

ABSTRACT
Major histocompatibility complex class II (MHC-II) molecules play a central role in adaptive antiviral immunity by presenting viral peptides to CD4+ T cells. Due to their key role in adaptive immunity, many viruses, including Kaposi's sarcoma-associated herpesvirus (KSHV), have evolved multiple strategies to inhibit the MHC-II antigen presentation pathway. The expression of MHC-II, which is controlled mainly at the level of transcription, is strictly dependent upon the binding of the class II transactivator (CIITA) to the highly conserved promoters of all MHC-II genes. The recruitment of CIITA to MHC-II promoters requires its direct interactions with a preassembled MHC-II enhanceosome consisting of cyclic AMP response element-binding protein (CREB) and nuclear factor Y (NF-Y) complex and regulatory factor X (RFX) complex proteins. Here, we show that KSHV-encoded latency-associated nuclear antigen (LANA) disrupts the association of CIITA with the MHC-II enhanceosome by binding to the components of the RFX complex. Our data show that LANA is capable of binding to all three components of the RFX complex, RFX-associated protein (RFXAP), RFX5, and RFX-associated ankyrin-containing protein (RFXANK), in vivo but binds more strongly with the RFXAP component in in vitro binding assays. Levels of MHC-II proteins were significantly reduced in KSHV-infected as well as LANA-expressing B cells. Additionally, the expression of LANA in a luciferase promoter reporter assay showed reduced HLA-DRA promoter activity in a dose-dependent manner. Chromatin immunoprecipitation assays showed that LANA binds to the MHC-II promoter along with RFX proteins and that the overexpression of LANA disrupts the association of CIITA with the MHC-II promoter. These assays led to the conclusion that the interaction of LANA with RFX proteins interferes with the recruitment of CIITA to MHC-II promoters, resulting in an inhibition of MHC-II gene expression. Thus, the data presented here identify a novel mechanism used by KSHV to downregulate the expressions of MHC-II genes.
IMPORTANCE Kaposi's sarcoma-associated herpesvirus is the causative agent of multiple human malignancies. It establishes a lifelong latent infection and persists in infected cells without being detected by the host's immune surveillance system. Only a limited number of viral proteins are expressed during latency, and these proteins play a significant role in suppressing both the innate and adaptive immunities of the host. Latency-associated nuclear antigen (LANA) is one of the major proteins expressed during latent infection. Here, we show that LANA blocks MHC-II gene expression to subvert the host immune system by disrupting the MHC-II enhanceosome through binding with RFX transcription factors. Therefore, this study identifies a novel mechanism utilized by KSHV LANA to deregulate MHC-II gene expression, which is critical for CD4+ T cell responses in order to escape host immune surveillance.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) is an oncogenic gammaherpesvirus that causes several malignancies, such as Kaposi's sarcoma (KS), primary effusion lymphomas (PELs), and multicentric Castleman's disease (MCD), in immunocompromised individuals (1, 2). The life cycle of KSHV consists of a predominant latent phase marked by restricted gene expression and a transient lytic replication phase characterized by the production of functional virions. KSHV maintains a lifelong persistent infection in susceptible hosts after primary infection (3, 4). One of the main factors contributing to the successful lifelong persistence of KSHV is its astounding ability to hide from host immune surveillance. During the course of evolution, KSHV has evolved multiple mechanisms to evade and modulate nearly all aspects of both the innate and adaptive immunities of infected hosts (5–7).
Latency-associated nuclear antigen (LANA or LANA-1) is the most abundantly expressed protein in all KSHV-infected cells (8–10). LANA is a large multifunctional protein that plays diverse roles in maintaining successful KSHV latency, such as the maintenance of viral episomes, the transcriptional regulation of many viral and cellular genes, and the progression of the cell cycle (1, 11, 12). Since latency is the immunologically silent stage of the KSHV life cycle and since LANA is the major latent protein, it has been speculated that LANA plays active roles in the modulation of the host immune response. Indeed, LANA has been shown to inhibit many aspects of the host's innate and adaptive immune pathways, including interference with neutrophil recruitment and tumor necrosis factor alpha (TNF-α) signaling (13), interference with interferon (IFN) signaling (14), and inhibition of major histocompatibility complex class I (MHC-I) peptide presentation (15, 16). Recently, LANA was also shown to inhibit the MHC-II antigen presentation pathway by inhibiting the transcription of the class II transactivator (CIITA) (17).
The effectiveness of adaptive immunity, which is a critical arm of the antiviral host defense, relies primarily on the activation of CD4+ T cells. Activation of CD4+ T cells seems to be particularly important for anti-KSHV immunity (18, 19). MHC-II molecules play a central role in the activation of CD4+ T cells by presenting antigenic peptides to these cells (20, 21). Since peptide presentation in conjunction with MHC-II molecules is indispensable for the activation of CD4+ T cells, downregulation of MHC-II molecules is a strategy frequently employed by many viruses (22). Reports from the past couple of years established that KSHV has the ability to downregulate MHC-II molecules (17, 23–25). Since the ability of KSHV to persist in infected hosts critically depends on being invisible to CD4+ T cells, KSHV most likely uses multiple simultaneous strategies to efficiently block the MHC-II antigen presentation pathway.
MHC-II molecules are highly polymorphic cell surface glycoproteins consisting of an alpha chain and a beta chain, which are constitutively expressed in professional antigen-presenting cells (26, 27). In humans, there are three “classical” isotypes, HLA-DR, HLA-DP, and HLA-DQ, that are present on the cell surface. The MHC-II gene family also includes the “nonclassical” cytoplasmic molecules HLA-DM and HLA-DO and the invariant chain (Ii) involved in peptide loading. Expressions of all the members of the MHC-II gene family are highly coordinated and regulated predominantly at the level of transcription (28–30). The promoters of all MHC-II genes contain highly conserved cis-acting DNA sequences consisting of S-X-X2-Y boxes, which are bound by various nuclear proteins. These proteins include regulatory factor X (RFX), nuclear factor Y (NF-Y), and cyclic AMP (cAMP) response element-binding protein (CREB) (29, 31). The RFX complex (RFX-Cx) is a heterotrimeric complex composed of RFX5, RFX-associated protein (RFXAP), and RFXANK that binds to the S and X boxes, and NF-Y and CREB bind to the Y and X2 boxes, respectively (32–39). The coordinated binding of these protein complexes to the conserved sequences of the MHC-II promoter makes the functional MHC-II enhanceosome (reviewed in references 22 and 40). Among these proteins, the RFX complex is the core DNA-binding component of the MHC-II enhanceosome. It facilitates the assembly of other enhanceosome proteins onto the MHC-II promoters (30, 41–43). All three components of the RFX complex are indispensable for constitutive and induced expression of MHC-II genes. Once assembled on MHC-II promoters, the enhanceosome complex recruits a transcriptional coactivator, CIITA, a master regulator of MHC-II expression. All the components of the MHC-II enhanceosome except CIITA are constitutively expressed in almost every cell type in humans (44). However, the expression of CIITA is constitutive only in antigen-presenting cells but can be induced in most other cell types in response to IFN-γ (44). Binding of CIITA to the MHC-II promoters is absolutely critical for the transcription of MHC-II genes. However, CIITA does not possess any DNA-binding domain to bind directly to the MHC-II promoter. Instead, it is recruited through the proteins of the enhanceosome complex, especially through a direct association with RFX5. Once bound to the MHC-II enhanceosome, CIITA orchestrates the transcription of MHC-II genes by recruiting the remaining transcriptional machinery to the promoters (40, 45–47).
So far, two KHSV latent proteins, viral interferon regulatory factor 3 (vIRF3) and LANA, have been reported to downregulate the MHC-II antigen presentation pathway (17, 18, 23, 25). A previous report by Cai et al. demonstrated that LANA downregulates MHC-II through the inhibition of CIITA transcription by suppressing PIII and PIV promoter activities (17). In this study, we identified an additional mechanism used by KSHV LANA to inhibit the expression of MHC-II genes. We show that LANA interferes with the assembly of a functional MHC-II enhanceosome by reducing the binding of CIITA to the HLA-DRA promoter. Immunoprecipitation (IP) experiments showed that LANA binds to all the three components of the RFX complex in vivo but preferentially binds to RFXAP in in vitro assays. Similarly, chromatin immunoprecipitation (ChIP) assays determined that LANA associates with chromatin of the HLA-DRA promoter. Electrophoretic mobility shift assays (EMSAs) further supported the association of LANA with the HLA-DRA promoter through its interactions with the components of the RFX complex. Additionally, the expression of LANA reduced the activity of the HLA-DRA core promoter in a dose-dependent manner even when CIITA was expressed through a constitutive cytomegalovirus (CMV) promoter, confirming that the presence of LANA prevents the binding of CIITA to the HLA-DRA promoter. The disruption of CIITA binding to the enhanceosome was further supported by a chromatin immunoprecipitation assay that showed a decreased association of CIITA at the HLA-DRA promoter in the presence of LANA. Taken together, these data identify a novel mechanism used by KSHV LANA to deregulate the expression of MHC-II genes.
MATERIALS AND METHODS
Cell culture.
The KSHV- and Epstein-Barr virus (EBV)-negative Burkitt lymphoma cell line BJAB, the KSHV-negative monocytic leukemia cell line THP-1, and the KSHV-positive but EBV-negative PEL cell lines BCBL-1 and BC-3 were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and penicillin-streptomycin (5 U/ml and 5 g/ml, respectively). The human embryonic kidney (HEK) cell lines HEK293T and HEK293L were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and penicillin-streptomycin (5 U/ml and 5 g/ml, respectively). All cell lines were grown at 37°C in a humidified environment supplemented with 5% CO2.
Antibodies.
The following commercial antibodies were used for this study: rat anti-LANA (Advanced Biotechnologies, Inc.), mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (U.S. Biological), mouse anti-Flag M2 (Sigma-Aldrich), mouse anti-Myc 9E10 (Sigma-Aldrich), rabbit polyclonal anti-MHC-II (Abcam), rabbit polyclonal anti-RFXANK (Abcam), mouse monoclonal anti-RFX5 (Santa Cruz Biotechnology), and mouse monoclonal anti-RFXAP (Santa Cruz Biotechnology). Mouse monoclonal anti-LANA antibody was a gift from Ke Lan (Institute Pasteur of Shanghai, Shanghai, China).
Plasmids.
To construct mammalian expression plasmids, RFXANK was PCR amplified from a Myc-DDK-tagged RFXANK clone (catalog number RC220744; Origene USA) and cloned into the Flag-tagged pA3F or Myc-tagged pA3M vector at BamHI and EcoRI restriction sites. RFXAP was PCR amplified from BJAB cell cDNA for cloning into vectors pA3F and pA3M at BamHI and EcoRI sites. RFX5 was PCR amplified from the RFX5-green fluorescent protein (GFP) construct (received from Jeremy Boss, Emory School of Medicine) and was subcloned into vector pA3M or pA3F at HindIII and EcoRV restriction sites. All the truncation mutants of RFXAP were PCR amplified from full-length RFXAP for subcloning into the pA3M or pA3F vector at BamHI and EcoRI restriction sites. CIITA was similarly PCR amplified from the CIITA plasmid (a gift from Jenny Ting, UNC) for cloning into vector pA3F using BglII and EcoRI restriction sites. In-frame positions of all the clones with the respective tags were confirmed by DNA sequencing performed at the Nevada Genomics Center, University of Nevada, Reno, NV. The HLA-DRA–Luc plasmid was a generous gift from Jenny Ting (UNC). Myc-tagged full-length LANA (pA3M-LANA), Flag-tagged full-length LANA (pA3F-LANA); deletion mutants containing the LANA N-terminal domain (LANA-N) (amino acids [aa] 1 to 340) and the LANA C-terminal domain (LANA-C) (aa 940 to 1162); GFP-nuclear localization signal (NLS)-Myc; GFP–LANA-N–Myc (aa 1 to 340) and its truncation mutants GFP–LANA-N250–Myc (aa 1 to 250), GFP–LANA-N150–Myc (aa 1 to 150), and GFP–LANA-N32–Myc (aa 1 to 32); and the lentiviral construct pLVX-LANA-YFP-Flag and its control construct pLVX-YFP-Flag were described previously (48). Short hairpin RNA (shRNA)-expressing plasmids, control shRNA (shControl), and LANA shRNA (shLANA) were described previously (49). LANA-N–glutathione S-transferase (GST) and LANA-C–GST were also described previously (50).
DNA transfections.
HEK293T cells were seeded at a density of 5 × 106 cells per 100-mm dish the day before transfection. Plasmids of interest were transfected by using polyethylenimine (PEI) (Polysciences, Inc.). Approximately 60 μg of total plasmid DNA was mixed with 150 mM NaCl and 70 μl of PEI solution (1 mg/ml, pH 7.0). The resulting transfection mix was incubated at room temperature for 15 min and then added dropwise onto the cells. For reporter assays, transfections were done by using Metafectene (Biontex Laboratories, GmbH) according to the manufacturer's protocol. B cells were transfected by electroporation as described previously (51).
Coimmunoprecipitation assays and Western blot analysis.
Approximately 20 million cells expressing the proteins of interest were washed with PBS (phosphate-buffered saline) (10 mM NaPO4, 137 mM NaCl, 2.5 mM KCl [pH 7.5]) and lysed in radioimmunoprecipitation assay (RIPA) cell lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, and 1% NP-40) supplemented with protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 10 μg/ml pepstatin, 10 μg/ml leupeptin, and 10 μg/ml aprotinin). Cellular lysates were then sonicated to shear DNA and centrifuged at 12,000 rpm for 10 min at 4°C to remove cellular debris. The supernatants were precleared with protein A- and G-conjugated Sepharose beads (GE Healthcare) for 30 min at 4°C and gently rotated overnight at 4°C with specific antibodies. The resulting immunocomplexes were captured by the addition of protein A- and G-conjugated Sepharose beads and rotation of the lysates for 2 h at 4°C. The immunocomplexes were collected by centrifugation at 2,000 rpm for 2 min at 4°C. The beads were washed three times with ice-cold RIPA buffer supplemented with protease inhibitors and boiled in 50 μl of SDS-PAGE sample loading buffer for 5 min. The immunoprecipitated proteins and the respective total cell lysates were resolved on a 9% SDS-polyacrylamide gel and transferred onto 0.45-μm nitrocellulose membranes (GE Healthcare) at 100 V for 75 min. The blots were blocked with 5% nonfat milk in TBST buffer (10 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.05% Tween 20) and washed three times with TBST buffer before incubation overnight at 4°C with specific primary antibodies. The blots were washed three times with TBST, followed by incubation with appropriate secondary antibodies conjugated with Alexa Fluor 680 or Alexa Fluor 800 (Molecular Probes, Carlsbad, CA) secondary antibodies at 1:10,000 dilutions. The membranes were scanned with the Odyssey scanner (Li-Cor, Lincoln, NE).
Yeast two-hybrid assay.
LANA-N (aa 1 to 340) was cloned in frame with the DNA-binding domain of GAL4 (GAL4-DBD) of a pAS1 vector (Clontech, Mountain View, CA) for use as bait. This plasmid was transformed into Saccharomyces cerevisiae strain Y190 (Clontech, Mountain View, CA) and selected on DOBA-Trp dropout medium (Clontech, Mountain View, CA). The expression level of LANA-N with a DBD fusion protein was determined by using an anti-GAL4 DBD antibody (Santa Cruz Biotechnology, Inc.) for Western blot analysis. Yeast strain Y190 containing LANA-N–DBD was transformed with a cDNA library (generous gift from Erle S. Robertson, University of Pennsylvania) cloned into the pACT vector and selected on DOBA-Trp, His, Leu dropout medium (Clontech, Mountain View, CA) in the presence of 30 mM 3-aminotriazole, as described previously (52). Selected colonies were subjected to plasmid isolation and were further electroporated into E. coli DH5α cells for their amplification. Plasmid DNAs from individual bacterial colonies were isolated and subjected to sequencing at the Nevada Genomics Center after determination of their restriction patterns. Identities of the cDNA clones were determined by matching sequences with the database sequence (NCBI).
Immunofluorescence assay.
KSHV-positive BCBL-1 and BC-3 cells were washed with PBS before they were spread onto coverslips. The cells were allowed to air dry for 10 min and fixed with 4% paraformaldehyde for 10 min at room temperature, followed by permeabilization with 0.2% Triton X-100 in PBS for 10 min at room temperature. Cells were blocked with PBS containing 0.4% fish skin gelatin and 0.05% Triton X-100 for 30 min at room temperature. The cells were then incubated with specific primary antibodies for 1 h at room temperature and washed with PBS before incubating them with Alexa Fluor-conjugated secondary antibodies (Molecular Probes) for 45 min at room temperature. The cells were washed three times with PBS to remove nonspecifically bound antibodies before staining with the nuclear stain To-Pro3 (Molecular Probes). Images were captured by using a confocal laser scanning microscope (Carl Zeiss, Inc.).
ChIP assay.
Chromatin immunoprecipitation was performed as described previously (53). Briefly, 15 million to 20 million cells were fixed with a final concentration of 1% formaldehyde for 10 min at room temperature, followed by the addition of glycine at a final concentration of 125 mM for 5 min to block cross-linking. The cells were rinsed three times with ice-cold PBS and lysed in cell lysis buffer [5 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) (pH 8.0), 85 mM KCl, and 0.5 mM NP-40] supplemented with protease inhibitors for 10 min on ice. The nuclei were enriched by low-speed centrifugation and resuspended in protease inhibitor-supplemented nuclear lysis buffer containing 50 mM Tris-HCl (pH 8.1), 10 mM EDTA, and 1% SDS. Chromatin was sonicated to an average length of 500 to 800 bp and centrifuged for 10 min at 13,000 rpm to remove the cell debris. The resulting supernatant was diluted 5-fold with ChIP dilution buffer containing 16.7 mM Tris-HCl (pH 8.1), 167 mM NaCl, 1.2 mM EDTA, 0.01% SDS, 1.1% Triton X-100, and protease inhibitors. The diluted chromatin was precleared with protein A- and G-conjugated Sepharose beads pretreated with 1 mg/ml bovine serum albumin (BSA) and 1 mg/ml sheared salmon sperm DNA for 30 min at 4°C with rotation, followed by incubation overnight with either control or specific antibodies at 4°C with rotation. Immune complexes were collected by incubation with protein A- and G-conjugated Sepharose beads for 1 to 2 h at 4°C. The beads were collected and washed subsequently with a low-salt buffer (0.1% SDS, 1.0% Triton X-100, 2 mM EDTA, 20 mM Tris [pH 8.1], 150 mM NaCl), a high-salt buffer (0.1% SDS, 1.0% Triton X-100, 2 mM EDTA, 20 mM Tris [pH 8.1], 500 mM NaCl), and an LiCl wash buffer (0.25 M LiCl, 1.0% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris [pH 8.0]). The beads were then washed twice with Tris-EDTA buffer, and chromatin was eluted by using an elution buffer (1% SDS, 0.1 M NaHCO3) and reverse cross-linked by the addition of 0.3 M NaCl at 65°C overnight. Eluted DNA was precipitated, treated with RNase and proteinase K at 45°C for 2 h, and purified by using a Min-Elute PCR purification kit (Qiagen, USA). The purified DNA was analyzed by PCR for the presence of the HLA-DRA promoter region.
Primers used for HLA-DRA promoter amplification were described previously (forward primer 5′-GTTGTCCTGTTTGTTTAAGAAC-3′ and reverse primer 5′-GCTCTTTTGGGAGTCAG-3′) (44). KSHV terminal repeat (TR) primers were also described previously (forward primer 5′-GGGGGACCCCGGGCAGCGAG-3′ and reverse primer 5′-GGCTCCCCCAAACAGGCTCA-3′) (51).
Dual-luciferase reporter assay.
A total of 5 × 105 HEK293L cells were seeded into 6-well plates the day before transfection. The cells were cotransfected with 0.5 μg of the DRA-Luc reporter plasmid, 0.2 μg of the CIITA-Flag plasmid, and increasing amounts of the LANA expression vector (1, 2, and 3 μg). The pA3F empty vector was used as filler DNA. Transfection efficiencies were monitored by transfection of the GFP-containing vector pEGFP. The Renilla luciferase-expressing plasmid (pRRLSV40) was transfected at 40 ng/well for data normalization. All the transfections for reporter assays were done by using Metafectene (Biontex Laboratories, GmbH) according to the manufacturer's protocol. At 36 h posttransfection, cells were lysed in cell lysis buffer (Promega), and 50 μl of the cell lysate was used for the reporter assay by using a dual-luciferase reporter assay kit (Promega, USA). Relative luciferase units (RLU) were calculated after normalization of the HLA-DRA luciferase readings with Renilla luciferase to account for the transfection efficiencies. A portion of the cell lysates was used for Western blotting to detect LANA, CIITA, and GAPDH. All experiments were repeated multiple times, and the data shown are means of data from three independent experiments.
In vitro translation and GST pulldown assay.
For the GST pulldown assays, GST fusion proteins were expressed in E. coli BL21 cells. Briefly, the cells were induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 4 h at 37°C, and the GST fusion proteins were extracted from the bacterial cell lysates by using glutathione-Sepharose beads (GE Healthcare, USA). Components of the RFX complex and its truncation mutants RFXAPΔ1-70, RFXAPΔ1-150, and RFXAPΔ1-240 were translated in vitro by using a TNT T7 Quick coupled transcription-translation system (Promega, Madison, WI). Nearly 3 μg of expression plasmids was translated per 50-μl reaction mixture containing 1 mM (32 μCi) [35S]methionine, according to the manufacturer's protocol. The translated proteins were rotated overnight at 4°C with either control GST, LANA-N–GST, or LANA-C–GST in NETN binding buffer (0.1% NP-40, 20 mM Tris, 1 mM EDTA, and 100 mM NaCl) supplemented with protease inhibitors. Control GST, LANA-N–GST, and LANA-C–GST proteins were described previously (54). The next day, GST-bound proteins were gently washed three times in ice-cold NETN binding buffer supplemented with protease inhibitors, and the precipitated protein complexes were resolved on SDS-PAGE gels and detected by autoradiography.
Electrophoretic mobility shift assay.
Electrophoretic mobility shift assays (EMSAs) were performed by using a double-stranded oligonucleotide containing the HLA-DRA X-box sequence as a probe. This sequence is known to bind to the RFX complex in gel shift assays (55). The probe was end labeled with α-32P and purified on a GE Illustra ProbeQuant G-50 microcolumn (GE Healthcare, USA). RFX complex proteins and LANA-N (aa 1 to 340) proteins were translated in vitro by using a TNT Quick coupled transcription-translation rabbit reticulocyte lysate system (Promega, USA). A 40-μl DNA-protein binding assay mixture contained 12 mM HEPES (pH 7.9), 12% glycerol, 60 mM KCl, 5 mM MgCl2, 0.12 mM EDTA, 0.3 mM dithiothreitol (DTT), 0.1 μg poly(dI/dC), and 0.05 μg of denatured salmon sperm DNA as a carrier (55). In vitro-translated proteins were preincubated on ice for 10 min before the addition of 1 μl of end-labeled DNA probe. The reaction mixture was incubated on ice for 30 min, and the bound complexes were resolved on 5% nondenaturing polyacrylamide gels in 0.5× TBE buffer (0.045 M Tris-borate [pH 8.2], 1.0 mM EDTA). The gel was run at 4°C at 100 V for 20 h. The signals were detected by autoradiography.
Quantitative real-time PCR (qRT-PCR) assays.
For quantitative reverse transcriptase PCR assays, total mRNA from the cells was extracted by using an Illustra RNAspin minikit (GE Healthcare) according to the manufacturer's instructions, and cDNA was made by using a High Capacity cDNA reverse transcription kit (Applied Biosystems, USA). Each PCR mixture consisted of 10 μl of 2× PCR master mix (Applied Biosystems, USA), 1 μM each forward and reverse primers, and 2 μl of cDNA. The cDNA was amplified on an ABI StepOne Plus real-time PCR machine (Applied Biosystems, USA), and relative gene copy numbers or transcript numbers were calculated by the ΔΔCT method. Each experiment included duplicate samples, and the data shown represent the means of data from three independent experiments. P values were calculated by two-tailed t tests using GraphPad (Prism 6) software. The primer sequences used for the amplification of different HLA genes were taken from a previous report and are listed in Table 1 (23).
TABLE 1.
Primers used in real-time PCR assays
| Gene | Forward primer | Reverse primer |
|---|---|---|
| β-Actin | 5′-ACAATGTGGCCGAGGACTTTGA-3′ | 5′-TGTGTGGACTTGGGAGAGGACT-3′ |
| LANA | 5′-TTGCCTATACCAGGAAGTCCCACA-3′ | 5′-GGAGGAAGACGTGGTTACGGG-3′ |
| HLA-DRA | 5′-TTTGAGGCTCAAGGTGCATTG-3′ | 5′-TGGAGGTACATTGGTGATCGG-3′ |
| HLA-DRB | 5′-CCGAGTACTGGAACAGCCAGAA-3′ | 5′-TGCACTGTGAAGCTCTCACCAA-3′ |
| HLA-DPA | 5′GCCCTGAAGACAGAATGTTCCA-3′ | 5′-GCGGCATAAGTTGACACATGG-3′ |
| HLA-DPB | 5′-ACAGTCTGATTCTGCCCGGAGT-3′ | 5′-CTTGCTCCTCCTGTGCATGAAG-3′ |
| HLA-DQA | 5′-ATCATCCAAGGCCTGCGTT-3′ | 5′-TCTTCTGCTCCTGTAGATGGCG-3′ |
| HLA-DQB | 5′-GCAGAGACTCTCCCGAGGATTT-3′ | 5′-CGCACGATCTCTTCTCGGTTAT-3′ |
RESULTS
LANA interacts with the components of the RFX complex.
Since LANA is a major latent protein, is expressed ubiquitously in all KSHV-infected cells, and modulates various cellular pathways, we sought to identify LANA-interacting proteins. Our yeast two-hybrid screen with the amino-terminal domain of LANA fused to the GAL4-DNA-binding domain (as bait) identified a number of cellular proteins, which are listed in Table 2. The clones of these identified proteins were detected at least twice in the screen. With the aim of determining the role of LANA in immune modulation, we focused further studies on RFXAP, a component of the RFX complex (RFX-Cx) critical for the transcription of HLA genes. RFX complex proteins are absolutely essential for the expression of MHC-II molecules. In order to determine whether RFX complex proteins interacted with LANA in KSHV-infected cells, we performed coimmunoprecipitation assays with the KSHV-infected primary effusion lymphoma (PEL) cell lines BCBL-1 and BC-3. The results of these coimmunoprecipitation experiments showed that all three components of the RFX complex, RFX5, RFXAP, and RFXANK, coimmunoprecipitated with LANA in KSHV-positive BCBL-1 and BC-3 PEL cells (Fig. 1A, lanes 3 and 6). The lack of RFX protein precipitation with control antibody (CoAb) confirmed the specificity of the LANA association with these proteins (Fig. 1A, lanes 2 and 5). In order to further validate the interactions between LANA and the components of the RFX complex, we examined the colocalization of LANA with RFX proteins in BC-3 and BCBL-1 cells by an immunofluorescence assay. As expected, LANA showed typical punctate staining in the nucleus (Fig. 1B, red). The components of the RFX complex, RFX5, RFXAP, and RFXANK, showed a distinct staining pattern in the nucleus (Fig. 1B, green). As shown in the merged images in Fig. 1B, all three proteins of the RFX complex showed various degrees of colocalization with LANA in the nuclear compartments of both PEL cell lines BC-3 and BCBL-1 (Fig. 1B, yellow dots), suggesting that some fraction of LANA is in the vicinity of the RFX proteins in the nucleus of KSHV-infected cells. To assess whether KSHV/LANA altered the normal localization patterns of RFX proteins, we examined the staining patterns of RFX proteins in KSHV-negative BJAB cells. Not surprisingly, BJAB cells also showed distinct staining patterns for RFX proteins, which were very similar to the patterns seen in KSHV-positive PEL cells (Fig. 1B). LANA was not detected in these BJAB cells, as expected. Nuclei of these PELs and BJAB cells were stained with To-Pro3, and the integrity of these cells was confirmed by differential interference contrast (DIC) microscopy (Fig. 1B).
TABLE 2.
LANA-interacting proteins identified by the yeast two-hybrid assay
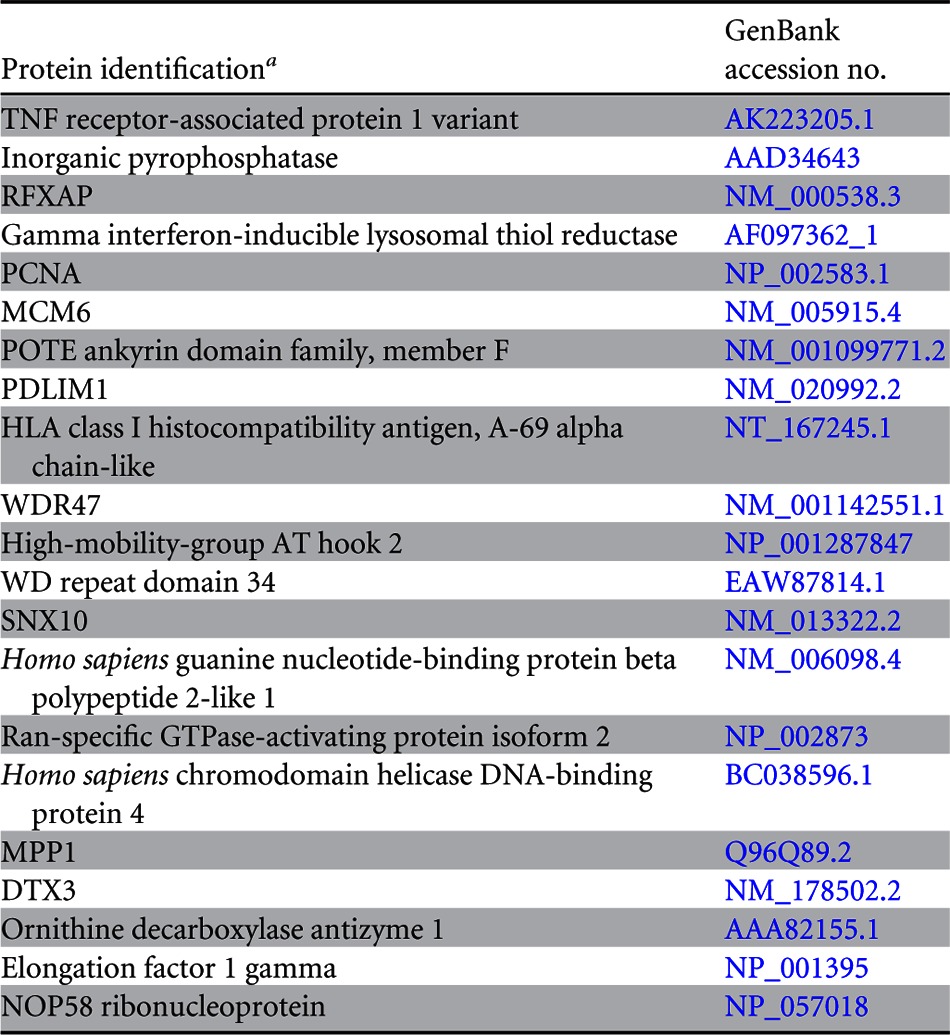
PCNA, proliferating cell nuclear antigen; MCM6, minichromosome maintenance complex component 6; PDLIM1, PDZ and LIM domain 1; WDR47, WD repeat domain 47; SNX10, Homo sapiens sorting nexin 10; MPP1, M-phase phosphoprotein 1; DTX3, Homo sapiens deltex homolog 3 (Drosophila).
FIG 1.
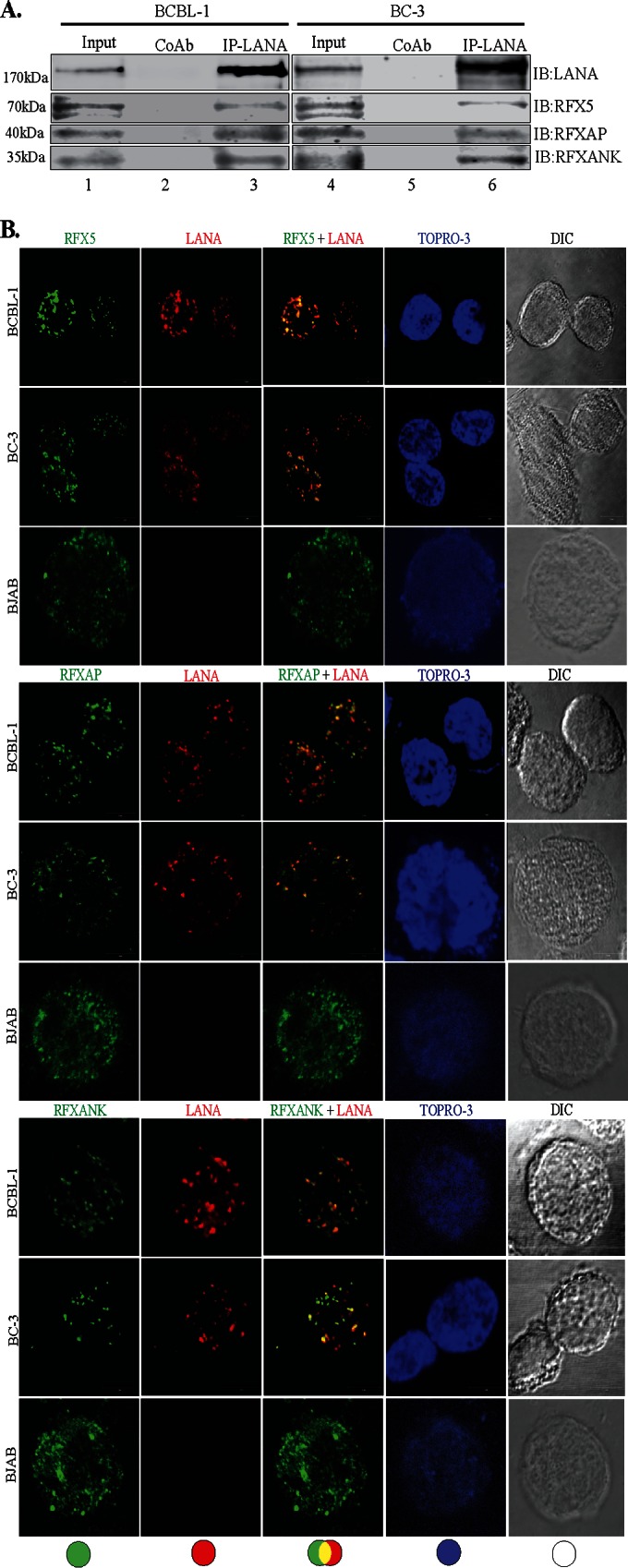

LANA interacts and colocalizes with the components of the RFX complex. (A) Coimmunoprecipitation of LANA with the components of the RFX complex, RFX5, RFXAP, and RFXANK, in KSHV-positive BCBL-1 and BC-3 cells. LANA was immunoprecipitated (IP) by using mouse anti-LANA antibody from precleared cellular lysates of nearly 20 million BCBL-1 or BC-3 cells. The coimmunoprecipitated proteins were analyzed by SDS-PAGE followed by immunodetection using antibodies specific for RFX5, RFXAP, or RFXANK. An isogenic antibody, mouse IgG (CoAb), was used as a control for immunoprecipitation in this assay. IB, immunoblotting. (B) Colocalization of LANA with individual components of the RFX complex, RFX5, RFXAP, and RFXANK. LANA was localized by using rat anti-LANA antibody, and the RFX proteins were detected with specific antibodies in BCBL-1, BC-3, and BJAB cells. Secondary antibodies for LANA and the RFX complex were Alexa Fluor 594 (red) and Alexa Fluor 488 (green), respectively. Nuclei were stained with To-Pro3 (blue). LANA colocalized with RFX5, RFXAP, and RFXANK in the nuclei of BCBL-1 and BC-3 cells as punctate dots. Staining of RFX5, RFXAP, and RFXANK in KSHV-negative BJAB cells showed distinct staining. DIC images were captured to show cell morphology. (C) Triple-immunofluorescence staining showing colocalization of RFXAP and RFX5 with LANA in BCBL-1, BC-3, and BJAB cells. LANA was stained with rat anti-LANA, RFXAP was stained with rabbit anti-RFXAP, and RFX5 was stained with mouse anti-RFX5. Host-matched secondary antibodies were used for their localization. The merged panels show the colocalization of all three proteins at certain foci in KSHV-positive cells.
In an attempt to determine whether all the components of the RFX complex were in the same nuclear compartment as LANA, we determined the colocalization of LANA with two RFX proteins (RFXAP and RFX5) in a triple-immunofluorescence assay. RFXAP and LANA in KSHV-positive BCBL-1 and BC-3 PEL cells (Fig. 1C, red) showed colocalization (based on the number of yellow dots from multiple cells) similar to that of RFX5 and LANA (Fig. 1C, sky blue dots). This finding suggested that LANA binds to the components of the RFX complex in KSHV-infected cells. BJAB cells showed perfect colocalization of RFXAP (Fig. 1C, red) and RFX5 (blue), which was also seen in KSHV-positive PEL cells (Fig. 1C, merged images), confirming that these RFX proteins are in the same nuclear compartments. We expect RFXANK to be in the same loci, but we were unable to perform colocalization experiments for all three RFX proteins with LANA because of a lack of host-specific antibody for all three RFX proteins.
LANA associates with the RFX complex independent of other viral components.
Next, we wanted to determine whether LANA alone is sufficient for its association with the components of the RFX complex or whether it requires any other viral latent proteins to associate with the components of the RFX complex. To this end, we performed coimmunoprecipitation assays of individual RFX proteins with LANA in an overexpression system in HEK293T cells. Lysates from cells cotransfected with plasmids expressing either control Flag or LANA-Flag and Myc-tagged RFX5, RFXAP, or RFXANK were subjected to immunoprecipitation with anti-Flag antibody. Detection of coprecipitating RFX proteins showed efficient binding of all three proteins of the RFX complex to LANA (Fig. 2A to C). The lack of coprecipitating RFX proteins in the control Flag lane confirmed the specificity of the assay. These results demonstrate that the presence of LANA is sufficient for association with the components of the RFX complex.
FIG 2.

LANA does not require other KSHV factors to associate with the components of the RFX complex. (A to C) Assays of protein-protein interactions of LANA with all three components of the RFX complex were performed with HEK293T cells by transiently cotransfecting cells with Flag-tagged wild-type LANA and Myc-tagged RFX5 (A), Myc-tagged RFXANK (B), or Myc-tagged RFXAP (C). The pA3F empty vector was used as a control. The cells were lysed 48 h after transfection, and LANA was immunoprecipitated by using anti-Flag antibody, followed by immunodetection with anti-Flag and anti-Myc antibodies. Overexpressed LANA efficiently coimmunoprecipitated RFX5, RFXAP, and RFXANK. (D) In vitro-translated full-length LANA-Flag protein was combined with in vitro-translated RFX5-Myc, RFXAP-Myc, and RFXANK-Myc and rotated overnight at 4°C after preclearing with protein A and G beads. LANA was immunoprecipitated with anti-Flag antibody, and the bound complexes were resolved on SDS-PAGE gels, followed by autoradiography to detect the coimmunoprecipitated proteins. Relative binding values (shown in parentheses) of RFX components were determined by taking the respective inputs as 1. (E) Schematic of LANA domains fused with GST used in pulldown assays. (F) GST pulldown assay showing binding of amino- and carboxy-terminal domains of LANA with the components of the RFX complex. Full-length in vitro-translated RFX5, RFXAP, and RFXANK proteins were rotated with either GST alone (Ctrl), LANA-N–GST, or LANA-C–GST after preclearing with GST beads. The bound proteins were resolved on SDS-PAGE gels and visualized by autoradiography. Expressions of GST and the GST fusion proteins were confirmed by SDS-PAGE and Coomassie staining (right).
Since all three components of the RFX complex, RFX5, RFXAP, and RFXANK, associated with LANA in PEL cell lines as well in the overexpression systems, we wanted to determine whether any of these three RFX proteins of the trimeric complex is the main interacting partner of LANA. To answer this, we performed an in vitro-binding assay by using in vitro-translated and [35S]methionine-labeled proteins. The sizes of these in vitro-translated proteins were confirmed by Western blotting with a specific antibody. The radiolabeled proteins of the RFX complex were incubated with in vitro-translated Flag-LANA of the empty vector for immunoprecipitation with anti-Flag antibody. The proteins were resolved on SDS-PAGE gels, and monitoring by autoradiography detected coimmunoprecipitating RFX proteins, which were also confirmed by specific antibodies. Consistent with the results of our endogenous and expressed protein immunoprecipitation assays, all three components of the RFX complex coimmunoprecipitated with LANA (Fig. 2D). Although all three components of RFX bound with LANA, the level of RFXAP binding was higher when normalized to the respective inputs. The binding efficiencies calculated by taking the input as 1 are presented in parentheses in Fig. 2D (with RFXAP at 0.81). These results again confirmed that LANA associates with the components of the RFX complex independent of other KSHV components. To validate these results by another experimental approach, we performed glutathione S-transferase (GST) fusion protein pulldown experiments. In this assay, in vitro-translated and [35S]methionine-labeled RFX5, RFXAP, or RFXANK was individually incubated with GST-fused LANA-N (aa 1 to 340), LANA-C (aa 940 to 1162), or control GST (Fig. 2E). The GST pulldown assay showed that while LANA-N–GST associated with RFXAP very strongly, it also showed weaker interactions with the other RFX components, RFX5 and RFXANK (Fig. 2F). Interestingly, LANA-C–GST showed a very weak interaction with RFXAP (Fig. 2F). The lack of any binding with control GST confirmed the specificity of the binding assay (Fig. 2F). A representative image of a Coomassie-stained gel for GST fusion proteins is shown in Fig. 2F. The binding of RFXAP to the N terminus of LANA was consistent with our yeast two-hybrid data, where the N terminus of LANA (bait) identified RFXAP as a potential LANA-interacting protein (Table 2). Since RFXAP was identified in the yeast two-hybrid assay, and it also bound most strongly to LANA in the GST pulldown assay, we focused the domain-mapping experiments on the RFXAP component of the RFX complex.
The LANA region spanning aa 1 to 150 interacts with RFXAP.
To further ensure that the amino terminus of LANA (aa 1 to 340) is responsible for binding to RFXAP, a coimmunoprecipitation experiment was performed with HEK293T cells, with RFXAP being transiently coexpressed with either the vector control, full-length LANA, LANA-N (aa 1 to 340), or LANA-C (aa 940 to 1162). Immune detection with anti-Myc antibody for RFXAP showed coimmunoprecipitation of RFXAP with both full-length LANA as well as the N terminus of LANA (aa 1 to 340) but not with LANA-C (aa 940 to 1162) (Fig. 3A). Immunoprecipitated LANA and its truncation mutants are indicated with an arrow in Fig. 3A. In line with yeast two-hybrid assay data and the GST pulldown results, this coimmunoprecipitation assay confirmed that RFXAP binds strongly to the amino-terminal domain of LANA. We were further interested in determining the regions in the amino-terminal domain of LANA sufficient to interact with RFXAP. To do this, LANA truncation regions spanning aa 1 to 340 (as a control for binding), aa 1 to 250, or aa 1 to 150 were transiently coexpressed with full-length RFXAP in HEK293T cells, and a coimmunoprecipitation assay was performed. Detection of coprecipitating RFXAP showed efficient binding with all three LANA truncations, suggesting that the LANA truncation mutant spanning aa 1 to 150 is sufficient for an interaction with RFXAP (Fig. 3B). LANA truncations were detected with anti-Myc antibody (Fig. 3B).
FIG 3.
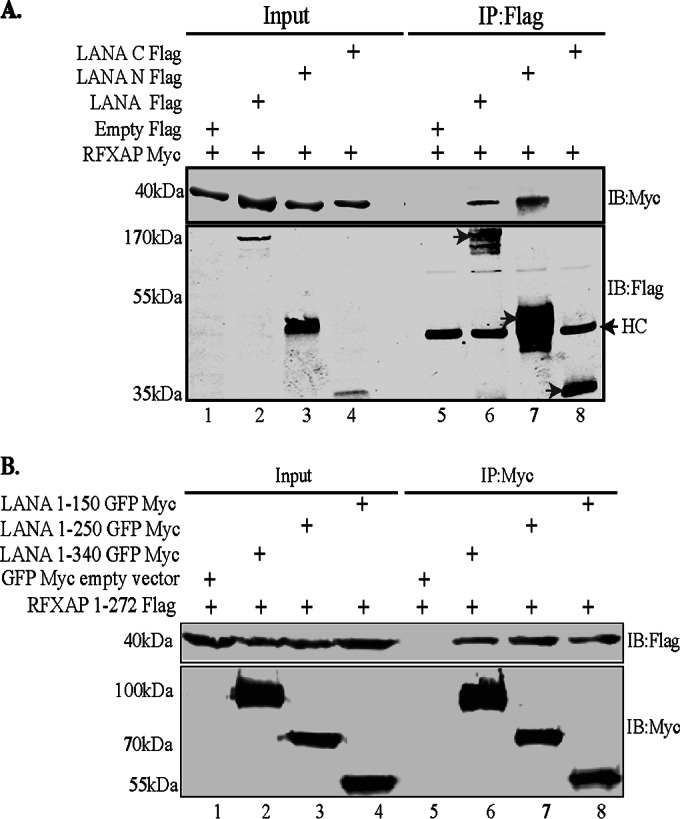
Coimmunoprecipitation of RFXAP with the LANA truncation spanning aa 1 to 150. (A) HEK293T cells were transiently transfected with the Flag-tagged pA3F vector (control), LANA-Flag, LANA-N–Flag, or LANA-C–Flag along with RFXAP-Myc. Cell lysates were subjected to immunoprecipitation with anti-Flag antibody, and the immunoprecipitated proteins were detected with anti-Flag and anti-Myc antibodies after resolving proteins on SDS-PAGE gels and Western blotting. HC represents the heavy chain of the antibody. (B) HEK293T cells were cotransfected with RFXAP-Flag and either NLS-GFP-Myc (vector control), the LANA truncation spanning aa 1 to 340 (LANA 1-340)-GFP-Myc, LANA 1-250–GFP–Myc, or LANA 1-150–GFP–Myc. Cell lysates were subjected to IP using rabbit anti-Myc antibody, and the immunoprecipitated proteins were resolved on SDS-PAGE gels, followed by detection with mouse anti-Myc and anti-Flag antibodies, in order to avoid detection of the heavy chain that comigrates with the LANA region spanning aa 1 to 150.
RFXAP interacts with LANA through its C terminus.
The RFXAP component of the RFX complex has distinct N-terminal acidic, C-terminal glutamine-rich, and middle basic domains. Previous studies have shown that the last 43 aa at the C-terminal glutamine-rich domain of RFXAP is sufficient for the expression of the HLA-DRA gene; however, the expression of other HLA genes (HLA-DQ and HLA-DP) requires a larger C-terminal segment of RFXAP (last 130 aa) (56). The last 43 aa in the C-terminal domain of RFXAP have been reported to interact with the other two components of the RFX complex, RFX5 and RFXANK (56–60). Hence, in order to understand the mechanism of how LANA binding may affect RFX complex formation and also the assembly of a functional enhanceosome to alter the expression of MHC-II genes, we sought to determine whether LANA binds to RFXAP in the C terminus or to the functionally dispensable N-terminal region of RFXAP. To this end, we constructed Flag-tagged deletion mutants of RFXAP, as shown in Fig. 4A, and performed in vitro GST binding and coimmunoprecipitation assays to determine the RFXAP-binding domain for LANA. Full-length RFXAP (aa 1 to 272) and its truncation mutants spanning aa 1 to 150, aa 151 to 272, and aa 1 to 210 were efficiently translated in vitro, detected as a distinct band by autoradiography. A binding assay with LANA-N–GST showed interactions with full-length RFXAP (aa 1 to 272), as expected, and with the C-terminal domain (aa 151 to 272) but not with the N-terminal domains (aa 1 to 150 or aa 1 to 210) (Fig. 4B). The GST fusion proteins (control GST or LANA-N–GST) used in this in vitro binding assay were detected by Coomassie staining (Fig. 4C). These results suggested that LANA binds to RFXAP in the C-terminal region of aa 210 to 272, which is also the region that mediates the interaction with the other two components of the RFX complex, RFXANK and RFX5. These binding results were further confirmed by a coimmunoprecipitation assay by coexpressing the LANA N terminus fused to GFP-Myc with either the empty Flag vector or Flag-tagged RFXAP (aa 1 to 272) and its truncation mutants (aa 1 to 172, aa 1 to 210, aa 150 to 272, and aa 210 to 272). The results of this coimmunoprecipitation assay also confirmed that the N-terminal domain of RFXAP (aa 1 to 210) was dispensable for its interaction with LANA. These binding assays determined that LANA binds in the C-terminal domain of RFXAP (aa 210 to 272), which is also the interaction domain for the other components of the RFX complex. We were unable to in vitro translate the smallest truncation of RFXAP (aa 210 to 272) despite repeated attempts; therefore, it was used only in the overexpression immunoprecipitation assay.
FIG 4.

The C terminus of RFXAP interacts with LANA. (A) Schematic showing RFXAP domains used for coimmunoprecipitation and GST pulldown assays. (B) In vitro-translated full-length RFXAP (aa 1 to 172) or the truncated RFXAP mutant spanning aa 1 to 150, aa 151 to 272, or aa 1 to 210 was pulled down with GST or LANA-N–GST and resolved on SDS-PAGE gels, followed by detection by autoradiography. (C) Expression of control GST and LANA-N–GST proteins that were used for GST pulldown of RFXAP truncation mutants. (D) Coimmunoprecipitation assay of the N terminus of LANA and different truncation mutants of RFXAP. HEK293T cells were transiently cotransfected with LANA-N–GFP–Myc and either the empty control vector pA3F, full-length Flag-tagged RFXAP (aa 1 to 272) or the truncated RFXAP protein spanning aa 1 to 210, aa 150 to 272, or aa 210 to 272. The cell lysates were immunoprecipitated with anti-Flag antibody, and immunoprecipitated proteins were immunoblotted with anti-Flag (mouse) and anti-Myc antibodies. HC represents the heavy chain detected because mouse anti-Flag was used for the detection of Flag-tagged proteins.
LANA associates with the HLA-DRA promoter.
The components of the RFX complex bind to the promoters of MHC-II molecules in order to promote the assembly of the multiprotein enhanceosome complex for the recruitment of CIITA (61). Since LANA bound with the RFX complex, we wanted to determine whether LANA associates with the promoters of MHC-II genes. In order to test this, we performed chromatin immunoprecipitation (ChIP) assays with anti-LANA antibody from the KSHV-infected PEL cell lines BCBL-1 and BC-3. The results determined by quantitative PCR (qPCR) showed copies of the HLA-DRA promoter in the chromatin precipitated with LANA antibody but not in the chromatin precipitated with control antibody in both BC-3 and BCBL-1 cells, indicating a specific association of LANA with the promoter of the HLA-DRA gene (Fig. 5Aa). Since LANA associates with the terminal repeat (TR) sequence of the KSHV genome, we used LANA-bound chromatin for the detection of the TR as a positive control for anti-LANA ChIP. As expected, the TR sequence amplified from chromatin immunoprecipitated with anti-LANA antibody but not with the control antibody, confirming the specificity of anti-LANA ChIP (Fig. 5Ab). A Western blot showing the efficiency of anti-LANA antibody immunoprecipitation is presented in Fig. 5Ac.
FIG 5.

LANA associates with the HLA-DRA promoter. (A) A ChIP assay was performed with anti-LANA antibody on the chromatins in BC-3 and BCBL-1 PEL cells. (a) Sonicated chromatin was immunoprecipitated with anti-LANA or control antibodies (IgG), and the chromatin-bound DNA was subjected to quantitation of HLA-DRA promoter activity by qPCR. (b) Amplification of the KSHV terminal region (TR), the binding site of LANA, was included as a positive control for LANA ChIP. (c) Immunoblot showing the specificity and efficiency of anti-LANA antibody in immunoprecipitation. (Ba) Chromatin immunoprecipitation with anti-Flag antibody from the sonicated chromatin of BJAB cells stably expressing YFP-LANA-Flag (BJAB-LYF) or control YFP (BJAB-YF). The presence of the HLA-DRA promoter sequence in the immunoprecipitated chromatin was detected by qPCR. The error bars represent standard deviations of the means from at least three experimental replicates. Relative binding (percent input) was determined by calculating the number of copies of the target sequence in the immunoprecipitated chromatin with respect to the number of copies in the inputs. P values were calculated by two-tailed t tests comparing LANA ChIP with the control antibody. ***, P < 0.001; ****, P < 0.0001. (b) Western blot showing the specificity and efficiency of anti-Flag antibody. (C) EMSA showing binding of LANA to the HLA-DRA promoter bound with RFX complex proteins. EMSA was performed with a 32P-labeled double-stranded X-box sequence of the HLA-DRA probe; in vitro-translated Flag-tagged RFX5, RFXAP, and RFXANK; and Myc-tagged LANA-N. Lane 1, X-box probe with rabbit reticulocyte lysate (RRL); lane 2, X-box probe with the RFX complex (RFX-Cx Flag) showing binding of RFX-Cx–Flag to the probe (arrow); lane 3, X-box probe with RFX-Cx–Flag and 200× specific cold competitor (SCC) probe, which competed for binding of the radiolabeled probe to RFX-Cx–Flag; lane 4, X-box probe with RFX-Cx–Flag and 200× nonspecific cold competitor (NSCC) probe, which was not able to compete for binding; lane 5, X-box probe with RFX-Cx–Flag and LANA-N–Myc showing reduced mobility of the probe due to binding of LANA-N–Myc with RFX-Cx–Flag (circle); lane 6, HLA-DRA probe with LANA-N–Myc without RFX-Cx–Flag showing no direct binding of LANA-N to the probe; lane 7, HLA-DRA probe with RFX-Cx–Flag, LANA-N–Myc, and anti-Myc antibody (the star shows the supershifted complex) (anti-Myc antibody further retarded the mobility of the probe bound to the RFX-Cx–LANA-N protein by binding to the Myc epitope tag of LANA-N); lane 8, HLA-DRA probe with RFX-Cx–Flag, LANA-N–Myc, and IgG control antibody. The IgG control antibody failed to supershift the mobility of the probe bound to the RFX-Cx–LANA-N protein complex.
The presence of LANA at the chromatin of the HLA-DRA promoter was also detected in cells stably expressing LANA as the only KSHV protein. A KSHV-negative B cell line, BJAB, was stably transduced with a lentivirus vector expressing LANA-yellow fluorescent protein (YFP)-Flag or control YFP-Flag and selected to obtain an enriched population of cells expressing YFP. ChIP was performed with anti-Flag antibody from both control YFP-Flag-expressing (BJAB-YF) and YFP-LANA-Flag-expressing (BJAB LYF) BJAB cells. DNA purified from the immunoprecipitated chromatin was subjected to qPCR to detect the HLA-DRA promoter sequence. Detection of the HLA-DRA promoter in the chromatin from BJAB cells with YFP-LANA-Flag but not with YFP-Flag (Fig. 5Ba) confirmed the specific association of LANA with the HLA-DRA promoter. Immunoprecipitating YFP-LANA-Flag and YFP-Flag were detected with anti-Flag antibody (Fig. 5Bb). Since LANA bound to the chromatin of the HLA-DRA promoter in BJAB cells expressing LANA as the sole KSHV protein, it was confirmed that LANA associates with the HLA-DRA promoter without the aid of any other KSHV episomes. These data further confirmed our findings that LANA can associate with RFX proteins independent of the involvement of other viral components.
Binding of LANA to the HLA-DRA promoter was further established by an electrophoretic mobility shift assay (EMSA). For EMSAs, the radiolabeled double-stranded RFX complex-binding sequence (X-box sequence) of the HLA-DRA promoter was used as a probe (55). The RFX complex proteins, RFX5, RFXAP, and RFXANK, as well as LANA-N were translated with cold methionine in vitro to ensure that these proteins were free of other cellular proteins. The radiolabeled X-box sequence (probe) showed binding with the RFX-Cx, as detected by a specific band (Fig. 5C, lane 2, arrow). The binding of the RFX-Cx with the radiolabeled probe was eliminated by the addition of a 200-fold excess of a specific cold competitor (SCC) (Fig. 5C, compare lanes 2 and 3) but not with a similar fold excess of a nonspecific (scrambled) probe (nonspecific cold competitor [NSCC]) (Fig. 5C, compare lanes 2 and 4). This confirmed that the RFX-Cx specifically binds to the X-box sequence of the probe. The addition of the in vitro-translated RFX-binding domain of LANA (LANA-N [aa 1 to 340]) to the RFX-Cx-bound probe further retarded the mobility of the complex (Fig. 5C, lane 5, circle), confirming the association of LANA-N with the RFX-Cx bound to the X-box sequence of the HLA-DRA promoter. Since LANA-N without the RFX complex was unable to alter the mobility of free probes (Fig. 5C, lane 6), it was confirmed that LANA-N cannot bind directly to the X-box sequence of the HLA-DRA promoter but rather binds through its association with the components of the RFX complex. The specificity of the association of LANA-N with the RFX-Cx bound to the radiolabeled probe was determined by adding anti-Myc antibody to the Myc epitope tag of LANA-N (Fig. 5C, lane 7). The mobility of the complex was further retarded (supershifted) in the presence of anti-Myc antibody but not with mouse IgG (control antibody) (Fig. 5C, star, and compare lanes 7 and 8). This confirmed that LANA-N forms a complex with the components of the RFX complex at the HLA-DRA promoter.
LANA reduces the transcriptional activity of the HLA-DRA promoter.
The expression of MHC-II genes is controlled mainly at the transcriptional level by binding of the MHC-II regulatory proteins, including the proteins of the RFX complex, at the MHC-II promoters (62). Since LANA interacted with the RFX complex bound to the HLA-DRA promoter, we wanted to investigate whether the presence of LANA affects the activity of the HLA-DRA promoter to alter the expression of MHC-II molecules. To determine the effect of LANA, dual-luciferase reporter assays were performed by using the core HLA-DRA promoter that contained the essential cis elements required for the transcription of the HLA-DRA gene (63, 64). An HLA-DRA reporter assay was performed with two different cell lines: HEK293L cells, non-antigen-presenting cells chosen because of ease of transfection, and DG75 cells, antigen-presenting B cells. The results of the promoter reporter assay revealed that CIITA was able to activate HLA-DRA promoter activity in both cell lines, as expected (Fig. 6A and B, lane 2). The coexpression of LANA in cells expressing CIITA reduced the luciferase activity, determined as relative luciferase units, in both HEK293L and DG75 cell lines. An increasing amount of the LANA expression vector subsequently reduced the HLA-DRA promoter activity in a dose-dependent manner (Fig. 6A and B, lanes 3 to 5). Importantly, the highest concentrations of LANA used in this assay completely abrogated CIITA-stimulated HLA-DRA promoter activity, making the level of activity comparable to basal levels (Fig. 6A and B, compare lanes 1 and 5). The finding that the amounts of overexpressed CIITA (by the CMV promoter) were comparable among those samples with increasing LANA amounts confirmed that the inhibition of the HLA-DRA promoter was not due to reduced levels of CIITA in these cells (Fig. 6A and B). Detection of LANA by anti-Myc immunoblotting showed increasing amounts of LANA in the lysate. Since the amino-terminal domain of LANA was able to bind to the RFX complex, we wanted to determine whether it would be sufficient to inhibit HLA-DRA promoter activity similarly to full-length LANA. To test this, we expressed increasing amounts of LANA-N in both HEK293L and DG75 cells, which showed effects on reducing the HLA-DRA promoter activity similar to those of full-length LANA (Fig. 6C and D, lanes 3 to 5). In contrast, the C-terminal domain of LANA, which did not bind to the CIITA complex, was unable to appreciably reduce HLA-DRA promoter activity even with high levels of LANA-C expression (Fig. 6E and F). Since comparable amounts of CIITA were overexpressed in all the test samples, the abrogation of promoter activity by full-length LANA and LANA-N is most likely due to the inhibition of the recruitment of CIITA to the enhanceosome because of LANA binding to the RFX complex. The dose-dependent inhibition of HLA-DRA promoter activity may suggest competition between LANA and CIITA to bind to the RFX complex.
FIG 6.

LANA inhibits transcriptional activity of the HLA-DRA promoter. Luciferase reporter assays show a dose-dependent reduction of the luciferase activity mediated by the HLA-DRA promoter when CIITA-expressing cells were cotransfected with full-length LANA or the amino-terminal region of LANA but not with the carboxy-terminal region of LANA. For the reporter assay, the HLA-DRA promoter fused with the luciferase reporter was transiently transfected into HEK293L cells (A, C, and E) or DG75 cells (B, D, and F). The cells were cotransfected with CIITA to stimulate the HLA-DRA promoter along with either full-length LANA (A and B), the RFXAP-interacting domain of LANA, the amino-terminal domain of LANA (C and D), or the carboxy-terminal domain of LANA (LANA-C) (E and F). The cell lysates were used for measuring luciferase activities using a dual-luciferase assay kit. Relative luciferase units were calculated relative to the basal promoter activity. Lane 1, basal DRA-luciferase activity; lane 2, CIITA-stimulated DRA-luciferase activity; lane 3, CIITA-stimulated DRA-luciferase activity with 1 μg of LANA or a truncation (LANA-N or LANA-C); lane 4, CIITA-stimulated DRA-luciferase activity with 2 μg of LANA or a truncation (LANA-N or LANA-C); lane 5, CIITA-stimulated DRA-luciferase activity with 3 μg of LANA or a truncation (LANA-N or LANA-C). The error bars represent standard deviations of the means from at least three experimental replicates. Equal amounts of cell lysates from each experimental set were used for the detection of overexpressed CIITA and LANA proteins. The GAPDH immunoblot shows equal loading of the cell lysates.
LANA downregulates the expression of MHC-II molecules.
Since RFX proteins are crucial for the expression of MHC-II molecules, and the expression of LANA inhibited the activity of one of the promoter (HLA-DRA) encoding MHC-II genes, we hypothesized that the binding of LANA to the components of the RFX complex may have a downstream effect on the transcription of MHC-II molecules. We therefore analyzed the levels of MHC-II transcripts in two antigen-presenting cell lines, BJAB and THP-1, stably expressing LANA through a lentiviral vector. Total RNAs extracted from control (YFP-Flag) and LANA (LANA-YFP-Flag [LYF]) cells were subjected to analysis of MHC-II genes by using a quantitative real-time PCR (qRT-PCR) assay. Since LANA also downregulates the expression of CIITA to consequently repress the expression of MHC-II molecules (17), we took lower levels of CIITA into account in determining the transcript levels of HLA genes by normalizing the levels of CIITA in LANA-expressing cells to those in control cells. While it was difficult to distinguish the percentages of MHC-II downregulation due to the binding of LANA with the RFX complex from the reduced expression of CIITA (LANA-mediated downregulation of the CIITA promoter) (17), we presumed that normalized levels of CIITA would yield comparable levels of MHC-II genes, if no other mechanism was involved. Therefore, we calculated the levels of MHC-II genes by determining the ratio of the level of CIITA in LANA-expressing cells to that in control cells, which was 0.6 ± 0.1. To compensate for the reduced levels of CIITA in LANA-expressing cells, the transcript levels of MHC-II genes were determined by multiplying the ratios of transcript levels of MHC-II genes by 1.66 (CIITA levels were reduced to 0.6-fold; therefore, the multiplication factor was 1/0.6 = 1.66). Importantly, the transcripts levels of all the classical MHC-II genes (HLA-DRA, HLA-DRB, HLA-DPA, HLA-DPB, HLA-DQA, and HLA-DQB) were downregulated significantly, even after normalization of the CIITA levels, in LANA-expressing BJAB as well as THP-1 cells (BJAB-LYF and THP-LYF, respectively) compared to their respective BJAB-YFP and THP-YFP control cells (Fig. 7A and C). The results of the reporter assays, where CIITA was expressed through a CMV promoter (the levels of CIITA were unaltered with LANA), showing a reduction in HLA promoter activity as well as the above-described CIITA levels normalized to MHC-II expression levels strongly advocate for an involvement of additional mechanisms in the downregulation of MHC-II genes by LANA along with the reduction of CIITA expression. Prevention of binding of CIITA to HLA promoters is one such plausible mechanism. Western blot analysis of MHC-II levels in these LANA-expressing BJAB and THP-1 cells showed reduced levels of MHC-II expression compared to those in control, YFP-expressing cells (Fig. 7B and D). Immunoblotting for the detection of LANA-YFP-Flag and YFP-Flag in these BJAB and THP-1 cells confirmed the expression of LANA (Fig. 7B and D). Furthermore, the expressions of MHC-II genes in the KSHV-infected PEL cell lines BC-3 and BCBL-1 showed significantly reduced levels compared to those in KSHV-negative BJAB cells, analyzed by normalizing the levels of CIITA (Fig. 7E). We were unable to detect mRNAs of HLA-DQB in KSHV-infected cells, even after multiple attempts; therefore, HLA-DQB was excluded from the relative mRNA calculation. Immunodetection of MHC-II expression in KSHV-infected cells showed very low levels compared to those in KSHV-negative BJAB cells (Fig. 7F). These results corroborate the results reported previously by Cai et al. (17), who showed reduced expression levels of MHC-II proteins during KSHV infection.
FIG 7.

LANA downregulates the expression of MHC-II molecules. (A, C, and E) Relative mRNA levels of different HLA genes in LANA-expressing BJAB (LYF) cells compared to control BJAB cells with YFP (YFP) (A), in LANA-expressing THP-1 (LYF) cells compared to control THP-1 cells with YFP (C), and in KSHV-infected BC-3 and BCBL-1 PEL cells compared to BJAB cells (E). The mRNA levels were quantified by using qRT-PCR, and the expression levels of HLA genes were normalized to CIITA levels, as explained in Results. The errors bars represent standard deviations of the means from at least three experimental replicates. P values were calculated by two-tailed t tests comparing LYF cells with control YFP cells. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (B, D, and F) Immune detection of MHC-II expression in BJAB-LYF and BJAB-YFP cells (B), in THP-1 LYF and YFP cells (D), and in BJAB, BC3, and BCBL1 cells (F). Flag-tagged LANA (YFP-LANA-Flag) and YFP were detected with an anti-Flag antibody, GAPDH, to show equal loading of the cell lysates. LANA in PEL cells was detected by using a rat anti-LANA antibody.
LANA interferes with the association of CIITA with RFXAP.
Since LANA interacted with the RFX complex at the HLA-DRA promoter and downregulated the expression of MHC-II molecules through other mechanisms in addition to CIITA downregulation, we hypothesized that LANA might interfere with the binding of CIITA to RFXAP to disrupt the formation of a functional enhanceosome. To test this hypothesis, we immunoprecipitated CIITA and analyzed coimmunoprecipitating RFXAP in the presence or absence of the RFXAP-binding region of LANA (LANA-N) in an overexpression system using HEK293T cells (Fig. 8A). As expected, RFXAP efficiently coimmunoprecipitated with CIITA in the absence of LANA-N but not with the control (Fig. 8A, lanes 5 and 6). However, the amounts of coimmunoprecipitating RFXAP were significantly reduced in the presence of LANA-N (Fig. 8A, lanes 7 and 8). The binding of RFXAP to CIITA was very weak at increased amounts of LANA-N (Fig. 8A, lane 8), confirming that the binding of LANA to RFXAP blocked the binding of CIITA to RFXAP. Furthermore, LANA did not coimmunoprecipitate with CIITA, indicating that LANA does not directly interact with CIITA (Fig. 8A, lanes 7 and 8).
FIG 8.

LANA interferes with the association of CIITA and RFXAP. (A) Coimmunoprecipitation assay showing reduced associations of RFXAP with CIITA in the presence of LANA. Flag-tagged CIITA, Myc-tagged RFXAP, and Myc-tagged LANA-N (aa 1 to 340) were used in this competitive coimmunoprecipitation assays after transfection into HEK293T cells. CIITA was immunoprecipitated from the cellular lysates by using an anti-Flag antibody. The proteins were resolved on SDS-PAGE gels, and coimmunoprecipitated proteins were detected by using anti-Myc and anti-Flag antibodies. HC, heavy chain. (B) Coimmunoprecipitation assay showing reduced association of RFX5 with CIITA in an in vitro binding assay. In vitro-translated and [35S]methionine-labeled LANA-N (aa 1 to 340)–Myc, RFXAP-Myc, RFX5-Myc, RFXANK-Myc, and CIITA-Flag were used for in vitro binding assays. The proteins were allowed to interact overnight at 4°C, followed by immunoprecipitation with anti-Flag antibody (CIITA). The bound protein complexes were resolved on SDS-PAGE gels, followed by detection by autoradiography. (Ca) ChIP assays showing reduced binding of CIITA to the HLA-DRA promoter in LANA-expressing BJAB cells compared to control YFP-expressing cells. (b and c) Relative binding of CIITA in LANA-depleted and control KSHV-infected BC-3 (b) and BCBL-1 (c) PEL cells. (d) Relative binding of CIITA on the chromatin HLA-DRA promoters in the expression system. The HLA-DRA-Luc vector and RFX components (Myc tagged) were transfected with the Flag vector (control), CIITA-Flag, or CIITA-Flag with LANA-Myc in HEK293L cells, followed by a ChIP assay with anti-Flag (CIITA) antibody. The amount of CIITA-bound HLA-DR was reduced in LANA-expressing cells. The error bars represent standard deviations of the means from at least three experimental replicates. P values were calculated by two-tailed t tests comparing treated to control cells. ***, P < 0.001; ****, P < 0.0001. (D) Immunoblot showing the expression of LANA and CIITA and the immunoprecipitation efficiency of anti-CIITA antibody used for ChIP assays. (a) LANA-expressing (LYF) and control YFP (YF) BJAB cells. (b and c) LANA depletion in KSHV-infected BC-3 (b) and BCBL-1 (c) PEL cells by the lentiviral vector showing efficient knockdown of LANA compared to shControl cells. (d) Immune detection of LANA and CIITA in inputs and immunoprecipitated cells with anti-Myc and anti-Flag antibodies, respectively.
A similar immunoprecipitation assay was also performed by using a cell-free system where CIITA, components of the RFX complex, and the LANA N terminus (aa 1 to 340) were translated in vitro for a coimmunoprecipitation assay with CIITA in the presence of LANA-N (aa 1 to 340). Immunoprecipitation of CIITA with anti-Flag antibody coprecipitated RFX5 (component of the RFX complex) (Fig. 8B, lane 6). The other components of the RFX complex did not coprecipitate with CIITA in this cell-free system, indicating that the direct interaction between CIITA and RFXAP/RFXANK is very weak when these proteins are translated in vitro. Interestingly, the coimmunoprecipitation of RFX5 was significantly reduced in the presence of LANA-N (Fig. 8B, lanes 7 and 8), confirming that the binding of LANA with the components of the RFX complex results in a reduced binding of CIITA to the RFX complex.
CIITA does not possess any direct DNA-binding domain but is recruited to the MHC-II promoters via interactions with the proteins of the MHC-II enhanceosome complex, including the proteins of the RFX complex (44). We speculated that a reduction in the binding of CIITA to the components of the RFX complex would result in a reduced binding of CIITA to the enhanceosome of the MHC-II promoter. To confirm this, we performed anti-CIITA ChIP assays with BJAB cells stably expressing LANA (BJAB-LYF) and compared the binding of CIITA to the MHC-II promoter in these cells to that in cells without LANA (BJAB-YF). qPCR analysis of the ChIP assay data revealed that the association of CIITA with the HLA-DRA promoter was significantly reduced in LANA-expressing BJAB cells compared to that in cells without LANA (Fig. 8Ca). The efficiency of the anti-CIITA antibody for immunoprecipitation in BJAB cells expressing LANA-YFP-Flag and YFP-Flag was analyzed, which showed comparable levels of CIITA in both cell types (Fig. 8D). LANA was detected with anti-Flag antibody in BJAB-LYF cells, as expected (Fig. 8Da). Furthermore, in the PEL cell lines BC-3 and BCBL-1, the shRNA-mediated reduction of LANA levels resulted in an increased association of CIITA with the chromatin of HLA-DRA (Fig. 8Cb and c), confirming a direct role of LANA in CIITA binding to the HLA-DRA promoter. shRNA for LANA significantly reduced the levels of LANA, which was confirmed by anti-LANA immunoblotting (Fig. 8Db and c). Anti-CIITA antibody precipitated comparable amounts of CIITA from shControl or shLANA cells (Fig. 8Db and c), confirming that the reduced levels of the HLA-DRA promoter were not due to reduced amounts of immunoprecipitated CIITA. As these results showed that the presence of LANA abrogates and the depletion of LANA facilitates CIITA binding to the promoter to regulate MHC-II expression, we performed a ChIP assay by expressing CIITA driven by the CMV promoter (to obtain equal expression levels of CIITA in all samples) to rule out the possibility that the reduction in CIITA binding to the HLA-DRA promoter was not due to reduced expression of CIITA by LANA. We overexpressed Flag-tagged CIITA with or without LANA in cells transfected with the HLA-DRA promoter plasmid and the components of the RFX complex in HEK293T cells by transient transfections. qPCR analysis of the HLA-DRA promoter from chromatin immunoprecipitated with anti-Flag (CIITA) showing reduced levels of the CIITA-bound HLA-DRA promoter in cells expressing LANA confirmed that LANA directly reduced the binding of CIITA to HLA-DRA chromatin (Fig. 8Cd). The expression and immunoprecipitation of LANA and CIITA were detected by anti-Myc and anti-Flag immunoblots, respectively (Fig. 8Dd).
Finally, to gain a better understanding of the mechanism by which LANA could be interfering with the recruitment of CIITA and the assembly of a functional enhanceosome, we investigated whether the binding of the components of the RFX complex to the enhanceosome is affected in the presence of LANA. To address this, we analyzed the binding of RFXAP and RFX5 on the HLA-DRA promoter by performing ChIP assays on cells stably expressing LANA and also on PEL cells depleted of LANA. We did not observe any significant difference in the association of RFXAP with the HLA-DRA promoter in BJAB cells without or with LANA expression, as BJAB-YF and LYF cells showed similar levels of the chromatin-bound HLA-DRA promoter (Fig. 9Aa). The depletion of LANA by shRNA did not significantly change the association of RFXAP with HLA-DRA in both PEL cell lines BC-3 and BCBL-1, as shControl and shLANA cells showed similar levels of the chromatin-bound HLA-DRA promoter (Fig. 9Ab and c). The efficiencies of RFXAP immunoprecipitations from BJAB and PEL cells showed comparable levels of RFXAP in the respective sets of cells (Fig. 9Ad to f). These results suggest that LANA interacts with RFXAP bound to the HLA-DRA promoter and does not interfere with the binding of RFXAP to the HLA-DRA promoter.
FIG 9.

Binding of RFXAP and RFX5 to the HLA-DRA promoter in the presence of LANA. (Aa) ChIP assays coupled with qPCR analysis showing binding of RFXAP to the HLA-DRA promoter in the presence of LANA in KSHV-negative BJAB cells stably expressing LANA (LYF) or control cells with YFP expression (YFP). (b and c) LANA depletion in KSHV-infected BC3 (b) and BCBL1 (c) PEL cells by a lentiviral vector. RFXAP ChIP in BJAB-YFP and LYF cells as well as with or without LANA knockdown in BC-3 and BCBL-1 cells showed similar binding of RFXAP. The error bars represents standard deviations of the means from at least three experimental replicates. P values calculated by two-tailed t tests showed no significant difference between analyzed samples. (d to f) Efficient immunoprecipitation with anti-RFXAP antibody. (Ba) ChIP assay with anti-RFX5 antibody showing reduced copy numbers of the HLA-DRA promoter bound to RFX5 in LANA-expressing BJAB cells. (b and c) The association of RFX5 with the HLA-DRA promoter was increased when LANA was knocked down in BC-3 (b) and BCBL1 (c) cells compared to cells without LANA knockdown. The error bars represent standard deviations of the means from at least three experimental replicates. P values were calculated by two-tailed t tests. *, P < 0.05; **, P < 0.01. (d to f) Efficient immunoprecipitation with anti-RFX5 antibody. (C) LANA-N disrupts the association of RFX5 with RFXANK. Immunoprecipitation of RFX5 (Flag tagged) in the presence of LANA-N (GFP–LANA-N–Myc) reduced the amounts of coprecipitating RFXANK compared to those in the cells without LANA-N (middle, compare lane 6 to lane 5).
Since the interaction of RFX5 with RFXAP is necessary for the binding of RFX5 to the MHC-II promoters (65), and LANA binds to the same domain of RFXAP that is required for the interaction of RFXAP with RFX5, we speculated that the association of LANA with RFXAP may result in a reduced binding of RFX5 to the HLA-DRA promoter. Interestingly, the association of RFX5 with the chromatin of the HLA-DRA promoter was significantly reduced in BJAB cells expressing LANA (BJAB-LYF) compared to control cells (BJAB-YF) (Fig. 9Ba). Additionally, RFX5 binding to chromatin of the HLA-DRA promoter was enhanced in LANA-depleted, KSHV-positive BC-3 and BCBL-1 cells (Fig. 9Bc and d). Immunoprecipitation with anti-RFX5 antibody showed comparable levels of RFX5 proteins in the respective sets of cells (Fig. 9Bd to f). Disrupted binding of RFX5 with the RFXAP component was further confirmed in a coimmunoprecipitation assay with the expression of LANA-N (RFXAP-binding domain of LANA). While RFX5 but not the control vector efficiently coimmunoprecipitated RFXAP (Fig. 9C, lanes 4 and 5), coimmunoprecipitation of RFXAP with RFX5 was reduced in the presence of LANA-N (Fig. 9C, compare lane 6 with lane 5). A detectable level of LANA-N coimmunoprecipitating with RFX5 (Fig. 9C, lane 6) further supported the binding of LANA with RFX components. Collectively, these data show that the binding of LANA with the components of the RFX complex inhibits the binding of RFX5 and CIITA without affecting the binding of RFXAP to the HLA-DRA promoter.
DISCUSSION
KSHV has evolved excellent immune evasion and immune modulation strategies that allow it to establish lifelong latency. Some of these strategies have been identified, but KSHV most likely has many more strategies up its sleeve to sabotage host immunity. Of these, targeting of the MHC-II antigen presentation pathway seems to be very crucial because MHC-II molecules can present virally derived antigens to CD4+ T lymphocytes (66) and turn on adaptive antiviral immune responses. In addition to coordinating adaptive immune responses, activated CD4+ T cells also play a direct cytolytic role in killing infected cells displaying viral antigens in conjugation with MHC-II molecules (67). So far, two KSHV latent proteins, vIRF3 and LANA, have been shown to inhibit the MHC-II antigen-presenting pathway (17, 23, 25). LANA was previously reported to inhibit the expression of MHC-II molecules via the suppression of CIITA transcripts (17). The data presented here establish yet another mechanism used by LANA to evade the host immune system. Here, we show that LANA disrupts the assembly of a functional MHC-II enhanceosome by interacting with the components of the RFX complex and thereby contributes to the inhibition of the MHC-II pathway. To the best of our knowledge, this is the first study that shows the disruption of a functional MHC enhanceosome by any virus to subvert host immunity.
The expression of MHC-II molecules is controlled primarily at the level of transcription by a set of highly conserved transcription factors (29). Of these, transcription factors of notable importance are CIITA and the three components of the RFX complex, RFX5, RFXAP, and RFXANK. The fact that defects in any of these four genes can result in bare lymphocyte syndrome (BLS), characterized by severely reduced expression of MHC-II genes, highlights the significance of these four proteins in the regulation of MHC-II genes (66, 68, 69). The transcription of all MHC-II genes is highly coordinated and is strictly contingent upon the binding of CIITA to the promoters of MHC-II. CIITA is a non-DNA-binding MHC-II transactivator that is recruited to the promoters of MHC-II genes via protein-protein interactions with the transcriptional factors preassembled at the promoters of MHC-II genes, termed the MHC-II enhanceosome (29). RFX proteins are very important components of the MHC-II enhanceosome, and binding of CIITA to the promoters of MHC-II requires its interactions with the components of the RFX complex. Binding of CIITA to MHC-II promoters is believed to be absolutely essential for the expression of MHC-II genes, as alternate pathways for MHC-II expression are not yet known to exist. Targeting of the RFX proteins and CIITA is therefore a very effective strategy for escaping host adaptive immunity. Cai et al. previously reported that LANA deregulates CIITA transcription, resulting in the downregulation of MHC-II expression (17). In this study, we have identified a novel mechanism by which KSHV LANA disrupts the association of a functional enhanceosome to suppress the transcription of MHC-II genes. Our report shows that LANA binds to the components of the RFX complex, which prevents the recruitment of the transactivator CIITA to the promoters of MHC-II genes to form a functional enhanceosome for downregulating the expression of MHC-II genes.
We identified RFX transcription factors as LANA-interacting proteins in a yeast two-hybrid screen. We further validated the interactions between LANA and RFX proteins using in vivo and in vitro assays. Our data show that LANA directly interacts with all three components of the RFX complex, RFX5, RFXAP, and RFXANK, and prevents the binding of CIITA to the promoters of MHC-II genes. While LANA can bind to all three components of the RFX complex, it has a higher affinity for RFXAP.
We demonstrated that the domain responsible for the interactions of LANA with RFX transcription factors is the N-terminal domain of LANA. Cai et al. reported that the C-terminal domain of LANA is involved in the downregulation of MHC-II by binding with IRF4 (17). The involvement of multiple domains of LANA in the downregulation of MHC-II emphasizes the importance of inhibiting the MHC-II pathway during KSHV latency. Since the amino-terminal domain of LANA is known to associate with host chromatin (51, 70), the possibility that LANA might be interacting with the RFX proteins indirectly through chromatin-binding factors was raised. However, the interactions between the N terminus of LANA and RFX proteins was verified in a cell-free system by using in vitro-translated LANA and RFX proteins, which confirmed that these interactions are direct and independent of any cellular or viral factors.
RFXAP contains three biochemically distinct regions in its primary sequence, including an acidic region, a basic region, and a glutamine-rich region. Of these regions, the C-terminal region, which includes the basic and the glutamine-rich regions, is a highly conserved region (71). This region is both necessary and sufficient for its interactions with the other components of the RFX complex, RFX5 and RFXANK (32, 41, 59, 60, 72). Since LANA bound to the C-terminal region of RFXAP, we hypothesized that binding of LANA to the C terminus of RFXAP may disrupt the assembly of a functional RFX complex by reducing its association with either RFX5, RFXANK, or both. In support of this hypothesis, when we pulled down in vitro-translated components of the RFX complex through LANA, we observed a distorted stoichiometry of the RFX complex, marked by a noticeably reduced association of RFX5 in the immunoprecipitated complex. This suggests that the binding of RFXAP to LANA partially excludes RFX5 from the RFX complex, which has important implications for recruiting CIITA to the MHC-II promoters, because RFX5 and RFXANK, but not RFXAP, are known to interact directly with CIITA (43, 59, 71, 73). Indeed, the association of CIITA at the HLA-DRA promoter was reduced in the presence of LANA in ChIP assays. We also demonstrated that coprecipitation of RFX5 was reduced in the presence of LANA when in vitro-translated components of the RFX complex were coimmunoprecipitated with CIITA. RFXAP did not directly coimmunoprecipitate with CIITA in a cell-free system, probably due to the lack of direct interactions between CIITA and RFXAP in the cell-free system or due to the lack of proper posttranslational modifications of these two proteins. However, when tested in vivo, coimmunoprecipitation of RFXAP with CIITA was reduced in a coimmunoprecipitation assay, affirming that the association of CIITA with RFX proteins was reduced in the presence of LANA.
We further showed that LANA associates with MHC-II promoters in order to disrupt the binding of CIITA with the RFX complex using an anti-LANA ChIP assay. However, LANA does not directly associate with the X-box DNA sequence, a conserved cis-acting RFX complex-binding domain on MHC-II promoters. Instead, LANA is recruited to the MHC-II promoters by binding to the RFX proteins, as evident by the results of electrophoretic mobility shift assays performed by using in vitro-translated RFX proteins and the amino-terminal domain of LANA. We anticipated that the association of LANA at MHC-II promoters inhibits MHC-II promoter activity, which subsequently results in reduced transcription and expression of MHC-II genes. As expected, the presence of full-length LANA as well as the N terminus of LANA, which binds to RFX complex proteins, efficiently and dose-dependently reduced the HLA-DRA promoter activity in luciferase reporter assays. While all the necessary components of the MHC-II transcription machinery are constitutively expressed in all cell types, the expression of CIITA is restricted to professional antigen-presenting cells only. However, the expression of CIITA can be induced in most cell types by IFN-γ treatment. Hence, in order to induce HLA-DRA promoter activity, we treated the cells with IFN-γ, which induced CIITA expression and HLA-DRA promoter activity but was suppressed in the presence of LANA (data not shown). While we observed an efficient inhibition of the HLA-DRA activity induced by IFN-γ treatment in the presence of LANA, the report by Cai et al. (17) raised the possibility that this inhibition could be because of reduced expression of CIITA, as LANA inhibits the activity of CIITA promoters PIII and PIV. In order to alleviate the effect of LANA on CIITA repression and the subsequent reduction of HLA promoter activity, we modified our luciferase reporter assays and expressed CIITA from a constitutively active CMV promoter, which showed comparable expression levels in the presence and absence of LANA. Our data showed that even when CIITA was expressed through a CMV promoter, the activity of the HLA-DRA promoter was efficiently and dose-dependently downregulated by full-length LANA as well as the amino-terminal domain of LANA. These results confirm that in order to ensure efficient inhibition of MHC-II transcription, LANA interferes with the binding of existing CIITA to the HLA promoters and disrupts the formation of a functional enhanceosome. In our coimmunoprecipitation experiments, we did not find CIITA interacting with LANA, suggesting that LANA does not directly associate with CIITA. Taken together, these data confirm that LANA binds to RFX proteins and thereby interferes with the recruitment of CIITA to MHC-II promoters. The eventual effect of LANA-mediated interference in CIITA binding to MHC-II promoters is reduced transcription and expression of MHC-II genes.
In an attempt to identify the mechanism by which LANA disrupts the MHC-II enhanceosome, we discovered that the association of LANA with RFXAP did not prevent the association of RFXAP with the HLA-DRA promoter, but it reduced the binding of RFX5 with RFXAP, as demonstrated by a ChIP assay. The reduced binding of RFX5 to the HLA-DRA promoter in the presence of LANA was most likely due to the binding of LANA to RFXAP in the same region as that of RFX5 binding, thus disrupting the association of RFXAP with the RFX complex. Notably, an association of RFXAP with RFX5 is required for relieving the autoinhibition of RFX5 that prevents the DNA-binding activity of RFX5 (65). Our ChIP data supported this hypothesis, as the association of RFX5 with the HLA-DRA promoter was reduced in the presence of LANA. Based on the overall results of this study, we propose a model (Fig. 10) in which LANA disrupts the assembly of the functional enhanceosome necessary for the expression of MHC-II genes.
FIG 10.

Proposed model of MHC-II transcriptional downregulation through interactions of LANA with RFX proteins. MHC-II expression is regulated predominantly at the transcriptional level, and transcription of MCH-II genes is strictly contingent upon the binding of a non-DNA-binding transactivator, CIITA, to the promoters of MHC-II genes. The promoters of all the MHC-II genes contain a highly conserved cis-acting module that consists of the S-, X1-, X2-, and Y-box sequences. A heterotrimeric RFX complex consisting of RFX5, RFXAP, and RFXANK occupies the X1 box. The X2 and Y boxes are occupied by CREB protein and trimeric NF-Y complexes, respectively. Proper assembly of these proteins on MHC-II promoters makes a functional MHC-II enhanceosome. In normal cells, the assembled MHC-II enhanceosome initiates the transcription of MHC-II genes by recruiting CIITA to the promoters of MHC-II genes. However, in KSHV-infected cells, the KSHV latent protein LANA, as shown previously, reduces the transcriptional activity of CIITA promoters PIII and PIV, resulting in reduced expression of CIITA, which in turn contributes to an inhibition of MHC-II genes. There is an additional mechanism to inhibit the binding of residual CIITA to the promoters of MHC-II. The results of the present study show that LANA binds to the components of the RFX complex at MHC-II promoters and disrupts the assembly of these factors. The disrupted RFX complex is unable to recruit CIITA and thus mitigates the efficiency of residual CIITA in stimulating MHC-II transcription. The culminating effect of these pathways leads to a reduction in the transcription of MHC-II genes.
Our findings on the association of LANA with the components of the RFX complex have implications for the expression of MHC-I molecules as well. The transcription of MHC-I genes is regulated by the binding of the NLRC5 transcriptional coactivator to the MHC-I promoter in a fashion similar to that of the regulation of MHC-II genes by CIITA (74). NLRC5-mediated expression of MHC-I requires its association with RFX proteins for the optimal transcription of MHC-I genes (74). Hence, a disruption of the RFX complex by LANA may result in a reduced binding of NLRC5 to the MHC-I enhanceosome, thus leading to reduced expression of MHC-I genes as well. MHC-I antigen presentation is critical for the activation of CD8+ T cells, which has also been shown to play an important role in controlling KSHV infections (75, 76). The role of LANA in inhibiting MHC-I through this mechanism remains to be explored, but targeting of the RFX complex by KSHV appears to help the virus dodge both MHC-I and MHC-II antigen-presenting pathways and consequently evade CD4+ T and CD8+ T cell immunities simultaneously. Given that collective T cell immunity is very critical in mounting an antiviral host response (18, 77, 78), targeting of the RFX complex gives an edge to KSHV in establishing lifelong persistence in the infected host.
In summary, we identified a new mechanism used by LANA to inhibit the expression of MHC-II molecules. LANA disrupts the functional MHC-II enhanceosome by binding to the RFX transcription factors and thus reduces the binding of the master regulator CIITA to the MHC-II promoters. The overall effect of the disruption of the functional enhanceosome is a reduction in the transcription of MHC-II genes, leading to a lower level of MHC-II expression. There may be an involvement of multiple strategies by KSHV to completely block the expression of MHC-II molecules on the cell surface, as demonstrated previously (25). Inhibition of the MHC-II antigen presentation pathway is very crucial for the establishment of effective lifelong KSHV latency, and hence, LANA as well as other latent proteins of KSHV are likely to target multiple immune pathways at multiple steps in order to efficiently block MHC-II expression.
ACKNOWLEDGMENTS
We thank Erle S. Robertson, University of Pennsylvania, for providing the cell lines and LANA expression plasmids; Jeremy Boss, Emory University School of Medicine, for providing the RFX5-GFP plasmid; and Jenny Ting, University of North Carolina, Chapel Hill, for providing the HLA-DRA-Luc promoter reporter and CIITA-Flag plasmids.
This work was supported by public health grants from the NIH ( CA174459 and AI105000) and the American Heart Association (13BGIA14520036) to S.C.V. S.T. was partially supported by the Reno Cancer Foundation and a Mick Hitchcock Fellowship, University of Nevada, Reno, NV.
REFERENCES
- 1.Cai Q, Verma SC, Lu J, Robertson ES. 2010. Molecular biology of Kaposi's sarcoma-associated herpesvirus and related oncogenesis. Adv Virus Res 78:87–142. doi: 10.1016/B978-0-12-385032-4.00003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cesarman E. 2014. Gammaherpesviruses and lymphoproliferative disorders. Annu Rev Pathol 9:349–372. doi: 10.1146/annurev-pathol-012513-104656. [DOI] [PubMed] [Google Scholar]
- 3.Toth Z, Brulois K, Jung JU. 2013. The chromatin landscape of Kaposi's sarcoma-associated herpesvirus. Viruses 5:1346–1373. doi: 10.3390/v5051346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verma SC, Robertson ES. 2003. Molecular biology and pathogenesis of Kaposi sarcoma-associated herpesvirus. FEMS Microbiol Lett 222:155–163. doi: 10.1016/S0378-1097(03)00261-1. [DOI] [PubMed] [Google Scholar]
- 5.Coscoy L. 2007. Immune evasion by Kaposi's sarcoma-associated herpesvirus. Nat Rev Immunol 7:391–401. doi: 10.1038/nri2076. [DOI] [PubMed] [Google Scholar]
- 6.Lee HR, Brulois K, Wong L, Jung JU. 2012. Modulation of immune system by Kaposi's sarcoma-associated herpesvirus: lessons from viral evasion strategies. Front Microbiol 3:44. doi: 10.3389/fmicb.2012.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alibek K, Baiken Y, Kakpenova A, Mussabekova A, Zhussupbekova S, Akan M, Sultankulov B. 2014. Implication of human herpesviruses in oncogenesis through immune evasion and suppression. Infect Agents Cancer 9:3. doi: 10.1186/1750-9378-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, van Marck E, Salmon D, Gorin I, Escande JP, Weiss RA, Alitalo K, Boshoff C. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc Natl Acad Sci U S A 96:4546–4551. doi: 10.1073/pnas.96.8.4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parravicini C, Chandran B, Corbellino M, Berti E, Paulli M, Moore PS, Chang Y. 2000. Differential viral protein expression in Kaposi's sarcoma-associated herpesvirus-infected diseases: Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease. Am J Pathol 156:743–749. doi: 10.1016/S0002-9440(10)64940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kellam P, Bourboulia D, Dupin N, Shotton C, Fisher C, Talbot S, Boshoff C, Weiss RA. 1999. Characterization of monoclonal antibodies raised against the latent nuclear antigen of human herpesvirus 8. J Virol 73:5149–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mesri EA, Cesarman E, Boshoff C. 2010. Kaposi's sarcoma and its associated herpesvirus. Nat Rev Cancer 10:707–719. doi: 10.1038/nrc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verma SC, Lan K, Robertson E. 2007. Structure and function of latency-associated nuclear antigen. Curr Top Microbiol Immunol 312:101–136. doi: 10.1007/978-3-540-34344-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li X, Liang D, Lin X, Robertson ES, Lan K. 2011. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen reduces interleukin-8 expression in endothelial cells and impairs neutrophil chemotaxis by degrading nuclear p65. J Virol 85:8606–8615. doi: 10.1128/JVI.00733-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cloutier N, Flamand L. 2010. Kaposi sarcoma-associated herpesvirus latency-associated nuclear antigen inhibits interferon (IFN) beta expression by competing with IFN regulatory factor-3 for binding to IFNB promoter. J Biol Chem 285:7208–7221. doi: 10.1074/jbc.M109.018838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaldumbide A, Ossevoort M, Wiertz EJ, Hoeben RC. 2007. In cis inhibition of antigen processing by the latency-associated nuclear antigen I of Kaposi sarcoma herpes virus. Mol Immunol 44:1352–1360. doi: 10.1016/j.molimm.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 16.Kwun HJ, da Silva SR, Qin H, Ferris RL, Tan R, Chang Y, Moore PS. 2011. The central repeat domain 1 of Kaposi's sarcoma-associated herpesvirus (KSHV) latency associated-nuclear antigen 1 (LANA1) prevents cis MHC class I peptide presentation. Virology 412:357–365. doi: 10.1016/j.virol.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai Q, Banerjee S, Cervini A, Lu J, Hislop AD, Dzeng R, Robertson ES. 2013. IRF-4-mediated CIITA transcription is blocked by KSHV encoded LANA to inhibit MHC II presentation. PLoS Pathog 9:e1003751. doi: 10.1371/journal.ppat.1003751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sabbah S, Jagne YJ, Zuo J, de Silva T, Ahasan MM, Brander C, Rowland-Jones S, Flanagan KL, Hislop AD. 2012. T-cell immunity to Kaposi sarcoma-associated herpesvirus: recognition of primary effusion lymphoma by LANA-specific CD4+ T cells. Blood 119:2083–2092. doi: 10.1182/blood-2011-07-366476. [DOI] [PubMed] [Google Scholar]
- 19.Zuo J, Rowe M. 2012. Herpesviruses placating the unwilling host: manipulation of the MHC class II antigen presentation pathway. Viruses 4:1335–1353. doi: 10.3390/v4081335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cresswell P. 1994. Assembly, transport, and function of MHC class II molecules. Annu Rev Immunol 12:259–293. doi: 10.1146/annurev.iy.12.040194.001355. [DOI] [PubMed] [Google Scholar]
- 21.Ting JP, Zhu XS. 1999. Class II MHC genes: a model gene regulatory system with great biologic consequences. Microbes Infect 1:855–861. doi: 10.1016/S1286-4579(99)00233-6. [DOI] [PubMed] [Google Scholar]
- 22.Hegde NR, Chevalier MS, Johnson DC. 2003. Viral inhibition of MHC class II antigen presentation. Trends Immunol 24:278–285. doi: 10.1016/S1471-4906(03)00099-1. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt K, Wies E, Neipel F. 2011. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 3 inhibits gamma interferon and major histocompatibility complex class II expression. J Virol 85:4530–4537. doi: 10.1128/JVI.02123-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butler LM, Jeffery HC, Wheat RL, Long HM, Rae PC, Nash GB, Blackbourn DJ. 2012. Kaposi's sarcoma-associated herpesvirus inhibits expression and function of endothelial cell major histocompatibility complex class II via suppressor of cytokine signaling 3. J Virol 86:7158–7166. doi: 10.1128/JVI.06908-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zuo J, Hislop AD, Leung CS, Sabbah S, Rowe M. 2013. Kaposi's sarcoma-associated herpesvirus-encoded viral IRF3 modulates major histocompatibility complex class II (MHC-II) antigen presentation through MHC-II transactivator-dependent and -independent mechanisms: implications for oncogenesis. J Virol 87:5340–5350. doi: 10.1128/JVI.00250-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siegrist CA, Martinez-Soria E, Kern I, Mach B. 1995. A novel antigen-processing-defective phenotype in major histocompatibility complex class II-positive CIITA transfectants is corrected by interferon-gamma. J Exp Med 182:1793–1799. doi: 10.1084/jem.182.6.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van den Elsen PJ, van der Stoep N, Vietor HE, Wilson L, van Zutphen M, Gobin SJ. 2000. Lack of CIITA expression is central to the absence of antigen presentation functions of trophoblast cells and is caused by methylation of the IFN-gamma inducible promoter (PIV) of CIITA. Hum Immunol 61:850–862. doi: 10.1016/S0198-8859(00)00159-2. [DOI] [PubMed] [Google Scholar]
- 28.Boss JM. 1997. Regulation of transcription of MHC class II genes. Current Opin Immunol 9:107–113. doi: 10.1016/S0952-7915(97)80166-5. [DOI] [PubMed] [Google Scholar]
- 29.Krawczyk M, Reith W. 2006. Regulation of MHC class II expression, a unique regulatory system identified by the study of a primary immunodeficiency disease. Tissue Antigens 67:183–197. doi: 10.1111/j.1399-0039.2006.00557.x. [DOI] [PubMed] [Google Scholar]
- 30.Reith W, Mach B. 2001. The bare lymphocyte syndrome and the regulation of MHC expression. Annu Rev Immunol 19:331–373. doi: 10.1146/annurev.immunol.19.1.331. [DOI] [PubMed] [Google Scholar]
- 31.Jabrane-Ferrat N, Nekrep N, Tosi G, Esserman LJ, Peterlin BM. 2002. Major histocompatibility complex class II transcriptional platform: assembly of nuclear factor Y and regulatory factor X (RFX) on DNA requires RFX5 dimers. Mol Cell Biol 22:5616–5625. doi: 10.1128/MCB.22.15.5616-5625.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durand B, Sperisen P, Emery P, Barras E, Zufferey M, Mach B, Reith W. 1997. RFXAP, a novel subunit of the RFX DNA binding complex is mutated in MHC class II deficiency. EMBO J 16:1045–1055. doi: 10.1093/emboj/16.5.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hasegawa SL, Boss JM. 1991. Two B cell factors bind the HLA-DRA X box region and recognize different subsets of HLA class II promoters. Nucleic Acids Res 19:6269–6276. doi: 10.1093/nar/19.22.6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masternak K, Barras E, Zufferey M, Conrad B, Corthals G, Aebersold R, Sanchez JC, Hochstrasser DF, Mach B, Reith W. 1998. A gene encoding a novel RFX-associated transactivator is mutated in the majority of MHC class II deficiency patients. Nat Genet 20:273–277. doi: 10.1038/3081. [DOI] [PubMed] [Google Scholar]
- 35.Moreno CS, Beresford GW, Louis-Plence P, Morris AC, Boss JM. 1999. CREB regulates MHC class II expression in a CIITA-dependent manner. Immunity 10:143–151. doi: 10.1016/S1074-7613(00)80015-1. [DOI] [PubMed] [Google Scholar]
- 36.Moreno CS, Emery P, West JE, Durand B, Reith W, Mach B, Boss JM. 1995. Purified X2 binding protein (X2BP) cooperatively binds the class II MHC X box region in the presence of purified RFX, the X box factor deficient in the bare lymphocyte syndrome. J Immunol 155:4313–4321. [PubMed] [Google Scholar]
- 37.Nagarajan UM, Louis-Plence P, DeSandro A, Nilsen R, Bushey A, Boss JM. 1999. RFX-B is the gene responsible for the most common cause of the bare lymphocyte syndrome, an MHC class II immunodeficiency. Immunity 10:153–162. doi: 10.1016/S1074-7613(00)80016-3. [DOI] [PubMed] [Google Scholar]
- 38.Reith W, Siegrist CA, Durand B, Barras E, Mach B. 1994. Function of major histocompatibility complex class II promoters requires cooperative binding between factors RFX and NF-Y. Proc Natl Acad Sci U S A 91:554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steimle V, Durand B, Barras E, Zufferey M, Hadam MR, Mach B, Reith W. 1995. A novel DNA-binding regulatory factor is mutated in primary MHC class II deficiency (bare lymphocyte syndrome). Genes Dev 9:1021–1032. doi: 10.1101/gad.9.9.1021. [DOI] [PubMed] [Google Scholar]
- 40.Ting JP, Trowsdale J. 2002. Genetic control of MHC class II expression. Cell 109Suppl:S21–S33. doi: 10.1016/S0092-8674(02)00696-7. [DOI] [PubMed] [Google Scholar]
- 41.Laird KM, Briggs LL, Boss JM, Summers MF, Garvie CW. 2010. Solution structure of the heterotrimeric complex between the interaction domains of RFX5 and RFXAP from the RFX gene regulatory complex. J Mol Biol 403:40–51. doi: 10.1016/j.jmb.2010.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emery P, Durand B, Mach B, Reith W. 1996. RFX proteins, a novel family of DNA binding proteins conserved in the eukaryotic kingdom. Nucleic Acids Res 24:803–807. doi: 10.1093/nar/24.5.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hake SB, Masternak K, Kammerbauer C, Janzen C, Reith W, Steimle V. 2000. CIITA leucine-rich repeats control nuclear localization, in vivo recruitment to the major histocompatibility complex (MHC) class II enhanceosome, and MHC class II gene transactivation. Mol Cell Biol 20:7716–7725. doi: 10.1128/MCB.20.20.7716-7725.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masternak K, Muhlethaler-Mottet A, Villard J, Zufferey M, Steimle V, Reith W. 2000. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev 14:1156–1166. doi: 10.1101/gad.14.9.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.LeibundGut-Landmann S, Waldburger JM, Krawczyk M, Otten LA, Suter T, Fontana A, Acha-Orbea H, Reith W. 2004. Mini-review: specificity and expression of CIITA, the master regulator of MHC class II genes. Eur J Immunol 34:1513–1525. doi: 10.1002/eji.200424964. [DOI] [PubMed] [Google Scholar]
- 46.Devaiah BN, Singer DS. 2013. CIITA and its dual roles in MHC gene transcription. Front Immunol 4:476. doi: 10.3389/fimmu.2013.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harton JA, Ting JP. 2000. Class II transactivator: mastering the art of major histocompatibility complex expression. Mol Cell Biol 20:6185–6194. doi: 10.1128/MCB.20.17.6185-6194.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Verma SC, Cai Q, Kreider E, Lu J, Robertson ES. 2013. Comprehensive analysis of LANA interacting proteins essential for viral genome tethering and persistence. PLoS One 8:e74662. doi: 10.1371/journal.pone.0074662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai QL, Knight JS, Verma SC, Zald P, Robertson ES. 2006. EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog 2:e116. doi: 10.1371/journal.ppat.0020116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaul R, Verma SC, Robertson ES. 2007. Protein complexes associated with the Kaposi's sarcoma-associated herpesvirus-encoded LANA. Virology 364:317–329. doi: 10.1016/j.virol.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Purushothaman P, McDowell ME, McGuinness J, Salas R, Rumjahn SM, Verma SC. 2012. Kaposi's sarcoma-associated herpesvirus-encoded LANA recruits topoisomerase IIbeta for latent DNA replication of the terminal repeats. J Virol 86:9983–9994. doi: 10.1128/JVI.00839-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Izumi KM. 2001. The yeast two-hybrid assay to identify interacting proteins. Methods Mol Biol 174:249–258. doi: 10.1385/1-59259-227-9:249. [DOI] [PubMed] [Google Scholar]
- 53.Gao J, Cai Q, Lu J, Jha HC, Robertson ES. 2011. Upregulation of cellular Bcl-2 by the KSHV encoded RTA promotes virion production. PLoS One 6:e23892. doi: 10.1371/journal.pone.0023892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krithivas A, Young DB, Liao G, Greene D, Hayward SD. 2000. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J Virol 74:9637–9645. doi: 10.1128/JVI.74.20.9637-9645.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hasegawa SL, Sloan JH, Reith W, Mach B, Boss JM. 1991. Regulatory factor-X binding to mutant HLA-DRA promoter sequences. Nucleic Acids Res 19:1243–1249. doi: 10.1093/nar/19.6.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peretti M, Villard J, Barras E, Zufferey M, Reith W. 2001. Expression of the three human major histocompatibility complex class II isotypes exhibits a differential dependence on the transcription factor RFXAP. Mol Cell Biol 21:5699–5709. doi: 10.1128/MCB.21.17.5699-5709.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nekrep N, Jabrane-Ferrat N, Peterlin BM. 2000. Mutations in the bare lymphocyte syndrome define critical steps in the assembly of the regulatory factor X complex. Mol Cell Biol 20:4455–4461. doi: 10.1128/MCB.20.12.4455-4461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Caretti G, Cocchiarella F, Sidoli C, Villard J, Peretti M, Reith W, Mantovani R. 2000. Dissection of functional NF-Y-RFX cooperative interactions on the MHC class II Ea promoter. J Mol Biol 302:539–552. doi: 10.1006/jmbi.2000.4028. [DOI] [PubMed] [Google Scholar]
- 59.DeSandro AM, Nagarajan UM, Boss JM. 2000. Associations and interactions between bare lymphocyte syndrome factors. Mol Cell Biol 20:6587–6599. doi: 10.1128/MCB.20.17.6587-6599.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Garvie CW, Stagno JR, Reid S, Singh A, Harrington E, Boss JM. 2007. Characterization of the RFX complex and the RFX5(L66A) mutant: implications for the regulation of MHC class II gene expression. Biochemistry 46:1597–1611. doi: 10.1021/bi6023868. [DOI] [PubMed] [Google Scholar]
- 61.Seguin-Estevez Q, De Palma R, Krawczyk M, Leimgruber E, Villard J, Picard C, Tagliamacco A, Abbate G, Gorski J, Nocera A, Reith W. 2009. The transcription factor RFX protects MHC class II genes against epigenetic silencing by DNA methylation. J Immunol 183:2545–2553. doi: 10.4049/jimmunol.0900376. [DOI] [PubMed] [Google Scholar]
- 62.Chang CH, Gourley TS, Sisk TJ. 2002. Function and regulation of class II transactivator in the immune system. Immunol Res 25:131–142. doi: 10.1385/IR:25:2:131. [DOI] [PubMed] [Google Scholar]
- 63.Cressman DE, Chin KC, Taxman DJ, Ting JP. 1999. A defect in the nuclear translocation of CIITA causes a form of type II bare lymphocyte syndrome. Immunity 10:163–171. doi: 10.1016/S1074-7613(00)80017-5. [DOI] [PubMed] [Google Scholar]
- 64.Radosevich M, Song Z, Gorga JC, Ksander B, Ono SJ. 2004. Epigenetic silencing of the CIITA gene and posttranscriptional regulation of class II MHC genes in ocular melanoma cells. Invest Ophthalmol Vis Sci 45:3185–3195. doi: 10.1167/iovs.04-0111. [DOI] [PubMed] [Google Scholar]
- 65.Garvie CW, Boss JM. 2008. Assembly of the RFX complex on the MHCII promoter: role of RFXAP and RFXB in relieving autoinhibition of RFX5. Biochim Biophys Acta 1779:797–804. doi: 10.1016/j.bbagrm.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 66.Schmid D, Pypaert M, Munz C. 2007. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brown DM. 2010. Cytolytic CD4 cells: direct mediators in infectious disease and malignancy. Cell Immunol 262:89–95. doi: 10.1016/j.cellimm.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Glimcher LH, Kara CJ. 1992. Sequences and factors: a guide to MHC class-II transcription. Annu Rev Immunol 10:13–49. doi: 10.1146/annurev.iy.10.040192.000305. [DOI] [PubMed] [Google Scholar]
- 69.Long AB, Boss JM. 2005. Evolutionary conservation and characterization of the bare lymphocyte syndrome transcription factor RFX-B and its paralogue ANKRA2. Immunogenetics 56:788–797. doi: 10.1007/s00251-004-0738-2. [DOI] [PubMed] [Google Scholar]
- 70.Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, Kaye KM. 2006. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311:856–861. doi: 10.1126/science.1120541. [DOI] [PubMed] [Google Scholar]
- 71.Long AB, Ferguson AM, Majumder P, Nagarajan UM, Boss JM. 2006. Conserved residues of the bare lymphocyte syndrome transcription factor RFXAP determine coordinate MHC class II expression. Mol Immunol 43:395–409. doi: 10.1016/j.molimm.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 72.Briggs L, Laird K, Boss JM, Garvie CW. 2009. Formation of the RFX gene regulatory complex induces folding of the interaction domain of RFXAP. Proteins 76:655–664. doi: 10.1002/prot.22379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhu XS, Linhoff MW, Li G, Chin KC, Maity SN, Ting JP. 2000. Transcriptional scaffold: CIITA interacts with NF-Y, RFX, and CREB to cause stereospecific regulation of the class II major histocompatibility complex promoter. Mol Cell Biol 20:6051–6061. doi: 10.1128/MCB.20.16.6051-6061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meissner TB, Liu YJ, Lee KH, Li A, Biswas A, van Eggermond MC, van den Elsen PJ, Kobayashi KS. 2012. NLRC5 cooperates with the RFX transcription factor complex to induce MHC class I gene expression. J Immunol 188:4951–4958. doi: 10.4049/jimmunol.1103160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bihl F, Berger C, Chisholm JV III, Henry LM, Bertisch B, Trojan A, Nadal D, Speck RF, Flepp M, Brander C, Mueller NJ, Swiss HIV Cohort Study . 2009. Cellular immune responses and disease control in acute AIDS-associated Kaposi's sarcoma. AIDS 23:1918–1922. doi: 10.1097/QAD.0b013e3283300a91. [DOI] [PubMed] [Google Scholar]
- 76.Lepone L, Rappocciolo G, Knowlton E, Jais M, Piazza P, Jenkins FJ, Rinaldo CR. 2010. Monofunctional and polyfunctional CD8+ T cell responses to human herpesvirus 8 lytic and latency proteins. Clin Vaccine Immunol 17:1507–1516. doi: 10.1128/CVI.00189-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Duman S, Toz H, Asci G, Alper S, Ozkahya M, Unal I, Celik A, Ok E, Basci A. 2002. Successful treatment of post-transplant Kaposi's sarcoma by reduction of immunosuppression. Nephrol Dial Transplant 17:892–896. doi: 10.1093/ndt/17.5.892. [DOI] [PubMed] [Google Scholar]
- 78.Kwun HJ, da Silva SR, Shah IM, Blake N, Moore PS, Chang Y. 2007. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mimics Epstein-Barr virus EBNA1 immune evasion through central repeat domain effects on protein processing. J Virol 81:8225–8235. doi: 10.1128/JVI.00411-07. [DOI] [PMC free article] [PubMed] [Google Scholar]