

Tissue-specific protein and phosphoprotein patterns underlie functional gradients along the maize primary root axis.
Abstract
A high-resolution proteome and phosphoproteome atlas of four maize (Zea mays) primary root tissues, the cortex, stele, meristematic zone, and elongation zone, was generated. High-performance liquid chromatography coupled with tandem mass spectrometry identified 11,552 distinct nonmodified and 2,852 phosphorylated proteins across the four root tissues. Two gradients reflecting the abundance of functional protein classes along the longitudinal root axis were observed. While the classes RNA, DNA, and protein peaked in the meristematic zone, cell wall, lipid metabolism, stress, transport, and secondary metabolism culminated in the differentiation zone. Functional specialization of tissues is underscored by six of 10 cortex-specific proteins involved in flavonoid biosynthesis. Comparison of this data set with high-resolution seed and leaf proteome studies revealed 13% (1,504/11,552) root-specific proteins. While only 23% of the 1,504 root-specific proteins accumulated in all four root tissues, 61% of all 11,552 identified proteins accumulated in all four root tissues. This suggests a much higher degree of tissue-specific functionalization of root-specific proteins. In summary, these data illustrate the remarkable plasticity of the proteomic landscape of maize primary roots and thus provide a starting point for gaining a better understanding of their tissue-specific functions.
The maize (Zea mays) root system consists of embryonic primary and seminal roots and postembryonic lateral and shoot-borne roots (Hochholdinger, 2009). The primary root is the first organ that emerges after germination, providing water and nutrients for the growing seedling. The longitudinal structure of maize roots is characterized by a root cap at the terminal end, a subterminal meristematic zone, followed by zones in which newly formed cells elongate and differentiate (Ishikawa and Evans, 1995). The differentiation zone, in which functionally distinct cell types are formed, can be tracked by the presence of epidermal root hairs. Hence, roots represent a gradient of cell differentiation along the longitudinal axis: young and undifferentiated cells are located at the distal end near the root tip, whereas differentiated cells are located toward the proximal end of the root (Ishikawa and Evans, 1995).
Radially, maize roots can be divided into the stele and the surrounding cortical parenchyma. The stele comprises vascular and ground tissue, which is enclosed by the single-layered pericycle as the outermost boundary. The multilayered cortical parenchyma comprises, from the center to the periphery, a single ring of endodermis cells, a multilayered cortical parenchyma, and single files of exodermis and epidermis cells connecting the root to the rhizosphere (Hochholdinger, 2009). The epidermis of the differentiation zone is densely populated by tubular root hairs, which are instrumental for the uptake of nutrients that are either transported into the shoot or metabolized in the cortical parenchyma (Marschner, 2011).
In recent years, soluble proteomes of whole maize roots were analyzed with respect to development (Hochholdinger et al., 2005), genotypic variation (Hochholdinger et al., 2004; Wen et al., 2005; Liu et al., 2006; Sauer et al., 2006; Hoecker et al., 2008), and environmental interactions (Chang et al., 2000; Li et al., 2007).
A major limitation of analyzing whole roots is their composite nature, being made up of distinct tissues. Each tissue provides unique protein accumulation patterns that have the potential to be masked when whole roots are analyzed. This constraint can be overcome by tissue-specific analyses. Thus far, only a few surveys have analyzed distinct tissues such as the primary root tip (Chang et al., 2000), the elongation zone (Zhu et al., 2006, 2007), and cortical parenchyma and stele (Saleem et al., 2010). The mechanical separation of maize root cortical parenchyma (hereafter referred to as cortex) and stele made the most abundant soluble proteins of these tissues accessible (Saleem et al., 2010). Combining two-dimensional gel electrophoresis and electrospray tandem mass spectrometry (MS/MS) identified 59 proteins preferentially expressed in the cortex and 11 proteins predominantly accumulated in the stele (Saleem et al., 2010). Among these proteins, a β-glucosidase functioning in the release of free bioactive cytokinin (Brzobohatý et al., 1993) was preferentially accumulated in the cortical parenchyma (Saleem et al., 2010). That study gave first insights into tissue-specific protein accumulation in the differentiation zone of maize roots. All these initial studies were based on the combination of two-dimensional SDS-PAGE with subsequent mass spectrometric analyses of selected proteins and thus were of limited resolution and depth.
More recently, gel-free shotgun proteomics further enhanced the depth of proteomic profiles by quantitative liquid chromatography-MS/MS analyses (Zhu et al., 2010). This technique allowed for the identification of more than 12,000 proteins in a maize leaf developmental time-course experiment (Facette et al., 2013) and a similar number of proteins isolated from developing maize seeds (Walley et al., 2013). These studies provided novel insights into protein accumulation patterns in these maize organs during development, which are instrumental for the better understanding of these biological systems.
In this study, we used gel- and label-free high-performance liquid chromatography (HPLC)-MS/MS to quantify the abundance of 11,552 nonmodified proteins and 2,852 phosphoproteins in four distinct tissues of the maize primary root. Proteins unique to certain root tissues or differentially accumulated between root tissues were functionally annotated and analyzed in detail. Moreover, the maize root proteome was compared with maize leaf and seed proteomes of similar size to identify conserved and organ-specific protein accumulation patterns. Furthermore, tissue-specific accumulation patterns of proteins encoded by classical maize genes were surveyed to reveal tissue-specific functions. Finally, proteome and RNA sequencing (RNA-Seq) data of primary root tissues were compared to determine the correlation of these classes of biomolecules.
RESULTS
Tissue-Specific Accumulation of 11,552 Nonmodified Proteins and 2,852 Phosphoproteins from Four Maize Primary Root Tissues
Maize primary roots of 2 to 4 cm in length were separated into four distinct tissues along the longitudinal axis of the primary root. These tissues were the meristematic zone, including the root cap; the elongation zone; and the differentiation zone, which was subdivided radially into the cortical parenchyma, including all cell types between the endodermis and epidermis, and the stele, comprising all cell types enclosed by the pericycle (Fig. 1A). The elongation zone was distinguished from the differentiation zone by root hairs present only in epidermis cells of the differentiation zone.
Figure 1.

Tissues of young maize primary roots surveyed in this study and tissue-specific protein accumulation patterns. A, Maize primary root manually dissected into four tissues (left) and color-coded schematic of the four tissues (right): meristematic zone (MZ), elongation zone (EZ), cortex (C; covering all radial tissues between the endodermis and epidermis), and stele (S; covering all central cell types covered by the pericycle). B and C, Tissue-specific accumulation of nonmodified (B) and phosphorylated (C) proteins in the four maize primary root tissues.
Proteins were extracted in four biological replicates per tissue and digested by trypsin. The resulting peptides were either directly analyzed by HPLC-MS/MS or first enriched for phosphopeptides. More than 8 million identified mass spectra were used to quantify protein (Supplemental Data Set S1) and phosphoprotein (Supplemental Data Set S2) abundance by spectral counting. The resulting spectra were identified using the protein database ZmB73 RefGen_v2 5a working gene set, downloaded from www.gramene.org. In total, 11,552 unique nonmodified proteins (protein groups) were identified from 73,938 distinct peptides (Supplemental Data Sets S1 and S3; see “Materials and Methods”). Approximately 96% of the identified proteins (11,042/11,552) mapped to the filtered gene set, a subset of the working gene set that consists of 39,656 high-confidence gene models. The remaining 510 proteins (4%) mapped exclusively to the working gene set and were annotated as pseudogenes, transposons, or other low-confidence gene models (Supplemental Data Sets S1 and S3).
Phosphorylated proteins were identified after phosphopeptide enrichment using cerium oxide and subsequent HPLC-MS/MS analyses. In total, 11,226 phosphorylated amino acids in 9,480 distinct peptides were detected, matching to a parsimonious set of 2,852 phosphorylated proteins (Supplemental Data Set S2). For 7,292 residues assigned to 6,080 phosphopeptides, the phosphorylated amino acid was localized (Supplemental Data Set S4). Specific Ser (84%; 6,150), Thr (15%; 1,070), and Tyr (1%; 72) phosphorylations were assigned using the variable modification localization (VML) score in the Spectrum Mill software version 3.03 (Agilent Technologies).
Pearson correlation coefficients of protein abundance were calculated between the distinct tissues by using averaged normalized spectral counts (SPCs) of each tissue. Correlation coefficients for the four tissues were on average 0.94 for the nonmodified and 0.76 for the phosphorylated proteins (Table I). The abundance of nonmodified and phosphorylated proteins showed higher correlation between adjacent tissues (e.g. meristematic zone versus elongation zone; 0.96 for nonmodified proteome and 0.76 for phosphorylated proteome) than between distant root tissues (e.g. meristematic zone versus cortex; 0.84 for nonmodified proteome and 0.42 for phosphorylated proteome).
Table I. Pearson correlation coefficients between meristematic zone (MZ), elongation zone (EZ), cortex (C), and stele (S) and between the four replicates of each tissue (in boldface) of the nonmodified (first number in each column) and phosphorylated (second number in each column) protein data sets based on averaged normalized SPCs.
| Tissue | MZ | EZ | C | S |
|---|---|---|---|---|
| MZ | 0.97/0.71 | |||
| EZ | 0.96/0.76 | 0.96/0.66 | ||
| C | 0.84/0.42 | 0.91/0.61 | 0.90/0.66 | |
| S | 0.85/0.43 | 0.89/0.65 | 0.91/0.71 | 0.95/0.60 |
A comparison of biological replicate correlations using averaged normalized SPCs of the four biological replicates was conducted to assess reproducibility. On average, Pearson correlation coefficients of 0.94 and 0.65 were determined between the replicates of the nonmodified proteomes and the phosphorylated proteomes (Table I).
Analysis of the 11,552 nonmodified proteins revealed that 61% (7,012/11,552) of these proteins were expressed in all four tissues (Fig. 1B). The remaining 39% (4,540/11,552) of proteins were either accumulated in two or three tissues (28%; 3,208/11,552) or only in one tissue (11%; 1,332/11,552). Among the 1,332 proteins accumulated in a single tissue, 36% (481/1,332) were identified exclusively in the meristematic zone, 12% (162/1,332) in the elongation zone, 29% (384/1,332) in the cortex, and 23% (305/1,332) in the stele. In contrast, among 2,852 phosphorylated proteins, only 30% (844/2,852) were present in all four tissues, whereas 39% (1,118/2,852) were accumulated in two or three tissues and 31% (890/2,852) showed tissue-specific expression (Fig. 1B).
Functional Analysis of Tissue-Specific Nonmodified and Phosphorylated Proteins
Tissue-specific proteins of the nonmodified and phosphorylated data sets were functionally analyzed using agriGO (http://bioinfo.cau.edu.cn/agriGO/analysis.php). agriGO is based on Gene Ontology (GO) terms describing gene product species in three independent categories: biological process, molecular function, and cellular component (http://www.geneontology.org). To identify significantly overrepresented (false discovery rate [FDR] < 5%) functional categories, a singular enrichment analysis (SEA) was performed. SEA compares each annotated protein class with all annotated expressed proteins in a tissue. Among 11,552 nonmodified proteins across all tissues, 8,017 (69%) were annotated by GO terms. Moreover, GO terms were available for 1,902 of 2,852 (67%) phosphorylated proteins identified in the different tissues. Overall, enrichment for 65 terms related to biological processes and 53 terms related to molecular functions were observed, whereas only two terms reflecting cellular components were identified (Supplemental Table S1).
Among the 481 nonmodified proteins specific for the meristematic zone (Fig. 1B), 279 were annotated, and 35 GO terms displayed overrepresentation (Supplemental Table S1). The majority of these proteins are involved in biological processes, particularly in the regulation of cellular and metabolic processes. Among the 292 meristematic zone-specific phosphorylated proteins, 172 were annotated by agriGO, and only four GO terms, GO:0006139, GO:0006259, GO:0003677, and GO:0003676, which are involved in DNA binding and DNA metabolic processes, were overrepresented. These four terms were also overrepresented among the nonmodified meristematic zone-specific proteins.
Only 162 nonmodified proteins (101 annotated) and 110 phosphorylated proteins (73 annotated) displayed elongation zone-specific accumulation. SEA revealed no overrepresented GO term.
Among 384 nonmodified cortex-specific proteins (274 annotated), nine overrepresented GO terms were identified. Eight of the nine GO terms belonged to the category molecular function, predominantly reflecting proteins with catalytic activity (e.g. GO:0016747 and GO:0016491) or binding function (e.g. GO:0046906 and GO:0020037; Supplemental Table S1). No GO term showed overrepresentation among the 248 phosphorylated cortex-specific proteins (171 annotated).
In total, 18 GO terms were overrepresented among the 305 stele-specific nonmodified proteins (187 annotated). Four of these GO terms were predominantly involved in lignin metabolism (Supplemental Table S1), while two GO terms were assigned to the category cellular component (GO: 0048046 and GO:0005576). Almost 50% (59/120) of all overrepresented GO terms were detected among the 241 phosphorylated stele-specific proteins (101 annotated). These proteins show, among others, transferase and transporter activities. The GO term GO:0009055, referring to proteins with electron carrier activity, was overrepresented among nonmodified proteins of the cortex and stele. This indicates that there are at least some conserved functions in these two primary root tissues.
In summary, only one of the 120 overrepresented GO terms was detected in two different tissues, whereas 119 GO terms showed tissue-specific overrepresentation, indicating highly regulated tissue-specific functions of some proteins.
Differential Expression of Protein Accumulation between the Four Primary Root Tissues
Differentially accumulated nonmodified and phosphorylated proteins were identified based on the six possible pairwise combinations between the four tissues: (1) elongation zone versus meristematic zone, (2) cortex versus meristematic zone, (3) stele versus meristematic zone, (4) cortex versus elongation zone, (5) stele versus elongation zone, and (6) stele versus cortex. Among 11,552 nonmodified proteins, between 326 (meristematic zone versus elongation zone) and 3,530 (meristematic zone versus cortex) were differentially expressed between two tissues (Supplemental Table S2). After functional annotation, between 318 (meristematic zone versus elongation zone) and 3,460 (meristematic zone versus cortex) were differentially expressed (Fig. 2A). In all comparisons, the elongation zone preferentially accumulated more differential proteins compared with the other zones, followed by the meristematic zone, cortex, and stele, leading to following protein accumulation trend: elongation zone > meristematic zone > cortex > stele.
Figure 2.

Number of up- and down-regulated nonmodified proteins in six pairwise tissue comparisons. A, Total number of up- and down-regulated proteins mapped to the 35 MapMan bins in the six comparisons. Blue and red bars in each comparison indicate the number of genes preferentially expressed in the tissue indicated on top of the bars. B, Functional classes in which the ratio of up- and down-regulated proteins differed significantly from the expected value (compare with A) in at least four of six pairwise tissue comparisons. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. C, Tissue-specific protein accumulation trends based on the results displayed in B. C, Cortex; EZ, elongation zone; MZ, meristematic zone; S, stele.
Among the 2,852 phosphorylated proteins, between 46 (meristematic zone versus elongation zone and cortex versus stele) and 919 (meristematic zone versus cortex) displayed differential accumulation (Supplemental Table S2), resulting in 44 (meristematic zone versus elongation zone) and 886 (meristematic zone versus cortex) differentially accumulated proteins after functional MapMan annotation (Supplemental Fig. S1A). Here, the following trend of protein abundance was observed: cortex > elongation zone > stele > meristematic zone. In comparisons 1 (meristematic zone versus elongation zone) and 6 (cortex versus stele), only a small number of differentially accumulated proteins were detected in both data sets, which might be the consequence of the close relation of the meristematic zone with the elongation zone and the cortex with the stele.
MapMan Annotation of Differentially Accumulated Root Tissue Proteins
The MapMan bin classification system was used to further analyze the function of the differentially accumulated nonmodified and phosphorylated proteins (Fig. 2A; Supplemental Fig. S1A) and to compare the distribution of up- and down-regulated proteins between tissues. For each comparison, the distribution of up- and down-regulated proteins across all MapMan bins was used as the expected value for further analyses. In a second step, the distribution of up- and down-regulated proteins in each of the 35 functional classes (MapMan bins) was compared with the expected value using Fisher’s exact test (two tailed). In eight classes, the distribution of up- and down-regulated proteins differed significantly from the expected value for at least four of six tissue comparisons (Fig. 2A), including cell wall, DNA, lipid metabolism, protein, RNA, secondary metabolism, stress, and transport (Fig. 2B; Supplemental Fig. S2). In the functional classes DNA, RNA, and protein, very similar distributions of protein up- and down-regulation were observed, resulting in the following trend of protein abundance: meristematic zone > elongation zone > stele > cortex, with the highest accumulation in the meristematic zone and gradually lower abundance in the elongation zone, the stele, and the cortex (Fig. 2C). Exactly the opposite protein accumulation trend was identified for the functional classes lipid metabolism, secondary metabolism, and transport: cortex > stele > elongation zone > meristematic zone, with the highest abundance in the cortex and the lowest abundance in the meristematic zone. Up- and down-regulated proteins among the six pairwise comparisons involved in cell wall and stress showed an expression trend of stele > cortex > elongation zone > meristematic zone (Fig. 2C).
The same analysis was conducted for the six sets of differentially accumulated phosphorylated proteins (Supplemental Figs. S1A and 2). Again, the assignment of all of these proteins to 35 functional classes was used as the expected distribution of up- and down-regulated proteins for subsequent analyses. Afterward, the bin-specific distribution of phosphorylated up- and down-regulated proteins was compared with the expected value. This comparison revealed numerical changes in the ratio of up- and down-regulated proteins in seven MapMan bins for at least one of six tissue comparisons (Supplemental Fig. S1B). For the differentially accumulated phosphorylated proteins in the classes DNA and RNA, the same expression trend as for nonmodified proteins was detected: meristematic zone > elongation zone > stele > cortex (Supplemental Fig. S1C). In contrast, proteins of the classes cell wall, major carbohydrate metabolism, protein, signaling, and transport showed a different expression trend: stele > cortex > elongation zone > meristematic zone (Supplemental Fig. S1C).
Classical Maize Genes among the Nonmodified Proteins
Classical maize genes are genes that were initially identified by visible mutant phenotypes before being cloned and characterized (Schnable and Freeling, 2011). To further characterize the tissue-specific nonmodified proteins, they were compared with the list of 468 classical maize genes (https://genomevolution.org/wiki/index.php/Classical_Maize_Genes; Schnable and Freeling, 2011). Among the nonmodified proteins, 242 proteins encoded by classical maize genes were detected (Fig. 3A; Supplemental Table S3). Similar numbers of classical maize proteins, ranging from 206 (stele) to 218 (meristematic zone), were identified in the distinct tissues. Among the nonmodified proteins, between two (elongation zone and stele) and 12 (meristematic zone) proteins encoded by genes of the classical maize gene list were present in only one tissue (Fig. 3A). Among the 10 cortex-specific proteins encoded by classical maize genes, six enzymes catalyze the initial steps of flavonoid biosynthesis, the synthesis of reduced glutathione (GSH)-anthocyanidin-glucoside from phenylpropanoids to GSH-anthocyanidin-glucoside (Fig. 3B).
Figure 3.

Maize proteins encoded by classical maize genes (i.e. genes that were first identified based on their mutant phenotypes). A, Tissue-specific accumulation of classical maize proteins identified among nonmodified proteins. *, Six of 10 cortex-specific proteins highlighted in red catalyze the initial steps of flavonoid biosynthesis. C, Cortex; EZ, elongation zone; MZ, meristematic zone; S, stele. B, Schematic of the initial steps of flavonoid biosynthesis including six of the 10 cortex-specific (asterisk in A) classical maize proteins (red): CHS, CHALCONE SYNTHASE; F3H, FLAVONONE 3-HYDROXYLASE; DFR, DIHYDROFLAVONOL-4-REDUCTASE; LDOX, LEUCOANTHOCYANIDIN DIOXYGENASE; UGT, UDP-GLUCOSYLTRANFERASE; GST, GLUTATHIONE S-TRANSFERASE. Classical gene names are highlighted in boldface.
Correlation of Protein and Transcript Abundance
Global correlations between mRNA and protein levels in the four tissues were surveyed by comparing the nonmodified proteome data set with RNA-Seq transcript profiling data from our laboratory (Paschold et al., 2014). For the meristematic zone, 7,092 accessions present in the transcriptome and proteome data set were identified, whereas 6,436 accessions for the elongation zone, 5,850 accessions for the cortex, and 5,542 accessions for the stele were determined (Supplemental Data Set S5). Pearson correlations revealed mRNA-protein correlation coefficients of 0.5 in the meristematic zone, 0.43 in the elongation zone, 0.38 in the cortex, and 0.38 in the stele (Table II). Spearman rank correlation coefficients were slightly higher for the meristematic zone (0.56), elongation zone (0.45), and 0.41 (stele) and similar for the cortex (0.38). In summary, Pearson and Spearman rank correlation coefficients displayed moderate positive correlation of RNA and protein abundance in the distinct primary root tissues. Tissue-specific comparison of transcripts and proteins demonstrated that, in all four tissues, a transcript was detected in 93% of proteins. Remarkably, for 7% of proteins, no transcript was found in the corresponding tissue (Supplemental Fig. S3).
Table II. Correlation between mRNA and protein levels within the four tissues: meristematic zone (MZ), elongation zone (EZ), cortex (C), and stele (S).
***P ≤ 0.0001.
| Tissue | No. of Genes | Pearson Correlation | Spearman Rank Correlation |
|---|---|---|---|
| MZ | 7,092 | 0.50*** | 0.56*** |
| EZ | 6,436 | 0.43*** | 0.45*** |
| C | 5,850 | 0.38*** | 0.38*** |
| S | 5,542 | 0.38*** | 0.41*** |
Comparison of Nonmodified Proteins between Roots, Seeds, and Leaves
Proteome data sets of seed and leaf tissues of the maize inbred line B73 were generated previously by the same analysis pipeline, revealing 12,453 and 12,032 nonmodified proteins from seeds (Walley et al., 2013) and leaves (Facette et al., 2013), respectively. Comparison of these protein sets with the 11,552 nonmodified proteins identified in primary root tissues in this study allowed for the identification of common and tissue-specific proteins. In total, 7,902 proteins, which correspond to 68% of all root proteins identified in this study, accumulated in all three maize organs (Fig. 4A). Organ-specific accumulation of nonmodified proteins was detected for 1,504 accessions in roots (13% of root proteins), 1,611 in leaf (13% of leaf proteins), and 2,110 in seed (20% of seed proteins). In summary, 16,680 different nonmodified proteins representing unique accessions were identified in roots, seeds, and leaves. Among 1,504 root-specific proteins, 342 proteins (23%) were detected in all four analyzed tissues (Fig. 4B), while 618 proteins (41%) accumulated in two or three tissues. Tissue specificity was observed for 544 proteins (36%), including 156 proteins in the meristematic zone, 68 in the elongation zone, 171 in the cortex, and 149 in the stele. agriGO SEA of the organ-specific proteins revealed 41 overrepresented GO terms among 873 annotated proteins identified exclusively in roots. These GO terms are mainly involved in response to chemical stimulus, oxidative stress, and lignin metabolic processes (FDR < 5%; Supplemental Table S4). In the seed-specific data set, 1,201 GO terms were annotated and 19 were overrepresented, reflecting, among others, defense proteins and biosynthesis-regulating proteins. Among 36 overrepresented GO terms (out of 892 annotated GO terms) of the leaf-specific data sets, mainly proteins involved in photosynthesis were detected, which is in line with their role in converting light energy in carbohydrates (Supplemental Table S4).
Figure 4.

A, Organ-specific accumulation of nonmodified proteins detected in primary root, seed, and leaf. B, Protein accumulation pattern of the 1,504 primary root-specific proteins in distinct primary root tissues. C, Cortex; EZ, elongation zone; MZ, meristematic zone; S, stele.
DISCUSSION
Analysis of four maize primary root tissues revealed that, among 11,552 identified nonmodified proteins, 61% were expressed in all four tissues (Fig. 1B), compared with 28% that were accumulated in two or three tissues and 11% active in only one tissue. Similarly, 10% of the accumulated proteins in developing maize seeds exhibited accumulation in only one tissue (Walley et al., 2013), whereas in different regions of the maize leaf, only 4% of nonmodified proteins showed zone-specific accumulation (Facette et al., 2013). In contrast to the nonmodified proteins, only 30% of phosphorylated proteins were detected in all four tissues (844/2,852 phosphoproteins), whereas 39% were present in two or three tissues and 31% were present in only one tissue. Similar results were reported from differentiating leaf zones (Facette et al., 2013) and differentiating human embryonic stem cells (Rigbolt et al., 2011), where the phosphoproteome displayed more dynamic changes than in the nonmodified proteome.
The identification of peptide phosphorylation sites in this study revealed the relative abundance of Ser (84%), Thr (15%), and Tyr (1%) phosphorylation. Recent studies showed similar distributions of Ser (86%), Thr (13%), and Tyr (1%) in maize leaves and seeds (Facette et al., 2013; Walley et al., 2013). This allowed for the reconstruction of a kinases-substrate regulatory network in maize seeds (Walley et al., 2013) and identified previously unknown phosphorylation sites in cell wall-related proteins of developing maize leaves (Facette et al., 2013). In general, in eukaryotes, Ser and Thr residues are predominantly phosphorylated, whereas phosphorylation of Tyr residues is less common (de la Fuente van Bentem and Hirt, 2009). This was observed not only in maize but also in Arabidopsis (Arabidopsis thaliana; Ser [85%], Thr [11%], and Tyr [4%]; Sugiyama et al., 2008) and human embryonic stem cells (Ser [80%], Thr [17%], and Tyr [3%]; Rigbolt et al., 2011).
Functional analysis of nonmodified proteins and phosphoproteins accumulated in only one tissue was based on the GO analysis toolkit (Du et al., 2010). In parallel, differentially accumulated proteins between six pairwise comparisons were functionally characterized using the MapMan bin classification system (Thimm et al., 2004). Among the nonmodified and phosphorylated proteins accumulated exclusively in the meristematic zone, the GO terms DNA binding and DNA metabolic processes were overrepresented. In line with that, proteins belonging to the MapMan classes DNA, RNA, and protein were up-regulated in the meristematic zone and decreased gradually in more differentiated tissues, underscoring the high mitotic activity of the meristematic zone (Ishikawa and Evans, 1995; Verbelen et al., 2006). The increased rate of mitosis in the meristematic zone was underscored by overrepresented GTP-binding proteins (G proteins; bin 30.5), cell division (bin 31.2), and cell cycle proteins (bin 31.3; Supplemental Fig. S4). G proteins are regulators for diverse signaling pathways in plants (e.g. flower, leaf, and fruit development and seed germination; for review, see Perfus-Barbeoch et al., 2004). Several studies showed that G proteins also modulate cell division in Arabidopsis roots (Ullah et al., 2003; Chen et al., 2006) and rice (Oryza sativa; Ashikari et al., 1999). Therefore, an overrepresentation of G proteins might lead to an increase of cell cycle proteins, resulting in higher cell division rates in the meristematic zone.
In contrast, differentially accumulated nonmodified proteins related to cell wall, lipid metabolism, secondary metabolism, stress, and transport were higher expressed in the differentiated cortex and stele versus the undifferentiated meristematic and elongation zones (Fig. 2C). In line with the differentiation status of the stele, several GO terms involved in lignin biosynthesis (related to the bins cell wall and secondary metabolism) were overrepresented in this tissue (Supplemental Table S1). Lignin is deposited in the xylem elements of the stele to provide structural support (Vermerris et al., 2010; Zhang et al., 2014). In line with the overrepresentation of lignin biosynthesis-related GO terms, six laccases (GRMZM2G146152_P01, GRMZM2G164467_P01, GRMZM5G814718_P01, GRMZM2G367668_P01, GRMZM2G388587_P01, and GRMZM2G305526_P01) were accumulated exclusively in the stele (Supplemental Data Sets S1 and S3). Laccases are multicopper-containing glykoproteins that are involved in lignin polymerization in plants (for review, see O’Malley et al., 1993). In a variety of plant species, laccases are expressed in lignified cells, as for instance in the vasculature of Arabidopsis (Zhao et al., 2013), the xylem of loblolly pine (Pinus taeda; Bao et al., 1993), and maize roots (Liang et al., 2006). Furthermore, stele-specific phosphoproteins related to the GO term transmembrane transporter activity were enriched, suggesting that transporters are more extensively regulated by phosphorylation. Accordingly, the MapMan analysis revealed an up-regulation of phosphorylated transport proteins (bin 34) in cortex and stele (Supplemental Fig. S1). Among such transport proteins, 50% of all ATP-binding cassette (ABC) transporters (11 of 22) identified in the phosphorylated data set preferentially accumulated in cortex or stele (Supplemental Fig. S5). ABC transporters are transmembrane proteins enabling the transport of hormones, lipids, metals, and other molecules (for review, see Higgins, 1992) via ATP hydrolysis at the ABC, representing a nucleotide-binding domain (Higgins and Linton, 2004). ABC transporters, at least in humans, are regulated by phosphorylation via protein kinases, which was recently reviewed (Stolarczyk et al., 2011). Among the stele-specific phosphoproteins, GO terms related to protein kinases were also enriched (Supplemental Table S1), which could be in line with the high ABC transporter activity and its activation by kinase-dependent phosphorylation in this tissue (Stolarczyk et al., 2011).
In a previous study based on two-dimensional gel electrophoresis, 70 differentially accumulated protein spots, representing 56 different GenBank accessions, were identified between cortex and stele of 2.5-d-old maize primary roots (Saleem et al., 2010). Only 18 of the 56 unique proteins (Supplemental Data Set S3) were also present in the B73 RefGen_v2 5a working gene set (www.gramene.org), while the remaining 38 proteins were identified based on homology with proteins of other plant species. Among these 18 cortex-specific maize proteins (Saleem et al., 2010), 15 (83%) were preferentially accumulated in the cortex versus the stele in the present data set. In the study by Saleem et al. (2010), preferential accumulation of a β-glucosidase (GRMZM2G016890; GenBank accession no. P49235) enzyme in the cortex was reported. This enzyme functions in the release of free bioactive cytokinin (Brzobohatý et al., 1993). This finding was underscored and extended by this study, as predominant accumulation of not only one but all 12 β-glucosidase enzyme accessions identified in this study were preferentially accumulated in the cortex (Supplemental Data Set S3). Furthermore, four enzymes involved in ammonium assimilation showed cortex-specific expression in the study by Saleem et al. (2010). Those authors suggested that the maize primary root cortex is the site of ammonium assimilation in young seedlings before exogenous nitrogen is assimilated from the soil. Similarly, in this study, most enzymes related to ammonium assimilation showed preferential accumulation in the cortex (Supplemental Data Set S3): two Glu dehydrogenases showed cortex-specific expression, among five Gln synthetases four revealed this trend, three of five Asp aminotransferases showed preferential accumulation in cortex compared with stele, and finally, among four Asn synthetases two were cortex specific.
To gain further functional insights, the tissue-specific nonmodified proteins were compared with the list of classical maize genes that were initially identified by their mutant phenotypes (Schnable and Freeling, 2011). This survey identified six cortex-specific proteins that are involved in the initial steps of flavonoid biosynthesis by catalyzing the synthesis of GSH-anthocyanidin-glucoside (Fig. 3B). Flavonoids belong to a large family of polyphenolic secondary metabolites, which are involved in a wide range of biological processes such as signaling in plant-microbe interactions and protection against ultraviolet light (Koes et al., 1994). Flavonoids accumulate in most plant tissues, often in the epidermis of plant organs, to protect subjacent tissues from radiation damage (Saslowsky and Winkel-Shirley, 2001). In contrast to aboveground tissue, where flavonoid accumulation can easily be traced by the coloring of flowers or seeds (Koes et al., 1994), there is little knowledge about flavonoid function in maize root tissues. Accumulation of flavonoids has been demonstrated in the endodermis, cortex, and epidermis of white clover (Trifolium repens; Hassan and Mathesius, 2012), which is in line with the accumulation of enzymes of flavonoid biosynthesis, as demonstrated here. ABC transporters, discussed above, are able to transport flavonoid glycosides such as GSH-anthocyanidin-glucoside to the vacuole (Goodman et al., 2004) or to export them through the plasma membrane for long-distance transport (Buer et al., 2007).
The analysis of three different maize organs, including root tissues surveyed in this study, seeds (Walley et al., 2013), and leaves (Facette et al., 2013), revealed between 1,504 and 2,110 organ-specific proteins (Fig. 4A) and nearly 8,000 proteins accumulated in diverse organs. SEA identified more general overrepresented GO terms, such as protein folding or cellular amine metabolic process, among the overlapping proteins (data not shown), whereas more specific overrepresented GO terms were identified among the organ-specific proteins (Supplemental Table S4). In line with their function, several GO terms related to lignin and cell wall biosynthesis were overrepresented among the root-specific proteins. In the endodermis and around xylem vessels of roots, the cell wall polymer lignin is deposited to a high extent to provide mechanical support for the plant body (Vermerris et al., 2010). In young developing roots, cell wall biosynthesis and, therefore, also lignification might be increased (e.g. because of rapid cell division). Among the leaf-specific proteins, many GO terms related to photosynthesis were overrepresented. This is related to the major function of leaves, which is the photosynthetic conversion of light energy into chemical energy in the form of carbohydrates. Compared with the relatively low number (11%) of tissue-specific nonmodified proteins identified among all 11,552 proteins identified in our data set (Fig. 1B), more than three times as many proteins (36%) were detected to be tissue specific among the 1,504 root-specific proteins (Fig. 4B). This suggests that root-specific proteins have more tissue-specific functions compared with general proteins, which are also expressed in nonroot tissues.
Traditionally, mRNA abundance measurements were used to infer protein abundance (for review, see Vogel and Marcotte, 2012). However, a growing body of research has found only moderate correlation between transcript and protein abundance (Gygi et al., 1999; Schwanhäusser et al., 2011; Petricka et al., 2012; Vogel and Marcotte 2012; Walley et al., 2013; Ponnala et al., 2014). In this study, Pearson and Spearman rank correlation coefficients revealed moderate positive relationships between transcript and protein abundance in primary root tissues (Table II). Both coefficients were used in previous studies to calculate mRNA-protein relationships (for review, see Maier et al., 2009; Vogel and Marcotte, 2012). In general, the relationship between mRNA and protein abundance is considered positive in a wide range of organisms but varies greatly between the studies (yeast [Lee et al., 2011], bacteria [Maier et al., 2011], maize endosperm and embryos [Walley et al., 2013], and maize leaf sections [Ponnala et al., 2014]). Variations in mRNA-protein correlation might be explained by many factors, such as bias in RNA and protein stability and movement, experimental measurement, and quantification methods (Ponnala et al., 2014). In addition, one transcript must not necessarily encode for only one protein but can give rise to many different proteins by alternative splicing or a wide range of protein modifications. These variants might have different half-lives; therefore, the observed protein stabilities reflect average stabilities of multiple isoforms rather than one-to-one protein-transcript relationships. Furthermore, the correlation of transcript and protein might vary as a result of translational mechanisms regulating and fine-tuning the translation of transcripts, such as by changes in the relative synthesis rate (Kristensen et al., 2013) or adjusting ribosome density (Liu et al., 2013). In this study, between 5,543 (stele) and 7,092 (meristematic zone) gene accessions were detected in both the transcriptome and proteome analyses (Table II). In all four tissues, 93% of all identified proteins were also found in the RNA-Seq data set. Remarkably, for 7% of proteins, no corresponding transcript was identified.
A similar observation was made previously in a correlation analysis of maize seeds, where many abundant proteins were not supported by a corresponding mRNA (Walley et al., 2013). The lack of transcripts for detected proteins can have different reasons. For instance, while proteins can be very stable, their corresponding transcript levels can oscillate because of circadian regulation of gene expression (Khan et al., 2010; Walley et al., 2013). In our data set, we identified three proteins where the corresponding genes are known to be regulated by the circadian rhythm (Khan et al., 2010): GRMZM2G055795 (elongation zone), GRMZM2G083841 (elongation zone and stele), and GRMZM2G097457 (cortical parenchyma). As a consequence, we only identified proteins but no transcripts of these accessions. Similarly, in maize endosperm and embryo, circadian-regulated proteins displayed a higher abundance than their transcripts (Walley et al., 2013). Another reason for the lack of transcripts is that proteins move into another tissue after translation. There are several examples in our data set that support this notion. For instance, a heat shock protein (GRMZM5G833699) was present in all tissues, whereas its transcript was detected only in the elongation zone, cortical parenchyma, and stele but not in the meristematic zone (Supplemental Data Set S3; Paschold et al., 2014). Similarly, a SUCROSE SYNTHASE2 (GRMZM2G318780) protein was detected in all four tissues, whereas the corresponding mRNA was lacking in the elongation zone. Similarly, a short-chain dehydrogenase/reductase (GRMZM2G066840) protein was present in all four tissues, whereas transcripts were detected only in the meristematic zone, elongation zone, and cortical parenchyma. In a recent study comparing endosperm versus whole seed proteomes and transcriptomes of maize, 15 proteins were detected in both tissues, while the RNA was detected in only one (Walley et al., 2013).
In summary, our data set represents a high-resolution proteome map of four distinct maize primary root tissues, revealing the functional diversity of the distinct root zones. Therefore, this data set will be a helpful resource and starting point for future in-depth analyses of metabolic functions of root tissues.
MATERIALS AND METHODS
Plant Material
Seeds of the maize (Zea mays) inbred line B73 were germinated in paper rolls as described previously (Hetz et al., 1996). Seedlings were grown for 5 d in the greenhouse without supplemental lighting. Then, primary roots of 2 to 4 cm in length were dissected into four distinct tissues (Fig. 1). The first 2 mm of the root tip, containing the root cap and the subterminal meristematic zone, was collected as the meristematic zone. Moreover, the proximal zone adjacent to the root tip up to the region where the root hairs emerge was sampled as the elongation zone. Finally, the differentiation zone, starting with the root hair zone up to the coleorhiza, was mechanically separated into cortex and stele according to Saleem et al. (2010). For each tissue, 200 roots were pooled for each of the four biological replicates. All samples were immediately frozen in liquid nitrogen and stored at −80°C until protein extraction.
Protein Extraction and Phosphopeptide Enrichment
Proteins were isolated from 2 g of frozen tissue per biological replicate. After grinding the tissue in frozen nitrogen, proteins were precipitated in methanol containing 0.2 mm Na3VO4 and washed three times in methanol followed by three washing steps in acetone. Precipitation and washing of proteins were performed at −20°C. Protein pellets were dried in a vacuum concentrator at 4°C. Subsequently, protein pellets were resuspended in 1 mL of extraction buffer (0.1% [w/v] SDS, 1 mm EDTA, and 50 mm HEPES buffer, pH 7). Cys residues were reduced using 1 mm Tris(2-carboxyethyl)phosphine at 95°C for 5 min and subsequently alkylated with 2.5 mm iodoacetamide at 37°C in the dark for 15 min. Proteins were digested overnight using trypsin (Roche; enzyme:substrate = 1:100, w/w). A second tryptic digestion (enzyme:substrate = 1:100, w/w) was performed the next day for 4 h. Digested peptides were purified on a Waters Oasis MCX cartridge to remove SDS. Peptides were eluted from the MCX column with 1 mL of 50% (v/v) isopropyl alcohol and 400 mm NH4HCO3 (pH 9.5) and then dried in a vacuum concentrator at 4°C. For proteome profiling of nonmodified proteins, peptides were resuspended in 1% (v/v) formic acid to a final pH of 3 and used for mass spectrometric analyses. For phosphoproteome profiling, peptides were resuspended in 3% (v/v) trifluoroacetic acid to a final pH of 1 and then subjected to phosphopeptide enrichment.
For phosphopeptide enrichment, 1% (w/v) colloidal CeO2 was added to the acidified peptide solution at 1:10 (w/w). CeO2 with captured phosphopeptides was briefly vortexed and then centrifuged at 1,000g for 1 min. After removing the supernatant, the CeO2 pellet was washed with 1 mL of 1% (v/v) trifluoroacetic acid. By adding eluting buffer [200 mm (NH4)2HPO4, 2 m NH3·H2O, and 10 mm EDTA, pH 9.5; identical volume as the added 1% [w/v] colloidal CeO2], phosphopeptides were eluted.
Mass Spectrometry and Peptide Identification
Mass spectrometry (MS) and peptide identification were performed as described previously (Facette et al., 2013; Walley et al., 2013). In brief, an 1100 HPLC system (Agilent Technologies) delivered a flow rate of 600 nL min−1 to a three-phase capillary chromatography column through a splitter. A custom pressure cell was used to pack 5 µm of Zorbax SB-C18 (Agilent Technologies) into fused silica capillary tubing (250-µm i.d., 360-µm o.d., 30 cm long) to form the first dimension reverse-phase (RP1) column. A 5-cm-long strong cation-exchange (SCX) column packed with 5 µm of polysulfoethyl (PolyLC) was connected to the RP1 column using a zero dead volume 1-µm filter (Upchurch; M548) attached to the exit of the RP1 column. As an analytical column, the fused silica capillary (200-µm i.d., 360-µm o.d., 20 cm in length) packed with 5 µm of Zorbax SB-C18 (Agilent Technologies) was connected to the SCX column. The electrospray tip of the fused silica tubing was pulled to a sharp tip with the i.d. smaller than 1 µm using a P-2000 (Sutter Instrument) laser puller. Using the custom pressure cell, the peptide mixtures were loaded onto the RP1 column. For each liquid chromatography-MS/MS analysis, a new set of columns was used. First, the peptides were eluted from the RP1 column to the SCX column using a 0% to 80% acetonitrile gradient for 150 min. The peptides were then fractionated by the SCX column using a series of salt gradients (from 10 mm to 1 m ammonium acetate for 20 min each), followed by high-resolution reverse-phase separation using an acetonitrile gradient of 0% to 80% for 120 min.
Spectra were analyzed on the LTQ Velos linear ion trap tandem mass spectrometer (Thermo Electron), which employed automated, data-dependent acquisition. The mass spectrometer was operated in positive ion mode with a source temperature of 250°C. As a final fractionation step, gas phase separation in the ion trap was employed to separate the peptides into three mass classes prior to scanning (300–800, 800–1,100, and 1,100–2,000 D). Each MS scan was followed by five MS/MS scans of the most intense ions from the parent MS scan. A dynamic exclusion of 1 min was used to improve the duty cycle.
Database Search
Spectrum Mill version 3.03 (Agilent Technologies) was used to extract and search the raw data. MS/MS spectra with a sequence tag length of 1 or less were discarded. The remaining MS/MS spectra were searched against the maize B73 RefGen_v2 5a working gene set downloaded from www.gramene.org. The enzyme parameter was limited to full tryptic peptides, allowing a maximum miscleavage of 1. All other search parameters were set to Spectrum Mill’s default settings (carbamidomethylation of Cys, ±2.5 D for precursor ions and ±0.7 D for fragment ions; and a minimum matched peak intensity of 50%). For the phosphoproteome data, oxidation of Met, N-terminal pyro-Gln, and phosphorylation on Ser, Thr, or Tyr were defined as variable modifications. A maximum of two modifications per peptide were used. A 1:1 concatenated forward:reverse database was constructed to calculate the FDR. The tryptic peptides in the reverse database were compared with the forward database and were shuffled if they matched to any tryptic peptides from the forward database. Peptide cutoff scores were dynamically assigned to each data set to maintain the FDR less than 0.1% at the peptide level. Maintaining a data set level FDR of 0.1% resulted in FDRs for the nonmodified proteome of 0.57 at the protein level (66 decoy hits among 11,618 protein group leaders) and 2.1 at the phosphoprotein level (61 decoy hits among 2,913 phosphoprotein group leaders). Phosphorylation sites were localized to a particular amino acid within a peptide using the VML score in Agilent’s Spectrum Mill software (Chalkley and Clauser, 2012). A sequence VML score of greater than 0.5 defined a particular amino acid as phosphorylated, whereas a VML score of 0.05 or less defined an ambiguous residue. Proteins that share common peptides were grouped using principles of parsimony to address the protein database redundancy. Thus, proteins within the same group shared the same set or subset of unique peptides. A specific protein of each protein group was assigned as a group leader if it was presented by the highest number of unique peptide identifications or showed the highest value of protein coverage. Protein abundance and phosphorylation levels were quantified by spectral counting. Spectral counts for each protein represent the total number of peptide spectral matches to that protein (Liu et al., 2004; Huttlin et al., 2010; Walley et al., 2013). Biological replicates were normalized as described previously (Marcon et al., 2010, 2013). Briefly, raw SPCs of each biological replicate were normalized by assigning 500,000 SPCs to the sum of all proteins detected in the distinct run. The resulting normalization factor (assigned sum of SPCs divided by the sum of SPCs of each run) was then multiplied with each protein SPC of each run to allocate the assigned sum of 500,000 SPCs. Expressed proteins were defined by at least one SPC in one of the four biological replicates.
GO Analyses
GO functional categories were assigned to tissue specifically accumulated proteins using the Web-based agriGO software (http://bioinfo.cau.edu.cn/agriGO/analysis.php). An SEA was used to compute enriched categories by comparing a list of tissue-specific proteins or tissue-specific phosphoproteins with all expressed proteins (11,552) or phosphoproteins (2,852), as described previously (Du et al., 2010). According to Benjamini and Yekutieli (2001), multiple comparison correction was performed and FDR was controlled at 5%. The SEACOMPARE tool allowed for combined cross comparisons of SEA results.
Functional Analysis of Differentially Accumulated Proteins
Raw data were normalized by the sum of the proteins detected in a sample and then converted to counts per million. A small value of 0.5 was added to avoid zero counts. Count data typically show nontrivial mean variance relationships. Raw counts show increasing variance with increasing count size, while log counts typically show a decreasing mean variance trend. We estimated the mean variance trend for log counts and then assigned a weight to each observation based on its predicted variance. The weights were then used in the linear modeling process to adjust for heteroscedasticity (Law et al., 2014).
To borrow strength across proteins in the estimation of the residual error variance, we used the empirical Bayes approach of Smyth (2004) implemented in the R package limma. Using Smyth’s moderated t statistic makes use of all estimated sample variances, which are shrunk to a pooled estimate. Especially in cases where few replicates are available, inference is far more stable in comparison with the ordinary t statistic. We fitted the following linear model for each gene:
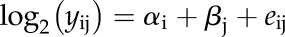 |
where log2 (yij) is the log2-transformed value of the normalized spectral read counts per million on protein level of the ith tissue in the jth sample. αi is the fixed effect of the tissue (i = 1, 2,…,4), βj is the random sample effect (j = 1, 2,…,4), and eij is the corresponding residual, assumed to be independently and identically distributed as N(0,σ2). P values over genes were then corrected for multiplicity using the FDR (Benjamini and Hochberg, 1995).
The MapMan bin classification system (Thimm et al., 2004) was used for functional annotations of proteins displaying differential accumulation in the following tissue comparisons: meristematic zone versus elongation zone, meristematic zone versus cortex, meristematic zone versus stele, elongation zone versus cortex, elongation zone versus stele, and cortex versus stele. The MapMan functional annotation file Zm_B73_5b_FGS_cds_2012 (http://mapman.gabipd.org) was used for the analysis.
Correlation Analysis
To assess the accuracy of the biological replicates, Pearson correlation coefficients of averaged normalized SPCs of the four biological replicates per tissue were calculated as described previously (Walley et al., 2013). To determine the relationship of mRNA and protein, RNA-Seq data from our laboratory (Paschold et al., 2014) were compared with the nonmodified protein data set of this study. The RNA-Seq data set was generated using the same tissues, in the same genotype (B73) at the same developmental stage, and mapped to the ZmB73 FGS_5B_filtered gene set version 2 (www.gramene.org). For global correlation analyses of transcript and protein abundance, the R package was used to calculate Pearson correlation and Spearman rank correlation of normalized log2-transformed data. To be included in the correlation analysis, we required that both the mRNA and protein were detected and that the mRNA and protein were present in at least three biological replicates. Pearson correlation measures a linear correlation between two variables, which is more appropriate if the variables have a jointly normal distribution, whereas the Spearman rank correlation quantifies monotonic correlation between two variables, which is less restrictive than Pearson correlation (Ponnala et al., 2014). Such a monotonic correlation could be the more appropriate assumption for our correlation analysis, since mRNA-protein abundance relationships may be nonlinear (de Sousa Abreu et al., 2009; Maier et al., 2009).
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Number of up- and down-regulated phosphorylated proteins resulting from six pairwise comparisons of the four maize primary root tissues.
Supplemental Figure S2. Distribution of differentially accumulated nonmodified and phosphorylated proteins between six tissue pairwise comparisons into 35 distinct MapMan bins.
Supplemental Figure S3. Number of gene accessions identified among nonmodified proteins and transcripts.
Supplemental Figure S4. Distribution of differentially accumulated nonmodified proteins in six pairwise comparisons of the four primary root tissues into distinct MapMan bins.
Supplemental Figure S5. Preferential accumulation of 11 ABC transporters in the cortex and stele of primary roots.
Supplemental Table S1. Enriched GO terms of tissue-specific proteins of nonmodified and phosphorylated protein data sets using SEA.
Supplemental Table S2. Number of differentially accumulated proteins (FDR = 5%) in the nonmodified and phosphorylated protein data set.
Supplemental Table S3. Classical maize protein identifications and their tissue-specific accumulation identified among nonmodified proteins.
Supplemental Table S4. A SEA of proteins being organ-specifically accumulated among nonmodified proteins of maize.
Supplemental Data Set S1. Raw spectral counts of nonmodified proteins.
Supplemental Data Set S2. Raw spectral counts of phosphorylated proteins.
Supplemental Data Set S3. Normalized spectral counts of nonmodified proteins.
Supplemental Data Set S4. Normalized spectral counts of phosphorylated proteins.
Supplemental Data Set S5. Raw and normalized reads and spectral counts for each gene which was observed in at least three biological replicates of the mRNA and nonmodified protein data set.
Supplementary Material
Acknowledgments
We thank Michelle Facette (University of California, San Diego) for helpful discussions and Philipp Zimmermann (University of Bonn) for helping to organize parts of the “Supplemental Data.”
Glossary
- MS/MS
tandem mass spectrometry
- RNA-Seq
RNA sequencing
- VML
variable modification localization
- SPC
spectral count
- GO
Gene Ontology
- FDR
false discovery rate
- SEA
singular enrichment analysis
- GSH
reduced glutathione
- ABC
ATP-binding cassette
- MS
mass spectrometry
- SCX
strong cation-exchange
- RP1
first dimension reverse-phase
- HPLC
high-performance liquid chromatography
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (grant no. HO2249/9–1 to F.H. and grant no. PI 377/12–1 to H.-P.P.) and by the National Science Foundation (grant no. 0924023 to S.P.B.).
Articles can be viewed without a subscription.
References
- Ashikari M, Wu J, Yano M, Sasaki T, Yoshimura A (1999) Rice gibberellin-insensitive dwarf mutant gene Dwarf 1 encodes the alpha-subunit of GTP-binding protein. Proc Natl Acad Sci USA 96: 10284–10289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao W, O’Malley DM, Whetten R, Sederoff RR (1993) A laccase associated with lignification in loblolly pine xylem. Science 260: 672–674 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57: 289–300 [Google Scholar]
- Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann Stat 29: 1165–1188 [Google Scholar]
- Brzobohatý B, Moore I, Kristoffersen P, Bako L, Campos N, Schell J, Palme K (1993) Release of active cytokinin by a beta-glucosidase localized to the maize root meristem. Science 262: 1051–1054 [DOI] [PubMed] [Google Scholar]
- Buer CS, Muday GK, Djordjevic MA (2007) Flavonoids are differentially taken up and transported long distances in Arabidopsis. Plant Physiol 145: 478–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkley RJ, Clauser KR (2012) Modification site localization scoring: strategies and performance. Mol Cell Proteomics 11: 3–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang WW, Huang L, Shen M, Webster C, Burlingame AL, Roberts JK (2000) Patterns of protein synthesis and tolerance of anoxia in root tips of maize seedlings acclimated to a low-oxygen environment, and identification of proteins by mass spectrometry. Plant Physiol 122: 295–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JG, Gao Y, Jones AM (2006) Differential roles of Arabidopsis heterotrimeric G-protein subunits in modulating cell division in roots. Plant Physiol 141: 887–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente van Bentem S, Hirt H (2009) Protein tyrosine phosphorylation in plants: more abundant than expected? Trends Plant Sci 14: 71–76 [DOI] [PubMed] [Google Scholar]
- de Sousa Abreu R, Penalva LO, Marcotte EM, Vogel C (2009) Global signatures of protein and mRNA expression levels. Mol Biosyst 5: 1512–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Zhou X, Ling Y, Zhang Z, Su Z (2010) agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 38: W64–W70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facette MR, Shen Z, Björnsdóttir FR, Briggs SP, Smith LG (2013) Parallel proteomic and phosphoproteomic analyses of successive stages of maize leaf development. Plant Cell 25: 2798–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman CD, Casati P, Walbot V (2004) A multidrug resistance-associated protein involved in anthocyanin transport in Zea mays. Plant Cell 16: 1812–1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gygi SP, Rochon Y, Franza BR, Aebersold R (1999) Correlation between protein and mRNA abundance in yeast. Mol Cell Biol 19: 1720–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan S, Mathesius U (2012) The role of flavonoids in root-rhizosphere signalling: opportunities and challenges for improving plant-microbe interactions. J Exp Bot 63: 3429–3444 [DOI] [PubMed] [Google Scholar]
- Hetz W, Hochholdinger F, Schwall M, Feix G (1996) Isolation and characterization of rtcs a mutant deficient in the formation of nodal roots. Plant J 10: 845–857 [Google Scholar]
- Higgins CF. (1992) ABC transporters: from microorganisms to man. Annu Rev Cell Biol 8: 67–113 [DOI] [PubMed] [Google Scholar]
- Higgins CF, Linton KJ (2004) The ATP switch model for ABC transporters. Nat Struct Mol Biol 11: 918–926 [DOI] [PubMed] [Google Scholar]
- Hochholdinger F. (2009) The maize root system: morphology, anatomy and genetics. InBennetzen J, Hake S, eds, Handbook of Maize. Springer, New York, pp 145–160 [Google Scholar]
- Hochholdinger F, Guo L, Schnable PS (2004) Lateral roots affect the proteome of the primary root of maize (Zea mays L.). Plant Mol Biol 56: 397–412 [DOI] [PubMed] [Google Scholar]
- Hochholdinger F, Woll K, Guo L, Schnable PS (2005) The accumulation of abundant soluble proteins changes early in the development of the primary roots of maize (Zea mays L.). Proteomics 5: 4885–4893 [DOI] [PubMed] [Google Scholar]
- Hoecker N, Keller B, Muthreich N, Chollet D, Descombes P, Piepho HP, Hochholdinger F (2008) Comparison of maize (Zea mays L.) F1-hybrid and parental inbred line primary root transcriptomes suggests organ-specific patterns of nonadditive gene expression and conserved expression trends. Genetics 179: 1275–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, Gygi SP (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143: 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Evans ML (1995) Specialized zones of development in roots. Plant Physiol 109: 725–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, Rowe SC, Harmon FG (2010) Coordination of the maize transcriptome by a conserved circadian clock. BMC Plant Biol 10: 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koes RE, Quattrocchio F, Mol JNM (1994) The flavonoid biosynthetic pathway in plants: function and evolution. BioEssays 16: 123–132 [Google Scholar]
- Kristensen AR, Gsponer J, Foster LJ (2013) Protein synthesis rate is the predominant regulator of protein expression during differentiation. Mol Syst Biol 9: 689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK (2014) voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15: R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MV, Topper SE, Hubler SL, Hose J, Wenger CD, Coon JJ, Gasch AP (2011) A dynamic model of proteome changes reveals new roles for transcript alteration in yeast. Mol Syst Biol 7: 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Xu C, Zhang K, Yang A, Zhang J (2007) Proteomic analysis of roots growth and metabolic changes under phosphorus deficit in maize (Zea mays L.) plants. Proteomics 7: 1501–1512 [DOI] [PubMed] [Google Scholar]
- Liang M, Haroldsen V, Cai X, Wu Y (2006) Expression of a putative laccase gene, ZmLAC1, in maize primary roots under stress. Plant Cell Environ 29: 746–753 [DOI] [PubMed] [Google Scholar]
- Liu H, Sadygov RG, Yates JR III (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem 76: 4193–4201 [DOI] [PubMed] [Google Scholar]
- Liu MJ, Wu SH, Wu JF, Lin WD, Wu YC, Tsai TY, Tsai HL, Wu SH (2013) Translational landscape of photomorphogenic Arabidopsis. Plant Cell 25: 3699–3710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lamkemeyer T, Jakob A, Mi G, Zhang F, Nordheim A, Hochholdinger F (2006) Comparative proteome analyses of maize (Zea mays L.) primary roots prior to lateral root initiation reveal differential protein expression in the lateral root initiation mutant rum1. Proteomics 6: 4300–4308 [DOI] [PubMed] [Google Scholar]
- Maier T, Güell M, Serrano L (2009) Correlation of mRNA and protein in complex biological samples. FEBS Lett 583: 3966–3973 [DOI] [PubMed] [Google Scholar]
- Maier T, Schmidt A, Güell M, Kühner S, Gavin AC, Aebersold R, Serrano L (2011) Quantification of mRNA and protein and integration with protein turnover in a bacterium. Mol Syst Biol 7: 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcon C, Lamkemeyer T, Malik WA, Ungrue D, Piepho HP, Hochholdinger F (2013) Heterosis-associated proteome analyses of maize (Zea mays L.) seminal roots by quantitative label-free LC-MS. J Proteomics 93: 295–302 [DOI] [PubMed] [Google Scholar]
- Marcon C, Schützenmeister A, Schütz W, Madlung J, Piepho HP, Hochholdinger F (2010) Nonadditive protein accumulation patterns in maize (Zea mays L.) hybrids during embryo development. J Proteome Res 9: 6511–6522 [DOI] [PubMed] [Google Scholar]
- Marschner P, editor (2011) Mineral Nutrition of Higher Plants. Academic Press, London [Google Scholar]
- O’Malley DM, Whetten R, Bao W, Chen CL, Sederoff R (1993) The role of laccase in lignification. Plant J 4: 751–757 [Google Scholar]
- Paschold A, Larson NB, Marcon C, Schnable JC, Yeh CT, Lanz C, Nettleton D, Piepho HP, Schnable PS, Hochholdinger F (2014) Nonsyntenic genes drive highly dynamic complementation of gene expression in maize hybrids. Plant Cell 26: 3939–3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfus-Barbeoch L, Jones AM, Assmann SM (2004) Plant heterotrimeric G protein function: insights from Arabidopsis and rice mutants. Curr Opin Plant Biol 7: 719–731 [DOI] [PubMed] [Google Scholar]
- Petricka JJ, Schauer MA, Megraw M, Breakfield NW, Thompson JW, Georgiev S, Soderblom EJ, Ohler U, Moseley MA, Grossniklaus U, et al. (2012) The protein expression landscape of the Arabidopsis root. Proc Natl Acad Sci USA 109: 6811–6818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnala L, Wang Y, Sun Q, van Wijk KJ (2014) Correlation of mRNA and protein abundance in the developing maize leaf. Plant J 78: 424–440 [DOI] [PubMed] [Google Scholar]
- Rigbolt KT, Prokhorova TA, Akimov V, Henningsen J, Johansen PT, Kratchmarova I, Kassem M, Mann M, Olsen JV, Blagoev B (2011) System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci Signal 4: rs3. [DOI] [PubMed] [Google Scholar]
- Saleem M, Lamkemeyer T, Schützenmeister A, Madlung J, Sakai H, Piepho HP, Nordheim A, Hochholdinger F (2010) Specification of cortical parenchyma and stele of maize primary roots by asymmetric levels of auxin, cytokinin, and cytokinin-regulated proteins. Plant Physiol 152: 4–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saslowsky D, Winkel-Shirley B (2001) Localization of flavonoid enzymes in Arabidopsis roots. Plant J 27: 37–48 [DOI] [PubMed] [Google Scholar]
- Sauer M, Jakob A, Nordheim A, Hochholdinger F (2006) Proteomic analysis of shoot-borne root initiation in maize (Zea mays L.). Proteomics 6: 2530–2541 [DOI] [PubMed] [Google Scholar]
- Schnable JC, Freeling M (2011) Genes identified by visible mutant phenotypes show increased bias toward one of two subgenomes of maize. PLoS ONE 6: e17855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M (2011) Global quantification of mammalian gene expression control. Nature 473: 337–342 [DOI] [PubMed] [Google Scholar]
- Smyth GK (2004) Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article 3 [DOI] [PubMed]
- Stolarczyk EI, Reiling CJ, Paumi CM (2011) Regulation of ABC transporter function via phosphorylation by protein kinases. Curr Pharm Biotechnol 12: 621–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama N, Nakagami H, Mochida K, Daudi A, Tomita M, Shirasu K, Ishihama Y (2008) Large-scale phosphorylation mapping reveals the extent of tyrosine phosphorylation in Arabidopsis. Mol Syst Biol 4: 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37: 914–939 [DOI] [PubMed] [Google Scholar]
- Ullah H, Chen JG, Temple B, Boyes DC, Alonso JM, Davis KR, Ecker JR, Jones AM (2003) The β-subunit of the Arabidopsis G protein negatively regulates auxin-induced cell division and affects multiple developmental processes. Plant Cell 15: 393–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbelen JP, De Cnodder T, Le J, Vissenberg K, Baluska F (2006) The root apex of Arabidopsis thaliana consists of four distinct zones of growth activities: meristematic zone, transition zone, fast elongation zone and growth terminating zone. Plant Signal Behav 1: 296–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermerris W, Sherman DM, McIntyre LM (2010) Phenotypic plasticity in cell walls of maize brown midrib mutants is limited by lignin composition. J Exp Bot 61: 2479–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, Marcotte EM (2012) Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet 13: 227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walley JW, Shen Z, Sartor R, Wu KJ, Osborn J, Smith LG, Briggs SP (2013) Reconstruction of protein networks from an atlas of maize seed proteotypes. Proc Natl Acad Sci USA 110: E4808–E4817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen TJ, Hochholdinger F, Sauer M, Bruce W, Schnable PS (2005) The roothairless1 gene of maize encodes a homolog of sec3, which is involved in polar exocytosis. Plant Physiol 138: 1637–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Paschold A, Marcon C, Liu S, Tai H, Nestler J, Yeh CT, Opitz N, Lanz C, Schnable PS, et al. (2014) The Aux/IAA gene rum1 involved in seminal and lateral root formation controls vascular patterning in maize (Zea mays L.) primary roots. J Exp Bot 65: 4919–4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Nakashima J, Chen F, Yin Y, Fu C, Yun J, Shao H, Wang X, Wang ZY, Dixon RA (2013) Laccase is necessary and nonredundant with peroxidase for lignin polymerization during vascular development in Arabidopsis. Plant Cell 25: 3976–3987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Alvarez S, Marsh EL, Lenoble ME, Cho IJ, Sivaguru M, Chen S, Nguyen HT, Wu Y, Schachtman DP, et al. (2007) Cell wall proteome in the maize primary root elongation zone: II. Region-specific changes in water soluble and lightly ionically bound proteins under water deficit. Plant Physiol 145: 1533–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Chen S, Alvarez S, Asirvatham VS, Schachtman DP, Wu Y, Sharp RE (2006) Cell wall proteome in the maize primary root elongation zone. I. Extraction and identification of water-soluble and lightly ionically bound proteins. Plant Physiol 140: 311–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Smith JW, Huang CM (2010) Mass spectrometry-based label-free quantitative proteomics. J Biomed Biotechnol 2010: 840518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.