

Abstract
Recent findings indicate that the ubiquitin–proteasome system is involved in the pathogenesis of cancer as well as autoimmune and several neurodegenerative diseases, and is thus a target for novel therapeutics. One disease that is related to aberrant protein degradation is multiple sclerosis, an autoimmune disorder involving the processing and presentation of myelin autoantigens that leads to the destruction of axons. Here, we show that brain-derived proteasomes from SJL mice with experimental autoimmune encephalomyelitis (EAE) in an ubiquitin-independent manner generate significantly increased amounts of myelin basic protein peptides that induces cytotoxic lymphocytes to target mature oligodendrocytes ex vivo. Ten times enhanced release of immunogenic peptides by cerebral proteasomes from EAE-SJL mice is caused by a dramatic shift in the balance between constitutive and β1ihigh immunoproteasomes in the CNS of SJL mice with EAE. We found that during EAE, β1i is increased in resident CNS cells, whereas β5i is imported by infiltrating lymphocytes through the blood–brain barrier. Peptidyl epoxyketone specifically inhibits brain-derived β1ihigh immunoproteasomes in vitro (kobs/[I] = 240 M−1s−1), and at a dose of 0.5 mg/kg, it ameliorates ongoing EAE in vivo. Therefore, our findings provide novel insights into myelin metabolism in pathophysiologic conditions and reveal that the β1i subunit of the immunoproteasome is a potential target to treat autoimmune neurologic diseases.—Belogurov Jr., A., Kuzina, E., Kudriaeva, A., Kononikhin, A., Kovalchuk, S., Surina, Y., Smirnov, I., Lomakin, Y., Bacheva, A., Stepanov, A., Karpova, Y., Lyupina, Y., Kharybin, O., Melamed, D., Ponomarenko, N., Sharova, N., Nikolaev, E., Gabibov, A. Ubiquitin-independent proteosomal degradation of myelin basic protein contributes to development of neurodegenerative autoimmunity.
Keywords: immunoproteasome, antigen presentation, oligodendrocytes, experimental autoimmune encephalomyelitis, multiple sclerosis
Numerous studies in animal models for multiple sclerosis, namely experimental autoimmune encephalomyelitis (EAE), have provided direct and univocal evidence that autoreactive Th1/Th17 cells are generally orchestrating a coordinated attack of the immune system on the CNS (1, 2). Nonetheless, it is now apparent that CD4+ T cells individually are unable to drive this multifactorial autoimmune pathology. Regardless of disease stage, the lymphocytes in multiple sclerosis plaques are biased toward the CD8 lineage—CD8 vs. CD4 T cells—by as much as 10 to 1 (3). Myelin-reactive cytotoxic T lymphocytes (CTLs) are thought to cause demyelination and therefore are potentially a major culprit in multiple sclerosis (4). Importantly, adoptively transferred CTLs are capable of inducing EAE in mice (5).
Major histocompatibility complex (MHC) class I–bound peptides that are recognized by CTLs are generated by a cryptic protease, the 26S proteasome, a massive 2.5 MDa molecular machine (6), strictly controlled by the ubiquitin system (7). There are 2 major types of proteasomes: constitutive proteasomes (CP) and immunoproteasomes (IP). During inflammation, the CP-IP balance is shifted toward IP, and 3 types of CP catalytic subunits, β1, β2, and β5, are substituted with β1i/low molecular mass polypeptide (LMP) 2, β2i/LMP10, and β5i/LMP7, respectively, generating the IP. These 2 types of proteasomes have different protein cleavage patterns as a result of the different specificity of catalytic subunits inside their cores (8).
Myelin basic protein (MBP), which is expressed exclusively in oligodendrocytes (ODCs) and Schwann cells, is one of the key neuroantigens involved in the development of EAE (9) and multiple sclerosis (10, 11). Proteasome-mediated degradation and presentation of myelin antigens, which recruit clonally expanded myelin-specific CTLs (12), is undoubtedly very important in the pathogenesis of multiple sclerosis. One study has shown direct lysis of human ODCs by MBP-specific CTLs (13). A study by Sobottka et al. (14) established the ability of ODCs to process and present antigens to autoreactive cytotoxic CD8+ T cells that directly target the myelin sheath and cause axonal loss due to “collateral bystander damage.” Nothing was known as to why ODCs fail to override presentation of MBP peptides using the highly evolved ubiquitination system to defend themselves from CTLs. Our previous report partially elucidated this enigmatic question by showing that 26S-mediated degradation of intracellular MBP is ubiquitin independent (15). We further reasoned that the ubiquitin independence of proteasomal MBP hydrolysis might have far-reaching pathophysiologic consequences because the spectrum of MBP peptides presented on the surface of ODCs is generally controlled by the catalytic subunits of the proteasome. In the present study, we elucidate the physiologic relevance of this finding and determine how ubiquitin-independent hydrolysis of MBP by β1i immunoproteasomes target ODCs for attack by CTLs.
MATERIALS AND METHODS
Materials
ATP and recombinant murine IL-2 were purchased from Sigma-Aldrich (St. Louis, MO, USA). The tissue culture sera and media were purchased from Gibco (Carlsbad, CA, USA). Mouse anti–β-actin antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); Q-Sepharose and monoQ columns were purchased from GE Healthcare (Waukesha, WI, USA). The proteasome inhibitor carbobenzoxy-l-leucyl-l-leucyl-l-leucinal (MG132) was purchased from Calbiochem (San Diego, CA, USA), and [(1R)-3-methyl-1-({(2S)-3-phenyl-2-[(pyrazin-2-ylcarbonyl)amino] propanoyl} amino)butyl] boronic acid (bortezomib, PS-341) was obtained from LC Laboratories (Woburn, MA, USA). The reagents for ECL were obtained from Bio-Rad (Hercules, CA, USA) and GE Healthcare. Bovine MBP was isolated as described (16). Most other chemicals were reagent grade and acquired from either Sigma-Aldrich or Helicon (Moscow, Russian Federation).
Induction and treatment of EAE in SJL mice
SJL and BALB/c mice were developed in specific-pathogen-free conditions in the animal facilities of the Assaf Harofeh Medical Center, Zerifin, Israel. The mice were maintained according to the guidelines of the Local Ethics Committee for Animal Experimentation, and the experimental protocols (#68-2009, 07-2010, 25-2010) were approved by the Ethics Committee of the Israeli Health ministry. EAE was induced in 6- to 8-week-old female SJL mice via subcutaneous immunization according to the following protocol: Mice were injected in all 4 footpads with 3.5 mg of spinal cord homogenate emulsified at a 1:1 ratio in complete Freund adjuvant supplemented with 4 mg/ml H37Ra. Pertussis toxin (0.25 ml, 250 ng; Sigma-Aldrich) was injected intravenously, immediately after and 48 hours later. Between 14 and 28 days after the immunization, mice with pronounced clinical symptoms (score from 2 to 4) were killed and their organs collected for later experiments. Treatment of mice with β1i-PEk and PS-341 was performed according to the following protocol. Each group of mice was treated with different combinations and doses of drugs or their respective controls (Table 1) twice weekly from day 0 until day 21 after immunization. Drugs were injected intravenously via the tail vein. After 7 injection cycles, clinical scoring was performed until day 25 after EAE induction.
TABLE 1.
Treatment of SJL mice with proteasome inhibitors PS-341 and β1i-PEK
| Group | n | Inhibitor | Dose, mg/kg | Injection volume i.v., μl | Including DMSO, μl | Administrationa | Immunization |
|---|---|---|---|---|---|---|---|
| 1 | 10 | PS-341 | 0.5 | 250 | 10 | + | + |
| 2 | 10 | β1i-PEk | 0.5 | 250 | 10 | + | + |
| 3 | 10 | — | — | 250 | 10 | + | + |
| 4 | 5 | — | — | 250 | 10 | + | – |
| 5 | 5 | — | — | — | 0 | – | + |
| 6 | 5 | — | — | — | 0 | – | – |
Twice weekly starting from day 0 after immunization, 7 times total.
Western blot analysis of tissues homogenates
Brain tissue from control and EAE-mice was collected, weighed, and placed immediately on ice. The frontal cortex, striatum, medial basal hypothalamus, cerebellum, and brain stem were homogenized separately (Braun Melsungen homogenizer, glass–glass; Braun, Melsungen, Germany) in 3V 50 mM Tris-HCl buffer (pH 7.5), containing 100 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol, 10% glycerol, and 10 mM Na2S2O5. The homogenates were centrifuged at 10,000 g for 30 minutes, and supernatants were used for further investigation. Supernatants were subjected to 13% SDS-PAGE, and proteins were transferred to Hybond C membranes. The membranes were blocked with ECL plus blocking reagent (GE Healthcare) and hybridized with one of the following antibodies: mouse anti–β-actin (1:1000, sc-81178; Santa Cruz Biotechnology); mouse mAb to α1, 2, 3, 5, 6, 7 subunits (1:2000, BML-PW8195); rabbit pAb to subunit β5 (1:2000, BML-PW8895); mouse mAb to subunit β1 (1:1000, BML-PW8140); mouse mAb to β5i (1:1000, BML-PW8845 1:1000); mouse mAb to β1i (1:1000, BML-PW8840) for 1 hour. All antibodies were obtained from Enzo Life Sciences (Farmingdale, NY, USA). Membranes were washed 3 times for 15 minutes with TNT buffer containing 0.05% Tween-20. Bound antibodies were visualized using a peroxidase-conjugated secondary antibody to mouse or rabbit IgG (1:2500; GE Healthcare) followed by detection using an ECL plus detection kit (GE Healthcare). Image analyses were performed using ImageJ software. For purified proteasomes, the Western blot procedure was similar, except that the buffer used was PBS, the normalizing marker was the band from mouse mAb to α1, 2, 3, 5, 6, 7 subunits (1:2000; Enzo Life Sciences), and the mouse mAb to the 19S subunit Rpt6 (1:2000; Enzo Life Sciences) was utilized to prove the integrity of the 26S proteasome. To detect β2i, the rabbit pAb to anti-β2i (1:2000; Santa Cruz Biotechnology) was used and for detection of β1i, the membrane was incubated with a rabbit pAb to β1i (1:1000; Biomol) for 48 hours at 4°C.
Immunohistochemistry and fluorescent microscopy
To examine the localization of β1i and β5i, control and EAE mice were perfused with 4% paraformaldehyde, and brains were frozen at −80°C before cryostat sectioning. Frozen brains were cut using a Leica CM-3050S cryostat (Leica Microsystems, Buffalo Grove, IL, USA), and 12 μm frontal brain slices of control and EAE mice were mounted on the same electrostatic Superfrost plus slides. Cryostat sections were subsequently incubated at room temperature either in PBS with (Fab)2 goat anti-mouse IgG (10% v/v) (for blocking nonspecific background in mouse tissues), 5% goat serum, and 0.3% Triton X-100 for 1 hour or with PBS containing 5% goat serum, 0.1% Triton X-100, and primary rabbit pAb β5i (1:1000; Biomol) or β1i (overnight, 1:200, Enzo Life Sciences). Colabeling of neurons (mouse anti-NeuN 1:1500; Millipore, Billerica, MA, USA), ODCs (rabbit anti-MBP, overnight, 1:100; Sigma-Aldrich), and T cells (rat anti-CD3, 1:500; Santa Cruz Biotechnology) was also conducted in conjunction with anti-β1i and anti-β5i staining. Secondary incubation with PBS containing Alexa Fluor 546 (red) goat anti-rabbit IgG or Alexa Fluor 488 (green) goat anti-mouse IgG (1:500, Invitrogen, Carlsbad, CA, USA) or FITC-conjugated goat anti-rat IgG (green) (1:100; Santa Cruz Biotechnology) for 2 hours was performed to detect the antigen. Stained samples were embedded in Mowiol mounting solution (Calbiochem), and images were taken on a Leica DM RXA 2 fluorescent microscope equipped with a ×20 air immersion objective (NA 0.70) or with a ×40 oil immersion objective (NA 1.0) or with confocal Leica SPE microscope equipped with an Ar-Kr laser at the Core Facility for Cell Technologies and Optical Research Methods in Developmental Biology of the Institute of Developmental Biology, Russian Academy of Sciences. Images were recorded and processed with Leica LCS software. To ensure equal illumination for all treatments, the same intensity and filter settings were used throughout. Images were recorded at a resolution of 1024 × 1024 pixels. Control experiments were performed by omitting primary or secondary antibodies.
Purification of proteasomes from mouse brains
Briefly, a BALB/c brain was homogenized (Dounce homogenizer; Thomas Scientific, Swedesboro, NJ, USA) in 3 volumes (w/w) of lysis buffer containing 30 mM Tris-HCl (pH 7.5), 2 mM ATP, 1 mM EDTA, 5 mM MgCl2, 1 mM DTT, 10% glycerol, 100 mM NaCl, and a protease inhibitor cocktail. The prepared brain homogenate was subjected to 3 repeated freeze–thaw cycles, and cell debris was removed by 2 consecutive centrifugations at 4°C (1500 g for 20 minutes and 13,000 g for 30 minutes). The supernatant (0.8 ml) was overlaid on top of a 24 ml glycerol gradient (10–55% glycerol in 25 mM Tris-HCl [pH 7.5], 1 mM DTT, and 4 mM ATP) and centrifuged at 125,000 g at 4°C for 16 hours. Fractions (1 ml each) were collected, and proteasome activity was quantified using Suc-LLVY-AMC as a substrate. To distinguish between the activity related to the 20S proteasome and the 26S proteasome, the assay was performed with or without 0.02% SDS. The buffer used for the measurement of the activity of the proteasomes contained 20 mM Tris pH 7.5, 1 mM ATP, 1 mM DTT, and 5 mM MgCl2. The fractions containing the 26S proteasome were subjected to ion-exchange chromatography on a MonoQ column using an NaCl gradient (100–500 mM in 15 column volumes) in buffer containing 20 mM Tris (pH 7.5), 1 mM ATP, 1 mM DTT, and 5 mM MgCl2. The fractions containing the 26S proteasome were dialyzed into storage buffer (25 mM Tris-HCl [pH 7.5], 1 mM DTT, 1 mM ATP, 5 mM MgCl2, and 10% glycerol). For long-term storage, up to 40% glycerol was added to the proteasome, and the purified proteasome was further stored at −20°C for 2 months.
In vitro 26S-mediated hydrolysis
The hydrolysis of proteins (1–3 μg) was performed in a volume of 12.5 μl. The reaction mixtures contained 20 mM Tris (pH 7.5), 1 mM DTT, 5 mM MgCl2, an ATP-regenerating system (phosphocreatine [10 mM] and creatine phosphokinase [0.5 μg]), and purified 26S proteasomes (0.25 μg). The mixtures were incubated for the indicated time at 37°C, and the reaction was terminated by the addition of sample buffer. The proteins were resolved by SDS-PAGE and visualized with Coomassie staining.
Enzymatic hydrolysis
Enzymes were incubated with 2 μg of MBP at 37°C for 3 hours at an enzyme–substrate molar ratio of 1:100 in a final volume of 20 μl of 50 mM Tris (pH 7.5) and 5 mM CaCl2.
Top-down analysis
For the top-down analysis, purified MBP was analyzed directly using electrospray ionization mass spectrometry (ESI-MS). ESI-MS was performed with a Bruker APEX Ultra Fourier transform ion cyclotron resonance mass spectrometer (Bruker Daltonics, Billerica, MA, USA) equipped with an actively shielded 7 T superconducting magnet, cylindrical infinity ion cyclotron resonance analyzer cell, and an Apollo II ion source. The following conditions were used for electrospray: flow rate 2 μl/min; positive ion mode; spray shield 3.5 kV; capillary voltage 4 kV. Ions were externally accumulated in the collision cell for 0.3 seconds. The spectra were acquired with an acquisition size of 2 megapoints with a mass range of 200 to 2000 m/z. ES tuning mix (Agilent Technologies, Palo Alto, CA, USA; reorder no. G2421A) was used for external mass calibration. A total of 100 time domain data were coadded, baseline zeroed, zero-filled once, and fast Fourier transformed using ApexControl 3.0 and Data Analysis 4.0 SP1 (Bruker Daltonics).
Liquid chromatography–mass spectrometry/mass spectrometry
Proteasome samples were mixed with nonubiquitinated MBP to final concentrations 27 μg/ml and 50 μg/ml, respectively, as determined by Bradford assay and further incubated at 37°C for 24 hours. The degree of MBP degradation was determined by 15% SDS-PAGE and further analyzed by liquid chromatography–mass spectrometry/mass spectrometry (LC-MS/MS). Peptide mixtures were separated using an Agilent 1100 system autosampler/nanoHPLC (Agilent Technologies). A sample volume of 2 μl was loaded by autosampler onto a capillary column (75 μm inner diameter × 12 cm, Reprosil-Pur Basic C18, 3 µm; Dr. Maisch HPLC GmbH, Ammerbuch-Entringen, Germany). Separation was performed at a flow rate of 0.3 μl/min using H2O/0.1% formic acid (v/v, solvent A) and acetonitrile/0.1% formic acid (v/v, solvent B). The column was preequilibrated with 3% (v/v) solvent B, then a linear gradient from 3 to 50% (v/v) of solvent B for 45 minutes, followed by isocratic elution (95%, v/v, of solvent B for 15 minutes), which was used for peptide separation. Mass spectrometric analysis was performed on a 7 T Finnigan LTQ-FT Ultra (Thermo Electron, Bremen, Germany) mass spectrometer equipped with a homemade nanoelectrospray ion source. Conditions for electrospray were as follows: positive ion mode; needle voltage 1.9 kV; no sheath and auxiliary gas flow; tube lens voltage 45 V; and capillary temperature 250°C. Data were acquired in data-dependent mode using Xcalibur (Thermo Finnigan, San Jose, CA, USA) software. The precursor ion scan MS spectra (m/z 300–1600) were acquired with resolution of R = 50.000 at m/z 400 (number of accumulated ions 5 × 105).
Quantitative LC-MS/MS analysis
Proteasome samples were mixed with nonubiquitinated MBP to final concentrations of 20 μg/ml and 100 μg/ml, respectively, as determined by Bradford assay, and further incubated at 37°C. Reactions were quenched by the addition of 5 μM PS-341 (1/25 v/v) in DMSO and frozen immediately. For 16O/18O quantitative LC-MS/MS analysis, immediately after ion-exchange chromatography, the fraction (100 μl) containing the 26S proteasome was dialyzed against 50 ml of proteasome buffer (20 mM Tris, 5 mM MgCl2, 1 mM ATP, 1 mM DTT, pH 7.5) prepared using H218O or H216O. After dialysis, proteasome activity was measured using the Suc-LLVY-AMC substrate, and all samples were diluted to equal activity per unit volume. Samples of lyophilized MBP were diluted in 20 mM Tris-HCl pH 7.2 prepared using 18O- or 16O-containing water. Directly before LC-MS/MS analysis, equivalent amounts (mol/mol) of 18O and 16O samples were mixed and further analyzed as described above. The peak intensities for the 18O-labeled peptides were corrected for the contribution of the nonlabeled 16O-containing peptide according to equation 1,
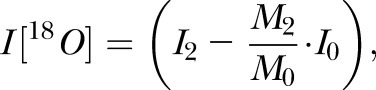 |
where I0 is the measured relative intensity for the monoisotopic peak, I2 is the measured relative intensity for the peak with mass + 2 Da, M0 is the theoretical relative intensity for the monoisotopic peak, and M2 is the theoretical relative intensity for the monoisotopic peaks with masses + 2 Da. For quantitative analysis using isotopically labeled peptide ENPVVHFF* (F* = 13C9H915NO) (Peptide Protein Research, Hampshire, United Kingdom), the labeled peptide was mixed with MBP hydrolyzed by various proteasomes in the same ratio (5 μl of labeled peptide + 5 μl of sample). Finally each sample (10 μl volume) contained 2.5 ng of ENPVVHFF* peptide. A sample volume of 2 μl was loaded by an autosampler and analyzed using the LC-MS/MS method. The amount of the peptide was determined as the ratio of the relative peak intensity of the labeled and nonlabeled peptide in mass spectra. The relative peak intensities were determined from extracted mass chromatograms using Xcalibur (Thermo Electron, San Jose, CA, USA) software.
Inhibition analysis
Inhibition analysis was performed using PS-341 (LC Laboratories), MG132 (Boston Biochem, Cambridge, MA, USA), and β1i-specific peptidyl epoxyketone, synthesized according to the following procedure (17). The range of inhibitor concentrations were: PS-341 0.1–1000 nM, MG132 0.1–1000 nM, β1i-specific peptidyl epoxyketone 2–100 μM. Reversible inhibition kinetics measurements were performed using the peptide fluorogenic substrate Suc-LLVY-MCA (Sigma-Aldrich) on a Tecan plate reader (GENios; Tecan, Männedorf, Switzerland). Inhibitor and substrate solutions in reaction a buffer containing 20 mM Tris-HCl pH 7.5, 1 mM ATP, 1 mM DTT, 5 mM MgCl2, and 1 mM EDTA were prepared from 50× stock solutions in DMSO. Isolated proteasomes were mixed with substrate solution (final concentration 50 μM) in wells and an equal volume of inhibitor solution was added immediately. After 20 minutes of incubation at 37°C, fluorescence of released 7-amino-4-methylcoumarin (excitation 360 nm, emission 465 nm) was measured continuously for 45 minutes. Reaction velocities were determined for all combinations of at least 4 substrate concentrations and at least 4 inhibitor concentrations. Inhibition constants were determined from data of 3 independent experiments using SigmaPlot software, version 8.0 (SigmaPlot, London, United Kingdom), Enzyme Kinetics module. Irreversible inhibition kinetic measurements were performed using the Proteasome-Glo Chymotrypsine-Like bioluminescent assay system (Promega, Madison, WI, USA) on a Varioskan Flash spectral scanning multimode reader (Thermo Scientific, Waltham, MA, USA). Reagents were prepared according to the manufacturer’s technical bulletin, and the final concentration of the peptide substrate Suc-Leu-Leu-Val-Tyr-aminoluciferin was 20 μM. Reagents were mixed with proteasomes (final concentration 0.1 nM) in Thermo Microfluor 384-well plates and incubated at 37°C. After 20 minutes, appropriate amounts of inhibitor were added from 20% DMSO 10× stock solutions; 20% DMSO without inhibitors was added as a control, and luminescence measurements was started immediately. kobs/[I] values were obtained using SigmaPlot 11.0 by nonlinear least-squares fit of the data to equation 2:
where L0 is the initial luminescence intensity that decays over time to a final luminescence intensity, Ls, with a rate constant, kobs. The kobs/[I] values given are an average of 3 independent experiments with at least 4 different inhibitor concentrations.
Mature ODC culture
Mature murine ODC cultures were prepared from brains of 3-day-old SJL mouse pups as described elsewhere (18). IFN-γ was added in the culture medium at a concentration of 1000 U/ml 48 hours before analysis, if indicated.
Multiple reaction monitoring (MRM) QTRAP analysis
Peripheral blood mononuclear cells (PBMCs) were isolated from BALB/c, C3H, and SJL mice peripheral blood by centrifugation over a Ficoll-Paque Plus solution (GE Healthcare Life Sciences). Small amounts of contaminating red blood cells were lysed with ACK lysis buffer (Life Technologies, Carlsbad, CA, USA). Alternatively mature ODCs culture was used. Cells (3 × 106 PBMCs per milliliter or 2 × 105 ODCs) were washed with PBS and loaded with ENPVVHFF peptide (50 μg/ml) for 3 hours at 37°C. Further cells were washed at 4°C twice with culture medium and once with PBS. The cell pellet was resuspended in water, and a further 3 volumes of cold acetonitrile supplemented with 0.3% formic acid was added. An insoluble pellet was removed by centrifugation, and supernatant was evaporated and further analyzed by MRM in a QTRAP mass spectrometer (AB Sciex, Framingham, MA, USA). To optimize MRM parameters the synthetic peptide was directly injected with a syringe. MS2 spectra for a doubly charged peptide peak with m/z 494.8 averaged for collision energy 5 to 40 EV was obtained and for the 5 best intensity MS2 peaks collision energy was manually optimized. The obtained MRM transition list was used to analyze experimental samples in LC-MS mode with a linear gradient from 5% to 50% acetonitrile in water in 20 minutes at a flow rate of 300 nl/min. Trap and analytical columns were C18 RP 0.5 × 0.3 mm and 150 × 0.075 mm correspondingly.
Generation of antigen-specific CD8+ T cells and cytotoxicity assay
CD8+ T cell lines were generated as described elsewhere (13). Briefly, the whole mononuclear cell fraction was isolated from the blood of naive SJL mice by centrifugation over a Ficoll-Paque Plus solution (GE Healthcare Life Sciences). CD8+ T cells were isolated from the mononuclear cell fraction using the Dynabeads Untouched Mouse CD8 Cells negative isolation kit (Life Technologies). After washing, CD8 T cells were suspended in culture medium at 106 cells/ml. Autologous dendritic cells were isolated from mouse spleens and pulsed with the peptides ENPVVHFF (MBP83–90) and FNFTAPFI (Theiler encephalomyelitis virus epitope was used as a control irrelevant peptide, GeneCust, Dudelange, Luxembourg) at 10 μg/ml for 1 hour at 37°C, and added to T-cell cultures at a T cell/APC ratio of 4:1. On day 3, 50 U/ml of recombinant mouse IL-2 (Sigma-Aldrich) was added to the T-cell cultures. On day 7 and for each subsequent week, T cells were provided with fresh peptide-pulsed dendritic cells, and 2 days after that, 50 U/ml of IL-2 was added. Cytotoxicity was determined using the Delphia assay (PerkinElmer, Waltham, MA, USA) as described in the manufacturer’s instructions. Briefly, ODCs were loaded with BATDA reagent for 30 minutes at 37°C and washed extensively directly before cytotoxic lymphocyte (CTL) addition. CD8 T cells were added to microwells at a 10:1 effector/target ratio with or without anti-mouse MHC class I antibody ER-MP42 (Abcam ab15680). The supernatant was collected after 2.5 hours and mixed with Europium solution. Time-resolved fluorescence was measured using a Varioskan Flash (Thermo Scientific) with the following parameters: exitation 340 nm, top emission 615 nm; time-resolved fluorescence delay 50 microseconds, time-resolved fluorescence integration time 800 microseconds, total measurement time 5000 milliseconds.
Immunocytochemistry ODC staining
Murine ODCs were cultured on poly-l-ornithine–coated coverslips, washed with PBS, fixed with 4% paraformaldehyde in PBS at room temperature for 20 minutes, washed twice with PBS, permeabilized for 1 hour at room temperature in PBS, 5% bovine serum albumin, and 0.2% Triton X-100 and blocked for 30 minutes at room temperature in 50% horse serum in PBS. Coverslips were incubated with O4 mouse mAb (Sigma-Aldrich), anti-β1i rabbit pAb, or anti-20S core rabbit pAb (Enzo Life Sciences) in reaction buffer (Tris-buffered saline, 1 mM CaCl2, 1% bovine serum albumin, 0.1% Triton X-100) for 2 hours at room temperature or at +4°C overnight, washed 4 times in wash buffer (Tris-buffered saline, 1 mM CaCl2, 0.2% Tween-20), and further incubated with secondary antibodies (donkey Alexa Fluor A594–conjugated anti-rabbit [IgG] pAb and donkey Alexa Fluor A488–conjugated anti-mouse [IgM] pAb). At the end of incubation, Hoechst 33342 was added to final concentration of 0.5 μg/ml for 2 minutes. Coverslips were washed 4 times for 5 to 10 min and mounted on microscope slides using ProLong Gold Antifade Reagent (Life Technologies).
Data processing and evaluation
All of the statistical analyses of experiments were performed by the SPSS 13 statistical package (IBM, Armonk, NY, USA). Calculations of enzyme kinetics were made by Enzyme Kinetics module 1.0 of the Sigma Plot 11.0 software.
RESULTS
Total proteasomes from the brains of EAE-SJL mice show a β1ihighβ5ilow phenotype
To determine if IPs accumulate in the CNS during EAE, we stained brain sections of BALB/c, nonimmunized SJL, and EAE-SJL mice for CP and IP subunits. Western blot analysis showed that both β1i and β5i expression was dramatically enhanced in all brain compartments of EAE-SJL mice compared to intact SJL and BALB/c mice (Fig. 1A). A statistically significant increase in the level of the β1i subunit was still evident in the cortex and striatum of intact SJL mice with an intact blood–brain barrier (BBB) (Fig. 1B). Immunization of BALB/c mice with MBP led to an elevated level of IP subunits only in compartments with increased BBB permeability (Fig. 1C). Therefore, both BALB/c and SJL mice showed induction of the IP upon MBP immunization; however, only SJL mice showed increased amounts of the IP across the BBB.
Figure 1.
CP-IP equilibrium is shifted to IP in brains of SJL mice in the course of autoimmune demyelination. A) Western blot analysis showing CP (β1/β5) and IP (β1i/β5i) subunit expression level in different brain regions of BALB/c, SJL, and EAE-SJL mice. B) Quantification of Western blot experiments (from A) by densitometry analysis. The median in each group of separate experiments (n = 3) is shown by a bold line; quartiles are displayed by bars; error bars represent 95% confidence interval. NS, not significant, *P < 0.05, **P < 0.01. C) Western blot analysis showing the expression level of IP subunits β1i and β5i in different brain regions of immunized and nonimmunized BALB/c mice. Data suggest an induction of inflammation and EAE upon MBP immunization in BALB/c and SJL mice, respectively.
During EAE, IP subunit β1i is increased in resident CNS cells whereas β5i is imported through the BBB
We then investigated the source of the IP in the mouse brain during autoimmune demyelination. Particularly, we wanted to determine whether IP-containing cells have migrated through the BBB or if they are resident CNS cells. Compared to nonimmunized SJL, EAE-SJL mice demonstrated more staining for β1i and less extensive staining for NeuN protein (Fig. 2A, B), a marker of neurons, linking neuronal damage to IP activity (19). Because the level of β1i was significantly increased compared to β5i in the brains of EAE mice, we stained brain sections of EAE-SJL mice for β1i and β5i immunosubunits, and we found that these proteins were not generally colocalized (Fig. 2C). Because the up-regulation of β1i was not linked to neurons, as seen in Fig. 2B, we stained brain sections with antibodies recognizing MBP and CD3, markers of ODCs and T cells, respectively (Fig. 2D). β1i was colocalized with MBP, indicating that β1i was up-regulated in ODCs. The β5i subunit was associated with infiltrating CD3-positive lymphocytes. Thus, the β1i subunit is predominantly expressed in ODCs in the CNS, while β5i appeared to be transported into the brain by lymphocytes.
Figure 2.

Accumulation of IP immunosubunits β1i and β5i in CNS has different etiology. A, B) Double immunostaining of cortex sections for IP subunit β1i (green) and marker of undamaged neurons, NeuN (red), in nonimmunized SJL (A) and EAE-SJL mice (B). Increased staining for β1i correlates with neuronal damage. C) Striatum sections of EAE-SJL mice immunostained for β1i (green)/β5i (red). D) Cortex sections of EAE-SJL mice immunostained for β1i (red)/MBP (green), β5i (red)/CD3 (green). β1i is colocalized with ODC marker MBP, whereas β5i is colocalized with T-cell marker CD3.
β1ihighβ5ilow IPs significantly alter MBP processing compared to CPs
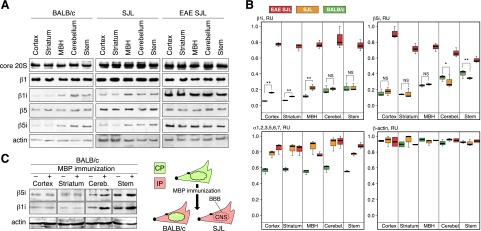
Next, we studied the pattern of highly purified bovine MBP (Supplemental Fig. S1A) processing by homogenous 26S proteasomes (Supplemental Fig. S1B) derived from total brain homogenates of BALB/c, nonimmunized SJL, and EAE-SJL mice. We also performed a comparative hydrolysis of MBP by number of multiple sclerosis–associated proteases (reviewed in ref. 20). MBP-derived hydrolyzates were analyzed with LC-ESI-MS (Fig. 3A) followed by label-free quantification. Neither enzymatic nor previously reported abzymatic (16, 21) patterns correlated with the observed proteasomal degradation. The enzymes generated peptides from the MBP region 110–130, whereas the proteasomes hydrolyzed MBP within all protein sequences. Among the enzymes tested, calpain displayed the closest specificity to the proteasomes, but it did not match the unique ability of the proteasomes to produce multiple MBP peptides. MS analysis revealed significant differences in the spectra of peptides produced by proteasomes from autoimmune and disease-free animals. Identified peptides generated by brain-derived proteasomes from EAE-SJL mice significantly overlapped with major T-cell epitopes of MBP, summarized in (20). Analysis of the length distribution demonstrated an enhanced ability of β1ihighβ5ilow IPs from EAE-SJL mice to generate 8 amino acid peptides (Fig. 3B), the length that is optimal for MHC class I loading. Label-free analysis showed a dramatic increase in the amount of the ENPVVHFF peptide in MBP hydrolyzates generated by proteasomes from EAE-SJL mice (Fig. 3C). A comparison of the patterns of MBP peptide degradation showed the absence of MBP fragment 46–62 in diseased and nonimmunized SJL mice, but not in normal mice. Interestingly, this peptide has previously been shown to have a therapeutic effect on rats developing EAE (22).
Figure 3.
Peptide patterns of MBP degradation by brain-derived CP/IP murine proteasomes and multiple sclerosis–associated proteases. A) Representative data from 2 LC-ESI-MS experiments showing MBP fragments generated by 26S proteasomes purified from BALB/c, SJL, and EAE-SJL mice versus trypsin, MMP-3, and MMP-9, and calpain. Arrows aligned according to MBP sequence represent the sequences of the respective peptide fragments. Width of the arrows represents amount of respective peptides according to label-free semiquantitative analysis. B) Length distribution of MBP peptides generated by various proteasomes. C) Label-free analysis of ENPVVHFF peptide generation by various proteasomes during in vitro MBP degradation.
β1ihighβ5ilow IPs from EAE-SJL mice generate elevated amounts of MBP peptides associated with pathogenic MHC class I molecules
We further quantitatively compared the spectrum of MHC class I restricted peptides generated during MBP hydrolysis without ubiquitin by brain-derived proteasomes isolated from BALB/c, intact SJL mice and SJL mice developing EAE. To accomplish this, we used quantitative comparative analysis of the peptides that resulted from the hydrolysis of MBP by various proteasome samples. This technique is based on the property of proteasomes to incorporate one oxygen atom from water into the C terminus of each peptide generated during protein digestion (Fig. 4A). We performed degradation of MBP by cerebral proteasomes from BALB/c mice in water containing oxygen with mass 18. Proteasomes from the brains of intact SJL and EAE-SJL mice hydrolyzed MBP in ordinary water. Concentration of proteasomes was adjusted in terms of LLVY-MCA activity equivalents. Digested samples of BALB/c-SJL and BALB/c-EAE-SJL were mixed pairwise in different molar ratios to compensate for the natural 13C/12C isotopic distribution and were analyzed by LC-MS/MS (Supplemental Fig. S2A–E). For each peptide generated by proteasomes from SJL mice, we detected the respective 18O-labeled peptide generated by proteasomes from BALB/c mice. The retention time and ionization efficiency of such peptides during LC-MS/MS analysis was equivalent as a result of their identical chemical structure. This approach resulted in quantitative analysis of more than 150 MBP-derived peptides generated by various proteasomes in a single experiment, from which we selected 5 peptides that matched MHC class I loading criteria (Fig. 4B). Among these peptides, we identified 2 octapeptides, DTGILDSL (MBP34–41) and ENPVVHFF (MBP83–90). The amount of these peptides in hydrolyzates generated by proteasomes from EAE-SJL mice was 10-fold greater compared to BALB/c proteasomes and 3 to 5 times greater compared to proteasomes from intact SJL mice.
Figure 4.

Brain-derived proteasomes from EAE-SJL mice generate MBP peptides recognized by MS-associated MHC class I molecules. A) Methodology of quantitative comparative analysis of peptides resulting from the hydrolysis of MBP by cerebral proteasomes from BALB/c mice in comparison with proteasomes from naive SJL and SJL mice developing EAE. MBP hydrolysis by cerebral proteasomes from BALB/c and SJL mice was performed in H218O and H216O, respectively, in order to quantitatively compare products of proteasome-mediated MBP degradation. B) Representative MS spectra (left) and quantitative estimation of the ratio of MHC class I–associated peptides in MBP hydrolysates (right) are shown. Data are shown as means ± sem (n = 3); **P < 0.01.
Peptide MBP83–90 is a part of the encephalitogenic MBP fragment (23). We thus performed a detailed study of its generation by cerebral proteasomes. In contrast to the majority of analyzed peptides, peptide MBP83–90 usually contained two C-terminal 18O atoms instead of one (Supplemental Fig. S2F). We assumed that this might be a result of a double hydrolytic reaction, possibly due to the enhanced affinity of this peptide to the active centers of the proteasome. This effect also complicated the calculations to compensate for the natural isotopic distribution. To overcome this difficulty, we synthesized a chemically identical peptide bearing a C-terminal phenylalanine in which all carbon and nitrogen atoms were of atomic weight 13 and 15, respectively, and used it as an internal standard in a time-course analysis of peptide MBP83–90 release (Fig. 5A and Supplemental Fig. S3). Remarkably, in line with the 16O/18O approach, quantitative LC-MS/MS analysis using the isotope-labeled peptide demonstrated an 8-fold increase of peptide MBP83–90 generation by cerebral proteasomes from EAE-SJL mice compared to nonimmunized SJL or BALB/c mice (Fig. 5B). Moreover, the proteasome from EAE-SJL mice produce up to 0.83 mol of this peptide per 1.0 mol of MBP in 24 hours, serving as a molecular machine to produce an MHC class I–restricted encephalitogenic MBP peptide epitope (Fig. 5C).
Figure 5.

Release of ENPVVHFF peptide is dramatically increased upon hydrolysis of MBP by cerebral proteasomes from EAE-SJL mice compared to nonimmunized SJL and BALB/c mice. Mass spectra (A), plotted time course (B) and 24-hour end point (C) of ENPVVHFF peptide release during MBP degradation by proteasomes isolated from control and autoimmune mice at the indicated time points monitored by an isotopically labeled internal standard. Data are presented as means ± sem (n = 2); **P < 0.01.
β1i-IP marks ODCs for CTL attack, and its inhibition ameliorates EAE in vivo
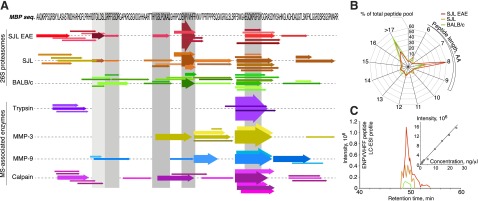
To clarify if peptide MBP83–90 has an ability to load on murine MHC class I molecules of H-2s haplotype, we isolated PBMCs from naive SJL (H-2s), C3H (H-2k), and BALB/c (H-2d) mice and incubated them with exogenous MBP83–90 peptide. According to the quantitative MRM analysis MBP83–90 was expectedly found in samples of cells of H-2k haplotype (5), whereas it was not detected in case of H-2d haplotype. Importantly, H-2s samples evidently contained MBP83–90 peptide (Fig. 6A). Further, we elucidated if there was IFN-γ–induced accumulation of MBP83–90 in mature ODCs culture from SJL mice. Using a similar mass spectrometric detection method, we showed that the content of MBP83–90 peptide in culture of mature ODCs is increased 5-fold upon IFN-γ treatment (Fig. 6A).
Figure 6.
IP subunit β1i is a specific target for treatment of autoimmune demyelination. A) Extracted ion chromatograms (monitoring of 5 transitions corresponding to the MBP83–90 peptide) obtained by MRM QTRAP mass spectrometry analysis of peptide extracts of SJL- and BALB/c-derived PBMCs loaded with MBP83–90 peptide are shown at left. Total ion current chromatography peak (18.3 min) obtained by MRM QTRAP mass spectrometry analysis of peptide extracts of SJL-, C3H-, and BALB/c-derived PBMCs loaded with MBP83–90 peptide (middle) and peptide extracts of mature ODCs cultures from C57BL/6 and SJL mice (right). B) Staining of mature ODC culture treated with or without IFN-γ for O4 ODC marker, core 20S subunits and β1i. HST, Hoechst 33342. C) Lysis of mature ODCs treated with or without IFN-γ by CTLs specific for MBP peptide ENPVVHFF (MBP83–90) and control peptide FNFTAPFI. Percentage of lysed target cells was estimated using DELFIA cytotoxicity assay. Anti–MHC class I Ab indicates ODCs treated with antibody specific for mouse MHC class I. Data are presented as means ± sem (n = 3); *P < 0.05, **P < 0.01. D) Activity of proteasomes from BALB/c, immunized, and nonimmunized SJL mice toward LLVY-MCA (top) and MBP (bottom) was monitored in the presence of proteasome inhibitors. Data are presented as means ± sem (n = 3). E) Mean disease scores of SJL mice developing EAE treated with β1i-PEk (0.5 mg/kg) and PS-341 (0.5 mg/kg) compared to untreated animals. Proteasome inhibitors or vehicle control were administrated twice weekly starting from day 0 after immunization, 7 times total. Error bars represent standard deviations.
To determine if the identified MBP peptides are recognized by CTLs ex vivo, we incubated mature ODCs with CTLs reactive to the peptide MBP83–90. In line with our in vivo studies, the IP catalytic subunit β1i was significantly up-regulated in ODCs subjected to IFN-γ treatment compared to untreated cells (Fig. 6B). CTLs reactive to peptide MBP83–90 were able to specifically lyse murine ODCs (Fig. 6C). The effectiveness of this attack was increased upon pretreatment of ODCs with IFN-γ and was inhibited by anti–MHC class I antibodies. To elucidate possible approaches to specifically inhibit the proteasome to suppress MBP proteolysis, we analyzed the effect of 3 inhibitors of proteolytic activity on isolated CP and IP in vitro: PS-341 (which affects all catalytic subunits of the CP and the IP), MG132 (an inhibitor of chymotrypsin-like activity), and a specific inhibitor of the β1i subunit, peptidyl epoxyketone (PEk), which irreversibly binds to the threonine in the active center of the β1i subunit. To determine the quantitative parameters of the proteasome-mediated proteolytic reaction, we analyzed the influence of these proteasome inhibitors on the hydrolysis of the model substrate Suc-LLVY-AMC (Supplemental Fig. S4). We found that MG132 and PS-341 proteasome inhibition activity was in line with previously published reports. We also determined the association constant kobs/[I] for β1i-PEk, which was found to be approximately 240 M−1s−1 (Table 2). PS-341 and MG132 showed similar effects that were not influenced by the CP/IP ratio, whereas proteasomes from EAE-SJL mice were significantly more inhibited by β1i-PEk than CPs purified from the brains of BALB/c mice (Fig. 6D). β1i-PEk blocked more than 70% of chymotrypsin-like activity of proteasomes from diseased SJL mouse brains, but only 15% of proteasome activity from nonimmunized SJL mice. No changes in chymotrypsin-like activity were observed in cerebral proteasomes from BALB/c mice treated with β1i-PEk at a concentration up to 100 μM. Further analysis revealed that the ability of proteasomes from brain tissues of EAE-SJL mice to process MBP peptide epitopes was significantly impeded by all 3 inhibitors (Fig. 6D). We further tested β1i-PEk and PS-341 in vivo on SJL mice developing EAE (Table 1). We found that β1i-PEk ameliorated ongoing autoimmune demyelination better than the previously tested PS-341 (26) at an equivalent dose (Fig. 6E).
TABLE 2.
Inhibition of CP and IP Chymotrypsine-Like activity
| Brain-derived 26S | PS-341 Ki, nMa | MG-132 Ki, nMb | β1i-PEk kobs/[I], M−1s−1 |
|---|---|---|---|
| BALB/c | 7 ± 2 | 28 ± 8 | Not defined |
| Naïve SJL | 10 ± 3 | 35 ± 8 | 230 ± 60 |
| EAE-SJL | 6 ± 2 | 26 ± 4 | 250 ± 40 |
DISCUSSION
Both multiple sclerosis and EAE are characterized by inflammation of the CNS, resulting in a massive release of IFN-γ (27), which increases the level of IPs in the diseased brain (28, 29). Our data suggest that resident proteasomes from EAE-SJL mice undergo structural changes, resulting in a different proteolytic activity that accounts for the distinct pattern of MBP degradation. The replacement of β1 by β1i leads to the enhancement of chymotrypsin-like activity (30) and to a reduction of caspase-like activity (31). Chymotrypsin-like activity of the proteasome results in the generation of peptides with hydrophobic C-terminal residues that facilitate MHC class I binding (32), whereas caspase-like activity may be responsible for destruction of CTL epitopes (33). The fact that the β1i subunit is highly expressed in ODCs indicates that the processing and presentation of neuroantigens may occur inside the brain, resulting in the activation of peripheral bystander antigen-experienced T cells that recognize neuroantigens and trigger neuronal autoimmunity. Importantly, our data are in agreement with recent findings showing that a mutated β1i IP subunit in an Italian female population alters MBP processing and reduces the risk of multiple sclerosis (28). Although Frausto and colleagues (34) reported that C57BL/6 β1i-knockout mice were still susceptible to EAE, it should be noted that a functional analysis of proteasome immunosubunits in the knockout mice was generally disappointing. Studies over the last 2 decades clearly revealed that the requirement for immunoproteasomes for pathogen elimination or autoimmune response varies markedly between animal models and further studying of immunoproteasome-deficient mice is necessary for complete understanding of the contribution of individual proteasome subunits to the immune response to infectious or self-antigens (35). The absence of H-2KS MHC class I in C57BL/6 mice and the induction of EAE using myelin oligodendrocyte glycoprotein (MOG) may decrease the role of the β1i subunit in EAE and preclude the presentation of MBP encephalitogenic peptides, even though they may be produced. Thus, development of EAE in β1i−/− C57BL/6 mice does not suggest that this particular immunosubunit plays no role in EAE development in EAE models other than C57BL/6.
In the present study, we showed that etiology of proteasome immunosubunits observed in CNS is different. The origin of β1i seems to be cerebral, whereas β5i is imported by lymphocytes and apparently other immune cells like macrophages through the BBB. β5i expression in lymphocytes stimulates their survival and controls their proliferative capacity, so the presence of β5i in brain-infiltrating lymphocytes likely facilitates T cell–mediated neuronal damage (36). One study showed a 20% to 30% reduced number of CD8+ compared with CD4+ T cells in lymphoid organs of β1i−/− mice (37). As was demonstrated later, these effects are thought to be a result of defective intrathymic development (38). It seems unlikely that administration of β1i inhibitor, which is evidently significantly less efficient compared to knockout of β1i, may change CD8+ T-cell repertoire, especially affect intrathymic development in adult mice, when autoreactive CD8+ T cell clones already exist. Thus, amelioration of EAE using β1i-PEk, successfully accomplished in the present study, looks very promising, as it touches ODCs but not immune cells. Nonetheless, a number of studies report that β1i−/− lymphocytes, compared to the β2i−/− or β5i−/− phenotype, proliferate normally but still may have defects in survival (39, 40). Therefore, we suggest that the therapeutic efficacy of β1i-PEk is mainly linked to an inhibition of β1i in CNS resident cells, but at least may be partially affected by systematic suppression of immune system.
Here, we showed that 2 peptides (Table 3), MBP83–90 and MBP34–41 (the numbering hereafter is given according to the 170 amino acid isoform of human MBP), increased 10-fold in MBP hydrolysates produced by proteasomes from EAE-SJL mice compared to those from BALB/c mice. Remarkably, adoptively transferred CTLs that induce severe EAE in C3H mice (5) showed a pronounced specificity toward MBP82–90. Both of these peptides have hydrophobic C-terminal residues and can be efficiently loaded directly into the peptide binding cleft of MHC class I molecules without additional processing by ERAAP/ERAP2 in the endoplasmic reticulum. The human multiple sclerosis–associated MHC class I molecules B44, B35, and A2 present very similar MBP-derived peptides, MBP81–89, MBP84–93, and MBP86–94, respectively (41). MBP34–44 and MBP35–44 were previously shown to be presented on MHC class I molecules in multiple sclerosis patients (42). Importantly, our data revealed that SJL-derived murine CTLs specific for MBP83–90 directly target IFN-γ–treated mature ODCs ex vivo, strongly suggesting a pathophysiologic relevance of our findings. Proteasomes used for quantitative MS studies of MBP degradation were adjusted in terms of chymotrypsin-like activity that should basically compensate recently reported difference in rate of processing of basic proteins by CP and IP (43). Moreover, we showed that the majority of peptides were equally presented in MBP hydrolysates regardless the source of proteasomes (Figs. 3 and 4), and that the amount of MBP peptide MBP83–90 was evidently less in dynamically monitored CP-mediated hydrolysates compared to IP even at the time of complete MBP hydrolysis (Fig. 5). Finally, both MBP peptides (MBP34–41 and MBP83–90) have hydrophobic C-terminal residues (leucine and phenylalanine, respectively). Thus, a more realistic explanation coming from the study by Raule et al. (43) is an 8-times increase in the Vmax of chymotrypsin-like activity of IP, measured using Ala-Ala-Phe-4-methylcoumaryl-7-amide substrate. We thus link our observations with an altered specificity of the proteasomal catalytic immunosubunits (44, 45), rather than with the recently suggested higher turnover of the immunoproteasome (46). However, at the present time, we cannot exclude the possibility that IP hydrolyzes basic proteins faster (43), especially in vivo (47).
TABLE 3.
Pathophysiologic significance of MBP peptides generated in elevated amounts by brain-derived proteasomes from EAE-SJL mice
| ENPVVHFF | ||
|---|---|---|
| Immunoproteasome generateda | ||
| Targeted by CTLb | DENPVVHFF | |
| Presented by multiple sclerosis–associated MHC class Ic | B44 | QDENPVVHF |
| B35 | NPVVHFFKNI | |
| A2 | VVHFFKNIV | |
| Immunoproteasome generateda | DTGILDSL | |
| Eluted from multiple sclerosis MHC class Id | DTGILDSIGRF | |
| TGILDSIGRF |
On the bases of these and other available experimental data, we propose the following sequence of events as a mechanism explaining how the IP is mechanistically involved in the pathogenesis of autoimmune demyelination (Fig. 7). The development of an inflammatory reaction in the brain, particularly the production of IFN-γ (27), induces the expression of the β1i subunit in ODCs and leads to efficient production of MBP peptides recognizable by CD8+ CTLs. Additionally, inflammation in CNS may prevent ODC-mediated purge of repertoire of autoreactive CD8+ T cells, as was recently shown by Na et al. (48). Further elevation of the IP mediated by increased production of IFN-γ and subsequent processing of MBP may function as a positive feedback loop, exacerbating disease progression. Experiments with ODC-OVA double-transgenic mice have shown that without an initial attack of CD8+ CTLs on ODCs that exclusively express the OVA antigen, the ODC-OVA cells remain invisible to CD4+ T cells. Furthermore, autoreactive CD8+ T cells may spontaneously and lethally attack neuronal cells without any cross-presentation (49). These observations suggest that even a slightly increased basal level of presentation of immunodominant MHC class I restricted peptides in the brain may trigger the priming of autoreactive CD8+ T cells. Therefore, according to our data, the predisposition to an autoimmune reaction in SJL mice can be explained, at least in part, by the increased basal IP expression in the CNS.
Figure 7.

Mechanistic explanation of IP involvement in the pathogenesis of autoimmune demyelination (see Discussion).
CONCLUSIONS
As we showed previously, no member of the ubiquitin family is capable of regulating MBP degradation by the proteasome (15). The pathophysiologic importance of our current findings lies in the fact that while uncontrolled by the ubiquitination system, IP-mediated MBP proteolysis under autoimmune conditions may be a cornerstone event that results in the overproduction of MHC class I–restricted peptides, marking ODCs for attack by CTLs. Therefore, the selective inhibition of the resident CNS β1i immunoproteasome by drugs may be a useful therapeutic intervention in multiple sclerosis.
Supplementary Material
Acknowledgments
The reported study was performed under the frame of Russian Scientific Foundation project Grant 14-14-00585 “Molecular mechanism and physiologic significance of the ubiquitin-independent proteasomal degradation of the proteins,” partially supported by Russian Foundation for Basic Research Grant 13-04-40277-H and Fellowship CΠ 2445.2013.4 (to A.B.). The work was performed according to the Russian Government Program of Competitive Growth of Kazan Federal University.
Glossary
- BBB
blood–brain barrier
- CP
constitutive proteasome
- CTL
cytotoxic lymphocyte
- EAE
experimental autoimmune encephalomyelitis
- ESI-MS
electrospray ionization mass spectrometry
- IP
immunoproteasome
- LMP
low molecular mass polypeptide
- MBP
myelin basic protein
- MHC
major histocompatibility complex
- ODC
oligodendrocyte
- PEk
peptidyl epoxyketone
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Langrish C. L., Chen Y., Blumenschein W. M., Mattson J., Basham B., Sedgwick J. D., McClanahan T., Kastelein R. A., Cua D. J. (2005) IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201, 233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Panitch H. S., Hirsch R. L., Haley A. S., Johnson K. P. (1987) Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet 1(8538), 893–895 [DOI] [PubMed] [Google Scholar]
- 3.Hauser S. L., Bhan A. K., Gilles F., Kemp M., Kerr C., Weiner H. L. (1986) Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann. Neurol. 19, 578–587 [DOI] [PubMed] [Google Scholar]
- 4.Steinman L. (2001) Myelin-specific CD8 T cells in the pathogenesis of experimental allergic encephalitis and multiple sclerosis. J. Exp. Med. 194, F27–F30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huseby E. S., Liggitt D., Brabb T., Schnabel B., Ohlén C., Goverman J. (2001) A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J. Exp. Med. 194, 669–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arrigo A. P., Tanaka K., Goldberg A. L., Welch W. J. (1988) Identity of the 19S “prosome” particle with the large multifunctional protease complex of mammalian cells (the proteasome). Nature 331, 192–194 [DOI] [PubMed] [Google Scholar]
- 7.Ciechanover A., Heller H., Katz-Etzion R., Hershko A. (1981) Activation of the heat-stable polypeptide of the ATP-dependent proteolytic system. Proc. Natl. Acad. Sci. USA 78, 761–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaczynska M., Rock K. L., Goldberg A. L. (1993) Gamma-interferon and expression of MHC genes regulate peptide hydrolysis by proteasomes. Nature 365, 264–267 [DOI] [PubMed] [Google Scholar]
- 9.Laatsch R. H., Kies M. W., Gordon S., Alvord E. C. Jr (1962) The encephalomyelitic activity of myelin isolated by ultracentrifugation. J. Exp. Med. 115, 777–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pette M., Fujita K., Wilkinson D., Altmann D. M., Trowsdale J., Giegerich G., Hinkkanen A., Epplen J. T., Kappos L., Wekerle H. (1990) Myelin autoreactivity in multiple sclerosis: recognition of myelin basic protein in the context of HLA-DR2 products by T lymphocytes of multiple-sclerosis patients and healthy donors. Proc. Natl. Acad. Sci. USA 87, 7968–7972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Webb C., Teitelbaum D., Abramsky O., Arnon R., Sela M. (1974) Lymphocytes sensitised to basic encephalitogen in patients with multiple sclerosis unresponsive to steroid therapy. Lancet 2(7872), 66–69 [DOI] [PubMed] [Google Scholar]
- 12.Oksenberg J. R., Panzara M. A., Begovich A. B., Mitchell D., Erlich H. A., Murray R. S., Shimonkevitz R., Sherritt M., Rothbard J., Bernard C. C., et al. (1993) Selection for T-cell receptor V beta-D beta-J beta gene rearrangements with specificity for a myelin basic protein peptide in brain lesions of multiple sclerosis. Nature 362, 68–70 [DOI] [PubMed] [Google Scholar]
- 13.Jurewicz A., Biddison W. E., Antel J. P. (1998) MHC class I–restricted lysis of human oligodendrocytes by myelin basic protein peptide-specific CD8 T lymphocytes. J. Immunol. 160, 3056–3059 [PubMed] [Google Scholar]
- 14.Sobottka B., Harrer M. D., Ziegler U., Fischer K., Wiendl H., Hünig T., Becher B., Goebels N. (2009) Collateral bystander damage by myelin-directed CD8+ T cells causes axonal loss. Am. J. Pathol. 175, 1160–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belogurov A. Jr., Kudriaeva A., Kuzina E., Smirnov I., Bobik T., Ponomarenko N., Kravtsova-Ivantsiv Y., Ciechanover A., Gabibov A. (2014) Multiple sclerosis autoantigen myelin basic protein escapes control by ubiquitination during proteasomal degradation. J. Biol. Chem. 289, 17758–17766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ponomarenko N. A., Durova O. M., Vorobiev I. I., Belogurov A. A. Jr., Kurkova I. N., Petrenko A. G., Telegin G. B., Suchkov S. V., Kiselev S. L., Lagarkova M. A., Govorun V. M., Serebryakova M. V., Avalle B., Tornatore P., Karavanov A., Morse H. C. III, Thomas D., Friboulet A., Gabibov A. G. (2006) Autoantibodies to myelin basic protein catalyze site-specific degradation of their antigen. Proc. Natl. Acad. Sci. USA 103, 281–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho Y. K. A., Bargagna-Mohan P., Wehenkel M., Mohan R., Kim K.-B. (2007) LMP2-specific inhibitors: chemical genetic tools for proteasome biology. Chem. Biol. 14, 419–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenberg P. A., Dai W., Gan X. D., Ali S., Fu J., Back S. A., Sanchez R. M., Segal M. M., Follett P. L., Jensen F. E., Volpe J. J. (2003) Mature myelin basic protein-expressing oligodendrocytes are insensitive to kainate toxicity. J. Neurosci. Res. 71, 237–245 [DOI] [PubMed] [Google Scholar]
- 19.Collombet J.-M., Masqueliez C., Four E., Burckhart M. F., Bernabé D., Baubichon D., Lallement G. (2006) Early reduction of NeuN antigenicity induced by soman poisoning in mice can be used to predict delayed neuronal degeneration in the hippocampus. Neurosci. Lett. 398, 337–342 [DOI] [PubMed] [Google Scholar]
- 20.Belogurov A. Jr., Kozyr A., Ponomarenko N., Gabibov A. (2009) Catalytic antibodies: balancing between Dr. Jekyll and Mr. Hyde. BioEssays 31, 1161–1171 [DOI] [PubMed] [Google Scholar]
- 21.Belogurov A. A. Jr., Kurkova I. N., Friboulet A., Thomas D., Misikov V. K., Zakharova M. Y., Suchkov S. V., Kotov S. V., Alehin A. I., Avalle B., Souslova E. A., Morse H. C. III, Gabibov A. G., Ponomarenko N. A. (2008) Recognition and degradation of myelin basic protein peptides by serum autoantibodies: novel biomarker for multiple sclerosis. J. Immunol. 180, 1258–1267 [DOI] [PubMed] [Google Scholar]
- 22.Belogurov A. A. Jr., Stepanov A. V., Smirnov I. V., Melamed D., Bacon A., Mamedov A. E., Boitsov V. M., Sashchenko L. P., Ponomarenko N. A., Sharanova S. N., Boyko A. N., Dubina M. V., Friboulet A., Genkin D. D., Gabibov A. G. (2013) Liposome-encapsulated peptides protect against experimental allergic encephalitis. FASEB J. 27, 222–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin R., Howell M. D., Jaraquemada D., Flerlage M., Richert J., Brostoff S., Long E. O., McFarlin D. E., McFarland H. F. (1991) A myelin basic protein peptide is recognized by cytotoxic T cells in the context of four HLA-DR types associated with multiple sclerosis. J. Exp. Med. 173, 19–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gardner R. C., Assinder S. J., Christie G., Mason G. G., Markwell R., Wadsworth H., McLaughlin M., King R., Chabot-Fletcher M. C., Breton J. J., Allsop D., Rivett A. J. (2000) Characterization of peptidyl boronic acid inhibitors of mammalian 20 S and 26 S proteasomes and their inhibition of proteasomes in cultured cells. Biochem. J. 346(Pt 2), 447–454 [PMC free article] [PubMed] [Google Scholar]
- 25.Rock K. L., Gramm C., Rothstein L., Clark K., Stein R., Dick L., Hwang D., Goldberg A. L. (1994) Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78:761–771 [DOI] [PubMed] [Google Scholar]
- 26.Fissolo N., Kraus M., Reich M., Ayturan M., Overkleeft H., Driessen C., Weissert R. (2008) Dual inhibition of proteasomal and lysosomal proteolysis ameliorates autoimmune central nervous system inflammation. Eur. J. Immunol. 38, 2401–2411 [DOI] [PubMed] [Google Scholar]
- 27.Steinman L. (1996) Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell 85, 299–302 [DOI] [PubMed] [Google Scholar]
- 28.Mishto M., Bellavista E., Ligorio C., Textoris-Taube K., Santoro A., Giordano M., D’Alfonso S., Listì F., Nacmias B., Cellini E., Leone M., Grimaldi L. M., Fenoglio C., Esposito F., Martinelli-Boneschi F., Galimberti D., Scarpini E., Seifert U., Amato M. P., Caruso C., Foschini M. P., Kloetzel P. M., Franceschi C. (2010) Immunoproteasome LMP2 60HH variant alters MBP epitope generation and reduces the risk to develop multiple sclerosis in Italian female population. PLoS ONE 5, e9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng J., Dasgupta A., Bizzozero O. A. (2012) Changes in 20S subunit composition are largely responsible for altered proteasomal activities in experimental autoimmune encephalomyelitis. J. Neurochem. 121, 486–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cardozo C., Kohanski R. A. (1998) Altered properties of the branched chain amino acid-preferring activity contribute to increased cleavages after branched chain residues by the “immunoproteasome.” J. Biol. Chem. 273, 16764–16770 [DOI] [PubMed] [Google Scholar]
- 31.Schmidtke G., Eggers M., Ruppert T., Groettrup M., Koszinowski U. H., Kloetzel P. M. (1998) Inactivation of a defined active site in the mouse 20S proteasome complex enhances major histocompatibility complex class I antigen presentation of a murine cytomegalovirus protein. J. Exp. Med. 187, 1641–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Groll M., Larionov O. V., Huber R., de Meijere A. (2006) Inhibitor-binding mode of homobelactosin C to proteasomes: new insights into class I MHC ligand generation. Proc. Natl. Acad. Sci. USA 103, 4576–4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Basler M., Lauer C., Moebius J., Weber R., Przybylski M., Kisselev A. F., Tsu C., Groettrup M. (2012) Why the structure but not the activity of the immunoproteasome subunit low molecular mass polypeptide 2 rescues antigen presentation. J. Immunol. 189, 1868–1877 [DOI] [PubMed] [Google Scholar]
- 34.Frausto R. F., Crocker S. J., Eam B., Whitmire J. K., Whitton J. L. (2007) Myelin oligodendrocyte glycoprotein peptide-induced experimental allergic encephalomyelitis and T cell responses are unaffected by immunoproteasome deficiency. J. Neuroimmunol. 192, 124–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Groettrup M., Kirk C. J., Basler M. (2009) Proteasomes in immune cells: more than peptide producers?. Nat. Rev. Immunol. 10, 73–78 [DOI] [PubMed] [Google Scholar]
- 36.Caudill C. M., Jayarapu K., Elenich L., Monaco J. J., Colbert R. A., Griffin T. A. (2006) T cells lacking immunoproteasome subunits MECL-1 and LMP7 hyperproliferate in response to polyclonal mitogens. J. Immunol. 176, 4075–4082 [DOI] [PubMed] [Google Scholar]
- 37.Van Kaer L., Ashton-Rickardt P. G., Eichelberger M., Gaczynska M., Nagashima K., Rock K. L., Goldberg A. L., Doherty P. C., Tonegawa S. (1994) Altered peptidase and viral-specific T cell response in LMP2 mutant mice. Immunity 1, 533–541 [DOI] [PubMed] [Google Scholar]
- 38.Chen W., Norbury C. C., Cho Y., Yewdell J. W., Bennink J. R. (2001) Immunoproteasomes shape immunodominance hierarchies of antiviral CD8(+) T cells at the levels of T cell repertoire and presentation of viral antigens. J. Exp. Med. 193, 1319–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moebius J., van den Broek M., Groettrup M., Basler M. (2010) Immunoproteasomes are essential for survival and expansion of T cells in virus-infected mice. Eur. J. Immunol. 40, 3439–3449 [DOI] [PubMed] [Google Scholar]
- 40.Hensley S. E., Zanker D., Dolan B. P., David A., Hickman H. D., Embry A. C., Skon C. N., Grebe K. M., Griffin T. A., Chen W., Bennink J. R., Yewdell J. W. (2010) Unexpected role for the immunoproteasome subunit LMP2 in antiviral humoral and innate immune responses. J. Immunol. 184, 4115–4122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berthelot L., Laplaud D. A., Pettré S., Ballet C., Michel L., Hillion S., Braudeau C., Connan F., Lefrère F., Wiertlewski S., Guillet J. G., Brouard S., Choppin J., Soulillou J. P. (2008) Blood CD8+ T cell responses against myelin determinants in multiple sclerosis and healthy individuals. Eur. J. Immunol. 38, 1889–1899 [DOI] [PubMed] [Google Scholar]
- 42.Fissolo N., Haag S., de Graaf K. L., Drews O., Stevanovic S., Rammensee H. G., Weissert R. (2009) Naturally presented peptides on major histocompatibility complex I and II molecules eluted from central nervous system of multiple sclerosis patients. Mol. Cell. Proteomics 8, 2090–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raule M., Cerruti F., Cascio P. (2014) Enhanced rate of degradation of basic proteins by 26S immunoproteasomes. Biochim. Biophys. Acta. 1843, 1942–1947 [DOI] [PubMed] [Google Scholar]
- 44.Nathan J. A., Spinnenhirn V., Schmidtke G., Basler M., Groettrup M., Goldberg A. L. (2013) Immuno- and constitutive proteasomes do not differ in their abilities to degrade ubiquitinated proteins. Cell 152, 1184–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guillaume B., Chapiro J., Stroobant V., Colau D., Van Holle B., Parvizi G., Bousquet-Dubouch M. P., Théate I., Parmentier N., Van den Eynde B. J. (2010) Two abundant proteasome subtypes that uniquely process some antigens presented by HLA class I molecules. Proc. Natl. Acad. Sci. USA 107, 18599–18604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seifert U., Bialy L. P., Ebstein F., Bech-Otschir D., Voigt A., Schröter F., Prozorovski T., Lange N., Steffen J., Rieger M., Kuckelkorn U., Aktas O., Kloetzel P. M., Krüger E. (2010) Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 142, 613–624 [DOI] [PubMed] [Google Scholar]
- 47.Kuzina E. S., Chernolovskaya E. L., Kudriaeva A. A., Zenkova M. A., Knorre V. D., Surina E. A., Ponomarenko N. A., Bobik T. V., Smirnov I. V., Bacheva A. V., Belogurov A. A., Gabibov A. G., Vlasov V. V. (2013) Immunoproteasome enhances intracellular proteolysis of myelin basic protein. Dokl. Biochem. Biophys. 453, 300–303 [DOI] [PubMed] [Google Scholar]
- 48.Na S. Y., Hermann A., Sanchez-Ruiz M., Storch A., Deckert M., Hünig T. (2012) Oligodendrocytes enforce immune tolerance of the uninfected brain by purging the peripheral repertoire of autoreactive CD8+ T cells. Immunity 37, 134–146 [DOI] [PubMed] [Google Scholar]
- 49.Na S. Y., Cao Y., Toben C., Nitschke L., Stadelmann C., Gold R., Schimpl A., Hünig T. (2008) Naive CD8 T-cells initiate spontaneous autoimmunity to a sequestered model antigen of the central nervous system. Brain 131, 2353–2365 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.