

Abstract
Histone post-translational modifications regulate transcription and other DNA-templated functions. This process is dynamically regulated by specific modifying enzymes whose activities require metabolites that either serve as co-substrates or act as activators/inhibitors. Therefore, metabolism can influence histone modification by changing local concentrations of key metabolites. Physiologically, the epigenetic response to metabolism is important for nutrient sensing and environment adaption. In pathologic states, the connection between metabolism and histone modification mediates epigenetic abnormality in complex disease. In this review, we summarize recent studies of the molecular mechanisms involved in metabolic regulation of histone modifications and discuss their biological significance.
Keywords: epigenetic, metabolism, histone, dynamic regulation, acetylation, methylation
Introduction
Post translational modifications (PTMs) occur on a wide range of proteins in every cellular compartment. These modifications are tightly regulated and highly dynamic, with many enzymes dedicated to their addition and removal. PTMs can reach maximal levels within minutes of extracellular stimulation (1), or persist even after cell division, enabling both fast-responding and long-lasting effects on cellular function. This dynamic regulation of PTMs in response to nutrient availability, hormone stimulation, cell differentiation, and cell cycle control serves as an important mechanism of cellular adaptation under different physiological conditions. Dysregulation of PTMs can have profound pathological effects and is related to a number of diseases including cancer, diabetes, and neurodegeneration (2–7).
Covalent PTMs take a variety of forms, including acetylation of lysine or N-termini, methylation of lysine and arginine; phosphorylation of serine threonine and tyrosine; fatty acylation, ADP-ribosylation, and many more recently discovered (8–10). As a general regulatory mechanism, these modifications can influence signal transduction, protein localization, enzyme activity, protein recognition and complex formation, or protein degradation.
Despite the variety in chemical nature, location, and time scale, PTMs share a common feature: the addition or removal is a function of the availability of central metabolites as co-substrates. For instance, the main phosphoryl donor for protein phosphorylation, ATP, is also the main energy currency in the cell. Acetyl-coA exists at the intersection of catabolic and anabolic metabolism and is the primary acetyl donor for protein acetylation. Direct participation of central metabolites in PTM enables cells to integrate information from metabolism into complex cellular decisions that ensures proper regulation of biological processes.
In this review, we will focus on histone PTMs and their regulation by changes in central metabolism. Histone PTMs impact cellular function by altering physical interactions with both DNA and other effector proteins, ultimately affecting not only replication and transcription, but also the ability of numerous chromatin-associated complexes to ‘read’, ‘erase’, and ‘write’ these modifications. The fundamental unit of eukaryotic chromatin is the nucleosome, which consists of ~147bp of superhelical DNA wrapped around an octamer of core histone proteins consisting of two copies each of H2A, H2B, H3 and H4. Ultimately, chromatin forms higher order structures that compact DNA by a factor of 30–40, restricting DNA access by other effector molecules (11).
Histones are small, highly basic, globular proteins with flexible N-terminal tails that protrude from the nucleosome core particle and are subject to a myriad of PTMs (reviewed in (12)). Histone PTMs, along with DNA methylation and differential deposition of histone core protein variants, comprise the so-called “histone code.” Acetylation of histones neutralizes positively charged lysine residues, which are highly abundant in histone proteins, ‘opening up’ chromatin and making DNA more accessible to other protein factors. Addition of methyl groups to histone lysine or arginine residues, however, does not neutralize their charge and is considered too small to induce large chromatin structural changes. This highlights a second role of histone PTMs as a signal integration platform. Modified histones, whose status is partly a function of cell metabolic state, serve as docking platforms for a myriad of chromatin associated proteins that possess exquisite specificity for histone modifications, including, but not limited to, acetylation and methylation.
This review will cover our current understanding of how small-molecule metabolites regulate histone PTM states and how these mechanisms ultimately influence cell function. Although many histone-modifying enzymes have non-histone substrates, we will focus our review primarily on transcriptional control via histone PTMs. Nevertheless, the observations and concepts that connect metabolism and histone PTM could provide universal principles that govern major cellular processes.
Reversible histone acetylation
Dual role of acetyl-coA as a central metabolite and a co-substrate in histone acetylation
Histone acetylation is catalyzed by various histone acetyltransferases (HATs), which transfer the acetyl group from acetyl-coA to the ε-amino group of lysine residues, producing coenzyme A (coA) as product. These enzymes often work in large multi-protein complexes that regulate specific chromatin targeting. Three main families of HATs, Gcn5, MYST, and p300/CBP, share a conserved acetyl-coA binding site (13). Comprehensive structural and kinetic studies have revealed catalytic and kinetic mechanisms, of these HATs, which are summarized in Table 1.
Table 1.
Mechanism and kinetic properties of histone lysine acetyltransferase and methyltransferase.
| Enzyme | Substrate | Inhibitor/Product | ||||
|---|---|---|---|---|---|---|
| Catalytic mechanism | Group* | Members* (species) | Km (Kd) [μM] | Kinetic Mechanism | Ki [μM]** | Mechanism** |
| Acetyltransferase | Acetyl-coA | coenzyme A | ||||
| N-ε-lysine activated by deprotonation (by Glu in Gcn5 and MYST); unprotonated lysine directly attacks the carbonyl of acetyl-CoA | Gcn5 | Gcn5 (yeast) | 2.5 (8.5) (14) | ordered bi-bi, acetyl-coA binds first | 6.7 (15) | competitive |
| Gcn5 (human) | 0.62 (0.56) (14) | |||||
| P/CAF (human) | 0.98 (0.64) (16), 0.64 (17) | 0.57, 0.44 (16) | competitive | |||
| MYST | Esa1 (yeast) | 0.9 (18) | ordered bi-bi, acetyl-coA binds first | 1.8 (18) | competitive | |
| p300/CBP | p300 (human) | 6.7 (19) | Theorell–Chance mechanism, acetyl-coA binds first | 21 (19) | competitive | |
|
| ||||||
| Methyltransferase | SAM | SAH | ||||
| Catalytic residue (Tyr in SET9, ? in Dot1) deprotonates histone lysine amino group, which attacks the methyl group of bound SAM | SET containing | SET9 (human) | 6.0 (20) | random bi-bi | ||
| SUV39H1 (human) | 12 (21) | 12 (21) | competitive | |||
| G9a (mouse) | 1.8, 2.7 (22) | 2.3 (22) | competitive | |||
| non-SET containing | DOT1L (human) | 0.67 (23) | 0.26 (23) | competitive | ||
Listed enzymes are examples; not all related enzymes shown here
Inhibition mechanisms and Ki values are for small metabolite co-substrate
Acetyl-coA occupies a hub of central carbon metabolism in eukaryotic cells, connecting catabolism, anabolism, and energy generation. In mitochondria, acetyl-coA is a product of catabolism either from glycolysis-generated pyruvate via oxidative decarboxylation by the pyruvate dehydrogenase complex (PDC), or from fatty acid β-oxidation or amino acid degradation. Acetyl-coA can also be synthesized from acetate by mitochondrial acetyl-coA synthetase (AceCS2). However, there is no known transporter to directly transport mitochondrial-derived acetyl-coA to the cytoplasm. To shuttle mitochondrial acetyl-coA to the cytoplasm, it is first converted to citrate by condensing with oxaloacetate via citrate synthase, which is then transported out of mitochondria, and subsequently converted back to oxaloacetate and acetyl-coA by ATP-citrate lyse (ACL). In the cytoplasm, acetyl-coA can also be produced from acetate by cytosolic acetyl-coA synthetase (AceCS1). The relative contribution of these pathways to acetyl-coA production depends on both cell type and environment. For example, in mammalian cells grown in high glucose tissue culture conditions, the majority of acetyl-coA used for histone acetylation is generated from glucose, transported into mitochondria as pyruvate, and shuttled as citrate into the cytosol where it is converted to acetyl-CoA by ACL (24). In contrast, yeast grown on acetate mainly produce acetyl-coA from acetate (25). Once in the cytosol, acetyl-coA can diffuse through nuclear pores into the nucleus, and is then available for histone acetylation.
The dual role of acetyl-coA as the acetyl-donor of histone acetylation and as a central metabolite suggests that HAT activity may connect metabolism to transcriptional regulation by sensing acetyl-coA level. To respond to cellular changes in acetyl-coA, the Km values of HATs should be near or greater than the levels of acetyl-coA. Indeed, some HATs appear to have the kinetic properties to respond to acetyl-coA fluctuations. In yeast, an evolutionarily conserved threonine/serine residue in Gcn5, which forms a hydrogen bond with the phosphate of coA, is replaced by alanine, making the Kd for acetyl-CoA of yeast Gcn5 more than 10-times higher (8.5 μM) than the human enzyme (14) and closer to acetyl-coA concentrations in yeast. Although the mechanism is unclear, cells expressing the mutant Gcn5 with higher affinity to acetyl-coA display a growth defect at 37°C on acetate as carbon source but not on glucose (26). One possibility is that binding affinity of Gcn5 is tuned to sense intracellular acetyl-coA concentration within a particular range, and outside this range, the mutant Gcn5 prevents a change in activity in adapting to nutrient conditions. In cultured human cells, the reported cellular concentration of acetyl-coA is 2–20μM (27) which is above the Km of human Gcn5 and P/CAF (14,16,17,28), but near the Km of p300 (19) (Table 1). Since Gcn5, P/CAF, and p300 display different site specificities and have distinct roles in transcriptional regulation (29), it is possible that these differences in Km may facilitate lysine acetylation at specific residues based on acetyl-coA availability. Additionally, coenzyme A is a competitive inhibitor of many HATs, whose Ki values are similar to their Km of acetyl-coA (Table 1) (18, 30). Given that the acetyl-coA/coA ratio ranges 0.1–2, the HATs that are not sensitive to acetyl-coA levels alone might be responsive to changes in acetyl-coA/coA ratio (27). Although more investigation is needed to fully determine how metabolically dynamic acetyl-coA/coA levels regulate epigenetic mechanisms of HAT function, there are compelling results to suggest the existence of these regulatory links.
Cells sense metabolic status and regulate histone acetylation though acetyl-coA
The rate of cellular acetyl-coA production is determined by many external and internal factors, the output of which can affect acetyl-coA-dependent histone acetylation. External factors include stimulation by hormones, such as insulin, and environmental conditions, such as oxygen tension and nutrient availability, particularly of carbon sources. In cultured mammalian cells, increasing glucose and acetate in media has been shown to increase intracellular acetyl-coA and histone acetylation, and subsequently affect gene expression (27). Internal factors influencing acetyl-coA levels include genetic perturbations that influence the activity of acetyl-coA-related metabolic pathways (27, 31). For example, oncogenes Myc, KRas and Akt, which enhance acetyl-coA production, were found to enhance histone acetylation in an acetyl-coA dependent manner(27, 31), and quiescent cells, which produce acetyl-coA at much lower rates than proliferating cells (32), exhibit lower rates of glucose-derived acetyl-coA deposition onto histones (33).
The ability of histone acetylation to respond to acetyl-coA levels may be particularly important in cellular assessment of metabolic states (34). Acetyl-coA, which fuels the TCA cycle and serves as an important precursor for biomass production, can serve as a “metabolic indicator”, reflecting the cell’s potential to generate energy and supply carbon. Regulation of histone acetylation under metabolic perturbation can provide a mechanism for cells to make appropriate decisions for cell fate or initiate metabolic adaption (35). When acetyl-coA is depleted by ACL silencing or glucose withdrawal, the differentiation of pre-adipocytes is blocked due to abolished histone acetylation (24). This regulation may represent an acetyl-coA-mediated “metabolic checkpoint” that limits adipocyte differentiation when acetyl-coA, the main substrate for fatty acid biosynthesis, is limiting. In yeast, acetyl-coA-dependent histone acetylation has been shown to be a likely nutrient-sensing mechanism. As yeast progress into stationary phase in response to nutrient depletion, overall histone acetylation declines. Glucose re-feeding induces overall acetylation of H3 and H4, and this induction is dependent on acetyl-coA production (36). Yeast grown under continuous glucose-limiting conditions in a chemostat spontaneously go through robust oscillations in metabolism and gene expression. Here, bursts of acetyl-coA production are concomitant with entry into growth and induction of Gcn5-mediated histone acetylation. Consistent with the idea that acetyl-coA-induced histone acetylation is required for growth, addition of acetate promotes cell entry into a growth phase (26). While both studies demonstrated that cells can sense nutrient availability via acetyl-coA, it is unclear whether the concomitant response of histone acetylation is loci-specific or global. Friis et al. reported that HAT complex members important for specific substrate targeting were not required for overall glucose-induced acetylation via western blot (36). However, using chromatin immunoprecipitation, Cai et al. reported increased histone acetylation in response to increased acetyl-coA at genetic loci important for growth (26). The seemingly different implications suggested that acetyl-coA-induced histone acetylation is likely achieved through both targeted and untargeted HAT complexes. Further, when acetyl-coA levels are high, it is known that HAT complexes undergo autoacetylation, which may affect their specificity even if complex composition remains the same (26, 36).
Histone deacetylases
The acetylation status of histones is regulated by a balance between the activities of HATs and HDACs (histone deacetylases). Mammalian HDACs are divided into 4 classes based on their homology to yeast orthologs and their cofactor dependence: Class I, IIa, IIb, III, and IV (37, 38). Classes I, II, and IV are zinc-dependent deacetylases and are generally inhibited by the HDAC inhibitors TSA and SAHA, both of which act by chelating Zn2+ at the enzyme active site. Known as sirtuins, class III HDACs are structurally distinct from Classes I, II, and IV, and share homology with yeast Sir2 (Silent information regulator 2).
Mammalian sirtuins, SIRT1-7, can deacetylate lysine residues on targeted proteins, utilizing a mechanism that requires NAD+ as a co-substrate, releasing nicotinamide (NAM), O-acetyl ADP ribose, and deacetylated protein (39). The discovery that yeast Sir2 histone deacetylase activity is regulated by NAD+ was one of the first reports linking small-molecule metabolites and gene regulation at the level of chromatin (40). Although all seven mammalian sirtuins possess highly conserved NAD+-binding and catalytic domains, they have divergent biological roles due to differences in subcellular location, tissue expression, and substrate specificity (39, 41). Sirtuins are involved in a variety of homeostatic processes, both at the organismal level and at the cellular level, playing roles in aging, calorie restriction, thermogenesis, malignancy, and in cellular stress responses, metabolic regulation, transcriptional regulation, and genome maintenance (42). There is mounting evidence that NAD+ levels affect sirtuin function. Here we will discuss the processes that generate and consume NAD+, and describe how these pathways regulate sirtuin function on chromatin.
NAD+ production affects sirtuin-dependent deacetylation
Although NAD+ can be synthesized de novo from tryptophan via the kynurenine pathway in the cytosol (reference (43) provides an excellent review of NAD+ metabolism), the major source of cellular NAD+ in mammalian cells is thought to be the canonical Preiss-Handler pathway, which salvages NAD+ from dietary niacin in all tissues (44, 45). Niacin (vitamin B3) is comprised of nicotinic acid (NA), NAM, and NAM riboside (NR), of which NAM is the major NAD+ precursor and also a general sirtuin inhibitor (46–48). In mammals, NAM is converted to NAM mononucleotide (NMN) by NAM phosphoribosyltransferase (NAMPT). This rate-limiting step in NAD+ salvage is dysregulated in several cancers, and cannot maintain nucleocytosolic NAD+ levels during genotoxic stress (49–51). NAMPT expression has also been shown to play a role in cellular senescence in smooth muscle and neural stem cells through mechanisms mediated by Sirtuins 1 and 2 (52, 53). Lastly, NAD+ salvage and de novo synthesis pathways converge, via the enzyme NMNAT (nicotinamide mononucleotide adenylyltransferase), which converts either NMN or NAMN to NAD+ in an ATP-dependent process (54).
NAD+ is an important redox cofactor required by many enzymes involved in catabolic or oxidative pathways including glycolysis, the TCA cycle, and β-oxidation of fatty acids. Thus NAD+ availability for nuclear sirtuins can become limited when these processes are perturbed. During multiple oxidative reactions of glycolysis and the TCA cycle, NAD+ serves as an electron acceptor, forming NADH. This NADH is then oxidized to NAD+ during oxidative phosphorylation, as it donates electrons to Complex I of the electron transport chain, ultimately generating a proton gradient across the inner mitochondrial membrane to drive ATP synthesis. Intracellular NAD+ levels are balanced between two main factors: structural NAD+ production (via NAD+ biosynthesis and salvage pathways) and consumption (e.g. by sirtuins and PARP), and redox conversion between NAD+ and NADH. In most cells under physiological conditions, the redox turnover flux between NAD+ and NADH greatly exceeds the structural turnover rate and is the major determinant of overall NAD+ levels. Thus, NAD+ is an important indicator of cellular redox state, and an optimal NAD+:NADH ratio is needed for homeostasis. The mitochondrial membrane is impermeable to NAD+, and although an NAD+-transporter has not been identified in mammals, a transporter has been identified in both yeast and Arabidopsis (55). Thus, mitochondrial and cytoplasmic NAD(H) stores are likely separate, regulated pools. Similarly, although nucleocytoplasmic NAD+ was initially thought to be a single, interchangeable pool, the fact that NMNAT1 (the nuclear NMNAT isoform) is the major form in mammals suggests separate regulation of nuclear and cytoplasmic NAD+ pools might exist (56).
During energy-restricted metabolic states, including calorie restriction (CR), starvation, and intermittent fasting, the NAD+:NADH ratio favors the oxidized form. To this affect, sirtuins (in particular Sirt1, 3, and 6) have been implicated in increased life- and health-span in nematodes, flies, yeast, and mammals (57). Notably, NAD+ salvage, and thus NAD+ availability, has been demonstrated as crucial for sirtuin-mediated effects on lifespan in human smooth muscle cells (Sirt1), yeast (Sir2), and C. elegans (Sirt1) (53, 58, 59).
Although expression of Sirt1 transcript and protein is not cyclic, the HDAC activity of Sirt1 has been demonstrated to follow a circadian rhythm in an enzymatic feedback loop (60). Within this loop, NAD+ levels oscillate in a circadian manner, which, in turn, activate Sirt1 in an oscillatory fashion. Sirt1 associates with CLOCK, recruiting Sirt1 to a chromatin complex containing BMAL1, which is then deacetylated at Lys537 in a cyclic manner by Sirt1 (61). BMAL1 deacetylation leads to recruitment of CRY1 to the complex and subsequent transcriptional repression of target genes (62). Such repression occurs at the NAMPT promoter, which contains two E-box elements known to bind CLOCK:BMAL1, thus completing the feedback loop by altering NAD+ synthesis via regulation of a key enzyme in the salvage pathway (61).
Assessment of NAD+ subcellular compartmentalization has been limited by both technical challenges and the fact that the majority of intracellular NAD+ is protein-bound, making it difficult to accurately measure or estimate free NAD+ levels. Additionally, there are likely tissue- and cell-type specific differences in total and compartment-specific levels. Nuclear NADH has been measured using 2-photon microscopy to be ~100 nM, from which NAD+ was estimated to be ~70 μM (63, 64). Mitochondrial NAD+ was measured to be 245.6 μM using mass spectrometry (51). While cytosolic NAD+ levels are unknown, whole cell NAD+ pools have been measured in HEK293 cells (365 ± 30.2 μM) and in mouse erythrocytes (368 μM) via mass spectrometry (51). Interestingly, the Km of yeast Sir2 for NAD+ is ~100μM (40). Similarly the mammalian nuclear sirtuin Sirt1 has a Km of ~150–170 μM (65). Unlike other sirtuins, Sirt6 can efficiently bind NAD+ (Kd of 27 μM) in the absence of acetylated substrate, suggesting Sirt6 may exist in a poised state with NAD+ already bound (66). With Km values ranging from ~100–300 μM (43), mitochondrial sirtuins like SIRT3 may exhibit smaller changes in activity in response to changes in mitochondrial NAD+, compared to non-mitochondrial sirtuins. However, circadian oscillations in NAD+ have been implicated in regulating SIRT3 activity, generating rhythmicity in acetylation and activity of mitochondrial oxidative metabolism enzymes across the circadian cycle (67). On the other hand, changes in nuclear NAD+ might be expected to yield more dramatic changes in SIRT1 deacetylation activity.
Since NAD+ homeostasis is important for a multitude of pathways, it is worth noting that effects of altered NAD+:NADH ratio are likely not limited to sirtuin catalysis. In light of this myriad of affected pathways, it is possible that some of the links between metabolic perturbations and chromatin modifications may be indirect, secondary affects of the initial metabolic aberration. Additionally, since chromatin modifying enzymes often function as members of large protein complexes, whose membership dictates not only recruitment to specific loci but also recruitment of other modifying factors that ultimately affect other enzyme activity, it is likely that the effects of altered metabolism on histone modifications is quite complex and will require continued investigation to identify key players in the response to such changes.
Other major NAD+ consumers include PARPs and CD38 (43). Particularly during DNA damage responses, PARP1 is a significant consumer of nuclear NAD+ and a potential competitor with nuclear sirtuins for the metabolite. Similarly, CD38, which is membrane-associated and uses NAD+ as a substrate to generate the signaling molecule cyclic ADP-ribose (cADPr), affects NAD+ levels through high amounts of NAD+ consumption (43). CD38 knockout mice have increased Sirt1 activity due to increased nuclear NAD+ and are also resistant to high fat diet-induced obesity via a Sirt1-PGC1α-mediated mechanism (68, 69).
Other Metabolite Effectors and Regulation of Deacetylation
In addition to substrate level regulation, small-molecule metabolites have been reported to regulate HDAC activity by either directly acting as an activator/inhibitor or influencing the PTM state of the HDAC itself. For example, oleic acid induces the cAMP/PKA signaling cascade, which leads to Ser-434 phosphorylation of SIRT1 and increased deacetylase activity (70). SIRT6 is grouped among the sirtuins that display low basal deacetylase activity in vitro. At physiological concentrations, several biologically relevant free long-chain fatty acids bind to and increase the catalytic efficiency of SIRT6 towards an H3K9Ac substrate in vitro (71). Omega-3 fatty acids decrease the expression of several glycolytic and lipogenic genes and reduce H3 and H4 acetylation at specific genomic loci (72, 73). The low basal activity of SIRT6 might be activated in response to elevated levels of certain fatty acids obtained through diet or fasting, leading to decreased acetylation and gene repression (Figure 1).
Figure 1. Proposed model for fatty acid stimulation of SIRT6 activity.
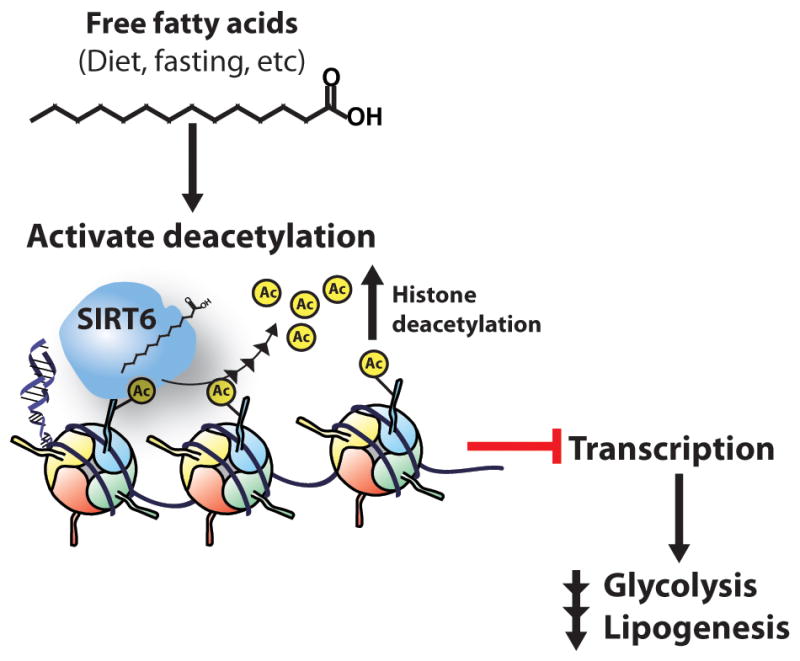
SIRT6-dependent deacetylation is stimulated by free long-chain fatty acids obtained through diet or from fasting, which leads to increased transcriptional repression and downregulation of glycolytic and lipogenic genes.
While class I and IIa HDACs are NAD+-independent, they are inhibited by the endogenous metabolite and ketone body, β-hydroxybutyrate (βOHB). In cultured cells and in vivo mouse studies, administration of βOHB induces histone acetylation to a similar extent as fasting and CR, both of which increase ketogenesis (74). βOHB increased histone acetylation at the promoters of two oxidative stress resistance factors, FOXO3a and MT2, whose expression correlated positively with treatment. Similarly, knockdown of HDAC1 or HDAC2 also induced expression of these genes, suggesting a direct role for βOHB as an HDAC inhibitor. Typical human serum βOHB levels are in the low micromolar range, but fasting, strenuous exercise, or a ketogenic diet can increase levels to 1–2 mM (fasting and strenuous exercise) or over 2 mM, respectively (75). Since βOHB inhibits members of Class I and IIa HDACs with a median IC50 of 2–5mM, it is likely that physiologic fluctuations in this metabolite play a significant regulatory role on histone deacetylation (74). The consequences of βOHB on HDAC inhibition are reviewed in further detail in reference (75).
Butyrate emerged as a potential oncometabolite in the mid-1970s, at which point it was shown to induce cell-cycle arrest and play a role in gene regulation and cell differentiation, but the mechanism was not understood (76). Then in 1977, sodium butyrate (n-butyrate) was shown to increase histone acetylation on H4 in both HeLa and Friend cells (77). Since then, butyrate has been shown to inhibit most HDACs except Class III members and HDACs 6 and 10. Although the detailed mechanism of butyrate inhibition is unknown, it is a noncompetitive inhibitor of HDACs with a Ki of~60 μM and an IC50 of~10–100 μM (78–80). Recently, interest has shifted toward butyrate as a product of gut bacterial fermentation of plant polysaccharides (fiber) and its role as an epigenetic regulator in colon cancer. Short-chain fatty acids, including acetate, propionate, and butyrate, are produced by bacteria in the lumen of the colon and are present at very high (mM) concentrations, and butyrate levels within dissected colon tumors have been estimated to be >100 μM (81, 82). Butyrate is the primary energy source for normal colonocytes, whereas acetate and propionate are primarily transported to muscle and liver, respectively (83). Normal colonocytes oxidize butyrate to acetyl-coA, which is then used as a substrate for the TCA cycle and HATs, resulting in upregulation of proliferative genes. However in cancerous colonocytes the Warburg effect shifts metabolism toward glycolysis and away from oxidation of acetyl-coA via the TCA cycle. Thus butyrate accumulates and functions as a HDAC inhibitor, increasing histone acetylation at pro-apoptotic genes (84). In a mouse model of colon cancer, butyrate has been shown to increase histone acetylation and drive apoptosis, reducing both the tumor burden and tumor grade (82). Although there is evidence suggesting acetate has no affect on HDAC activity, propionate and polyphenol metabolites, though much less potent than butyrate, have been found to inhibit HDAC activity (80). Thus, it remains to be determined whether any other bacterially-derived metabolites will affect host histone PTMs, and in which tissues, via HDAC inhibition or other mechanisms.
Reversible histone methylation
SAM and SAH levels regulate histone methylation
Methylation on the ε-amino group of lysine residues is the most prevalent form of histone methylation, and can give rise to mono-, di- and tri-methylated states. This reaction is catalyzed by a large number of histone methyltransferases (HMTs). The specificity, mechanism, and kinetics of these HMTs have been intensively studied in recent years, and are summarized in Table 1. Although all known HMTs have different specificity toward histone substrates, all use S-adenosyl-methionine (SAM, also known as AdoMet) as a methyl donor and produce S-adenosyl-homocycsteine (SAH, also known as AdoHcy) as a product. SAH was found to be a competitive inhibitor of SAM and uncompetitive or mixed inhibitor of peptide substrate with Ki values generally on the same order as the Km values for SAM (Table 1).
Under normal physiological conditions, tissue SAM concentrations range from 10–100 μM, while SAH levels are generally lower, at 0.1 to 20 μM, with actual concentrations and SAM/SAH dependent on tissue type, development stage, and age (85–88). These concentrations are in the range of Km and Ki values of SAM and SAH, respectively, for HMTs. Thus, it is possible for histone methylation to respond to fluctuating levels of either SAM or SAH, with sensitivity dependent on cell type, condition, and enzyme.
SAM is originally derived from amino acids through a vitamin-dependent metabolic cycle, as shown in Figure 2. Methionine adenosyltransferase (MAT) catalyzes the SAM synthesis reaction from methionine and ATP, which is conserved across all branches of life (89). After donating the methyl-group on SAM to histones, the product SAH is hydrolyzed to homocysteine (Hcy), which can be either remethylated to methionine, made available for a new cycle of SAM production, or degraded and removed from the methylation cycle via two vitamin B6-dependent enzymes. While methionine can be synthesized de novo in some species like yeast, humans require methionine from the diet (90). However, even in humans, where methionine is an essential amino acid, methionine can be regenerated from Hcy via methionine synthase, which depends on vitamin B12, using 5-methyl-THF as methyl donor, or via betaine-homocysteine methyltransferase, using betaine as methyl donor. Both 5-methyl-THF and betaine are produced in folate-dependent one-carbon metabolism that generates active one-carbon units for cellular methylation and nucleotide synthesis, mainly from the hydroxymethyl group of serine and the α-carbon of glycine.
Figure 2. Metabolic pathways generating methyl-groups for histone methylation.

SAM is produced via an amino acid- and vitamin- dependent metabolic cycle. Active one-carbon units that ultimately methylate histones are labeled in red. Metabolite structures are shown in brackets.
The SAM cycle integrates input from many parts of cellular metabolism, where dietary and environmental factors significantly influence the levels of SAM, SAH, and their ratio. These include: 1) vitamins, particularly, folate and vitamins B6 and B12, which are required to drive the SAM cycle either by re-methylation or degradation of Hcy, 2) amino acids, particularly methionine, serine, glycine and threonine, and 3) other factors that influence the activity of methylation cycle enzymes such as dietary fat intake, alcohol consumption, and oxidative stress.
A growing number of studies provide evidence that these factors influence SAM/SAH levels and affect histone methylation. In mouse embryonic stem cells, threonine is an important precursor for glycine (distinct from human), which contributes the active methyl group on SAM. Threonine is metabolized to SAM through a threonine dehydrogenase-dependent pathway. Restriction of threonine in the culture medium or threonine dehydrogenase knockdown decreased the ratio of SAM/SAH and significantly decreased H3K4 di- and tri-methylation, causing reduced growth and increased differentiation (91). This decrease in methylation of H3K4 was rescued by the addition of glycine and pyruvate, which restored SAM/SAH (91). In yeast, Sadhu et al. measured histone methylation while manipulating cellular SAM levels by varying methionine levels in a methionine-dependent Δmet strain or by varying folate levels in a Δfol strain. In both cases, increased one-carbon metabolism led to a significant increase in H3K4me2/me3 and altered expression of target genes (92). Interestingly, both studies found the sensitivity of histone methylation to methyl group availability was site-specific: although H3K4 methylation (Set1 methyltransferase) responded to one carbon limitation, H3K79 methylation (Dot1 methyltransferase) was much more resistant to methyl-group restriction. The difference in response to this metabolic challenge could result from different kinetic properties of site-specific HMTs, and/or because some methylation sites are turned over more actively. To test whether the lower Km of Dot1, relative to Set1, was responsible for the lower susceptibility of H3K79 to methyl-group restriction, Sadhu et al. examined the response in a yeast strain expressing hypomorphic Dot1, which presumably has lower affinity to SAM, and found that in this strain H3K79 methylation was now more susceptible to folate limitation (92). Additionally, expression of different HMTs has been demonstrated to vary during development, and folate limitation during these stages leads to distinct outcomes (93, 94). Therefore, the specificities and kinetic parameters of different HMTs not only impacts the site-specific response of these enzymes to changes in one-carbon metabolism, but may also determine the sensitivity of histone methylation to environmental and nutritional perturbations throughout development and in mature organisms.
Histone demethylation is closely linked to mitochondrial oxidative metabolism
Histone lysine methylation is removed by two main classes of histone demethylases: the LSD family of FAD-dependent demethylases and the JmjC family of α-ketoglutarate-dependent demethylases. Here we will focus on their connection to metabolism, while the structure, mechanism, specificity and interaction with other proteins have been reviewed elsewhere (95, 96). The demethylation reaction is a redox reaction, and while both classes of demethylases remove the methyl group to produce formaldehyde, they catalyze the reaction through different intermediates using different electron accepters. LSD family demethylases are proposed to catalyze the reaction through an amine oxidation reaction coupled with FAD reduction to FADH2. JmjC family demethylases catalyze the reaction via a hydroxymethyl intermediate, using oxygen and α-ketoglutarate as co-substrates, producing CO2 and succinate (95).
FAD, FADH2, α-ketoglutarate, and succinate are all components of the TCA cycle. Additionally, FAD(H2) is a required cofactor for fatty acid β-oxidation and oxidative phosphorylation, and α-ketoglutarate is the main amino-acceptor for amino-acid metabolism (Figure 3). The close connection of these histone demethylation co-substrates to mitochondrial oxidation and energy metabolism provides cells an opportunity to communicate mitochondrial status to the nucleus to regulate transcription of genes involved in metabolism.
Figure 3. Metabolism is tightly connected with histone acetylation and methylation.

Addition and removal of histone acetylation and methylation are catalyzed by several classes of enzymes, whose activity requires metabolites that either serve as co-substrates or act as activators/inhibitors. The reactions are color-coded by enzymes (color code indicated in side box), with the involved metabolites as substrates and products connected by solid arrows, and metabolites as activators and inhibitors connected by dash lines. Simplified metabolic pathways are shown in blue arrows, indicating the position of key histone modifying compounds in the metabolic network.
Highlighting the correlation between histone methylation and energy metabolism, a recent study in 3T3-L1 cells showed that histone demethylation by LSD1 regulates energy-expenditure genes, inhibition of LSD1 increases cell oxidation rate (97). And inhibition of FAD synthesis increased the expression of energy-expenditure LSD1-target genes, which the authors suggested is due to loss of LSD1-mediated repression of these genes (97). Oxygen availability has also been shown to impact histone methylation by regulating demethylase activity. Hypoxia has been reported to increase H3K4me3 in mammalian cells, which is likely due to decreased activity of the oxygen-dependent demethylase JARID1A (KDM5A). This idea is supported by the observation that the effect was dependent on JARID1A but not methionine, and in vitro assays confirmed a lower activity of JARID1A during hypoxia (98). Several other studies also noted that hypoxia generally increases histone methylation (99–101). However, the mechanism by which histone PTM senses oxygen levels likely involves many additional layers, including the effects of hypoxia on oxidative metabolism, and thus the concentration of FAD/FADH2, succinate and α-ketoglutarate. In addition, hypoxia can regulate the levels of histone modifying enzymes or their associated proteins. For example, an important oxygen sensing protein, hypoxia-inducible factor (HIF), is a transcription factor targeting some histone demethylases and is associated with p300 as a coactivator (102, 103). Interestingly, HIF is regulated by a hydroxylation reaction that shares a similar Fe(II)-dependent mechanism with JmjC-dependent demethylation, requiring oxygen and α-ketoglutarate as substrate and producing succinate (88, 104). Thus, changes in histone PTMs in response to oxygen availability are likely multifactorial, and there is some evidence that these oxygen-regulated histone PTMs are both site- and cell type- specific (99, 100). To tease out the direct effect of oxygen on demethylases, in vitro study on the kinetics of different demethylases under various oxygen levels would provide useful insight.
Mutations of metabolic enzymes disrupt histone demethylation in cancer
Recent studies have highlighted the importance of other metabolites that regulate histone demethylase activity, particularly in the context of cancer. Human and yeast α-ketoglutarate-dependent histone demethylases are reported to be inhibited by the product succinate, presumably due to competitive inhibition with α-ketoglutarate. The downstream product and structural analog of succinate, fumarate, is also an inhibitor of Jmjc-family demethylases (86, 88). TCA cycle enzymes that metabolize these compounds, fumarate dehydrogenase (FH) and succinate dehydrogenase (SDH), are frequently mutated in a subset of human cancers. These mutations result in decreased or complete loss of enzymatic activity, causing robust accumulation of succinate or fumurate (86). This accumulation leads to significant hypermethylation of both histones and DNA, and greatly alters the expression of the genes associated with hypermethylation (85, 86, 105).
Another example of the effects of dysfunctional metabolism on epigenetic mechanisms involves 2-hydroxyglutarate (2HG), which has received significant attention in recent years as the first oncometabolite identified. 2HG is significantly elevated in some cancers, including glioma and melanoma, and is mainly produced as an aberrant product by mutated isocitrate dehydrogenase (106, 107). Structural and kinetic studies of Jmjc-family demethylases identified 2HG as a competitive inhibitor of α-ketoglutarate (104, 108). High levels of 2HG were demonstrated to cause histone hypermethylation, blocking differentiation and promoting tumorigenesis (108–110).
The potential of metabolic side-products to influence epigenetic programs and cause disease is very likely not unique to 2HG or cancer. It is likely that similar mechanisms can occur in other cases, particularly in response to environmental toxicants, since metabolism of xenobiotics results in a variety of compounds from which cells are not evolutionarily protected. It is also tempting to speculate that molecular aging may be due to accumulation of metabolic side-products that impact the epigenome.
Other histone PTMs
It is important to note that histones can be modified by other chemical moieties derived from metabolites. Histone glycosylation via O-linked N-acetylglucosamine (O-GlcNAc) of serine and threonine has been observed in C. elegans, D. melanogaster, mouse, and human. This PTM reflects changes in glucose metabolism, and has been linked to cancer (reviewed in (35, 111, 112)). NAD+ consuming PARPs have been shown to ADP-ribosylate all four core histone proteins in vitro, although less than 1% of histone proteins are actually ADP-ribosylated and histone H1 is the main ADP-ribose acceptor (113). Other histone PTMs include ubiquitination, SUMOylation, carbonylation, 2-hydroxyisobutyrylation, crotonylation, and more (10). The molecular basis and biological significance of these newly discovered modifications is still under investigation.
PTM synergy and antagonism
Although histone tail PTMs can be regulated and interpreted independently, some are regulated through synergetic and antagonistic mechanisms. Different histone PTMs can compete with each other for modification sites and for small molecule substrates. For instance, acetylation and methylation of the same lysine site are mutually exclusive, thus the methylation of one site can act as a placeholder to prevent activation by acetylation. Interestingly, methylation and acetylation of the same site are often associated with opposite effects on gene expression. For example, H3K9 or H3K27 methylation generally results in repression, while acetylation at the same site has an activating effect. During the G0 to G1/S transition, H3K9 on the DHFR (dihydrofolate reductase) promoter changes from a methylated to an acetylated state. This observation suggests that the balance and transition between different histone modifications at a particular site is important for regulating gene expression at different phases during cell cycle (114). Competition among PTMs can occur for the histone modifying metabolites as well. As discussed above, NAD+-dependent PARP activity and sirtuin activity compete for the same nuclear NAD+ pool.
Besides direct competition, PTMs at a given site can also regulate the modification of neighboring sites. It has been observed that different histone PTMs at the same loci often show significant correlation with each other (115). This interplay influences both modification states and modification turnover rates. For example, ubiquitination of H2BK123 is reported to be a prerequisite for H3K4 methylation (116), and methylation of H4R3 is important for H3 and H4 acetylation (117). The acetylation or methylation state of H3K9 affects the acetylation turnover rate of H3K14 (33). These correlations among different histone PTMs can be the result of co-regulation by a common mechanism, chromatin structure and accessibility, or the interaction or recruitment of specific histone modifying enzymes. As demonstrated in a growing number of studies, modification at one site can greatly affect binding and activity of histone modifying complexes that act on other sites.
For example, phosphorylation of H3T6 prevents LSD1 from demethylating H3K4 (118), while phosphorylation of H3S10 increases the affinity to the acetyltransferase Gcn5, promoting H3K14 acetylation (119). Similarly, H3T11 phosphorylation may promote acetylation in a Gcn5-dependent manner (120). The interplay between PTMs not only occurs among different histone sites, but also between DNA and histones (121). At sites of DNA methylation, methyl-CpG-binding proteins play an important role in recruiting histone methyltranferase or histone deacetylase complexes that promote further histone modification (122, 123). Together, these PTMs on various sites constitute a “histone code” that collectively regulates cellular function (reviewed and discussed in (124, 125)). This interplay may be an important mechanism for integrating regulatory information from multiple pathways, regulating gene expression according to current epigenetic state, or sustaining either long-term activation or silencing.
The enzymes and other members of histone modifying complexes are often regulated by PTM as well (126). One well-studied example involves members of the histone acetyltransferase complex. Acetyltransferase P/CAF can undergo autoacetylation and is also acetylated by p300. This acetylation induces its translocation to the nucleus and enhances its activity (127). Histone acetylation activity of CBP/p300 is also activated by acetylation of key residues (128, 129). Other members of histone acetyltransferase complex, like SPT7, SGF73 and ADA3 of SAGA, are dynamically acetylated as well (26). Additionally, HATs p300 and CBP can be phosphorylated. Similar to acetylation, phosphorylation of these HATs regulates histone acetylation by modifying enzyme activity, complex assembly and recruitment to histones (130, 131). Histone methylation activity is also controlled by PTM state of relevant enzyme complexes. SUV39H1, a H3K9 methyltransferase, can be acetylated on its SET domain, inhibiting its activity. SIRT1 can affect H3K9 trimethylation levels through interaction with and deacetylation of SUV39H1 (132). Beyond regulatory PTMs on the protein itself, the levels of histone modifying enzymes can be regulated by PTMs. For instance, EZH2 mediated H3K27 trimethylation can lead to elevated protein levels of MMSET, which dimethylates H3K36, by repression of MMSET-regulating miRNAs (133).
Similar to histone modifying enzymes, the metabolic enzymes catalyzing the production of key metabolites can also be regulated by PTMs. An important acetyl-coA producing enzyme, acetyl-coA synthetase, is controlled by reversible acetylation. Different sirtuins are able to deacetylate cytosolic or mitochondrial acetyl-coA synthetases to promote their activity (134, 135). S-adenosylhomocysteine hydrolase (SAHH) can also be acetylated in vivo. Acetylation leads to inhibition of activity, which may result in build up of intracellular SAH, inhibiting methylation activity (136). Similarly, it has been long recognized that another main acetyl-coA producing enzyme, pyruvate dehydrogenase, is tightly regulated by its phosphorylation (137). These examples demonstrate a common mechanism wherein the activities of enzymes responsible for histone modification are also regulated by post-translational modification. As a result of this crosstalk among different PTMs, fluctuation in the levels of one key metabolite may not only directly affect the PTM-state of histones, but also the state of histone modifying complexes or the production of other key metabolites.
Perspectives
Does protein modification affect the availability of key metabolites?
While metabolism has been shown to impact the epigenome through direct participation of key metabolites in histone modification, the question has recently been raised whether this connection might also operate in reverse, i.e. whether turnover of PTMs may influence the availability of relevant metabolites for metabolic processes. In support of this hypothesis, excessive PARP1 activation during genotoxic stress has been demonstrated to cause NAD+-depletion (138), and in some cases, bioenergetic collapse and cell death, which was thought to be solely due to NAD+-depletion, however, recent studies also suggest a potential role for PAR-mediated inhibition of hexokinase (139, 140).
Considering that the majority of biomass is protein, with small metabolites accounting for a minor fraction, it is likely that protein PTMs might influence the material supply chain for metabolism. Martinez-Pastor, et al. explored this idea in-depth and compared the pool size of acetyl or methyl groups versus the acetyl-coA or active one-carbon metabolite levels. For acetylation, the authors estimated ≥ 3.4×106 potential acetylation sites on yeast histones, which can store 6.5–65-fold more acetyl-groups than free acetyl-coA (141). Similarly, considering lysine frequency and average acetylation occupancy, in mammalian cells, acetyl groups in protein-bound form can be estimated to be ~ 50 pmole/μg protein. At this level, the acetate from protein acetylation is ~ 100-fold higher than free acetyl-coA, and could serve as a buffer for the two carbon unit. This comparison points to the suggestion that during transient disruption of carbon metabolism, cells could potentially restore acetyl-coA levels by utilizing the acetyl-group from modified protein. To explore this possibility, it is important to consider not only how the two pool sizes compare, but also how the turnover rates of acetyl-coA for metabolism versus protein modification differ. Using isotopic tracing approaches, the acetyl-coA production rate from glucose was 0.2–4 nmole/h/μg protein in several cultured cell lines (32, 142, 143). The acetyl-coA consumption rate for protein modification depends on the number of acetylation sites, their stoichiometry, and turnover rate. Depending on the site, a histone acetylation half-life of 50–90 min was measured by proteomic analysis (33). Assuming an average half-life of 1h, the acetylation turnover is 0.035 nmole/h/μg protein, which is ~ 10-fold slower than the rate of metabolic acetyl-coA production. Therefore, under normal tissue culture conditions, histone acetylation itself does not appear to cost a significant fraction of the acetyl-coA supply. Conversely, “acetyl-storage” on protein is not sufficient to sustain the normal level of cellular metabolic activity when other substrates are not available. However, under metabolic stress, such a reservoir might be important for immediate survival, which would require acetyl-coA at a much lower rate than the metabolic flux under normal nutrient conditions. It is also worth noting that this analysis utilized rates from highly proliferative cells, where a majority of acetyl-coA is rapidly consumed for biomass. This demand for net synthesis is not present in most non-proliferating cells, suggesting the possibility that histone acetylation may, in fact, contribute a larger fraction of acetyl-coA consumption.
To understand the net consumption of acetyl groups for histone/protein acetylation, it is essential to understand the fate of acetyl groups resulting from deacetylation. Whether this acetate is efficiently returned to metabolism or disposed through other pathways will greatly influence the overall cellular demands for acetyl-groups. Class I and II HDACs produce acetate as the deacetylation product. The metabolic utilization of acetate is dependent on the activity of AceCS. In contrast, sirtuins remove protein acetyl-groups by cleaving NAD+ and transferring the acetyl-group to the ADP-ribose of NAD+, yielding O-acetyl-ADP-ribose (OAADPr). Although some progress has been made (144), the exact metabolic fate of OAADPr is not yet clear. It is tempting to speculate that the acetyl-group on OAADPr returns to metabolism as an activated acetyl-donor, similar to acetyl-coA.
Histone acetylation and deacetylation reactions may also participate in other metabolic processes. Recently, McBrian et al. reported that under low pH, global histone acetylation decreases as a result of deacetylation, while acetyl-coA levels remain unchanged. The acetate produced by HDAC reactions was co-secreted from cells along with protons, thereby preventing a decrease in intracellular pH. This suggests a role for reversible histone acetylation as a rheostat to regulate intracellular pH (145).
Localized metabolite production regulates histone PTMs
Dynamic histone PTM occurs in the nucleus and therefore requires access to local concentrations of metabolites. Since eukaryotic metabolism is compartmentalized, the trafficking and distribution of metabolites is particularly important here. Microscopic measurements have provided valuable information on organelle-specific concentration of some key metabolites, such as NAD(H). Additionally, recently developed intracellular metabolite sensors hold special promise in detecting localized metabolite level change. However, accurate measurement of nuclear concentration of most histone modifying metabolites, including SAM, SAH, and acetyl-coA, remains challenging.
Interestingly, many enzymes involved in the metabolism of these compounds have a nuclear fraction. For example, acetyl-coA producing enzymes ACL and AceCS1 have been shown to locate to both the cytoplasm and nucleus (24), and the predominant form of NMNAT in mammals is NMNAT1, the nuclear isoform (56). The presence of these enzymes in the nucleus suggests that local generation of their products can provide an efficient means to specifically regulate these nuclear processes.
Additional evidence for such ‘moonlighting’ by metabolic enzymes has recently emerged. Sutendra et al. found that pyruvate dehydrogenase complex (PDC), the main acetyl-coA producing enzyme complex, is translocated from mitochondria into the nucleus during S phase. The presence of catalytically active pyruvate dehydrogenase in the nucleus enables cells to produce acetyl-coA directly from pyruvate in the nucleus, bypassing the mitochondria-to-cytosol shuttling system. This seemingly costly effort can dynamically affect the local acetyl-coA concentration and subsequent histone acetylation. The authors showed that nuclear PDC is important for histone acetylation and S phase entry, and that its translocation can be stimulated by growth factor (146). Similarly, a recent study revealed that the SAM synthesizing enzyme, methionine adenosyltransferase (MAT), directly interacts with histone methyltransferase SETDB1. MAT is recruited to the promoter of the COX-2 gene together with associated proteins including SETDB1 to specifically repress COX-2 expression. The physical interaction between the metabolic enzyme and HMT demonstrates the coupling of local SAM synthesis with histone methylation (147). With these examples, it is appealing to suggest a general model in which co-substrates of chromatin enzymes are produced ‘on site’ to elicit a specific regulatory PTM.
Acknowledgments
The authors acknowledge the support of NIH grant GM059785-15/P250VA. Kimberly A. Krautkramer is supported by Molecular and Applied Nutrition Training Program (NIH grant 5T32DK007665-22). We apologize for the omissions of relevant publications due to space limitation.
References
- 1.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 2.Santer FR, Hoschele PP, Oh SJ, Erb HH, Bouchal J, Cavarretta IT, Parson W, Meyers DJ, Cole PA, Culig Z. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol Cancer Ther. 2011;10:1644–1655. doi: 10.1158/1535-7163.MCT-11-0182. [DOI] [PubMed] [Google Scholar]
- 3.Berdasco M, Esteller M. Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Developmental cell. 2010;19:698–711. doi: 10.1016/j.devcel.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Mellor KM, Brimble MA, Delbridge LM. Glucose as an agent of post-translational modification in diabetes - New cardiac epigenetic insights. Life Sci. 2014 doi: 10.1016/j.lfs.2014.03.020. [DOI] [PubMed] [Google Scholar]
- 5.Leroy G, Dimaggio PA, Chan EY, Zee BM, Blanco MA, Bryant B, Flaniken IZ, Liu S, Kang Y, Trojer P, Garcia BA. A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics Chromatin. 2013;6:20. doi: 10.1186/1756-8935-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pena-Altamira LE, Polazzi E, Monti B. Histone post-translational modifications in Huntington’s and Parkinson’s diseases. Curr Pharm Des. 2013;19:5085–5092. doi: 10.2174/13816128113199990355. [DOI] [PubMed] [Google Scholar]
- 7.Miao F, Chen Z, Genuth S, Paterson A, Zhang L, Wu X, Li SM, Cleary P, Riggs A, Harlan DM, Lorenzi G, Kolterman O, Sun W, Lachin JM, Natarajan R. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes. 2014;63:1748–1762. doi: 10.2337/db13-1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dai L, Peng C, Montellier E, Lu Z, Chen Y, Ishii H, Debernardi A, Buchou T, Rousseaux S, Jin F, Sabari BR, Deng Z, Allis CD, Ren B, Khochbin S, Zhao Y. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat Chem Biol. 2014;10:365–370. doi: 10.1038/nchembio.1497. [DOI] [PubMed] [Google Scholar]
- 9.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arnaudo AM, Garcia BA. Proteomic characterization of novel histone post-translational modifications. Epigenetics Chromatin. 2013;6:24. doi: 10.1186/1756-8935-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 12.Arnaudo AM, Garcia BA. Epigenetics Chromatin. BioMed Central Ltd; 2013. Proteomic characterization of novel histone post-translational modifications; p. 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annual review of biochemistry. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 14.Langer MR, Fry CJ, Peterson CL, Denu JM. Modulating acetyl-CoA binding in the GCN5 family of histone acetyltransferases. The Journal of biological chemistry. 2002;277:27337–27344. doi: 10.1074/jbc.M203251200. [DOI] [PubMed] [Google Scholar]
- 15.Tanner KG, Langer MR, Kim Y, Denu JM. Kinetic mechanism of the histone acetyltransferase GCN5 from yeast. The Journal of biological chemistry. 2000;275:22048–22055. doi: 10.1074/jbc.M002893200. [DOI] [PubMed] [Google Scholar]
- 16.Tanner KG, Langer MR, Denu JM. Kinetic mechanism of human histone acetyltransferase P/CAF. Biochemistry. 2000;39:11961–11969. doi: 10.1021/bi001272h. [DOI] [PubMed] [Google Scholar]
- 17.Lau OD, Courtney AD, Vassilev A, Marzilli LA, Cotter RJ, Nakatani Y, Cole PA. p300/CBP-associated factor histone acetyltransferase processing of a peptide substrate. Kinetic analysis of the catalytic mechanism. The Journal of biological chemistry. 2000;275:21953–21959. doi: 10.1074/jbc.M003219200. [DOI] [PubMed] [Google Scholar]
- 18.Berndsen CE, Albaugh BN, Tan S, Denu JM. Catalytic mechanism of a MYST family histone acetyltransferase. Biochemistry. 2007;46:623–629. doi: 10.1021/bi602513x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Wang L, Zhao K, Thompson PR, Hwang Y, Marmorstein R, Cole PA. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature. 2008;451:846–850. doi: 10.1038/nature06546. [DOI] [PubMed] [Google Scholar]
- 20.Trievel RC, Beach BM, Dirk LM, Houtz RL, Hurley JH. Structure and catalytic mechanism of a SET domain protein methyltransferase. Cell. 2002;111:91–103. doi: 10.1016/s0092-8674(02)01000-0. [DOI] [PubMed] [Google Scholar]
- 21.Chin HG, Patnaik D, Esteve PO, Jacobsen SE, Pradhan S. Catalytic properties and kinetic mechanism of human recombinant Lys-9 histone H3 methyltransferase SUV39H1: participation of the chromodomain in enzymatic catalysis. Biochemistry. 2006;45:3272–3284. doi: 10.1021/bi051997r. [DOI] [PubMed] [Google Scholar]
- 22.Patnaik D, Chin HG, Esteve PO, Benner J, Jacobsen SE, Pradhan S. Substrate specificity and kinetic mechanism of mammalian G9a histone H3 methyltransferase. The Journal of biological chemistry. 2004;279:53248–53258. doi: 10.1074/jbc.M409604200. [DOI] [PubMed] [Google Scholar]
- 23.Richon VM, Johnston D, Sneeringer CJ, Jin L, Majer CR, Elliston K, Jerva LF, Scott MP, Copeland RA. Chemogenetic analysis of human protein methyltransferases. Chem Biol Drug Des. 2011;78:199–210. doi: 10.1111/j.1747-0285.2011.01135.x. [DOI] [PubMed] [Google Scholar]
- 24.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Virgilio C, Burckert N, Barth G, Neuhaus JM, Boller T, Wiemken A. Cloning and disruption of a gene required for growth on acetate but not on ethanol: the acetyl-coenzyme A synthetase gene of Saccharomyces cerevisiae. Yeast. 1992;8:1043–1051. doi: 10.1002/yea.320081207. [DOI] [PubMed] [Google Scholar]
- 26.Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Molecular cell. 2011;42:426–437. doi: 10.1016/j.molcel.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S, Jackson E, Aiello NM, Haas NB, Rebbeck TR, Judkins A, Won KJ, Chodosh LA, Garcia BA, Stanger BZ, Feldman MD, Blair IA, Wellen KE. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell metabolism. 2014;20:306–319. doi: 10.1016/j.cmet.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leemhuis H, Nightingale KP, Hollfelder F. Directed evolution of a histone acetyltransferase--enhancing thermostability, whilst maintaining catalytic activity and substrate specificity. Febs J. 2008;275:5635–5647. doi: 10.1111/j.1742-4658.2008.06689.x. [DOI] [PubMed] [Google Scholar]
- 29.Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, Wang C, Brindle PK, Dent SY, Ge K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. The EMBO journal. 2011;30:249–262. doi: 10.1038/emboj.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wiktorowicz JE, Campos KL, Bonner J. Substrate and product inhibition initial rate kinetics of histone acetyltransferase. Biochemistry. 1981;20:1464–1467. doi: 10.1021/bi00509a009. [DOI] [PubMed] [Google Scholar]
- 31.Morrish F, Noonan J, Perez-Olsen C, Gafken PR, Fitzgibbon M, Kelleher J, VanGilst M, Hockenbery D. Myc-dependent mitochondrial generation of acetyl-CoA contributes to fatty acid biosynthesis and histone acetylation during cell cycle entry. The Journal of biological chemistry. 2010;285:36267–36274. doi: 10.1074/jbc.M110.141606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lemons JM, Feng XJ, Bennett BD, Legesse-Miller A, Johnson EL, Raitman I, Pollina EA, Rabitz HA, Rabinowitz JD, Coller HA. Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol. 2010;8:e1000514. doi: 10.1371/journal.pbio.1000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evertts AG, Zee BM, Dimaggio PA, Gonzales-Cope M, Coller HA, Garcia BA. Quantitative dynamics of the link between cellular metabolism and histone acetylation. The Journal of biological chemistry. 2013;288:12142–12151. doi: 10.1074/jbc.M112.428318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cai L, Tu BP. Driving the cell cycle through metabolism. Annu Rev Cell Dev Biol. 2012;28:59–87. doi: 10.1146/annurev-cellbio-092910-154010. [DOI] [PubMed] [Google Scholar]
- 35.Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nature reviews Molecular cell biology. 2012;13:270–276. doi: 10.1038/nrm3305. [DOI] [PubMed] [Google Scholar]
- 36.Friis RM, Wu BP, Reinke SN, Hockman DJ, Sykes BD, Schultz MC. A glycolytic burst drives glucose induction of global histone acetylation by picNuA4 and SAGA. Nucleic Acids Res. 2009;37:3969–3980. doi: 10.1093/nar/gkp270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mihaylova MM, Shaw RJ. Metabolic reprogramming by class I and II histone deacetylases. Trends Endocrinol Metab. 2013:48–57. doi: 10.1016/j.tem.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parra M. Class IIa HDACs: New insights into their functions in physiology and pathology. FEBS J. 2014 doi: 10.1111/febs.13061. [DOI] [PubMed] [Google Scholar]
- 39.Feldman JL, Dittenhafer-Reed KE, Denu JM. Sirtuin catalysis and regulation. J Biol Chem. 2012:42419–42427. doi: 10.1074/jbc.R112.378877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 41.Feldman JL, Baeza J, Denu JM. J Biol Chem. American Society for Biochemistry and Molecular Biology; 2013. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins; pp. 31350–31356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haigis MC, Sinclair DA. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu Rev Pathol Mech Dis. 2010:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Houtkooper RH, Cantó C, Wanders RJ, Auwerx J. The Secret Life of NAD +: An Old Metabolite Controlling New Metabolic Signaling Pathways. Endocrine Reviews. 2010:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Preiss J, Handler P. Biosynthesis of diphosphopyridine nucleotide. I. Identification of intermediates. Journal of Biological Chemistry. 1958:488–492. [PubMed] [Google Scholar]
- 45.Preiss J, Handler P. Biosynthesis of diphosphopyridine nucleotide. II. Enzymatic aspects. Journal of Biological Chemistry. 1958:493–500. [PubMed] [Google Scholar]
- 46.Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends in Biochemical Sciences. 2007:12–19. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 47.de Figueiredo LF, Gossmann TI, Ziegler M, Schuster S. Pathway analysis of NAD+ metabolism. Biochem J. 2011:341–348. doi: 10.1042/BJ20110320. [DOI] [PubMed] [Google Scholar]
- 48.Sporty J, Lin S-J, Kato M, Ognibene T, Stewart B, Turteltaub K, Bench G. Yeast. John Wiley & Sons, Ltd; 2009. Quantitation of NAD +biosynthesis from the salvage pathway in Saccharomyces cerevisiae; pp. 363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duarte-Pereira S, Silva SS, Azevedo L, Castro L, Amorim A, Silva RM. NAMPT and NAPRT1: novel polymorphisms and distribution of variants between normal tissues and tumor samples. Sci Rep. 2014:6311. doi: 10.1038/srep06311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou T, Wang T, Garcia JGN. Expression of Nicotinamide Phosphoribosyltransferase-Influenced Genes Predicts Recurrence-Free Survival in Lung and Breast Cancers. Sci Rep. 2014:6107. doi: 10.1038/srep06107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, de Cabo R, Sauve AA, Sinclair DA. Nutrient-Sensitive Mitochondrial NAD+ Levels Dictate Cell Survival. Cell. 2007:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wiley C, Campisi J. NAD+ controls neural stem cell fate in the aging brain. EMBO J. 2014 doi: 10.15252/embj.201488969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ho C, van der Veer E, Akawi O, Pickering JG. FEBS letters. Federation of European Biochemical Societies; 2009. SIRT1 markedly extends replicative lifespan if the NAD+ salvage pathway is enhanced; pp. 3081–3085. [DOI] [PubMed] [Google Scholar]
- 54.Sorci L, Cimadamore F, Scotti S, Petrelli R, Cappellacci L, Franchetti P, Orsomando G, Magni G. Initial-Rate Kinetics of Human NMN-Adenylyltransferases: Substrate and Metal Ion Specificity, Inhibition by Products and Multisubstrate Analogues, and Isozyme Contributions to NAD +Biosynthesis †. Biochemistry. 2007:4912–4922. doi: 10.1021/bi6023379. [DOI] [PubMed] [Google Scholar]
- 55.Stein LR, Imai S-i. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab. 2012:420–428. doi: 10.1016/j.tem.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. Journal of Biological Chemistry. 2005:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- 57.Giblin W, Skinner ME, Lombard DB. Trends in Genetics. Elsevier Ltd; 2014. Sirtuins: guardians of mammalian healthspan; pp. 271–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Belenky P, Racette FG, Bogan KL, McClure JM, Smith JS, Brenner C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+ Cell. 2007:473–484. doi: 10.1016/j.cell.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 59.Moroz N, Carmona JJ, Anderson E, Hart AC, Sinclair DA, Blackwell TK. Dietary restriction involves NAD +-dependent mechanisms and a shift toward oxidative metabolism. Aging Cell. 2014 doi: 10.1111/acel.12273. pp n/a-n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, Guarente LP, Sassone-Corsi P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008:329–340. doi: 10.1016/j.cell.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sassone-Corsi P. Minireview: NAD +, a Circadian Metabolite with an Epigenetic Twist*. Endocrinology. 2012:1–5. doi: 10.1210/en.2011-1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hirayama J, Sahar S, Grimaldi B, Tamaru T, Takamatsu K, Nakahata Y, Sassone-Corsi P. CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature. 2007:1086–1090. doi: 10.1038/nature06394. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Q, Piston DW, Goodman RH. Regulation of corepressor function by nuclear NADH. Science. 2002:1895–1897. doi: 10.1126/science.1069300. [DOI] [PubMed] [Google Scholar]
- 64.Fjeld CC, Birdsong WT, Goodman RH. Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc Natl Acad Sci USA. 2003:9202–9207. doi: 10.1073/pnas.1633591100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith BC, Hallows WC, Denu JM. Anal Biochem. Elsevier Inc; 2009. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes; pp. 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pan PW, Feldman JL, Devries MK, Dong A, Edwards AM, Denu JM. J Biol Chem. American Society for Biochemistry and Molecular Biology; 2011. Structure and biochemical functions of SIRT6; pp. 14575–14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, Ilkayeva O, Marcheva B, Kobayashi Y, Omura C, Levine DC, Bacsik DJ, Gius D, Newgard CB, Goetzman E, Chandel NS, Denu JM, Mrksich M, Bass J. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science. 2013;342:1243417. doi: 10.1126/science.1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aksoy P, Escande C, White TA, Thompson M, Soares S, Benech JC, Chini EN. Regulation of SIRT 1 mediated NAD dependent deacetylation: a novel role for the multifunctional enzyme CD38. Biochemical and Biophysical Research Communications. 2006:353–359. doi: 10.1016/j.bbrc.2006.08.066. [DOI] [PubMed] [Google Scholar]
- 69.Barbosa MTP, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P, Chini EN. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007:3629–3639. doi: 10.1096/fj.07-8290com. [DOI] [PubMed] [Google Scholar]
- 70.Lim JH, Gerhart-Hines Z, Dominy JE, Lee Y, Kim S, Tabata M, Xiang YK, Puigserver P. Oleic acid stimulates complete oxidation of fatty acids through protein kinase A-dependent activation of SIRT1-PGC1alpha complex. The Journal of biological chemistry. 2013;288:7117–7126. doi: 10.1074/jbc.M112.415729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feldman JL, Baeza J, Denu JM. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. The Journal of biological chemistry. 2013;288:31350–31356. doi: 10.1074/jbc.C113.511261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jump DB, Clarke SD, Thelen A, Liimatta M. Coordinate regulation of glycolytic and lipogenic gene expression by polyunsaturated fatty acids. J Lipid Res. 1994;35:1076–1084. [PubMed] [Google Scholar]
- 73.Xu J, Christian B, Jump DB. Regulation of rat hepatic L-pyruvate kinase promoter composition and activity by glucose, n-3 polyunsaturated fatty acids, and peroxisome proliferator-activated receptor-alpha agonist. The Journal of biological chemistry. 2006;281:18351–18362. doi: 10.1074/jbc.M601277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV, de Cabo R, Ulrich S, Akassoglou K, Verdin E. Science. American Association for the Advancement of Science; 2013. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor; pp. 211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Newman JC, Verdin E. Diabetes Research and Clinical Practice. Elsevier Ireland Ltd; 2014. β-hydroxybutyrate: Much more than a metabolite; pp. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr. 2003:2485S–2493S. doi: 10.1093/jn/133.7.2485S. [DOI] [PubMed] [Google Scholar]
- 77.Riggs MG, Whittaker RG, Neumann JR, Ingram VM. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature. 1977:462–464. doi: 10.1038/268462a0. [DOI] [PubMed] [Google Scholar]
- 78.Cousens LS, Gallwitz D, Alberts BM. Different accessibilities in chromatin to histone acetylase. Journal of Biological Chemistry. 1979:1716–1723. [PubMed] [Google Scholar]
- 79.Sekhavat A, Sun J-M, Davie JR. Competitive inhibition of histone deacetylase activity by trichostatin A and butyrate. Biochem Cell Biol. 2007:751–758. doi: 10.1139/o07-145. [DOI] [PubMed] [Google Scholar]
- 80.Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem. 2008:587–593. doi: 10.1016/j.jnutbio.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 81.Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Gut. BMJ Publishing Group Ltd and British Society of Gastroenterology; 1987. Short chain fatty acids in human large intestine, portal, hepatic and venous blood; pp. 1221–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Donohoe DR, Holley D, Collins LB, Montgomery SA, Whitmore AC, Hillhouse A, Curry KP, Renner SW, Greenwalt A, Ryan EP, Godfrey V, Heise MT, Threadgill DS, Han A, Swenberg JA, Threadgill DW, Bultman SJ. Cancer Discovery. American Association for Cancer Research; 2014. A Gnotobiotic Mouse Model Demonstrates that Dietary Fiber Protects Against Colorectal Tumorigenesis in a Microbiota- and Butyrate-Dependent Manner; p. CD-14-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bultman SJ. Molecular pathways: gene-environment interactions regulating dietary fiber induction of proliferation and apoptosis via butyrate for cancer prevention. Clin Cancer Res. 2014:799–803. doi: 10.1158/1078-0432.CCR-13-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Donohoe Dallas R, Collins Leonard B, Wali A, Bigler R, Sun W, Bultman Scott J. The Warburg Effect Dictates the Mechanism of Butyrate-Mediated Histone Acetylation and Cell Proliferation. Molecular cell. 2012:612–626. doi: 10.1016/j.molcel.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, Buffet A, Marcaillou C, Bertherat J, Amar L, Rustin P, De Reynies A, Gimenez-Roqueplo AP, Favier J. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer cell. 2013;23:739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 86.Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, Guan KL. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes & development. 2012;26:1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Forneris F, Binda C, Vanoni MA, Mattevi A, Battaglioli E. Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett. 2005;579:2203–2207. doi: 10.1016/j.febslet.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 88.Smith EH, Janknecht R, Maher LJ., 3rd Succinate inhibition of alpha-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Hum Mol Genet. 2007;16:3136–3148. doi: 10.1093/hmg/ddm275. [DOI] [PubMed] [Google Scholar]
- 89.Thomas D, Surdin-Kerjan Y. The synthesis of the two S-adenosyl-methionine synthetases is differently regulated in Saccharomyces cerevisiae. Mol Gen Genet. 1991;226:224–232. doi: 10.1007/BF00273607. [DOI] [PubMed] [Google Scholar]
- 90.Townsend DM, Tew KD, Tapiero H. Sulfur containing amino acids and human disease. Biomed Pharmacother. 2004;58:47–55. doi: 10.1016/j.biopha.2003.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, Zhang J, Onder T, Unternaehrer JJ, Zhu H, Asara JM, Daley GQ, Cantley LC. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science. 2013;339:222–226. doi: 10.1126/science.1226603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sadhu MJ, Guan Q, Li F, Sales-Lee J, Iavarone AT, Hammond MC, Cande WZ, Rine J. Nutritional control of epigenetic processes in yeast and human cells. Genetics. 2013;195:831–844. doi: 10.1534/genetics.113.153981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 94.Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2011;13:97–109. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- 95.Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nature reviews Molecular cell biology. 2007;8:307–318. doi: 10.1038/nrm2143. [DOI] [PubMed] [Google Scholar]
- 96.Del Rizzo PA, Trievel RC. Molecular basis for substrate recognition by lysine methyltransferases and demethylases. Biochimica et biophysica acta. 2014 doi: 10.1016/j.bbagrm.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 97.Hino S, Sakamoto A, Nagaoka K, Anan K, Wang Y, Mimasu S, Umehara T, Yokoyama S, Kosai K, Nakao M. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nature communications. 2012;3:758. doi: 10.1038/ncomms1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhou X, Sun H, Chen H, Zavadil J, Kluz T, Arita A, Costa M. Hypoxia induces trimethylated H3 lysine 4 by inhibition of JARID1A demethylase. Cancer Res. 2010;70:4214–4221. doi: 10.1158/0008-5472.CAN-09-2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Park SJ, Kim JG, Son TG, Yi JM, Kim ND, Yang K, Heo K. The histone demethylase JMJD1A regulates adrenomedullin-mediated cell proliferation in hepatocellular carcinoma under hypoxia. Biochem Biophys Res Commun. 2013;434:722–727. doi: 10.1016/j.bbrc.2013.03.091. [DOI] [PubMed] [Google Scholar]
- 100.Tausendschon M, Dehne N, Brune B. Hypoxia causes epigenetic gene regulation in macrophages by attenuating Jumonji histone demethylase activity. Cytokine. 2011;53:256–262. doi: 10.1016/j.cyto.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 101.Chen H, Yan Y, Davidson TL, Shinkai Y, Costa M. Hypoxic stress induces dimethylated histone H3 lysine 9 through histone methyltransferase G9a in mammalian cells. Cancer Res. 2006;66:9009–9016. doi: 10.1158/0008-5472.CAN-06-0101. [DOI] [PubMed] [Google Scholar]
- 102.Melvin A, Rocha S. Chromatin as an oxygen sensor and active player in the hypoxia response. Cell Signal. 2012;24:35–43. doi: 10.1016/j.cellsig.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Watson JA, Watson CJ, McCann A, Baugh J. Epigenetics, the epicenter of the hypoxic response. Epigenetics. 2010;5:293–296. doi: 10.4161/epi.5.4.11684. [DOI] [PubMed] [Google Scholar]
- 104.Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, McDonough MA, King ON, Clifton IJ, Klose RJ, Claridge TD, Ratcliffe PJ, Schofield CJ, Kawamura A. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO reports. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cervera AM, Bayley JP, Devilee P, McCreath KJ. Inhibition of succinate dehydrogenase dysregulates histone modification in mammalian cells. Mol Cancer. 2009;8:89. doi: 10.1186/1476-4598-8-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, Sharp KA, Levine RL, Thompson CB. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2010;465:966. doi: 10.1038/nature09132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG., Jr (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell metabolism. 2012;16:9–17. doi: 10.1016/j.cmet.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013:489–498. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]
- 112.Slawson C, Hart GW. O-GlcNAc signalling: implications for cancer cell biology. Nature reviews Cancer. 2011;11:678–684. doi: 10.1038/nrc3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hottiger MO. ADP-ribosylation of histones by ARTD1: an additional module of the histone code? FEBS Lett. 2011:1595–1599. doi: 10.1016/j.febslet.2011.03.031. [DOI] [PubMed] [Google Scholar]
- 114.Nicolas E, Roumillac C, Trouche D. Balance between acetylation and methylation of histone H3 lysine 9 on the E2F-responsive dihydrofolate reductase promoter. Molecular and cellular biology. 2003;23:1614–1622. doi: 10.1128/MCB.23.5.1614-1622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lasserre J, Chung HR, Vingron M. Finding associations among histone modifications using sparse partial correlation networks. PLoS computational biology. 2013;9:e1003168. doi: 10.1371/journal.pcbi.1003168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sun ZW, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002;418:104–108. doi: 10.1038/nature00883. [DOI] [PubMed] [Google Scholar]
- 117.Huang S, Litt M, Felsenfeld G. Methylation of histone H4 by arginine methyltransferase PRMT1 is essential in vivo for many subsequent histone modifications. Genes & development. 2005;19:1885–1893. doi: 10.1101/gad.1333905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Metzger E, Imhof A, Patel D, Kahl P, Hoffmeyer K, Friedrichs N, Muller JM, Greschik H, Kirfel J, Ji S, Kunowska N, Beisenherz-Huss C, Gunther T, Buettner R, Schule R. Phosphorylation of histone H3T6 by PKCbeta(I) controls demethylation at histone H3K4. Nature. 2010;464:792–796. doi: 10.1038/nature08839. [DOI] [PubMed] [Google Scholar]
- 119.Cheung P, Tanner KG, Cheung WL, Sassone-Corsi P, Denu JM, Allis CD. Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Molecular Cell. 2000:905–915. doi: 10.1016/s1097-2765(00)80256-7. [DOI] [PubMed] [Google Scholar]
- 120.Shimada M, Niida H, Zineldeen DH, Tagami H, Tanaka M, Saito H, Nakanishi M. Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell. 2008;132:221–232. doi: 10.1016/j.cell.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 121.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nature reviews Genetics. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 122.Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes & development. 2001;15:2343–2360. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- 123.Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. The Journal of biological chemistry. 2003;278:4035–4040. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- 124.Gardner KE, Allis CD, Strahl BD. Operating on chromatin, a colorful language where context matters. J Mol Biol. 2011;409:36–46. doi: 10.1016/j.jmb.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Karch KR, Denizio JE, Black BE, Garcia BA. Identification and interrogation of combinatorial histone modifications. Front Genet. 2013;4:264. doi: 10.3389/fgene.2013.00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Katada S, Imhof A, Sassone-Corsi P. Connecting threads: epigenetics and metabolism. Cell. 2012;148:24–28. doi: 10.1016/j.cell.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 127.Santos-Rosa H, Valls E, Kouzarides T, Martinez-Balbas M. Mechanisms of P/CAF auto-acetylation. Nucleic Acids Res. 2003;31:4285–4292. doi: 10.1093/nar/gkg655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Karukurichi KR, Wang L, Uzasci L, Manlandro CM, Wang Q, Cole PA. Analysis of p300/CBP histone acetyltransferase regulation using circular permutation and semisynthesis. Journal of the American Chemical Society. 2010;132:1222–1223. doi: 10.1021/ja909466d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Thompson PR, Wang D, Wang L, Fulco M, Pediconi N, Zhang D, An W, Ge Q, Roeder RG, Wong J, Levrero M, Sartorelli V, Cotter RJ, Cole PA. Regulation of the p300 HAT domain via a novel activation loop. Nat Struct Mol Biol. 2004;11:308–315. doi: 10.1038/nsmb740. [DOI] [PubMed] [Google Scholar]
- 130.Huang WC, Chen CC. Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Molecular and cellular biology. 2005;25:6592–6602. doi: 10.1128/MCB.25.15.6592-6602.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yang W, Hong YH, Shen XQ, Frankowski C, Camp HS, Leff T. Regulation of transcription by AMP-activated protein kinase: phosphorylation of p300 blocks its interaction with nuclear receptors. The Journal of biological chemistry. 2001;276:38341–38344. doi: 10.1074/jbc.C100316200. [DOI] [PubMed] [Google Scholar]
- 132.Vaquero A, Scher M, Erdjument-Bromage H, Tempst P, Serrano L, Reinberg D. SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature. 2007;450:440–444. doi: 10.1038/nature06268. [DOI] [PubMed] [Google Scholar]
- 133.Sarris M, Nikolaou K, Talianidis I. Context-specific regulation of cancer epigenomes by histone and transcription factor methylation. Oncogene. 2014;33:1207–1217. doi: 10.1038/onc.2013.87. [DOI] [PubMed] [Google Scholar]
- 134.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sahar S, Masubuchi S, Eckel-Mahan K, Vollmer S, Galla L, Ceglia N, Masri S, Barth TK, Grimaldi B, Oluyemi O, Astarita G, Hallows WC, Piomelli D, Imhof A, Baldi P, Denu JM, Sassone-Corsi P. Circadian control of fatty acid elongation by SIRT1 protein-mediated deacetylation of acetyl-coenzyme A synthetase 1. The Journal of biological chemistry. 2014;289:6091–6097. doi: 10.1074/jbc.M113.537191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang Y, Kavran JM, Chen Z, Karukurichi KR, Leahy DJ, Cole PA. Regulation of s-adenosylhomocysteine hydrolase by lysine acetylation. The Journal of biological chemistry. 2014;289:31361–31372. doi: 10.1074/jbc.M114.597153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Holness MJ, Sugden MC. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochemical Society transactions. 2003;31:1143–1151. doi: 10.1042/bst0311143. [DOI] [PubMed] [Google Scholar]
- 138.Grady SL, Hwang J, Vastag L, Rabinowitz JD, Shenk T. Herpes simplex virus 1 infection activates poly(ADP-ribose) polymerase and triggers the degradation of poly(ADP-ribose) glycohydrolase. J Virol. 2012;86:8259–8268. doi: 10.1128/JVI.00495-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Andrabi SA, Umanah GK, Chang C, Stevens DA, Karuppagounder SS, Gagne JP, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci U S A. 2014;111:10209–10214. doi: 10.1073/pnas.1405158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ha HC, Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A. 1999;96:13978–13982. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Martinez-Pastor B, Cosentino C, Mostoslavsky R. A tale of metabolites: the cross-talk between chromatin and energy metabolism. Cancer discovery. 2013;3:497–501. doi: 10.1158/2159-8290.CD-13-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, Rabinowitz JD. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol. 2013;9:712. doi: 10.1038/msb.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G, Chiaradonna F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523. doi: 10.1038/msb.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tong L, Denu JM. Function and metabolism of sirtuin metabolite O-acetyl-ADP-ribose. Biochimica et biophysica acta. 2010;1804:1617–1625. doi: 10.1016/j.bbapap.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.McBrian MA, Behbahan IS, Ferrari R, Su T, Huang TW, Li K, Hong CS, Christofk HR, Vogelauer M, Seligson DB, Kurdistani SK. Histone acetylation regulates intracellular pH. Molecular cell. 2013;49:310–321. doi: 10.1016/j.molcel.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E, Michelakis ED. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell. 2014;158:84–97. doi: 10.1016/j.cell.2014.04.046. [DOI] [PubMed] [Google Scholar]
- 147.Kera Y, Katoh Y, Ohta M, Matsumoto M, Takano-Yamamoto T, Igarashi K. Methionine adenosyltransferase II-dependent histone H3K9 methylation at the COX-2 gene locus. The Journal of biological chemistry. 2013;288:13592–13601. doi: 10.1074/jbc.M112.429738. [DOI] [PMC free article] [PubMed] [Google Scholar]