

Abstract
The robustness and safety of liver-directed gene therapy can be substantially improved by enhancing expression of the therapeutic transgene in the liver. To achieve this, we developed a new approach of rational in silico vector design. This approach relies on a genome-wide bio-informatics strategy to identify cis-acting regulatory modules (CRMs) containing evolutionary conserved clusters of transcription factor binding site motifs that determine high tissue-specific gene expression. Incorporation of these CRMs into adeno-associated viral (AAV) and non-viral vectors enhanced gene expression in mice liver 10 to 100-fold, depending on the promoter used. Furthermore, these CRMs resulted in robust and sustained liver-specific expression of coagulation factor IX (FIX), validating their immediate therapeutic and translational relevance. Subsequent translational studies indicated that therapeutic FIX expression levels could be attained reaching 20–35% of normal levels after AAV-based liver-directed gene therapy in cynomolgus macaques. This study underscores the potential of rational vector design using computational approaches to improve their robustness and therefore allows for the use of lower and thus safer vector doses for gene therapy, while maximizing therapeutic efficacy.
Introduction
Convincing evidence continues to emerge from clinical trials that gene therapy is yielding therapeutic effects in patients suffering from a wide range of diseases.1,2 In particular, liver-directed gene therapy is becoming a promising modality to obtain sustained hepatocyte-specific expression of secreted factors into the circulation. This has implications for other liver-borne genetic and complex diseases. Despite these successes, there have been concerns regarding the efficacy and safety of some gene delivery approaches. The major limiting factors are insufficient and/or transient transgene expression levels and inappropriate expression of the transgene in unwanted cell types. Higher vector doses are typically used in gene therapy clinical trials to improve therapeutic efficacy. However, this often triggers T-cell–mediated immune responses against the vector capsid antigens displayed by transduced cells, particularly hepatocytes, in the context of MHC class I.3,4,5 This contributes to the elimination of the gene-modified cells and liver toxicity, resulting in short-term gene expression. Moreover, inadvertent transgene expression in antigen-presenting cells (APCs), increases the risk of untoward immune responses against the gene-modified hepatocytes and/or the therapeutic transgene product.6,7 Hence, there is a need to generate improved gene therapy vectors allowing the use of lower and safer vector doses that enable sustained hepatocyte-specific expression of the therapeutic gene.
Typically, conventional methods of vector design rely on haphazard trial-and-error approaches whereby transcriptional enhancers are combined with promoters to boost expression levels. Though this can sometimes be effective,6,8 it often results in non-productive combinations that result in either modest or no increased expression levels of the gene of interest in hepatocytes and/or loss of liver specificity.6 An intrinsic bias associated with the design of gene therapy vectors is that it frequently relies on the in vitro characteristics of its regulatory elements in cell lines, which is usually not predictive of their in vivo performance.6,9 Moreover, these conventional approaches in vector design do not take into account the importance of including evolutionary conserved regulatory motifs into the expression modules, which is particularly relevant for clinical translation.
In silico design offers unique opportunities to generate robust liver-specific gene therapy vectors and overcome some of the limitations of conventional gene therapy vector development. Though data-mining has been used to identify cis-regulatory modules (CRMs) and analyze expression patterns in vivo,10,11 surprisingly however, this area of research remains largely unexplored in the context of vector development and gene therapy. To address this caveat and overcome some of the limitations of conventional vector design, we have developed and validated an alternative strategy to generate efficient, liver-specific gene therapy vectors using computational methods. In this study, we relied on a genome-wide in silico identification of CRMs that contained clusters of evolutionarily conserved transcription factor binding site (TFBS) motifs associated with robust hepatocyte-specific expression. We employed a comprehensive computational strategy that takes into account not only the over-representation of a given TFBS but also its context-dependent co-occurrence with other TFBS.12 Such an approach has not yet been used to improve the performance of gene therapy vectors, which underscores the novelty of this study. This computational approach has broad implications for liver-directed gene therapy and allows for the use of lower and thus safer vector doses, while maximizing therapeutic efficacy in pre-clinical mouse models and in non-human primates. Furthermore, the validation of these liver-specific CRMs provides new insights into the molecular determinants underlying transcriptional control in hepatocytes.
Results
Computational de novo design of liver-specific CRMs
Rational de novo design of robust liver-specific gene therapy vectors relies on the identification of tissue-specific CRMs associated with high hepatocyte-specific expression. To identify these critical hepatocyte-specific CRMs (i.e., HS-CRMs), we used a multi-step computational approach. This bio-informatics approach was originally used to identify CRMs associated with differential gene expression in response to in vitro stimuli.12 In this study, we extended this computational approach to identify evolutionary conserved CRMs associated with highly expressed liver-specific promoters. One of the unique features of this computational strategy is that it takes into account the over-representation of a given TFBS and its context-dependent co-occurrence with other TFBS on a genome-wide scale.12
A total of 14 different HS-CRMs were identified, ranging in size from 41 bp to 551 bp (Figure 1 and Table 1 and Supplementary Figure S1 and Table S2). The HS-CRM comprised putative binding sites for eight different TFs: HNF1α, C/EBP, LEF1, FOX, IRF, LEF1/TCF, Tal1β/E47, and MyoD. These HS-CRMs contain a “molecular signature” that represents a hallmark of highly expressed genes in the liver. We observed that most HS-CRMs contain multiple TFBS that are similar but the uniqueness of each HS-CRM is dictated by its specific arrangement of the TFBS. Since all HS-CRMs exhibited a high degree of phylogenetic conservation among 44 divergent species, this suggests a strong evolutionary selective pressure to maintain these particular TFBS combinations for high tissue-specific expression. Phylogenetic conservation also increases the likelihood that the performance of the HS-CRMs is preserved across species in the process of clinical translation.
Figure 1.

Nucleotide sequence of the HS-CRM8 element located with the promoter of human Serpina1. This HS-CRM8 element is the most potent HS-CRM that was identified by the hydrodynamic transfection screen (see Figure 3). The evolutionary conservation is highlighted and the TFBS are shown. The TFBS include binding sites for LEF-1 (brown), LEF-1/TCF (dark green), CEBP (yellow), FOXA1 (light blue), HNF1 (light green), and MyoD (purple).
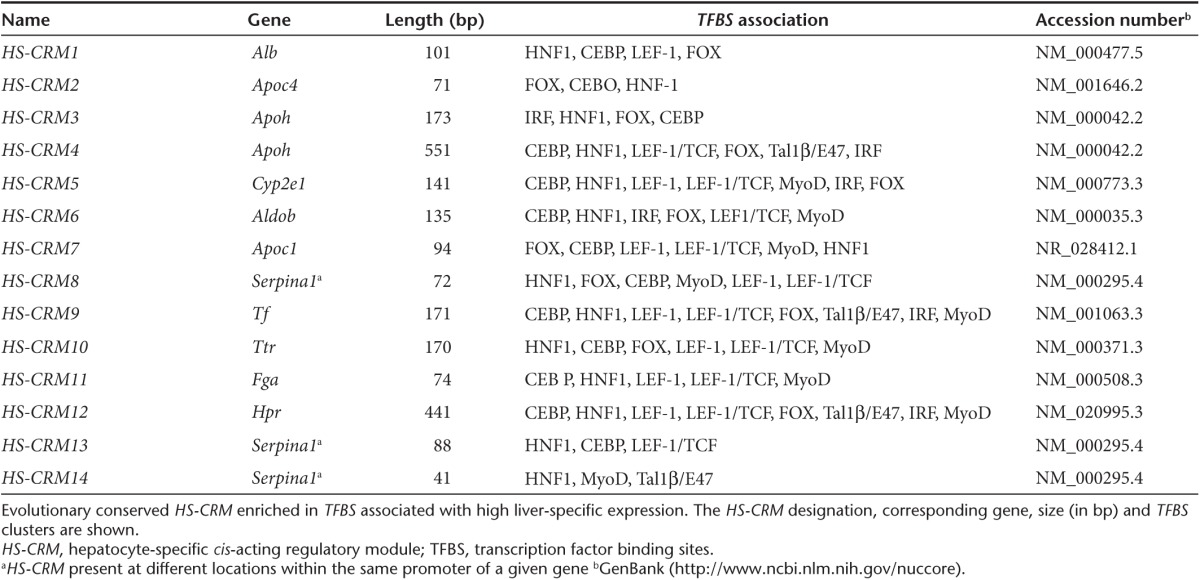
Table 1. TFBS strongly associated with high liver-specific expression.
In vivo validation of hepatocyte-specific CRMs
We subsequently assessed the efficiency and specificity of these novel HS-CRMs in liver. We first expressed human clotting factor IX (hFIX) from a chimeric promoter, composed of a potent liver-specific minimal transthyretin promoter (TTR) in conjunction with the different HS-CRM (Figure 2a). We choose hydrodynamic delivery to screen the 14 different hepatocyte-specific cis-regulatory modules (HS-CRMs) since it allows for a rapid and reliable assessment of their in vivo performance, without having to produce AAV vectors for each of them. Only the best HS-CRM was then selected for a subsequent comprehensive characterization in an AAV vector context (see below). Semi-high throughput hydrodynamic transfection of these expression plasmids into C57BL/6 mice revealed that the large majority (i.e., 85%) of the de novo designed HS-CRMs resulted in a significant increase in hFIX expression (Figure 3a). In particular, the most potent HS-CRM8 resulted in a robust improvement in hFIX expression, yielding up to 2.5 µg/ml. We subsequently validated the HS-CRM8 element in AAV9-based vectors in which the hFIX gene was driven from either a moderately active (Palm) (Figure 3b) or potent liver promoter (TTR) (Figure 3c). The de novo designed HS-CRM8 element in the AAV vector resulted in a robust and sustained 100-fold increase in hFIX expression from the moderately active Palm promoter (P < 0.0001) (5 × 1011 vg, Figure 3b). Moreover, this HS-CRM8 element significantly improved hFIX expression seven- to tenfold (P < 0.00005), even when a potent liver-specific promoter was used like TTR (Figure 3c), consistent with the increased hFIX expression after hydrodynamic transfection (Figure 3a). This resulted in stable therapeutic hFIX expression levels of about 100%, even at relatively low vector doses (5 × 109 vg). The HS-CRM8 element increased transcription from both TTR and Palm promoters (Supplementary Figure S2a,b). The increased hFIX antigen levels correlated strongly with the FIX mRNA levels (Supplementary Figure S2c). There was no significant difference in vector copy number in mice injected with the vectors with or without the HS-CRM8 element (Supplementary Figure S2d). The vector copy number correlated with the vector doses.
Figure 2.

Schematic representation of vectors used. (a) AAV9-HS-CRM-TTR-FIX, (b) AAV9-HS-CRM-PALM-FIX, (c) AAV9-HS-CRM8-TTR-FIX, and (d) AAV9-HS-CRM8-TTR-FIXco. The expression cassettes in a–b were packaged in a single-stranded AAV9, flanked by the 5′ and 3′ AAV inverted terminal repeats (ITR). (a) The liver-specific minimal transthyretin (TTR) promoter drives the human FIX minigene. Alternatively, (b) a Palm (paralemmin) promoter was used that was only weakly expressed in liver to drive the FIX mini-gene. The hepatocyte-specific CRMs (i.e., HS-CRM1 to HS-CRM14) are located upstream of the (a) TTR promoter. Similarly, HS-CRM8 is located upstream of the (b) Palm promoter. The FIX first intron (intron A) and polyadenylation sites (pA) are also indicated. The control vectors AAV9-TTR-FIX and AAV9-Palm-FIX are identical to AAV9-HS-CRM8-TTR-FIX and AAV9-HS-CRM8-Palm-FIX, respectively, except for the absence of any HS-CRM elements. The vectors (c) AAV9-HS-CRM8-TTR-FIX and (d) AAV9-HS-CRM8-TTR-FIXco were packaged in a self-complimentary AAV9. Both vectors had a small MVM (minute virus of mice) intron cloned upstream of the hFIX gene.
Figure 3.

In vivo validation of HS-CRMs. (a) Semi-high throughput HS-CRM screening in vivo after intravenous hydrodynamic liver-directed injection with pAAV-HS-CRM-TTR-FIX and pAAV-TTR-FIX plasmids at a dose of 2 µg DNA. Significant differences compared to the control without HS-CRM were indicated (t-test, *P ≤ 0.05, mean ± SD). (b) FIX expression levels after intravenous administration of AAV9-HS-CRM8-Palm-FIX and AAV9-Palm-FIX (1 × 1011 vg/mouse) (n = 5 per cohort, C57Bl/6). The difference in FIX expression levels was significant (t-test, *P ≤ 0.0001). (c) FIX expression levels after intravenous administration of AAV9-HS-CRM8-TTR-FIX and AAV9-TTR-FIX control vectors (5 × 109 vg/mouse) (n = 5 per cohort, C57Bl/6). The difference in FIX expression levels was significant (t-test, *P ≤ 0.00005). FIX levels were determined using a hFIX-specific ELISA. (d) Hepatocyte-specificity of AAV9 containing HS-CRM8. RT-PCR analysis on 20 ng total RNA from different organs of C57/Bl6 mice (n = 3) injected intravenously with AAV9-HS-CRM8-TTR-FIX vectors (3 × 1012 vg/mouse); RNA liver samples were amplified with and without RT to exclude genomic DNA amplification. Amplification of hFIX mRNA was not detectable in these control samples without RT (data not shown). (e) Corresponding qRT-PCR analysis of hFIX mRNA levels in the different organs expressed relative to hFIX mRNA levels in the liver. Expression levels (mean ± SD) relative to liver are shown. H2O, water control; MW, molecular weight marker; PBS liver, liver sample of PBS-injected control mice; RT, reverse transcription.
The potency of the HS-CRM8 element was subsequently evaluated through assessment of phenotypic correction in hemophilic FIX-deficient mice following a tail-clip injury. The treated mice survived the injury in contrast to naive controls, consistent with a significant dose-dependent reduction in blood loss (naive mice: 1,167 ± 215 µl; 5 × 109 vg/mouse: 596 ± 92 µl (P < 0.0005); 2 × 1010 vg/mouse: 495 ± 82 µl (P < 0.00005). hFIX mRNA expression was restricted to the liver (Figure 3d,e), even upon administration of extremely high vector doses (3 × 1012 vg), despite the detection of viral genomes in non-hepatic tissues (i.e., heart, spleen, muscle, etc.) (data not shown).
To further enhance hFIX expression from the HS-CRM8-TTR-FIX construct, we subsequently replaced hFIX Intron A with an MVM intron upstream of the hFIX transgene (Figure 2c) or its codon-optimized equivalent (hFIXco) (Figure 2d). Each of these changes led to a significant increase in hFIX expression (Figure 4a,b). This optimized HS-CRM8-TTR-MVM-FIXco construct was then packaged into scAAV for subsequent validation in a non-human primate model.
Figure 4.

Optimization of the HS-CRM8-TTR-hFIX construct. The (a) pAAV-HS-CRM8-TTR-MVM-hFIX or (b) pAAV-HS-CRM8-TTR-FIXIA and (a,b) pAAV-HS-CRM8-TTR-MVM-hFIXco plasmids were hydrodynamically transfected into C57BL/6 mice at doses of (a) 2 µg/mouse or (b) 0.5 µg/mouse. FIX expression was measured using a hFIX-specific ELISA (n = 4) on plasma samples collected 1 or 2 days after transfection (**P < 0.01; ***P < 0.001).
Preclinical validation of efficacy and safety in non-human primates
The scAAV9-HS-CRM8-TTR-MVM-hFIXco vector was injected into two cynomolgus macaques (OUG6 and OBEV7) at a dose of 9 × 1011 vg/kg. There was a steady increase in the circulating hFIX protein in both animals that reached a peak by week 4, corresponding to 1,718 ng/ml and 885 ng/ml for OUG6 and OBEV7, respectively (Figure 5a). These levels correspond to therapeutic FIX levels in the 20–35% range of normal FIX levels. After a few weeks, the hFIX transgene expression began to decline (Figure 5a), which could be ascribed to the induction of anti-hFIX antibodies (Figure 5c,d), often seen in non-human primates following AAV liver gene transfer,13,14 despite the use of liver-specific promoters. The AAV9 vector administration also triggered anti-AAV9 neutralizing antibodies (Figure 5b). Both animals had consistent coagulation parameters despite the occurrence of hFIX inhibitory antibodies (Supplementary Figure S3). This indicates that the anti-hFIX antibodies did not cross-react with the macaque FIX. The animals did not develop any adverse events or toxicity (Supplementary Figures S4 and S5). Human FIX mRNA expression was restricted to the liver whereas only background levels were apparent in any other organ or tissue tested (Figure 6a). This confirms the liver-specificity of the synthetic in silico designed HS-CRM8-TTR promoter, consistent with the mouse data (Figure 3d,e). The AAV9 vector resulted predominately in liver transduction though significant gene transfer was also apparent in heart, lungs, spleen, kidney and brain (Figure 6b).
Figure 5.
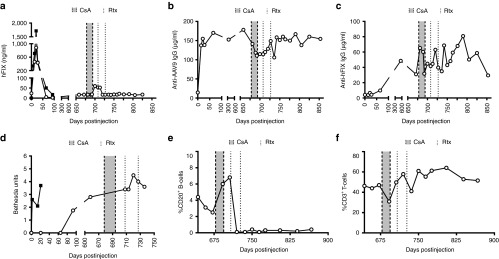
Preclinical validation of efficacy and safety in non-human primates. The scAAV9-HS-CRM8-TTR-MVM-hFIXco vector was injected in two cynomolgus macaques at a dose of 9 × 1011 vg/kg. (a) hFIX antigen expression, (b) anti-AAV9 capsid antibodies, and (c) anti-hFIX antibodies were measured by ELISA. (b) The drop in anti-AAV9 IgG during the immunosuppressive treatment corresponded to a drop in anti-AAV9 neutralization titer (from 1:1,000 to 1:316). (d) Inhibitor anti-hFIX antibodies were determined using Bethesda assays. The effect of the immunosuppressive regimen was monitored by determining the percentage of (e) CD20+ B cells and (f) CD3+ T cells. Open circles: animal OBEV7; filled squares: animal OUG6; gray area between dashed lines: cyclosporine A (CsA) administration; dotted line: rituximab (Rtx) administration.
Figure 6.

Expression biodistribution and transduction efficiency analysis in non-human primates. (a) Expression was analyzed by qRT-PCR and (b) biodistribution and transduction efficiency in different organs was determined by qPCR. Background signals from non-injected animals were subtracted from the experimental values.
One of our animals (OUG6) was euthanized due to unexpected complications unrelated to the gene therapy, which occurred after a surgical procedure to obtain a liver biopsy. We subsequently tried to eliminate the anti-hFIX inhibitors in the remaining animal (OBEV7) following, transient immunosuppression with cyclosporin (CsA) and Rituximab (Rtx). This led to a modest increase in circulating FIX levels (Figure 5a), consistent with a decrease in anti-hFIX antibodies (Figure 5c,d). This was consistent with the profound decrease in CD20+ B cells (Figure 5e), whereas the percentage of CD3+ T cells remained unaffected (Figure 5f). Unfortunately, only partial and transient immunosuppression could be achieved, since the animal refused to ingest CsA after the initial treatment. Transient immunosuppression also led to transient and partial decline in anti-AAV9 neutralizing antibodies (Figure 5b).
Discussion
In this study, we have validated a computational approach to improve the performance of current gene therapy vectors. Our data provide evidence that evolutionary conserved clusters of TFBS motifs corresponding to hepatocyte-specific CRMs could be identified in silico that are strongly associated with robust liver-specific expression in vivo. We discovered that the selected HS-CRMs identified contain a “molecular signature” composed of TFBS clusters that represent a hallmark of highly expressed genes in the liver. We demonstrated that these HS-CRMs resulted in a substantial increase in transgene expression in the liver after gene therapy, while maintaining a high degree of tissue-selectivity. In particular, a significant 10- to 100-fold increase in gene expression could be obtained depending on the type of HS-CRM and promoter used. The increased protein expression levels were consistent with an increased transcriptional activity. We subsequently validated the potential of these hepatocyte-specific HS-CRMs in a clinically relevant therapeutic setting and demonstrated robust and sustained supra-physiologic expression levels of coagulation FIX following hepatic transduction with AAV vectors. The use of these potent HS-CRMs therefore allows for the use of lower and thus safer vector doses in gene therapy clinical trials, that may in turn reduce the risk of developing T-cell–mediated immune responses against the vector antigens displayed on the transduced target cells.1,2,3,4,5 In silico vector design may therefore pave the way towards a more rational and comprehensive strategy for vector optimization with broad implications to improve the efficacy and safety of gene therapy. Indeed, we have confirmed the superiority of the HS-CRM8 expression module for liver-directed gene therapy using other transgenes (i.e., factor VIII, phenylalanine hydroxylase; ref. 15 and data not shown) Another advantage of these CRMs is their small size so that they can be readily accommodated into viral vectors in conjunction with relatively large therapeutic transgenes. The use of hyper-active FIX transgenes can further enhance vector performance.16,17
We subsequently validated the efficacy and safety of the AAV-FIX vectors containing the most potent HS-CRM8 expression module in a non-human primate model. Systemic vector administration resulted in robust therapeutic FIX levels in the 20–35% range at clinically relevant vector doses (9 × 1011 vg/kg). This is consistent with the high vector copy number in the liver biopsies. Exhaustive safety testing revealed no significant adverse effects. The performance of our vector compared favorably with that of one of the most potent AAV vectors that was used in a clinical trial in patients suffering from severe hemophila B.3 Subjects injected with 6 × 1011 vg/kg, comparable to the dose used in our primate study, resulted in ~1–3% FIX. In those subjects injected with higher vectors doses (i.e., 2 × 1012 vg/kg), ~10% FIX could be obtained.3 Previously, we had assessed the performance of the same clinical trial vector in cynomolgus macaques.13 When calibrating the vector doses, it would appear that our current vector design is about tenfold more efficient. Since a human FIX transgene was employed, it is known that this could potentially evoke neutralizing and non-neutralizing anti-hFIX antibodies in non-human primate models.13 This is consistent with our observations that anti-hFIX antibodies were detected in the recipients which accounted for the subsequent decline in FIX expression. This provided us with an opportunity to test whether we could restore FIX by transient immunosuppression using rituximab and CsA. FIX could be detected after immune-suppression, even up to nearly 2 years after gene transfer. This suggests that the FIX-transduced hepatocytes persisted long-term and that the short-term FIX expression was primarily due to a humoral immune response. Our results caution that rituximab by itself may not suffice to treat patients that develop inhibitory antibodies after gene therapy and that more complex combination therapies may be required to achieve optimal immune suppression.
In this study, we choose to employ AAV9 instead of AAV8. Previously, Gao et al. evaluated the relationship of phylogenetic relatedness of distinct AAV serotypes and serologic activity.18 Polyclonal antisera generated against AAV2 cross-reacted to a lesser extent with AAV9 than with AAV8. Conversely, polyclonal antisera generated against AAV9 cross-reacted to a lesser extent with AAV2 than when the polyclonal antisera were generated against AAV8. These experiments suggest that it may be desirable to select AAV9 over AAV8 to overcome neutralization by pre-existing anti-AAV2 antibodies in human subjects, which represents an important bottleneck in clinical trials. Moreover, we choose to explore AAV9 since the bio-distribution had not systematically been analyzed in non-human primates. Finally, we had previously conducted a side-by-side comparison between AAV8 and AAV9 and found that AAV9 transduced the liver just as efficiently as AAV8 in mice.19
The advantage of this particular computational approach used to identify the tissue-specific CRMs compared to other in silico approaches was discussed previously.12 One particularly distinctive feature is that it identifies not only the TFBS that are over-represented in liver-specific CRMs but also allows for the identification of frequently co-occurring TFBS. Consequently, this in silico analysis is more comprehensive and allows for the identification of TFBS elements that tend to cluster together in CRMs of genes that are highly expressed in a given target tissue (in casu liver). It therefore takes into account the actual context-dependent TFBS interactions from a broad genome-wide perspective, instead of just relying on the over-representation of a single TFBS element.12 Our previous studies had shown that the present computational approach was more reliable, compared to other data-mining strategies that typically rely solely on the over-representation of a given TFBS, regardless of its context. The current in silico strategy that defines the HS-CRMs has been further improved by taking into account cross-species evolutionary conservation of the TFBS clusters. This approach is more comprehensive and increases the likelihood that the superior performance of the HS-CRMs is maintained in patients undergoing gene therapy, consistent with the robust FIX levels obtained in the non-human primate model. In addition, this study goes beyond the initial description and validation of the algorithm12 and establishes a bona fide causal relationship between the presence of these evolutionary conserved TFBS clusters and high liver-specific expression. In contrast, the previously published bioinformatics approach was applied in a different context and merely established correlations between TFBS clusters and differentially expressed genes following a given physiologic stimulus. However, that study did not provide direct proof of the impact of such TFBS clusters on gene expression levels. This study addresses this caveat by direct in vivo gene transfer, hereby providing experimental in vivo validation of the bioinformatics algorithm. Though the majority of HS-CRMs resulted in a significant increase in gene expression, a few elements had only limited impact. This suggests that other unknown factors may influence gene expression, such as the spacing between a given HS-CRM and the promoter/TATA box or epigenetic factors and chromatin remodeling. This study sets the stage to further refine the in silico analysis to improve gene therapy vectors. Moreover, the most potent HS-CRM8 could be used in conjunction with other liver-specific promoters, beyond those characterized in this study.
This study has broad implications for the design of better gene therapy vectors that target different diseases. The same principle could be applied to other vectors24 or to other organs, tissues or promoters, which we have validating experimentally in heart and skeletal muscle (Rincon MY, Sarcar S, Danso-Abeam D, Keyaerts M, Matrai J, Samara-Kuko E, et al. unpublished data). This may ultimately impact not only on the clinical translation of gene therapy but also on fundamental biological and transgenic studies that rely on the use of robust tissue-specific gene expression.
Materials and Methods
Extended methods are available in the Supplementary Methods. The accession numbers of the genes from which the hepatocyte-specific CRMs (i.e., HS-CRM1 to 14) are derived are listed in Table 1.
Identification of hepatocyte-specific CRMs. To identify the hepatocyte-specific (HS)-CRMs (Figure 1 and Table 1 and Supplementary Figure S1 and Table S1), we used a step-wise in silico strategy: (i) we first identified liver-specific genes that are highly expressed in the liver based on statistical analysis of micro-array expression data of normal human tissues;20,21 (ii) we then extracted the corresponding promoter sequences up to 800 bp upstream of the transcriptional start site (TSS) from public databases (NCBI36/hg18 genome assembly); (iii) using the TRANSFAC database we subsequently mapped TFBS elements to these promoters. We then identified the tissue-specific CRM using a multidimensional scaling (MDS)/ differential distance matrix (DDM) approach, as described;12 (iv) Finally, the cross-species conservation was taken into consideration to identify highly conserved HS-CRMs.
Generation of CRM constructs. The different HS-CRMs (Table 1 and Supplementary Table S1) were synthesized by conventional oligonucleotide synthesis and cloned upstream of the liver-specific minimal transthyretin (TTR) (Figure 2a) or paralemmin (Palm) promoters (Figure 2b), driving the expression of the factor IX (FIX) mini-gene.9 Alternatively, the TTR promoter coupled to the HS-CRM8 element was used to drive wild-type FIX (Figure 2c) or its codon-optimized counterpart (Figure 2d) in the context of a self-complementary (sc) AAV backbone.22 These vectors also contained a minute virus of mice (MVM) mini-intron. Cloning details are available upon request.
AAV vector production and purification. Hepatotropic AAV9 vectors were prepared and purified, as previously described.19 AAV vectors were purified by cesium chloride gradient ultracentrifugation. Vector titers were determined by quantitative (q) PCR (see Supplementary Methods).
Animal procedures. Wild-type or hemophilic mice23 were transfected hydrodynamically with plasmids or injected intravenously with purified AAV9 vectors. hFIX antigen levels were assayed in citrated mouse plasma using a hFIX-specific enzyme-linked immunosorbent assay (ELISA). Hemophilic mice were subjected to a tail-clipping assay.7,19 The scAAV9-HS-CRM8-TTR-MVM-hFIXco vector (Figure 2d) (9 × 1011 vg/kg) was intravenously injected in cynomolgus macaques. Animals were subjected to transient immune suppression by cyclosporine A (CsA) and rituximab treatment.13 Immunological analysis, hFIX antigen, anti-AAV9 capsid IgG, anti-hFIX IgG and Bethesda assays are described in Supplementary Methods. Gene expression levels were evaluated by qRT-PCR and normalized to mRNA levels of the endogenous monkey GAPDH gene. Transduction efficiency was determined by qPCR and transgene copy numbers were determined as described previously.19
SUPPLEMENTARY MATERIAL Figure S1. Nucleotide sequence of the HS-CRM elements (HS-CRM1 to HS-CRM14). Figure S2. Hepatic hFIX mRNA levels and AAV copy numbers following intravenous injection of AAV9-FIX vectors in C57/Bl6 mice (n = 3). Figure S3. Safety analysis of scAAV9-HS-CRM8-TTR-MVM-hFIXco gene therapy in non-human primates: analysis of coagulation parameters. Figure S4. Safety analysis of scAAV9-HS-CRM8-TTR-MVM-hFIXco gene therapy in non-human primates: analysis of hematological parameters. Figure S5. Safety analysis of scAAV9-HS-CRM8-TTR-MVM-hFIXco gene therapy in non-human primates: blood chemistry. Table S1. Evolutionary conserved HS-CRMs enriched in TFBS associated with high liver-specific expression. Methods.
Acknowledgments
This work was supported by grants from the European Union Framework Programme 7 for Research and Technological Development (222878, PERSIST); Association Française contre les Myopathies; Fonds Wetenschappelijk Onderzoek – Vlaanderen (FWO); European Hematology Association (EHA); Bayer-Schering, Geconcerteerde Onderzoeksacties (GOA) – EPIGEN (VUB); Strategic Research Program “Grower” – GENEFIX (VUB); Industrieel Onderzoeksfonds (IOF) – Groups of Applied Research (GEAR) – GENECURE (VUB); Industrieel Onderzoeksfonds (IOF) – Proof of Concept grant (PoC) (VUB); Willy Gepts Fund (VUB); Stichting tegen Kanker, to M.K.C. and T.V. M.Y.R. is a recipient of a Fundacion Cardiovascular de Colombia/Colciencias Fellowship and a PhD Fellowship from the Association Française contre les Myopathies. M.D.M. and J.W. are recipients of an FWO PhD fellowship and I.P. received an IWT fellowship. N.N. is also supported by FWO. F.M. is a recipient of a Bayer Early Cancer Investigator Award and a Marie Curie PCiG12-GA-2012-333628 NoSmod grant. The authors thank Ma Ling for technical assistance. They also thank A. Srivastava for the self-complementary AAV backbone and I. Verma, L. Wang, and M. Kay for the hemophilic mice. The authors thank all the personnel of the Boisbonne Center for Gene Therapy (ONIRIS, Atlantic Gene Therapies, Nantes, France) for the handling and care of the nonhuman primates. The authors declare no conflict of interest.
Supplementary Material
Nucleotide sequence of the HS-CRM elements (HS-CRM1 to HS-CRM14).
Hepatic hFIX mRNA levels and AAV copy numbers following intravenous injection of AAV9-FIX vectors in C57/Bl6 mice (n = 3).
Safety analysis of scAAV9-HS-CRM8-TTR-MVM-hFIXco gene therapy in non-human primates: analysis of coagulation parameters.
Safety analysis of scAAV9-HS-CRM8-TTR-MVM-hFIXco gene therapy in non-human primates: analysis of hematological parameters.
Safety analysis of scAAV9-HS-CRM8-TTR-MVM-hFIXco gene therapy in non-human primates: blood chemistry.
Evolutionary conserved HS-CRMs enriched in TFBS associated with high liver-specific expression.
References
- Kay MA. State-of-the-art gene-based therapies: the road ahead. Nat Rev Genet. 2011;12:316–328. doi: 10.1038/nrg2971. [DOI] [PubMed] [Google Scholar]
- Seymour LW, Thrasher AJ. Gene therapy matures in the clinic. Nat Biotechnol. 2012;30:588–593. doi: 10.1038/nbt.2290. [DOI] [PubMed] [Google Scholar]
- Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, Rasko JE, et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med. 2007;13:419–422. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BD, Cantore A, Annoni A, Sergi LS, Lombardo A, Della Valle P, et al. A microRNA-regulated lentiviral vector mediates stable correction of hemophilia B mice. Blood. 2007;110:4144–4152. doi: 10.1182/blood-2007-03-078493. [DOI] [PubMed] [Google Scholar]
- Mátrai J, Cantore A, Bartholomae CC, Annoni A, Wang W, Acosta-Sanchez A, et al. Hepatocyte-targeted expression by integrase-defective lentiviral vectors induces antigen-specific tolerance in mice with low genotoxic risk. Hepatology. 2011;53:1696–1707. doi: 10.1002/hep.24230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Eastman EM, Schwartz RJ, Draghia-Akli R. Synthetic muscle promoters: activities exceeding naturally occurring regulatory sequences. Nat Biotechnol. 1999;17:241–245. doi: 10.1038/6981. [DOI] [PubMed] [Google Scholar]
- Miao CH, Ohashi K, Patijn GA, Meuse L, Ye X, Thompson AR, et al. Inclusion of the hepatic locus control region, an intron, and untranslated region increases and stabilizes hepatic factor IX gene expression in vivo but not in vitro. Mol Ther. 2000;1:522–532. doi: 10.1006/mthe.2000.0075. [DOI] [PubMed] [Google Scholar]
- Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444:499–502. doi: 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- Portales-Casamar E, Swanson DJ, Liu L, de Leeuw CN, Banks KG, Ho Sui SJ, et al. A regulatory toolbox of MiniPromoters to drive selective expression in the brain. Proc Natl Acad Sci USA. 2010;107:16589–16594. doi: 10.1073/pnas.1009158107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bleser P, Hooghe B, Vlieghe D, van Roy F. A distance difference matrix approach to identifying transcription factors that regulate differential gene expression. Genome Biol. 2007;8:R83. doi: 10.1186/gb-2007-8-5-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi F, Chen Y, Murphy SL, Edmonson SC, Tai A, Price SD, et al. Pharmacological modulation of humoral immunity in a nonhuman primate model of AAV gene transfer for hemophilia B. Mol Ther. 2012;20:1410–1416. doi: 10.1038/mt.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani AC, Gray JT, Ng CY, Zhou J, Spence Y, Waddington SN, et al. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood. 2006;107:2653–2661. doi: 10.1182/blood-2005-10-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viecelli HM, Harbottle RP, Wong SP, Schlegel A, Chuah MK, Vandendriessche T, et al. 2014Treatment of phenylketonuria using minicircle-based naked-DNA gene transfer to murine liver Hepatology (epub ahead of print). [DOI] [PMC free article] [PubMed]
- Cantore A, Nair N, Della Valle P, Di Matteo M, Màtrai J, Sanvito F, et al. Hyperfunctional coagulation factor IX improves the efficacy of gene therapy in hemophilic mice. Blood. 2012;120:4517–4520. doi: 10.1182/blood-2012-05-432591. [DOI] [PubMed] [Google Scholar]
- Nair N, Rincon MY, Evens H, Sarcar S, Dastidar S, Samara-Kuko E, et al. Computationally designed liver-specific transcriptional modules and hyperactive factor IX improve hepatic gene therapy. Blood. 2014;123:3195–3199. doi: 10.1182/blood-2013-10-534032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, et al. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandendriessche T, Thorrez L, Acosta-Sanchez A, Petrus I, Wang L, Ma L, et al. Efficacy and safety of adeno-associated viral vectors based on serotype 8 and 9 vs. lentiviral vectors for hemophilia B gene therapy. J Thromb Haemost. 2007;5:16–24. doi: 10.1111/j.1538-7836.2006.02220.x. [DOI] [PubMed] [Google Scholar]
- Liu X, Yu X, Zack DJ, Zhu H, Qian J. TiGER: a database for tissue-specific gene expression and regulation. BMC Bioinformatics. 2008;9:271. doi: 10.1186/1471-2105-9-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son CG, Bilke S, Davis S, Greer BT, Wei JS, Whiteford CC, et al. Database of mRNA gene expression profiles of multiple human organs. Genome Res. 2005;15:443–450. doi: 10.1101/gr.3124505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Sun J, Zhang T, Yin C, Yin F, Van Dyke T, et al. Optimization of self-complementary AAV vectors for liver-directed expression results in sustained correction of hemophilia B at low vector dose. Mol Ther. 2008;16:280–289. doi: 10.1038/sj.mt.6300355. [DOI] [PubMed] [Google Scholar]
- Wang L, Zoppè M, Hackeng TM, Griffin JH, Lee KF, Verma IM. A factor IX-deficient mouse model for hemophilia B gene therapy. Proc Natl Acad Sci USA. 1997;94:11563–11566. doi: 10.1073/pnas.94.21.11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Matteo M, Samara-Kuko E, Ward N, Waddingon S, McVey JH, Chuah MK, et al. Hyperactive PIGGYBAC transposons for sustained and robust liver-targeted gene therapy Mol Ther(in press). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Nucleotide sequence of the HS-CRM elements (HS-CRM1 to HS-CRM14).
Hepatic hFIX mRNA levels and AAV copy numbers following intravenous injection of AAV9-FIX vectors in C57/Bl6 mice (n = 3).
Safety analysis of scAAV9-HS-CRM8-TTR-MVM-hFIXco gene therapy in non-human primates: analysis of coagulation parameters.
Safety analysis of scAAV9-HS-CRM8-TTR-MVM-hFIXco gene therapy in non-human primates: analysis of hematological parameters.
Safety analysis of scAAV9-HS-CRM8-TTR-MVM-hFIXco gene therapy in non-human primates: blood chemistry.
Evolutionary conserved HS-CRMs enriched in TFBS associated with high liver-specific expression.