

ABSTRACT
The placenta mediates maternal-fetal exchange and has historically been regarded as a passive conduit for nutrients. However, emerging evidence suggests that the placenta actively responds to nutritional and metabolic signals from the mother and the fetus. We propose that the placenta integrates a multitude of maternal and fetal nutritional cues with information from intrinsic nutrient-sensing signaling pathways to match fetal demand with maternal supply by regulating maternal physiology, placental growth, and nutrient transport. This process, which we have called placental nutrient sensing, ensures optimal allocation of resources between the mother and the fetus to maximize the chances for propagation of parental genes without jeopardizing maternal health. We suggest that these mechanisms have evolved because of the evolutionary pressures of maternal undernutrition, which result in decreased placental growth and down-regulation of nutrient transporters, thereby limiting fetal growth to ensure maternal survival. These regulatory loops may also function in response to maternal overnutrition, leading to increased placental growth and nutrient transport in cases of maternal obesity or gestational diabetes. Thus, placental nutrient sensing modulates maternal-fetal resource allocation to increase the likelihood of reproductive success. This model implies that the placenta plays a critical role in mediating fetal programming and determining lifelong health.
Keywords: fetal demand, fetal growth, imprinted genes, maternal supply, placental signaling pathways, placental transport, syncytiotrophoblast
INTRODUCTION
Reproductive success is dependent on fine-tuned sharing of resources between the mother and her fetus, allowing optimal fetal growth without jeopardizing maternal survival, in particular when resources are limited. Maternal physiology undergoes extensive adaptations to pregnancy, which are essential for the redistribution of nutrients and oxygen to the growing fetus. These maternal physiological changes include an increased cardiac output and a redistribution of blood flow to the developing placental circulation, maternal hyperventilation facilitating exchange of respiratory gases across the placental barrier, and insulin resistance, which mobilizes glucose, amino acids, and lipids in the second half of pregnancy for transfer to the fetus. The maternal physiological adaptations to pregnancy are, in part, controlled by the placenta.
In eutherian mammals, the placenta constitutes the main interface between mother and fetus and represents the primary site for maternal-fetal exchange. The syncytiotrophoblast, a highly specialized multinucleated epithelial cell layer covering the surface of the chorionic villi, produces a multitude of hormones, mediates nutrient transport, and forms a physical and immunological barrier between the maternal and fetal circulations. Because of the syncytial nature of this epithelium, most solutes transferred between the mother and the fetus must be actively or passively transported across the two polarized plasma membranes of the syncytiotrophoblast, an apical microvillous membrane in direct contact with the maternal blood, and a basal membrane facing the fetal capillaries [1]. Thus, the syncytiotrophoblast is strategically positioned as a large maternal-fetal interface, which determines nutrient supply to the fetus. Moreover, a wide array of cellular signaling pathways in the syncytiotrophoblast modulates and integrates placental growth and function in response to maternal and fetal cues [2].
A large body of epidemiological and animal experimental data suggests that adverse influences during early development, in particular during fetal life, increase the risk of developing disease in adult life [3]. This paradigm, referred to as “fetal programming” or “developmental origins of health and disease,” has a profound impact on public health strategies for the prevention of major illnesses. The adverse influences that program the fetus for later disease often originate in the maternal compartment. These perturbations include impaired utero-placental blood flow leading to decreased oxygenation of the intervillous space, diabetes, obesity, toxins, altered maternal nutrition (overnutrition or undernutrition), and inflammation, among others. The placenta plays an essential role in developmental programming, because changes in, for example, maternal metabolism and nutrient availability are transmitted to the fetus via the placenta. The placenta responds to these perturbations by changing its structure and function, which influences nutrient and oxygen supply and secretion of hormones and circulating factors to the mother and fetus.
In this brief review we will discuss the mechanisms by which the placenta integrates maternal nutritional cues with fetal demand signals and allocates resources between the mother and her fetus to ensure reproductive success. First, we will review some of the basic mechanisms involved in placental control of maternal physiological adaptations to pregnancy, which are essential to allow maternal resources to be redistributed for fetal growth. We will then discuss how placental function is regulated by maternal supply and fetal demand and explore placental signaling pathways that may integrate these diverse signals in a process called placental nutrient sensing to regulate placental function, thereby controlling the allocation of resources between the mother and fetus. In addition, the role of imprinted genes will be discussed. Finally, we will propose an integrated model of how maternal-fetal resource allocation is controlled and discuss the evolutionary significance of these processes.
MATERNAL PHYSIOLOGICAL ADAPTATIONS TO PREGNANCY
Cardiovascular Adaptation
Pregnancy is associated with increased venous return and cardiac preload secondary to increased maternal total blood volume [4]. Cardiac output increases gradually during early pregnancy and reaches a plateau by the beginning of the second trimester, which is largely maintained throughout pregnancy [5]. In parallel, there is a gradual and substantial increase in heart rate [6] and a decrease in vascular resistance [5, 7] to allow for increased blood flow in numerous vascular beds including the liver, the kidney, and the utero-placental circulation. In rats, it has been shown that the levels of relaxin, a polypeptide hormone produced by the corpus luteum, increase gradually and reach a peak at the end of pregnancy [7]. The treatment of nonpregnant rats with relaxin reproduces the hemodynamic adaptations in response to pregnancy, suggesting that relaxin is a major contributor to pregnancy-associated decreased vascular resistance and increased maternal cardiac output [7].
Following involution of sex steroid hormone production by the corpus luteum, secretion of estrogen and progesterone by the placenta into the maternal circulation increases exponentially throughout pregnancy. Among various effects, estrogen increases utero-placental blood flow [8]. In vitro experiments on uterine and placental blood vessels [9] and animal studies [10, 11] have shown that 17β-estradiol or agonists of estrogen receptors exert specific localized vasodilatory actions. The placenta lacks autonomic innervation [12], thus highlighting the importance of the role of estrogen and/or local placental vasoactive factors for adequate utero-placental blood perfusion.
Metabolic Adaptation
During early to mid gestation maternal food intake increases. There is a 60% increase in insulin secretion, which stimulates lipogenesis and reduces fatty acid oxidation, thereby promoting maternal lipid accumulation [8]. As a result, maternal leptin levels start to rise [13, 14]. Maternal serum adiponectin is increased in early gestation [15], which may contribute to the increased insulin sensitivity that is observed in the first trimester. During mid-late gestation, maternal food intake and lipid accumulation continue to increase, but maternal insulin resistance develops as reflected by a decrease in insulin sensitivity by 45%–70% [16]. These changes occur concomitantly to a reduction in plasma levels of adiponectin, which increases the leptin to adiponectin ratio [14, 17–19].
Maternal insulin resistance in the second half of pregnancy promotes increases hepatic gluconeogenesis, reduces glucose uptake and energy storage in maternal skeletal muscle and adipose tissue, and increases lipolysis in adipose tissue, thereby making glucose and lipids available for transfer to the fetus [8, 20]. A multitude of factors contribute to the development of insulin resistance in pregnancy. Placental growth hormone (pGH), a variant of growth hormone produced by the placenta, is believed to be of particular importance [17, 18, 21]. Placental growth hormone is a potent insulin antagonist that stimulates maternal lipolysis [22, 23] and hepatic gluconeogenesis [24].
Overall, the maternal metabolic adaptations in early pregnancy are characterized by accumulation of nutrients, in particular fat stores. In contrast, during the second half of pregnancy placental and maternal hormones promote allocation of nutrients to the fetus.
PLACENTAL FUNCTION AND MATERNAL-FETAL RESOURCE ALLOCATION
The placenta controls maternal-fetal resource allocation by influencing maternal supply or by altering nutrient delivery to the fetus. First, and as discussed above, the placenta regulates maternal supply by altering the secretion of hormones and signaling factors into the maternal circulation. For example, in intrauterine growth restriction (IUGR), pGH secretion is reduced [25], and because this is a key factor responsible for maternal insulin resistance in pregnancy, lower pGH levels may explain the observation that mothers of IUGR babies are more insulin sensitive than mothers giving birth to a normal-sized infant. In addition, because pGH is the primary factor regulating the release of insulin-like growth factor I (IGF-I) from the maternal liver, and IGF-I in turn stimulates placental growth and transport functions [8, 26–28], the decreased pGH secretion in IUGR may function as a negative feedback loop contributing to the restricted fetal growth in this condition. We propose that this regulatory loop constitutes an adaptive mechanism serving the purpose to down-regulate placental nutrient transport in response to, for example, reduced utero-placental blood flow, thereby matching fetal growth to the decreased maternal oxygen and nutrient supply.
Insulin-like growth factor 2 (IGF-II) is abundantly expressed by the placenta in many species, including humans and rodents. Recent studies have shown that deletion of the placenta-specific promoter (P0; [29]) of the Igf2 gene (Igf2P0; not expressed in humans), markedly affects maternal physiology and these changes may contribute to the placental and fetal phenotype [30]. Specifically, Sferruzzi-Perri et al. [30] report that deletion of Igf2P0 alters maternal metabolic and endocrine profiles and their response to undernutrition. In particular, alpha-amino nitrogen (a measure of total amino acid levels) in the maternal circulation is lower in the dams carrying Igf2P0 knockout fetuses, consistent with the possibility that placenta-specific IGF-II knockout may affect maternal adaptations to pregnancy. In addition, maternal corticosterone levels are markedly higher in dams carrying Igf2P0 embryos and these dams were hyperinsulinemic [30]. Although the relevance of these observations for humans remain to be established, these findings suggest that placental IGF-II modulates signaling from the placenta to the mother, which adapts maternal metabolism possibly in response to changes in nutrient supply to the placenta.
Nutrient transport is the primary function of the syncytiotrophoblast and determines not only placental growth but also fetal nutrient availability and growth. Mechanisms for regulation of placental nutrient transport, changes in placental nutrient transport systems in pregnancy complications, and placental nutrient transport in response to changes in maternal nutrition have been reviewed in detail elsewhere [31, 32]. Although normal fetal growth is dependent on the availability of a number of different classes of nutrients, for the purpose of this brief review we will discuss only placental amino acid transport. Amino acid availability is a key determinant for fetal growth [33] and fetal concentrations of amino acids are generally higher in the fetus compared with maternal levels [34]. Although at least 25 different amino acid transporter systems are expressed in the placenta [35–37], only a few have been studied in detail. System A is a Na+-dependent amino acid transporter that mediates the uptake of nonessential neutral amino acids against their concentration gradient into the syncytiotrophoblast [32, 35, 38]; the high intracellular concentration of nonessential amino acids that is generated serves as a driving force for exchange of extracellular essential amino acids through the system L amino acid transporter, a Na+-independent exchanger. System A activity is highly polarized to the syncytiotrophoblast apical microvillous plasma membrane [39], whereas system L is expressed in both syncytiotrophoblast membranes in an isoform-dependent manner [40, 41].
Placental amino acid transporters are regulated by hormones, cytokines, and nutrients [32]. The apical microvillous membrane of the syncytiotrophoblast expresses numerous hormone receptors, such as insulin [42, 43], IGF-I [44], and leptin [45] receptors, consistent with regulation of placental function by maternal hormones. It has been shown that insulin [46–49], IGF-I [48, 50], leptin [51], and cytokines such as interleukin 6 (IL-6) [52] and tumor necrosis factor alpha [52] are positive regulators of system A amino acid transporters. Moreover, the activation of trophoblast system A activity by IL-6, leptin, and oleic acid appears to involve the transcription factor STAT-3 [49, 52, 53], indicating that there are common pathways to integrate and “sense” different maternal metabolic cues. On the contrary, interleukin 1beta, a proinflammatory cytokine with increased placental expression in obesity [54], inhibits insulin signaling and prevents insulin-stimulated amino acid transport in cultured primary human trophoblasts [55]. In addition, maternal glucocorticoid treatment during midgestation reduces system A transport by late gestation in the mouse placenta [56] and results in reduced fetal weight [57].
PLACENTAL RESPONSES TO CHANGES IN MATERNAL SUPPLY
Evidence from experimental animals shows that placental structure, growth, and function are directly affected by maternal nutrient availability and/or perturbations to the maternal environment [29, 58–63]. These changes will directly influence placental transfer of solutes and oxygen to the fetus and will alter hormone secretion and the release of signaling molecules into the fetal circulation [2].
There are a multitude of signals that provide information with respect to the ability of the maternal supply line to support pregnancy. These include utero-placental blood flow, which must increase rapidly during pregnancy to support normal fetal growth and oxygenation [64, 65]. The importance of adequate oxygen supply for normal fetal growth is illustrated by the increased incidence of fetal growth restriction at high altitude [66, 67]. Furthermore, circulating maternal levels of nutrients and metabolic hormones such as cortisol, insulin, leptin, and IGF-I are key metabolic cues informing the placenta about the nutritional status of the mother. For example, maternal nutrient restriction is typically associated with low circulating insulin, IGF-I, and leptin and elevated cortisol levels [30, 58], whereas conditions characterized by overnutrition, such as maternal obesity, often are associated with elevated serum insulin, IGF-I, and leptin [19, 68]. In addition, adipokines secreted by maternal adipose tissue may carry important information about maternal fat stores and the proinflammatory milieu. In particular, maternal serum levels of adiponectin are inversely correlated with birth weight in healthy pregnant women with varying early pregnancy body mass index [19, 69], as well as in women with gestational diabetes mellitus (GDM) [70]. In contrast to maternal adiponectin, fetal adiponectin levels are positively correlated to birth weight [71] and studies using genetic approaches to manipulate adiponectin gene expression in mice have demonstrated a link between elevated fetal adiponectin and increased size of fat stores in early life [72]. This suggests that maternal and fetal adiponectin may have opposite and independent roles in regulating fetal growth. We have recently shown that maternal adiponectin functions as an endocrine link between maternal adipose stores, placental function, and fetal growth [20]. Specifically, maternal adiponectin inhibits placental insulin signaling mediated by generation of ceramide, resulting in inhibition of mechanistic target of rapamycin (mTOR) signaling and reduced trafficking of specific amino acid transporter isoforms to the trophoblast plasma membrane, thereby reducing fetal amino acid availability and limiting fetal growth [46, 73, 74] (Fig. 1). Thus, in contrast to skeletal muscle and the liver, adiponectin causes insulin resistance in the placenta. We propose that maternal adiponectin signals to the placenta on the status of maternal fat stores. In a lean mother, high maternal adiponectin tends to limit fetal growth by inhibition of placental insulin signaling and nutrient transport. On the other hand, in obesity and GDM, low maternal adiponectin removes the inhibition of placental insulin signaling, contributing to increased fetal growth (Fig. 2).
FIG. 1.
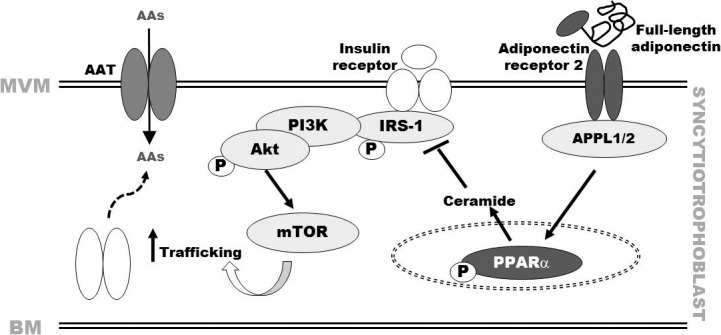
Maternal adiponectin inhibits trophoblast insulin signaling and amino acid transport. Based on studies in cultured primary human trophoblast cells [46, 73, 74] and in vivo experiments in mice [46, 73, 74], we propose that maternal adiponectin activates transcription factor peroxisome proliferator-activated receptor alpha (PPARα) and increases ceramide production, which inhibits insulin signaling (insulin receptor substrate 1 [IRS-1]) and subsequently Akt and insulin-stimulated amino acid transport. Insulin regulates amino acid transport in primary trophoblasts by both transcriptional mechanisms and posttranslational processes, which involve mTOR-dependent trafficking of specific amino acid transporter (AAT) isoforms to the plasma membrane. Consequently, adiponectin inhibition of insulin signaling is likely to impact upon insulin-dependent mTOR activation and regulation of amino acid transport, thereby reducing fetal amino acid availability and limiting fetal growth. MVM, apical microvillous membrane; BM, basal membrane; AAs, amino acids; APPL, adaptor protein, phosphotyrosine interaction, PH domain and leucine zipper containing 1–2; Akt, protein kinase B; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; P, phosphorylation/activation.
FIG. 2.
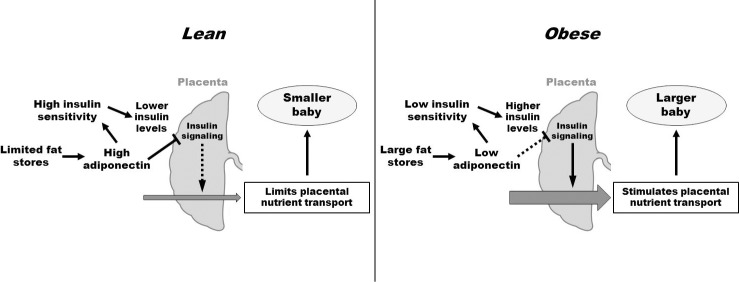
Maternal adiponectin functions as an endocrine link between maternal adipose stores, placental function, and fetal growth. In a lean woman, high maternal adiponectin tends to limit fetal growth by inhibition of placental nutrient transport. In obesity and GDM, low adiponectin levels may promote fetal growth.
The placental response to changes in maternal oxygen and nutrient supply includes alterations in the expression and/or function of nutrient transporters [31, 58, 62, 75, 76]. For instance, maternal undernutrition or restricted utero-placental blood flow results in down-regulation of placental nutrient transporters and, consequently, decreased fetal nutrient availability. Thus, these changes will directly contribute to the development of fetal growth restriction, reflecting a placental adaptation to reduced maternal supply. The placental responses to changes in maternal supply therefore represent a mechanism by which fetal growth is matched to the ability of the maternal supply line to allocate resources to the fetus.
PLACENTAL RESPONSES TO FETAL DEMAND
Most of the evidence for control of placental function by fetal demand has been obtained in experiments using mouse models where it is possible to control the fetal environment [77–79]. Based on these studies, it is proposed that the fetus signals the placenta to homeostatically regulate growth and nutrient transport. Thus, in response to maternal undernutrition or impaired utero-placental blood flow, which results in decreased fetal oxygen and/or nutrient availability, the fetus signals to up-regulate placental growth and nutrient transport [29, 78, 80]. This suggests that the regulation of placental function by fetal demand signals represents a compensatory rather than an adaptive mechanism to maintain fetal growth within a normal range.
There are some observations in the human in support of fetal demand signals controlling placental function. Amino acid transporter activity in the syncytiotrophoblast apical microvillous membrane has been reported to inversely correlate to fetal size at birth, within the normal birth weight range [81]. In addition, the fetal hormone parathyroid hormone-related peptide (PTHrp) regulates placental Ca2+ pump activity in the syncytiotrophoblast basal plasma membrane [82, 83]. Indeed, in IUGR pregnancies, the elevated fetal circulating levels of PTHrp [83] and increased placental Ca2+ pump activity [82] may reflect regulation of placental function by a fetal demand signal. In support of this hypothesis, elegant mouse studies show that deletion of the PTHrp gene abolishes the positive fetal-maternal Ca2+ gradient [84, 85] and that placental Ca2+ transport is up-regulated in mouse fetuses with growth restriction induced by placental Igf2P0 deletion [86].
Key nutrient transporters in the syncytiotrophoblast are down-regulated in human fetal growth restriction [39, 87, 88] and placental transport capacity is increased in association to fetal overgrowth in some [89, 90] but not all studies [91]. These findings are at odds with the notion that fetal demand signals are important regulators of placental nutrient transport. Instead, these observations support a strong influence of maternal signals, which adapts placental function according to the availability of resources that can be allocated to the growing fetus. However, most of these studies were performed at term and it is therefore possible that compensatory changes due to fetal demand signals may be present in the placenta at earlier stages of gestation.
The nature of fetal demand signals regulating placental function is largely unknown. Based on evidence from mouse models of placenta-specific and global Igf2 gene deletions, it has been proposed that IGF-II is a key fetal demand signal. In mice with placental-specific deletion of Igf2, placentas are growth restricted but the expression and activity of specific nutrient transporters has been reported to be up-regulated [78]. In pups with global Igf2 gene knockout, placental and fetal growth restriction develops, but in contrast to the animals with placental-specific deletion of Igf2, no up-regulation of placental nutrient transporters is observed [78]. Conversely, in humans, fetal circulating levels of IGF-II are reduced in fetal growth restriction [92] and increased in association with fetal overgrowth [93], which is not consistent with IGF-II as a fetal demand signal.
PLACENTAL SIGNALING PATHWAYS RESPONDING TO ALTERED METABOLISM AND NUTRITION
A multitude of nutrient-sensing pathways have been identified in the syncytiotrophoblast that may participate in the integration of maternal and fetal signals and the regulation of fetal nutrient availability by modulating placental growth and nutrient transport [76]. These placental nutrient sensors include adenosine monophosphate-activated protein kinase (AMPK), amino acid response-signal transduction pathway (AAR), glycogen synthase 3 (GSK-3), the hexosamine signaling pathway [2], and mTOR complex 1 (mTORC1) [76]. Of these signaling pathways, the evidence supporting a role of mTORC1 in placental nutrient sensing is particularly compelling and is discussed in a subsequent section.
The AAR adapts the cell to nutrient stress and is activated by limitation or imbalance of essential amino acids [94]. In these situations, there is an increase of uncharged tRNA species, which become bound by the general control nonderepressible 2 kinase, leading to phosphorylation of the eukaryotic translation initiation factor 2-subunit alpha (eIF2α). This factor decreases global translation but increases the translation of activating transcription factor (ATF) 4, resulting in increased transcription of a subset of specific genes, a process collectively known as the unfolded protein response [94]. Many ATF4 target genes have been shown to be involved in transport, metabolism, and oxidative stress. In the placenta, the AAR pathway has been reported to be activated in a rat model of maternal protein restriction [95]. In addition, phosphorylation of eIF2α is increased in placentas from growth-restricted fetuses [96], whereas in pregnant rats fed with a high-fat diet leading to fetal overgrowth, eIF2α phosphorylation is decreased [97]. Interestingly, in pregnancies at high altitude, which are associated with chronic maternal hypobaric hypoxia and low birth weight, there is increased phosphorylation of eIF2α, indicative of inhibition of protein synthesis [98].
AMPK is well established as the global energy sensor of the cell [99]. A reduction in ATP levels, and thus an increase in AMP levels, phosphorylates and activates AMPK and promotes ATP-generating catabolic pathways such as an increase in glucose uptake and fatty acid oxidation. Moreover, AMPK activation inhibits ATP-consuming anabolic pathways, including protein synthesis, in order to maintain cellular energy balance and homeostasis. Placental AMPK phosphorylation has been reported to be increased in sheep subjected to 50% calorie restriction [100] and decreased in overnourished, obese pregnant ewes [101], high-fat diet-fed pregnant rats [97], and obese women giving birth to large babies [89]. In contrast, placental AMPK activity is unaltered in response to maternal protein restriction in the rat [62]. Although AMPK phosphorylation constitutes a proxy for tissue ATP levels, more detailed studies are needed to better understand placental energy sensing and the link between placental energy metabolism and fetal growth.
GSK-3 is a serine/threonine protein kinase that is inhibited by insulin/IGF-I/Akt signaling [102] and functions as a glucose sensor [103]. In the human placenta, GSK-3 is activated in pregnancies complicated by fetal growth restriction [96], but the underlying mechanisms remain unclear [96, 104, 105]. In addition, placental GSK-3 is activated in maternal nutrient restriction in the baboon, which is associated with down-regulation of placental nutrient transporters, decreased circulating fetal levels of amino acids, and fetal growth restriction [27].
Nuclear and cytoplasmic proteins are dynamically modified at their serine and threonine hydroxyl groups by the attachment of O-linked N-acetylglucosamine (O-GlcNAc) monosaccharides. The enzymes of the O-GlcNAc cycle (O-GlcNAc transferase/O-GlcNAcase) couple nutrient-dependent synthesis of UDP-GlcNAc to O-GlcNAc modification of serine/threonine amino acid residues [106]. These nutrients include glucose, glutamine, and acetyl-CoA. This series of reactions culminating in O-GlcNAcylation of targets is known as the hexosamine signaling pathway. This pathway is a cellular regulator of signaling cascades influencing growth, metabolism, and cellular stress. Although there is little evidence supporting a role of this pathway as a placental nutrient sensor, preliminary studies have demonstrated that the hexosamine signaling pathway is active in first-trimester human placenta and influences hormone production and IGF signaling [107].
THE ROLE OF mTOR IN PLACENTAL NUTRIENT SENSING
Mechanistic target of rapamycin is a ubiquitously expressed serine/threonine kinase that is present as two complexes differing in both their regulation and function. In particular, mTORC1 is a master regulator of the cellular translational machinery; it controls cell growth, proliferation, and metabolism in response to nutrient availability and growth factor signaling. Mechanistic target of rapamycin complex 1 promotes protein synthesis via downstream targets including ribosomal S6 kinases (S6K1 and S6K2), eukaryotic initiation factor 4E-binding proteins (4E-BP1 and 4E-BP2), and eukaryotic initiation factor 4G (eIF4G) [108, 109], and thereby affects overall cellular translation rates. The mTORC1 signaling pathway also influences the transcription of genes involved in amino acid, lipid, and nucleotide metabolism and immune modulation [110, 111]. Placental mTOR signaling has been proposed to play an important role as an integrator of diverse signals in the placenta and to regulate fetal nutrient availability by modulating placental growth and nutrient transport [76].
The first line of evidence consistent with an important role for mTORC1 in placental nutrient sensing is that mTORC1 has a multitude of upstream regulators, including free fatty acids, amino acids, glucose, ATP, and oxygen. It is likely that the placental concentrations of some of these nutrients are changed in conditions such as placental insufficiency, maternal undernutrition, or obesity. Indeed, some growth-restricted fetuses are hypoglycemic and hypoxemic, and have low circulating levels of essential amino acids [112, 113], suggesting that nutrients and oxygen are also low in the placenta. Furthermore, phosphorylation of placental AMPK, an upstream regulator of mTOR activity, is markedly decreased in maternal obesity [89], indicative of high ATP levels. In the placenta, mTORC1 is highly expressed in the syncytiotrophoblast [114], and evidence indicates that mTORC1 is regulated by glucose and amino acid concentrations in cultured primary human trophoblasts [48]. In addition to nutrients, mTORC1 is also regulated by an array of growth factors and hormones. Both maternal undernutrition and overnutrition alter circulating levels of hormones and factors including insulin, IGF-I, adiponectin, and leptin. Mechanistic target of rapamycin complex 1 is activated by insulin and IGF-I, and placental insulin/IGF-I signaling is inhibited in IUGR [96, 104, 105]. In addition, leptin signaling activates and cortisol inhibits mTORC1 [2]. Thus, mTORC1 signaling links maternal metabolic hormones and local nutrient levels to placental growth and function.
The second argument to support an important role for mTORC1 in placental nutrient sensing is that mTORC1 regulates trophoblast amino acid transport. Mechanistic target of rapamycin complex 1 stimulates system A and L amino acid transporters [48, 114, 115], which are critical for transport of both essential and nonessential amino acids to the fetus. Mechanistic target of rapamycin complex 1 regulates trophoblast amino acid transport by posttranslational mechanisms involving modulation of the trafficking of specific amino acid transporter isoforms to the plasma membrane [116]. Thus, placental mTORC1 signaling links maternal nutrient supply to fetal growth by influencing the transfer of amino acids across the placenta.
A third line of evidence consistent with an important role for mTORC1 signaling in placental nutrient sensing is that the activity of this signaling pathway is altered in pregnancy complications associated with abnormal fetal growth and in animal models where maternal nutrient availability has been altered experimentally. Placental mTORC1 activity is inhibited in human IUGR [96, 114] and activated in placentas of large babies born to obese mothers [97]. Furthermore, placental mTORC1 activity has been reported to be decreased in hyperthermia-induced IUGR in the sheep [117] and in response to a maternal low-protein diet in the rat [62] and maternal calorie restriction in the baboon [27].
IMPRINTED GENES IN MATERNAL-FETAL RESOURCE ALLOCATION
The placenta is of fetal origin, thus sharing the fetal genotype. From an evolutionary perspective, the conflict theory (also known as kinship theory of genomic imprinting) proposes that imprinted genes (genes that are expressed in a parent-specific manner) have evolved because of the need to allocate maternal resources to the offspring [118]. Indeed, paternally derived genes are thought to support the extraction of maternal resources for the benefit of a unique offspring, and, on the contrary, maternally derived genes tend to counteract this effect in order to prevent depletion of resources by allocating them equally among different offspring [78, 119]. Several studies have demonstrated that placental signals originating from imprinted genes regulate nutrient transport in the mouse placenta [120] and thereby control fetal growth [78, 79, 120].
More than 70 imprinted genes have been identified in the mouse placenta. Importantly, subgroups of these genes are imprinted only in the placenta and are involved in regulation of fetal and placental growth [120]. One of the most studied paternally expressed/maternally repressed imprinted genes in the placenta is Igf2 [29]. In mice, Igf2P0 knockout results in placental growth restriction at Embryonic Day 12 (e12) onwards, but fetal growth restriction does not occur until e16, where there is a moderate but significant reduction in fetal growth; moreover, at e19 fetal growth restriction is more pronounced [29, 78]. Therefore, the Igf2P0 knockout mouse is a model where the growth potential of the placenta has been genetically manipulated independently of that of the fetus. In this model, the fetal:placental weight ratio at e16 is 50% higher in Igf2P0 knockout fetuses compared to wild type. Furthermore, system A amino acid transport activity and expression shows a 50% increase compared to wild-type mice, indicating an up-regulation of nutrient transport. In contrast, at e19 the fetal:placental weight ratio and system A activity/expression were not different between Igf2P0 knockout and wild-type mice. Therefore, nutrient transporters can be up-regulated to increase placental efficiency when the placental size is small to meet the demands of the normally growing fetus, suggesting a compensatory mechanism to match fetal-placental growth. However, this up-regulation and increased efficiency are temporary and are not maintained in late pregnancy in the face of prolonged insult because fetal growth restriction ultimately develops in Igf2P0 knockout mice [29, 78].
H19 is an example of a maternally imprinted gene in the placenta [121]. Deletion of H19 results in overgrowth of the placenta, decreased gene expression of nutrient transporters, and reduced transport capacity [120, 122, 123]. The H19 gene encodes a highly expressed, growth-regulating, noncoding RNA that shares regulatory elements with IGF-II [124]. Interestingly, the H19 gene controls imprinting of the Igf2 locus [125], so its deletion results in biallelic expression of Igf2 and hence a double dosage of the growth factor [119]. Overall, studies in mice have demonstrated that altered imprinting of H19 and Igf2 are associated with placental and fetal growth abnormalities [29, 124], where changes in the expression and/or function of nutrient transporters are proposed to be due to fetal demand signals in an attempt to halt (H19) or increase (Igf2) fetal growth.
Despite extensive experimental evidence from gene-targeting approaches in the mouse, a physiological role of imprinted genes in the active day-to-day regulation of maternal-fetal resource allocation remains to be established. It is possible that placental imprinted genes influence the long-term functional capacity of the placenta, thereby establishing a trajectory for placental function for the entire pregnancy.
PLACENTAL CONTROL OF MATERNAL-FETAL RESOURCE ALLOCATION: AN INTEGRATED MODEL
We have proposed a model (placental nutrient sensing) in which the placenta integrates a multitude of maternal and fetal nutritional cues with information from intrinsic nutrient-sensing signaling pathways to match fetal demand with maternal supply by regulating maternal physiology, placental growth, and nutrient transport [31] (Fig. 3). As discussed previously, there is compelling evidence to suggest that trophoblast mTOR signaling is one important component of the placental nutrient sensor. We propose that these mechanisms have evolved because of the evolutionary pressures of maternal undernutrition. Although these regulatory loops may also function in response to overnutrition, it is possible that these responses may not be as readily apparent in maternal obesity or diabetes as in response to maternal undernutrition. Differential expression of placental imprinted genes influences the growth and transport capacity of the placenta, with paternally imprinted genes allocating nutrients to the fetus whereas maternally expressed genes allocate nutrients to the mother and limit fetal growth. We propose that, whereas imprinted genes are involved in determining the overall long-term functional capacity of the placenta for the entire pregnancy, placental nutrient sensing mediates dynamic regulation of placental function in response to changes in the maternal and fetal compartments (Fig. 3). It is important to note that our current model of placental control of maternal-fetal resource allocation is largely based on associations rather than cause-and-effect studies and much more work is needed to better understand the mechanistic links between placental responses to changes in nutrition and placental function, fetal growth, and the long-term health of the offspring.
FIG. 3.

Placental nutrient-sensing model. The placenta integrates a multitude of maternal and fetal cues with information from intrinsic nutrient-sensing signaling pathways to match fetal demand with maternal supply by regulating maternal physiology, placental growth, and nutrient transport. In addition, the expression of imprinted genes will determine the overall long-term functional capacity of the placenta for the entire pregnancy.
The relative importance of maternal supply and fetal demand signals for the regulation of placental function may differ between species, and depend on the type, duration, and severity of the nutritional perturbation [31]. It is plausible, for instance, that regulation by fetal demand signals is predominant when the nutritional challenge is moderate or brief, whereas regulation by maternal supply may override fetal demand signals if the nutritional challenge is more severe or prolonged. Matching fetal growth to maternal resources in situations of significant maternal undernutrition will produce an offspring that is smaller in size but that, in most instances, will survive and reproduce. Thus, rather than enhanced nutrient extraction from the already-deprived mother, which will jeopardize the survival of both the mother and her fetus, restricted fetal growth is the trade-off to avoid reproductive failure.
Footnotes
Supported by grants from NIH (HD065007, HD068370, DK089989, and HD071306).
REFERENCES
- Sideri M, Virgiliis G, Rainoldi R, Remotti G. The ultrastructural basis of the nutritional transfer: evidence of different patterns in the plasma membranes of the multilayered placental barrier In Miller R, Thiede H. (eds.), Fetal Nutrition, Metabolism, and Immunology, vol. 1 Rochester, New York: Springer US; 1984. 15 25 [Google Scholar]
- Jansson T, Powell TL. Role of placental nutrient sensing in developmental programming. Clin Obstet Gynecol. 2013;56:591–601. doi: 10.1097/GRF.0b013e3182993a2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science. 2004;305:1733–1736. doi: 10.1126/science.1095292. [DOI] [PubMed] [Google Scholar]
- Longo LD. Maternal blood volume and cardiac output during pregnancy: a hypothesis of endocrinologic control. Am J Physiol. 1983;245:R720–R729. doi: 10.1152/ajpregu.1983.245.5.R720. [DOI] [PubMed] [Google Scholar]
- Clapp JF, Capeless E. Cardiovascular function before, during, and after the first and subsequent pregnancies. Am J Cardiol. 1997;80:1469–1473. doi: 10.1016/s0002-9149(97)00738-8. [DOI] [PubMed] [Google Scholar]
- Barron WM, Mujais SK, Zinaman M, Bravo EL, Lindheimer MD. Plasma catecholamine responses to physiologic stimuli in normal human pregnancy. Am J Obstet Gynecol. 1986;154:80–84. doi: 10.1016/0002-9378(86)90397-2. [DOI] [PubMed] [Google Scholar]
- Conrad KP. Maternal vasodilation in pregnancy: the emerging role of relaxin. Am J Physiol Regul Integr Comp Physiol. 2011;301:R267–R275. doi: 10.1152/ajpregu.00156.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbern D, Freemark M. Placental hormones and the control of maternal metabolism and fetal growth. Curr Opin Endocrinol Diabetes Obes. 2011;18:409–416. doi: 10.1097/MED.0b013e32834c800d. [DOI] [PubMed] [Google Scholar]
- Corcoran JJ, Nicholson C, Sweeney M, Charnock JC, Robson SC, Westwood M, Taggart MJ. Human uterine and placental arteries exhibit tissue-specific acute responses to 17beta-estradiol and estrogen-receptor-specific agonists. Mol Hum Reprod. 2014;20:433–441. doi: 10.1093/molehr/gat095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott PA, Tremblay A, Brochu M, St-Louis J. Vasorelaxant action of 17β-estradiol in rat uterine arteries: role of nitric oxide synthases and estrogen receptors. Am J Physiol Heart Circ Physiol. 2007;293:H3713–H3719. doi: 10.1152/ajpheart.00736.2007. [DOI] [PubMed] [Google Scholar]
- Pastore MB, Jobe SO, Ramadoss J, Magness RR. Estrogen receptor-alpha and estrogen receptor-beta in the uterine vascular endothelium during pregnancy: functional implications for regulating uterine blood flow. Semin Reprod Med. 2012;30:46–61. doi: 10.1055/s-0031-1299597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myatt L. Control of vascular resistance in the human placenta. Placenta. 1992;13:329–341. doi: 10.1016/0143-4004(92)90057-z. [DOI] [PubMed] [Google Scholar]
- Highman TJ, Friedman JE, Huston LP, Wong WW, Catalano PM. Longitudinal changes in maternal serum leptin concentrations, body composition, and resting metabolic rate in pregnancy. Am J Obstet Gynecol. 1998;178:1010–1015. doi: 10.1016/s0002-9378(98)70540-x. [DOI] [PubMed] [Google Scholar]
- Masuzaki H, Ogawa Y, Sagawa N, Hosoda K, Matsumoto T, Mise H, Nishimura H, Yoshimasa Y, Tanaka I, Mori T, Nakao K. Nonadipose tissue production of leptin: leptin as a novel placenta-derived hormone in humans. Nat Med. 1997;3:1029–1033. doi: 10.1038/nm0997-1029. [DOI] [PubMed] [Google Scholar]
- Mazaki-Tovi S, Kanety H, Pariente C, Hemi R, Wiser A, Schiff E, Sivan E. Maternal serum adiponectin levels during human pregnancy. J Perinatol. 2007;27:77–81. doi: 10.1038/sj.jp.7211639. [DOI] [PubMed] [Google Scholar]
- Freemark M. Regulation of maternal metabolism by pituitary and placental hormones: roles in fetal development and metabolic programming Horm Res 2006. 65 (suppl 3): 41 49 [DOI] [PubMed] [Google Scholar]
- Kirwan JP. Hauguel-De Mouzon S, Lepercq J, Challier JC, Huston-Presley L, Friedman JE, Kalhan SC, Catalano PM. TNF-alpha is a predictor of insulin resistance in human pregnancy. Diabetes. 2002;51:2207–2213. doi: 10.2337/diabetes.51.7.2207. [DOI] [PubMed] [Google Scholar]
- Catalano PM, Hoegh M, Minium J, Huston-Presley L, Bernard S, Kalhan S. Hauguel-De Mouzon S. Adiponectin in human pregnancy: implications for regulation of glucose and lipid metabolism. Diabetologia. 2006;49:1677–1685. doi: 10.1007/s00125-006-0264-x. [DOI] [PubMed] [Google Scholar]
- Jansson N, Nilsfelt A, Gellerstedt M, Wennergren M, Rossander-Hulthen L, Powell TL, Jansson T. Maternal hormones linking maternal body mass index and dietary intake to birth weight. Am J Clin Nutr. 2008;87:1743–1749. doi: 10.1093/ajcn/87.6.1743. [DOI] [PubMed] [Google Scholar]
- Aye ILMH, Powell TL, Jansson T. Review: Adiponectin—the missing link between maternal adiposity, placental transport and fetal growth? Placenta 2013. 34 (suppl): S40 S45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastorakos G, Ilias I. Maternal and fetal hypothalamic-pituitary-adrenal axes during pregnancy and postpartum. Ann N Y Acad Sci. 2003;997:136–149. doi: 10.1196/annals.1290.016. [DOI] [PubMed] [Google Scholar]
- Barbour LA, Shao J, Qiao L, Pulawa LK, Jensen DR, Bartke A, Garrity M, Draznin B, Friedman JE. Human placental growth hormone causes severe insulin resistance in transgenic mice. Am J Obstet Gynecol. 2002;186:512–517. doi: 10.1067/mob.2002.121256. [DOI] [PubMed] [Google Scholar]
- Barbour LA, Shao J, Qiao L, Leitner W, Anderson M, Friedman JE, Draznin B. Human placental growth hormone increases expression of the p85 regulatory unit of phosphatidylinositol 3-kinase and triggers severe insulin resistance in skeletal muscle. Endocrinology. 2004;145:1144–1150. doi: 10.1210/en.2003-1297. [DOI] [PubMed] [Google Scholar]
- Lacroix MC, Guibourdenche J, Frendo JL, Muller F, Evain-Brion D. Human placental growth hormone—a review Placenta 2002. 23 (suppl A): S87 S94 [DOI] [PubMed] [Google Scholar]
- Mirlesse V, Frankenne F, Alsat E, Poncelet M, Hennen G, Evain-Brion D. Placental growth hormone levels in normal pregnancy and in pregnancies with intrauterine growth retardation. Pediatr Res. 1993;34:439–442. doi: 10.1203/00006450-199310000-00011. [DOI] [PubMed] [Google Scholar]
- Karl PI. Insulin-like growth factor-1 stimulates amino acid uptake by the cultured human placental trophoblast. J Cell Physiol. 1995;165:83–88. doi: 10.1002/jcp.1041650111. [DOI] [PubMed] [Google Scholar]
- Kavitha JV, Rosario FJ, Nijland MJ, McDonald TJ, Wu G, Kanai Y, Powell TL, Nathanielsz PW, Jansson T. Down-regulation of placental mTOR, insulin/IGF-I signaling, and nutrient transporters in response to maternal nutrient restriction in the baboon. FASEB J. 2014;28:1294–1305. doi: 10.1096/fj.13-242271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CT, Owens JA, Sferruzzi-Perri AN. Distinct actions of insulin-like growth factors (IGFs) on placental development and fetal growth: lessons from mice and guinea pigs Placenta 2008. 29 (suppl A): S42 S47 [DOI] [PubMed] [Google Scholar]
- Constancia M, Hemberger M, Hughes J, Dean W, Ferguson-Smith A, Fundele R, Stewart F, Kelsey G, Fowden A, Sibley C, Reik W. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature. 2002;417:945–948. doi: 10.1038/nature00819. [DOI] [PubMed] [Google Scholar]
- Sferruzzi-Perri AN, Vaughan OR, Coan PM, Suciu MC, Darbyshire R, Constancia M, Burton GJ, Fowden AL. Placental-specific Igf2 deficiency alters developmental adaptations to undernutrition in mice. Endocrinology. 2011;152:3202–3212. doi: 10.1210/en.2011-0240. [DOI] [PubMed] [Google Scholar]
- Gaccioli F, Lager S, Powell TL, Jansson T. Placental transport in response to altered maternal nutrition. J Dev Orig Health Dis. 2013;4:101–115. doi: 10.1017/S2040174412000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lager S, Powell TL. Regulation of nutrient transport across the placenta. J Pregnancy. 2012;2012:179827. doi: 10.1155/2012/179827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown LD, Green AS, Limesand SW, Rozance PJ. Maternal amino acid supplementation for intrauterine growth restriction. Front Biosci (Schol Ed) 2011;3:428–444. doi: 10.2741/s162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetin I, Marconi AM, Corbetta C, Lanfranchi A, Baggiani AM, Battaglia FC, Pardi G. Fetal amino acids in normal pregnancies and in pregnancies complicated by intrauterine growth retardation. Early Hum Dev. 1992;29:183–186. doi: 10.1016/0378-3782(92)90136-5. [DOI] [PubMed] [Google Scholar]
- Jansson T. Amino acid transporters in the human placenta. Pediatr Res. 2001;49:141–147. doi: 10.1203/00006450-200102000-00003. [DOI] [PubMed] [Google Scholar]
- Cleal JK, Lewis RM. The mechanisms and regulation of placental amino acid transport to the human foetus. J Neuroendocrinol. 2008;20:419–426. doi: 10.1111/j.1365-2826.2008.01662.x. [DOI] [PubMed] [Google Scholar]
- Kudo Y, Boyd CA. Human placental amino acid transporter genes: expression and function. Reproduction. 2002;124:593–600. doi: 10.1530/rep.0.1240593. [DOI] [PubMed] [Google Scholar]
- Desforges M, Sibley C. Placental nutrient supply and fetal growth. Int J Dev Biol. 2010;54:377–390. doi: 10.1387/ijdb.082765md. [DOI] [PubMed] [Google Scholar]
- Jansson T, Ylven K, Wennergren M, Powell TL. Glucose transport and system A activity in syncytiotrophoblast microvillous and basal plasma membranes in intrauterine growth restriction. Placenta. 2002;23:392–399. doi: 10.1053/plac.2002.0826. [DOI] [PubMed] [Google Scholar]
- Okamoto Y, Sakata M, Ogura K, Yamamoto T, Yamaguchi M, Tasaka K, Kurachi H, Tsurudome M, Murata Y. Expression and regulation of 4F2hc and hLAT1 in human trophoblasts. Am J Physiol Cell Physiol. 2002;282:C196–C204. doi: 10.1152/ajpcell.2002.282.1.C196. [DOI] [PubMed] [Google Scholar]
- Kudo Y, Boyd CA. Characterisation of L-tryptophan transporters in human placenta: a comparison of brush border and basal membrane vesicles. J Physiol. 2001;531:405–416. doi: 10.1111/j.1469-7793.2001.0405i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavare JM, Holmes CH. Differential expression of the receptors for epidermal growth factor and insulin in the developing human placenta. Cell Signal. 1989;1:55–64. doi: 10.1016/0898-6568(89)90020-x. [DOI] [PubMed] [Google Scholar]
- Desoye G, Hartmann M, Blaschitz A, Dohr G, Hahn T, Kohnen G, Kaufmann P. Insulin receptors in syncytiotrophoblast and fetal endothelium of human placenta. Immunohistochemical evidence for developmental changes in distribution pattern. Histochemistry. 1994;101:277–285. doi: 10.1007/BF00315915. [DOI] [PubMed] [Google Scholar]
- Fang J, Furesz TC, Lurent RS, Smith CH, Fant ME. Spatial polarization of insulin-like growth factor receptors on the human syncytiotrophoblast. Pediatr Res. 1997;41:258–265. doi: 10.1203/00006450-199702000-00017. [DOI] [PubMed] [Google Scholar]
- Bodner J, Ebenbichler CF, Wolf HJ, Muller-Holzner E, Stanzl U, Gander R, Huter O, Patsch JR. Leptin receptor in human term placenta: in situ hybridization and immunohistochemical localization. Placenta. 1999;20:677–682. doi: 10.1053/plac.1999.0431. [DOI] [PubMed] [Google Scholar]
- Jones HN, Jansson T, Powell TL. Full-length adiponectin attenuates insulin signaling and inhibits insulin-stimulated amino acid transport in human primary trophoblast cells. Diabetes. 2010;59:1161–1170. doi: 10.2337/db09-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson N, Greenwood SL, Johansson BR, Powell TL, Jansson T. Leptin stimulates the activity of the system A amino acid transporter in human placental villous fragments. J Clin Endocrinol Metab. 2003;88:1205–1211. doi: 10.1210/jc.2002-021332. [DOI] [PubMed] [Google Scholar]
- Roos S, Lagerlof O, Wennergren M, Powell TL, Jansson T. Regulation of amino acid transporters by glucose and growth factors in cultured primary human trophoblast cells is mediated by mTOR signaling. Am J Physiol Cell Physiol. 2009;297:C723–C731. doi: 10.1152/ajpcell.00191.2009. [DOI] [PubMed] [Google Scholar]
- von Versen-Hoynck F, Rajakumar A, Parrott MS, Powers RW. Leptin affects system A amino acid transport activity in the human placenta: evidence for STAT3 dependent mechanisms. Placenta. 2009;30:361–367. doi: 10.1016/j.placenta.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Mao D, Smith CH, Fant ME. IGF regulation of neutral amino acid transport in the BeWo choriocarcinoma cell line (b30 clone): evidence for MAP kinase-dependent and MAP kinase-independent mechanisms. Growth Horm IGF Res. 2006;16:318–325. doi: 10.1016/j.ghir.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Araujo JR, Correia-Branco A, Ramalho C, Goncalves P, Pinho MJ, Keating E, Martel F. L-methionine placental uptake: characterization and modulation in gestational diabetes mellitus. Reprod Sci. 2013;20:1492–1507. doi: 10.1177/1933719113488442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones HN, Jansson T, Powell TL. IL-6 stimulates system A amino acid transporter activity in trophoblast cells through STAT3 and increased expression of SNAT2. Am J Physiol Cell Physiol. 2009;297:C1228–C1235. doi: 10.1152/ajpcell.00195.2009. [DOI] [PubMed] [Google Scholar]
- Lager S, Gaccioli F, Ramirez VI, Jones HN, Jansson T, Powell TL. Oleic acid stimulates system A amino acid transport in primary human trophoblast cells mediated by toll-like receptor 4. J Lipid Res. 2013;54:725–733. doi: 10.1194/jlr.M033050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts KA, Riley SC, Reynolds RM, Barr S, Evans M, Statham A, Hor K, Jabbour HN, Norman JE, Denison FC. Placental structure and inflammation in pregnancies associated with obesity. Placenta. 2011;32:247–254. doi: 10.1016/j.placenta.2010.12.023. [DOI] [PubMed] [Google Scholar]
- Aye IL, Jansson T, Powell TL. Interleukin-1beta inhibits insulin signaling and prevents insulin-stimulated system A amino acid transport in primary human trophoblasts. Mol Cell Endocrinol. 2013;381:46–55. doi: 10.1016/j.mce.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audette MC, Challis JR, Jones RL, Sibley CP, Matthews SG. Antenatal dexamethasone treatment in midgestation reduces system A-mediated transport in the late-gestation murine placenta. Endocrinology. 2011;152:3561–3570. doi: 10.1210/en.2011-0104. [DOI] [PubMed] [Google Scholar]
- Vaughan OR, Sferruzzi-Perri AN, Fowden AL. Maternal corticosterone regulates nutrient allocation to fetal growth in mice. J Physiol. 2012;590:5529–5540. doi: 10.1113/jphysiol.2012.239426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson N, Pettersson J, Haafiz A, Ericsson A, Palmberg I, Tranberg M, Ganapathy V, Powell TL, Jansson T. Down-regulation of placental transport of amino acids precedes the development of intrauterine growth restriction in rats fed a low protein diet. J Physiol. 2006;576:935–946. doi: 10.1113/jphysiol.2006.116509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malandro MS, Beveridge MJ, Kilberg MS, Novak DA. Effect of low-protein diet-induced intrauterine growth retardation on rat placental amino acid transport. Am J Physiol Cell Physiol. 1996;271:C295–C303. doi: 10.1152/ajpcell.1996.271.1.C295. [DOI] [PubMed] [Google Scholar]
- Zhu MJ, Du M, Hess BW, Nathanielsz PW, Ford SP. Periconceptional nutrient restriction in the ewe alters MAPK/ERK1/2 and PI3K/Akt growth signaling pathways and vascularity in the placentome. Placenta. 2007;28:1192–1199. doi: 10.1016/j.placenta.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Zhu MJ, Ma Y, Long NM, Du M, Ford SP. Maternal obesity markedly increases placental fatty acid transporter expression and fetal blood triglycerides at midgestation in the ewe. Am J Physiol Regul Integr Comp Physiol. 2010;299:R1224–R1231. doi: 10.1152/ajpregu.00309.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario FJ, Jansson N, Kanai Y, Prasad PD, Powell TL, Jansson T. Maternal protein restriction in the rat inhibits placental insulin, mTOR, and STAT3 signaling and down-regulates placental amino acid transporters. Endocrinology. 2011;152:1119–1129. doi: 10.1210/en.2010-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkacemi L, Nelson DM, Desai M, Ross MG. Maternal undernutrition influences placental-fetal development. Biol Reprod. 2010;83:325–331. doi: 10.1095/biolreprod.110.084517. [DOI] [PubMed] [Google Scholar]
- Jauniaux E, Watson AL, Hempstock J, Bao YP, Skepper JN, Burton GJ. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am J Pathol. 2000;157:2111–2122. doi: 10.1016/S0002-9440(10)64849-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton GJ, Woods AW, Jauniaux E, Kingdom JCP. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta. 2009;30:473–482. doi: 10.1016/j.placenta.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamudio S, Moore LG. Altitude and fetal growth: current knowledge and future directions. Ultrasound Obstet Gynecol. 2000;16:6–8. doi: 10.1046/j.1469-0705.2000.00155.x. [DOI] [PubMed] [Google Scholar]
- Mehta AR, Mehta PR. The hypoxia of high altitude causes restricted fetal growth in chick embryos with the extent of this effect depending on maternal altitudinal status. J Physiol. 2008;586:1469–1471. doi: 10.1113/jphysiol.2008.151332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay JE, Ferrell WR, Crawford L, Wallace AM, Greer IA, Sattar N. Maternal obesity is associated with dysregulation of metabolic, vascular, and inflammatory pathways. J Clin Endocrinol Metab. 2002;87:4231–4237. doi: 10.1210/jc.2002-020311. [DOI] [PubMed] [Google Scholar]
- Lowe LP, Metzger BE, Lowe WL, Jr, , Dyer AR, McDade TW, McIntyre HD; HAPO Study Cooperative Research Group. Inflammatory mediators and glucose in pregnancy: results from a subset of the Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study. J Clin Endocrinol Metab. 2010;95:5427–5434. doi: 10.1210/jc.2010-1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ategbo JM, Grissa O, Yessoufou A, Hichami A, Dramane KL, Moutairou K, Miled A, Grissa A, Jerbi M, Tabka Z, Khan NA. Modulation of adipokines and cytokines in gestational diabetes and macrosomia. J Clin Endocrinol Metab. 2006;91:4137–4143. doi: 10.1210/jc.2006-0980. [DOI] [PubMed] [Google Scholar]
- Sivan E, Mazaki-Tovi S, Pariente C, Efraty Y, Schiff E, Hemi R, Kanety H. Adiponectin in human cord blood: relation to fetal birth weight and gender. J Clin Endocrinol Metab. 2003;88:5656–5660. doi: 10.1210/jc.2003-031174. [DOI] [PubMed] [Google Scholar]
- Qiao L, Yoo HS, Madon A, Kinney B, Hay WW, Jr, , Shao J. Adiponectin enhances mouse fetal fat deposition. Diabetes. 2012;61:3199–3207. doi: 10.2337/db12-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aye ILMH, Gao X, Weintraub ST, Jansson T, Powell TL. Adiponectin inhibits insulin function in primary trophoblasts by pparα-mediated ceramide synthesis. Mol Endocrinol. 2014;28:512–524. doi: 10.1210/me.2013-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario FJ, Schumacher MA, Jiang J, Kanai Y, Powell TL, Jansson T. Chronic maternal infusion of full-length adiponectin in pregnant mice down-regulates placental amino acid transporter activity and expression and decreases fetal growth. J Physiol. 2012;590:1495–1509. doi: 10.1113/jphysiol.2011.226399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson T, Powell TL. IFPA. 2005 Award in Placentology Lecture. Human placental transport in altered fetal growth: does the placenta function as a nutrient sensor?—a review Placenta 2006. 27 (suppl A): S91 S97 [DOI] [PubMed] [Google Scholar]
- Jansson T, Aye IL, Goberdhan DC. The emerging role of mTORC1 signaling in placental nutrient-sensing Placenta 2012. 33( suppl 2): e23- e29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley CP, Brownbill P, Dilworth M, Glazier JD. Review: adaptation in placental nutrient supply to meet fetal growth demand: implications for programming Placenta 2010. 31 (suppl): S70 S74 [DOI] [PubMed] [Google Scholar]
- Constancia M, Angiolini E, Sandovici I, Smith P, Smith R, Kelsey G, Dean W, Ferguson-Smith A, Sibley CP, Reik W, Fowden A. Adaptation of nutrient supply to fetal demand in the mouse involves interaction between the Igf2 gene and placental transporter systems. Proc Natl Acad Sci U S A. 2005;102:19219–19224. doi: 10.1073/pnas.0504468103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angiolini E, Coan PM, Sandovici I, Iwajomo OH, Peck G, Burton GJ, Sibley CP, Reik W, Fowden AL, Constancia M. Developmental adaptations to increased fetal nutrient demand in mouse genetic models of Igf2-mediated overgrowth. FASEB J. 2011;25:1737–1745. doi: 10.1096/fj.10-175273. [DOI] [PubMed] [Google Scholar]
- Sferruzzi-Perri AN, Vaughan OR, Haro M, Cooper WN, Musial B, Charalambous M, Pestana D, Ayyar S, Ferguson-Smith AC, Burton GJ, Constancia M, Fowden AL. An obesogenic diet during mouse pregnancy modifies maternal nutrient partitioning and the fetal growth trajectory. FASEB J. 2013;27:3928–3937. doi: 10.1096/fj.13-234823. [DOI] [PubMed] [Google Scholar]
- Godfrey KM, Matthews N, Glazier J, Jackson A, Wilman C, Sibley CP. Neutral amino acid uptake by the microvillous plasma membrane of the human placenta is inversely related to fetal size at birth in normal pregnancy. J Clin Endocrinol Metab. 1998;83:3320–3326. doi: 10.1210/jcem.83.9.5132. [DOI] [PubMed] [Google Scholar]
- Strid H, Bucht E, Jansson T, Wennergren M, Powell TL. ATP dependent Ca2+ transport across basal membrane of human syncytiotrophoblast in pregnancies complicated by intrauterine growth restriction or diabetes. Placenta. 2003;24:445–452. doi: 10.1053/plac.2002.0941. [DOI] [PubMed] [Google Scholar]
- Strid H, Care A, Jansson T, Powell T. Parathyroid hormone-related peptide (38–94) amide stimulates ATP-dependent calcium transport in the Basal plasma membrane of the human syncytiotrophoblast. J Endocrinol. 2002;175:517–524. doi: 10.1677/joe.0.1750517. [DOI] [PubMed] [Google Scholar]
- Kovacs CS, Lanske B, Hunzelman JL, Guo J, Karaplis AC, Kronenberg HM. Parathyroid hormone-related peptide (PTHrP) regulates fetal-placental calcium transport through a receptor distinct from the PTH/PTHrP receptor. Proc Natl Acad Sci U S A. 1996;93:15233–15238. doi: 10.1073/pnas.93.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond H, Dilworth MR, Baker B, Cowley E. Requena Jimenez A, Boyd RD, Husain SM, Ward BS, Sibley CP, Glazier JD. Increased maternofetal calcium flux in parathyroid hormone-related protein-null mice. J Physiol. 2008;586:2015–2025. doi: 10.1113/jphysiol.2007.149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilworth MR, Kusinski LC, Cowley E, Ward BS, Husain SM, Constancia M, Sibley CP, Glazier JD. Placental-specific Igf2 knockout mice exhibit hypocalcemia and adaptive changes in placental calcium transport. Proc Natl Acad Sci U S A. 2010;107:3894–3899. doi: 10.1073/pnas.0911710107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson T, Scholtbach V, Powell TL. Placental transport of leucine and lysine is reduced in intrauterine growth restriction. Pediatr Res. 1998;44:532–537. doi: 10.1203/00006450-199810000-00011. [DOI] [PubMed] [Google Scholar]
- Glazier JD, Cetin I, Perugino G, Ronzoni S, Grey AM, Mahendran D, Marconi AM, Pardi G, Sibley CP. Association between the activity of the system A amino acid transporter in the microvillous plasma membrane of the human placenta and severity of fetal compromise in intrauterine growth restriction. Pediatr Res. 1997;42:514–519. doi: 10.1203/00006450-199710000-00016. [DOI] [PubMed] [Google Scholar]
- Jansson N, Rosario FJ, Gaccioli F, Lager S, Jones HN, Roos S, Jansson T, Powell TL. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J Clin Endocrinol Metab. 2013;98:105–113. doi: 10.1210/jc.2012-2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson T, Ekstrand Y, Bjorn C, Wennergren M, Powell TL. Alterations in the activity of placental amino acid transporters in pregnancies complicated by diabetes. Diabetes. 2002;51:2214–2219. doi: 10.2337/diabetes.51.7.2214. [DOI] [PubMed] [Google Scholar]
- Kuruvilla AG, D'Souza SW, Glazier JD, Mahendran D, Maresh MJ, Sibley CP. Altered activity of the system A amino acid transporter in microvillous membrane vesicles from placentas of macrosomic babies born to diabetic women. J Clin Invest. 1994;94:689–695. doi: 10.1172/JCI117386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smerieri A, Petraroli M, Ziveri MA, Volta C, Bernasconi S, Street ME. Effects of cord serum insulin, IGF-II, IGFBP-2, IL-6 and cortisol concentrations on human birth weight and length: pilot study. PLoS One. 2011;6:e29562. doi: 10.1371/journal.pone.0029562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christou H, Connors JM, Ziotopoulou M, Hatzidakis V, Papathanassoglou E, Ringer SA, Mantzoros CS. Cord blood leptin and insulin-like growth factor levels are independent predictors of fetal growth. J Clin Endocrinol Metab. 2001;86:935–938. doi: 10.1210/jcem.86.2.7217. [DOI] [PubMed] [Google Scholar]
- Kilberg MS, Balasubramanian M, Fu L, Shan J. The transcription factor network associated with the amino acid response in mammalian cells. Adv Nutr. 2012;3:295–306. doi: 10.3945/an.112.001891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strakovsky RS, Zhou D, Pan YX. A low-protein diet during gestation in rats activates the placental mammalian amino acid response pathway and programs the growth capacity of offspring. J Nutr. 2010;140:2116–2120. doi: 10.3945/jn.110.127803. [DOI] [PubMed] [Google Scholar]
- Yung HW, Calabrese S, Hynx D, Hemmings BA, Cetin I, Charnock-Jones DS, Burton GJ. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am J Pathol. 2008;173:451–462. doi: 10.2353/ajpath.2008.071193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaccioli F, White V, Capobianco E, Powell TL, Jawerbaum A, Jansson T. Maternal overweight induced by a diet with high content of saturated fat activates placental mTOR and eIF2alpha signaling and increases fetal growth in rats Biol Reprod 2013. 89 4: 96 [DOI] [PubMed] [Google Scholar]
- Yung HW, Cox M. Tissot van Patot M, Burton GJ. Evidence of endoplasmic reticulum stress and protein synthesis inhibition in the placenta of non-native women at high altitude. FASEB J. 2012;26:1970–1981. doi: 10.1096/fj.11-190082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Kim J, Guan KL. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol. 2012;52:381–400. doi: 10.1146/annurev-pharmtox-010611-134537. [DOI] [PubMed] [Google Scholar]
- Ma Y, Zhu MJ, Uthlaut AB, Nijland MJ, Nathanielsz PW, Hess BW, Ford SP. Upregulation of growth signaling and nutrient transporters in cotyledons of early to mid-gestational nutrient restricted ewes. Placenta. 2011;32:255–263. doi: 10.1016/j.placenta.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu MJ, Du M, Nijland MJ, Nathanielsz PW, Hess BW, Moss GE, Ford SP. Down-regulation of growth signaling pathways linked to a reduced cotyledonary vascularity in placentomes of over-nourished, obese pregnant ewes. Placenta. 2009;30:405–410. doi: 10.1016/j.placenta.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, Woodgett JR. GSK-3: Functional insights from cell biology and animal models. Front Mol Neurosci. 2011;4:40. doi: 10.3389/fnmol.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Altman BJ, Coloff JL, Herman CE, Jacobs SR, Wieman HL, Wofford JA, Dimascio LN, Ilkayeva O, Kelekar A, Reya T, Rathmell JC. Glycogen synthase kinase 3alpha and 3beta mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Mol Cell Biol. 2007;27:4328–4339. doi: 10.1128/MCB.00153-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street ME, Viani I, Ziveri MA, Volta C, Smerieri A, Bernasconi S. Impairment of insulin receptor signal transduction in placentas of intra-uterine growth-restricted newborns and its relationship with fetal growth. Eur J Endocrinol. 2011;164:45–52. doi: 10.1530/EJE-10-0752. [DOI] [PubMed] [Google Scholar]
- Laviola L, Perrini S, Belsanti G, Natalicchio A, Montrone C, Leonardini A, Vimercati A, Scioscia M, Selvaggi L, Giorgino R, Greco P, Giorgino F. Intrauterine growth restriction in humans is associated with abnormalities in placental insulin-like growth factor signaling. Endocrinology. 2005;146:1498–1505. doi: 10.1210/en.2004-1332. [DOI] [PubMed] [Google Scholar]
- Hanover JA, Krause MW, Love DC. The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim Biophys Acta. 2010;1800:80–95. doi: 10.1016/j.bbagen.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya G, Albrecht C, Benton SJ, Cotechini T, Dechend R, Dilworth MR, Duttaroy AK, Grotmol T, Heazell AE, Jansson T, Johnstone ED, Jones HN, et al. IFPA Meeting 2011 workshop report I: placenta: predicting future health; roles of lipids in the growth and development of feto-placental unit; placental nutrient sensing; placental research to solve clinical problems—a translational approach Placenta 2012. 33 (suppl): S4 S8 [DOI] [PubMed] [Google Scholar]
- Proud CG. Signalling to translation: how signal transduction pathways control the protein synthetic machinery. Biochem J. 2007;403:217–234. doi: 10.1042/BJ20070024. [DOI] [PubMed] [Google Scholar]
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- Peng T, Golub TR, Sabatini DM. The immunosuppressant rapamycin mimics a starvation-like signal distinct from amino acid and glucose deprivation. Mol Cell Biol. 2002;22:5575–5584. doi: 10.1128/MCB.22.15.5575-5584.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Economides DL, Nicolaides KH. Blood glucose and oxygen tension levels in small-for-gestational-age fetuses. Am J Obstet Gynecol. 1989;160:385–389. doi: 10.1016/0002-9378(89)90453-5. [DOI] [PubMed] [Google Scholar]
- Cetin I, Corbetta C, Sereni LP, Marconi AM, Bozzetti P, Pardi G, Battaglia FC. Umbilical amino acid concentrations in normal and growth-retarded fetuses sampled in utero by cordocentesis. Am J Obstet Gynecol. 1990;162:253–261. doi: 10.1016/0002-9378(90)90860-a. [DOI] [PubMed] [Google Scholar]
- Roos S, Jansson N, Palmberg I, Saljo K, Powell TL, Jansson T. Mammalian target of rapamycin in the human placenta regulates leucine transport and is down-regulated in restricted fetal growth. J Physiol. 2007;582:449–459. doi: 10.1113/jphysiol.2007.129676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos S, Kanai Y, Prasad PD, Powell TL, Jansson T. Regulation of placental amino acid transporter activity by mammalian target of rapamycin. Am J Physiol Cell Physiol. 2009;296:C142–C150. doi: 10.1152/ajpcell.00330.2008. [DOI] [PubMed] [Google Scholar]
- Rosario FJ, Kanai Y, Powell TL, Jansson T. Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. J Physiol. 2013;591:609–625. doi: 10.1113/jphysiol.2012.238014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo JA, Brown LD, Galan HL. Placental mammalian target of rapamycin and related signaling pathways in an ovine model of intrauterine growth restriction. Am J Obstet Gynecol. 2009;201:616.e1–616.e7. doi: 10.1016/j.ajog.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowden AL, Moore T. Maternal-fetal resource allocation: co-operation and conflict Placenta 2012. 33 (suppl 2): e11 e15 [DOI] [PubMed] [Google Scholar]
- Burton GJ, Fowden AL. Review: the placenta and developmental programming: balancing fetal nutrient demands with maternal resource allocation Placenta 2012. 33 (suppl): S23 S27 [DOI] [PubMed] [Google Scholar]
- Coan PM, Burton GJ, Ferguson-Smith AC. Imprinted genes in the placenta—a review Placenta 2005. 26 (suppl A): S10 S20 [DOI] [PubMed] [Google Scholar]
- Leighton PA, Ingram RS, Eggenschwiler J, Efstratiadis A, Tilghman SM. Disruption of imprinting caused by deletion of the H19 gene region in mice. Nature. 1995;375:34–39. doi: 10.1038/375034a0. [DOI] [PubMed] [Google Scholar]
- Reik W, Constancia M, Fowden A, Anderson N, Dean W, Ferguson-Smith A, Tycko B, Sibley C. Regulation of supply and demand for maternal nutrients in mammals by imprinted genes. J Physiol. 2003;547:35–44. doi: 10.1113/jphysiol.2002.033274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko B, Morison IM. Physiological functions of imprinted genes. J Cell Physiol. 2002;192:245–258. doi: 10.1002/jcp.10129. [DOI] [PubMed] [Google Scholar]
- Gabory A, Ripoche MA, Yoshimizu T, Dandolo L. The H19 gene: regulation and function of a non-coding RNA. Cytogenet Genome Res. 2006;113:188–193. doi: 10.1159/000090831. [DOI] [PubMed] [Google Scholar]
- Thorvaldsen JL, Duran KL, Bartolomei MS. Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev. 1998;12:3693–3702. doi: 10.1101/gad.12.23.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]