

Summary
Background
Red blood cell (RBC) variants protect African children from severe Plasmodium falciparum malaria. Their individual and interactive impacts on mild disease and parasite density, and their modification by age-dependent immunity, are poorly understood.
Methods
We conducted a 4-year, prospective cohort study of children aged 0.5–17 years in Maliin 2008-2011. Exposures were haemoglobin S (HbS), HbC, α-thalassaemia, ABO blood groups, and glucose-6-phosphate dehydrogenase (G6PD)deficiency encoded by the X-linked A- allele. Primary and secondary outcomes were malaria incidence and parasite density. Incidence rate ratios (IRRs) were modeled with quasi-Poisson regression; parasite densities were analyzed with Generalized Estimating Equations.
Findings
We diagnosed 4091 malaria episodes in 1543 children over 2656 child-years of follow-up (cyfu). RBC variants were common: HbAS 14.2%, HbAC 6.7%, α-thalassaemia 28.4%, type O blood group 40.2%, and G6PD deficiency9.4% (boys) and 20.4% (girls). Malaria incidence was 1.54 episodes/cyfu, ranged from 2.78 at age 3 to 0.40 at age 16 years, was reduced 34% in HbAS vs HbAA children (adjusted IRR [aIRR] 0.66; 95% CI 0.59-0.75) and 49% in G6PD A-/A- vs A+/A+ girls (aIRR 0.51; 95% CI 0.29-0.90), but was increased 15% in HbAC children (aIRR 1.15; 95% CI 1.01-1.32). Parasite density was reduced in HbAS vs HbAA children (median 10,550 vs 15, 150 parasites/μL; p=0.0004). HbAS-associated reductions in malaria risk and parasite density were greatest in early childhood.
Interpretation
Individual and interactive impacts of HbAS, HbAC, and G6PD A-/A-on malaria risk and parasite density define clinical and cellular correlates of protection. Further identification of the molecular mechanisms of these protective effects may uncover novel targets for intervention.
Funding
Intramural Research Program, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Introduction
Human red blood cell (RBC) variants are encoded by common genetic mutations that alter the structure of β-globins (haemoglobin S [HbS] and HbC), reduce the expression of α- or β-globins (thalassaemias), or decrease the activity of essential enzymes (glucose-6-phosphate dehydrogenase [G6PD] deficiency). RBCs are further diversified by variation in surface antigens, including those that define the ABO, Duffy, and Rhesus blood groups.
This RBC diversity is partially driven by malaria caused by Plasmodium falciparum, which invades and matures within RBCs. To varying degrees, several RBC variants protect African children from life-threatening falciparum malaria. In clinical studies across multiple populations, sickle-cell trait (HbAS) and Hb Chomozygosity (HbCC) reduce this risk by 70-90%; lesser reductions in risk are reported for Hb Cheterozygosity (HbAC), α-thalassaemia, type O blood group, and G6PDdeficiency.1-4 These RBC variants confer protection by molecular mechanisms that remain under active investigation. Since these mechanisms reproducibly attenuate malaria pathogenesis, RBC variants constitute a naturally-occurring, in-vivo model of protection that can be used to investigate how to antagonize the detrimental effects of malaria parasites. In doing so, we may identify novel targets for preventive measures and adjunct therapies to reduce the estimated 437,000 African children who die annually of falciparum malaria.5
To investigate the individual and interactive effects of RBC variants on the clinical epidemiology of falciparum malaria, we conducted the Kenieroba Innate Defense Study for Malaria (KIDS-Malaria). In this 4-year, prospective cohort study of 1543Malian children, we hypothesized that RBC variants – alone and in combination – differentially impact malaria risk and parasite densities. We tested these hypotheses using multivariate models including each RBC variant and adjusting for age, sex, ethnicity, and year. Furthermore, we anticipated that the effects of RBC variants on these outcomes are modified by age, which is a strong surrogate for naturally-acquired immunity in malaria-hyperendemic areas of Africa.
Methods
Participants and setting
The KIDS-Malaria cohort comprises children enrolled in a prospective study between 2008 and 2011 in the adjacent villages of Kenieroba, Fourda, and Bozokin in southern Mali, where P. falciparumis transmitted from June to December. Written informed consent was obtained from parents or guardians. The study was approved by the Ethics Committee of the Faculty of Medicine, Pharmacy, and Dentistry at the University of Bamako, and the Institutional Review Board at the National Institute of Allergy and Infectious Diseases (NIAID), US National Institutes of Health (NIH). The study is registered with Clinicaltrials.gov, number NCT00669084.
Beginning May 1, 2008, 1312 children were initially enrolled; children were subsequently enrolled into the cohort at age 6 months and removed at age 18 years (figure 1). Inclusion criteria were age 6 months to 17 years, lifelong residency in the three villages, and no plans to relocate before 2012; exclusion criteria were conditions that rendered the child unable to comply with the protocol (e.g., psychiatric disease) or posed unnecessary risks to the child (e.g., severe malnutrition). No sample size was predefined, although a priori power calculations were done assuming 1000 children would be included (appendix); we recruited as many children as possible without a formal census.
Figure 1.
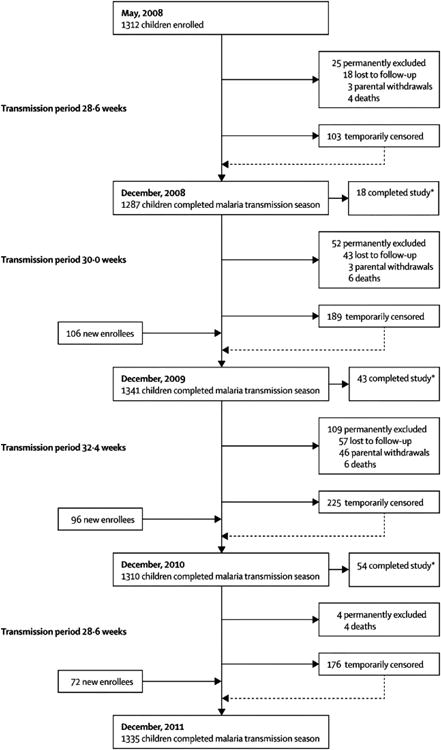
Child enrollment and follow-up. We enrolled a total of 1586 children in the KIDS-Malaria cohort: 1312 children during initial enrollment in May 2008, and 274 who aged into the study in subsequent years. Of these 1586 children, 1335 (84.2%) completed follow-up through the end of the 2011 transmission season. The major reasons for 251 children not completing follow-up were relocation (47.0%), attaining 18 years of age (24.3%), parental withdrawal (20.7%), and death from any cause (8.0%).
Outcome assessment
Case detection was passive; all parents were routinely encouraged to attend clinic for evaluation of childhood fever or other malaria symptoms. Outside our study clinic, health care options for evaluating fever and other malaria symptoms were essentially confined to visiting traditional healers, who worked closely with us to identify malaria patients and refer them to our study. Giemsa-stained thick blood films were prepared and examined on site, and asexual parasites were counted while also counting 300 leukocytes. Parasite density was defined as the number of parasites per 300 leukocytes multiplied by 25 (which assumes 7500 leukocytes/μL in whole blood).
We defined falciparum malaria as axillary temperature >37.5°C (or history of fever within 24 h) and a sexual P. falciparum parasitaemia, without other obvious causes of fever. We used World Health Organization criteria6 to define episodes as “major-severe” if the child had cerebral malaria, severe malarial anaemia, or respiratory distress; or “minor-severe” if the child had a parasite density >100,000 parasites/μL or needed parenteral therapy due to prostration, repetitive vomiting, or inability to tolerate oral therapy (appendix). Children without parasitemia were managed at the discretion of the study physician; these maneuvers and outcomes in children without malaria were not captured by our protocol. Children with malaria were treated with either oral artesunate-amodiaquine or parenteral quinine (appendix).
Laboratory procedures
At enrollment, all children provided a finger-prick blood sample for phenotyping and genotyping RBC variants. ABO blood groups were identified in the villages by agglutination assay (Cardinal Health, Dublin, OH); β-globin variants were determined at the University of Bamako by HPLC (D-10 instrument, Bio-Rad, Hercules, CA); and α-globin deletions (-α3.7kb) and G6PD A- alleles were detected at the NIH using PCR-based assays, as described.7, 8
Statistical analyses
Time at risk for each study participant was quantified only during the malaria season, accounted for temporary or permanent censoring (appendix), and is expressed as child-years of follow-up (cyfu). We investigated malaria incidence rates using quasi-Poisson regression models and parasite densities using a log-transformed linear model with Generalized Estimating Equations (GEE). For each type of model, we tested covariates after adjusting for either age alone or a multivariate model. We investigated effect modification of HbAS on incidence and density by age by stratifying the models by age and re-calculating effect estimates. Absolute risk reductions (ARRs) of incidence in age-stratified groups by HbAS were computed using difference in incidence rates using Wald confidence intervals based on Poisson models, and events averted by HbAS were computed as Events avertedAS = IRAA×cyfuAS – Events observedAS. We assessed for effect modification of HbAS and HbAC on incidence and density by α-thalassaemia by stratifying the quasi-Poisson regression models and GEEs of HbAA, HbAS, and HbAC children by α+-thalassaemia (-α3.7/α α) and α0-thalassaemia (-α3.7/-α3.7) and re-calculating effect estimates. The overall epistasis effect was tested by F test for quasi-Poisson or score test for GEE for comparing the model with only β-globin and α-thalassaemia main effects to one that additionally included the interaction effect. Inferences for severe vs mild malaria used GEE logistic regression. Software and additional details are found in the appendix.
Role of the funding source
The sponsor had no role in study design, data collection, data analysis, data interpretation, or writing of the report. The corresponding author had full access to all data in the study and the final responsibility for the decision to submit for publication.
Results
We successfully typed all five RBC variants for 1543 (97.3%) children, who thus constituted the analyzable population (table 1). Boys and girls were represented equally and the mean age at enrollment was 6 years, reflecting the recurring enrollment of newborns at age 6 months. Most children were Malinke (86.3%);relatively few were Fulani (7.8%). The prevalences of RBC variants were: HbAS 14.2%, HbAC 6.7%, HbCC 0.1%, HbSC 0.7%, α+-thalassaemia 26.2%, α0-thalassaemia2.2%, type O blood group40.2%, and G6PD A- deficiency 9.4% in boys and 20.4% in girls. The gender balance, ethnic makeup, and prevalence of these RBC variants in our study population were broadly reflective of southern Malian populations.
Table 1. Characteristics of 1543 child participants.
| Village, n (%) | |
| Kenieroba | 1178 (76.3) |
| Fourda | 180 (11.7) |
| Bozokin | 185 (12.0) |
|
| |
| Ethnicity*, n (%) | |
| Malinke | 1332 (86.3) |
| Fulani | 121 (7.8) |
| Bambara | 62 (4.0) |
| Sarakole | 24 (1.6) |
| Dogon | 4 (0.3) |
|
| |
| Sex, n (%) | |
| Boys | 767 (49.7) |
| Girls | 776 (50.3) |
|
| |
| Age at enrollment, y, mean (SD) | 6 (5.1) |
|
| |
| β-globin variant, n (%) | |
| HbAA | 1206 (78.2) |
| HbAS | 220 (14.2) |
| HbAC | 103 (6.7) |
| HbCC | 1 (0.1) |
| HbSC | 11 (0.7) |
| HbSS | 2 (0.1) |
|
| |
| α-globin variant, n (%) | |
| Normal (αα/αα) | 1105 (71.6) |
| α+-thalassaemia (-α3.7/αα) | 404 (26.2) |
| α0-thalassaemia (-α3.7/-α3.7) | 34 (2.2) |
|
| |
| Blood group, n (%) | |
| A | 465 (30.1) |
| B | 334 (21.7) |
| AB | 123 (8.0) |
| O | 621 (40.2) |
|
| |
| G6PD A- genotype, n (%) | |
| Boys | |
| Normal | 695 (45.0) |
| A- hemizygotes | 72 (4.7) |
| Girls | |
| Normal | 618 (40.1) |
| A-/A+ heterozygotes | 145 (9.4) |
| A-/A- homozygotes | 13 (0.8) |
Self-reported.
Over 2656 child-years of follow-up [cyfu; median years per child 2.01, interquartile range (IQR 1.15-2.24)], we recorded 4091 episodes of malaria: 3697 (90.4%) were mild and 394(9.6%) were severe (either minor- or major-severe) (appendix). The overall incidence rate was 1.540 episodes/cyfu (table 2). This rate was lower in 2008 (1.188 episodes/cyfu) than in each of the next 3 years (1.691, 1.650, 1.611; p<0.0001), but did not differ significantly between sexes or villages. Ethnicity was associated with malaria risk in adjusted analyses: relative to Malinke children, incidence was higher in Bambara (adjusted incidence rate ratio [aIRR] 1.355; 95% CI 1.145-1.604; p=0.0004) and Dogon (aIRR 1.841; 95% CI 1.173-2.891; p=0.0080), unchanged in Fulani, and lower in Sarakole children (aIRR 0.571; 95% CI 0.403-0.809; p=0.0016).
Table 2. Impact of year, ethnicity, and red blood cell variants on malaria incidence.
| cyfu | No. events | Incidence rate (events/cyfu) | IRR* (95% CI) | p-value* | Adjusted IRR** (95% CI) | p-value** | |
|---|---|---|---|---|---|---|---|
| Total | 2655.7 | 4091 | 1.540 | NA | NA | NA | NA |
| Year | |||||||
| 2008 | 636.4 | 756 | 1.188 | REF | NA | REF | NA |
| 2009 | 694.3 | 1174 | 1.691 | 1.455 (1.304, 1.624) | <0.0001 | 1.456 (1.307, 1.621) | <0.0001 |
| 2010 | 673.3 | 1111 | 1.65 | 1.438 (1.287, 1.606) | <0.0001 | 1.434 (1.286, 1.599) | <0.0001 |
| 2011 | 651.6 | 1050 | 1.611 | 1.415 (1.265, 1.582) | <0.0001 | 1.408 (1.261, 1.572) | <0.0001 |
| Village | |||||||
| Kenieroba | 2021.7 | 3145 | 1.556 | REF | NA | NA | NA |
| Fourda | 328.7 | 480 | 1.46 | 0.906 (0.807, 1.017) | 0.0950 | NA | NA |
| Bozokin | 305.2 | 466 | 1.527 | 0.937 (0.833, 1.053) | 0.2736 | NA | NA |
| Ethnicity | |||||||
| Malinke | 2306.4 | 3474 | 1.506 | REF | NA | REF | NA |
| Fulani | 206.2 | 344 | 1.668 | 1.089 (0.953, 1.243) | 0.2092 | 1.038 (0.909, 1.184) | 0.5845 |
| Bambara | 92.0 | 201 | 2.184 | 1.418 (1.196, 1.681) | 0.0001 | 1.355 (1.145, 1.604) | 0.0004 |
| Sarakole | 44.6 | 45 | 1.009 | 0.585 (0.411, 0.832) | 0.0029 | 0.571 (0.403, 0.809) | 0.0016 |
| Dogon | 6.4 | 27 | 4.243 | 1.954 (1.240, 3.078) | 0.0039 | 1.841 (1.173, 2.891) | 0.0080 |
| Sex | |||||||
| Girls | 1303.5 | 2057 | 1.578 | REF | NA | NA | NA |
| Boys | 1352.2 | 2034 | 1.504 | 1.015 (0.943, 1.093) | 0.6864 | NA | NA |
| β-globin variant | |||||||
| HbAA | 2092.2 | 3343 | 1.598 | REF | NA | REF | NA |
| HbAS | 378.0 | 408 | 1.079 | 0.656 (0.580, 0.742) | <0.0001 | 0.662 (0.586, 0.747) | <0.0001 |
| HbAC | 167.8 | 321 | 1.913 | 1.185 (1.033, 1.359) | 0.0152 | 1.154 (1.007, 1.321) | 0.0390 |
| HbSC | 14.0 | 12 | 0.855 | 0.577 (0.293, 1.136) | 0.1116 | 0.557 (0.286, 1.085) | 0.0853 |
| HbCC | 2.2 | 4 | 1.815 | 0.759 (0.235, 2.455) | 0.6457 | 0.776 (0.245, 2.464) | 0.6677 |
| HbSS | 1.5 | 3 | 2.06 | 1.994 (0.513, 7.748) | 0.3188 | 2.142 (0.564, 8.143) | 0.2635 |
| α-globin variant | |||||||
| Normal (αα/αα) | 1904.1 | 2876 | 1.51 | REF | NA | REF | NA |
| α+-thalassaemia (-α3.7/αα) | 699.9 | 1127 | 1.61 | 1.041 (0.958, 1.132) | 0.3414 | 1.051 (0.969, 1.141) | 0.2306 |
| α0-thalassaemia (-α3.7/-α3.7) | 51.7 | 88 | 1.702 | 1.244 (0.963, 1.607) | 0.0949 | 1.194 (0.929, 1.533) | 0.1658 |
| Blood type | |||||||
| A | 804.2 | 1266 | 1.574 | REF | NA | REF | NA |
| B | 574.8 | 936 | 1.628 | 1.062 (0.960, 1.176) | 0.2434 | 1.053 (0.953, 1.164) | 0.3095 |
| AB | 198.1 | 327 | 1.651 | 1.139 (0.983, 1.319) | 0.0828 | 1.132 (0.980, 1.307) | 0.0927 |
| O | 1078.6 | 1562 | 1.448 | 0.926 (0.847, 1.012) | 0.0900 | 0.918 (0.840, 1.002) | 0.0555 |
| G6PD genotype | |||||||
| Boys | |||||||
| Normal | 1226.0 | 1837 | 1.498 | REF | NA | REF | NA |
| A- hemizygotes | 126.2 | 197 | 1.561 | 0.938 (0.786, 1.119) | 0.4779 | 0.945 (0.791, 1.128) | 0.5294 |
| Girls | |||||||
| Normal | 1044.3 | 1621 | 1.552 | REF | NA | REF | NA |
| A-/A+ heterozygotes | 235.6 | 419 | 1.778 | 1.132 (0.995, 1.288) | 0.0597 | 1.124 (0.989, 1.277) | 0.0736 |
| A-/A- homozygotes | 23.6 | 17 | 0.721 | 0.472 (0.266, 0.839) | 0.0105 | 0.513 (0.292, 0.900) | 0.0200 |
cyfu, child-years of follow-up; REF, reference group; CI, confidence interval; IRR, incidence rate ratio; NA, not applicable; G6PD, glucose-6-phosphate dehydrogenase
Adjusted for age only
Adjusted for age, year, ethnicity, β-globin and α-globin variants, ABO blood group, and G6PD A- genotype
In multivariate analyses that included RBC variants, age, ethnicity, and year, malaria incidence was reduced by 34% in HbAS (aIRR 0.662; 95% CI 0.586-0.747; p<0.0001) compared to HbAA children, and 49% in G6PD A-/A- girls (aIRR 0.513; 95% CI 0.292-0.900; p=0.0200) relative to G6PD A+/A+ girls. Malaria risk was not significantly altered by α-thalassaemia or ABO blood groups, but was increased by 15% in HbAC children (aIRR 1.154; 95% CI 1.007-1.321; p=0.0390)compared to HbAA children. In models adjusted only for age, the IRR estimates were similar (table 2).
As reported in other studies,7, 9 we found suggestions that α-thalassaemia modified the HbAS effect on malaria risk. Relative to αα/αα children with HbAA, children with HbAS were protected from malaria (aIRR 0.662; 95% CI 0.572-0.767; p<0.0001), but there was no protection when -α3.7/-α3.7 was coinherited with HbAS (aIRR 1.133; 95% CI 0.473-2.72)(figure 2A; appendix). Additionally, an increased risk for HbAC children with αα/αα was non-significantly increased even more for HbAC children with coinherited -α3.7/-α3.7 (figure 2A; appendix). Despite these suggestions, the overall test that the β-globin effects change based on α-thalassaemia type is not significant (appendix, adjusted p=0.4821), likely due to the small numbers of HbAS or HbAC children with coinherited -α3.7/-α3.7. Nevertheless, the trend agrees with earlier reports, collectively suggesting a common mechanism by which α°-thalassaemia may increase malaria risk when coinherited with β-globin variants.
Figure 2.
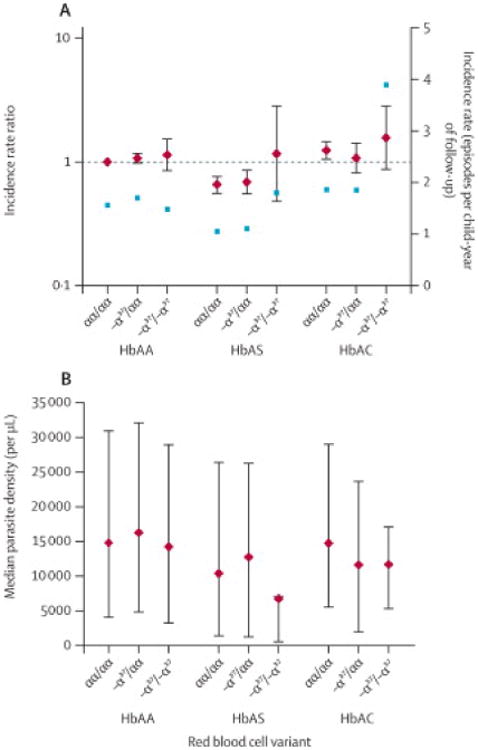
Modification of HbAS and HbAC effects on malaria incidence (A) and parasite density (B) by α-thalassaemia. A. Diamonds and bars indicate incidence rate ratios adjusted for age (relative to HbAA, αα/αα children)and 95% confidence intervals (left y-axis). Squares indicate crude incidence rates (right y-axis).cyfu, child-years of follow-up. B. Diamonds and bars indicate median parasite density and interquartile range.
Peak malaria incidence (2.78 episodes/cyfu) occurred at age 3 years (all ages in years except where indicated) and was nearly 7-fold higher (IRR 6.853; 95% CI 4.221-11.13) than at age 17 (figure 3A; appendix). From age 4 to 13, malaria risk decreased 13.8% (95% CI 11.6-16.0) per year. Age modified the HbAS effect on malaria incidence: relative to HbAA, HbAS conferred the greatest protection at age <1 (IRR 0.392; 95% CI 0.185-0.833; p=0.0155) with generally higher IRRs for older ages (figure 3B; appendix 6). A test of this trend (better HbS protection for younger children) based on the multivariate quasi-Poisson model was significant (p=0.02). From age <1 to 17, the IRR of HbAS relative to HbAA increased (i.e., protection decreased) on average 7.6% per year.
Figure 3.
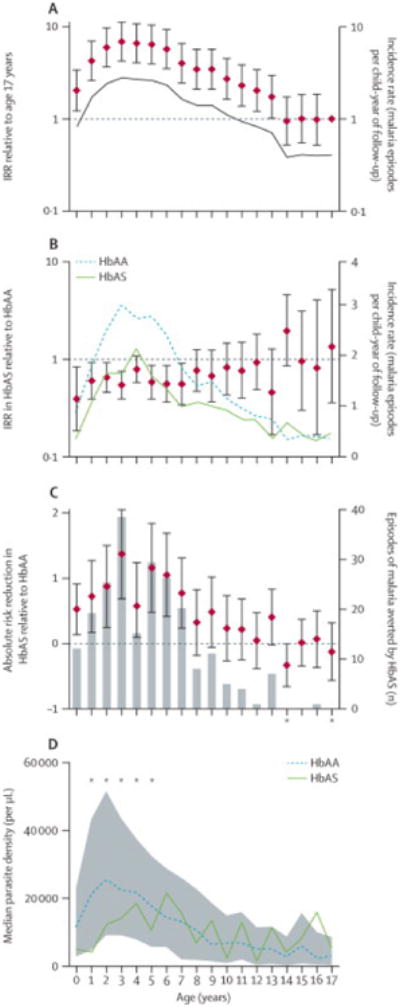
Impact of age on (A) malaria incidence in all children, (B) relative malaria protection by HbAS, (C) absolute malaria risk in HbAS children, and (D) relative reduction in parasite density by HbAS. A. Diamonds and bars indicate incidence rate ratios (IRRs) relative to age 17 and 95% confidence intervals (CI; left y-axis). Line indicates incidence rate (right y-axis). B. Diamonds and bars indicate IRRs in HbAS relative to HbAA children and 95% CIs (left y-axis). Lines indicate incidence rate in HbAA (dotted) and HbAS (dashed) children. C. Diamonds and bars indicate absolute risk reductions (ARRs) in HbAS children and 95% CIs (left y-axis). Gray bars indicate the estimated number of episodes averted by HbAS. Asterisks indicate years with negative ARR and therefore negative averted cases. D. Lines indicate median parasite densities in HbAA (dotted) and HbAS (dashed) children. Gray area indicates interquartile ranges for parasite densities in all children irrespective of β-globin variant. Asterisks indicate ages at which parasite densities were significantly different (p<0.05) between HbAA and HbAS children. In all panels, age=0 indicates age<1 year.
To identify age groups where HbAS protects children from malaria most effectively, we computed absolute risk reductions (ARRs) and the number of episodes averted by HbAS in each age group. All ARRs were significant through age 7 (figure 3C). Both measures were greatest at age 3, when an ARR of 1.372 (95% CI 0.688-2.05) was estimated to have prevented 38episodes in HbAS children. Because older age groups eventually acquire protection irrespective of β-globin variant, 98.6% of the episodes averted by HbAS were at age ≤11 (figure 3C).
Overall, median parasite density at clinical presentation was 14,700 parasites/μL (IQR3850-30,100) (table 3). Of the covariates listed in table 3, only β-globin variant was significantly associated with parasite density. In analyses adjusted for age, ethnicity, and other RBC variants, densities were lower in HbAS vs HbAA children (median 10,550 vs 15,150 parasites/μL; p=0.0004). We tested whether the effect of β-globin variant differed by α-thalassaemia genotype and found no significant epistasis (figure 2B; appendix).
Table 3. Impact of ethnicity and red blood cell variants on P. falciparum density.
| cyfu | No. events | Median parasites/μL (IQR) | p-value* | p-value** | |
|---|---|---|---|---|---|
| Total | 2655.7 | 4091 | 14700(3850, 30100) | NA | NA |
|
| |||||
| Ethnicity | |||||
| Malinke | 2306.4 | 3474 | 14900(3900, 30600) | REF | REF |
| Fulani | 206.2 | 344 | 13275(3000, 27650) | 0.2164 | 0.0769 |
| Bambara | 92.0 | 201 | 14700(5300, 29950) | 0.1426 | 0.1395 |
| Sarakole | 44.6 | 45 | 16025(3375, 24950) | 0.3091 | 0.2429 |
| Dogon | 6.4 | 27 | 8550(1900, 18700) | 0.1911 | 0.1943 |
|
| |||||
| Sex | |||||
| Girls | 1303.5 | 2057 | 14100(3900, 29475) | REF | NA |
| Boys | 1352.2 | 2034 | 15000(3800, 31000) | 0.9419 | NA |
|
| |||||
| β-globin variant | |||||
| HbAA | 2092.2 | 3343 | 15150(4250, 31050) | REF | REF |
| HbAS | 378.0 | 408 | 10550(1350, 26250) | 0.0004 | 0.0004 |
| HbAC | 167.8 | 321 | 13950(5100, 27050) | 0.6759 | 0.7757 |
| HbSC | 14.0 | 12 | 750(75, 1700) | 0.1126 | 0.1029 |
| HbCC | 2.2 | 4 | 5100(425, 6300) | 0.3165 | 0.3154 |
| HbSS | 1.5 | 3 | 22125(100, 22125) | 0.3154 | 0.3150 |
|
| |||||
| α-globin variant | |||||
| Normal (αα/αα) | 1904.1 | 2876 | 14250(3800, 29900) | REF | REF |
| α+ trait (-α3.7/αα) | 699.9 | 1127 | 15675(4050, 30550) | 0.7769 | 0.7273 |
| α0 trait (-α3.7/-α3.7) | 51.7 | 88 | 12775(3950, 26850) | 0.6932 | 0.6874 |
|
| |||||
| ABO blood group | |||||
| A | 804.2 | 1266 | 14400(3900, 28125) | REF | REF |
| B | 574.8 | 936 | 14250(3100, 32325) | 0.8027 | 0.7580 |
| AB | 198.1 | 327 | 15750(4650, 32450) | 0.1503 | 0.1843 |
| O | 1078.6 | 1562 | 14915(4175, 29975) | 0.5881 | 0.9525 |
|
| |||||
| G6PD A- genotype | |||||
| Boys | |||||
| Normal | 1226.0 | 1837 | 15000(3725, 30600) | REF | REF |
| A- hemizygotes | 126.2 | 197 | 15100(4350, 33075) | 0.5188 | 0.4440 |
| Girls | |||||
| Normal | 1044.3 | 1621 | 14025(3900, 28950) | REF | REF |
| A- heterozygotes | 235.6 | 419 | 14750(3900, 30600) | 0.6897 | 0.8397 |
| A- homozygotes | 23.6 | 17 | 10950(450, 36650) | 0.1091 | 0.1843 |
REF, reference group.
Adjusted for age only (bivariate analyses)
Adjusted from multivariate analyses that included age, ethnicity, β-globin and α-globin variants, ABO blood group, and G6PD A- genotype
Median parasite density peaked at age 2 (24,300 parasites/μL; IQR 9225-51,700) and declined substantially by age 17 (3100 parasites/μL; IQR 350-8600) (figure 3D; appendix). We used stratified analyses to investigate the interaction between HbAS and age on parasite density. At ages 1 through 5, parasite densities were significantly lower in HbAS than HbAA children (all p<0.04) (figure 3D; appendix); this effect was most pronounced at age 1, when median parasite density was 21,150 parasites/μL (IQR 7275-47,600) in HbAA and 4200 parasites/μL (IQR 250-8250; p=0.005) in HbAS children. At ages >5, parasite densities were not significantly different between HbAA and HbAS children. In multivariate models that included ethnicity and other RBC variants as covariates, these age-stratified differences in parasite densities between HbAA and HbAS children remained significant (appendix), and a test on the age by HbS interaction from a multivariate model showed that the HbS effect was significantly more protective for younger ages (p<0.0001).
Of 4091 episodes of malaria, only 394 (9.6%) were treated as severe (appendix). Of these, 38 (10%) were major-severe defined by cerebral malaria, severe malarial anaemia, or respiratory distress, 19 (5%) were characterized by parasite densities >100,000 parasites/μL, and 337 (85%) were minor-severe because they necessitated parenteral therapy owing to severe prostration, repetitive vomiting, or inability to tolerate oral therapy. The proportion of severe cases varied significantly between ages (p=0.002), and was highest at age <1 (17) and lowest at age 16 (3.3%) (appendix). This proportion was significantly lower in HbAS (5.4%) than HbAA (10.3%) children (age-adjusted p=0.002). Other RBC variants did not affect the distribution of mild and severe cases. Four severe cases died from: severe prostration; severe malarial anaemia; cerebral malaria, respiratory distress, and shock; and severe prostration and respiratory distress. These fatalities occurred in children aged 6 months or 2 years who lacked β-globin mutations, α-globin deletions, and G6PD A-alleles.
Discussion
To investigate the effects of common RBC variants on malaria risk and parasite density, we enrolled 1543 children into a prospective cohort study over 4 years and recorded 4091 malaria episodes in 2656cyfu in southern Mali. In this area of intense, seasonal P. falciparum transmission, 71.1% of children carried at least one RBC variant shown to reduce malaria risk in Africa in prior studies. HbAS most clearly conferred protection from malaria; moreover, age modified this effect, which was attenuated after early childhood and absent in teenagers. Similarly, HbAS reduced the density of parasites at clinical presentation, and did so most substantially before age 5. Of the other RBC variants, only G6PD A-/A- in girls reduced malaria risk.
Relative to HbAA, HbAC increased malaria risk and α0-thalassaemiamay have augmented this risk. These findings are surprising because HbCC homozygotes experience substantial protection from severe malaria.10 While one case-control study found reduced malaria risk in HbAC children,10 two prior prospective studies in West Africa have suggested a small increase in risk.7, 11 This increased risk may sufficiently impair fitness in malaria-endemic areas to account for the very limited geographic distribution of HbC, despite the clear protection it affords to homozygotes, the mild clinical sequelae associated with it, and the greater age of HbC compared with HbS alleles.12 The molecular pathology of HbAC on RBCs is minimal, manifesting as slightly reduced RBC lifespan, mild anaemia (with normal reticulocytaemia),13 and increased mean corpuscular haemoglobin concentration.14 In-vitro experiments indicate that HbAC RBCs support normal invasion and parasite maturation15, 16 but reduce the expression of parasite cytoadherence proteins on the RBC surface;17 this latter finding, also present in HbAS RBCs,18 suggests a common mechanism of protection where in these β-globin variants attenuate cytoadherence and modulate innate immune activation.19 Our finding of increased malaria risk in HbAC children suggests that these cellular phenotypes may not correlate strongly with mild disease risk, or may be modified by other factors in vivo.
As expected based on prior prospective studies (reviewed in1), HbAS reduced malaria risk by 34% relative to HbAA. Additionally, this protection was unapparent when HbAS was coinherited with α0-thalassaemia. Although the interaction between variants was not statistically significant, this observation supports epistasis, or effect modification, by α-thalassaemia on malaria risk. Epistasis has been reported in earlier prospective studies,7, 9, 11 likely due to reduced production of HbS in HbAS RBCs that harbor α-globin deletions,20 and in an in-vitro study21 showing that α-thalassaemia antagonizes HbAS-mediated reductions in cytoadherence. This finding may explain the clinical observations that α-thalassaemia exerts negative epistatic effects on HbAS-mediated protection from malaria, as reported in prior studies7, 9 and suggested by our data.
Age significantly modified malaria risk, which peaked at age 3 and remained significantly elevated through age 13 (relative to age 17). We also studied age modification of the HbAS effect on risk. Our study shows that protection of HbAS relative to HbAA children was significantly better at younger ages with respect to both malaria incidence (p=0.02) and parasite density (p<0.0001). This suggests that the biological effects of HbAS on P. falciparum progressively decline with advancing age in early childhood. This adds to a previousstudy22 that found non-significant trends for age-modifying effects on HbS protection. Several lines of evidence support the notion that naturally-acquired immunity is partly dependent on intrinsic age-related factors that are independent of parasite exposure (reviewed in 23).
Of all the RBC traits we investigated, only HbAS was associated with reduced parasite densities at the time of presentation. Whether this reduction is clinically significant is not known, but elucidating the mechanism of this effect may provide insights into HbAS-mediated protection from mild and severe malaria. Intriguingly, this effect was significant only through age 5, after which densities were comparable between HbAS and HbAA children. Infected HbAS RBCs demonstrate reduced cytoadherence in vitro;18 this phenotype, which may be enhanced in vivo by RBC sickling in relatively hypoxic environments of post-capillary venules and the cytoadherence-blocking effects of naturally-acquired IgG responses, may contribute to the reduced parasite densities we observed in HbAS children. The coincident reduction in malaria risk attributed to HbAS before age 5 supports the premise that reduced parasite density contributes directly to this protection. Indeed, several in-vitro studies have reported that HbAS impairs parasite growth because of reduced oxygen tension24, 25 or the action of RBC microRNAs,26 and these phenomena may limit parasite propagation in vivo. However, it seems increasingly clear that attenuation of parasite growth alone is insufficient to prevent disease.27 Also, although α0-thalassaemiaappeared to antagonize HbAS protection from malaria, it did not lessen the reduction in parasite density in HbAS children. It therefore seems unlikely that HbAS protects principally by suppressing parasite density. Additional hypotheses are that HbAS modulates the strength of cytoadherence18 or intensity of innate immune responses, enabling children to tolerate parasitemia without developing symptoms.28
HbAS also reduced the proportion of malaria cases with severe manifestations. As often seen in cohort studies, classic manifestations of life-threatening malaria syndromes – including cerebral malaria, severe malarial anaemia, and respiratory distress – were uncommon, and most cases required treatment as severe due to an inability to tolerate oral therapy. Nevertheless, HbAS reduced these “minor-severe” syndromes as well, indicating that it generally attenuates the pathophysiology of malaria. The mechanisms of this effect require further exploration as a basis for preventive or therapeutic intervention.
In our cohort, neither α-thalassaemias, ABO blood groups, nor G6PD A- hetero- or hemizygosities protected from malaria. Prior studies have reported that α-thalassaemia protects from severe but not mild malaria (reviewed in 1) and that type O blood group weakly protects from severe malaria.29 Given the high prevalence of these traits in our cohort, our study was well-powered to detect appreciable differences in risk. Based on our data, these protective impacts appear limited to severe disease and are insufficient to substantially prevent mild malaria.
To our knowledge, this study is the first to report that G6PD A-/A- girls are protected from malaria: relative to G6PD A+/A+ girls, malaria risk was slightly higher for A+/A-(aIRR 1.124) but significantly lower for A-/A-girls (aIRR 0.513). G6PD A-/A- girls also had lower parasite densities than A+/A- girls, suggesting a significant biological impact of a uniform population of G6PD-deficient RBCs within a host. We interpret these data with some caution, however, as they derive from 17 episodes in 13 G6PD A-/A- girls, and are not corroborated by a similar trend in data from G6PD A+/A- girls. Both the overlapping geographical distributions of G6PD deficiency and falciparum malaria, as well as in vitro experiments,30, 31 suggest that G6PD deficiency confers some protection; however, prior clinical studies of G6PD deficiency and malaria risk are inconsistent, reporting increased disease risk in heterozygous girls,32 decreased disease risk in deficient girls,33 decreased severe disease risk in hemizygous boys,8 and an absence of effect.34 Our study benefits from a prospective design, prolonged follow-up period, and relatively high prevalence of A-/A- homozygotes; also, the absence of alternative alleles encoding the A- form of G6PDdeficiency in Mali35 enabled us to accurately assign G6PD status to children based only on the 202A mutation, thus avoiding misclassifications that have bedeviled other studies in western Africa.36 Despite the protection in G6PD A-/A-girls, G6PD A- boys were clearly not protected, perhaps due to the more marked phenotypic deficiency in hemizygotes than homozygotes in Africa.35 Future work is needed to explore the relative impacts of phenotypic and genotypic G6PD deficiency on falciparum malaria in high-transmission settings.
Finally, ethnicity was associated with malaria risk independent of other measured traits. Surprisingly, Fulani children were not protected from malaria, though they had slightly lower parasite densities than Malinke children in adjusted analyses. Prior studies have reported lower parasite prevalences in Fulani (relative to Mossi in Burkina Faso37 and Dogon in Mali38), which was subsequently correlated with deficits of regulatory T cells (relative to Mossi39) and enhanced early interferon-γ production in response to parasite antigens (relative to Dogon40). Our contradictory findings may result from two key differences: our designation of Malinke children as the comparator group, and our use of a prospective study design that measured malaria incidence instead of parasite prevalence. Interestingly, malaria risk differed significantly between Malinke, Bambara, and Sarakole children despite all three groups belonging to the larger Mande ethnic group, which population genetics analyses have distinguished from other groups in the region.41
Our study has several potential limitations. It is possible that we did not capture all malaria episodes, but other options for health care are limited in the study area, and it is unlikely that the incidence of missed episodes would vary between participants with different RBC variants. We enrolled few children with the putatively protective variants of homozygous HbC, G6PD A-, or α-thalassemia, limiting our ability to quantify their effects on malaria. Since we provided rapid access to effective therapy, we cannot directly estimate the impact of these mutations on survival in malaria-endemic areas. Finally, unmeasured covariates – including bed net use, socioeconomic status, co-infections, or heretofore unknown mediators of protection – could have biased our protective estimates.
Our prospective study of children carrying a high prevalence of RBC variants in an area of intense, seasonal P. falciparum transmission clearly defines the clinical impact of these traits on malaria risk and parasite density. Investigating natural patterns of disease susceptibility in endemic areas may uncover mechanisms of pathogenesis that can be targeted to prevent and treat malaria.
Supplementary Material
Research in context panel.
Evidence before this study
We previously conducted a systematic review and meta-analysis of the influence of haemoglobinopathies on the risks of severe and mild malaria on September 9, 2011[Taylor SM et al. Lancet Infect Dis 2012; 12(6): 457-68]. We repeated the search in PubMed using identical search criteria on January 12, 2015; this search returned 949 papers, of which we reviewed the full texts of 15, of which seven contained relevant information. Four of these papers [Timmann C et al. Nature 2012; 489(7416): 443-6; Atkinson SH et al. Blood 2014; 123(13): 2008-16; Manjurano A et al. Plos One 2012; 7(10): e47463; Toure O et al. Plos One 2012; 7(9): e43987] confirmed that HbAS substantially reduces the risk of severe falciparum malaria (by 88-97%), and other studies confirmed that HbAS mildly reduces the risk of mild malaria. The risk of severe malaria was also decreased by heterozygous α-thalassaemia, heterozygous G6PDdeficiency in girls, and type O blood group. This search also returned a meta-analysis of case-control studies of the impact of ABO blood group type on malaria [Panda AK et al. Malar J 2011; 10: 309]. This meta-analysis of six studies reported that type O blood group reduces the risk of severe malaria by 50%. Finally, we searched PubMed on January 12, 2015 using the search terms (“malaria” [Title/Abstract] OR “malaria/blood” [MAJR] OR “malaria/genetics” [MeSH Terms]) AND (“G6PD” [Title/Abstract] OR “glucose-6-phosphate-dehydrogenase” [Title/Abstract] OR “glucose phosphate dehydrogenase deficiency/genetics” [MeSH Terms] OR “glucose phosphate dehydrogenase deficiency/blood” [MeSH Terms]). This search returned 572 papers. There exists no consensus on the influence of G6PD deficiency on malaria; investigations of this relationship have been complicated by the genotypic and phenotypic diversity of G6PD deficiency, diverse parasite epidemiology, and varied study designs and control selection. There remains a paucity of prospective cohort studies of G6PD deficiency on falciparum malaria.
Added value of this study
HbAS reduces both the risk of falciparum malaria and the density of parasites at the time of clinical presentation with malaria; the greatest degree of protection is conferred in the early years of childhood, when malaria incidence in children overall is highest and before immunity to clinical disease is acquired. Homozygous G6PD-deficiency encoded by the X-linked A- allele reduced the risk of malaria in girls, while HbAC increased the risk of malaria in children.
Implications of all the available evidence
By reducing the risk of falciparum malaria, suppressing parasite density, or both, RBC variants offer models of attenuated pathogenesis. These models can be exploited by future cellular, molecular, and immunologic studies to define mechanisms by which these RBC variants counteract the detrimental effects of P. falciparum parasites. Defining these mechanisms will provide targets for future antiparasitic or adjunct therapies for children with falciparum malaria.
Acknowledgments
This research was supported in part by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID) of the US National Institutes of Health (NIH). JMA, MAK, MPF, CAL, and RMF are currently employees of NIH. SMT was supported by the NIAID under award number K08AI100924. This project was also supported in part with federal funds from the National Cancer Institute, NIH (Contract No. HHSN261200800001E). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. We thank Jaymin Patel and Steven Meshnick (University of North Carolina, Chapel Hill, NC, USA) for helpful discussions; Robert Gwadz, Dick Sakai, and Thomas Wellems (NIAID/NIH, Rockville, MD, USA) for supporting this work; the children and parents in Kenieroba, Fourda, and Bozokin for their participation; and the village communities and health district authorities of Bancoumana and Kati for their long-term commitment to this study.
Footnotes
Contributors: TML-M, JMA, MPF, CAL, MD, and RMF contributed to study design. TML-M, SD, DK, JMA, MD, ASK, SASD, KT, MAK, and AD acquired data. TML-M, KM, WG, MPF, SMT, CAL, MD, and RMF analyzed and interpreted data. WG, MPF, SMT, and RMF performed statistical analysis. JMA, AD, SEM, and GST provided administrative or technical support. CAL, MD, and RMF supervised the study. MPF, SMT, and RMF wrote the report.
Conflicts of Interest: We declare that we have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Taylor SM, Parobek CM, Fairhurst RM. Haemoglobinopathies and the clinical epidemiology of malaria: a systematic review and meta-analysis. Lancet Infect Dis. 2012;12(6):457–68. doi: 10.1016/S1473-3099(12)70055-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clark TG, Fry AE, Auburn S, Campino S, Diakite M, Green A, et al. Allelic heterogeneity of G6PD deficiency in West Africa and severe malaria susceptibility. Eur J Hum Genet. 2009;17(8):1080–5. doi: 10.1038/ejhg.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rowe JA, Handel IG, Thera MA, Deans AM, Lyke KE, Kone A, et al. Blood group O protects against severe Plasmodium falciparum malaria through the mechanism of reduced rosetting. Proc Nat Acad Sci U S A. 2007;104(44):17471–6. doi: 10.1073/pnas.0705390104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malaria Genomic Epidemiology Network. Reappraisal of known malaria resistance loci in a large multicenter study. Nat Genet. 2014;46(11):1197. doi: 10.1038/ng.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization. World Malaria Report 2014. [Accessed February 18, 2015];2014 Available from: http://www.who.int/malaria/publications/world_malaria_report_2014/en/
- 6.Severe falciparum malaria. World Health Organization, Communicable Diseases Cluster. Trans Roy soc Trop Med Hyg. 2000;94 Suppl 1:S1–90. [PubMed] [Google Scholar]
- 7.Crompton PD, Traore B, Kayentao K, Doumbo S, Ongoiba A, Diakite SA, et al. Sickle cell trait is associated with a delayed onset of malaria: implications for time-to-event analysis in clinical studies of malaria. J Infect Dis. 2008;198(9):1265–75. doi: 10.1086/592224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guindo A, Fairhurst RM, Doumbo OK, Wellems TE, Diallo DA. X-linked G6PD deficiency protects hemizygous males but not heterozygous females against severe malaria. PLoS Med. 2007;4(3):e66. doi: 10.1371/journal.pmed.0040066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williams TN, Mwangi TW, Wambua S, Peto TE, Weatherall DJ, Gupta S, et al. Negative epistasis between the malaria-protective effects of alpha+-thalassemia and the sickle cell trait. Nat Genet. 2005;37(11):1253–7. doi: 10.1038/ng1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Modiano D, Luoni G, Sirima BS, Simpore J, Verra F, Konate A, et al. Haemoglobin C protects against clinical Plasmodium falciparum malaria. Nature. 2001;414(6861):305–8. doi: 10.1038/35104556. [DOI] [PubMed] [Google Scholar]
- 11.Kreuels B, Kreuzberg C, Kobbe R, Ayim-Akonor M, Apiah-Thompson P, Thompson B, et al. Differing effects of HbS and HbC traits on uncomplicated falciparum malaria, anemia, and child growth. Blood. 2010;115(22):4551–8. doi: 10.1182/blood-2009-09-241844. [DOI] [PubMed] [Google Scholar]
- 12.Modiano D, Bancone G, Ciminelli BM, Pompei F, Blot I, Simpore J, et al. Haemoglobin S and haemoglobin C: ‘quick but costly’ versus ‘slow but gratis’ genetic adaptations to Plasmodium falciparum malaria. Hum Mol Genet. 2008;17(6):789–99. doi: 10.1093/hmg/ddm350. [DOI] [PubMed] [Google Scholar]
- 13.Prindle KH, Jr, McCurdy PR. Red cell lifespan in hemoglobin C disorders (with special reference to hemoglobin C trait) Blood. 1970;36(1):14–9. [PubMed] [Google Scholar]
- 14.Bunn HF, Noguchi CT, Hofrichter J, Schechter GP, Schechter AN, Eaton WA. Molecular and cellular pathogenesis of hemoglobin SC disease. Proc Nat Acad Sci U S A. 1982;79(23):7527–31. doi: 10.1073/pnas.79.23.7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedman MJ, Roth EF, Nagel RL, Trager W. The role of hemoglobins C, S, and Nbalt in the inhibition of malaria parasite development in vitro. Am J Trop Med Hyg. 1979;28(5):777–80. [PubMed] [Google Scholar]
- 16.Fairhurst RM, Fujioka H, Hayton K, Collins KF, Wellems TE. Aberrant development of Plasmodium falciparum in hemoglobin CC red cells: implications for the malaria protective effect of the homozygous state. Blood. 2003;101(8):3309–15. doi: 10.1182/blood-2002-10-3105. [DOI] [PubMed] [Google Scholar]
- 17.Fairhurst RM, Baruch DI, Brittain NJ, Ostera GR, Wallach JS, Hoang HL, et al. Abnormal display of PfEMP-1 on erythrocytes carrying haemoglobin C may protect against malaria. Nature. 2005;435(7045):1117–21. doi: 10.1038/nature03631. [DOI] [PubMed] [Google Scholar]
- 18.Cholera R, Brittain NJ, Gillrie MR, Lopera-Mesa TM, Diakite SA, Arie T, et al. Impaired cytoadherence of Plasmodium falciparum-infected erythrocytes containing sickle hemoglobin. Proc Nat Acad Sci U S Aa. 2008;105(3):991–6. doi: 10.1073/pnas.0711401105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor SM, Cerami C, Fairhurst RM. Hemoglobinopathies: Slicing the Gordian knot of Plasmodium falciparum malaria pathogenesis. PLoS Pathog. 2013 doi: 10.1371/journal.ppat.1003327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steinberg MH, Embury SH. Alpha-thalassemia in blacks: genetic and clinical aspects and interactions with the sickle hemoglobin gene. Blood. 1986;68(5):985–90. [PubMed] [Google Scholar]
- 21.Opi DH, Ochola LB, Tendwa M, Siddondo BR, Ocholla H, Fanjo H, et al. Mechanistic Studies of the Negative Epistatic Malaria-protective Interaction Between Sickle Cell Trait and α+thalassemia. EBioMedicine. 2014;1(1):29–36. doi: 10.1016/j.ebiom.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gong L, Maiteki-Sebuguzi C, Rosenthal PJ, Hubbard AE, Drakeley CJ, Dorsey G, et al. Evidence for both innate and acquired mechanisms of protection from Plasmodium falciparum in children with sickle cell trait. Blood. 2012;119(16):3808–14. doi: 10.1182/blood-2011-08-371062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doolan DL, Dobano C, Baird JK. Acquired immunity to malaria. Clin Microbiol Rev. 2009;22(1):13–36. doi: 10.1128/CMR.00025-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedman MJ. Erythrocytic mechanism of sickle cell resistance to malaria. Proc Nat Acad Sci U S A. 1978;75(4):1994–7. doi: 10.1073/pnas.75.4.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pasvol G, Weatherall DJ, Wilson RJ. Cellular mechanism for the protective effect of haemoglobin S against P. falciparum malaria. Nature. 1978;274(5672):701–3. doi: 10.1038/274701a0. [DOI] [PubMed] [Google Scholar]
- 26.LaMonte G, Philip N, Reardon J, Lacsina JR, Majoros W, Chapman L, et al. Translocation of sickle cell erythrocyte microRNAs into Plasmodium falciparum inhibits parasite translation and contributes to malaria resistance. Cell Host Microbe. 2012;12(2):187–99. doi: 10.1016/j.chom.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goncalves BP, Huang CY, Morrison R, Holte S, Kabyemela E, Prevots DR, et al. Parasite burden and severity of malaria in Tanzanian children. New Engl J Med. 2014;370(19):1799–808. doi: 10.1056/NEJMoa1303944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferreira A, Marguti I, Bechmann I, Jeney V, Chora A, Palha NR, et al. Sickle hemoglobin confers tolerance to Plasmodium infection. Cell. 2011;145(3):398–409. doi: 10.1016/j.cell.2011.03.049. [DOI] [PubMed] [Google Scholar]
- 29.Fry AE, Griffiths MJ, Auburn S, Diakite M, Forton JT, Green A, et al. Common variation in the ABO glycosyltransferase is associated with susceptibility to severe Plasmodium falciparum malaria. Hum Mol Genet. 2008;17(4):567–76. doi: 10.1093/hmg/ddm331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cappadoro M, Giribaldi G, O'Brien E, Turrini F, Mannu F, Ulliers D, et al. Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)-deficient erythrocytes parasitized by Plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood. 1998;92(7):2527–34. [PubMed] [Google Scholar]
- 31.Luzzatto L, Usanga FA, Reddy S. Glucose-6-phosphate dehydrogenase deficient red cells: resistance to infection by malarial parasites. Science. 1969;164(3881):839–42. doi: 10.1126/science.164.3881.839. [DOI] [PubMed] [Google Scholar]
- 32.Parikh S, Dorsey G, Rosenthal PJ. Host polymorphisms and the incidence of malaria in Ugandan children. The American journal of tropical medicine and hygiene. 2004;71(6):750–3. [PubMed] [Google Scholar]
- 33.Clark TD, Greenhouse B, Njama-Meya D, Nzarubara B, Maiteki-Sebuguzi C, Staedke SG, et al. Factors determining the heterogeneity of malaria incidence in children in Kampala, Uganda. J Infect Dis. 2008;198(3):393–400. doi: 10.1086/589778. [DOI] [PubMed] [Google Scholar]
- 34.Martin SK, Miller LH, Alling D, Okoye VC, Esan GJ, Osunkoya BO, et al. Severe malaria and glucose-6-phosphate-dehydrogenase deficiency: a reappraisal of the malaria/G-6-P.D. hypothesis. Lancet. 1979;1(8115):524–6. doi: 10.1016/s0140-6736(79)90946-2. [DOI] [PubMed] [Google Scholar]
- 35.Carter N, Pamba A, Duparc S, Waitumbi JN. Frequency of glucose-6-phosphate dehydrogenase deficiency in malaria patients from six African countries enrolled in two randomized anti-malarial clinical trials. Malaria J. 2011;10:241. doi: 10.1186/1475-2875-10-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sirugo G. Reassessing an old claim: natural selection of hemizygotes and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Am J Hematol. 2013;88(5):436. doi: 10.1002/ajh.23424. [DOI] [PubMed] [Google Scholar]
- 37.Modiano D, Petrarca V, Sirima BS, Nebie I, Diallo D, Esposito F, et al. Different response to Plasmodium falciparum malaria in west African sympatric ethnic groups. Proc Nat Acad Sci U S A. 1996;93(23):13206–11. doi: 10.1073/pnas.93.23.13206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dolo A, Modiano D, Maiga B, Daou M, Dolo G, Guindo H, et al. Difference in susceptibility to malaria between two sympatric ethnic groups in Mali. Am J Trop Med Hyg. 2005;72(3):243–8. [PubMed] [Google Scholar]
- 39.Torcia MG, Santarlasci V, Cosmi L, Clemente A, Maggi L, Mangano VD, et al. Functional deficit of T regulatory cells in Fulani, an ethnic group with low susceptibility to Plasmodium falciparum malaria. Proc Nat Acad Sci U S A. 2008;105(2):646–51. doi: 10.1073/pnas.0709969105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCall MB, Hopman J, Daou M, Maiga B, Dara V, Ploemen I, et al. Early interferon-gamma response against Plasmodium falciparum correlates with interethnic differences in susceptibility to parasitemia between sympatric Fulani and Dogon in Mali. J Infect Dis. 2010;201(1):142–52. doi: 10.1086/648596. [DOI] [PubMed] [Google Scholar]
- 41.Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, Froment A, et al. The genetic structure and history of Africans and African Americans. Science. 2009;324(5930):1035–44. doi: 10.1126/science.1172257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.