

Abstract
Deficiencies in DNA repair due to inherited germ-line mutations in DNA repair genes cause increased risk of gastrointestinal (GI) cancer. In sporadic GI cancers, mutations in DNA repair genes are relatively rare. However, epigenetic alterations that reduce expression of DNA repair genes are frequent in sporadic GI cancers. These epigenetic reductions are also found in field defects that give rise to cancers. Reduced DNA repair likely allows excessive DNA damages to accumulate in somatic cells. Then either inaccurate translesion synthesis past the un-repaired DNA damages or error-prone DNA repair can cause mutations. Erroneous DNA repair can also cause epigenetic alterations (i.e., epimutations, transmitted through multiple replication cycles). Some of these mutations and epimutations may cause progression to cancer. Thus, deficient or absent DNA repair is likely an important underlying cause of cancer. Whole genome sequencing of GI cancers show that between thousands to hundreds of thousands of mutations occur in these cancers. Epimutations that reduce DNA repair gene expression and occur early in progression to GI cancers are a likely source of this high genomic instability. Cancer cells deficient in DNA repair are more vulnerable than normal cells to inactivation by DNA damaging agents. Thus, some of the most clinically effective chemotherapeutic agents in cancer treatment are DNA damaging agents, and their effectiveness often depends on deficient DNA repair in cancer cells. Recently, at least 18 DNA repair proteins, each active in one of six DNA repair pathways, were found to be subject to epigenetic reduction of expression in GI cancers. Different DNA repair pathways repair different types of DNA damage. Evaluation of which DNA repair pathway(s) are deficient in particular types of GI cancer and/or particular patients may prove useful in guiding choice of therapeutic agents in cancer therapy.
Keywords: Epigenetic, DNA damage, DNA repair, DNA repair deficiency disorders, Epimutation, Genomic instability, Germ-line mutation, MicroRNAs, Precancerous conditions, Gastrointestinal cancer
Core tip: The primary cause of cancer is DNA damage. DNA damage leads to replication errors and erroneous repair, and can result in driver mutations and epimutations. While germ-line mutations in DNA repair genes cause cancer-prone syndromes, mutations in DNA repair genes are infrequent in sporadic gastrointestinal cancers. However, reduction of DNA repair proteins due to epigenetic repression of DNA repair genes is very frequent and can cause early steps in sporadic cancers. Evaluation of which DNA repair pathway(s) are deficient in particular types of GI cancer and/or particular patients may prove useful in guiding choice of therapeutic agents.
REDUCED DNA REPAIR INCREASES CANCER RISK
Germ-line mutations in DNA repair genes cause increased risk of GI cancers. Examples are given in Table 1.
Table 1.
Inherited mutations in DNA repair genes that increase the risk of gastrointestinal cancer
| DNA repair gene(s) | Repair pathway(s) affected | Cancers with increased risk |
| BLM | HRR[1] | Leukemia, lymphoma, colon, breast, skin, lung, auditory canal, tongue, esophagus, stomach, tonsil, larynx, uterus[2] |
| WRN | HRR, NHEJ, long patch BER[3] | Soft tissue sarcoma, colorectal, skin, thyroid, pancreatic[4] |
| Fanconi's anemia genes FANC A, B, C, D1, D2, E, F, G, I, J, L, M, N | HRR and TLS[5] | Leukemia, liver tumors, solid tumors in many areas including esophagus, stomach and colon[6] |
| MSH2, MSH6, MLH1, PMS2 | MMR[7] | Colorectal, endometrial[7] |
| MUTYH | BER of A mispaired with 8-OHdG[8] | Colon[8] |
| P53 | HRR, BER, NER, NHEJ, MMR[9] | Sarcoma, breast, osteo-sarcoma, brain, adreno-cortical carcinomas[10] and colon and pancreas[11] |
HRR: Homologous recombinational repair; NHEJ: Non-homologous end joining; BER: Base excision repair; TLS: Translesion synthesis; MMR: Mismatch repair; DDR: DNA damage response.
About 5% to 10% of all types of cancers are due to hereditary cancer syndromes[12]. Two reviews on hereditary cancer syndromes list 48 and 55 such syndromes[12,13]. Mutation in any of 37 DNA repair genes, including those listed in Table 1, can cause an hereditary cancer syndrome[14]. That hereditary cancer syndromes are frequently caused by mutations in DNA repair genes indicates that reduction in DNA repair gene expression can be a crucial early event in progression to cancer. If DNA repair gene expression is reduced in a somatic tissue by epigenetic repression, this is also likely to be a crucial early event in progression to cancer in that tissue.
Epimutations in DNA repair genes are frequent during progression to cancer
Vogelstein et al[15], reviewing evidence from sequencing 3284 tumors and the 294881 mutations found in those cancers, noted that germ-line mutations that give rise to hereditary cancer syndromes are infrequent in sporadic tumors.
More in depth studies of defects in DNA repair genes O-6-methylguanine-DNA methyltransferase (MGMT) and PMS2, important in progression to GI cancer, are consistent with the observations of Vogelstein et al[15]. In the case of MGMT, 113 sequential colorectal cancers were evaluated and only four had a missense mutation in the DNA repair gene MGMT, while most had reduced MGMT expression due to methylation of the MGMT promoter region[16]. Other laboratories, quantifying their results, reported that 40% to 90% of colorectal cancers have reduced MGMT expression due to methylation of the MGMT promoter region[17-21].
In the case of PMS2, when 119 colorectal cancers deficient in DNA mismatch repair gene PMS2 expression were examined, mutation in PMS2 was present in 6 cases while in 103 cases the pairing partner of PMS2, MLH1 was repressed due to promoter methylation (PMS2 protein is unstable in the absence of MLH1)[22]. In the remaining 10 cases it was likely that epigenetic over-expression of the miRNA, miR-155, which down-regulates MLH1 messenger RNA (mRNA), caused the loss of PMS2 expression[23].
These findings suggest that, if an early step in progression to sporadic GI cancer is reduction in function of a DNA repair gene, that reduction is likely due to an epigenetic alteration rather than to a mutation in that gene.
DNA DAMAGES ARE VERY FREQUENT AND AN IMPORTANT CAUSE OF CANCER
An average of more than 60000 endogenous DNA damages occur per cell per day in humans (Table 2). These are largely caused by hydrolytic reactions, interactions with reactive metabolites such as lipid peroxidation products, endogenous alkylating agents and reactive carbonyl species, and exposure to reactive oxygen molecules[28].
Table 2.
Endogenous DNA damages/cell/day for humans
However, more important still in causing cancer, are DNA damages caused by exogenous agents. Doll et al[29] compared cancer rates for 37 specific cancers in the United States to rates for these cancers in countries where there is low incidence for these cancers. The populations for comparison included Norwegians, Nigerians, Japanese, British and Israeli Jews. They concluded that 75%-80% of the cases of cancer in the United States were likely avoidable. They indicated that the avoidable sources of cancer included tobacco, alcohol, diet (especially meat and fat), food additives, occupational exposures (including aromatic amines, benzene, heavy metals, vinyl chloride), pollution, industrial products, medicines and medical procedures, UV light from the sun, exposure to medical X-rays, and infection. Many of these sources of cancer are DNA damaging agents.
One example of diet-related DNA damaging agents likely important in human GI cancer are bile acids. Bernstein et al[30] summarized 14 published reports showing that the secondary bile acids deoxycholic acid and lithocholic acid, formed by bacterial action in the colon, cause DNA damage. Bile acids are increased in the colon after the gall bladder releases bile acids into the digestive tract in response to consumption of fatty foods to aid in their digestion. Bile acids in the colon were doubled in the colonic contents of humans in the United States who were on typical diets and then were experimentally fed a high fat diet[31]. Cancer rate comparisons can be made between two similar populations, one with low levels and one with high levels of colonic bile acids. For instance, deoxycholic acid (DOC) in the feces of Native Africans in South Africa is present at 7.30 nmol/g wet weight stool while for African Americans DOC is present at 37.51 nmol/g wet weight stool, a 5.14 fold higher concentration[32]. Native Africans in South Africa have a colon cancer rate of < 1:100000[33] compared to the incidence rate for male African Americans of 72:100000[34], a more than 72-fold difference in rates of colon cancer.
The likely role of bile acids as causative agents in colon cancer is further illustrated by experiments with mice. When mice were fed a diet supplemented with the bile acid deoxycholate (DOC) for 10 mo, raising their colonic level of DOC to that of humans on a high fat diet, 45% to 56% of these mice developed colon cancers, while mice fed the standard diet alone, with 1/10 the level of colonic DOC, developed no colon cancers[35,36].
Another indication that diet is important in colon cancer is observed in populations migrating from low-incidence to high-incidence countries. Cancer rates change rapidly, and within one generation reach the rate in the high-incidence country. This has been observed, for instance, in the colon cancer incidence of migrants from Japan to Hawaii[37].
MANY GENES INVOLVED IN DNA REPAIR
At least 169 enzymes are either directly employed in DNA repair or influence DNA repair processes[38]. Of these, 139 are directly employed in DNA repair processes including base excision repair (BER), nucleotide excision repair (NER), homologous recombinational repair (HRR), non-homologous end joining (NHEJ), mismatch repair (MMR) and direct reversal of lesions (DR). The other 30 enzymes are employed in the DNA damage response (DDR) needed to initiate DNA repair; chromatin structure modification required for repair; reactions needed for the reversible, covalent attachment of ubiquitin and small ubiquitin-like modifier proteins to DDR factors that facilitate DNA repair; or modulation of nucleotide pools.
When the incidence of endogenous and exogenous DNA damages is high, decreases in expression of DNA repair genes or DDR genes lead to a build-up of DNA damage within a cell. These excessive damages provide more opportunities for replication errors and erroneous repair to occur (see mechanisms below) and cause higher rates of mutation and epimutation. Higher numbers of mutations and epimutations increase the chance of including selectively advantageous driver mutations and epimutations that, in turn, promote progression to cancer.
DNA DAMAGES GIVE RISE TO MUTATIONS AND EPIGENETIC ALTERATIONS
Translesion synthesis (TLS) past a single-stranded DNA damage introduces mutations.
Single-strand DNA damages are the most frequent endogenous DNA damages (Table 2). TLS is a DNA damage tolerance process that allows the DNA replication machinery to replicate past single-strand DNA lesions in the template strand. This permits replication to be completed, rather than blocked (which may kill the cell or cause a translocation or other chromosomal aberration)[39].
Humans have four translesion polymerases in the Y family of polymerases [REV1, Pol κ (kappa), Pol η (eta), and Pol ι (iota)] and one in the B family of polymerases [Pol ζ (zeta)][39]. The temporary tolerance of DNA damage during chromosome replication may allow DNA repair processes to remove the damage later[40], and avoid immediate genome instability[41]. However, translesion synthesis is less accurate than the replicative polymerases δ (delta) and ε (epsilon) and tends to introduce mutations[39].
Deficiency in expression of a DNA repair gene can allow excessive DNA damages to accumulate. Some of the excess damages will likely be processed by translesion synthesis, causing increased mutation.
Kunz et al[42] summarized numerous experiments in yeast, in which forward mutations were measured (by sequence analyses of a few selected genes) in cells carrying either wild-type alleles or one of 11 inactivated DNA repair genes. Their results indicated that DNA repair deficient cells accumulate excess DNA damages that then give rise to mutations after error-prone translesion synthesis. The 11 inactivated DNA repair genes were distributed among MMR, NER, BER and HRR genes. Deficiencies in DNA repair increased mutation frequencies by factors between 2- and 130-fold, but most often by double digit-fold increases. Overall, the authors concluded that 60% or more of spontaneous single base pair substitutions and deletions are likely caused by translesion synthesis.
Stuart et al[43] examined spontaneous mutation frequencies in a lacI transgene (in a Big Blue mutation assay[44]) in either replicating tissues or in largely non-replicating tissues of mice. If most mutations occur during translesion synthesis, then non-replicating brain tissue, which has little or no synthesis once maturity is reached, would have little or no further mutation accumulation. In mouse brain, after 6 mo of age, there was no increase in mutation frequency, even at 25 mo of age. In bladders of mice, with replicating tissues, mutation frequency increased with age, almost tripling between ages of 1.5 mo and 12 mo of age. The authors concluded that the age related increases in spontaneous mutation frequencies reflect endogenous DNA damages that subsequently gave rise to mutations following DNA replication. This indicates that translesion synthesis is a major source of mutation in mouse replicating tissues.
Mutations are frequently caused by error-prone repair of double-strand breaks
While only a minority of endogenous DNA damages in the average cell are double-strand breaks (Table 2), this type of lesion appears to contribute substantially to the mutation rate as well. As indicated by Vilenchik and Knudson[27], the doubling dose for ionizing radiation (IR) induced double-strand breaks is similar to the doubling dose for mutation and induction of carcinomas by IR. Thus, double-strand breaks likely lead frequently to mutations.
As described by Bindra et al[45], non-homologous end-joining (NHEJ) and HRR comprise the two major pathways by which double-strand breaks (DSBs) are repaired in cells. NHEJ processes and re-ligates the exposed DNA termini of DSBs without the use of significant homology, whereas HRR uses homologous DNA sequences as a template for repair. HRR predominates in S-phase cells, when a sister chromatid is available as a template for repair, and is a high-fidelity process. NHEJ is thought to be active throughout the cell cycle, and it is more error-prone than HRR. NHEJ repair comprises both canonical NHEJ and non-canonical pathways. The former pathway results in minimal processing of the DSB during repair, whereas the latter pathway, with or without the use of sequence microhomology for re-ligation, typically results in larger insertions or deletions. Mutagenic NHEJ repair is a robust process, yielding percentages of mutated sites at the position of a DSB ranging from 20% to 60%.
As pointed out by Vilenchik et al[27], about 1% of single-strand DNA damages escape repair and are not bypassed, and some of these become converted to double-strand breaks. This may contribute to the impact of double-strand breaks in causing mutations and carcinogenesis.
Epigenetic alterations occur due to DNA damage
Epigenetic alterations can arise due to incomplete repair of DNA double-strand breaks. As an example, O’Hagan et al[46] used a cell line stably transfected with a plasmid containing a consensus I-SceI cut site inserted into a copy of the E-cad promoter. This promoter contained a CpG island. O’Hagan et al[46] induced a defined double-strand break in the E-cadherin CpG island, and the CpG island was not currently hypermethylated. As the repair of the break began, they observed that key proteins involved in establishing and maintaining transcriptional repression were recruited to the site of damage, to allow repair of the break. Most cells examined after the DNA break was repaired showed that DNA repair occurred faithfully, with the promoter not hypermethylated and the silencing factors removed. However, a small percentage of the cells retained heritable silencing. In these cells the chromatin around the break site was enriched for most of the silencing chromatin proteins and histone marks, and the region had increased DNA methylation in the CpG island of the promoter. Thus, repair of a DNA break can occasionally cause heritable silencing of a CpG island-containing promoter. Such CpG island methylation is frequently associated with tight gene silencing in cancer.
Morano et al[47] also showed that epigenetic alterations can arise as a consequence of DNA damage. They studied a system in which recombination between partial duplications of a chromosomal green fluorescent protein (GFP) gene is initiated by a DSB in one copy. Two cell types were generated after recombination: clones expressing high levels of GFP and clones expressing low levels of GFP, referred to as H and L clones, respectively. Relative to the parental gene, the repaired GFP gene was hypomethylated in H clones and hypermethylated in L clones. The altered methylation pattern was largely restricted to a segment 3’ to the DSB. Although it is 2000 base pairs distant from the strong cytomegalovirus promoter that drives GFP expression, hypermethylation of this tract significantly reduced transcription. The ratio of L (hypermethylated) to H (hypomethylated) clones was 1:2 or 1:4, depending on the insertion site of the GFP reporter. These experiments were performed in mouse embryonic (ES) or human cancer (Hela) cells. HRR-induced methylation depended on DNA methyltransferase I. These data, taken together, argue for a cause-effect relationship between double-strand DNA damage-repair and altered DNA methylation.
The main function of the proteins in the BER pathway is to repair DNA single-strand breaks and deamination, oxidation, and alkylation-induced DNA base damage. In addition, Li et al[48] reviewed studies indicating that one or more BER proteins also participate(s) in epigenetic alterations involving DNA methylation, demethylation or reactions coupled to histone modification. Franchini et al[49] also showed that DNA demethylation can be mediated by BER and other DNA repair pathways requiring processive DNA polymerases. Another form of epigenetic silencing also appears to occur during DNA repair. PARP1 [poly(ADP)-ribose polymerase 1] and its product poly(ADP)-ribose (PAR) accumulate at sites of DNA damage as intermediates of a DNA repair process[50]. This directs recruitment and activation of the chromatin remodeling protein ALC1, which can cause nucleosome remodeling[51]. Nucleosome remodeling, in one case, has been found to cause epigenetic silencing of DNA repair gene MLH1[52]. These reports, overall, indicate that DNA damages needing repair can cause epigenetic alterations by a number of different mechanisms.
Other causes of epigenetic alterations
Heavy metals and other environmental chemicals cause many epigenetic alterations, including DNA methylation, histone modifications and miRNA alterations[53]. DNA damage itself causes programmed changes in non-coding RNAs, and a large number of miRNAs are transcriptionally induced upon DNA damage[54]. However, it is not clear what proportion of these alterations are reversed or are retained as epimutations after the external sources of damage are removed upon repair of the DNA damages[55].
Mutations in isocitrate dehydrogenase 1 (IDH1) and 2 (IDH2) are frequent in several types of cancer and they can cause epigenetic alterations. As reviewed by Wang et al[56], IDH1 and IDH2 mutations represent the most frequently mutated metabolic genes in human cancer. These mutations occur in more than 75% of low grade gliomas and secondary glioblastoma multiforme, 20% of acute myeloid leukemias, 56% of chondrosarcomas, over 80% of Ollier disease and Maffucci syndrome, and 10% of melanomas. IDH1 is also mutated in 13% of inflammatory bowel disease-associated intestinal adenocarcinoma with low-grade tubuloglandular histology but not in sporadic intestinal adenocarcinoma[57]. The IDH1 and IDH2 mutations that give rise to epimutations usually occur in the hotspot codons Arg132 of IDH1, or the analogous codon Arg172 of IDH2. These mutations allow accumulation of the metabolic intermediate 2-hydroxyglutarate (2-HG), and 2-HG inhibits the activity of alpha ketoglutarate (α-KG) dependent dioxygenases, including α-KG-dependent histone demethylases and the TET family of 5-methylcytosine hydroxylases.
Wang et al[56] found that histone H3K79 dimethylation levels were significantly elevated in cholangiocarcinoma samples that harbored IDH1 or IDH2 mutations (80.8%) compared to tumors with wild-type IDH1 and IDH2 (45.0%).
In addition, they surveyed over 462000 CpG sites in CpG islands, CpG shores and intragenic regions, and found that 2309 genes had significantly increased methylation in the presence of IDH1 or IDH2 mutations. In particular, Sanson et al[58] found that methylation of the DNA repair gene MGMT was associated with IDH1 mutation, since 81.3% of IDH1-mutated gliomas were MGMT methylated compared with 58.3% methylated in IDH1 non-mutated tumors.
DNA REPAIR GENES WITH EPIGENETICALLY REDUCED EXPRESSION ARE LIKELY PASSENGERS IN A SPREADING FIELD DEFECT
A DNA repair gene that is epigenetically silenced or whose expression is reduced would not likely confer any selective advantage upon a stem cell. However, reduced or absent expression of a DNA repair gene would cause increased rates of mutation, and one or more of the mutated genes may cause the cell to have a strong selective advantage. The expression-deficient DNA repair gene could then be carried along as a selectively neutral or only slightly deleterious passenger (hitch-hiker) gene when there is selective expansion of the mutated stem cell. The continued presence of a DNA repair gene that is epigenetically silenced or has reduced expression would continue to generate further mutations and epigenetic alterations.
The spread of a clone of cells with a selective advantage, but carrying along a gene with epigenetically reduced expression of a DNA repair protein would be expected to generate a field defect, from which smaller clones with still further selective advantages would arise. This is consistent with the finding of field defects in colonic resections, that have both a cancer and multiple small polyps, such as the one shown in Figure 1.
Figure 1.
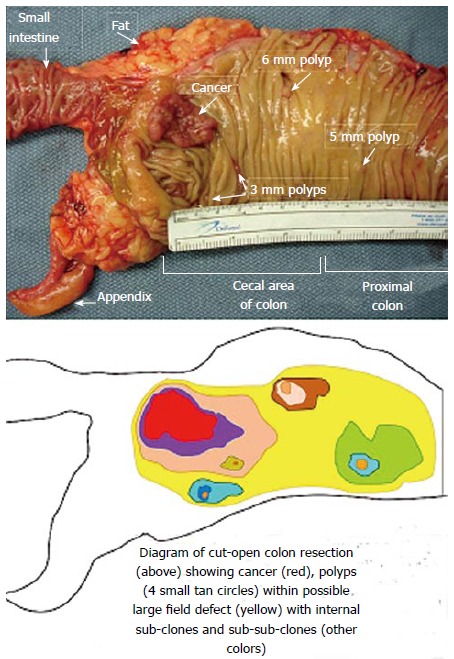
Cut open gross specimen of proximal human colon showing multiple tumors[59].
For any particular type of GI cancer, an epigenetic reduction in expression of a specific DNA repair gene may be common. In cases where a specific epigenetic reduction of expression of a DNA repair gene occurs in a cancer, it is also likely to be evident in the field defect surrounding the cancer (Table 3). The lower frequency in the surrounding field defect that is usually found (Table 3) likely reflects the process whereby the expanding clone laterally displaces the more normal epithelium. This displacement may be only partial. Thus, areas with the DNA repair deficiency would be present at a lower frequency in the field defect than in the cancer. In the cancer, the cells carrying the DNA repair deficiency are members of a founding clone. Thus, in the cancer, the DNA repair defect, along with other accumulated mutations and epigenetic alterations, would be seen at a relatively higher frequency than in the surrounding field defect.
Table 3.
Epigenetic deficiency of DNA repair genes in gastrointestinal cancers and field defects
| Cancer | Gene | Frequency in cancer | Frequency in adjacent field defect |
| Colorectal[17] | MGMT | 46% | 34% |
| Colorectal[19] | MGMT | 47% | 11% |
| Colorectal[60] | MGMT with MSI | 70% | 60% |
| Colorectal[19] | MSH2 | 13% | 5% |
| Colorectal[61] | MBD4 | Frequent | Frequent |
| Colorectal[62] | ERCC1 | 100% | 40% |
| Colorectal[62] | PMS2 | 88% | 50% |
| Colorectal[62] | XPF | 55% | 40% |
| Colorectal[63] | WRN | 29% | 13% |
| Stomach[64] | MGMT | 88% | 78% |
| Stomach[65] | MLH1 | 73% | 20% |
| Esophagus[66] | MLH1 | 77%-100% | 23%-79% |
MSI: Microsatellite instability.
DECREASED EXPRESSION OF MULTIPLE DNA REPAIR GENES IN GI CANCERS
The protein expressions of three DNA repair genes within a 20 cm colon resection were evaluated at six different locations within the resection (Figure 2)[62]. One of the DNA repair proteins, KU86, was only deficient infrequently, with the deficiencies occurring in small patches (up to three crypts). These KU86 defects are not likely important in progression to colon cancer. However, two of the evaluated DNA repair proteins, ERCC1 and PMS2, were often deficient in patches of tens to hundreds of adjacent crypts at each of the locations evaluated (see Nguyen et al[68] at minutes 18 to 24 of a 28 min video of crypts immunostained for ERCC1 or PMS2).
Figure 2.
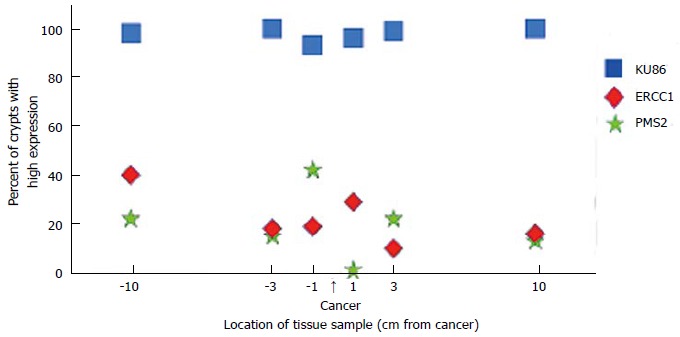
Expression of three DNA repair proteins, KU86, ERCC1 and PMS2, at locations sampled along the 20 cm length of a colon resection that had a cancer at the indicated location[67].
Overall, ERCC1 (NER) was deficient in 100% of 49 colon cancers evaluated, and in an average of 40% of the crypts within 10 cm on either side of the cancer. PMS2 (MMR) was deficient in 88% of the 49 cancers and in an average of 50% of the crypts within 10 cm of the cancer. As reported by Facista et al[62], the pattern of expression of ERCC1 in the crypts within 10 cm of a colon cancer indicated that when the ERCC1 protein was deficient, this deficiency was due to an epigenetic reduction in expression of the ERCC1 gene. When the PMS2 protein is deficient, it is usually due to the epigenetic repression of its pairing partner, MLH1, and the instabilty of PMS2 in the absence of MLH1[22]. In the study of Facista et al[62], ERCC1 and PMS2 were also deficient in all 10 tubulovillous adenomas evaluated (precursors to colonic adenocarcinomas). Thus ERCC1 and PMS2 are deficient at early times (in the field defect), at intermediate times (in tubulovillus polyps), and at late times (within the cancer) during progression to colon cancer. Another DNA repair protein, XPF, was deficient in 55% of the cancers, as well[62]. The majority of cancers were simultaneously deficient for ERCC1, PMS2 and XPF.
Deficiencies in multiple DNA repair genes were also observed in gastric cancers. Kitajima et al[69] evaluated MGMT (direct reversal repair), MLH1 (MMR) and MSH2 (MMR) and found that synchronous losses of MGMT and MLH1 increase during progression and stage of differentiated-type cancers. In un-differentiated-type gastric cancers, the frequency of MGMT deficiency increased from early to late stages of the cancer, while frequencies of MLH1 and MSH2 deficiencies were between 48% and 74% at both early and late stages. Thus, in un-differentiated-type gastric cancers, MLH1 or MSH2 deficiency, if it is present, is an early step, while MGMT deficiency is often a later step in progression of this cancer.
Farkas et al[70] evaluated 160 genes in 12 paired colorectal tumors and adjacent histologically normal mucosal tissues for differential promoter methylation. They found aberrant methylation in 23 genes, including six DNA repair genes. These DNA repair genes (with DNA repair pathways indicated) were NEIL1 (BER), NEIL3 (BER), DCLRE1C (NHEJ), NHEJ1 (NHEJ), GTF2H5 (NER), and CCNH (NER).
Lynam-Lennon et al[71] found that miR-31 is over-expressed in 47% of esophageal cancers and examined the consequences of over-expression of miR-31 in these cancers. Using a cell line, they first tested the effect of over-expression of miR-31 on the expression of 84 DNA repair genes. They found that 11 DNA repair genes were repressed by over-expression of miR-31. They then evaluated the expression of the five most altered DNA repair genes in 10 esophageal cancers that had high expression of miR-31 and low resistance to radiation treatment (likely low levels of DNA repair). These 10 cancers showed significantly reduced mRNA levels of DNA repair genes PARP1, SMUG1, MLH1 and MMS19. Asangani et al[72] showed that miR-31 is an epigenetically regulated microRNA. This microRNA is encoded in an intron of MIR31HG (miR-31 host gene). The transcriptional regulatory region of MIR31HG is enriched for histone 3 that could be acetylated on lysine (K) 27 (this is designated H3K27Ac), and H3K27Ac causes an epigentic “mark” that is associated with transcriptionally active genes. If, instead, this histone 3 has triple methylation on lysine 27 (H3K27me3), this causes gene silencing. The regulatory region of MIR31HG also has 77 CpG islands surrounding the transcription start site. These observations indicate that miR-31 transcription could be up-regulated by H3K27Ac or silenced by CpG island methylation or by histone H3K27me3. It appears that DNA repair genes PARP1 (BER and HRR), SMUG1 (BER), MLH1 (MMR) and MMS19 (NER) are epigenetically repressed by over-expressed miR-31 in esophageal cancers.
Based on the examples above, decreased expression of multiple DNA repair genes likely occurs often in GI neoplasia.
EFFECTS LIKELY DUE TO DNA REPAIR DEFECTS
Regression of early lesions
If DNA repair defects are present early in progression to cancer, this should result in increased mutation frequency in those neoplastic lesions. Most new mutations are expected to be deleterious to the cells in which they arise, and thus would cause negative selection of those cells. This expectation is consistent with the observations of Hofstad et al[73] who showed that when colonic polyps were identified during a colonoscopy and followed but not removed, between 11% and 46% of polyps smaller than 5 mm diameter were not detectable in the succeeding one to three years. For polyps between 5 and 9 mm in diameter, between 4% and 24% became undetectable in the succeeding one to three years. Of the remaining 68 polyps that were followed for three years, 35% decreased in diameter, 25% remained the same size and 40% increased in diameter. Similarly, Stryker et al[74] followed 226 patients with colonic polyps that were ≥ 1 cm in size for an average of 5.7 years (though some patients were followed for as long as 19 years). Stryker et al[74] found that 37% of polyps ≥ 1 cm enlarged (at least doubled in volume) during the study while 4% of the polyps that had been observed at least twice, previously, were later not found. The risk of these polyps ≥ 1 cm producing an invasive carcinoma within 20 years was 24%. The data of Hofstad et al[73] and Stryker et al[74] are also consistent with statistics showing more frequent occurrence of adenomas during colonoscopy and autopsy compared to the frequency of colon cancer, indicating there must be a significant regression rate for adenomas[75].
Subclones in cancers
When infrequent positively selected mutations arise in a cell, this can provide the cell with a competitive advantage that promotes its preferential clonal proliferation, leading to cancer. The continued presence of epigenetically repressed DNA repair genes, carried along as passengers in the development of cancers, also predicts that cancers will contain heterogeneous genotypes (multiple subclones). For instance, as a test for the presence of subclones, in one primary renal carcinoma with multiple metastases, 101 non-synonymous point mutations and 32 indels (insertions and deletions) were identified[76]. Five mutations were not validated and excluded from the study. Of the remaining 128 mutations, 40 were “ubiquitous” and present in each region of the tumor sampled. There were 59 “shared” mutations, present in several but not all regions, and 29 “private” mutations, unique to a specific region evaluated. The authors constructed a phylogenetic tree and concluded that the evolution in the tumor and its metastases was branching, and not linear.
A deficiency of DNA repair would likely produce genetic clonal diversity, through generation and selection for new mutational variants. In a study by Maley et al[77], 268 patients with Barrett’s esophagus were followed for an average of 4.4 years during which 37 esophageal adenocarcinomas (EACs) developed. Genetic clonal diversity within Barrett’s esophagus proved to be a better predictor of EAC than the presence of specific mutations in genes associated with EAC, such as mutation in P53. This finding suggests that DNA repair deficiency is of primary importance in progression to cancer.
EPIGENETIC REPRESSION OF DNA REPAIR GENES, DUE TO ALTERATIONS IN CPG ISLAND METHYLATION IN GI CANCERS
Table 4 gives examples of reports of DNA repair genes repressed by CpG island hypermethylation (or with increased expression due to CpG hypomethylation, which may cause unbalanced repair processes) in GI cancers (this is only a partial list). Nine different DNA repair genes (all listed among the 169 DNA repair and DDR genes previously identified[38]) were often hyper- (or sometimes hypo-) methylated in one or more GI cancer. Such alterations in methylation of promoter regions of DNA repair genes can cause deficient repair of DNA damages. Thus, hyper- (or hypo-) methylations of DNA repair genes are frequently important factors responsible for lack of appropriate repair of DNA damages. Faulty DNA repair leads to increased mutation and epigenetic alteration, central to progression to cancer.
Table 4.
CpG island hyper- (and hypo-) methylation of DNA repair genes in cancers
| Cancer | Gene | Frequency of promoter hyper- (or hypo-) methylation in cancer |
| Colorectal | LIG4 | 82%[78] |
| MGMT | 40%-90%[17-21] | |
| ERCC1 | 38%[79] | |
| WRN | 29%-38%[63,80] | |
| MLH1 | 9%-10%[22,81] | |
| FEN1 | Frequent (hypo-)[82] | |
| MBD4 | Frequent (hyper-)[61] | |
| Esophageal | MGMT | 23%-79%[65,83,84] |
| MLH1 | 43%[82], 64%[85] | |
| MSH2 | 29%[83], 75%[84] | |
| Stomach | MGMT | 88%[60] |
| MLH1 | 73%[64] | |
| WRN | 24%-25%[80,86] | |
| FEN1 | Frequent (hypo-)[82] | |
| Gastric lymphoma | ATM | 11%[87] |
DNA REPAIR GENE EXPRESSION MAY BE REPRESSED BY MULTIPLE PROCESSES
A number of the DNA repair genes with reduced expression due to CpG island hypermethylation are also epigenetically repressed by other means. Many protein coding genes are repressed by microRNAs. MicroRNAs (miRNAs) are small noncoding endogenously produced RNAs that play key roles in controlling the expression of many cellular proteins. Once they are recruited and incorporated into a ribonucleoprotein complex, they can target specific messenger RNAs (mRNAs) in a miRNA sequence-dependent process and interfere with the translation into proteins of the targeted mRNAs via several mechanisms (see detailed review by Lages et al[88]).
As discussed above, when mismatch DNA repair protein PMS2 is deficient in colorectal cancer, this may be due to hypermethylation of its pairing partner MLH1, or due to over-expression of the miRNA miR-155 which targets the MLH1 gene for repression.
While only 38% of cancers have CpG island methylation of the ERCC1 promoter (Table 4), Facista et al[62] found that 100% of colon cancers have significantly reduced levels of ERRC1 protein expression. In the 49 cancers examined, ERCC1 protein expression varied from 0% to 45% (with a median value of 28%) of the level of ERCC1 expression of neoplasm-free individuals. It is likely that ERCC1 can be repressed by more than one mechanism. A second mechanism of repression of ERCC1 may be due to the combined effects of epigenetically deficient miRNA let-7a and resulting over-expression of HMGA2 protein, which then represses ERCC1, as discussed below.
As indicated by Motoyama et al[89], the let-7a miRNA normally represses the HMGA2 gene, and in normal adult tissues, almost no HMGA2 protein is present. In breast cancers, for instance, the promoter region controlling let-7a-3/let-7b miRNA is frequently repressed by hypermethylation[90]. Reduction or absence of let-7a miRNA allows high expression of the HMGA2 protein. Regulation of gene expression by HMGA2 is achieved by binding to AT-rich regions in the DNA and/or direct interaction with several transcription factors[91].
HMGA2 targets and modifies the chromatin architecture at the ERCC1 gene, reducing its expression[92]. As shown by Mayr et al[93], using an artificial construct, the lack of let-7a miRNA repression of HMGA2 could occur through translocation of HMGA2, disrupting the 3’UTR of HMGA2 which is the target of let-7a miRNA, and this can lead to an oncogenic transformation. However, the promoter controlling let-7a miRNA also can be strongly regulated by hypermethylation in intact cells. When human lung cells are exposed to cigarette smoke condensation, the promoter region controlling let-7a becomes highly hypermethylated[94]. It is likely that hypermethylation of the promoter for let-7a miRNA reduces its expression. This allows hyperexpression of HMGA2. Hyperexpression of HMGA2 can then reduce expression of ERCC1. The combined effects of reduced let-7a miRNA and hyperexpressed HMGA2 or other possible epigenetic mechanism(s) may cause the reduced protein expression of ERCC1 in colorectal cancers in addition to the 38% of colorectal cancers in which the ERCC1 gene is directly hypermethylated.
DNA REPAIR PROTEINS AND MIRNAS
A review by Wouters et al[95] lists 74 DNA repair genes that are potentially targeted by miRNAs, and two additional reviews[96,97] list, combined, 30 miRNAs known to target DNA repair genes. The review by Wouters et al[95] used “in silico” computer programs (Targetscan and Mirbase) to identify likely miRNAs that could target their 74 DNA repair genes of interest, and, for each of these genes, indicated between 1 and 19 “conserved” miRNAs that were predicted to repress those genes. They define “conserved” miRNAs as miRNAs found in at least five mammalian species. However, about half of the miRNAs they found “in silico” were inducible by UV irradiation, and may have been controlled by transcriptional regulation and not by an epigenetic mechanism. Tessitore et al[96] and Vincent et al[97] each list about 20 miRNAs that are altered in cancers and which control expression of DNA repair genes. However, they did not indicate how these miRNAs are deregulated.
Deregulation of miRNA expression in cancers has been found to occur by epigenetic as well as non-epigenetic mechanisms[88,98]. One non-epigenetic mechanism includes alterations in genomic miRNA copy numbers and location. Some of these are deletions that include the miRNA clusters 15a/16-1 or let-7g/mir-135-1, or else amplification or translocation of the mir-17-92 cluster. In some cancers miRNAs were deregulated because of defects in the biogenesis mechanism (the process of creating miRNAs, which has a number of steps). Some cancers have deregulated miRNAs due to single nucleotide polymorphisms (SNPs) in the genes coding for the miRNAs, or SNPs in the target gene area to which the miRNA is targeted. Some miRNAs, that target DNA repair genes, are regulated by oncogenes. For instance ATM is down-regulated by miR-421, but miR-421 is regulated by N-Myc[99]. Thus, not all instances of deregulation of DNA repair genes or DDR genes by miRNAs are due to epigenetic alterations affecting expression of the miRNAs.
EPIGENETIC REPRESSION OF DNA REPAIR GENES DUE TO ALTERATIONS OF METHYLATION OF PROMOTERS OF MIRNAS IN VARIOUS CANCERS
Table 5 lists nine miRNAs that have three characteristics: (1) their expression is epigenetically controlled by the methylation level of the promoter regions coding for the miRNAs; (2) they control expression of DNA repair genes; and (3) their level of expression was frequently epigenetically altered in one or more types of GI cancer. This list is not exhaustive. Many of the 30 miRNAs listed by Tessitore et al[96] or Vincent et al[97] might also meet these criteria upon further examination. Four of the miRNAs on this list are not noted by Tessitore et al[96] or Vincent et al[97]. Most of the studies of these epigenetically controlled miRNAs have not noted the frequencies with which their alterations occur in cancers. Thus, these studies are somewhat less systematic than those detailing methylation of DNA repair genes in Table 4. However, the nine epigenetically controlled miRNAs listed in Table 5 can repress the 16 DNA repair genes listed in Table 5 and these genes are repressed in various GI cancers.
Table 5.
Epigenetic ↑ or ↓ miRNAs, altered in cancers, targeting DNA repair genes
| Specific miRNA | DNA repair gene targets | Cancers affected (frequency if measured) | References indicating epigenetic control of miRNA | References indicating target gene(s) of miRNAs | References indicating cancer type(s) affected |
| miR-103 miR-107 | RAD51, RAD51D | Osteosarcoma, lung, endometrial, stomach | [100] | [101] | [101] |
| miR-34c | UNG | Gastric (70%) field defect gastric (27%) colon (98%) field defect colon (60%) chronic lymphocytic leukemia (18%) small-cell lung cancer (67%) NSCLC (26%) | [102,104] | [103] | [102,105,106] |
| miR-31 | PARP1 MLH1 SMUG1 MMS19 | Esophagus (47%) colon | [72] | [21] | [71,107,108] |
| miR-124 | KU70 | Colon | [109] | [110] | [109] |
| miR-155 | RAD51 MLH1 MSH2 MSH6 | Breast Colon | [90,111] | [23,112] | [23,90] |
| let-7a repression increases HMGA2; HMGA2 alters chromatin architecture of and represses ERCC1) | ERCC1 | (Colon) Anaplastic astrocytoma | [90] | [92,113] | [113] |
| Let-7b repression increases HMGA1; HMGA1 targets P53 | P53 | Prostate Colon | [90] | [114,115] | [114,115] |
| miR-182 | BRCA1 NBN RAD17 | Breast Colon | [116] | [117,118] | [107,117,119] |
WHOLE GENOME SEQUENCING INDICATES A HIGH LEVEL OF MUTAGENESIS IN GI CANCERS
Almost 3000 pairs of tumor/normal tissues were analyzed for mutations by whole exome sequencing (sequencing the protein coding parts of whole genomes) and more than a hundred pairs of tumor/normal tissues were analyzed for mutations by whole genome sequencing by Lawrence et al[120]. Median mutation frequencies for 27 different types of cancer were found to vary by 1000-fold. When there was a particular median mutation frequency for a type of cancer, the scatter of values (in individual cancers) for that type of cancer, above and below that median value, sometimes also varied by as much as 1000-fold. Some mutation frequencies in GI cancers, given as numerical values of median numbers of mutations per megabase in a review of the literature by Tuna et al[121], and recent values for esophageal cancers by Weaver et al[122], are shown in Table 6. The values were also converted to mutation frequency per whole diploid genome.
Table 6.
Median mutation frequencies and ranges
| Parent/child per generation or cancer type | Mutation frequency per million bases | Mutation frequency per diploid genome |
| Parent/child per generation | 0.00000023 | 30-70 |
| Colorectal carcinoma | Approximately 5 | Approximately 30000 |
| MSS colon cancer | 2.8 | 16800 |
| MSI colon cancer (mismatch DNA repair deficient) | 47 | 282000 |
| Hepatocellular carcinoma | 4.2 | 25200 |
| Esophageal carcinoma (single nucleotide variants) | 2.8 | 16994 |
| Range 0.7-9.3 | Range 4516-56528 | |
| Esophageal carcinoma (small insertions and deletions) | 994 Range 262-3573 |
MSS: Microsatellite stable; MSI: Microsatellite instable.
The mutation frequency in the whole genome [not just the exome (protein coding regions)] between generations for humans (parent to child) is about 30-70 new mutations per generation[123-125]. For protein coding regions of the genome in individuals without cancer, Keightley[126] estimated there would be 0.35 mutations per parent to child generation. Whole genome sequencing was also performed in blood cells for a pair of monozygotic (identical twin) 100 years old centenarians[127]. Only 8 somatic differences were found between the twins, though somatic variation occurring in less than 20% of blood cells would be undetected. These findings, as well as the data summarized in Table 6, indicate that cancer cell lineages experience substantially higher mutation rates than non-cancer cell lineages.
EPIGENETICALLY REDUCED EXPRESSION OF DNA REPAIR GENES IN GI CANCERS OCCUR IN DIFFERENT REPAIR PATHWAYS
Figure 3[128] indicates some types of DNA damaging agents that may be encountered by cells in the GI tract, some of the DNA lesions they cause and the pathways used to repair these lesions. Many of the genes active in these pathways are included in Figure 3 and are indicated by their acronyms. The acronyms listed in red represent genes whose expression is frequently reduced due to epigenetic alterations in various types of GI cancers, as discussed above. Such reduced expression could be a substantial source of the genomic instability that is characteristic of these cancers.
Figure 3.
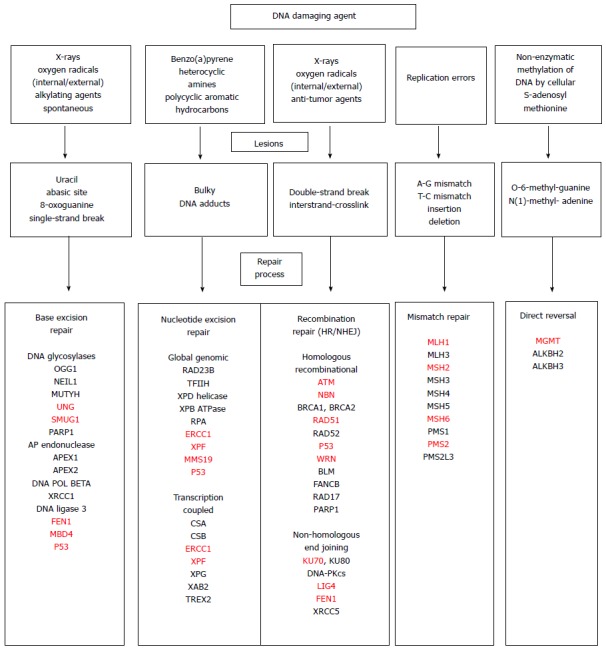
DNA damaging agents, the lesions they produce and the repair pathways that deal with the DNA damages, including acronyms for many of the genes in each of the pathways. Acronyms in red represent genes indicated in the text that have altered (usually reduced) expression due to an epigenetic alteration in one or more types of gastrointestinal cancer[128].
THE CENTRAL ROLE OF DNA DAMAGE AND EPIGENETIC DEFECTS IN DNA REPAIR DURING PROGRESSION TO GI CANCER
The central role of DNA damage and epigenetic defects in DNA repair are illustrated in Figure 4[129]. When DNA damage results in epigenetic reduction in expression of one or more DNA repair genes, the resulting DNA repair deficiency can allow DNA damage to accumulate at a much increased rate. As indicated in Figure 3, at least 18 DNA repair genes that are frequently epigenetically deficient in one or more GI cancers have been identified. These epigenetic defects in DNA repair are often found to be present in field defects from which the cancers arose, so that such epigenetic reductions in DNA repair are likely early events in progression to cancer. A large increase in unrepaired DNA damage, due to an epigenetic reduction in DNA repair, can then lead to the large increase in mutation frequencies found in GI cancers (Table 6).
Figure 4.
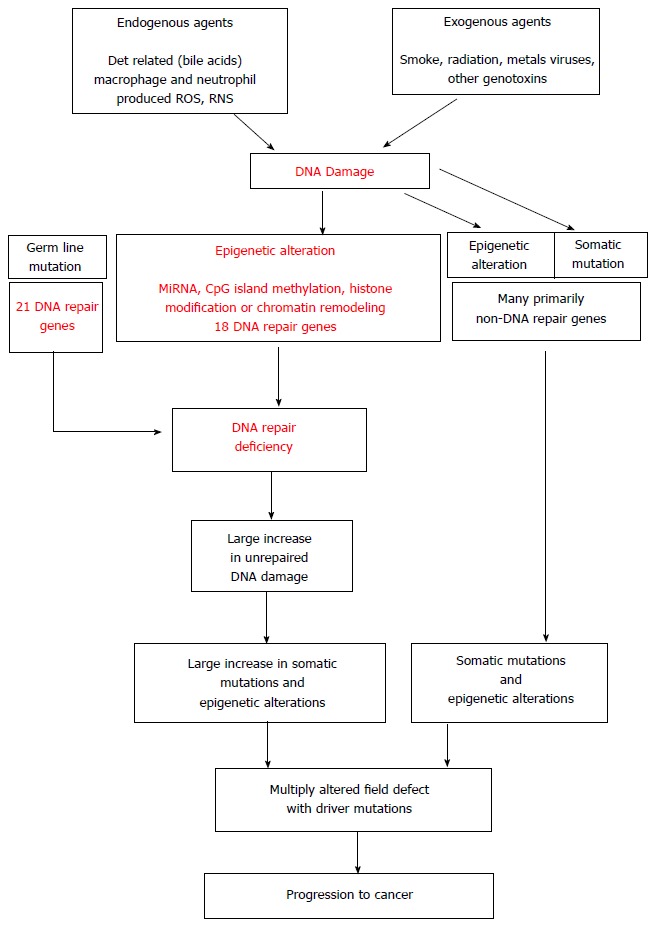
The central role of DNA damage and epigenetic alteration in DNA repair genes in gastrointestinal carcinogenesis[129].
An epigenetic reduction of DNA repair may be the key early event that accelerates progression to cancer.
SELECTIVE TUMOR KILLING
DNA-damaging agents have a long history of use in cancer chemotherapy. As pointed out by Cheung-Ong et al[130], and indicated in the text earlier in this article, cancer cells are typically deficient in DNA damage-sensing/repair capabilities. That makes them more susceptible to DNA damage than normal cells. As Cheung-Ong et al[130] describe, both the earliest as well as the most frequent current cancer chemotherapeutic agents are DNA damaging agents.
A recently developing strategy for more effective and selective treatment of cancer is to inhibit one of the tumor’s remaining DDR or DNA repair pathways. This can hyper-sensitize a tumor to radiation or chemotherapeutic agents, compared to the sensitivity of a tumor treated with a DNA damaging agent alone. This strategy is called synthetic lethality.
An early effort to implement synthetic lethality was the successful trial of Fong et al[131], in which a PARP inhibitor was given to germ-line mutated BRCA carriers. In this case, 12 of 19 (63%) of these patients in a Phase I trial had a clinical benefit from treatment with the PARP inhibitor olaparib alone, with no other chemotherapy. The patients in this Phase I trial had tumors that had been refractory to the 1 - ≥ 4 therapies that had been tried previously. As noted by O’Sullivan et al[132], the BRCA proteins are active in the HRR pathway, and PARP is largely active in BER, though it is also important in HRR. O’Sullivan et al[132] indicated that PARP inhibition appears to have synthetic lethality for both BRCA mutation-associated and “BRCA-like” solid tumors. As reviewed by O’Sullivan et al[132], PARP inhibitors are currently being evaluated in Phase I and Phase II trials of many different cancers, including GI cancers in pancreas, liver, colorectum, stomach and esophagus. They summarize some early quantitative results (in the range of 14% to 23% tumor regression or delayed progression) in pancreatic and colorectal cancers. McLornan et al[133] summarize positive results (tumor regression or delayed progression), often in the range of about 40% to 50%, with PARP inhibitors used in treatment of advanced solid tumors in other Phase I and II trials, including one on recurrent or metastatic gastric cancer.
Hosoya et al[134] listed a large number of synthetic lethality Phase I and Phase II trials that included not only PARP inhibitors but also inhibitors of DDR elements CHK1 and CHK2 and inhibitors of DNA repair elements DNA-PK and APE1. In addition they discuss interesting pre-clinical, potentially useful, synthetic lethal experiments with inhibitors of ATM/ATR and the MRN complex, DNA ligases, RAD51, RAD52 and histone deactylases.
Clinical applications of synthetic lethality are just beginning, as Phase I and II trials, but appear to be a new and potentially effective avenue for cancer therapy. How synthetic lethality may relate to epigenetically repressed DNA repair genes is currently unclear. The epigenetic repression of DNA repair genes appears to be important for progression for many types of cancer, for cancer susceptibility to DNA damaging agents, and for increased cancer susceptibility to synthetic lethality.
When Phase III trials indicate which efforts at synthetic lethality are beneficial therapeutically, synthetically lethal down regulation of DNA repair pathways should be incorporated into standard medical treatments of cancers.
Evaluation of which DNA repair pathway(s) are epigenetically deficient in particular types of GI cancer and/or particular patients may prove useful in guiding choice of radiation, chemotherapeutic and/or synthetic lethality agent.
Footnotes
Conflict-of-interest: The authors have no conflicts of interest. Carol Bernstein has no conflicts of interest. Harris Bernstein has no conflicts of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: December 16, 2014
First decision: March 6, 2015
Article in press: April 20, 2015
P- Reviewers: Amornyotin S, Hsu LS S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
References
- 1.Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci USA. 2008;105:16906–16911. doi: 10.1073/pnas.0809380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.German J. Bloom’s syndrome. I. Genetical and clinical observations in the first twenty-seven patients. Am J Hum Genet. 1969;21:196–227. [PMC free article] [PubMed] [Google Scholar]
- 3.Bohr VA. Deficient DNA repair in the human progeroid disorder, Werner syndrome. Mutat Res. 2005;577:252–259. doi: 10.1016/j.mrfmmm.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 4.Monnat RJ. Human RECQ helicases: roles in DNA metabolism, mutagenesis and cancer biology. Semin Cancer Biol. 2010;20:329–339. doi: 10.1016/j.semcancer.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson LH, Hinz JM. Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: mechanistic insights. Mutat Res. 2009;668:54–72. doi: 10.1016/j.mrfmmm.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alter BP. Cancer in Fanconi anemia, 1927-2001. Cancer. 2003;97:425–440. doi: 10.1002/cncr.11046. [DOI] [PubMed] [Google Scholar]
- 7.Meyer LA, Broaddus RR, Lu KH. Endometrial cancer and Lynch syndrome: clinical and pathologic considerations. Cancer Control. 2009;16:14–22. doi: 10.1177/107327480901600103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Markkanen E, Dorn J, Hübscher U. MUTYH DNA glycosylase: the rationale for removing undamaged bases from the DNA. Front Genet. 2013;4:18. doi: 10.3389/fgene.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viktorsson K, De Petris L, Lewensohn R. The role of p53 in treatment responses of lung cancer. Biochem Biophys Res Commun. 2005;331:868–880. doi: 10.1016/j.bbrc.2005.03.192. [DOI] [PubMed] [Google Scholar]
- 10.Testa JR, Malkin D, Schiffman JD. Connecting molecular pathways to hereditary cancer risk syndromes. Am Soc Clin Oncol Educ Book. 2013:81–90. doi: 10.1200/EdBook_AM.2013.33.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruijs MW, Verhoef S, Rookus MA, Pruntel R, van der Hout AH, Hogervorst FB, Kluijt I, Sijmons RH, Aalfs CM, Wagner A, et al. TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet. 2010;47:421–428. doi: 10.1136/jmg.2009.073429. [DOI] [PubMed] [Google Scholar]
- 12.Garber JE, Offit K. Hereditary cancer predisposition syndromes. J Clin Oncol. 2005;23:276–292. doi: 10.1200/JCO.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 13.Lindor NM, McMaster ML, Lindor CJ, Greene MH. Concise handbook of familial cancer susceptibility syndromes - second edition. J Natl Cancer Inst Monogr. 2008;(38):1–93. doi: 10.1093/jncimonographs/lgn001. [DOI] [PubMed] [Google Scholar]
- 14.Bernstein C, Prasad AR, Nfonsam V, Bernstein H. DNA Damage, DNA Repair and Cancer, New Research Directions in DNA Repair, Prof. Clark Chen (Ed.). InTech. 38. 2013. Available from: http://www.intechopen.com/books/new-research-directions-in-dna-repair/dna-damage-dna-repair-and-cancer. [Google Scholar]
- 15.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halford S, Rowan A, Sawyer E, Talbot I, Tomlinson I. O(6)-methylguanine methyltransferase in colorectal cancers: detection of mutations, loss of expression, and weak association with G: C& gt; A: T transitions. Gut. 2005;54:797–802. doi: 10.1136/gut.2004.059535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR, Einspahr JG, et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst. 2005;97:1330–1338. doi: 10.1093/jnci/dji275. [DOI] [PubMed] [Google Scholar]
- 18.Psofaki V, Kalogera C, Tzambouras N, Stephanou D, Tsianos E, Seferiadis K, Kolios G. Promoter methylation status of hMLH1, MGMT, and CDKN2A/p16 in colorectal adenomas. World J Gastroenterol. 2010;16:3553–3560. doi: 10.3748/wjg.v16.i28.3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee KH, Lee JS, Nam JH, Choi C, Lee MC, Park CS, Juhng SW, Lee JH. Promoter methylation status of hMLH1, hMSH2, and MGMT genes in colorectal cancer associated with adenoma-carcinoma sequence. Langenbecks Arch Surg. 2011;396:1017–1026. doi: 10.1007/s00423-011-0812-9. [DOI] [PubMed] [Google Scholar]
- 20.Amatu A, Sartore-Bianchi A, Moutinho C, Belotti A, Bencardino K, Chirico G, Cassingena A, Rusconi F, Esposito A, Nichelatti M, et al. Promoter CpG island hypermethylation of the DNA repair enzyme MGMT predicts clinical response to dacarbazine in a phase II study for metastatic colorectal cancer. Clin Cancer Res. 2013;19:2265–2272. doi: 10.1158/1078-0432.CCR-12-3518. [DOI] [PubMed] [Google Scholar]
- 21.Mokarram P, Zamani M, Kavousipour S, Naghibalhossaini F, Irajie C, Moradi Sarabi M, Hosseini SV. Different patterns of DNA methylation of the two distinct O6-methylguanine-DNA methyltransferase (O6-MGMT) promoter regions in colorectal cancer. Mol Biol Rep. 2013;40:3851–3857. doi: 10.1007/s11033-012-2465-3. [DOI] [PubMed] [Google Scholar]
- 22.Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO, Bannwart F, Yurtsever H, Neuweiler J, Riehle HM, et al. Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer. Gastroenterology. 2005;128:1160–1171. doi: 10.1053/j.gastro.2005.01.056. [DOI] [PubMed] [Google Scholar]
- 23.Valeri N, Gasparini P, Fabbri M, Braconi C, Veronese A, Lovat F, Adair B, Vannini I, Fanini F, Bottoni A, et al. Modulation of mismatch repair and genomic stability by miR-155. Proc Natl Acad Sci USA. 2010;107:6982–6987. doi: 10.1073/pnas.1002472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helbock HJ, Beckman KB, Shigenaga MK, Walter PB, Woodall AA, Yeo HC, Ames BN. DNA oxidation matters: the HPLC-electrochemical detection assay of 8-oxo-deoxyguanosine and 8-oxo-guanine. Proc Natl Acad Sci USA. 1998;95:288–293. doi: 10.1073/pnas.95.1.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakamura J, Walker VE, Upton PB, Chiang SY, Kow YW, Swenberg JA. Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer Res. 1998;58:222–225. [PubMed] [Google Scholar]
- 26.Tice RR, Setlow RB. DNA repair and replication in aging organisms and cells. In: Finch EE and Schneider EL (eds., editor. ) Handbook of the Biology of Aging. New York: Van Nostrand Reinhold; 1985. pp. 173–224. [Google Scholar]
- 27.Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci USA. 2003;100:12871–12876. doi: 10.1073/pnas.2135498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Bont R, van Larebeke N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004;19:169–185. doi: 10.1093/mutage/geh025. [DOI] [PubMed] [Google Scholar]
- 29.Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J Natl Cancer Inst. 1981;66:1191–1308. [PubMed] [Google Scholar]
- 30.Bernstein H, Bernstein C, Payne CM, Dvorak K. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J Gastroenterol. 2009;15:3329–3340. doi: 10.3748/wjg.15.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reddy BS, Hanson D, Mangat S, Mathews L, Sbaschnig M, Sharma C, Simi B. Effect of high-fat, high-beef diet and of mode of cooking of beef in the diet on fecal bacterial enzymes and fecal bile acids and neutral sterols. J Nutr. 1980;110:1880–1887. doi: 10.1093/jn/110.9.1880. [DOI] [PubMed] [Google Scholar]
- 32.Ou J, DeLany JP, Zhang M, Sharma S, O’Keefe SJ. Association between low colonic short-chain fatty acids and high bile acids in high colon cancer risk populations. Nutr Cancer. 2012;64:34–40. doi: 10.1080/01635581.2012.630164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Keefe SJ, Kidd M, Espitalier-Noel G, Owira P. Rarity of colon cancer in Africans is associated with low animal product consumption, not fiber. Am J Gastroenterol. 1999;94:1373–1380. doi: 10.1111/j.1572-0241.1999.01089.x. [DOI] [PubMed] [Google Scholar]
- 34.American Cancer Society. Cancer Facts and Figures. New York: Van Nostrand Reinhold; 2009. Available from: http://www.cancer.org/Research/CancerFactsFigures/cancer-facts-figures-2009. [Google Scholar]
- 35.Bernstein C, Holubec H, Bhattacharyya AK, Nguyen H, Payne CM, Zaitlin B, Bernstein H. Carcinogenicity of deoxycholate, a secondary bile acid. Arch Toxicol. 2011;85:863–871. doi: 10.1007/s00204-011-0648-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prasad AR, Prasad S, Nguyen H, Facista A, Lewis C, Zaitlin B, Bernstein H, Bernstein C. Novel diet-related mouse model of colon cancer parallels human colon cancer. World J Gastrointest Oncol. 2014;6:225–243. doi: 10.4251/wjgo.v6.i7.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maskarinec G, Noh JJ. The effect of migration on cancer incidence among Japanese in Hawaii. Ethn Dis. 2004;14:431–439. [PubMed] [Google Scholar]
- 38.Human DNA Repair Genes. This is an update of the table cited in Wood RD, Mitchell M, & Lindahl T Mutation Research, 2005, in Science, 2001, in the reference book DNA Repair and Mutagenesis, 2nd ed, 2006, and in Nature Reviews Cancer, 2011. Available from: http://sciencepark.mdanderson.org/labs/wood/dna_repair_genes.html.
- 39.Sharma S, Helchowski CM, Canman CE. The roles of DNA polymerase ζ and the Y family DNA polymerases in promoting or preventing genome instability. Mutat Res. 2013;743-744:97–110. doi: 10.1016/j.mrfmmm.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, Walker GC. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev. 2009;73:134–154. doi: 10.1128/MMBR.00034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Temviriyanukul P, Meijers M, van Hees-Stuivenberg S, Boei JJ, Delbos F, Ohmori H, de Wind N, Jansen JG. Different sets of translesion synthesis DNA polymerases protect from genome instability induced by distinct food-derived genotoxins. Toxicol Sci. 2012;127:130–138. doi: 10.1093/toxsci/kfs074. [DOI] [PubMed] [Google Scholar]
- 42.Kunz BA, Ramachandran K, Vonarx EJ. DNA sequence analysis of spontaneous mutagenesis in Saccharomyces cerevisiae. Genetics. 1998;148:1491–1505. doi: 10.1093/genetics/148.4.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stuart GR, Oda Y, de Boer JG, Glickman BW. Mutation frequency and specificity with age in liver, bladder and brain of lacI transgenic mice. Genetics. 2000;154:1291–1300. doi: 10.1093/genetics/154.3.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kohler SW, Provost GS, Fieck A, Kretz PL, Bullock WO, Sorge JA, Putman DL, Short JM. Spectra of spontaneous and mutagen-induced mutations in the lacI gene in transgenic mice. Proc Natl Acad Sci USA. 1991;88:7958–7962. doi: 10.1073/pnas.88.18.7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bindra RS, Goglia AG, Jasin M, Powell SN. Development of an assay to measure mutagenic non-homologous end-joining repair activity in mammalian cells. Nucleic Acids Res. 2013;41:e115. doi: 10.1093/nar/gkt255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4:e1000155. doi: 10.1371/journal.pgen.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morano A, Angrisano T, Russo G, Landi R, Pezone A, Bartollino S, Zuchegna C, Babbio F, Bonapace IM, Allen B, et al. Targeted DNA methylation by homology-directed repair in mammalian cells. Transcription reshapes methylation on the repaired gene. Nucleic Acids Res. 2014;42:804–821. doi: 10.1093/nar/gkt920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li J, Braganza A, Sobol RW. Base excision repair facilitates a functional relationship between Guanine oxidation and histone demethylation. Antioxid Redox Signal. 2013;18:2429–2443. doi: 10.1089/ars.2012.5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Franchini DM, Chan CF, Morgan H, Incorvaia E, Rangam G, Dean W, Santos F, Reik W, Petersen-Mahrt SK. Processive DNA demethylation via DNA deaminase-induced lesion resolution. PLoS One. 2014;9:e97754. doi: 10.1371/journal.pone.0097754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malanga M, Althaus FR. The role of poly(ADP-ribose) in the DNA damage signaling network. Biochem Cell Biol. 2005;83:354–364. doi: 10.1139/o05-038. [DOI] [PubMed] [Google Scholar]
- 51.Gottschalk AJ, Timinszky G, Kong SE, Jin J, Cai Y, Swanson SK, Washburn MP, Florens L, Ladurner AG, Conaway JW, et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc Natl Acad Sci USA. 2009;106:13770–13774. doi: 10.1073/pnas.0906920106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin JC, Jeong S, Liang G, Takai D, Fatemi M, Tsai YC, Egger G, Gal-Yam EN, Jones PA. Role of nucleosomal occupancy in the epigenetic silencing of the MLH1 CpG island. Cancer Cell. 2007;12:432–444. doi: 10.1016/j.ccr.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hou L, Zhang X, Wang D, Baccarelli A. Environmental chemical exposures and human epigenetics. Int J Epidemiol. 2012;41:79–105. doi: 10.1093/ije/dyr154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sharma V, Misteli T. Non-coding RNAs in DNA damage and repair. FEBS Lett. 2013;587:1832–1839. doi: 10.1016/j.febslet.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O’Hagan HM. Chromatin modifications during repair of environmental exposure-induced DNA damage: a potential mechanism for stable epigenetic alterations. Environ Mol Mutagen. 2014;55:278–291. doi: 10.1002/em.21830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang P, Dong Q, Zhang C, Kuan PF, Liu Y, Jeck WR, Andersen JB, Jiang W, Savich GL, Tan TX, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013;32:3091–3100. doi: 10.1038/onc.2012.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hartman DJ, Binion D, Regueiro M, Schraut W, Bahary N, Sun W, Nikiforova M, Pai RK. Isocitrate dehydrogenase-1 is mutated in inflammatory bowel disease-associated intestinal adenocarcinoma with low-grade tubuloglandular histology but not in sporadic intestinal adenocarcinoma. Am J Surg Pathol. 2014;38:1147–1156. doi: 10.1097/PAS.0000000000000239. [DOI] [PubMed] [Google Scholar]
- 58.Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, El Hallani S, Boisselier B, Mokhtari K, Hoang-Xuan K, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009;27:4150–4154. doi: 10.1200/JCO.2009.21.9832. [DOI] [PubMed] [Google Scholar]
- 59.This file is licensed under the Creative Commons Attribution-Share Alike 3. 0 Unported license. This image is free for re-use as long as the Wikimedia file is referred to. Available from: http://commons.wikimedia.org/wiki/File: Image_of_resected_colon_segment_with_cancer_&_4_nearby_polyps_plus_schematic_of_field_defects_with_sub-clones.jpg.
- 60.Svrcek M, Buhard O, Colas C, Coulet F, Dumont S, Massaoudi I, Lamri A, Hamelin R, Cosnes J, Oliveira C, et al. Methylation tolerance due to an O6-methylguanine DNA methyltransferase (MGMT) field defect in the colonic mucosa: an initiating step in the development of mismatch repair-deficient colorectal cancers. Gut. 2010;59:1516–1526. doi: 10.1136/gut.2009.194787. [DOI] [PubMed] [Google Scholar]
- 61.Howard JH, Frolov A, Tzeng CW, Stewart A, Midzak A, Majmundar A, Godwin A, Heslin M, Bellacosa A, Arnoletti JP. Epigenetic downregulation of the DNA repair gene MED1/MBD4 in colorectal and ovarian cancer. Cancer Biol Ther. 2009;8:94–100. doi: 10.4161/cbt.8.1.7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Facista A, Nguyen H, Lewis C, Prasad AR, Ramsey L, Zaitlin B, Nfonsam V, Krouse RS, Bernstein H, Payne CM, et al. Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer. Genome Integr. 2012;3:3. doi: 10.1186/2041-9414-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kawasaki T, Ohnishi M, Suemoto Y, Kirkner GJ, Liu Z, Yamamoto H, Loda M, Fuchs CS, Ogino S. WRN promoter methylation possibly connects mucinous differentiation, microsatellite instability and CpG island methylator phenotype in colorectal cancer. Mod Pathol. 2008;21:150–158. doi: 10.1038/modpathol.3800996. [DOI] [PubMed] [Google Scholar]
- 64.Zou XP, Zhang B, Zhang XQ, Chen M, Cao J, Liu WJ. Promoter hypermethylation of multiple genes in early gastric adenocarcinoma and precancerous lesions. Hum Pathol. 2009;40:1534–1542. doi: 10.1016/j.humpath.2009.01.029. [DOI] [PubMed] [Google Scholar]
- 65.Wani M, Afroze D, Makhdoomi M, Hamid I, Wani B, Bhat G, Wani R, Wani K. Promoter methylation status of DNA repair gene (hMLH1) in gastric carcinoma patients of the Kashmir valley. Asian Pac J Cancer Prev. 2012;13:4177–4181. doi: 10.7314/apjcp.2012.13.8.4177. [DOI] [PubMed] [Google Scholar]
- 66.Agarwal A, Polineni R, Hussein Z, Vigoda I, Bhagat TD, Bhattacharyya S, Maitra A, Verma A. Role of epigenetic alterations in the pathogenesis of Barrett’s esophagus and esophageal adenocarcinoma. Int J Clin Exp Pathol. 2012;5:382–396. [PMC free article] [PubMed] [Google Scholar]
- 67. Available from: http://commons.wikimedia.org/wiki/File: Expression_of_DNA_repair_proteins_ERCC1,_PMS2_&_KU86_in_field_defect.jpg.
- 68.Nguyen H, Loustaunau C, Facista A, Ramsey L, Hassounah N, Taylor H, Krouse R, Payne CM, Tsikitis VL, Goldschmid S, et al. Deficient Pms2, ERCC1, Ku86, CcOI in field defects during progression to colon cancer. J Vis Exp. 2010;(41) doi: 10.3791/1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kitajima Y, Miyazaki K, Matsukura S, Tanaka M, Sekiguchi M. Loss of expression of DNA repair enzymes MGMT, hMLH1, and hMSH2 during tumor progression in gastric cancer. Gastric Cancer. 2003;6:86–95. doi: 10.1007/s10120-003-0213-z. [DOI] [PubMed] [Google Scholar]
- 70.Farkas SA, Vymetalkova V, Vodickova L, Vodicka P, Nilsson TK. DNA methylation changes in genes frequently mutated in sporadic colorectal cancer and in the DNA repair and Wnt/β-catenin signaling pathway genes. Epigenomics. 2014;6:179–191. doi: 10.2217/epi.14.7. [DOI] [PubMed] [Google Scholar]
- 71.Lynam-Lennon N, Reynolds JV, Marignol L, Sheils OM, Pidgeon GP, Maher SG. MicroRNA-31 modulates tumour sensitivity to radiation in oesophageal adenocarcinoma. J Mol Med (Berl) 2012;90:1449–1458. doi: 10.1007/s00109-012-0924-x. [DOI] [PubMed] [Google Scholar]
- 72.Asangani IA, Harms PW, Dodson L, Pandhi M, Kunju LP, Maher CA, Fullen DR, Johnson TM, Giordano TJ, Palanisamy N, et al. Genetic and epigenetic loss of microRNA-31 leads to feed-forward expression of EZH2 in melanoma. Oncotarget. 2012;3:1011–1025. doi: 10.18632/oncotarget.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hofstad B, Vatn MH, Andersen SN, Huitfeldt HS, Rognum T, Larsen S, Osnes M. Growth of colorectal polyps: redetection and evaluation of unresected polyps for a period of three years. Gut. 1996;39:449–456. doi: 10.1136/gut.39.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stryker SJ, Wolff BG, Culp CE, Libbe SD, Ilstrup DM, MacCarty RL. Natural history of untreated colonic polyps. Gastroenterology. 1987;93:1009–1013. doi: 10.1016/0016-5085(87)90563-4. [DOI] [PubMed] [Google Scholar]
- 75.Loeve F, Boer R, Zauber AG, Van Ballegooijen M, Van Oortmarssen GJ, Winawer SJ, Habbema JD. National Polyp Study data: evidence for regression of adenomas. Int J Cancer. 2004;111:633–639. doi: 10.1002/ijc.20277. [DOI] [PubMed] [Google Scholar]
- 76.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, Paulson TG, Blount PL, Risques RA, Rabinovitch PS, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38:468–473. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- 78.Kuhmann C, Li C, Kloor M, Salou M, Weigel C, Schmidt CR, Ng LW, Tsui WW, Leung SY, Yuen ST, et al. Altered regulation of DNA ligase IV activity by aberrant promoter DNA methylation and gene amplification in colorectal cancer. Hum Mol Genet. 2014;23:2043–2054. doi: 10.1093/hmg/ddt599. [DOI] [PubMed] [Google Scholar]
- 79.Chen HY, Shao CJ, Chen FR, Kwan AL, Chen ZP. Role of ERCC1 promoter hypermethylation in drug resistance to cisplatin in human gliomas. Int J Cancer. 2010;126:1944–1954. doi: 10.1002/ijc.24772. [DOI] [PubMed] [Google Scholar]
- 80.Agrelo R, Cheng WH, Setien F, Ropero S, Espada J, Fraga MF, Herranz M, Paz MF, Sanchez-Cespedes M, Artiga MJ, et al. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proc Natl Acad Sci USA. 2006;103:8822–8827. doi: 10.1073/pnas.0600645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koinuma K, Kaneda R, Toyota M, Yamashita Y, Takada S, Choi YL, Wada T, Okada M, Konishi F, Nagai H, et al. Screening for genomic fragments that are methylated specifically in colorectal carcinoma with a methylated MLH1 promoter. Carcinogenesis. 2005;26:2078–2085. doi: 10.1093/carcin/bgi184. [DOI] [PubMed] [Google Scholar]
- 82.Singh P, Yang M, Dai H, Yu D, Huang Q, Tan W, Kernstine KH, Lin D, Shen B. Overexpression and hypomethylation of flap endonuclease 1 gene in breast and other cancers. Mol Cancer Res. 2008;6:1710–1717. doi: 10.1158/1541-7786.MCR-08-0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ling ZQ, Li P, Ge MH, Hu FJ, Fang XH, Dong ZM, Mao WM. Aberrant methylation of different DNA repair genes demonstrates distinct prognostic value for esophageal cancer. Dig Dis Sci. 2011;56:2992–3004. doi: 10.1007/s10620-011-1774-z. [DOI] [PubMed] [Google Scholar]
- 84.Su Y, Yin L, Liu R, Sheng J, Yang M, Wang Y, Pan E, Guo W, Pu Y, Zhang J, et al. Promoter methylation status of MGMT, hMSH2, and hMLH1 and its relationship to corresponding protein expression and TP53 mutations in human esophageal squamous cell carcinoma. Med Oncol. 2014;31:784. doi: 10.1007/s12032-013-0784-4. [DOI] [PubMed] [Google Scholar]
- 85.Vasavi M, Ponnala S, Gujjari K, Boddu P, Bharatula RS, Prasad R, Ahuja YR, Hasan Q. DNA methylation in esophageal diseases including cancer: special reference to hMLH1 gene promoter status. Tumori. 2006;92:155–162. doi: 10.1177/030089160609200212. [DOI] [PubMed] [Google Scholar]
- 86.Wang L, Xie L, Wang J, Shen J, Liu B. Correlation between the methylation of SULF2 and WRN promoter and the irinotecan chemosensitivity in gastric cancer. BMC Gastroenterol. 2013;13:173. doi: 10.1186/1471-230X-13-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huang Q, Su X, Ai L, Li M, Fan CY, Weiss LM. Promoter hypermethylation of multiple genes in gastric lymphoma. Leuk Lymphoma. 2007;48:1988–1996. doi: 10.1080/10428190701573224. [DOI] [PubMed] [Google Scholar]
- 88.Lages E, Ipas H, Guttin A, Nesr H, Berger F, Issartel JP. MicroRNAs: molecular features and role in cancer. Front Biosci (Landmark Ed) 2012;17:2508–2540. doi: 10.2741/4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Motoyama K, Inoue H, Nakamura Y, Uetake H, Sugihara K, Mori M. Clinical significance of high mobility group A2 in human gastric cancer and its relationship to let-7 microRNA family. Clin Cancer Res. 2008;14:2334–2340. doi: 10.1158/1078-0432.CCR-07-4667. [DOI] [PubMed] [Google Scholar]
- 90.Vrba L, Muñoz-Rodríguez JL, Stampfer MR, Futscher BW. miRNA gene promoters are frequent targets of aberrant DNA methylation in human breast cancer. PLoS One. 2013;8:e54398. doi: 10.1371/journal.pone.0054398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cleynen I, Van de Ven WJ. The HMGA proteins: a myriad of functions (Review) Int J Oncol. 2008;32:289–305. [PubMed] [Google Scholar]
- 92.Borrmann L, Schwanbeck R, Heyduk T, Seebeck B, Rogalla P, Bullerdiek J, Wisniewski JR. High mobility group A2 protein and its derivatives bind a specific region of the promoter of DNA repair gene ERCC1 and modulate its activity. Nucleic Acids Res. 2003;31:6841–6851. doi: 10.1093/nar/gkg884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lyn-Cook L, Word B, George N, Lyn-Cook B, Hammons G. Effect of cigarette smoke condensate on gene promoter methylation in human lung cells. Tob Induc Dis. 2014;12:15. doi: 10.1186/1617-9625-12-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wouters MD, van Gent DC, Hoeijmakers JH, Pothof J. MicroRNAs, the DNA damage response and cancer. Mutat Res. 2011;717:54–66. doi: 10.1016/j.mrfmmm.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 96.Tessitore A, Cicciarelli G, Del Vecchio F, Gaggiano A, Verzella D, Fischietti M, Vecchiotti D, Capece D, Zazzeroni F, Alesse E. MicroRNAs in the DNA Damage/Repair Network and Cancer. Int J Genomics. 2014;2014:820248. doi: 10.1155/2014/820248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vincent K, Pichler M, Lee GW, Ling H. MicroRNAs, genomic instability and cancer. Int J Mol Sci. 2014;15:14475–14491. doi: 10.3390/ijms150814475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Farazi TA, Spitzer JI, Morozov P, Tuschl T. miRNAs in human cancer. J Pathol. 2011;223:102–115. doi: 10.1002/path.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hu H, Du L, Nagabayashi G, Seeger RC, Gatti RA. ATM is down-regulated by N-Myc-regulated microRNA-421. Proc Natl Acad Sci USA. 2010;107:1506–1511. doi: 10.1073/pnas.0907763107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee KH, Lotterman C, Karikari C, Omura N, Feldmann G, Habbe N, Goggins MG, Mendell JT, Maitra A. Epigenetic silencing of MicroRNA miR-107 regulates cyclin-dependent kinase 6 expression in pancreatic cancer. Pancreatology. 2009;9:293–301. doi: 10.1159/000186051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Huang JW, Wang Y, Dhillon KK, Calses P, Villegas E, Mitchell PS, Tewari M, Kemp CJ, Taniguchi T. Systematic screen identifies miRNAs that target RAD51 and RAD51D to enhance chemosensitivity. Mol Cancer Res. 2013;11:1564–1573. doi: 10.1158/1541-7786.MCR-13-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Suzuki H, Yamamoto E, Nojima M, Kai M, Yamano HO, Yoshikawa K, Kimura T, Kudo T, Harada E, Sugai T, et al. Methylation-associated silencing of microRNA-34b/c in gastric cancer and its involvement in an epigenetic field defect. Carcinogenesis. 2010;31:2066–2073. doi: 10.1093/carcin/bgq203. [DOI] [PubMed] [Google Scholar]
- 103.Hegre SA, Sætrom P, Aas PA, Pettersen HS, Otterlei M, Krokan HE. Multiple microRNAs may regulate the DNA repair enzyme uracil-DNA glycosylase. DNA Repair (Amst) 2013;12:80–86. doi: 10.1016/j.dnarep.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 104.Tanaka N, Toyooka S, Soh J, Kubo T, Yamamoto H, Maki Y, Muraoka T, Shien K, Furukawa M, Ueno T, et al. Frequent methylation and oncogenic role of microRNA-34b/c in small-cell lung cancer. Lung Cancer. 2012;76:32–38. doi: 10.1016/j.lungcan.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 105.Wu XD, Song YC, Cao PL, Zhang H, Guo Q, Yan R, Diao DM, Cheng Y, Dang CX. Detection of miR-34a and miR-34b/c in stool sample as potential screening biomarkers for noninvasive diagnosis of colorectal cancer. Med Oncol. 2014;31:894. doi: 10.1007/s12032-014-0894-7. [DOI] [PubMed] [Google Scholar]
- 106.Wang LQ, Kwong YL, Wong KF, Kho CS, Jin DY, Tse E, Rosèn A, Chim CS. Epigenetic inactivation of mir-34b/c in addition to mir-34a and DAPK1 in chronic lymphocytic leukemia. J Transl Med. 2014;12:52. doi: 10.1186/1479-5876-12-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ito M, Mitsuhashi K, Igarashi H, Nosho K, Naito T, Yoshii S, Takahashi H, Fujita M, Sukawa Y, Yamamoto E, et al. MicroRNA-31 expression in relation to BRAF mutation, CpG island methylation and colorectal continuum in serrated lesions. Int J Cancer. 2014;135:2507–2515. doi: 10.1002/ijc.28920. [DOI] [PubMed] [Google Scholar]
- 108.Sarver AL, French AJ, Borralho PM, Thayanithy V, Oberg AL, Silverstein KA, Morlan BW, Riska SM, Boardman LA, Cunningham JM, et al. Human colon cancer profiles show differential microRNA expression depending on mismatch repair status and are characteristic of undifferentiated proliferative states. BMC Cancer. 2009;9:401. doi: 10.1186/1471-2407-9-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Harada T, Yamamoto E, Yamano HO, Nojima M, Maruyama R, Kumegawa K, Ashida M, Yoshikawa K, Kimura T, Harada E, et al. Analysis of DNA methylation in bowel lavage fluid for detection of colorectal cancer. Cancer Prev Res (Phila) 2014;7:1002–1010. doi: 10.1158/1940-6207.CAPR-14-0162. [DOI] [PubMed] [Google Scholar]
- 110.Zhu F, Liu JL, Li JP, Xiao F, Zhang ZX, Zhang L. MicroRNA-124 (miR-124) regulates Ku70 expression and is correlated with neuronal death induced by ischemia/reperfusion. J Mol Neurosci. 2014;52:148–155. doi: 10.1007/s12031-013-0155-9. [DOI] [PubMed] [Google Scholar]
- 111.Chang S, Wang RH, Akagi K, Kim KA, Martin BK, Cavallone L, Haines DC, Basik M, Mai P, Poggi E, et al. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nat Med. 2011;17:1275–1282. doi: 10.1038/nm.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gasparini P, Lovat F, Fassan M, Casadei L, Cascione L, Jacob NK, Carasi S, Palmieri D, Costinean S, Shapiro CL, et al. Protective role of miR-155 in breast cancer through RAD51 targeting impairs homologous recombination after irradiation. Proc Natl Acad Sci USA. 2014;111:4536–4541. doi: 10.1073/pnas.1402604111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang MY, Chen MT, Huang PI, Wang CY, Chang YC, Yang YP, Lo WL, Sung WH, Liao YW, Lee YY, et al. Nuclear Localization Signal-enhanced Polyurethane-Short Branch Polyethylenimine-mediated Delivery of Let-7a Inhibited Cancer Stem-like Properties by Targeting the 3’UTR of HMGA2 in Anaplastic Astrocytoma. Cell Transplant. 2014:Jun 3; Epub ahead of print. doi: 10.3727/096368914X682107. [DOI] [PubMed] [Google Scholar]
- 114.Puca F, Colamaio M, Federico A, Gemei M, Tosti N, Bastos AU, Del Vecchio L, Pece S, Battista S, Fusco A. HMGA1 silencing restores normal stem cell characteristics in colon cancer stem cells by increasing p53 levels. Oncotarget. 2014;5:3234–3245. doi: 10.18632/oncotarget.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schubert M, Spahn M, Kneitz S, Scholz CJ, Joniau S, Stroebel P, Riedmiller H, Kneitz B. Distinct microRNA expression profile in prostate cancer patients with early clinical failure and the impact of let-7 as prognostic marker in high-risk prostate cancer. PLoS One. 2013;8:e65064. doi: 10.1371/journal.pone.0065064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tang H, Wang Z, Liu Q, Liu X, Wu M, Li G. Disturbing miR-182 and -381 inhibits BRD7 transcription and glioma growth by directly targeting LRRC4. PLoS One. 2014;9:e84146. doi: 10.1371/journal.pone.0084146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Moskwa P, Buffa FM, Pan Y, Panchakshari R, Gottipati P, Muschel RJ, Beech J, Kulshrestha R, Abdelmohsen K, Weinstock DM, et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol Cell. 2011;41:210–220. doi: 10.1016/j.molcel.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Krishnan K, Steptoe AL, Martin HC, Wani S, Nones K, Waddell N, Mariasegaram M, Simpson PT, Lakhani SR, Gabrielli B, et al. MicroRNA-182-5p targets a network of genes involved in DNA repair. RNA. 2013;19:230–242. doi: 10.1261/rna.034926.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Perilli L, Vicentini C, Agostini M, Pizzini S, Pizzi M, D’Angelo E, Bortoluzzi S, Mandruzzato S, Mammano E, Rugge M, et al. Circulating miR-182 is a biomarker of colorectal adenocarcinoma progression. Oncotarget. 2014;5:6611–6619. doi: 10.18632/oncotarget.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tuna M, Amos CI. Genomic sequencing in cancer. Cancer Lett. 2013;340:161–170. doi: 10.1016/j.canlet.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Weaver JM, Ross-Innes CS, Shannon N, Lynch AG, Forshew T, Barbera M, Murtaza M, Ong CA, Lao-Sirieix P, Dunning MJ, et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat Genet. 2014;46:837–843. doi: 10.1038/ng.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Roach JC, Glusman G, Smit AF, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Campbell CD, Chong JX, Malig M, Ko A, Dumont BL, Han L, Vives L, O’Roak BJ, Sudmant PH, Shendure J, et al. Estimating the human mutation rate using autozygosity in a founder population. Nat Genet. 2012;44:1277–1281. doi: 10.1038/ng.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Keightley PD. Rates and fitness consequences of new mutations in humans. Genetics. 2012;190:295–304. doi: 10.1534/genetics.111.134668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ye K, Beekman M, Lameijer EW, Zhang Y, Moed MH, van den Akker EB, Deelen J, Houwing-Duistermaat JJ, Kremer D, Anvar SY, et al. Aging as accelerated accumulation of somatic variants: whole-genome sequencing of centenarian and middle-aged monozygotic twin pairs. Twin Res Hum Genet. 2013;16:1026–1032. doi: 10.1017/thg.2013.73. [DOI] [PubMed] [Google Scholar]
- 128.This file is licensed under the Creative Commons Attribution-Share Alike 3. 0 Unported license. This image is free for re-use as long as the Wikimedia file is referred to. This figure was adapted from a Wikimedia image and only includes DNA repair genes epigenetically modified in one or more gastrointestinal cancers. Available from: http://commons.wikimedia.org/wiki/File: DNA_damage,_repair,_epigenetic_alteration_of_repair_in_cancer.jpg.
- 129.This file is licensed under the Creative Commons Attribution-Share Alike 3. 0 Unported license. This image is free for re-use as long as the Wikimedia file is referred to. Available from: http://commons.wikimedia.org/wiki/File: Diagram_Damage_to_Cancer_Wiki_300dpi. svg.
- 130.Cheung-Ong K, Giaever G, Nislow C. DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol. 2013;20:648–659. doi: 10.1016/j.chembiol.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 131.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 132.O’Sullivan CC, Moon DH, Kohn EC, Lee JM. Beyond Breast and Ovarian Cancers: PARP Inhibitors for BRCA Mutation-Associated and BRCA-Like Solid Tumors. Front Oncol. 2014;4:42. doi: 10.3389/fonc.2014.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.McLornan DP, List A, Mufti GJ. Applying synthetic lethality for the selective targeting of cancer. N Engl J Med. 2014;371:1725–1735. doi: 10.1056/NEJMra1407390. [DOI] [PubMed] [Google Scholar]
- 134.Hosoya N, Miyagawa K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014;105:370–388. doi: 10.1111/cas.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]