

Abstract
Background
Plastid genomes, also known as plastomes, are shaped by the selective forces acting on the fundamental cellular functions they code for and thus they are expected to preserve signatures of the adaptive path undertaken by different plant species during evolution. To identify molecular signatures of positive selection associated to adaptation to contrasting ecological niches, we sequenced with Solexa technology the plastomes of two congeneric Brassicaceae species with different habitat preference, Cardamine resedifolia and Cardamine impatiens.
Results
Following in-depth characterization of plastome organization, repeat patterns and gene space, the comparison of the newly sequenced plastomes between each other and with 15 fully sequenced Brassicaceae plastomes publically available in GenBank uncovered dynamic variation of the IR boundaries in the Cardamine lineage. We further detected signatures of positive selection in ten of the 75 protein-coding genes of the examined plastomes, identifying a range of chloroplast functions putatively involved in adaptive processes within the family. For instance, the three residues found to be under positive selection in RUBISCO could possibly be involved in the modulation of RUBISCO aggregation/activation and enzymatic specificty in Brassicaceae. In addition, our results points to differential evolutionary rates in Cardamine plastomes.
Conclusions
Overall our results support the existence of wider signatures of positive selection in the plastome of C. resedifolia, possibly as a consequence of adaptation to high altitude environments. We further provide a first characterization of the selective patterns shaping the Brassicaceae plastomes, which could help elucidate the driving forces underlying adaptation and evolution in this important plant family.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1498-0) contains supplementary material, which is available to authorized users.
Keywords: Cardamine, Molecular adaptation, Large single copy region (LSC), Small single copy region (SSC), Plastomes, Positive selection, Repeats, Codon usage
Background
Chloroplast genomes, hereafter referred to as plastomes, have been widely used as models for elucidating the patterns of genetic variation in space and time, ranging from colonization to speciation and phylogeny, encompassing both micro- and macro-evolutionary events across all lineages of plants [1]. Understanding the phyletic patterns of chloroplast evolution can also potentially layout the basis of species discrimination [2], as indicated by the fact that the core DNA barcode chosen for plants is composed by the two plastomic regions rbcL and matK [3]. In fact, the presence of a high number of plastomes per cell, ease of amplification across the angiosperm phylogeny, and good content in terms of phylogenetic information explain the popularity of these and other plastidial markers for both species identification and phylogenetic reconstruction. The organization of the plastome is remarkably conserved in higher plants, and it is characterized by two usually large inverted repeat regions (IRA and IRB) separated by single copy regions of different lengths, called large single copy region (LSC) and small single copy region (SSC; [4]). Both traditional Sanger sequencing and next generation sequencing approaches have been widely employed to elucidate the dynamic changes of these four plastome regions, revealing patterns of evolutionary expansion and contraction in different plant lineages [5,6]. The genes present in plastomes play fundamental functions for the organisms bearing them: they encode the core proteins of photosynthetic complexes, including Photosystem I, Photosystem II, Cytochrome b6f, NADH dehydrogenase, ATP synthase and the large subunit of RUBISCO, tRNAs and ribosomal RNAs and proteins necessary for chloroplast ribosomal assembly and translation, and sigma factors necessary for transcription of chloroplast genes [7]. Plastomes of seed plants typically encode four rRNAs, around 30 tRNAs and up to 80 unique protein-coding genes [6-8]. With the notable exception of extensive photosynthetic gene loss in parasitic plants [9], genic regions are generally conserved across the plastomes of higher plants reported so far; inversions and other rearrangements, however, are frequently reported [5]. In line with the higher conservation of genic versus inter-genic regions, a recent report of plastome from basal asterids indicates the conservation of the repeat patterns in the coding regions, whereas the evolution of the repeats in the non-coding regions is lineage-specific [10]. Due to the endosymbiotic origin of plastomes, several of the genes are coordinately transcribed in operons (e.g. the psbB operon) [11,12]. Additionally, chloroplast transcripts undergo RNA editing, especially in ancient plant lineages like ferns and hornworts [13,14].
The Cardamine genus represents one of the largest and most polyploid-rich genera of the Brassicaceae, and underwent several recent and rapid speciation events contributing to the divergent evolution of its species [15]. The diversification of Cardamine has been driven by multiple events of polyploidization and hybridization, which, together with the high number of species, has till now hindered the obtainment of a comprehensive phylogeny of the genus [16]. Using cpDNA regions, patterns of extensive genetic variation have been previously reported in Cardamine flexuosa and related species [17]. The high seed production characterizing several Cardamine taxa makes them highly invasive species, which can become noxious in both wild habitats and cultivation. C. flexuosa and C. hirsuta, for instance, are among the most common weeds in cultivation [17]. C. impatiens is rapidly colonizing North America, where it is considered as one of the most aggressive invaders of the understory given its high adaptability to low light conditions [18]. Several Cardamine species have been object of growing interest as models for evolutionary adaptive traits and morphological development. C. hirsuta, a cosmopolitan weed with fast life cycle, is now a well established model for development of leaf dissection in plants [19]. C. flexuosa has been recently used to elucidate the interplay between age and vernalization in regulating flowering [20]. Earlier, in a pioneering study with cross-species microarray hybridization, the whole transcriptome of C. kokaiensis provided insights on the molecular bases of cleistogamy and its relationship with environmental conditions, especially chilling temperatures [21].
More recently, using the Cardamine genus as a model we demonstrated transcriptome-wide patterns of molecular evolution in genes pertaining to different environmental habitat adaptation by comparative analysis of low altitude, short lived, nemoral species C. impatiens to high altitude, perennial, open-habitat dweller C. resedifolia, suggesting contrasting patterns of molecular evolution in photosynthetic and cold-tolerance genes [22]. The results explicitly demonstrated faster evolution of the cold-related genes exclusively in the high altitude species C. resedifolia [22]. To extend the understanding of positive selection signatures observed in the aforementioned transcriptome-wide analysis to organelles, in this study we carried out the complete sequencing with Solexa technology of the plastome of both species and characterized their gene space and repeat patterns. The comparison of the newly sequenced plastomes between each other and with 15 fully sequenced Brassicaceae plastomes publically available in GenBank uncovered dynamic variation of the IR boundaries in the Cardamine lineage associated to generation of lineage-specific pseudogenic fragments in this region. In addition, we could detect signatures of positive selection in ten of the 75 protein-coding genes of the plastomes examined as well as specific rbcL residues undergoing intra-peptide co-evolution. Overall our results support the existence of wider signatures of positive selection in the plastome of C. resedifolia, possibly as a consequence of adaptation to high altitude environments.
Results and discussion
Genome assembly and validation
In order to further our understanding of selective patterns associated to contrasting environmental adaptation in plants, we obtained and annotated the complete plastome sequence of two congeneric species, high altitude Cardamine resedifolia (GenBank accession number KJ136821) and low altitude C. impatiens (accession number KJ136822). The primers used amplified an average of 6,2 Kbp, with a minimum and maximum amplicon length of 3,5 and 9,0 Kbp, respectively (Additional file 1: Table S1). In this way, a total of 650335 x100 bp paired-end (PE) reads with a Q30 quality value and mean insert size of 315 bp were obtained for C. resedifolia, while 847076 x100 bp PE reads with 325 bp insert size were obtained for C. impatiens. Velvet de-novo assembly resulted in 36 and 48 scaffolds in C. resedifolia and C. impatiens, respectively (Table 1). To validate the accuracy of the assembled plastome we carried out Sanger sequencing of PCR amplicons spanning the junction regions (LSC/IRA, LSC/IRB, SSC/ IRA, SSC/IRB). The perfect identity of the sequences to those resulting from assembly confirmed the reliability of assembled plastomes (data not shown). Additionally, we Sanger-sequenced selected regions of the plastome genic space to verify the correct translational frame of the coding regions and to eliminate any Ns still present in the assembly. The finished, high quality organelle genome sequences thus obtained were used for downstream analyses.
Table 1.
Sequencing statistics and general characteristics of C. resedifolia and C. impatiens plastome assembly
| C. resedifolia | C. impatiens | |
|---|---|---|
| PE reads with a Q > 30 | 650335 (315 bp*) | 847076 (325 bp*) |
| Type of Assembler | de-bruijn Graph | de-bruijn Graph |
| K-mer used | 63 | 63 |
| Number of scaffolds | 36 | 48 |
| Reference species | Nasturtium officinale | Nasturtium officinale |
| Assembled plastome size | 155036 bp | 155611 bp |
| Number of genes | 85(79unique) | 85(79unique) |
| Number of t-RNA | 37(30unique) | 37(30unique) |
| Number of r-RNA | 8(4unique) | 8(4unique) |
| Length of IRa and IRb | 26502 bp | 26476 bp |
| Length of SSC | 17867 bp | 17948 bp |
| Length of LSC | 84165 bp | 84711 bp |
| Annotation | cpGAVAS, DOGMA | CpGAVAS, DOGMA |
*Number in parenthesis indicate the insert size of the PE library.
Plastome structural features and gene content
The finished plastomes of C. resedifolia and C. impatiens have a total length of 155036 bp and 155611 bp and a GC content of 36.30% and 36.33%, respectively. These values of GC content suggest an AT-rich plastome organization, which is similar to the other Brassicaceae plastomes sequenced so far (Figures 1 and 2). Quadripartite organization of plastomes, characterized by two large inverted repeats, plays a major role in the recombination and the structural diversity by gene expansion and gene loss in chloroplast genomes [8]. Each plastome assembly displayed a pair of inverted repeats (IRA and IRB) of 26502 bp and 26476 bp respectively in C. resedifolia and C. impatiens, demarking large single copy (LSC) regions of 84165 bp and 84711 bp and small single copy (SSC) regions of 17867 bp and 17948 bp in C. resedifolia and C. impatiens respectively (Table 1, Additional file 2: Table S2). The assembled plastomes contained a total of 85 protein-coding genes, 37 t-RNAs, and 8 r-RNAs in both C. resedifolia and C. impatiens. We observed a total of 12 protein-coding regions and 6 t-RNAs containing one or more introns (Table 2), which is similar to Nicotiana tabacum, Panax ginseng and Salvia miltiorrhiza [23] but higher than the basal plastomes of the Asterid lineage, where only ycf3 and clpP have been reported to be protein-coding genes with introns [10]. Of the observed gene space in C. resedifolia and C. impatiens, 79 protein-coding genes, 30 t-RNA and 4 r-RNAs were found to be unique while 6 protein-coding (ndhB, rpl23, rps7, rps12, ycf2, rpl2), 7 t-RNAs (trnA-UGC, trnI-CAU, trnI-GAU, trnL-CAA, trnN-GUU, trnR-ACG and trnV-GAC) and 4 r-RNA genes (rrn4.5, rrn5, rrn16, rrn23) were found be duplicated in IRA and IRB (Table 2). GC content analysis of the IR, SSC and LSC showed no major fluctuations, with SSC regions accounting for 29.26%/29.16% GC, LSC 34.06%/34.00%, IRA and IRB each accounting for 42.36%/42.36% GC in C. impatiens and C. resedifolia, respectively. Of the observed intron-containing genes, clpP and ycf3 contained two introns. In rps12 a trans-splicing event was observed with the 5′ end located in the LSC region and the duplicated 3′ end in the IR region as previously reported in Nicotiana [24]. In the trnK-UUU gene was located the largest intron, harboring the matK gene and accounting for 2552 bp in C. resedifolia and 2561 bp in C. impatiens (Additional file 3: Table S3).
Figure 1.
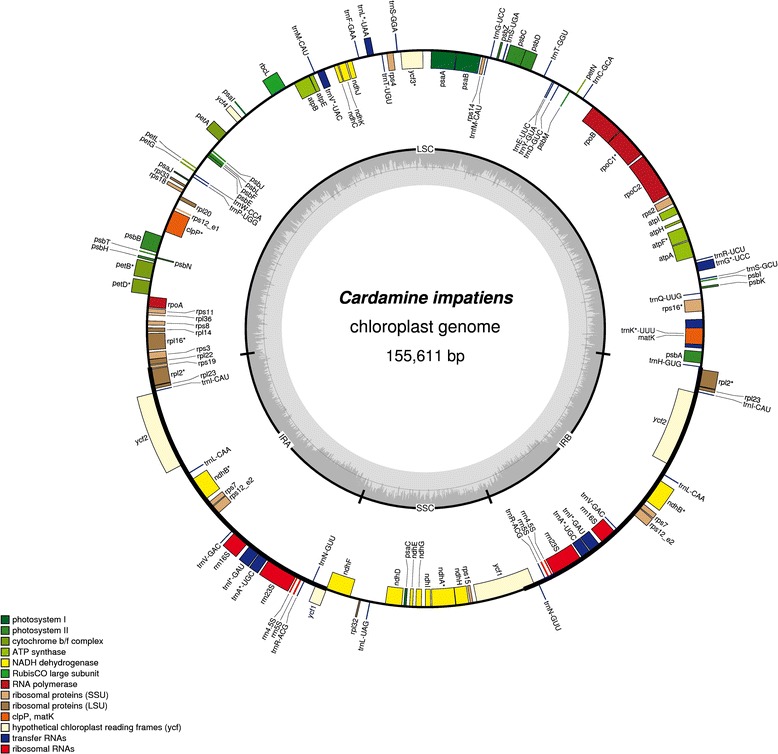
Plastome map of C. resedifolia. Genes shown outside of the larger circle are transcribed clockwise, while genes shown inside are transcribed counterclockwise. Thick lines of the smaller circle indicate IRs and the inner circle represents the GC variation across the genic regions.
Figure 2.
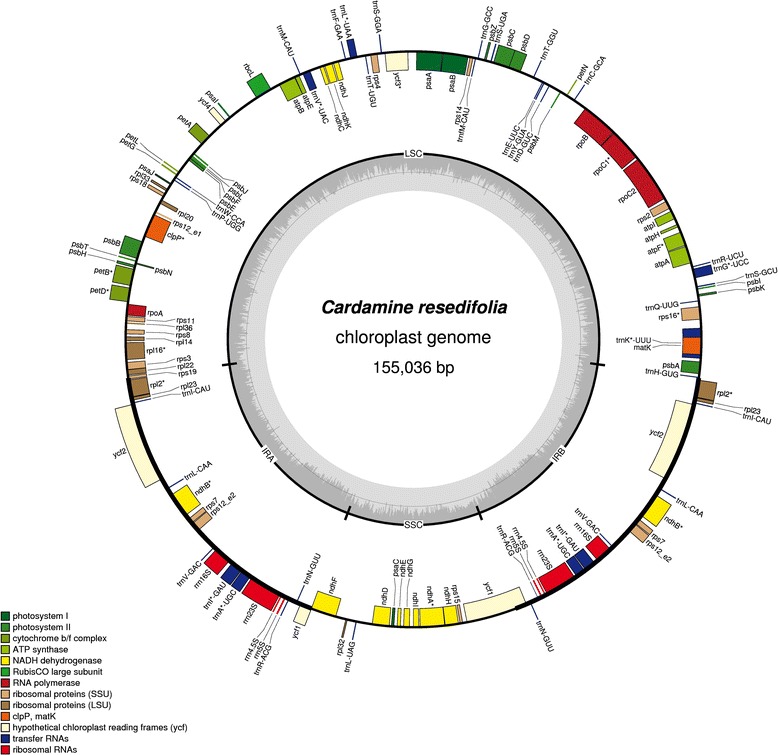
Plastomic map of C. impatiens. Genes shown outside of the larger circle are transcribed clockwise, while genes shown inside are transcribed counterclockwise. Thick lines of the smaller circle indicate IRs and the inner circle represents the GC variation across the genic regions.
Table 2.
List of genes encoded in C. impatiens and C. resedifolia plastomes
| Gene Category | Genes |
|---|---|
| ribosomal RNAS | § rrn4.5, § rrn5, § rrn16, § rrn23 |
| transfer RNAs | § *trnA-UGC, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, *trnG-UCC, trnG-UCC, trnH-GUG, § trnI-CAU, § *trnI-GAU, *trnK-UUU, § trnL-CAA, *trnL-UAA, trnL-UAG, trnM-CAU, § trnN-GUU, trnP-UGG, trnQ-UUG, § trnR-ACG, trnR-UCU, trnS-GCU, trnS-UGA, trnS-GGA, trnT-UGU, trnT-GGU, *trnV-UAC, § trnV-GAC, trnW-CCA, trnY-GUA |
| Photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ |
| Cytochrome | petA, *petB, *petD, petG, petL, petN |
| ATP synthase | atpA, atpB, atpE, *atpF, atpH, atpI |
| Rubisco | rbcL |
| NADH dehydrogenase | *ndhA, § *ndhB, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK |
| Ribosomal protein (large subunit) | § *rpl2, rpl14, *rpl16, rpl20, rpl22, § rpl23, rpl32, rpl33, rpl36 |
| Ribosomal protein (small subunit) | rps2, rps3, rps4, § rps7, rps8, rps11, § *rps12, rps14, rps15, *rps16, rps18, rps19 |
| RNA polymerase | rpoA, rpoB, *rpoC1, rpoC2 |
| ATP-dependent protease | *clpP |
| Cytochrome c biogenesis | ccsA |
| Membrane protein | cemA |
| Maturase | matK |
| Conserved reading frames | ycf1_short, ycf1_long, § ycf2, *ycf3, ycf4 |
§Gene completely duplicated in the inverted repeat. *Gene with intron(s).
Pseudogenization events (gene duplication followed by loss of function) have been reported in several plant lineages, e.g., in the plastomes of Anthemideae tribe within the Asteraceae family and Cocus nucifera, which belongs to the Arecaceae family [8,25]. Among the genes that underwent pseudogenization there are ycf68, ycf1 and rps19, which showed incomplete duplication in the IRA/IRB and LSC junction regions with loss of function due to accumulation of premature stop codons or truncations. In both Cardamine species a partial duplication (106 bp) of the full-length copy of the rps19 gene (279 bp) located at the IRA/LSC boundary is found in the IRB/LSC region. The fact that only one gene copy is present in the outgroup N. officinale indicates that the duplication event leading to rps19 pseudogenization occurred after the split between Nasturtium and Cardamine. Sequencing of IRB/LSC regions from additional Cardamine species and closely related outgroups will be required to ascertain whether the psedogenization event is genus-specific or not. The conservation of pseudogene length and the close phylogenetic proximity of Nasturtium to Cardamine [26], however, point to a relatively recent origin of the causal duplication. The basal position of the clade comprising C. resedifolia further corroborates the view that the duplication possibly happened early during the radiation of the Cardamine genus [15].
Among the coding regions of the sequenced plastomes, the majority of genes have canonical ATG as bona-fide start codons. Only 3 genes (ndhD, psbC, rps19) had non-canonical or conflicting starting codon annotations compared to those in the reference plastomes deposited in GenBank, thus requiring manual curation. Previously, RNA editing events of the AUG initiation site to GUG have been reported for psbC [27] and rps19 [8,25]. Analogously (but not observed in our study), RNA editing events contributing to the change of the translational initiation codon to GUG have been reported also in cemA [28]. Previous studies on non-canonical translational mechanisms suggest that translational efficiency of GUG codons is relatively high as compared to canonical AUG as initiation codon [29]. It is, therefore, possible that the GTG start codons observed in Brassicaceae psbC and rps19 are required to ensure enhanced translational efficiency for these genes. Also in the case of ndhD we identified a bona fide non-canonical start codon (ACG), analogously to what observed in other dicotyledonous and monocotyledonous species [8,30,31]. The reported lack of conservation among congeneric Nicotiana species [32] and the ability of unedited ndhD mRNA to associate to polysomes [33], however, renders the adaptive relevance of this non-canonical start codon in Brassicaceae elusive.
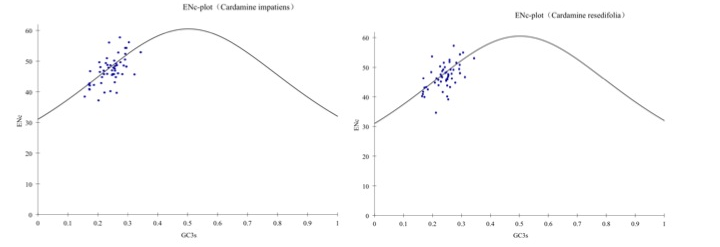
We further analyzed the codon usage frequency and the relative synonymous codon usage frequency (RSCU) in the two Cardamine plastomes. Mutational bias has been reported as an important force shaping codon usage in both animal and plant nuclear genomes [34,35]. Only few studies addressed the role of mutational bias in plant organelles, and earlier evidence pointed to a comparativley larger effect of natural selection in organellar biased usage of codons [36-38]. More recent studies, however, challenge this view and convincingly show that mutational bias can also be a dominant force in shaping the coding capacity of plant organelles and especially of Poaceace plastomes [39,40]. We, therefore, evaluated Nc plots to estimate the role of mutational bias in shaping the codon usage frequency in C. resedifolia and C. impatiens and found that most of the genes falls below the expected line of Nc, suggesting a relevant role of mutational bias in C. resedifolia and C. impatiens (Additional file 4: Figure S1). To provide support for the observed mutational bias, statistical analysis invoking Spearman-rank correlations (ρ) were further implemented between Nc and GC3s and were found to be significant in case of C. resedifolia (ρ = 0.557, p < 0.01) and C. impatiens (ρ = 0.595, p < 0.01). We also evaluated (ρ) between Nc and G3s and positive correlations (ρ = 0.620; C. impatiens, ρ = 0.597, C. resedifolia) were observed, which demonstrates the role of mutational bias in the biased codon usage frequency in C. resedifolia and C. impatiens. Taken together, these results indicate that in the two Cardamine plastomes sequenced in this study a major role is played by mutational bias, analogously to what suggested in the case of the Coffea arabica plastome [41]. Currently we do not have any data on translational efficiency in Cardamine, but we cannot exclude it as a possible factor contributing to codon bias in their plastomes as previously suggested in the case of O. sativa [42]. Our data, on the other hand, indicate a small fraction of positively selected amino acids (see below), suggesting only marginal contributions of natural selection to codon usage bias in Cardamine.
Distribution of repeat content and SSRs analysis
In addition to the larger repeats constituted by IRA and IRB, plastid genomes encompass a number of other repeated sequences. We employed REPUTER for the identification of the repeats, which are > 30 bp using a Hamming distance of 90. A total of 49 and 43 repeats were classified in the C. impatiens and the C. resedifolia plastome (Additional file 5: Table S4), values which are intermediate between those in Poaceae and Arecaceae and the one in Orchidaceae [8]. Among the perfect repeats, we detected four forward repeats, which are located in the LSC (spacer between trnL and trnF), and two palindromic repeats also localized in the LSC (spacer between psbT and psbN; Additional file 5: Table S4). Among the imperfect repeats, we annotated a total of 29 forward tandem repeats with a prevalence of them in the spacer between trnL and trnF and additional 14 palindromic repeats distributed throughout the plastome of C. impatiens. In C. resedifolia, we observed only two perfect repeats, both palindromic, located in the LSC (spacer between petN and psbM and spacer between psbE and petL; Additional file 5: Table S4). All others were imperfect repeats: 15 forward, two reverse and one compound tandem repeats. Interestingly, in C. resedifolia we did not observe the large number of repeats found in the trnL/trnF spacer of C. impatiens. As repeat organization and expansion in plastomes may induce recombination and rearrangements (e.g. in Poaceae and Geraniaceae) [8], the trnL/trnF spacer appears to be a particularly interesting region to reconstruct micro- and macro-evolutionary patterns in C. impatiens and closely related species like C. pectinata [43].
We further analyzed the distribution of the simple sequence repeats (SSRs), repetitive stretches of 1-6 bp distributed across nuclear and cytoplasmatic genomes, which are prone to mutational errors in replication. Previously, SSRs have been described as a major tool to unravel genome polymorphism across species and for the identification of new species on the basis of the repeat length polymorphism [44]. Since SSRs are prone to slip-strand mispairing, which is demonstrated as a primary source of microsatellite mutational expansion [45], we applied a length threshold greater than 10 bp for mono-, 4 bp for di- and tri- and 3 minimum repetitive units for tetra-, penta- and hexa-nucleotide repeats patterns. We observed a total of 169 SSRs in C. resedifolia and 145 SSRs stretches in C. impatiens (Additional file 6: Table S5). The observed number of repetitive stretches is in line with the previous results obtained in Brassicaceae [44,46] and other plastomes [23]. Among the observed repeats, the most abundant pattern was found to be stretches of mononucleotides (A/T) accounting for a total of 81 and 61 stretches of polyadenine (polyA) or polythymine (polyT) (A/T) followed by di-nucleotide patterns accounting for a total of 77 and 71 repetitive units in C. resedifolia and C. impatiens. Interestingly, we observed a higher tendency of longer repeats to occur species-specifically (see e.g. motifs such as AATAG/ATTCT in C. resedifolia and AACTAT/AGTTAT in C. impatiens; Additional file 6: Table S5), a possible consequence of their rarity [44,46]. Based on the identified SSR stretches, we provide a total of 127 and 114 SSR primer pairs in C. resedifolia and in C. impatiens, respectively (Additional file 6: Table S5), which can be used for future in-depth studies of phylogeography and population structure in these species.
Synteny conservation and phylogeny of sequenced Brassicaceae plastomes
Among the Brassicaceae species whose plastomes have been fully sequenced so far (a total of 15 at the time of the analyses), only Nasturtium officinale and Barbarea verna belong to the Cardamineae tribe like C. impatiens and C. resedifolia. As Nasturtium has been indicated as putative sister genus to Cardamine [26], the plastome of N. officinale was used as reference to calculate average nucleotide identity (ANI) plots using a window size of 1000 bp, step size of 200 bp and a alignment length of 700 bp, 70% identity. As expected by their close relatedness, a high degree of synteny conservation with the reference plastome was observed (Additional file 7: Figure S2). Average nucleotide identity value based on 748 and 568 fragments using one-way and two-way ANI indicated a similarity of 97.76% (SD 2.25%) and 97.55% (SD 2.17%) between C. resedifolia and N. officinale. Similarly, one-way and two-way ANI values of 98.19% (SD 1.88%) and 98.03% (SD 1.78%) based on 759 fragments and 603 fragments were observed in case of C. impatiens and N. officinale. Syntenic analysis of the coding regions across Brassicaceae and one outgroup belonging to the Caricaceae family (Carica papaya) revealed perfect conservation of gene order along the plastome of the analyzed species (Figure 3). Similarity among plastomes was a function of plastome organization and gene content, with IR and coding regions of fundamental genes being the most highly conserved, as indicated by analysis of pairwise mVISTA plots using C. impatiens as reference (Additional file 8: Figure S3).
Figure 3.
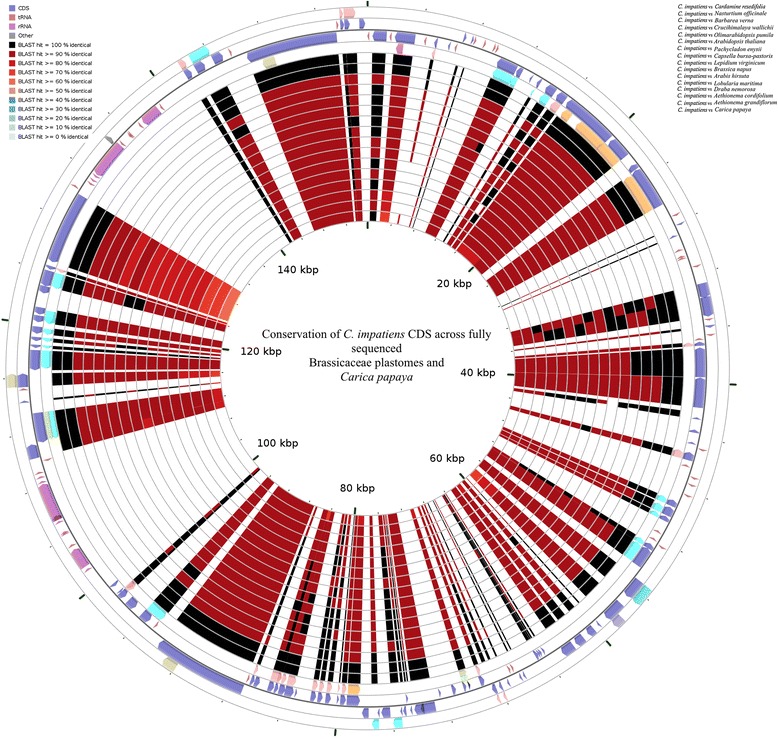
Circular map displaying the conservation of the coding regions across the Brassicacae, the Cardamine plastomes sequenced in this study and the outgroup Carica papaya.
To precisely determine the phylogenetic position and distance of C. resedifolia and C. impatiens with respect to the other Brassicaceae with fully sequenced plastome, we performed a concatenated codon-based sequence alignment of the 75 protein coding genes, representing a total of 67698 nucleotide positions. The GTR + I + G model resulted the best fitting model for the matrix according to the JModelTest program using the Akakie information criterion (AIC) and Bayesian information criterion (BIC). Phylogenetic reconstruction was carried out using maximum parsimony (MP), Maximum likelihood (ML) and Bayesian inference (BI). MP analysis resulted in a tree length of 15739, a consistency index of 0.819 and retention index of 0.646. ML analysis revealed a phylogenetic tree with the -lnL of 186099.2 using the GTR + I + G model as estimated using JModelTest. For MP and ML analysis, 1000 bootstrap replicates were evaluated and all the trees obtained were rooted using Carica papaya as an outgroup (Figure 4). All phylogenetic methods provided consistent topologies, indicating good reproducibility of the recovered phylogeny. The tree positioning of Lepidium virginicum, which lacked resolution in the MP tree, constituted the only exception. As expected, the four taxa from the Cardamineae tribe (genera Cardamine, Nasturtium and Barbarea) formed a well-supported, monophyletic clade with B. verna as most basal species. Our phylogenetic reconstruction is in agreement with previous reports on the relationships among Brassicaeacea tribes [47,48], thus indicating that it can be used as a reliable framework for assessment of protein coding gene evolution in the Brassicaeae family in general and Cardamine species in particular.
Figure 4.
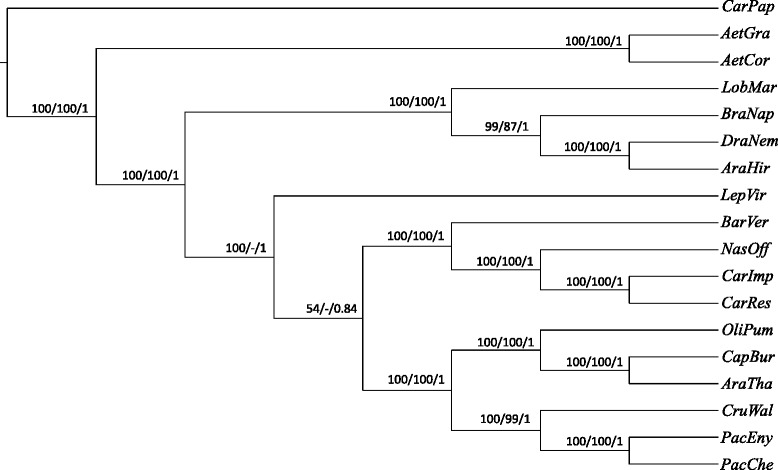
Cladogram of the phylogenetic relationships among Brassicaceae species with fully sequenced plastome used in this study. The cladogram represents the consensus topology of the maximum likelihood (ML), maximum parsimony (MP) and bayesian inference (BI) phylogenetic reconstructions using the concatenated alignment of 75 protein coding genes. Numbers on branches indicate ML/MP/BI support values (bootstrap proportion > 50%). Dashes indicate lack of statistical support. Abbreviation of species names can be found in Additional file 10: Table S7. Phylogenetic tree visualization was done using FigTree.
Molecular evolution of Brassicaceae plastomes
Understanding the patterns of divergence and adaptation among the members of specific phylogenetic clades can offer important clues about the forces driving its evolution [49,50]. To pinpoint whether any genes underwent adaptive evolution in Brassicaceae plastomes in general and in the Cardamine genus in particular, we carried out the identification of genes putatively under positive selection using Selecton. At the family level, we observed signatures of positive selection in 10 genes (ycf1, rbcL, rpoC2, rpl14, matK, petD, ndhF, ccsA, accD, and rpl20) at a significance level of 0.01 (Table 3). Two of these genes, namely ycf1 and accD, have been reported to undergo fast evolution in other plant lineages as well. ycf1 is one of the largest plastid genes and it has been classified as the most divergent one in plastomes of tracheophytes [5]. Despite it has been reported to be essential in tobacco [51], it has been lost from various angiosperm groups [52]. Recently, ycf1 was identified as one of the core proteins of the chloroplast inner envelope membrane protein translocon forming a complex (called TIC) with Tic100, Tic56, and Tic20-I [53]. None of the 24 amino acids putatively under positive selection in Brassicaceae are located in predicted transmembrane domains [53], indicating that in Brassicaceae evolution of predicted channel-forming residues is functionally constrained. Analogously to what found for Brassicaceae in our study, in the asterid lineage recent studies also show accelerated rates of evolution in accD, a plastid-encoded beta-carboxyl transferase subunit of acetyl-CoA carboxylase (ACCase) [54], which has been functionally re-located to nucleus in the Campanulaceae [55]. As in none of the fully sequenced Brassicaceae re-location of plastidial accD to the nuclear genome has been observed, it is likely that the fast evolution of this gene is independent from the genome from which it is expressed. On the other hand, accD has been demonstrated to be essential for proper chloroplast and leaf development [54]. Plastidial accD together with three nucleus-encoded subunits form the ACCase complex, which been reported to produce the large majority of malonyl CoA required for de novo synthesis of fatty acids [56,57] under the regulatory control of the PII protein [58]. Most importantly, there are direct evidences that accD can affect plant fitness and leaf longevity [59]. The signatures of positive selection observed in both Brassicaceae (our study) and asterids [55], therefore, indicate that this gene may have been repeatedly involved in the adaptation to specific ecological niches during the radiation of dicotyledonous plants.
Table 3.
Positive selection sites identified with selecton with d.f. =1
| Gene | Null | Positive | Putative sites under positive selection * |
|---|---|---|---|
| ycf1 | -21668,5 | -21647,6 | 24(343 P, 424 A, 533 D, 565 H, 970 L, 1293 L, 1313 N, 1399 R, 1400 N, 1414 R, 459 W, 564 I, 738 K, 922 F, 928 L, 1081 F, 1113 T, 1235 K, 1259 P, 1343 R, 1428 F, 1475 S, 1477 R, 1533 Y) |
| rbcL | -3000,07 | -2984,64 | 3(326 V, 472 V, 477 A) |
| rpoC2 | -11431,8 | -11423,5 | 7(490 F, 527 L, 540 P, 541 H, 981 A, 998 L, 1375 Y) |
| rpl14 | -631,147 | -623,836 | 2(18 K, 33 K) |
| matK | -5014,38 | -5007,21 | 1(51 V) |
| petD | -1052,21 | -1045,47 | 2(138 V, 139 V) |
| ndhF | -6497,59 | -6491,61 | 4(65 I, 509 F, 594 Q, 734 M) |
| ccsA | -3031,79 | -3026,12 | 5(97 H, 100 H, 176 L, 182 E, 184 F) |
| accD | -4142,84 | -4137,43 | 3(112 F, 167 H, 485 E) |
| rpl20 | -834,791 | -831,556 | 2(80 R, 117 E) |
*lower bound > 1.
“Null” and “Positive” columns list likelihood values obtained under the models M8a (null model) and M8 (positive selection), respectively.
Given the prominent role that plastid proteins play in the constitution of cores of photosynthetic complexes [60], one could expect that some photosynthetic genes would also be targeted by positive selection. Previous analyses in leptosporangiates, for instance, uncovered a burst of putatively adaptive changes in the psbA gene, which is coding for a core subunit of Photosystem II (PSII). Extensive residue co-evolution along with positive Darwinian selection was also detected [61]. However, we did not observe such burst of high rate of evolution in Brassicaceae psbA. We instead observed co-evolving residues along with positive signatures of Darwinian selection in rbcL (ribulose-1, 5-bisphosphate carboxylase/oxygenase), which codes for RUBISCO, the enzyme catalyzing photosynthetic assimilation of CO2 and one of the major rate-limiting steps in this process. Positive rates of selection were observed at three sites across Brassicacae. The observed rates of positive selection on neutral hydrophobic residues such A (alanine) and V (valine) are consistent with previous estimates of selection sites across land plants [62]. As compared to RUBISCO adaptive selection in gymnosperms, where previous reports suggest 7 sites under positive selection (A11V, Q14K, K30Q, S95N, V99A, I133L, and L225I) [63], the low frequency of the sites under positive selection observed in Brassicaceae, which belongs to Angiosperms, could be a consequence of the more recent origin of the latter group. The fact that the long series of geological variations of atmospheric CO2 concentrations experienced by gymnosperms seem to parallel adaptive bursts of co-evolution between RUBISCO and RUBISCO activase lend support to this view [63]. Recent studies across Amaranthaceae sensu lato identified multiple parallel replacements in both monocotyledonous and dicotyledonous C4 species at two residues (281 and 309), suggesting their association with selective advantages in terms of faster and less specific enzymatic activity (e.g. in C4 taxa or C3 species from cold habitats) [64]. We found no evidence of selection in these or other residues in their proximity in the crystal structure of RUBISCO, indicating that in the Brassicaceae species analyzed (including high altitude C. resedifolia) this kind of adaptation possibly did not occur. The three residues under positive selection in our study belong to RUBISCO loop 6 (amino acid 326 V) and C-terminus (amino acids 472 V and 477 A). None of these aminoacids belong to the set of highly conserved residues identified among RUBISCO and RUBISCO-like proteins, which are likely under strong purifying selection [65,66]. This result is in agreement with the observation that in monocotyledons adaptive mutations preferentially affect residues not directly involved in catalysis, but either aminoacids in proximity of the active site or at the interface between RUBISCO subunits [67]. The C-terminus of RUBISCO is involved in interactions between large subunits (intra-dimer) and with RUBISCO activase, and amino acid 472 was previously identified among rbcL residues evolving under positive selection [64]. It is, therefore, possible that the mutation in residues 472 and 477 could contribute to modulate the aggregation and/or activation state of the enzyme in Brassicaceae. Also amino acid 326 has consistently been identified as positively selected in different studies, although in relatively few plant groups [64]. This residue is in close proximity to the fourth among the most often positively selected RUBISCO residues in plants (amino acid 328), which has been associated to adaptive variation of RUBISCO active site possibly by modifying the position of H327, the residue coordinating the P5 phosphate of ribulose-1,5-bisphosphate [64,67]. Such “second shell mutations” in algae and cyanobacteria are known to be able to modulate RUBISCO catalytic parameters [68], and were recently shown to be implicated in the transition from C3 to C4 photosynthesis in monocotyledons by enhancing conformational flexibility of the open-closed transition [67]. Taken together, these data indicate that in Brassicaceae residue 326 could affect RUBISCO discrimination between CO2 and O2 fixation, analogously to what suggested for residue 328 in several other plant groups.
The other genes displaying signature of positive selection in our study belong to 4 main functional classes: transcription and transcript processing (rpoC2, matK), translation (rps14 and rpl20), photosynthetic electron transport and oxidoreduction (petD, ndhF), cytochrome biosynthesis (ccsA). The broad spectrum of candidate gene functional classes affected indicate that natural selection target different chloroplast functions, supporting the possible involvement of plastid genes in adaptation and speciation processes in the Brassicaceae family [69].
To obtain a more precise picture of the phylogenetic branch(es), where the putatively adaptive changes took place, the rate of substitution mapping on each individual branch was estimated by the MapNH algorithm [70]. Focusing on the Cardamineae tribe and using a branch length threshold to avoid bias towards shorther branches, we found that genes under positive selection in the Cardamine lineage (accD, ccsA, matK, ndhF, rpoC2) evolved faster in C. resedifolia as compared to C. impatiens, suggesting that adaptive changes may have occurred more frequently in response to the highly selective conditions of high altitude habitats (Additional file 9: Table S6). These results are in line with the accelerated evolutionary rates of cold-related genes observed for C. resedifolia in the transcriptome-wide comparison of its transcriptome to that of C. impatiens [22]. Given the different genomic inheritance and low number of genes encoded in the chloroplast, it is unfortunately difficult to directly compare the evolutionary patterns observed for photosynthetic plastid genes in this study with the strong purifying selection identified for nuclear-encoded photosynthetic genes of C. resedifolia [22]. It is, however, worth of note that the genes with larger differences in evolutionary rates between C. resedifolia and C. impatiens are not related to photosynthetic light reactions, suggesting that this function is likely under intense purifying selection also for plastidial subunits in Cardamine species (Additional file 9: Table S6). Given the relatively few studies available and the complex interplay among the many factors potentially affecting elevational adaptation in plants [71,72], however, additional studies will be needed to specifically address this point.
Conclusion
In conclusion, the comparative analysis of the de-novo sequences of Cardamine plastomes obtained in our study identified family-wide molecular signatures of positive selection along with mutationally biased codon usage frequency in Brassicaceae chloroplast genomes. We additionally found evidence that the plastid genes of C. resedifolia experienced more intense positive selection than those of the low altitude C. impatiens, possibly as a consequence of adaptation to high altitude environments. Taken together, these results provide a series of candidate plastid genes to be functionally tested for elucidating the driving forces underlying adaptation and evolution in this important plant family.
Methods
Illumina sequencing, plastome assembly, comparative plastomics and plastome repeats
Genomic DNA was extracted from young leaves of Cardamine impatiens and C. resedifolia using the DNeasy Plant Mini kit (Qiagen GmbH, Hilden, Germany) and Long PCR amplification with a set of 22 primer pairs was carried out using Advantage 2 polymerase mix (Clontech Laboratories Inc., Mountain View, CA, USA) according to manufacturer’s instructions. We chose to use a long-PCR whole plastome amplification approach to maximize the number of reads to be used for assembly. The primer pairs used are listed in Additional file 1: Table S1. Amplicons from each species were pooled in equimolar ratio, sheared with Covaris S220 (Covaris Inc., Woburn, MA, USA) to the average size of 400 bp and used for illumina sequencing library preparation. Each library was constructed with TruSeq DNA sample preparation kits V2 for paired-end sequencing (Illumina Inc., San Diego, CA) and sequenced on a HiSeq 2000 at The Genome Analysis Centre (Norwich, UK). Subsequently, the reads were quality filtered using a Q30 quality value cutoff using FASTX_Toolkit available from http://hannonlab.cshl.edu/fastx_toolkit/. After subsequent quality mapping on the Brassicaceae plastomes, contaminating reads were filtered off. Specifically, raw reads were mapped on the publicly available Brassicaceae plastomes (Additional file 10: Table S7) using the Burrows-Wheeler Aligner (BWA) programusing -n 2, -k 5 and -t 10. SAM and BAM files obtained as a result were consecutively filtered for the properly paired end (PE) reads using SAMtools [73].
To obtain the de novo plastome assembly, properly PE reads were assembled using Velvet assembler [74]. In Velvet, N50 and coverage were evaluated for all K-mers ranging from 37 to 73 in increments of 4. Finally, the plastome assembly with K-mer = 65 was used for all subsequent analyses in both species. The selected Velvet assembly was further scaffolded using optical read mapping as implemented in Opera [75]. Assembled scaffolds were further error corrected using the SEQUEL software by re-mapping the reads and extending/correcting the ends of the scaffolded regions [76]. Gap filling was performed using the GapFiller program with parameters –m 80 and 10 rounds of iterative gap filling [77]. All the given computational analysis was performed on a server equipped with 128 cores and a total of 512 GB.
Following scaffolding and gap filling, C. resedifolia and C. impatiens scaffolds were systematically contiguated based on the Nasturtium officinale plastome (AP009376.1, 155,105 bp) using the nucmer and show-tiling programs of the MUMmer package [78]. Finally, mummer plot from the same package was used to evaluate the syntenic plots and the organization of the inverted repeats by pairwise comparison between the N. officinale and C. resedifolia and C. impatiens plastomes. Due to assembler’s insufficient accuracy in assembly of repeat regions, manual curations of the IRs were carried out using the BLAST2Seq program by comparison of the scaffolded regions with the N. officinale plastome. To test assembly quality and coverage, average nucleotide identity plots were calculated. Additionally, the junctions of the IRs and all remaining regions containing Ns were amplified by PCR using the primers listed in Additional file 1: Table S1 and Sanger sequenced. The finished C. resedifolia and C. impatiens chloroplast sequences have been deposited to GenBank with accession numbers KJ136822 and KJ136821, respectively.
To assess the levels of plastid syntenic conservation, the assembled plastomes of C. resedifolia and C. impatiens were compared to all publicly available plastomes of Brassicaceae using CGview by computing pairwise similarity [79]. Additionally, mVISTA plots were constructed using the annotated features of C. resedifolia and C. impatiens plastomes with a rank probability of 0.7 (70% alignment conservation) to estimate genome-wide conservation profiles [80]. To identify the stretches of the repetitive units, the REPUTER program was used with parameters -f –p –r –c –l 30 –h 3 –s and the repeat patterns along with the corresponding genomic co-ordinates were tabulated [81]. Additionally, we mined the distribution of perfect and compound simple sequence repeats using MISA (http://pgrc.ipk-gatersleben.de/misa/). In our analysis, we defined a minimum repetitive stretch of 10 nucleotides as mono-nucleotide, a consecutive stretch of 4 repeats units to be classified as di- and tri-nucleotide, and a stretch of 3 repeat units for each tetra-, penta- and hexa-nucleotide stretches as simple sequence repeats (SSRs).
Chloroplast genome annotation and codon usage estimation
The assembled plastome of C. resedifolia and C. impatiens was annotated using cpGAVAS [82] and DOGMA (Dual Organellar GenoMe Annotator) [83]. Manual curation of start and stop codons was carried out using the 20 available reference Brassicaceae plastomes. The predicted coding regions were manually inspected and were re-sequenced with Sanger chemistry whenever large differences in conceptually translated protein sequences were detected compared with the reference plastome of N. officiale (Additional file 10: Table S7). GenomeVx [84] was used for visualization of plastome maps. Transfer-RNAs (t-RNAs) were identified using the t-RNAscan-SE software using the plastid genetic code and the covariance models of RNA secondary structure as implemented in cove algorithm [85]. Only coding regions longer than 300 bp from Cardamine and the other Brassicaceae plastomes were used for estimation of codon usage in CodonW with translational table = 11 (available from codonw.sourceforge.net). We further tabulated additional codon usage measures such as Nc (effective number of codons), GC3s (frequency of the GC at third synonymous position). GC, GC1, GC2 and GC3 were calculated with in-house Perl scripts. Estimation of the standard effective number of codon (Nc) was tabulated using the equation N(c) = 2 + s + 29/(s(2) + (1-s)(2)), where s denotes GC3s [86].
Molecular evolution in Cardamine plastomes
For evaluating the patterns of molecular evolution, codon alignment of the coding regions was created using MACSE, which allows the identification of frameshift events [87]. Model selection was performed using the JmodelTest 2 [88]. Phylogenetic reconstruction was performed using PhyML with 1000 bootstrap replicates [89]. To identify the role of selection on the evolution of plastid genes, MACSE codon alignments were analysed using Selecton [90] allowing for two models: M8 (model of positive selection) and M8a (null model) and likelihood scores were compared for each gene set followed by a chi-square test with 1 degree of freedom. Only tests with probability lower than 0.01 were considered significant and were classified as genes under positive selection. We further mapped the substitution rate on the phylogeny of the Brassicaceae species using MapNH [70] with a threshold of 10 to provide a reliable estimation of the braches under selection.
Availability of supporting data
The data set supporting the results of this article are available in the GenBank repository, Cardamine resedifolia plastome (GenBank accession number KJ136821) and C. impatiens (accession number KJ136822). The phylogenetic matrix and trees are available from Treebase (http://purl.org/phylo/treebase/phylows/study/TB2:S17255).
Acknowledgements
This work was supported by: the Autonomous Province of Trento (Italy) through core funding of the Ecogenomics group (EB, GS, ML, RV and CV) and the ACE-SAP project (regulation number 23, 12 June 2008, of the Servizio Universita’ e Ricerca Scientifica); the China Scholarship Council (BW, DQ, HS).
Abbreviations
- RUBISCO
Ribulose-1, 5-bisphosphate carboxylase/oxygenase
- IR
Inverted repeat region
- LSC
Large single copy region
- SSC
Small single copy region
- Bp
Base pair
- Nc
Effective number of codons used in a gene
- GC
Guanine-cytosine
- SSR
Simple sequence repeat
- ANI
Average nucleotide identity
Additional files
Long-range PCR primers used for tiled whole-plastome amplification.
Summary of distribution and localization of genes in the C. resedifolia and C. impatiens plastomes.
Genes with introns in C. resedifolia (a) and C. impatiens (b) plastome and length of exons and introns.
{kind=link}
Nc plot showing the distribution of the genes >300 bp in C. resedifolia and C. impatiens. The black line in the curve represents the standard effective number of codons (Nc) calculated using the equation N(c) = 2 + s + 29/(s(2) + (1-s)(2)), where s denotes GC3s (Wright [86]).
Distribution and localization of repeat sequences in cpDNA of C. impatiens and C. resedifolia.
Cumulative SSR frequency and corresponding primer pairs in C. resedifolia and C. impatiens.
{kind=link}
Average nucleotide identity plots of the C. resedifolia and C. impatiens against Nasturtium officinale.
{kind=link}
mVISTA plots showing genome-wise similarity between C. resedifolia, C. impatiens and N. officinale with rank probability of 70% and window size of 100 bp. The annotations displayed are derived from the C. impatiens plastome.
Phylogenetic distribution map of substitution rates using probabilistic substitution mapping under the homogenous model of sequence evolution.
Accessions and references for fully sequenced plastomes used in phylogenetic reconstruction and genome comparison in this study.
Footnotes
Shiliang Hu and Gaurav Sablok contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BW, DQ and EB helped to carry out lab work and draft the manuscript. ML contributed to conceive and design of the study, carried out all the phases of lab work, helped to draft the manuscript. GS carried out data analyses, drafted the manuscript. HS carried out data analyses and helped to draft the manuscript. RV helped to draft the manuscript. CV conceived, designed and coordinated the study, finalized the manuscript. All authors read and approved the final manuscript.
Contributor Information
Shiliang Hu, Email: shilang.hu@fmach.it.
Gaurav Sablok, Email: gaurav.sablok@fmach.it.
Bo Wang, Email: bo.wang@fmach.it.
Dong Qu, Email: qudong@gmail.com.
Enrico Barbaro, Email: enrico.barbaro@fmach.it.
Roberto Viola, Email: roberto.viola@fmach.it.
Mingai Li, Email: mingai.li@fmach.it.
Claudio Varotto, Email: claudio.varotto@fmach.it.
References
- 1.Wu J, Liu B, Cheng F, Ramchiary N, Choi SR, Lim YP, et al. Sequencing of chloroplast genome using whole cellular DNA and Solexa sequencing technology. Front Plant Sci. 2012;3:243. doi: 10.3389/fpls.2012.00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waters DLE, Nock CJ, Ishikawa R, Rice N, Henry RJ. Chloroplast genome sequence confirms distinctness of Australian and Asian wild rice. Ecol Evol. 2012;2:211–7. doi: 10.1002/ece3.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Plant C, Group W. A DNA barcode for land plants. Proc Natl Acad Sci U S A. 2009;106:12794–7. doi: 10.1073/pnas.0905845106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sugiura M. The chloroplast genome. Plant Mol Biol. 1992;19:149–68. doi: 10.1007/BF00015612. [DOI] [PubMed] [Google Scholar]
- 5.Kim K-J, Lee H-L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004;11:247–61. doi: 10.1093/dnares/11.4.247. [DOI] [PubMed] [Google Scholar]
- 6.Yang M, Zhang X, Liu G, Yin Y, Chen K, Yun Q, et al. The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.) PLoS One. 2010;5:e12762. doi: 10.1371/journal.pone.0012762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green BR. Chloroplast genomes of photosynthetic eukaryotes. Plant J. 2011;66:34–44. doi: 10.1111/j.1365-313X.2011.04541.x. [DOI] [PubMed] [Google Scholar]
- 8.Huang Y-Y, Matzke AJM, Matzke M. Complete sequence and comparative analysis of the chloroplast genome of coconut palm (Cocos nucifera) PLoS One. 2013;8:e74736. doi: 10.1371/journal.pone.0074736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braukmann T, Kuzmina M, Stefanović S. Plastid genome evolution across the genus Cuscuta (Convolvulaceae): two clades within subgenus Grammica exhibit extensive gene loss. J Exp Bot. 2013;64:977–89. doi: 10.1093/jxb/ers391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ku C, Hu JM, Kuo CH. Complete plastid genome sequence of the basal asterid Ardisia polysticta Miq. and comparative analyses of asterid plastid genomes. PLoS One. 2013;8:e62548. doi: 10.1371/journal.pone.0062548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Westhoff P, Herrmann RG. Complex RNA maturation in chloroplasts: the psbB operon from spinach. Eur J Biochem. 1988;171:551–64. doi: 10.1111/j.1432-1033.1988.tb13824.x. [DOI] [PubMed] [Google Scholar]
- 12.Barkan A. Expression of plastid genes: organelle-specific elaborations on a prokaryotic scaffold. Plant Physiol. 2011;155:1520–32. doi: 10.1104/pp.110.171231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kugita M. RNA editing in hornwort chloroplasts makes more than half the genes functional. Nucleic Acids Res. 2003;31:2417–23. doi: 10.1093/nar/gkg327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolf PG, Hasebe M, Rowe CA. High levels of RNA editing in a vascular plant chloroplast genome: analysis of transcripts from the fern Adiantum capillus-veneris. Gene. 2004;339:89–97. doi: 10.1016/j.gene.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 15.Carlsen T, Bleeker W, Hurka H, Elven R, Brochmann C. Biogeography and phylogeny of Cardamine (Brassicaceae) Ann Missouri Bot Gard. 2009;96:215–36. doi: 10.3417/2007047. [DOI] [Google Scholar]
- 16.Marhold K, Lihová J. Polyploidy, hybridization and reticulate evolution: lessons from the Brassicaceae. Plant Syst Evol. 2006;259:143–74. doi: 10.1007/s00606-006-0417-x. [DOI] [Google Scholar]
- 17.Lihová J, Marhold K. Worldwide phylogeny and biogeography of Cardamine flexuosa (Brassicaceae) and its relatives. Am J Bot. 2006;93:1206–21. doi: 10.3732/ajb.93.8.1206. [DOI] [PubMed] [Google Scholar]
- 18.Huffman KM. Investigation into the potential invasiveness of the exotic narrow-leaved bittercress, (Cardamine impatiens L.), Brassicaceae. Master’s Thesis. Virginia Polytechnic Institute and State University, Biological Sciences Department. 2008. [Google Scholar]
- 19.Canales C, Barkoulas M, Galinha C, Tsiantis M. Weeds of change: Cardamine hirsuta as a new model system for studying dissected leaf development. J Plant Res. 2010;123:25–33. doi: 10.1007/s10265-009-0263-3. [DOI] [PubMed] [Google Scholar]
- 20.Zhou C-M, Zhang T-Q, Wang X, Yu S, Lian H, Tang H, et al. Molecular basis of age-dependent vernalization in Cardamine flexuosa. Science. 2013;340:1097–100. doi: 10.1126/science.1234340. [DOI] [PubMed] [Google Scholar]
- 21.Morinaga SI, Nagano AJ, Miyazaki S, Kubo M, Demura T, Fukuda H, et al. Ecogenomics of cleistogamous and chasmogamous flowering: genome-wide gene expression patterns from cross-species microarray analysis in Cardamine kokaiensis (Brassicaceae) J Ecol. 2008;96:1086–97. doi: 10.1111/j.1365-2745.2008.01392.x. [DOI] [Google Scholar]
- 22.Ometto L, Li M, Bresadola L, Varotto C. Rates of evolution in stress-related genes are associated with habitat preference in two Cardamine lineages. BMC Evol Biol. 2012;12:7. doi: 10.1186/1471-2148-12-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qian J, Song J, Gao H, Zhu Y, Xu J, Pang X, et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS One. 2013;8:e57607. doi: 10.1371/journal.pone.0057607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hildebrand M, Hallick RB, Passavant CW, Bourque DP. Trans-splicing in chloroplasts: the rps 12 loci of Nicotiana tabacum. Proc Natl Acad Sci U S A. 1988;85:372–6. doi: 10.1073/pnas.85.2.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Huo N, Dong L, Wang Y, Zhang S, Young HA, et al. Complete Chloroplast genome sequences of Mongolia medicine Artemisia frigida and phylogenetic relationships with other plants. PLoS One. 2013;8:e57533. doi: 10.1371/journal.pone.0057533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sweeney PW, Price RA. Polyphyly of the genus Dentaria (Brassicaceae): evidence from trnL intron and ndhF sequence data. Syst Bot. 2000;25:468–78. doi: 10.2307/2666690. [DOI] [Google Scholar]
- 27.Kuroda H, Suzuki H, Kusumegi T, Hirose T, Yukawa Y, Sugiura M. Translation of psbC mRNAs starts from the downstream GUG, not the upstream AUG, and requires the extended Shine-Dalgarno sequence in tobacco chloroplasts. Plant Cell Physiol. 2007;48:1374–8. doi: 10.1093/pcp/pcm097. [DOI] [PubMed] [Google Scholar]
- 28.Moore MJ, Bell CD, Soltis PS, Soltis DE. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc Natl Acad Sci U S A. 2007;104:19363–8. doi: 10.1073/pnas.0708072104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rohde W, Gramstat A, Schmitz J, Tacke E, Prüfer D. Plant viruses as model systems for the study of non-canonical translation mechanisms in higher plants. J Gen Virol. 1994;75:2141–9. doi: 10.1099/0022-1317-75-9-2141. [DOI] [PubMed] [Google Scholar]
- 30.Neckermann K, Zeltz P, Igloi GL, Kössel H, Maier RM. The role of RNA editing in conservation of start codons in chloroplast genomes. Gene. 1994;146:177–82. doi: 10.1016/0378-1119(94)90290-9. [DOI] [PubMed] [Google Scholar]
- 31.Hirose T, Sugiura M. Both RNA editing and RNA cleavage are required for translation of tobacco chloroplast ndhD mRNA: a possible regulatory mechanism for the expression of a chloroplast operon consisting of functionally unrelated genes. EMBO J. 1997;16:6804–11. doi: 10.1093/emboj/16.22.6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sasaki T, Yukawa Y, Miyamoto T, Obokata J, Sugiura M. Identification of RNA editing sites in chloroplast transcripts from the maternal and paternal progenitors of tobacco (Nicotiana tabacum): comparative analysis shows the involvement of distinct trans-factors for ndhB editing. Mol Biol Evol. 2003;20:1028–35. doi: 10.1093/molbev/msg098. [DOI] [PubMed] [Google Scholar]
- 33.Zandueta-Criado A, Bock R. Surprising features of plastid ndhD transcripts: addition of non-encoded nucleotides and polysome association of mRNAs with an unedited start codon. Nucleic Acids Res. 2004;32:542–50. doi: 10.1093/nar/gkh217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawabe A, Miyashita NT. Patterns of codon usage bias in three dicot and four monocot plant species. Genes Genet Syst. 2003;78:343–52. doi: 10.1266/ggs.78.343. [DOI] [PubMed] [Google Scholar]
- 35.Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet. 2011;12:32–42. doi: 10.1038/nrg2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Q, Feng Y, Xue Q. Analysis of factors shaping codon usage in the mitochondrion genome of Oryza sativa. Mitochondrion. 2004;4:313–20. doi: 10.1016/j.mito.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 37.Liu Q, Xue Q. Comparative studies on codon usage pattern of chloroplasts and their host nuclear genes in four plant species. J Genet. 2005;84:55–62. doi: 10.1007/BF02715890. [DOI] [PubMed] [Google Scholar]
- 38.Zhang W, Zhou J, Li Z, Wang L, Gu X, Zhong Y. Comparative analysis of codon usage patterns among mitochondrion, chloroplast and nuclear genes in Triticum aestivum L. Acta Botanica Sinica. 2007;49:246–54. [Google Scholar]
- 39.Sablok G, Nayak KC, Vazquez F, Tatarinova TV. Synonymous codon usage, GC3, and evolutionary patterns across plastomes of three pooid model species: emerging grass genome models for monocots. Mol Biotechnol. 2011;49:116–28. doi: 10.1007/s12033-011-9383-9. [DOI] [PubMed] [Google Scholar]
- 40.Zhou M, Li X. Analysis of synonymous codon usage patterns in different plant mitochondrial genomes. Mol Biol Rep. 2009;36:2039–46. doi: 10.1007/s11033-008-9414-1. [DOI] [PubMed] [Google Scholar]
- 41.Nair RR, Nandhini MB, Monalisha E, Murugan K, Nagarajan S, Surya N, et al. Synonymous codon usage in chloroplast genome of Coffea arabica. Bioinformation. 2012;8:1096–104. doi: 10.6026/97320630081096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morton BR, So BG. Codon usage in plastid genes is correlated with context, position within the gene, and amino acid content. J Mol Evol. 2000;50:184–93. doi: 10.1007/s002399910020. [DOI] [PubMed] [Google Scholar]
- 43.Kučera J, Lihová J, Marhold K. Taxonomy and phylogeography of Cardamine impatiens and C. pectinata (Brassicaceae) Bot J Linn Soc. 2006;152:169–95. doi: 10.1111/j.1095-8339.2006.00559.x. [DOI] [Google Scholar]
- 44.Sablok G, Mudunuri SB, Patnana S, Popova M, Fares MA, La Porta N. Chloromitossrdb: open source repository of perfect and imperfect repeats in organelle genomes for evolutionary genomics. DNA Res. 2013;20:127–33. doi: 10.1093/dnares/dss038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schlötterer C, Harr B. Microsatellite instability. Encycl life Sci. 2001:1–4.
- 46.Gandhi SG, Awasthi P, Bedi YS. Analysis of SSR dynamics in chloroplast genomes of Brassicaceae family. Bioinformation. 2010;5:1–5. doi: 10.6026/97320630005016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Couvreur TLP, Franzke A, Al-shehbaz IA, Bakker FT, Koch A, Mummenhoff K. Molecular phylogenetics, temporal diversification, and principles of evolution in the mustard family (Brassicaceae) Mol Biol Evol. 2010;27:55–71. doi: 10.1093/molbev/msp202. [DOI] [PubMed] [Google Scholar]
- 48.Franzke A, Lysak MA, Al-Shehbaz IA, Koch MA, Mummenhoff K. Cabbage family affairs: the evolutionary history of Brassicaceae. Trends Plant Sci. 2011;16:108–16. doi: 10.1016/j.tplants.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 49.Duchene D, Bromham L. Rates of molecular evolution and diversification in plants: chloroplast substitution rates correlated with species-richness in the Proteaceae. BMC Evol Biol. 2013;13:65. doi: 10.1186/1471-2148-13-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wicke S, Schäferhoff B, Depamphilis CW, Müller KF. Disproportional plastome-wide increase of substitution rates and relaxed purifying selection in genes of carnivorous Lentibulariaceae. Mol Biol Evol. 2014;31:529–45. doi: 10.1093/molbev/mst261. [DOI] [PubMed] [Google Scholar]
- 51.Drescher A, Stephanie R, Calsa T, Carrer H, Bock R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 2000;22:97–104. doi: 10.1046/j.1365-313x.2000.00722.x. [DOI] [PubMed] [Google Scholar]
- 52.Huang JL, Sun GL, Zhang DM. Molecular evolution and phylogeny of the angiosperm ycf2 gene. J Syst Evol. 2010;48:240–8. doi: 10.1111/j.1759-6831.2010.00080.x. [DOI] [Google Scholar]
- 53.Kikuchi S, Bédard J, Hirano M, Hirabayashi Y, Oishi M, Imai M, et al. Uncovering the protein translocon at the chloroplast inner envelope membrane. Science. 2013;339:571–4. doi: 10.1126/science.1229262. [DOI] [PubMed] [Google Scholar]
- 54.Kode V, Mudd EA, Iamtham S, Day A. The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 2005;44:237–44. doi: 10.1111/j.1365-313X.2005.02533.x. [DOI] [PubMed] [Google Scholar]
- 55.Rousseau-Gueutin M, Huang X, Higginson E, Ayliffe M, Day A, Timmis JN. Potential functional replacement of the plastidic acetyl-CoA carboxylase subunit (accD) gene by recent transfers to the nucleus in some angiosperm lineages. Plant Physiol. 2013;161:1918–29. doi: 10.1104/pp.113.214528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohlrogge J, Browse J. Lipid biosynthesis. Plant Cell. 1995;7:957–70. doi: 10.1105/tpc.7.7.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sasaki Y, Nagano Y. Plant acetyl-CoA carboxylase: structure, biosynthesis, regulation, and gene manipulation for plant breeding. Biosci Biotechnol Biochem. 2004;68:1175–84. doi: 10.1271/bbb.68.1175. [DOI] [PubMed] [Google Scholar]
- 58.Feria Bourrellier AB, Valot B, Guillot A, Ambard-Bretteville F, Vidal J, Hodges M. Chloroplast acetyl-CoA carboxylase activity is 2-oxoglutarate-regulated by interaction of PII with the biotin carboxyl carrier subunit. Proc Natl Acad Sci U S A. 2010;107:502–7. doi: 10.1073/pnas.0910097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Madoka Y, Tomizawa K, Mizoi J, Nishida I, Nagano Y, Sasaki Y. Chloroplast transformation with modified accD operon increases acetyl- CoA carboxylase and causes extension of leaf longevity and increase in seed yield in tobacco. Plant Cell Physiol. 2002;43:1518–25. doi: 10.1093/pcp/pcf172. [DOI] [PubMed] [Google Scholar]
- 60.Allen JF, de Paula WBM, Puthiyaveetil S, Nield J. A structural phylogenetic map for chloroplast photosynthesis. Trends Plant Sci. 2011;16:645–55. doi: 10.1016/j.tplants.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 61.Sen L, Fares M, Su Y-J, Wang T. Molecular evolution of psbA gene in ferns: unraveling selective pressure and co-evolutionary pattern. BMC Evol Biol. 2012;12:145. doi: 10.1186/1471-2148-12-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang M, Kapralov MV, Anisimova M. Coevolution of amino acid residues in the key photosynthetic enzyme Rubisco. BMC Evol Biol. 2011;11:266. doi: 10.1186/1471-2148-11-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sen L, Fares MA, Liang B, Gao L, Wang B, Wang T, et al. Molecular evolution of rbcL in three gymnosperm families: identifying adaptive and coevolutionary patterns. Biol Direct. 2011;6:29. doi: 10.1186/1745-6150-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kapralov MV, Smith JAC, Filatov DA. Rubisco evolution in C4 eudicots: an analysis of Amaranthaceae sensu lato. PLoS One. 2012;7:e52974. doi: 10.1371/journal.pone.0052974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tabita FR, Hanson TE, Satagopan S, Witte BH, Kreel NE. Phylogenetic and evolutionary relationships of RubisCO and the RubisCO-like proteins and the functional lessons provided by diverse molecular forms. Philos Trans R Soc Lond B Biol Sci. 2008;363:2629–40. doi: 10.1098/rstb.2008.0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tabita FR, Hanson TE, Li H, Satagopan S, Singh J, Chan S. Function, structure, and evolution of the RubisCO-like proteins and their RubisCO homologs. Microbiol Mol Biol Rev. 2007;71:576–99. doi: 10.1128/MMBR.00015-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Studer RA, Christin P-A, Williams MA, Orengo CA. Stability-activity tradeoffs constrain the adaptive evolution of RubisCO. Proc Natl Acad Sci U S A. 2014;111:2223–8. doi: 10.1073/pnas.1310811111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Parry MAJ. Manipulation of Rubisco: the amount, activity, function and regulation. J Exp Bot. 2003;54:1321–33. doi: 10.1093/jxb/erg141. [DOI] [PubMed] [Google Scholar]
- 69.Greiner S, Bock R. Tuning a ménage à trois: co-evolution and co-adaptation of nuclear and organellar genomes in plants. Bioessays. 2013;35:354–65. doi: 10.1002/bies.201200137. [DOI] [PubMed] [Google Scholar]
- 70.Romiguier J, Figuet E, Galtier N, Douzery EJP, Boussau B, Dutheil JY, et al. Fast and robust characterization of time-heterogeneous sequence evolutionary processes using substitution mapping. PLoS One. 2012;7:e33852. doi: 10.1371/journal.pone.0033852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gale J. Plants and altitude–revisited. Ann Bot. 2004;94:199. doi: 10.1093/aob/mch143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shi Z, Liu S, Liu X, Centritto M. Altitudinal variation in photosynthetic capacity, diffusional conductance and δ13C of butterfly bush (Buddleja davidii) plants growing at high elevations. Physiol Plant. 2006;128:722–31. doi: 10.1111/j.1399-3054.2006.00805.x. [DOI] [Google Scholar]
- 73.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao S, Sung WK, Nagarajan N. Opera: reconstructing optimal genomic scaffolds with high-throughput paired-end sequences. J Comput Biol. 2011;18:1681–91. doi: 10.1089/cmb.2011.0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ronen R, Boucher C, Chitsaz H, Pevzner P. sEQuel: improving the accuracy of genome assemblies. Bioinformatics. 2012;28:i188–96. doi: 10.1093/bioinformatics/bts219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boetzer M, Pirovano W. Toward almost closed genomes with GapFiller. Genome Biol. 2012;13:R56. doi: 10.1186/gb-2012-13-6-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Delcher AL, Phillippy A, Carlton J, Salzberg SL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002;30:2478–83. doi: 10.1093/nar/30.11.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grant JR, Arantes AS, Stothard P. Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genomics. 2012;13:202. doi: 10.1186/1471-2164-13-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32:W273–9. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29:4633–42. doi: 10.1093/nar/29.22.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu C, Shi L, Zhu Y, Chen H, Zhang J, Lin X, et al. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics. 2012;13:715. doi: 10.1186/1471-2164-13-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20:3252–5. doi: 10.1093/bioinformatics/bth352. [DOI] [PubMed] [Google Scholar]
- 84.Conant GC, Wolfe KH. GenomeVx: simple web-based creation of editable circular chromosome maps. Bioinformatics. 2008;24:861–2. doi: 10.1093/bioinformatics/btm598. [DOI] [PubMed] [Google Scholar]
- 85.Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005;33:W686–9. doi: 10.1093/nar/gki366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wright F. The “effective number of codons” used in a gene. Gene. 1990;87:23–9. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- 87.Ranwez V, Harispe S, Delsuc F, Douzery EJP. MACSE: Multiple Alignment of Coding SEquences accounting for frameshifts and stop codons. PLoS One. 2011;6:e22594. doi: 10.1371/journal.pone.0022594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 90.Stern A, Doron-Faigenboim A, Erez E, Martz E, Bacharach E, Pupko T. Selecton 2007: advanced models for detecting positive and purifying selection using a Bayesian inference approach. Nucleic Acids Res. 2007;35:W506-11. doi: 10.1093/nar/gkm382. [DOI] [PMC free article] [PubMed] [Google Scholar]