

ABSTRACT
Genetically based diseases constitute a major human health burden, and de novo germline mutations represent a source of heritable genetic alterations that can cause such disorders in offspring. The availability of transgenic rodent systems with recoverable, mutation reporter genes has been used to assess the occurrence of spontaneous point mutations in germline cells. Previous studies using the lacI mutation reporter transgenic mouse system showed that the frequency of spontaneous mutations is significantly lower in advanced male germ cells than in somatic cell types from the same individuals. Here we used this same mutation reporter transgene system to show that female germ cells also display a mutation frequency that is lower than that in corresponding somatic cells and similar to that seen in male germ cells, indicating this is a common feature of germ cells in both sexes. In addition, we showed that statistically significant differences in mutation frequencies are evident between germ cells and somatic cells in both sexes as early as mid-fetal stages in the mouse. Finally, a comparison of the mutation frequency in a general population of early type A spermatogonia with that in a population enriched for Thy-1-positive spermatogonia suggests there is heterogeneity among the early spermatogonial population such that a subset of these cells are predestined to form true spermatogonial stem cells. Taken together, these results support the disposable soma theory, which posits that genetic integrity is normally maintained more stringently in the germ line than in the soma and suggests that this is achieved by minimizing the initial occurrence of mutations in early germline cells and their subsequent gametogenic progeny relative to that in somatic cells.
Keywords: mutations, oogenesis, spermatogenesis, spermatogonial stem cells
INTRODUCTION
In mammals and other sexually reproducing species, the primary function of the gametes is to transmit genetic information from parents to their offspring so that this information can direct the formation of a new individual following fertilization. In 1977, TB Kirkwood proposed the “disposable soma theory,” which suggests that because germ cells give rise to the entire subsequent generation of individuals, it is evolutionarily advantageous for these cells to use mechanisms to maintain an enhanced level of genetic integrity, even if this requires the expenditure of additional energy [1]. That there is indeed a low frequency of spontaneous mutations in mammalian germline cells was suggested by early studies of mice [2, 3]. The specific locus test was used to examine spontaneous and induced germline mutation frequencies to evaluate the effects of suspected genotoxins [4–8]. These and related studies estimated the spontaneous mutation frequencies for female and male germ cells to be approximately 1.4 × 10−6 and 6.6 × 10−6, respectively [9]. However, this technique involved examining progeny for readily assessed phenotypic deviation from expected frequencies of homozygous recessive phenotypes encoded at multiple loci and was therefore limited to data derived from offspring rather than providing any sort of direct analysis of a specific cell type such as germ cells. Additionally, the specific locus test could not be used to determine mutation frequencies in somatic tissues, which precluded using this method to assess differences in the frequencies of mutations in germ cells and somatic cells, respectively. Finally, this method provided an overall indication of the frequency of germline mutations and relied on the timing of a genotoxic challenge to estimate developmental stage-specific acquisition of spontaneous mutations during gametogenesis.
An alternative approach to the assessment of frequency and spectrum of point mutations in the mouse was facilitated by the development of a novel transgenic reporter system termed the “Big Blue mouse” [10]. This system uses the prokaryotic lacI gene as a mutation reporter transgene, which can be selectively recovered from genomic DNA from any tissue or cell type as part of a lambda shuttle vector and packaged into infectious phage particles that can then be plated on Escherichia coli host cells. Because the lacI gene encodes the repressor of the lac operon, if the transgene has undergone a mutation while resident in the mouse cells, the repressor will fail to inhibit production of beta galactosidase in the E coli cells, and this can be detected by a colorimetric assay upon addition of the correct substrates to the plating medium [10–14]. This system therefore facilitates a direct assessment of frequencies and types of spontaneous point mutations in any cell type [10].
In a study published in 1998, Walter et al. [15] used the lacI mutation reporter transgene system to show that the frequency of spontaneous mutations in postnatal male germ cells is 5- to 10-fold lower than that in developmentally matched somatic cells from the same animal. Many studies have indicated that germ cells and pluripotent cells use mechanisms to specifically limit the initial occurrence of spontaneous point mutations based on elevated levels of DNA repair pathways that ameliorate most potentially mutagenic DNA damage [16–29]. These findings all support the disposable soma theory in that they suggest that a more stringent mechanism to maintain genetic integrity operating in germ cells or pluripotent cells leads to a lower mutational load than that found in somatic cells or differentiated cells, respectively. However, the disposable soma theory predicts that genetic integrity will be more stringently maintained in the germ lines of both males and females, and there have been no direct studies of mutation frequencies in female germ cells by using a mutation reporter transgene. This is due largely to the fact that many fewer germ cells can typically be recovered from female mice than from male mice. However, we were able to recover populations of primary oocytes at two developmental time points, one prenatal at 15.5 days postcoitum (dpc) and one postnatal at 2–6 days postpartum (dpp), in sufficient quantities to facilitate analysis using the lacI mutation reporter transgene system. In addition, we also recovered a population of type T1 prospermatogonia [30, 31] from fetal male mice at 15.5 dpc so that we could compare frequencies of mutations in male with those in female germ cells at this stage and the extent to which these frequencies differed from those in somatic cells from the same fetuses. Finally, we determined the frequency of mutations in a population of Thy1-positive (Thy-1+) spermatogonia from prepuberal male mice at 6 dpp, because the Thy1+ population is known to be enriched for true spermatogonial stem cells (SSCs).
Our results confirm that the disposable soma theory does indeed apply to both sexes and that this is a fundamental characteristic of the germ line that distinguishes it from the soma during most of the life cycle, from as early as the mid-fetal stages through adulthood. Furthermore, our results provide a unique perspective on the development of SSCs, suggesting that the early perinatal prospermatogonial and spermatogonial populations are heterogeneous with respect to the potential to form true SSCs and that the progenitors of these cells appear to be predetermined at a very early stage.
MATERIALS AND METHODS
Animals
“Big Blue” male and female mice homozygous for the lacI transgene on a C57BL/6 background were obtained from Taconic Farms, Inc. (Hudson, NY) and used for natural mating and as a source of male and female germ cells and somatic tissues. All procedures involving animals were approved in advance by the University of Texas at San Antonio Institutional Animal Care and Use Committee. Following euthanasia, gonads were rapidly removed and used immediately for germ cell separation procedures, and/or somatic tissues were recovered and frozen in liquid nitrogen and stored at −80°C until used for genomic DNA isolation.
Preparation of Cells and Genomic DNA Isolation
Isolations of male and female germ cells were performed on a mini-Sta Put 2%–4% bovine serum albumen gradient at unit gravity as previously described [32, 33]. Type T1 prospermatogonia were isolated from testes of 15.5 dpc male mice, and primary oocytes were isolated from ovaries of 15.5 dpc and 2–6 dpp female mice. The sex of each animal was determined on the basis of gonadal morphology. Purities of populations of each recovered germ cell type were ≥85%, based on cellular morphology as viewed with phase contrast microscopy. Samples of purified germ cells were frozen in liquid nitrogen and stored at −80°C.
Enrichment for True Spermatogonial Stem Cells from Testes of Male Mice at 6 dpp Based on Selection for Thy-1+ Cells
Testes were digested using 0.18 mg/ml trypsin plus 0.6 mg/ml DNase in minimal essential media alpha medium at pH 7.4 for 10 min at 37°C, fetal bovine serum was added to 10% (v/v), and cells were dispersed by gentle pipetting, centrifuged, resuspended in Hanks buffered salt solution and then centrifuged thru 30% Percoll solution (Sigma, St. Louis, MO) in Dulbecco phosphate buffered saline (DPBS) at 600 × g for 7 min. Cells were resuspended in DPBS and incubated with Thy-1 magnetic cell sorting (MACS) microbeads. Thy-1+ cells were selectively recovered using MACS separation columns (Miltenyi Biotec, Auburn, CA) and then snap frozen in liquid nitrogen and stored at −80°C. Importantly, it has previously been shown that Thy-1+ spermatogonia are enriched for true SSCs as assessed by transplanting these cells into recipient testes and monitoring subsequent spermatogenesis emanating from the transplanted cells [34].
Preparation of Genomic DNA from Cell or Tissue Samples
We previously developed a miniaturized version of the standard protocol for recovering high-molecular-weight genomic DNA from cell or tissue samples carrying the lacI transgene [35], and we followed that method for preparation of genomic DNA from germ cell and somatic tissue samples used in this study. Approximately 700 000–2 000 000 cells of each germ cell type were pooled for isolation of each genomic DNA sample. Male and female somatic tissues were collected from both sexes at 15.5 dpc and from female pups at 2–6 dpp, frozen in liquid nitrogen, and stored at −80°C. Genomic DNA was isolated from each germ cell population and from somatic tissues as previously described [35, 36]. Frozen germ cells were resuspended in 50 μl of ice-cold lysis buffer and pipetted up and down several times to lyse the cells and liberate genomic DNA. For somatic tissues, 50 μl of ice-cold lysis buffer was added to each 1-cm2 piece of tissue, and the mixture was transferred to a 2-ml Kontes Dounce tissue homogenizer (Sigma) and carefully homogenized approximately five times with each of two different sized plungers. The homogenate was filtered through a 100-μm Millipore filter (Millipore, Billerica, MA) fitted onto a 1.5-ml microcentrifuge tube. Germ cell or somatic cell samples were then centrifuged at 13 200 rpm for 15 min at 4°C, and the supernatant was removed, and the pellet was resuspended in digestion buffer containing RNace-it (Stratagene) and incubated at 50°C for 5 min. Twenty microliters of proteinase K solution was then added, and the sample was incubated for approximately 1.5 h at 50°C. Fifty microliters of 1× tris-EDTA (TE) was then added to each sample, followed by drop dialysis using a 0.025-μm Millipore dialysis membrane floating on 500 ml of 1× TE for 2 days at room temperature.
Analysis of Mutation Frequency
The lambda shuttle vector containing the lacI mutation reporter was selectively recovered from genomic DNA using Transpack packaging extract (Stratagene/Agilent) according to the manufacturer's instructions as previously described [35, 36]. Packaged phage were mixed with SCS-8 E coli strain cells (Stratagene/Agilent Technologies) and plated at ≤17 500 plaque-forming units per 25 × 25 cm NZY agar (Stratagene/Agilent Technologies) assay tray. Following incubation at 37°C for 16–18 h, mutant plaques were identified by their blue color, counted, cored, and replated on fresh 5-bromo-4-chloro-indolyl-β-D-galactopyranoside/NZY plates to confirm and plaque-purify phage displaying the lacI mutant phenotype. A representative portion of each plate was counted and used to determine the total number of plaque-forming units recovered from each packaging reaction. After adjustment for “clonal mutations” (see explanation in Analysis of Mutation Types following), the final mutation frequency was determined by dividing the number of confirmed, independent (nonclonal) mutant plaques by the total number of plaque-forming units screened.
Analysis of Mutation Types
DNA samples from isolated mutant phage were sent to the Center for Biomedical Research DNA Sequencing Facility, University of Victoria, Canada, for sequence analysis. In each case, a 15-μl aliquot of phage stock (cored mutant plaque stored in suspension medium buffer) was used as template for PCR amplification of a fragment spanning the lacI gene. Numbering of base positions in the amplified sequence was in accordance with that described by Farabaugh [37]. Amplification of each mutant phage was initially performed using lacI AF and lacI AR primers to produce a 2114-base pair (bp) “standard” PCR product (Table 1). However, several mutant phage from the 2–6 dpp primary oocyte sample failed to amplify with this primer set and so were subjected to amplification with a second primer set that included the lacI UP primer plus the previously used lacI AR downstream primer (Table 1). These primers, which would normally be expected to amplify a 3949-bp product from the unmutated lambda shuttle vector transgene were able to amplify a 2011-bp PCR product from mutants that carried a 1938-bp deletion that ablated the binding site for the lacI AF primer, thereby explaining the inability to amplify a product from these mutants by using the lacI AF and lacI AR primers. As with other incidences of recovery of more than one mutant plaque from the same original cell sample carrying the same mutation at the same position in the transgene, this multiplicity was assumed to be caused by clonal expansion of a single original mutation and so was scored as a single mutation when we determined the mutation frequency in that cell sample regardless of how many copies of the same mutant plaque were recovered, as previously described [35, 36, 38–42].
TABLE 1.
Primers used for sequence analysis.

Each PCR product was sequenced using a LI-COR Long ReadIR 4200 DNA sequencer to examine samples prepared by cycle sequencing using the SequiTherm EXCEL II DNA Sequencing Kit-LC (Epicentre, Madison, WI) according to the manufacturer's instructions. The sequence was analyzed using e-Seq version 3.0 base-calling software (LI-COR, Lincoln, NE) and SeqMan II version 6.1 analysis software (DNAStar, Madison, WI). A comparison of the lacI gene sequence in mutant phage with the wild-type lacI gene sequence revealed and confirmed each individual mutation. Mutations were categorized as transitions (pyrimidine-to-pyrimidine or purine-to-purine substitutions), transversions (pyrimidine-to-purine substitutions or vice versa), insertions, or deletions.
Statistical Analyses
Mutation frequency data were analyzed by a Poisson regression model with parameter estimates obtained by the method of maximum likelihood [43]. Statistical tests of differences used the likelihood ratio test. All computations were performed using SAS PROC GENMOD software (version 9.1; SAS Institute, Cary, NC). Mutation type data were analyzed using a chi-square test for independence [44]. Because of low expected frequencies, the exact P value was calculated (using SAS version 9.1 software).
RESULTS
Mutation Frequencies in Germ Cells and Somatic Cells
To determine the spontaneous mutation frequencies for female germ cells at two developmental stages, prenatal and postnatal, we enriched primary oocytes from female mouse fetuses at 15.5 dpc and from female pups at 2–6 dpp. We also purified type T1 prospermatogonia from male fetuses at 15.5 dpc. For each sample, we determined the frequency of mutations in the lacI reporter gene as described previously [15, 35, 36]. The spontaneous mutation frequencies determined for male and female germ cells at 15.5 dpc (0.99 × 10−5 and 0.82 × 10−5, respectively) were not significantly different from one another (P = 0.75) but were significantly lower than the frequencies found in male and female somatic cells at this same stage (2.82 × 10−5 and 2.91 × 10−5, respectively; P = 0.05) (Table 2). This suggests that the basic tenet of the disposable soma theory, which predicts that germ cells are likely to accumulate fewer spontaneous mutations than somatic cells, is a fundamental characteristic of the germ line in both males and females even during the early stages of germ cell development.
TABLE 2.
Spontaneous mutation frequencies in murine germ and somatic cells.
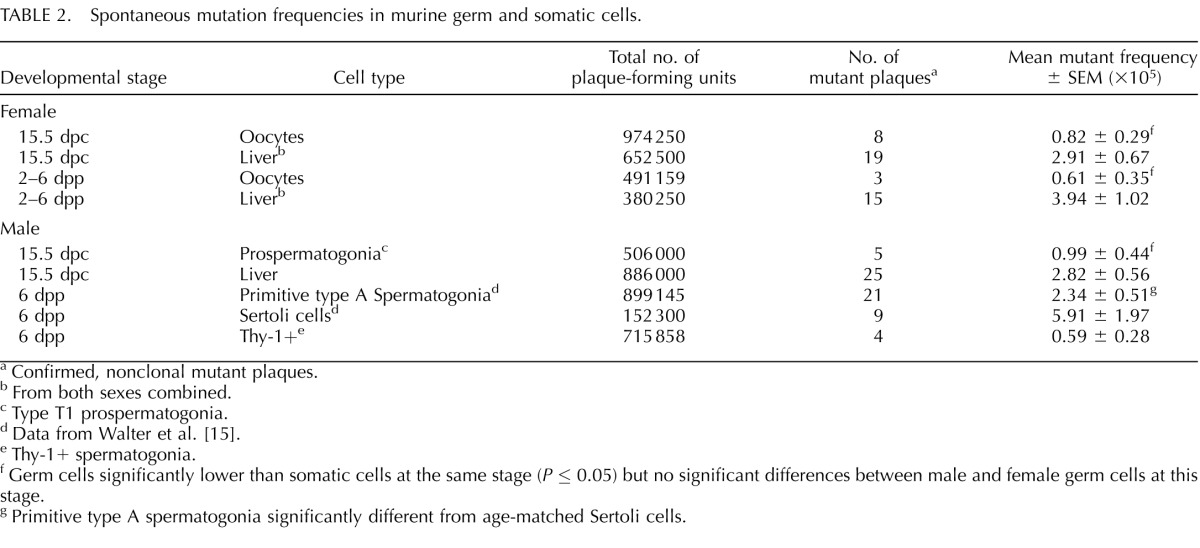
We next examined the frequency of spontaneous mutations in female germ cells and somatic cells at 2–6 dpp and compared this value to the mutation frequency found for primary oocytes at 15.5 dpc and that previously determined for male germ cells at 6 dpp [15]. We found that the spontaneous mutation frequency in female germ cells at 2–6 dpp (0.61 × 10−5) was similar to that in male and female germ cells at 15.5 dpc (0.82 × 10−5 and 0.99 × 10−5; P > 0.51) but was significantly lower than that reported for primitive type A spermatogonia from males at 6 dpp (2.34 × 10−5; P = 0.03). We suspect the difference in mutation frequency between male and female germ cells at 6 dpp may reflect the more active proliferation of male germ cells compared to the quiescent state of female germ cells at this stage. Nevertheless, the data shown in Table 2 reveal a consistent pattern of lower frequencies of spontaneous point mutations in germ cells relative to that in somatic cells in both sexes at every age investigated. Thus, the frequency of mutations in liver cells in both sexes combined at 15.5 dpc and 2–6 dpp was statistically significantly different from that in germ cells from both sexes at these same stages (P = 0.0001). This lends further support to the fundamental difference between germ cells and somatic cells in this regard as predicted by the disposable soma theory [1].
Mutation Types in Germ Cells and Somatic Cells
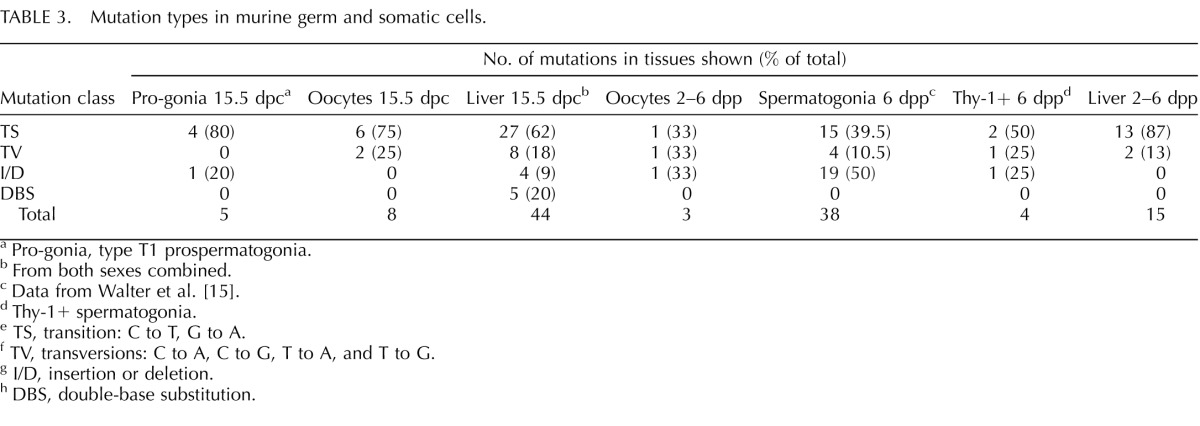
We sequenced each of the mutant lacI genes recovered from each cell or tissue sample to confirm that a mutation had occurred in each case and to determine the range of types of mutations associated with each cell sample (Table 3). Transition mutations were the most common type of point mutation detected in each cell type, with the exception of postnatal female germ cells. C-to-T transitions are typically the most common spontaneous mutation found in mammalian cells because of deamination of methylated cytosines to form uracils, which are then converted to thymines during DNA replication [11, 45]. Although we observed statistically significant differences between liver and germ cells of both sexes combined in the incidence of double-base substitutions at 15.5 dpc and 2–6 dpp (P = 0.0001) and certain other minor differences of borderline statistical significance in the relative proportion of different types of mutations in different cell types (e.g., between male and female germ cells at these same stages), we did not observe any extensive differences that could be ascribed to known biological distinctions among these cell types.
TABLE 3.
Mutation types in murine germ and somatic cells.
Developmental Kinetics of Accumulation of Spontaneous Mutations
Our new data acquired from primary oocytes at 15.5 dpc and 2–6 dpp and from type T1 prospermatogonia and male and female somatic cells at 15.5 dpc were combined with that previously determined for somatic cells at 10.5 dpc [35, 36] and for somatic cells and male germ cells at juvenile and adult stages [15] to reveal the developmental kinetics of accumulation of spontaneous mutations during prenatal and postnatal development of the germ line and various somatic lineages, respectively (Fig. 1). Because of a “bottleneck effect,” the frequency of mutations in the lacI mutation reporter transgene typically returns to zero at the beginning of each generation, and new spontaneous point mutations then accumulate thereafter [36]. During prenatal stages, we observed a relatively rapid accumulation of new mutations, resulting in a mutation frequency ranging from 2 × 10−5 to 3 × 10−5 in somatic cells by birth. This rapid accumulation of mutations likely reflects the extensive cellular proliferation activity that occurs during the prenatal stages of development. Following birth, we observed a slower increase in the frequency of spontaneous mutations in somatic cell lineages. Mutation frequencies in adult somatic cell types ranged from 3 × 10−5 to ≥6 × 10−5, with lineage-specific differences that may be based on either differential rates of cellular proliferation and/or differential exposure to mutagenic effects. No significant differences were observed in the frequencies of spontaneous mutations in male and female somatic cells, respectively, at any stage of development.
FIG. 1.

Accumulation of spontaneous mutations in germ and somatic cells during murine development. Spontaneous point mutations begin to accumulate in the lacI mutation reporter transgene following fertilization. The frequency of mutations rises quickly in somatic cells between fertilization and birth and then continues to rise at a lower rate after birth depending on specific somatic cell lineage. By 15.5 dpc, both male and female germ cells have accumulated significantly lower frequencies of mutations than developmentally matched somatic cells. Female germ cells then maintain a relatively low frequency of mutations until 2–6 dpp and are presumed to maintain this relatively low mutation frequency into adulthood (dashed line and dashed square). Male germ cells, on the other hand, show an increase in mutation frequency between 15.5 dpc and 6 dpp but then undergo a dramatic decrease in the frequency of mutations coincident with a known wave of apoptosis in spermatogonia. This model suggests that a subset of early spermatogonia are predestined to form the population of functional SSCs in the adult testis and that these cells maintain a low frequency of mutations throughout their development (dashed/dotted line). Thy-1+ spermatogonia, which are known to be enriched for true SSCs, show a significantly lower frequency of mutations than does the general population of spermatogonia at 6 dpp. Closed circles, somatic cells; closed triangles, male germ cells; closed squares, female germ cells; open diamond, Thy-1+ spermatogonia; dpc, days postcoitum; dpp, days postpartum; dashed line, extrapolated mutant frequency in female germ cells; dashed/dotted line, proposed mutant frequency in precursors of true SSCs. Based on data from this study and those from Walter et al. [15].
However, during these same stages, germ cells were found to accumulate significantly fewer mutations than somatic cells. Thus, as noted above, by as early as 15.5 dpc, the frequency of spontaneous mutations in germ cells of both sexes was significantly lower than that in somatic cells in the same individuals. As shown in Figure 1, this trend continued into adulthood. In females, the frequency of spontaneous mutations in primary oocytes reached a level of 0.82 × 10−5 by 15.5 dpc and remained essentially unchanged in oocytes recovered at 2–6 dpp (the frequency of 0.61 × 10−5 detected in oocytes at 2–6 dpp was not significantly different from the frequency of 0.82 × 10−5 detected in oocytes at 15.5 dpc; P = 0.66). In males, the frequency of mutations reached 0.99 × 10−5 in type T1 prospermatogonia at 15.5 dpc and then rose to 2.30 × 10−5 in primitive type A spermatogonia at 6 dpp [15]. However, the mutation frequency then dropped in later spermatogenic cell types, reaching a low of 0.3 to 0.4 × 10−5 in primary spermatocytes by 18 dpp. From this point on, the frequency of mutations in spermatogenic cell types remained fairly constant, reaching 0.8 × 10−5 in spermatozoa [15].
Thy-1-Positive Spermatogonia Carry Fewer Mutations than Thy-1-Negative Spermatogonia
To test the hypothesis that true SSCs preferentially derive from a subset of developing prospermatogonia/spermatogonia that carry fewer mutations than other early spermatogonia, we recovered a population of Thy-1+ spermatogonia at 6 dpp. Previous studies have shown that selection for Thy-1+ cells from a population of early spermatogonia leads to an enrichment of functional SSCs as assessed by transplantation of these cells into recipient testes and subsequent quantification of donor-derived spermatogenesis [34, 46]. We then determined the frequency of mutations in the Thy-1+ spermatogonial population and compared this to the frequency of mutations previously found in a general population of purified type A spermatogonia. We found a mutation frequency of 0.59 × 10−5 in the Thy-1+ spermatogonia, which was significantly lower than the mutation frequency of 2.3 × 10−5 observed in the general spermatogonial population (P = 0.01).
DISCUSSION
From a genetic standpoint, the life of any somatic cell lineage lasts a single generation, whereas the germ line is essentially immortal. Therefore, to ensure faithful transmission of genetic information between generations, it is much more important for germline cells to maintain genetic integrity than it is for somatic cells to do so. This is the rationale on which the disposable soma theory was based [1]. In the study described here, we examined the frequency of spontaneous point mutations in germ cells and somatic cells from the same individuals at various stages of the lifecycle and/or development of the germ line. Results from this study, in conjunction with those from earlier studies [15, 35, 36], corroborate the disposable soma theory by showing that the mutational load is significantly lower in germ cells than in somatic cells from the same individuals at all stages of the life cycle investigated in both males and females.
Our study provides the first direct assessment of mutation frequencies in the female germ line and shows that, as in males, germline mutation frequencies are lower than somatic cell mutation frequencies in females as well. Although we were not able to directly assess the frequency of mutations in mature oocytes from adult females due to a lack of sufficient numbers of cells to facilitate the lacI mutation reporter assay, Figure 1 shows that if the same mutation frequency found in primary oocytes at 2–6 dpp (0.61 × 10−5) is maintained into adult stages in ovulated oocytes, the frequency of mutations in female gametes will be statistically similar to that found in spermatozoa (0.8 × 10−5; P = 0.73) (Fig. 1, dashed line). This suggests that, as expected, the disposable soma theory appears to apply equally well to males and females. In addition, we show that a significant difference in mutation frequency between germ cells and somatic cells is manifest as early as 15.5 dpc. This indicates that enhanced genetic integrity is a fundamental characteristic of germline cells throughout the lifecycle.
The frequency at which genetic mutations accumulate in any cell lineage is related, in part, to the exposure of cells to mutagenic influences, the rate of proliferation of each particular lineage, the level at which DNA repair pathways function to reverse potentially mutagenic damage, and/or the function of cell death pathways to eliminate genetically damaged cells from the lineage. The rate of proliferation of a cell lineage is a contributing factor because the potential to incur new spontaneous mutations is normally increased during DNA replication [47]. This represents the most likely explanation for the rapid increase in mutations we observed during embryonic and fetal development when proliferation rates are generally high. This would also explain the slower accumulation of new mutations following birth, when proliferation rates are generally reduced (Fig. 1). Similarly, the sex-specific divergence in rates of accumulation of new mutations in male and female germ cells between 15.5 dpc and 6 dpp may also reflect a difference in proliferation rates. Thus, following entry of female germ cells into first meiotic prophase between 13.5 and 15.5 dpc, these cells cease replication and remain quiescent until they undergo oocyte maturation and ovulation in the adult female. In contrast, male germ cells remain mitotically active between 13.5 and 15.5 dpc, then enter a mitotic arrest until just after birth, and then undergo a wave of active mitosis again between 3 and 6 dpp. Thus, we would suggest that the more active history of proliferation of male germ cells relative to female germ cells during this developmental period may be at least partially responsible for the higher frequency of mutations we observed in male germ cells relative to female germ cells at 6 dpp.
Differential proliferation rates, however, cannot explain the difference in mutation frequencies observed between germ cells and somatic cells at 15.5 dpc, given that both germline and somatic cell types derive from lineages that are mitotically active prior to this stage. Nor can differential proliferative activity explain the significantly lower frequency of mutations found in spermatozoa than in somatic cells from the same adult males, given that spermatozoa are the product of the highly proliferative seminiferous epithelium, but show a mutation frequency that is approximately sixfold lower than the average frequency observed in adult male somatic cells [15]. Indeed, even somatic tissues composed of predominantly nondividing, terminally differentiated cells, such as brain, carry mutation frequencies that are significantly higher than those found in proliferating seminiferous epithelium [15]. Thus, germ cells must use unique mechanisms to maintain enhanced genetic integrity relative to somatic cells. The most likely candidates for such mechanisms are elevated activities of various DNA repair pathways as well as higher levels of certain cell death pathways.
Several studies of DNA repair and cell death pathways have confirmed that these activities are elevated in germ cells relative to somatic cells [16–24]. Spontaneous DNA damage in spermatogenic cells is largely repaired through the base excision repair pathway or during replication by the mismatch repair system. Base excision repair activity has been shown to be high in the male germ line [16, 17, 48], and in the absence of this pathway as demonstrated by experimentally induced haploinsufficiency of the base excision repair genes Ape1 and Polb, the spontaneous mutation frequency becomes elevated in spermatogenic cells in young adults [19, 24]. In contrast, studies have shown that nucleotide excision repair [19] and double-strand break repair [49] activities are typically not elevated in the male germ line.
Elevated DNA repair activities mitigate the effects of DNA damage and minimize the formation of new mutations in germline cells. Thus, this mechanism can explain why germ cells tend to accumulate new mutations at a lower rate than somatic cells. However, this does not explain the observed decrease in the frequency of mutations as spermatogonia proceed from the undifferentiated type A stage to the differentiating type B stage. Thus, while it is straight forward to explain an increase in the frequency of spontaneous mutations as a function of development due to the ongoing accumulation of mutations in a particular cell lineage, it is more difficult to explain a decline in mutation frequency as we observed during the transition between type A and type B spermatogonia, where the mutation frequency dropped from 2.3 × 10−5 to 0.8 × 10−5 [15]. The timing of this decline is correlated with a known wave of cell death in spermatogenic cells [50], suggesting there is a nonrandom loss of spermatogonia bearing relatively higher mutational loads and progression of cells carrying fewer mutations to become functional SSCs in the adult testis. Indeed, studies show lacI transgenic mice deficient in the proapoptotic BAX protein display reduced cell death among spermatogenic cells and fail to undergo the decline in mutant frequency observed during the first wave of spermatogenesis in wild type mice [22].
These observations suggest that programmed cell death selectively eliminates spermatogonia carrying relatively higher mutant frequencies. However, there is no known mechanism that can actively distinguish cells carrying different frequencies of point mutations to facilitate this distinction, unless one or more of those mutations leads to a specific phenotypic defect that impacts cell function. An alternative explanation is that the population of early spermatogonia (type T1 prospermatogonia [type A spermatogonia]) [30] is heterogeneous and contains a subset of cells that are predestined to form SSCs and that these cells maintain a particularly low level of mutations throughout their development. This theoretical possibility is shown in Figure 1 (dotted/dashed line) and suggests that a certain subpopulation of cells within the early spermatogonial population maintains a particularly low level of mutations because these cells are predestined to become functional SSCs and are therefore the key cells among the early male germ cell population that must adhere to the disposable soma theory to maintain enhanced genetic integrity for the future of the species.
Importantly, the spermatogonial cell types in which we observed the significant drop in mutation frequency were all from the initial wave of spermatogenesis, whereas the pachytene spermatocytes, round spermatids, and spermatozoa in which we observed significantly lower frequencies of point mutations came from subsequent waves of spermatogenesis. It has been reported that the first wave of spermatogenesis derives from a subpopulation of non-self-renewing spermatogonia that is distinct from the self-renewing SSCs that give rise to all subsequent waves of spermatogenesis [51].
Evidence for heterogeneity among spermatogonia during the first wave of spermatogenesis has been provided by Yoshida and colleagues [51–54] and Shinohara and colleagues [55, 56], among others [57, 58]. That work demonstrated that distinct subpopulations of As-al spermatogonia can be distinguished on the basis of marker gene expression and that these different subpopulations show differences in their potential to become functional SSCs. In addition, Yoshida et al. [51–53] demonstrated that the initial wave of spermatogenesis appears to derive directly from a spermatogonial population that does not include a self-renewing SSC, whereas later waves of spermatogenesis derive from self-renewing SSCs. Within this context, our data are consistent with the suggestion that the non-self-renewing spermatogonia that give rise to the first wave of spermatogenesis carry higher frequencies of mutations on average than do the self-renewing SSCs that give rise to subsequent waves of spermatogenesis.
It remains to be determined whether the self-renewing SSCs that take up residence at the base of the seminiferous epithelium derive from a subpopulation that is determined stochastically or as the result of some sort of selection mechanism or is predetermined from an earlier stage in the male germ line. Given that reverse mutations are extremely rare, the advanced spermatogenic cell types carrying low mutation frequencies that we observed in the adult testis must derive from SSCs that carry low frequencies of mutations, and these SSCs must, in turn, nonrandomly derive from a subpopulation of early spermatogonia carrying lower frequencies of mutations than other early spermatogonia. This would argue against a completely stochastic process determining which cells form functional SSCs and leaves two other possibilities. One possibility is that there is some type of selection process that ensures that either only early spermatogonia with low mutational loads will form SSCs or that only immediate progeny of SSCs with low mutational loads will progress thru spermatogenesis. The second possibility is that no active selection takes place but rather that one of the subpopulations of early spermatogonia is predestined to form functional SSCs and that because of this fate, this subpopulation maintains a low mutational load.
We favor the interpretation that functional SSCs derive from a predestined subset of early spermatogonia that have maintained differentially enhanced genetic integrity throughout their developmental history. The point mutations we measured in the lacI mutation reporter transgene are indicative of mutations in regions of the genome that do not lead to either positive or negative selection, given that the lacI transgene is not expressed. While the occurrence of mutations detected in this transgene is believed to reflect the genome-wide occurrence of point mutations in any particular cell type, such spontaneous mutations are normally rare, and most that do occur are not likely to negatively impact cellular function in a manner that could provide a phenotypic basis for the extensive selection mechanism that would be required to nonrandomly cull the majority of early spermatogonial cells to yield the ultimate, small subpopulation of functional SSCs. Thus, we believe our data are compatible with the notion of heterogeneous subpopulations of early spermatogonia with nonrandom differences in cell fate, as demonstrated by Yoshida and others [51–58], but we would extend this notion to suggest that one of these subpopulations is preferentially fated to form SSCs. Our previous studies indicated that enhanced maintenance of genetic integrity is a phenotype regulated by epigenetic mechanisms in germ cells and early embryos [35, 36]. We would suggest that epigenetic programming in the subset of early spermatogonia (and their precursors) that gives rise to SSCs also favors elevated expression of DNA repair and related genes that minimize the occurrence of spontaneous point mutations in these cells to yield a population of SSCs that displays enhanced maintenance of genetic integrity.
ACKNOWLEDGMENT
This work is dedicated to the memory of Patricia Murphey. We thank Jacey Hornecker and Jeanene de Avila for technical assistance and Dr. Brian Hermann for reading the manuscript and participating in very helpful discussions.
Footnotes
Deceased.
Supported in part by National Institutes of Health grant HD42772 to J.R.M. and grant HD046521 to D.J.M.
REFERENCES
- Kirkwood TB. Evolution of ageing. Nature. 1977;270:301–304. doi: 10.1038/270301a0. [DOI] [PubMed] [Google Scholar]
- Ehling UH, Neuhauser A. Procarbazine-induced specific-locus mutations in male mice. Mutat Res. 1979;59:245–256. doi: 10.1016/0027-5107(79)90163-5. [DOI] [PubMed] [Google Scholar]
- Russell LB, Russell WL, Kelly EM. Analysis of the albino-locus region of the mouse. I. Origin and viability. Genetics. 1979;91:127–139. doi: 10.1093/genetics/91.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell LB, Russell WL. Frequency and nature of specific-locus mutations induced in female mice by radiations and chemicals: a review. Mutat Res. 1992;296:107–127. doi: 10.1016/0165-1110(92)90035-8. [DOI] [PubMed] [Google Scholar]
- Russell LB, Russell WL. Spontaneous mutations recovered as mosaics in the mouse specific-locus test. Proc Natl Acad Sci U S A. 1996;93:13072–13077. doi: 10.1073/pnas.93.23.13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell WL, Bangham JW, Russell LB. Differential response of mouse male germ-cell stages to radiation-induced specific-locus and dominant mutations. Genetics. 1998;148:1567–1578. doi: 10.1093/genetics/148.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell LB. Effects of male germ-cell stage on the frequency, nature, and spectrum of induced specific-locus mutations in the mouse. Genetica. 2004;122:25–36. doi: 10.1007/s10709-004-1443-7. [DOI] [PubMed] [Google Scholar]
- Russell LB, Hunsicker PR, Russell WL. Comparison of the genetic effects of equimolar doses of ENU and MNU: while the chemicals differ dramatically in their mutagenicity in stem-cell spermatogonia, both elicit very high mutation rates in differentiating spermatogonia. Mutat Res. 2007;616:181–195. doi: 10.1016/j.mrfmmm.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby PB. Discovery of numerous clusters of spontaneous mutations in the specific-locus test in mice necessitates major increases in estimates of doubling doses. Genetica. 1998;102–103:463–487. [PubMed] [Google Scholar]
- Kohler SW, Provost GS, Fieck A, Kretz PL, Bullock WO, Putman DL, Sorge JA, Short JM. Analysis of spontaneous and induced mutations in transgenic mice using a lambda ZAP/lacI shuttle vector. Environ Mol Mutagen. 1991;18:316–321. doi: 10.1002/em.2850180421. [DOI] [PubMed] [Google Scholar]
- Kohler SW, Provost GS, Fieck A, Kretz PL, Bullock WO, Sorge JA, Putman DL, Short JM. Spectra of spontaneous and mutagen-induced mutations in the lacI gene in transgenic mice. Proc Natl Acad Sci U S A. 1991;88:7958–7962. doi: 10.1073/pnas.88.18.7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapp RW, Jr,, Marino DJ, Gardiner TH, Masten LW, McKee RH, Tyler TR, Ivett JL, Young RR. In vitro and in vivo assays of isopropanol for mutagenicity. Environ Mol Mutagen. 1993;22:93–100. doi: 10.1002/em.2850220207. [DOI] [PubMed] [Google Scholar]
- Provost GS, Kretz PL, Hamner RT, Matthews CD, Rogers BJ, Lundberg KS, Dycaico MJ, Short JM. Transgenic systems for in vivo mutation analysis. Mutat Res. 1993;288:133–149. doi: 10.1016/0027-5107(93)90215-2. [DOI] [PubMed] [Google Scholar]
- Dycaico MJ, Provost GS, Kretz PL, Ransom SL, Moores JC, Short JM. The use of shuttle vectors for mutation analysis in transgenic mice and rats. Mutat Res. 1994;307:461–478. doi: 10.1016/0027-5107(94)90257-7. [DOI] [PubMed] [Google Scholar]
- Walter CA, Intano GW, McCarrey JR, McMahan CA, Walter RB. Mutation frequency declines during spermatogenesis in young mice but increases in old mice. Proc Natl Acad Sci U S A. 1998;95:10015–10019. doi: 10.1073/pnas.95.17.10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intano GW, McMahan CA, Walter RB, McCarrey JR, Walter CA. Mixed spermatogenic germ cell nuclear extracts exhibit high base excision repair activity. Nucleic Acids Res. 2001;29:1366–1372. doi: 10.1093/nar/29.6.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intano G, McMahan CA, McCarrey JR, Walter RB, McKenna AE. Base excision repair is limited by different proteins in male germ cell nuclear extracts prepared from young and old mice. Mol Cell Biol. 2002;22:2410–2418. doi: 10.1128/MCB.22.7.2410-2418.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter CA, Intano GW, McMahan CA, Kelner K, McCarrey JR, Walter RB. Mutation spectral changes in spermatogenic cells obtained from old mice. DNA Repair (Amst) 2004;3:495–504. doi: 10.1016/j.dnarep.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Huamani J, McMahan CA, Herbert DC, Reddick R, McCarrey JR, MacInnes MI, Chen DJ, Walter CA. Spontaneous mutagenesis is enhanced in Apex heterozygous mice. Mol Cell Biol. 2004;24:8145–8153. doi: 10.1128/MCB.24.18.8145-8153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Spivak G, Mitchell DL, Mori T, McCarrey JR, McMahan CA, Walter RB, Hanawalt PC, Walter CA. Nucleotide excision repair activity varies among murine spermatogenic cell types. Biol Reprod. 2005;73:123–130. doi: 10.1095/biolreprod.104.039123. [DOI] [PubMed] [Google Scholar]
- Xu G, Intano GW, McCarrey JR, Walter RB, McMahan CA, Walter CA. Recovery of a low mutant frequency after ionizing radiation-induced mutagenesis during spermatogenesis. Mutat Res. 2008;654:150–157. doi: 10.1016/j.mrgentox.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Vogel KS, McMahan CA, Herbert DC, Walter CA. BAX. and tumor suppressor TRP53 are important in regulating mutagenesis in spermatogenic cells in mice. Biol Reprod. 2010;83:979–987. doi: 10.1095/biolreprod.110.085415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, McMahan CA, Hildreth K, Garcia RA, Herbert DC, Walter CA. Ionizing radiation-induced mutant frequencies increase transiently in male germ cells of older mice. Mutat Res. 2012;744:135–139. doi: 10.1016/j.mrgentox.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel KS, Perez M, Momand JR, Acevedo-Torres K, Hildreth K, Garcia RA, Torres-Ramos CA, Ayala-Torres S, Prihoda TJ, McMahan CA, Walter CA. Age-related instability in spermatogenic cell nuclear and mitochondrial DNA obtained from Apex1 heterozygous mice. Mol Reprod Dev. 2011;78:906–919. doi: 10.1002/mrd.21374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambrook PJ. An ageing question: do embryonic stem cells protect their genomes? Mech Ageing Dev. 2007;128:31–35. doi: 10.1016/j.mad.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Tichy ED, Stambrook PJ. DNA repair in murine embryonic stem cells and differentiated cells. Exp Cell Res. 2008;314:1929–1936. doi: 10.1016/j.yexcr.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes RB, Stringer JR, Shao C, Tischfield JA, Stambrook PJ. Embryonic stem cells and somatic cells differ in mutation frequency and type. Proc Natl Acad Sci U S A. 2002;99:3586–3590. doi: 10.1073/pnas.062527199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waard H, Sonneveld E, de Wit J, Esveldt-van Lange R, Hoeijmakers JH, Vrieling H, van der Horst GT. Cell-type-specific consequences of nucleotide excision repair deficiencies: embryonic stem cells versus fibroblasts. DNA Repair. 2008;7:1659–1669. doi: 10.1016/j.dnarep.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Maynard S, Swistowska AM, Lee JW, Liu Y, Liu ST, Da Cruz AB, Rao M, de Souza-Pinto NC, Zeng X, Bohr VA. Human embryonic stem cells have enhanced repair of multiple forms of DNA damage. Stem Cells. 2008;26:2266–2274. doi: 10.1634/stemcells.2007-1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarrey JR. Development of the germ cell In Desjardins C, Ewing LL. (eds.), Cell and Molecular Biology of the Testis Oxford University Press: Oxford; 1993. 58 89 [Google Scholar]
- Burgoyne PS. The role of the mammalian Y chromosome in spermatogenesis. Development. 1987;101((suppl)):133–141. doi: 10.1242/dev.101.Supplement.133. [DOI] [PubMed] [Google Scholar]
- McCarrey JR, Berg WM, Paragioudakis SJ, Zhang PL, Dilworth DD, Arnold BL, Rossi JJ. Differential transcription of Pgk genes during spermatogenesis in the mouse. Dev Biol. 1992;154:160–168. doi: 10.1016/0012-1606(92)90056-m. [DOI] [PubMed] [Google Scholar]
- Kafri T, Ariel M, Brandeis M, Shemer R, Urven L, McCarrey J, Cedar H, Razin A. Developmental pattern of gene-specific DNA methylation in the mouse embryo and germ line. Genes Dev. 1992;6:705–714. doi: 10.1101/gad.6.5.705. [DOI] [PubMed] [Google Scholar]
- Kubota H, Avarbock MR, Brinster RL. Culture conditions and single growth factors affect fate determination of mouse spermatogonial stem cells. Biol Reprod. 2004;71:722–31. doi: 10.1095/biolreprod.104.029207. [DOI] [PubMed] [Google Scholar]
- Caperton L, Murphey P, Yamazaki Y, McMahan CA, Walter CA, Yanagimachi R, McCarrey JR. Assisted reproductive technologies do not alter mutation frequency or spectrum. Proc Natl Acad Sci U S A. 2007;104:5085–5090. doi: 10.1073/pnas.0611642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphey P, Yamazaki Y, McMahan CA, Walter CA, Yanagimachi R, McCarrey JR. Epigenetic regulation of genetic integrity is reprogrammed during cloning. Proc Natl Acad Sci U S A. 2009;106:4731–4735. doi: 10.1073/pnas.0900687106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farabaugh PJ. Sequence of the Laci gene. Nature. 1978;274:765–769. doi: 10.1038/274765a0. [DOI] [PubMed] [Google Scholar]
- Nishino H, Buettner VL, Haavik J, Schaid DJ, Sommer SS. Spontaneous mutation in Big Blue transgenic mice: analysis of age, gender, and tissue type. Environ Mol Mutagen. 1996;28:299–312. doi: 10.1002/(SICI)1098-2280(1996)28:4<299::AID-EM2>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Heddle JA. On clonal expansion and its effects on mutant frequencies, mutation spectra and statistics for somatic mutations in vivo. Mutagenesis. 1999;14:257–260. doi: 10.1093/mutage/14.3.257. [DOI] [PubMed] [Google Scholar]
- Hill KA, Buettner VL, Glickman BW, Sommer SS. Spontaneous mutations in the Big Blue transgenic system are primarily mouse derived. Mutat Res. 1999;436:11–19. doi: 10.1016/s1383-5742(98)00024-6. [DOI] [PubMed] [Google Scholar]
- Hill KA, Buettner VL, Halangoda A, Kunishige M, Moore SR, Longmate J, Scaringe WA, Sommer SS. Spontaneous mutation in Big Blue mice from fetus to old age: tissue-specific time courses of mutation frequency but similar mutation types. Environ Mol Mutagen. 2004;43:110–120. doi: 10.1002/em.20004. [DOI] [PubMed] [Google Scholar]
- Thybaud V, Dean S, Nohmi T, de Boer J, Douglas GR, Glickman BW, Gorelick NJ, Heddle JA, Heflich RH, Lambert I, et al. In vivo transgenic mutation assays. Mutat Res. 2003;540:141–151. doi: 10.1016/j.mrgentox.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Agresti A. Categorical Data Analysis New York: Wiley; 2002. [Google Scholar]
- Maxwell AE. Analysing Qualitative Data London: Methuen & Co Ltd.; 1971. [Google Scholar]
- Cooper DN, Youssoufian H. The CpG dinucleotide and human genetic disease. Hum Genet. 1988;78:151–155. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- Kubota H, Avarbock MR, Brinster RL. Spermatogonial stem cells share some, but not all, phenotypic and functional characteristics with other stem cells. Proc Natl Acad Sci U S A. 2003;100:6487–92. doi: 10.1073/pnas.0631767100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RE, Washington MT, Prakash S, Prakash L. Fidelity of human DNA polymerase eta. J Biol Chem. 2000;274:36835–36838. doi: 10.1074/jbc.274.52.36835. [DOI] [PubMed] [Google Scholar]
- Olsen AK, Bjortuft H, Wiger R, Holme J, Seeberg E, Bjoras M, Brunborg G. Highly efficient base excision repair (BER) in human and rat male germ cells. Nucl Acids Res. 2003;31:1351–1363. doi: 10.1093/nar/29.8.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava N, Raman MJ. Homologous recombination-mediated double-strand break repair in mouse testicular extracts and comparison with different germ cell stages. Cell Biochem Funct. 2007;25:75–86. doi: 10.1002/cbf.1375. [DOI] [PubMed] [Google Scholar]
- Coucouvanis EC, Sherwood SW, Carswell-Crumpton C, Spack EG, Jones PP. Evidence that the mechanism of prenatal germ cell death in the mouse is apoptosis. Exp Cell Res. 1993;209:238–247. doi: 10.1006/excr.1993.1307. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Sukeno M, Nakagawa T, Ohbo K, Nagamatsu G, Suda T, Nabeshima Y. The first round of mouse spermatogenesis is a distinctive program that lacks the self-renewing spermatogonia stage. Development. 2006;133:1495–1505. doi: 10.1242/dev.02316. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Nabeshima Y, Nakagawa T. Stem cell heterogeneity: actual and potential stem cell compartments in mouse spermatogenesis. Ann N Y Acad Sci. 2007;1120:47–58. doi: 10.1196/annals.1411.003. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Sada A, Yoshida S, Saga Y. The heterogeneity of spermatogonia is revealed by their topology and expression of marker proteins including the germ cell-specific proteins Nanos2 and Nanos3. Dev Biol. 2009;336:222–231. doi: 10.1016/j.ydbio.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Sharma M, Nabeshima Y, Braun RE, Yoshida S. Functional hierarchy and reversibility within the murine spermatogenic stem cell compartment. Science. 2010;328:62–67. doi: 10.1126/science.1182868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto H, Kanatsu-Shinohara M, Takashima S, Chuma S, Nakatsuji N, Takehashi M, Shinohara T. Phenotypic plasticity of mouse spermatogonial stem cells. PLoS One. 2009;4:e7909. doi: 10.1371/journal.pone.0007909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara T, Ishii K, Kanatsu-Shinohara M. Unstable side population phenotype of mouse spermatogonial stem cells in vitro. J Reprod Dev. 2011;57:288–295. doi: 10.1262/jrd.10-168n. [DOI] [PubMed] [Google Scholar]
- Grisanti L, Falciatori I, Grasso M, Dovere L, Fera S, Muciaccia B, Fuso A, Berno V, Boitani C, Stefanini M, Vicini E. Identification of spermatogonial stem cell subsets by morphological analysis and prospective isolation. Stem Cells. 2009;27:3043–3052. doi: 10.1002/stem.206. [DOI] [PubMed] [Google Scholar]
- Zheng K, Wu X, Kaestner KH, Wang PJ. The pluripotency factor LIN28 marks undifferentiated spermatogonia in mouse. BMC Dev Biol. 2009;9:38. doi: 10.1186/1471-213X-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]