jmm.0.023051-0. [DOI] [PubMed] [Google Scholar]
- Knetsch CW, Hensgens MP, Harmanus C, van der Bijl MW, Savelkoul PH, Kuijper EJ, et al. Genetic markers for Clostridium difficile lineages linked to hypervirulence. Microbiology. 2011;157:3113–3123. doi: 10.1099/mic.0.051953-0. [DOI] [PubMed] [Google Scholar]
- Le Roux F, Davis BM, Waldor MK. Conserved small RNAs govern replication and incompatability of a diverse new plasmid family from marine bacteria. Nucleic Acids Res. 2011;39:1004–1013. doi: 10.1093/nar/gkq852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenhart JS, Schroeder JW, Walsh BW, Simmons LA. DNA repair and genome maintenance in Bacillus subtilis. Microbiol Mol Biol Rev. 2012;76:530–564. doi: 10.1128/MMBR.05020-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, Wang A. UV mutagenesis in Escherichia coli K-12: cell survival and mutation frequency of the chromosomal genes lacZrpoBompF, and ampA. J Exp Microbiol Immunol. 2001;1:32–46. [Google Scholar]
- McKenzie GJ, Harris RS, Lee PL, Rosenberg SM. The SOS response regulates adaptive mutation. Proc Natl Acad Sci USA. 2000;97:6646–6651. doi: 10.1073/pnas.120161797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meibom KL, Blokesch M, Dolganov NA, Wu C-Y, Schoolnik GK. Chitin induces natural competence in Vibrio cholerae. Science. 2005;310:1824–1827. doi: 10.1126/science.1120096. [DOI] [PubMed] [Google Scholar]
- Murphy RA, Boyd EF. Three pathogenicity islands of Vibrio cholerae can excise from the chromosome and form circular intermediates. J Bacteriol. 2008;190:636–647. doi: 10.1128/JB.00562-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M, Jiang Q, Woodgate R, Cox MM, Goodman MF. A new model for SOS-induced mutagenesis: how RecA protein activates DNA polymerase V. Crit Rev Biochem Mol Biol. 2010;45:171–184. doi: 10.3109/10409238.2010.480968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Permina EA, Mironov AA, Gelfand MS. Damage-repair error-prone polymerases of eubacteria: association with mobile genome elements. Gene. 2002;293:133–140. doi: 10.1016/s0378-1119(02)00701-1. [DOI] [PubMed] [Google Scholar]
- Piddock LJ, Wise R. Induction of the SOS response in Escherichia coli by 4-quinolone antimicrobial agents. FEMS Microbiol Lett. 1987;41:289–294. [Google Scholar]
- Polosina YY, Cupples CG. Wot the ‘L-Does MutL do? Mutat Res. 2010;705:228–238. doi: 10.1016/j.mrrev.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Pope CF, O'Sullivan DM, McHugh TD, Gillespie SH. A practical guide to measuring mutation rates in antibiotic resistance. Antimicrob Agents Chemother. 2008;52:1209–1214. doi: 10.1128/AAC.01152-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamurthy T, Yamasaki S, Takeda Y, Nair GB. Vibrio cholerae O139 Bengal: odyssey of a fortuitous variant. Microbes Infect. 2003;5:329–344. doi: 10.1016/s1286-4579(03)00035-2. [DOI] [PubMed] [Google Scholar]
- Rattray AJ, Strathern JN. Error-prone DNA polymerases: when making a mistake is the only way to get ahead. Annu Rev Genet. 2003;37:31–66. doi: 10.1146/annurev.genet.37.042203.132748. [DOI] [PubMed] [Google Scholar]
- Safa A, Bhuiya AN, Murphy D, Bates J, Nusrin S, Kong RYC, et al. Multilocus genetic analysis reveals that the Australian strains of Vibrio cholerae O1 are similar to the pre-seventh pandemic strains of the El Tor biotype. J Med Microbiol. 2009;58:105–111. doi: 10.1099/jmm.0.004333-0. [DOI] [PubMed] [Google Scholar]
- Saltikov CW, Newman DK. Genetic identification of a respiratory arsenate reductase. Proc Natl Acad Sci USA. 2003;100:10983–10988. doi: 10.1073/pnas.1834303100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: A laboratory manual.
- Sanchez-Alberola N, Campoy S, Barbé J, Erill I. Analysis of the SOS response of Vibrio and other bacteria with multiple chromosomes. BMC Genomics. 2012;13:58. doi: 10.1186/1471-2164-13-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine OC, Sozhamannan S, Gou Q, Zheng S, Morris JG, Jr, Johnson JA. Phylogeny of Vibrio cholerae based on recA sequence. Infect Immun. 2000;68:7180–7185. doi: 10.1128/iai.68.12.7180-7185.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino A, Peebles CL, Kreuzer KN, Cozzarelli NR. Mechanism of action of nalidixic acid: purification of Escherichia coli nalA gene product and its relationship to DNA gyrase and a novel nicking-closing enzyme. Proc Natl Acad Sci USA. 1977;74:4767–4771. doi: 10.1073/pnas.74.11.4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tark M, Tover A, Tarassova K, Tegova R, Kivi G, Hõrak R, Kivisaar M. A DNA polymerase V homologue encoded by TOL plasmid pWW0 confers evolutionary fitness on Pseudomonas putida under conditions of environmental stress. J Bacteriol. 2005;187:5203–5213. doi: 10.1128/JB.187.15.5203-5213.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thi TD, Lopez E, Rodriguez-Rojas A, Rodriguez-Beltran J, Couce A, Guelfo JR, et al. Effect of recA inactivation on mutagenesis of Escherichia coli exposed to sublethal concentrations of antimicrobials. J Antimicrob Chemother. 2011;66:531–538. doi: 10.1093/jac/dkq496. [DOI] [PubMed] [Google Scholar]
- Thompson CC, Thompson FL, Vandemeulebroecke K, Hoste B, Dawyndt P, Swings J. Use of recA as an alternative phylogenetic marker in the family Vibrionaceae. Int J Syst Evol Microbiol. 2004;54:919–924. doi: 10.1099/ijs.0.02963-0. [DOI] [PubMed] [Google Scholar]
- Wertman KF, Mount DW. Nucleotide sequence binding specificity of the LexA repressor of Escherichia coli K-12. J Bacteriol. 1985;163:376–384. doi: 10.1128/jb.163.1.376-384.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeiser B, Pepper ED, Goodman MF, Finkel SE. SOS-induced DNA polymerases enhance long-term survival and evolutionary fitness. Proc Natl Acad Sci USA. 2002;99:8737–8741. doi: 10.1073/pnas.092269199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ysern P, Clerch B, Castaňo M, Gilbert I, Barbé J, Llagostera M. Induction of SOS genes in Escherichia coli and mutagenesis in Salmonella typhimurium by fluoroquinolones. Mutagenesis. 1990;5:63–66. doi: 10.1093/mutage/5.1.63. [DOI] [PubMed] [Google Scholar]
- Yu X, VanLoock MS, Yang S, Reese JT, Egelman EH. What is the structure of the RecA-DNA filament? Curr Protein Pept Sci. 2004;5:73–79. doi: 10.2174/1389203043486883. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Possible insertion and homologous recombination events of the recA genomic island with pOriVn700-recAS22. Production of merodiploids because of homologous recombination between recAS22 and recAS24 could result in two genetic structures (crossover 1 and crossover 2). In both instances, PCR products could be generated using the vector-specific and RME-specific primers used to detect insertion of the RME into recAS22 (broken lines) in the orientation found in the V. cholerae S24 genome (insertion 1). Insertion of the RME in the inverse orientation (insertion 2) would generate products using inverse primer pairs. The inverse insertion cannot be explained by homologous recombination and indicates an integration event.
Appendix S1. Genbank file of the recA mobile element (RME); Accession Number KJ123688.