

Abstract
Frankia strains are nitrogen-fixing soil actinobacteria that can form root symbioses with actinorhizal plants. Phylogenetically, symbiotic frankiae can be divided into three clusters, and this division also corresponds to host specificity groups. The strains of cluster II which form symbioses with actinorhizal Rosales and Cucurbitales, thus displaying a broad host range, show suprisingly low genetic diversity and to date can not be cultured. The genome of the first representative of this cluster, Candidatus Frankia datiscae Dg1 (Dg1), a microsymbiont of Datisca glomerata, was recently sequenced. A phylogenetic analysis of 50 different housekeeping genes of Dg1 and three published Frankia genomes showed that cluster II is basal among the symbiotic Frankia clusters. Detailed analysis showed that nodules of D. glomerata, independent of the origin of the inoculum, contain several closely related cluster II Frankia operational taxonomic units. Actinorhizal plants and legumes both belong to the nitrogen-fixing plant clade, and bacterial signaling in both groups involves the common symbiotic pathway also used by arbuscular mycorrhizal fungi. However, so far, no molecules resembling rhizobial Nod factors could be isolated from Frankia cultures. Alone among Frankia genomes available to date, the genome of Dg1 contains the canonical nod genes nodA, nodB and nodC known from rhizobia, and these genes are arranged in two operons which are expressed in D. glomerata nodules. Furthermore, Frankia Dg1 nodC was able to partially complement a Rhizobium leguminosarum A34 nodC::Tn5 mutant. Phylogenetic analysis showed that Dg1 Nod proteins are positioned at the root of both α- and β-rhizobial NodABC proteins. NodA-like acyl transferases were found across the phylum Actinobacteria, but among Proteobacteria only in nodulators. Taken together, our evidence indicates an Actinobacterial origin of rhizobial Nod factors.
Introduction
Actinorhizal root nodule symbioses are formed between nitrogen-fixing actinobacteria from the genus Frankia and a diverse group of mostly woody dicotyledonous plants from 23 genera and eight different families [1]. These families group in three orders, Fagales (Betulaceae, Casuarinaceae and Myricaceae), Rosales (Elaeagnaceae, Rhamnaceae and Rosaceae) and Cucurbitales (Coriariaceae and Datiscaceae). These plants, together with the legumes and Parasponia (Cannabaceae) which establish root nodule symbioses with rhizobia, belong to a clade of angiosperms known as the nitrogen-fixing clade (NFC; [2]). The scattered distribution of nitrogen-fixing plants within this relatively small clade supports a common origin of the predisposition to evolve root nodule symbioses assumed to have arisen ca. 100 million years ago (mya; [3,4]). This presumed gain-of-function evolved only once; however, among the plants with the predisposition, subsequently nitrogen-fixing root nodule symbioses evolved several times independently, with common themes but differences in infection process, nodule structure, nodule physiology and microsymbiont specificity. Using a database of nearly 3500 species within the NFC and the angiosperm phylogeny of Zanne et al. [5], Werner et al. [4] tested a series of models and came to the conclusion that the earliest root-nodule symbiosis arose in the common ancestors of actinorhizal Cucurbitales, followed by those of legumes, followed by actinorhizal Casuarinaceae and Rosaceae, and then by the other groups of actinorhizal plants.
Based on 16S rDNA phylogeny, the genus Frankia has been divided into four clusters. The basal group consists of so-called ‘atypical’ or ‘Frankia-like’ strains that have been isolated from nodules but cannot induce nodule formation, also called Cluster IV. This group seems to be closely related to rhizosphere strains previously detected only by direct amplification of 16S rDNA [6], and forms a heterologous group of Frankia strains with the highest diversity [7]. Symbiotic Frankia strains make up the other three clusters, which also correspond in general to host specificity groups. While cross inoculation may be possible within a cluster, generally not all strains of a cluster can colonize all of the cluster’s host plants [7]. Cluster I is the largest and most divergent one; its members are capable of colonizing species of the three actinorhizal families of the order Fagales, Betulaceae, Casuarinaceae and Myricaceae. Cluster III strains form nitrogen-fixing root nodule symbioses with the host-plant genus Gymnostoma (Casuarinaceae, in Fagales), Elaeagnaceae (Rosales), and all actinorhizal genera of the Rhamnaceae (Rosales) except Ceanothus. Cluster II strains nodulate members of the families Coriariaceae and Datiscaceae (Cucurbitales); as well as all the actinorhizal members of the Rosaceae and Ceanothus. In contrast with strains of Clusters I and III, Cluster II strains could so far not be cultured despite numerous attempts, leading to the suggestion that they might be obligate symbionts or at least have lower saprotrophic capabilities than the strains from the other clusters [8]. The relative phylogenetic positions of the three Frankia clusters within the genus has fluctuated, in that different genes used for phylogenetic analysis led to different topologies [7,9]. However a recent study using a concatenation of 54 proteins and other measures shows that Cluster II is basal to the other two clusters [10].
The genomes of three strains of Cluster I (Frankia alni ACN14a, QA3 and Frankia sp. CcI3) and of four strain of Cluster III (Frankia sp. EAN1pec, EUN1f, BCU110501 and BMG5.12) have been published [11,12,13,14,15]). The large genome size variation between 9.3 Mb (EUN1f) and 5.43 Mb (CcI3) has been suggested to reflect differences in saprotrophic potential [11].
In spite of their large host range, Cluster II strains display a remarkably low level of genetic diversity [16] which in an ancient lineage is evocative of a recent evolutionary bottleneck. Such a bottleneck could be associated with previous abundance of certain host plants, followed by a catastrophic event leading to loss or reduction of autonomy of the microsymbiont. Interestingly, one of the host plant genera nodulated by Cluster II strains is Dryas (Rosaceae, Rosales), shown to initiate colonization of gravel till following glacial retreat, due to its capacity for biological nitrogen fixation [17], and which once was so abundant that a stadial during an interglacial age was named after it. This genus appears to be losing its symbiotic potential since one of the three known species, the Eurasian D. octopetala is systematically found devoid of nodules [18]. Two other Cluster II host genera, Datisca and Coriaria (Cucurbitales), also have a disjunct distribution evocative of taxa threatened by extinction [19,20]. In the case of Datisca, one species (Datisca glomerata) is found in California and northern Baja California, Mexico, while the other species (Datisca cannabina) is found in the east Mediterranean and in the foothills of the Himalayas in Pakistan and northwestern India [21].
The genome of the first representative of Cluster II, Candidatus Frankia datiscae Dg1 was sequenced using bacteria isolated from the infected cells of nodules (Dg1; NC_015656.1 (chromosome) and NC_015664.1 (plasmid); [21]). Consistent with the hypothesis that genome size reflects saprotrophic, not symbiotic potential, the genome of Dg1 has, at 5.32 Mb, one of the smallest Frankia genome known thus far, suggesting a genomic reduction that could correspond to a reduced saprotrophic lifestyle. At the same time, the difference in genome size between Dg1 and the Cluster I strain CcI3, which can be cultured, is not large. In this study, the Frankia genomes that were first published—ACN14a, CcI3 and EAN1pec [11] will be used for comparisons.
The capability to form root nodule symbioses evolved in part by recruiting mechanisms adapted from the evolutionarily older arbuscular mycorrhizal (AM) symbioses [22]. In legume/rhizobia symbioses, flavonoids exuded by plant roots induce expression of bacterial nodulation (nod) genes leading to the synthesis of lipo-chito-oligosaccharide (LCO) signal molecules called Nod factors. These LCOs are perceived by plant kinases of the LysM-RLK family and activate a signaling pathway, the common symbiotic pathway, that controls both legume/rhizobia and AM symbioses. Also AM fungi produce LCO signal factors [23], although in their case, there is no evidence to link these factors to host specificity. Results obtained using the only non-legume nodulated by rhizobia, Parasponia sp., show that the establishment of an AM symbiosis requires a LysM receptor kinase [24]. The common symbiotic pathway, the known components of which include the receptor kinase SymRK and the calcium- and calmodulin dependent kinase CCaMK [25] is also used for microsymbiont signaling in actinorhizal symbioses, as shown for SymRK in D. glomerata (Cucurbitales; [26]), for SymRK and CCaMK in Casuarina glauca (Fagales; [27,28]) and supported by the presence of homologs of the whole symbiotic cascade in Alnus glutinosa and C. glauca [29] as well as D. glomerata [30]. Since LCOs or, alternatively, short-chain chitin oligomers (COs) are thought to be involved in signaling via the common symbiotic pathway in AM symbioses [23,31], the ancestral symbiosis from which features were recruited for both legume and actinorhizal symbioses, it seems likely that the symbiotic signals produced by Frankia strains were also LCO-like or CO-like compounds that are perceived by LysM receptor kinases.
However, no signaling substances with the chemical properties of LCOs have been detected to date in Frankia culture supernatants [32]. At the genome level, synthesis of the acylated chitin oligomer backbone of Nod factors requires the activity of three specific enzymes, encoded by the so-called “canonical nod genes” nodABC (NodA—acyl transferase, NodB—chitin deacetylase, NodC—chitin synthase), which form a cluster present in all rhizobia characterized thus far except for one subgroup that nodulates stems of plants of the genus Aeschynomene [33]. No homologs of these canonical nod genes were found in the genomes of the Frankia strains from Clusters I and III [11], aside from genes encoding polysaccharide deacetylases (but with low similarity to the NodB subfamily) and chitin synthases (but not of the NodC subfamily). As a result of whole-genome sequencing, finishing, and annotation [21], we identified homologs of the canonical nod genes in the genome of the Cluster II Frankia strain, Dg1 (Fig 1) and thus set out to assess their phylogeny and function.
Fig 1. Circular map of the Dg1 genome.

The outer circle shows the GC content (above 50%), the next circle shows the positions of IS elements (for detailed positions, see S2 Table). The next circle shows the results of mapping the transcriptome of a closely related cluster II strain on the Dg1 genome: the red peaks represent the genes expressed in the top 5% of expression levels, the grey peaks represent genes represented by at least one read per kb. The next circle shows the operons of genes encoding enzymes involved in the biosynthesis of secondary metabolites (S4 Table): blue color denotes genes which are represented in the symbiotic transcriptome, grey color operons that are not. The last circle shows the positions of the two operons of canonical nod genes (nodA’B1A and nodB2C) and the nif gene operon. The nod operons of Dg1 are depicted in detail. The nodA’B1A operon is at position 2,490,158–2,489,200 of the genome, the nodB2C operon at position 3,662,361–3,664,562. The latter is followed by the nltIJ genes at position 3,664,716–3,666,622.
Materials and Methods
Frankia sp. phylogeny based on 50 housekeeping genes
A set of 50 housekeeping genes was identified in Candidatus Frankia datiscae Dg1 and then used in a BLASTP search as the query. The corresponding BLAST searches were restricted to Frankia alni ACN14a, Frankia sp. Ccl3, Frankia sp. EAN1pec, Acidothermus cellulolyticus 11B, Geodermatophilus obscurus DSM 43160, Nakamurella multipartita DSM 44233, Stackebrandtia nassauensis DSM 44728 and Thermobifida fusca YX. All alignments were created using MUSCLE (multiple sequence comparison by log- expectation; [34]) at the EMBL-EBI website. Maximum parsimony analyses were performed using the software package PAUP* version 4.0b10 [35]. All characters were weighted equally and gaps in the alignment were treated as missing. A heuristic search strategy with 10 random replicates, TBR branch-swapping and the MULTREES optimization was used. MAXTREES parameter was set to 10,000 per replicate. Support for branches was evaluated using bootstrap analysis [36] and random sequence addition for 100 replicates, using the same parameters.
Nod protein sequence phylogenies
NodA, NodB, NodC, NodI and NodJ protein sequences were obtained from GenBank (NCBI) [37] by a multistep BLAST procedure using the protein sequence from Dg1 NodA, NodB, NodC, NodI and NodJ (GenBank accession numbers AEH09514, AEH10396, AEH10398 and AEH10399, respectively) as query. A BLASTP search of the non-redundant protein sequence (nr) database using default parameters (word size = 3, Expect threshold = 10, limiting to 100 target sequences, BLOSUM matrix, and an 11:1 gap cost/extension) generated 100 protein sequences of high similarity. To identify more distantly related potential homologs within the Actinobacteria and Proteobacteria, five iterations of PSI-BLAST were conducted using default parameters (word size = 3, Expect threshold = 10, limiting to 500 target sequences, BLOSUM matrix, and an 11:1 gap cost/extension and a threshold for inclusion of 0.0001). Between 30 and 40 sequences were evaluated for analyses that maximized both sequence and taxonomic diversity. Some NodA sequences from diverse taxa in GenBank were eliminated due to short sequence length and/or origin in a nodule metagenome. For Dg1 NodA (AEH09513) homologs in actinobacteria, a BLASTP search for Actinobacterial sequences with similarity of 1e-10 or better in the NCBI and additionally in the JGI databases. Nineteen sequences were found in each database (not including Dg1). From these 38, sequences with query coverage of less than 75% were removed. Also, some of the remaining sequences were identical or a partial sequence of another. In such cases, only the longest sequence was kept. This way, altogether 18 unique actinobacterial sequences of NodA homologs were identified.
All alignments were created using MUSCLE [34] at the EMBL-EBI website, followed by manual adjustment. Selection of the appropriate amino acid substitution model was determined using PROTTEST 2.4 [38] through the server housed at the University of Vigo (http://darwin.uvigo.es/software/prottest2_server.html). Maximum Likelihood analyses for all alignments (NodA, NodB, NodC, NodI, and NodJ) were performed using PhyML 3.0 [39] on the ATGC bioinformatics server. The settings for the NodA run included the JTT model and rate variation sampled from a gamma distribution (JTT+G). The settings for the NodB run included the WAG model, estimated invariant sites, and rate variation sampled from a gamma distribution (WAG+I+G). NodC settings included the LG model, estimated invariant sites, and rate variation sampled from a gamma distribution (LG+I+G). NodI and NodJ settings were LG+G. Support for branches was evaluated using bootstrap analysis [40] and random sequence addition for 100 replicates, using the same parameters.
Transcriptome analysis
Seedlings of Datisca glomerata (Presl.) Baill were grown in a greenhouse (UC Davis) and inoculated with rhizosphere soil from previously-nodulated Ceanothus spp. Nodules from two batches of four plants each were snap-frozen in liquid nitrogen at 4 or 6 weeks after inoculation, and pooled by weight. Total nodule RNA was extracted after grinding tissue in liquid nitrogen, using the Plant RNeasy kit (Qiagen, Valencia, CA, USA). Ribosomal RNA was twice subtracted from total RNA with the RiboMinus Plant Kit for RNAseq (Invitrogen, Carlsbad, CA, USA). A cDNA library template was constructed, fragmented, and gel-purified, and the resulting 250 bp average fragments were amplified, according to Illumina (San Diego, CA, USA) protocols. RNA quality and length were checked at each step with a Bioanalyzer (Agilent, Santa Clara, CA, USA). Total RNA was sequenced in a single lane on an Illumina GAIIx system producing ca. 27 million reads (40 bp single reads). A total of 3,712,391 reads mapped to the complete genome of the Frankia strain Dg1 (GenBank: CP002801- CP002803) using Bowtie [41]. Ribosomal RNA (16S, 23S and 5S) accounted for 96% of the total mapped reads (3,566,390 reads). The remaining 146,001 reads were filtered for exact duplicate sequences (likely PCR artifacts), sorted and converted to BAM files using the samtools package [42], leaving a final 66,962 reads which were analyzed.
Data visualization and browsing were conducted with the Integrative Genomics Viewer software [43]. Gene expression was calculated as the number of mapped reads per kilobase of gene sequence (rpkb) using custom R scripts [44]. Standard database fields in NCBI were used for gene annotation.
The rpkb data were fitted with a negative binomial distribution (S1 Fig) using the fitdistrplus package in R [45]. Expression quantiles (99%, 95%, 90%, 80%, 70%, and 60%) were computed. Expression of complete or nearly complete metabolic pathways was determined by importing the genome and transcriptome into BioCyc [46], and using Pathway Tools [47]. Pathways, gene and protein annotations, and functional gene clusters were cross-checked in the KEGG Pathway Database supplemented with other sequence databases. A subset of total pathways was selected for further analysis based on high relative expression values, and a minimum of three genes per pathway. Statistically significant overrepresentation of key biosynthetic pathways in the active transcriptome was assessed for genes in each of the quantile bins using a hypergeometric test.
Quantitative reverse transcription-PCR (qPCR) analysis
For analysis of nod gene expression in symbiosis, total RNA was isolated from D. glomerata nodules after grinding in liquid nitrogen and five passages of 1 min each in a Tissuelyzer (Qiagen), using the RNeasy Plant Mini Kit with on-column DNase digestion (Qiagen). Reverse transcription was performed on 1 μg total RNA from three biological replicates in a final volume of 20 μl, using the TATAA GrandScript cDNA Synthesis Kit (TATAA Biocenter, Gothenburg, Sweden) following the protocol provided by the manufacturer. All qPCR assays contained 1x Maxima SYBR Green qPCR Master Mix (Fermentas, Vilnius, Lithuania), 300 nM of each primer, 2 μl of 10x diluted cDNA in a total reaction volume of 10 μl. qPCR was conducted under the following conditions: 10 min of initial denaturation at 95°C, 40 cycles of 15 s at 95°C, 30 s at 60°C and 30 s at 72°C, followed by steps for dissociation curve generation (15 s at 95°C, 15 s at 60°C and 15 s at 95°C). Primer sequences are given in S1 Table. Assay performance was evaluated with a standard curve. The Eco Real-Time PCR system (Illumina, San Diego, CA, USA) was used for data collection and standard curve generation. PCR products were validated by dissociation curve analysis and agarose gel electrophoresis. Assays were analyzed in triplicate with the standard curve method [48]. PCR efficiency was calculated with the Eco software with data obtained from the exponential phase of each amplification plot. Primers were designed using Beacon Designer software (PREMIER Biosoft, Palo Alto, USA; S1 Table). Gene expression data was normalized against expression of the translation initiation factor gene IF-3. Data analysis including data pre-processing and normalization was performed with GenEx (version 5.4.1, MultiD Analyses, Gothenburg, Sweden).
For qPCR analysis of nitrogen assimilation genes, reverse transcription was performed on 1 μg total RNA from three biological replicates in a final volume of 25 μl, using the MMLV-RT Promega M170A kit following the protocol provided by the manufacturer [49]. All qPCR assays contained 1x Maxima SYBR Green qPCR Master Mix (Applied Biosystems), 5 μM of each primer, 1 μl of 10x diluted cDNA in a total reaction volume of 20 μl. qPCR was conducted under the following conditions: 10 min of initial denaturation at 95°C, 40 cycles of 15 s at 95°C, 30 s at 58°C in a Termociclador ABI Prism 7500. Primer sequences for: asl, 5’-GAACACCGGCTCCTTGTCCTC-3’ and 5’- CGTCGAGCTGGGTTTCGACTC-3’; for gogatFD, 5’ GATGGTGGCGGTGTACTTCT-3’ and 5’- TGCTGAAGGTCATGTCCAAG-5’; for glnI, 5’-TTCGCTTCTGTGACCTTCCT-3’ and 5’- GTCGTAGCGTACGTCGTCAA-3’; for glnII, 5’-CAGGCCTACGAGAAGTACGC-3’ and 5’- CCTGGTGGAGAAGTTGGTGT-3’; for argJ, 5’-GTTCGTCCAGACCGTCAGTT-3’ and 5’- GGTGCAACCTGCTCAAGTG-3’. Primers for the reference gene, 16S rRNA, were 5’-GGGGTCCGTAAGGGTC-3´ and 5´-CCGGGTTTCCCCATTCGG-3´.
Genome analyses
The Dg1 genome consisted of a chromosome (CP002801) and two plasmids, pFSYMDG01 (CP002802) and pFSYMDG02 (CP002803). Since pFSYMDG01 is represented in the transcriptome while pFSYMDG02 is not, and pFSYMDG02 shows 100% homology with yeast transposons (see e.g., AP012213.1), we can conclude that pFSYMDG02 represents a contamination.
The analysis of the genome sequences with regard to biochemical pathways in Dg1 was performed using Pathway tools [40], MAGE, and IMG/ER. Secondary metabolism (of ACN14a, CcI3 EAN1pec, CcI3, and Dg1) was analyzed using antiSMASH (http://antismash.secondarymetabolites.org; [50]). The core genome between four sequenced Frankia strains (ACN14a, EAN1pec, CcI3, and Dg1) was determined using EDGAR (http://edgar.cebitec.uni-bielefeld.de; [51]). The Phyloprofile function of the MAGE platform [52] was used to extract those genes present in the ACN14a, CcI3 and EAN1pec but absent in Dg1 at a sequence similarity of 30% over a length of 80% of the shortest sequence. Palindromic Repeats were analyzed with the palindrome tool from EMBOSS (http://bips.u-strasbg.fr/EMBOSS/) with no mismatches and the following parameters: 1. Repeat units between 8 and 11 bases with up to a 3 base gap. 2. Repeat units between 12 and 19 bases with up to a 7 base gap. 3. Repeat units between 20 and 90 bases with up to a 20 base gap. 4. Repeat units of less than 12 bases must occur at least 10 times in the genome. 5. Repeat units of less than 20 bases must occur twice in the genome. Tandem repeats was analyzed with the MUMmer 3.13 package (http://www.tigr.org/software/mummer/) with the following parameters: Minimum match length = 20 bases. 2. It is assumed that one copy of a tandem repeat in a genome is not very significant unless it is long. Therefore, a genome-wide screen for the repeat used was added. The total number of bases incorporated into repeats for a particular repeat unit must total 50 or more bases.
Pseudogene counts were based on analysis by the IMG (Integrated Microbial Genomes) platform of JGI.
Finding and identifying repeats
To identify ISs in the investigated organisms, a database of potential ISs was assembled from two sources: First, the program RepeatScout [53] was used to identify all repeated nucleotide sequences with >500 nt length in the investigated organisms. Repeats associated to non-mobile repeated genes, e.g. rRNA, photosynthesis genes and other regular genes present in multiple copies in the genome, were removed. This yielded 173 putative IS elements.
Second, 4512 likely ISs identified in a previous investigation [54] were added to the database. This database included 3377 repeats from the ISfinder site [55], a web repository of known ISs, as well as 1135 repeats from cyanobacteria, Frankia genomes available in 2012 and other known symbiotic bacteria. The repeats found by RepeatScout were named with an abbreviation of the name of the originating organism, the letter ‘R’ and a number, e.g. ‘Npun_R_21’ is a repeat from the organism N. punctiforme, the twenty-first found by RepeatScout. The ISs from ISfinder retained their original names, all starting with ‘IS’. ISfinder and RepeatScout sequences were collected into a single Fasta file. Redundancies, generated when ISfinder sequences were also detected by RepeatScout, were removed, with the ISfinder name taking precedence. Each of the collected nucleotide sequences was used as queries in a BlastN search against each of the genomes, one at a time. All areas of the genome that received hits with a BlastN expect value <1e-6 were collected. Often several queries scored hits on the same region of a genome, together making up a “footprint” in the genome.
In the next step, each footprint was analysed to determine what repeats it consisted of. This was performed by using the footprints as queries against the database of repeats. The repeat sequence that received the highest score with the footprint as query was chosen as the most likely IS to occupy the footprint. In some cases, the best scoring repeat didn’t cover the entire footprint. The search was then repeated with the remaining footprint as query. The process was repeated until all parts of the footprints had been identified. The whole process yielded a GenBank file for each of the investigated genomes, where the verified and putative ISs are described with position, kind and quality of identification (S2 Table).
Analysis of nodule occupancy
Seedlings of Datisca glomerata were grown and nodulated under controlled conditions in two locations: at the Department of Plant Ecology, Stockholm University (SU) and the Department of Plant Sciences, University of California, Davis (UCD). The inoculum source for the SU plants was a ground nodule suspension containing Frankia originally from Pakistan [56]; the inoculum source for the UCD plants was soil from the rhizosphere of nodulated Ceanothus griseus in California. DNA was extracted from several nodules from a combination of at least four plants, using the QIAGEN DNeasy plant mini kit following the manufacturer’s instructions. Two replicates from each source were amplified by PCR using universal primers 27F (5'-AGAGTTTGATCCTGGCTCAG-3') and 338R (5'-TGCTGCCTCCCGTAGGAGT-3') and sequenced on a GS FLX + Sequencing System (454 Life Science/Roche), Kansas State University.
92,880 sequencing reads from the 454 were initially generated from the four DNA samples, and after filtering for barcodes, adapters, length, and quality, 54,361 remained. Sequences were trimmed and denoised [57] to avoid artificial inflation of rare diversity from homopolymer read errors, and processed in QIIME 1.5.0 [58] to assign operational taxonomic units (OTUs) using a 97% identity threshold, assign taxonomy to OTUs (identifications of >80% confidence), and calculate abundances of OTUs in samples. Finally, using BLAST [59] and QIIME, reads were curated to remove sequences from mitochondria or chloroplast, and to merge OTUs not reaching 0.2% abundance in any sample into a single category. Finally, 2,833 and 4,209 bacterial reads from the two SU nodule samples (with mean 3521 and standard deviation 973), and 1,994 and 4,822 bacterial reads from the two UCD nodule samples (with a mean of 3408 and a standard deviation of 2000) were included in analysis.
Phylogenetic analysis was conducted on five v1-2 region of 16S rDNA sequences (S3 Table) of Frankia OTUs together with seven v1-2 region sequences and two v2 region sequences (Cn endophyte; Dc endophyte) of Cluster II Frankia strains and three v1-2 region sequences of Cluster I or III Frankia strains available in GenBank [37]; S3 Table). Among the added Cluster II Frankia sequences, three originated in Pakistan (Dg1, Cn endophyte, Dc endophyte), one originated in New Zealand (FE37), and all others originated in North America. Sequence alignment was done using MAFFT [60]; the length of the aligned sequences was 321bp. A neighbor-joining tree was constructed based on a pairwise distance matrix of percent nucleotide difference with 1000 boot strap replicates using PAUP* version 4.0b10 [61]. All positions were weighted equally and all gaps were treated as missing data.
Rhizobium leguminosarum nodC complementation
As a positive control, a cluster comprising nodD, nodA, nodB and nodC genes was amplified using DNA from Rhizobium leguminosarum strain A34 [62], Pfu (Promega, Charbonnière-Les-Bains, France) polymerase and primers F7800 5’-GTGCTGCATGCGTGCCGCTTACGACGTACAACTT-3’ and F7799 5’-TATAGGAATTCCTGCAGTGACGCGTTCATCACT-3’. PCR conditions were: 2 min at 95°C followed by 35 cycles at 95°C 1 min; 63°C 30 s; 72°C 7 min, followed by 72°C for 5 min. The PCR fragment was cloned into a pGEM-T easy vector (Promega) and the DNA insert verified by sequencing. The plasmid containing a PCR insert was then digested using restrictions enzymes SphI and EcoR1 and ligated into a SphI /EcoRI digested pBBR1 MCS5 vector [63]. It was then introduced first into E. coli DH10B (Invitrogen) and then into R. leguminosarum A56 (nodC128::Tn5) [62] by electroporation [64].
The nodC gene from Dg1 (nodC_Dg1) was amplified using primers F7308 5’-ACCAGGATCCTCACGATGACAGCGGGG-3’ and F7350 5’-AAACCCATATGTCGACCGCGGTGAG-3’ and Taq (Invitrogen, Villebon sur Yvette, France) in 1 x Taq buffer containing 5% DMSO (vol/vol). PCR conditions were: 5 min at 95°C followed by 40 cycles at 94°C, 45 s; 59°C, 45 s; 72°C, 45 s, followed by 72°C for 10 min. The PCR fragment was cloned into the pGEM vector, which was transformed into E coli DH10B and confirmed by sequencing. Plasmids containing the nodC_Dg1 gene were used as DNA template for further amplification and cloning.
The R. leguminosarum nodC gene replacement by Candidatus Frankia datiscae Dg1 nodC_Dg1 gene was done in three PCR steps. nodDnodAB genes from R. leguminosarum A56 were amplified using primers F8029 5’-CATGGTTTCTCGTTTGTCCAGTGTTTC-3’ and F7800 5’-GTGCTGCATGC GTGCCGCTTACGACGTACAACTT-3’ using Pfu polymerase. PCR conditions were: 2 min at 95°C followed by 35 cycles at 95°C, 1 min; 60°C, 30 s; 72°C, 5 min, followed by 72°C for 10 min. The nodC_Dg1 gene from Frankia Dg1 was amplified using primers F8030 3’-GAAACACTGGACAAACGAGAAACCATGTCGACCGCGGTGAG-3’ and F8210 5’-ACCAGGAATTCTCACGATGACAGCGGGG-3’ and Taq polymerase (Invitrogen). One μL of each PCR product were mixed and amplified using primers F7800, F8210 and Taq polymerase (Invitrogen). PCR conditions were: 4 min at 95°C followed by 35 cycles at 95°C, 1 min; 51°C, 30 s; 72°C, 7 min, followed by 72°C for 10 min. A 3.7 kb DNA fragment containing fusion nodDABC_Dg1 was eluted from the agarose gel using a micro elute column (Qiagen, Courtaboeuf, France), ligated into the pGEM-T easy vector (Promega, Madison, WI, USA) and transformed into E. coli DH10B by electroporation. Plasmid DNA isolation were done from several colonies. The correct gene fusion was confirmed by sequencing. The DNA insert was then cloned in the EcoRI and SphI sites of the pBBR1 MCS5 vector and transformed into E. coli DH5α. Transfer to the R. leguminosarum A56 nodC mutant strain was performed by tripartite conjugation using E. coli containing the pRK2013 plasmid. The construct in R. leguminosarum A56 with the Frankia Dg1 nodC_Dg1 gene was verified after conjugation by amplification of nodC-Dg1 gene and of the 16S rDNA and confirmed by sequencing, plasmid DNA preparation and enzyme restriction analysis.
Seeds of Pisum sativum (Wisconsin perfection) were obtained from INRA Dijon (France), surface sterilized with ethanol 70% (vol/vol) for 1 minute, rinsed and treated for 12 minutes in NaClO 10% (w/vol), rinsed and then germinated on agar. The seedlings were then transferred into 500 ml glass flasks containing 250 ml of nitrogen-free FP medium solution [65] gelified with 0.5% agar and capped with cotton wool. When the seedlings were 2 weeks old, they were inoculated with wild-type R. leguminosarum A34 (WT), R. leguminosarum A56 (nodC128::Tn5) with recombinant nodDnodABC, or with R. leguminosarum A56 with recombinant nodDnodABC_Dg1 with nodC from Dg1, or not inoculated, grown for 72 h at 30°C on TY agar with 0.6 mM Calcium Chloride [66] and resuspended in distilled water. The plants were grown in the greenhouse at a 21°C/16°C day/night cycle. The effects of the rhizobia on the plant roots was followed under a stereomicroscope. After infection, the roots were observed twice per week over a period of two months, looking for root hair deformation and nodule formation, on two separate sets of sextuplicates for all genetic constructs. After 31 days, photographs were taken of deformed root hairs. First, plants were observed under a Leica MZ8 stereomicroscope using lateral illumination to avoid light reflections. For photography, a Zeiss Axioskop was used with an Axiocam MRC5 camera, and the root systems with agar were removed from the glass flasks.
Results
Does the genome of Dg1 show auxotrophies or symptoms of reduction typical of obligate symbionts?
The genome of Dg1 has a coding density of 78% which is slightly lower than those of the other sequenced Frankia strains. The core genome (at a threshold level of 30% amino acid identity over 80% of the length of the shortest sequence) of the four studied genomes of Frankia strains consisted of 1616 coding sequences (CDSs; S2 Fig). The CcI3 genome displayed the highest similarity to the core genome, with only 30% (1351 CDSs) of its CDSs not shared with the other Frankia strains. In contrast, 39% (1598 CDSs) of the CDSs of Dg1, 45% (3051 CDSs) of ACN14a and 50% (3606 CDSs) of EAN1pec were not shared with any other Frankia strain.
A comparison of the metabolic pathways encoded in the genomes of the sequenced Frankia strains using MicroCyc (http://www.genoscope.cns.fr/agc/microscope/metabolism/microcyc.php) and Phyloprofile (https://www.genoscope.cns.fr/agc/website/spip.php?article600) showed several putative auxotrophies in Dg1 that could impede saprotrophic growth. However, since similar auxotrophies were found in the genomes of cultured Frankia strains, the only safe conclusion to be drawn is that not all actinobacterial versions of common metabolic enzymes are characterized yet. For this reason, it is also difficult to compare the saprotrophic potential—i.e., capability of growing on different nutrient sources—of different Frankia strains based on their genome sequences.
In comparison with CcI3, EAN1pec and ACN14a which have a high capacity for the production of secondary metabolites [67], Dg1 had a lower potential for the biosynthesis of polyketides, non-ribosomal peptide synthases and terpenoids (S4 Table). Several operons for lantibiotic biosynthesis that are present in CcI3, EAN1pec and ACN14a are absent in Dg1, and genes for bacteriocins present in ACN14a and EAN1pec are not present in Dg1 (S4 Table). The gvp operon, responsible for the synthesis of gas vesicle proteins that are supposed to maintain bacteria at the top of the water table in the soil [68], which is present partly in CcI3, and completely in ACN14a and EAN1pec, is missing in Dg1. These features indicate that Dg1 possesses a comparatively low potential for the biosynthesis of siderophores for iron scavenging, and of compounds used against competitors in the soil habitat.
In bacterial symbionts, proliferation of the mobilome is a key feature of early stages of genome reduction [69,70,71]. The mobilome encompasses mobile genetic elements—genetic units that can move within a genome or from cell to cell. Intracellular mobile genetic elements include transposons and insertion sequenes (ISs). The latter are defined as genomic sequences of mobile DNA, typically 800–1300 bp in length, that encode the enzyme transposase. E.g., clusters of ISs have been shown to correlate with areas of gene loss and genomic recombination in Frankia strains [72]. In this context, it is interesting that a larger number of transposases was observed in Dg1 compared with the other studied Frankia genomes (S5 Table). To further our understanding on genome reduction in Frankia strains, we examined the identity and distribution, as well as length distribution, of IS elements in the Frankia genomes published to date. We defined a particular DNA sequence encoding a transposase as an IS when it was present in multiple copies in a genome, taking into account that IS elements may have accumulated mutations leading to differences in sequence and length. The results showed that Dg1 clearly contains the highest relative amount of IS elements, followed by the Elaeagnus-infective strain EAN1pec and the Casuarina-infective strain CcI3 (S3A Fig). The latter, like Dg1, has a small genome which has been attributed to IS-mediated reductive evolution [11].
A study of the length distribution of the IS elements in each strain showed that only in Dg1, full length IS elements dominated, while in all other strains examined, full length IS elements represented a minority among the IS elements (in CcI3 and EAN1pec, they made up less than 30%; S3B Fig). The distribution of IS elements in the genome is depicted in Fig 1 and S2 Table.
Many degrading genomes display an elevated level of pseudogenes [73]. The genome of Dg1 contains 325 pseudogenes, 7.01% of the total number of genes. In comparison, CcI3 has 50 pseudogenes (1%), ACN14a 12 (0.18%), and EAN1pec has 128 (1.78%; S5 Table). As an extreme, 31.2% of the CDSs in the obligate symbiont Nostoc azollae are pseudogenes [74].
Evolution of symbiotic Frankia strains: which Cluster is basal?
In the framework of a re-examination of the phylogeny of actinobacteria using 54 house-keeping genes from 100 sequenced genomes, Sen et al. [10] determined Frankia Cluster II (represented by Dg1) as basal Frankia clade, followed by the non-symbiotic Cluster IV, and then the symbiotic Cluster III and finally Cluster I as the most derived. This is consistent with the evolution of nitrogen-fixing symbioses as examined by Werner et al. [4] in that the oldest symbioses—of Cucurbitales, since the evolution of the symbiotic capabilities must have preceded speciation of Coriaria sp. [20]—are those involving Cluster II strains. However, given the wide range of the analysis, a new, more focussed analysis seemed in order to confirm the basal position of Cluster II among the symbiotic Frankia strains.
The phylogenies of 50 different housekeeping genes were analyzed for the first four published Frankia genomes, using Geodermatophilus obscurus G20 (NC_013757 [75]), Acidothermus cellulolyticus 11B (NC_008578 [76]), Stackebrandtia nassauensis (NC_013947 [77]) and Nakamurella multipartita Y-104 (NC_013235 [78]) as outgroups. The results are depicted in S4 Fig and summarized in Table 1. For 42 out of 50 genes, Dg1 was basal to the other Frankia strains, while EAN1pec (Cluster III) and ACN14a/CcI3 (Cluster I) formed derived sister groups. Eight genes (dapB, dihydrodipicolinate reductase; dnaA, chromosomal replication initiation protein; folC, dihydrofolate synthase; glpX, fructose 1,6-bisphosphatase II; idi, isopentenyldiphosphate isomerase; ispA, intracellular septation protein; murC, UDP-N-acetylmuramate-L-alanine ligase; rplA, ribosomal protein L1) showed different phylogenies; for four of them (dnaA, folC, glpX, idi) Dg1/EAN1pec (Clusters II/III) and ACN14a/CcI3 (Cluster I) appeared as sister groups, and for two of them (murC, rplA), EAN1pec (Cluster III) was basal. In summary, the results predominantly show that Cluster II is the basal group of symbiotic Frankia strains. Those genes that yielded a topology with Dg1 at a derived position were shorter (with a mean length of 464 aa, 1392 bp) than those (with a mean length of 521 aa, 1563 bp) that yielded a topology with Dg1 at the base of the Frankia radiation. The smaller length is correlated with an a priori smaller phylogenetic weight.
Table 1. Frankia phylogeny was analysed using 50 housekeeping genes and the four published genomes of strains ACN14a and CcI3 (Cluster I), Dg1 (Cluster II) and EAN1pec (Cluster III).
| Gene | DNA or Protein | Length of Alignment | Score of best tree found | # of most pars. Trees | Frankia Dg1 first branching? | Frankia mono-phyletic? |
|---|---|---|---|---|---|---|
| 16S 23S | DNA | 4944 | 4505 | 1 | Y | Y |
| aceE | Protein | 500 | 988 | 1 | Y | Y |
| aroK | Protein | 256 | 513 | 5 | Y | Y |
| atpA | Protein | 556 | 705 | 1 | Y | Y |
| bioA | Protein | 486 | 1138 | 1 | Y | Y |
| bioB | Protein | 458 | 966 | 1 | Y | Y |
| clpP | Protein | 237 | 366 | 1 | Y | Y |
| dapA | Protein | 356 | 627 | 1 | Y | Y |
| dapB | Protein | 278 | 449 | 1 | N | Y |
| dnaA | Protein | 466 | 574 | 1 | n | Y |
| dnaB | Protein | 547 | 629 | 1 | Y | Y |
| dxr | Protein | 460 | 686 | 1 | Y | Y |
| dxs | Protein | 660 | 965 | 1 | Y | Y |
| eno | Protein | 465 | 491 | 3 | Y | Y |
| folC | Protein | 525 | 1153 | 1 | n | Y |
| ftsZ | Protein | 590 | 784 | 1 | Y | Y |
| fusA | Protein | 705 | 735 | 1 | Y | Y |
| glmU | Protein | 540 | 1126 | 1 | Y | Y |
| glpX | Protein | 375 | 517 | 4 | n | Y |
| gltA | Protein | 489 | 1009 | 1 | Y | Y |
| gyrA | Protein | 850 | 970 | 1 | Y | Y |
| htpX | Protein | 338 | 604 | 1 | Y | Y |
| idI | Protein | 236 | 572 | 1 | n | n |
| ispA | Protein | 359 | 678 | 1 | Y | Y |
| ksgA | Protein | 339 | 591 | 2 | Y | Y |
| lipA | Protein | 360 | 447 | 2 | Y | n |
| lytB | Protein | 355 | 472 | 2 | Y | Y |
| metK | Protein | 418 | 380 | 1 | Y | Y |
| mfd | Protein | 780 | 1014 | 1 | Y | Y |
| mraY | Protein | 368 | 611 | 2 | Y | Y |
| murA | Protein | 516 | 1004 | 1 | Y | Y |
| murC | Protein | 668 | 1320 | 1 | n | Y |
| murG | Protein | 396 | 767 | 1 | n | Y |
| nth | Protein | 289 | 476 | 2 | Y | Y |
| pgk | Protein | 420 | 639 | 1 | Y | Y |
| recA | Protein | 355 | 261 | 3 | Y | Y |
| ribA | Protein | 480 | 627 | 1 | Y | Y |
| ribE | Protein | 175 | 288 | 1 | Y | Y |
| ribF | Protein | 355 | 758 | 1 | Y | Y |
| ribH | Protein | 175 | 289 | 1 | Y | Y |
| rplA | Protein | 260 | 262 | 1 | n | Y |
| rplB | Protein | 279 | 184 | 3 | Y | Y |
| rplC | Protein | 245 | 291 | 1 | Y | Y |
| rplD | Protein | 306 | 458 | 1 | Y | Y |
| rplE | Protein | 206 | 205 | 3 | Y | Y |
| rplF | Protein | 179 | 227 | 6 | Y | n |
| rpoB | Protein | 1209 | 948 | 1 | Y | Y |
| shc | Protein | 751 | 923 | 1 | Y | n |
| tpiA | Protein | 288 | 469 | 1 | Y | Y |
| trpE | Protein | 450 | 1367 | 1 | Y | Y |
| uppS | Protein | 293 | 585 | 2 | Y | Y |
A. cellulolyticus 11B, S. nassauensis DSM 44728, G. obscurus DSM 43160, N. multipartita DSM 44233, and T. fusca YX were used as outgroups. The statistical evaluation is given in S4 Fig.
Dg1 contains two operons of canonical nod genes
Two operons of homologs of the canonical nod genes were found in the Dg1 genome: 1) an operon containing nodA’B1A, coding for: NodA’, GenBank accession number AEH09515 (a truncated NodA, only 36 amino acids); NodB1, AEH09514; and a full-length NodA, AEH09513; and 2) a nodB2CIJ operon, coding for NodB2, AEH10396; and NodC, AEH10397 (Fig 1). The proteins encoded by these genes display very high amino acid sequence similarity to the canonical nodABC genes (Figs 2, 3 and 4; S5 Fig; S6 Table), and their arrangement in the Dg1 chromosome suggests that they are expressed as operons, similar to their counterparts in rhizobia. Based on these findings of amino acid sequence similarity and synteny, they are referred to as nod genes throughout this manuscript. The structure of the nodA’B1A operon indicates that it is the result of at least two transposition events, though it is not clear whether nodA inserted into a functional nodAB1C operon from which nodC and the largest part of the original nodA gene were subsequently lost, or whether the operon was truncated before the insertion.
Fig 2. Maximum Likelihood trees of NodA proteins.

All rhizobial sequences are given in green, all sequences from Dg1 are given in red, sequences from Streptomyces bottropensis are given in purple. The Dg1 NodA sequence is indicated in blue. All sequences used for the phylogenetic analysis are given in S6 Table.
Fig 3. Maximum Likelihood trees NodB proteins.

All rhizobial sequences are given in green, all sequences from Dg1 are given in red, sequences from Streptomyces bottropensis are given in purple. Dg1 NodB1 and NodB2 sequences are indicated in blue. All sequences used for the phylogenetic analysis are given in S6 Table.
Fig 4. Maximum Likelihood trees of NodC proteins.

All rhizobial sequences are given in green, all sequences from Dg1 are given in red, sequences from Streptomyces bottropensis are given in purple. The Dg1 NodC sequence is indicated in blue. All sequences used for the phylogenetic analysis are given in S6 Table.
The NodA proteins belong to a family of acyl transferases previously only described in rhizobia; however, several NodA homologs are also present in a diverse group of Actinobacteria that includes Dg1 (Fig 2; S6 Table), phylogenetically distant from the rhizobia. While these actinobacterial genera with NodA homologs span across the whole phylum [10], only the nodulating taxa within the Proteobacteria have NodA homologs. Furthermore, as shown by the longer branch lengths (Fig 2), the NodA sequences in actinobacteria are more diverse than those of α- or β-Proteobacteria.
In order to ensure that all potential gene familes were represented in the NodA tree, a set of more dissimilar potential NodA sequences with lower e-values in BLAST (two sequences per genus within all proteobacteria) was analyzed by maximum likelihood. In this analysis, even the sequences with low similarities still were nested within the rhizobial clade (data not shown). Thus, no NodA paralogs were found in rhizobia.
In the NodB protein phylogeny (Fig 3), the Dg1 NodB1 is at the base of the rhizobial NodB clade with Dg1 NodB2 at a slightly more derived position, following the insertion of a pair of sequences from Azorhizobium caulinodans ORS571 [79]. In the NodC protein phylogeny, NodC is basal to all the rhizobial NodC proteins with strong support. However, sister to this clade is the clade of Sphingomonas and Parvibaculum, non-nodulating Proteobacteria (Fig 4).
Particularly notable across all the trees are the positions of the NodABC homologs from Streptomyces bottropensis. Sequences of S. bottropensis were found as sister to Dg1 sequences in the NodA, NodB, and NodC trees (Figs 2, 3 and 4). Moreover, although S. bottropensis so far has not been known to induce nodules, it is the only actinobacterial taxon beside Dg1 that has homologs of all three canonical nod genes nodABC. Intriguingly, the nodA-nodB-nodC genes of S. bottropensis are present in a single operon. This positioning could suggest that the common ancestor of Dg1 and S. bottropensis specifically among the actinobacteria was the source of the rhizobial canonical nod genes.
Directly downstream from the nodB2C operon an ABC transporter operon with significant amino acid sequence similarity (>45%) to rhizobial nodIJ is found (encoding AEH10398 and AEH10399). NodJ represents an ABC transporter and NodI its ATPase subunit. It is interesting to note that the position of this operon with respect to nodB2C is identical to the position of the nodIJ genes in the canonical nod gene operon of rhizobia (nodABCIJ; [80]). However, the Dg1 genes with homology to nodIJ located near nodB2C are expressed in symbiosis at very low levels, if at all (see below, transcriptome); therefore, we have termed the genes nltIJ for “nod-linked transporters I and J”. Intriguingly, in S. bottropensis the homologs of the rhizobial canonical nod genes are present in a nodBCnltIJnodA operon, and the NltI/NltJ protein encoded in this operon show high amino acid similarity with Dg1 NltI/NltJ. Our phylogenetic analysis showed that the actinobacterial NltI and NltJ proteins encoded by nodBC-linked genes—as opposed to other ABC transporter/ATPase pairs in the same genomes—have very low amino acid similarity with rhizobial NodI/NodJ proteins, obviously coming from a different ABC transporter/ATPase lineage (S6 Fig).
The Dg1 genome has an overall GC content of 70.04%, and the GC contents of the nod genes, most strikingly nodA, are clearly below that (nodA, 58.4%; nodB1, 65.2%; nodB2, 66.8%; nodC; 64.5%). This pattern is also true for the ABC transporter operon linked to nodB2C (nltI, 65.4%; nltJ, 62.4%). Rapid gene evolution is known to be accompanied by AT enrichment, so this may be one explanation for the lower %GC in this gene region [81]. A similar pattern of low %GC of the nod genes relative to the genome is found in rhizobia [82], and in the nodA homolog of S. bottropensis (S6 Table). By contrast, the %GC of the more distant nodA homologs within the actinobacteria is higher than the %GC of the respective genomes (S6 Table), implying a relatively slow rate of gene evolution [83]. Alternately, another explanation for the lower GC% of the nod operons could be their position at the replication terminus, as has been shown to occur in several bacterial genomes [83].
Both Dg1 nod operons are expressed in symbiosis
The expression of the two nod operons in Dg1 in nodules of D. glomerata was examined by quantitative RT-PCR, using the Dg1 translation initiation factor gene IF-3 (GenBank accession number AEH10595) as constitutive control (see [84]). Transcription of nodA, nodB1, nodB2 and nodC in nodules (Table 2) was confirmed through transcriptome analysis (see below).
Table 2. Expression of the Dg1 genes nodA, nodB1, nodB2, and nodC in D. glomerata nodules given in relative units.
| Gene | Expression |
|---|---|
| nodA | 64.72 ± 23.29 |
| nodB1 | 2.23 ± 1.55 |
| nodB2 | 1.69 ± 0.45 |
| nodC | 5.62 ± 2.20 |
The expression values represent means ± SD (n = 3). The expression of nodA is significantly higher than those of nodB1, nodB2 or nodC. (One-Way ANOVA with Tukey's multiple comparison test, p ≤ 0.05)
Dg1 nodC can partially complement a Rhizobium leguminosarum nodC mutant strain
The R. leguminosarum A34 (wt), the A56 nodC mutant (nodC128::Tn5; [62]) can be complemented by the homologous nodC_Dg1 gene. When the rhizobial nodC gene in the nodABC operon was replaced by Frankia Dg1 (nodC_Dg1), the ability of the strain to induce root hair deformation on pea was restored, but the ability to nodulate was not (Fig 5). While the restoration of root hair deformation shows that Dg1 NodC_Dg1 does complement R. leguminosarum A56 nodC mutant, the lack of restoration of nodulation indicates that the either activity of NodC_Dg1 in R. leguminosarum A56 is too low to provide enough Nod factors for this process or that there is incompatibility with the other Nod proteins, formation of an oligomer of a length different from the optimal, etc. Nevertheless, this result shows that Dg1 NodC can fulfill the function of a chitin synthase in Nod factor biosynthesis.
Fig 5. Analysis of the ability of Frankia Dg1 nodC (nodC_Dg1) to complement a rhizobial nodC mutant.
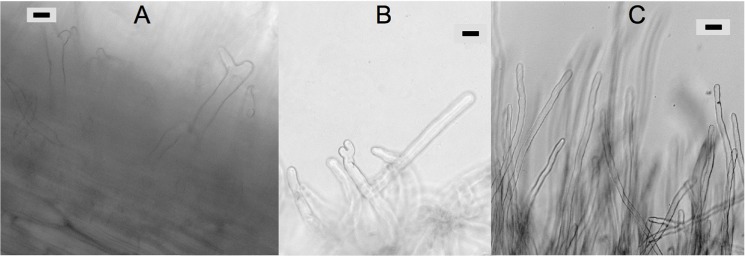
Roots of P. sativum show root hair deformation 31 days after inoculation with (A) R. leguminosarum A56 nodC::Tn5 mutant complemented by Frankia Dg1 nodC_Dg1 and after inoculation with (B) wild type R. leguminosarum A34. (C) Roots inoculated with the original R. leguminosarum A56 nodC::Tn5 mutant do not show root hair deformation. Root hair deformation was more pronounced after the infection with wild type rhizobium (B) or with the nodC::Tn5 mutant complemented by the homologous nodDnodABC operon (data not shown), than with the strain complemented by Frankia Dg1 nodC_Dg1 (A). Size bars indicate 10 μm.
Transcriptome analysis
The total transcriptome of Frankia in young nodules of D. glomerata encompasses genes expressed in an indeterminate developmental pattern that would correspond to and encompass the infection and nitrogen fixation zones described by Pawlowski and Demchenko [85]. Thus, the calibration of relative transcript abundance for specialized processes such as Nod factor biosynthesis or nitrogen assimilation depends on a spatio-temporally heterogeneous source, and intact biosynthetic pathways are a stronger metric for interpreting metabolic expression patterns than single-gene scores.
All the biosynthetic pathways listed in Table 3 were statistically overrepresented (p < 0.05) in the 80th quantile (top 20% most abundant) of rpkb-ranked transcriptome data. Genes coding for the pathway for nitrogenase biosynthesis (nifHDK, nifV, nifENXWZBS) were the most highly expressed, with significant overrepresentation in the top 1% (99th quantile (p = 1.2e-5). The TCA pathway, iron-sulfur cluster biosynthesis (SUF pathway), and NADH quinone oxidoreductase synthesis were significantly enriched in the top 5% (p < 0.02). In the top 10% (90th quantile), gene clusters coding for cytochrome c oxidase biosynthesis, isoprene biosynthesis, and branched-chain amino acids pathway (ILV pathway) were enriched significantly (p< 0.05). Nod factor biosynthesis (nodA´B1A and nodB2C operons) and the arginine biosynthesis complete pathway were significant in the 80th quantile (p<0.03). The ABC transporter operon downstream to the nodB2C operon (nltIJ) was expressed at low confidence, in the 70th and 60th quantile, respectively (not shown in table).The expression of the genes for enzymes involved in nitrogen assimilation and amino acid biosynthesis was confirmed by qPCR (Table 4).
Table 3. Statistically significant overrepresentation of key biosynthetic pathways in the active nodule transcriptome.
| Pathway/product | # genes in pathway | # genes expressed >99th quantile (/69 genes) | p-hyper | # genes expressed >95th quantile (/196 genes) | p-hyper | # genes expressed >90th quantile (/334 genes) | p-hyper | # genes expressed >80th quantile (/680 genes) | p-hyper | # genes expressed >70th quantile (/1146 genes) | p-hyper | # genes expressed >60th quantile (/1805 genes) | p-hyper |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nitrogenase | 10 | 4 | 1.2E-05 | 7 | 4.0E-08 | 8 | 4.9E-08 | 10 | 9.6E-09 | 10 | 1.8E-06 | 10 | 1.7E-04 |
| TCA pathway | 12 | 1 | 1.5E-01 | 5 | 1.2E-04 | 10 | 4.2E-10 | 12 | 2.4E-10 | 12 | 1.3E-07 | 12 | 3.1E-05 |
| FeS cluster assembly (SUF) | 6 | 0 | 9.3E-02 | 3 | 1.7E-03 | 5 | 1.6E-05 | 6 | 1.6E-05 | 6 | 3.6E-04 | 6 | 5.6E-03 |
| NADH quinone reductase | 13 | 0 | 1.9E-01 | 3 | 1.9E-02 | 8 | 1.1E-06 | 11 | 8.6E-08 | 12 | 1.3E-06 | 13 | 1.3E-05 |
| Isoprene pathway (MEP) | 9 | 1 | 1.4E-01 | 1 | 3.4E-01 | 4 | 3.3E-03 | 6 | 8.5E-04 | 7 | 2.1E-03 | 8 | 5.5E-03 |
| Nod factor synthesis (nodABC) | 5 | 0 | 7.8E-02 | 0 | 2.1E-01 | 1 | 3.3E-01 | 3 | 3.1E-02 | 5 | 1.4E-03 | 5 | 1.3E-02 |
| Arginine pathway | 7 | 0 | 1.1E-01 | 0 | 2.8E-01 | 2 | 9.8E-02 | 6 | 9.5E-05 | 7 | 9.7E-05 | 7 | 2.3E-03 |
| ILV pathway | 11 | 0 | 1.6E-01 | 1 | 4.0E-01 | 3 | 4.8E-02 | 9 | 2.5E-06 | 10 | 1.5E-05 | 11 | 7.3E-05 |
TCA is the tricarboxylic acid cycle (respiration), FeS clusters are iron-sulfer clusters (used in nitrogenase, hydrogenase etc.), MEP is the non-mevalonate pathway or 2-C-methyl-D-erythritol 4-phosphate pathway (hopanoids), ILV pathway is the isoleucine, leucine and valine pathway (biosynthesis of branched chain amino acids).
Table 4. Expression of asl, gogatFD, glnII, glnI and argJ in D. glomerata nodules using relative quantitation of the Comparative CT Method (ABI Prism user bulletin #2).
| asl | gogatFD | glnII | glnI | argJ | |
|---|---|---|---|---|---|
| average | 1.26 | 4.41 | 2.4 | 1.33 | 0.5 |
| SD | 0.17 | 0.11 | 0.15 | 0.18 | 0.19 |
The expression level of gogatFD is significantly higher than those of asl, glnI, glnII and argJ, but all the genes encoding N-assimilatory enzymes are expressed. The reference gene used was 16S rRNA.
Nodule occupancy
Since the microsymbionts of D. glomerata cannot be cultured, it cannot easily be ascertained how many strains occupy an individual nodule, whether nodules house Frankia exclusively or in combination with other bacteria. In addition, it was important to detect any major contribution to nodule occupancy by a non-Frankia taxon that could carry accessory nodulation genes (e.g. nodABC). To shed light on these questions, D. glomerata nodules induced by Frankia inoculant originating in Pakistan [21] and grown at Stockholm University (SU), and D. glomerata nodules induced by a California source of Frankia, and grown at the University of California in Davis (UCD), respectively, were used for DNA isolation and analysis of operational taxonomic units (OTUs) via PCR amplification with universal 16S rDNA primers 27F and 388R and 454 sequencing (SRA BioProject PRJNA258479).
Most reads were identified as one of five OTUs of Frankia, which represented the majority of bacterial reads (68% of the bacterial reads from SU nodules and 84% of the bacterial reads from UCD nodules; see Table 5; S7 Table). Two Frankia OTUs contributed most of the Frankia reads: OTU 51 from SU nodules (Pakistan inoculant) and OTU 770 from UCD nodules (California inoculant), representing 96–98% and 99–99.6% of Frankia reads within each sample, respectively. As shown in the neighbor-joining tree (Fig 6), all the Frankia OTUs formed a clade within Cluster II. OTU 51 and OTU 770 belonged to two subclades that were weakly-supported statistically; however these sequences differed by 12 base-pairs (>3.7%; Fig 6). The other Frankia OTUs belonged to a clade either with OTU 51 or with OTU 770.
Table 5. Processed bacterial sequences and taxonomy assignments by sample source.
| Taxonomy | Dg_UCD b | Dg_SU c | #OTUs e | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Phylum | Class | Order | Family | Genus | Mean | SD d | Mean | SD d | |
| Actinobacteria | Actinobacteria | Frankiales | Frankiaceae | Frankia | 85.96% | 3.99% | 68.29% | 1.27% | 9 |
| Mycobacteriales | Mycobacteriaceae | Mycobacterium | 0.01% | 0.01% | 1.34% | 0.39% | 2 | ||
| Streptomycetales | Streptomycetaceae | Streptomyces | 0.26% | 0.19% | 0.03% | 0.01% | 1 | ||
| Bacteroidetes | Sphingobacteria | Sphingobacteriales | Flexibacteraceae | Cytophaga | 0.06% | 0.06% | 1.81% | 0.18% | 2 |
| Dyadobacter | 0.32% | 0.33% | 0.15% | 0.06% | 1 | ||||
| Proteobacteria | Alpha-proteobacteria | Caulobacterales | Caulobacteraceae | Caulobacter | 0.11% | 0.06% | 0.46% | 0.05% | 1 |
| Rhizobiales | Bradyrhizobiaceae | Afipia | 0.32% | 0.11% | 0.05% | 0.03% | 1 | ||
| Sphingomonadales | Sphingomonadaceae | Novosphingobium | 0.28% | 0.31% | 0.28% | 0.14% | 1 | ||
| Sphingobium | 0.23% | 0.17% | 0.04% | 0.01% | 1 | ||||
| Beta- proteobacteria | Burkholderiales | 0.21% | 0.20% | 0.48% | 0.01% | 1 | |||
| Methylophilales | Methylophilaceae | Methylophilus | 0.25% | 0.21% | 0.23% | 0.18% | 1 | ||
| Delta-proteobacteria | Myxococcales | 0.04% | 0.02% | 0.65% | 0.18% | 1 | |||
| TM7 | TM7-3 | EW055 | 0.0% | 0.0% | 0.39% | 0.15% | 1 | ||
| Unassigned a | 5.69% | 0.15% | 6.87% | 1.37% | 5 | ||||
| Low abundance a | 6.27% | 2.49% | 18.94% | 0.05% | 789 | ||||
| Total bacteria reads and OTUs | 3424 | 2006 | 3535 | 974 | 817 | ||||
aLow abundance OTUs (less than 0.1% of bacterial sequences), and OTUs not assignable to any taxonomy are summed across OTUs.
bDg_UCD are D. glomerata nodule samples from University of California Davis, inoculated with a Californian source.
cDg_SU are D. glomerata nodule samples from Stockholm University, inoculated with a Pakistani source.
dStandard deviations are based on n = 2.
e# OTUs refers to the number of OTUs at 97% identity in each taxonomic category.
Fig 6. Neighbor-joining tree of Frankia OTUs observed in Datisca glomerata plants inoculated with material from either Pakistan or California combined with other Cluster II Frankia strains.
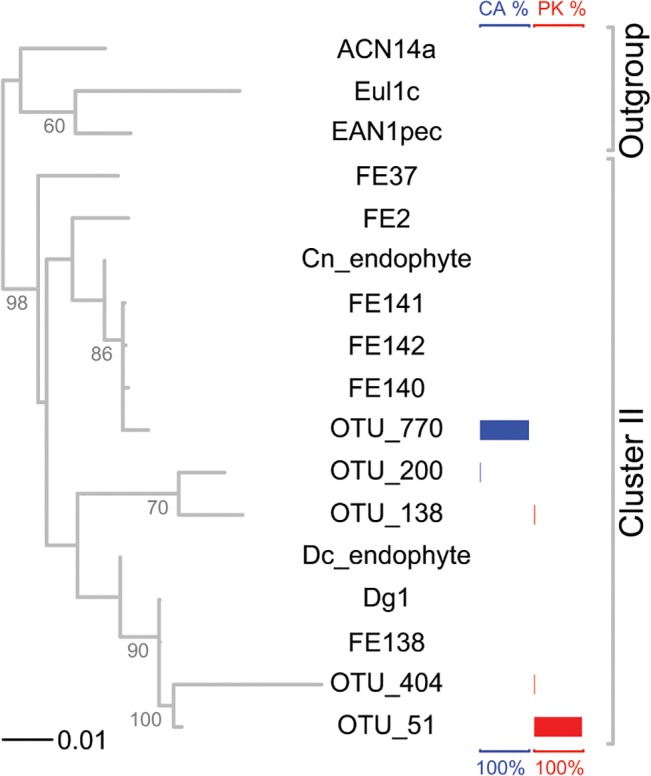
Cluster I and III Frankia strains are used as outgroups. Branches with bootstrap values of 50 or higher are indicated in the figure. *: Cn endophyte sequence only contains v2 region. Relative abundances of each Frankia OTU are shown in colored bars to the right; blue indicates each OTU's percent of Frankia reads in nodules of plants inoculated with material from California (CA), red indicates percent in nodules of plants inoculated with material from Pakistan (PK).
While Frankia dominated the nodule bacterial component, other taxa were minor components (less than 2% of bacterial reads from any individual sample; Table 5). Of the taxa besides Frankia that were detected in nodules, only Mycobacterium, Cytophaga, and an OTU in the Myxococcales exceeded 0.2% of the total reads in any sample (Table 5). Mycobacterium and Cytophaga are known plant endophytes [86,87]. Some low-abundance OTUs (<0.2% of total reads) were also assigned to genera that include known plant endophytes or rhizosphere bacteria, e.g. Streptomyces, Dyadobacter, Caulobacter, Novosphingobium, Sphingobium, and Methylophilus [88,89,90,91,92]. Identification to the order level only for the OTU in the Myxococcales is too broad to infer any ecological characters, however this clade does include species commonly found in soil. None of the OTUs in the nodule samples were assigned to genera reported to possess nodABC genes.
Discussion
Is Candidatus Frankia datiscae Dg1 an obligate symbiont?
The first genome sequence of a representative of the non-cultured Cluster II Frankia strains was expected to answer the question of whether these strains were obligate symbionts or just had (an) auxotrophy/auxotrophies that made attempts at isolation very difficult. The data obtained in this study do not point to any obvious auxotrophy, however the comparatively low capacity for the synthesis of secondary metabolites (S4 Table) and the loss of the gas vesicle protein (gvp) genes indicate a reduced ability to thrive in the highly competitive soil biotope.
The genome sizes of Frankia strains have been suggested to indicate the strains’ saprotrophic capacities [11]. The genome size of Dg1, which is smaller than, but close to that of the Casuarina-infective strain CcI3, suggests a low saprotrophic potential but does not make as definitive a case for genome reduction as might be expected in comparison with animal symbionts [71,93]. However, when comparing the reduction of the mitochondrial genomes (14–42 kB vs. 184–2400 kB; [94]), animal symbionts seem to be more impacted with regard to genome reduction than plant symbionts. Similarly, genome reduction in evolutionary younger plant symbionts seems to be much less dramatic than in animal symbionts; e.g., the obligate endophyte Rhizophagus irregularis, the symbiosis of which goes back at least 460 My [95] still has a genome size of more than 140 Mb [96]. The fact that Glomalean fungi, while obligate endophytes, are not transmitted vertically, but have a pre-symbiotic growth phase in the soil, might explain the comparable lack of genome reduction. In this context, it should be pointed out that Cluster II Frankia strains are not transmitted vertically either. The fact that soil from underneath plants containing nodules induced by Cluster II Frankia strains can be used to infect new plants indicates that Cluster II Frankia strains, like Glomalean fungi, have a pre-symbiotic growth phase in soil. Furthermore, the identification of a Cluster II Frankia strain in soil devoid of any compatible host plant species [97] points at some saprotrophic capability. Also the fact that the Dg1 genome contains several operons for the biosynthesis of siderophores and other secondary metabolites that are not expressed in nodules (S4 Table), one of them containing a gene encoding protein of 6077 amino acids (WP_013873316.1) indicates some selection pressure to maintain these operons, and thus, (a) non-symbiotic growth phase(s).
Host plants of Cluster II strains are distributed in the Americas, the Mediterranean, Asia, Oceania and New Zealand. The low genetic diversity of Cluster II strains, indicating the likelihood of an evolutionary bottleneck, was first suggested by Vanden Heuvel et al. [16]. Our findings add further support to this interpretation. The transcriptome of the Cluster II strain from California presented here (represented by OTU 770) mapped with relatively few polymorphisms to the genome of Dg1, which originated in Pakistan (represented by OTU 51), despite the disjunct geographic distribution of the host plant genera of Cluster II strains, most strikingly of Coriaria.
Yokoyama et al. [20] came to the conclusion that the disjunct distribution pattern of Coriaria spp. was the result of several geographical migrations and separations in the Cenozoic. Since Frankia propagules can be distributed by birds [98], by waterways [99], and potentially by other vectors, a compatible microsymbiont might have been distributed along with the host plant seeds. Thus, Cluster II strains might have been spread together with their host plants (Datiscaceae and Coriariaceae), while the stability of their ecological niche prevented further genome diversification.
Why is the basal clade of symbiotic Frankia strains saprotrophically challenged?
The result that Cluster II is the basal clade of symbiotic Frankia strains is consistent with plant phylogenetic results implying that the oldest actinorhizal symbioses are those of Cucurbitales [4].
Non-symbiotic, non-nitrogen fixing so-called Frankia-like strains or atypical Frankia strains (Cluster IV) were considered to be basal to symbiotic Frankia strains [7,56,100]; however, this is contradicted by more recent analysis by Sen et al. [10] who find that cluster II, as represented by Dg1, is basal to all other Frankia strains including the atypical ones. So far, most atypical strains were isolated from nodules induced by Cluster II strains. Since these strains are unable to induce nodules on their own, they may have represented contaminations from the nodule periderm. The fact that like Cluster I and Cluster III Frankia strains, these Cluster IV Frankia strains could be cultured suggests that their saprotrophic capabilities are broader than those of Cluster II strains. The genome sequence of one of these strains, CN3 [101] comprises 10 MB and represents the largest Frankia genome known to date, while being still in the range of the genome sizes of cluster III strains.
Small genomes (5.27–5.6 MB) also are found among the Casuarina-infective subgroup of Cluster I [11,102] which have a narrow host range of plants with a narrow native distribution, and the small genome size of the Casuarina-infective strains are assumed to be due to genome shrinkage [11]. This phytogeographic context does not represent the situation of Cluster II strains which have a broad host range, and a broader geographic distribution. Dg1 has more than 800 genes not present in any other Frankia genome available in the JGI database. Most strikingly, only Dg1 among sequenced Frankia genomes contains a copy (AEH09479-AEH09484) of the mammalian cell entry (mce) gene cluster from Mycobacterium tuberculosis, assumed to be involved in the uptake of sterols, which in Streptomyces coelicolor is required for plant root colonization [103]. So we speculate that the ancestors of all Frankia strain were rhizosphere bacteria with large genomes and broad saprotrophic potential when one subgroup—the progenitors of Cluster II—acquired the gene set for nitrogen fixation and evolved symbiotic capabilities. Symbiosis led to genome reduction in this subgroup. In due time, two other subgroups (the progenitors of Clusters I and III, respectively) developed symbiotic capabilities; later, a subgroup of Cluster I—the Casuarina-infective strains—underwent genome shrinkage.
Interestingly, several of the Dg1 operons involved in the synthesis of secondary metabolites contain genes that are expressed in nodules of D. glomerata (S4 Table), so even though secondary metabolism loci are reduced overall, the corresponding functions do seem to play a role in symbiosis and are not only relevant during saprotrophic growth.
Nodule occupancy
Cluster II Frankia were the predominant bacteria detected in root nodules of D. glomerata inoculated with sources from two geographically distinct locations, Pakistan and California, in separate experiments. Other bacterial taxa occurred at extremely low levels, indicating that there are no major co-symbionts in the root nodules.
The topology of the neighbor-joining tree for the nodule Frankia OTUs is broadly consistent with the findings of Vanden Heuvel et al. [16]. Two distinct strains of Cluster II Frankia (represented by OTU 51 (Dg1) and OTU 770) were detected in nodules induced by the Pakistan and the California source, respectively. Interestingly, OTU 51 (Pakistan) clusters strongly with FE138, which was detected in field nodules in California [16]; and OTU 770 groups phylogenetically with Cn endophyte, a strain detected in soil beneath Coriaria nepalensis from Pakistan [104], and with strains detected in western North America [16] (Fig 6). These findings indicate a phylogenetic overlap in the composition of the respective communities of Cluster II Frankia strains.
Transcriptome analysis
Taken together, the analysis of the most abundantly-expressed Frankia biosynthetic pathways in young root nodules indicate that nitrogen fixation and associated processes comprise the predominant nodule activity of Frankia. At the same time, the data suggest that Nod factor biosynthesis occurs in these young nodules. It has been shown that rhizobia express their nod genes inside the nodule until bacteroid differentiation [105], and components of the common symbiotic signal transduction pathway are expressed in the apices of nitrogen-fixing nodules [106].
Genes for enzymes involved in nitrogen assimilation and amino acid biosynthesis, including GS-GOGAT, branched-chain amino acid biosynthesis, and the complete arginine biosynthesis pathway, were expressed in Dg1 in symbiosis. These findings provide further evidence for a model of novel partitioning of nitrogen assimilation in D. glomerata nodules in which the expression of plant glutamine synthetase in the uninfected cells surrounding the infected cells, not in the infected cells themselves, indicated that the microsymbiont likely exports amino acids, not ammonium [107,108]. Physiological studies had suggested that the nitrogen export form was arginine [107]. The high expression of the arginine biosynthetic pathway confirms an important role for arginine biosynthesis as a major intermediate nitrogen storage pathway in cucurbitoid nodule N assimilation [107]. The abundance of transcripts encoding enzymes from the branched chain amino acid pathway was surprising since it has been shown that at least in pea and bean nodules, synthesis of branched chain amino acids is downregulated in symbiosis and they are provided by the host plant [109], although this is not the case for all rhizobial symbioses [110]. The expression of these complex amino acid biosynthetic pathways is a likely further example of a degree of metabolic independence of Frankia Dg1 in this symbiosis, compared with legume-rhizobia symbioses or other Frankia symbioses [85].
Active expression of the isoprene biosynthetic pathway, with mRNAs in the 90% quantile, was expected as it is responsible for the synthesis of hopanoids, bacterial steroid lipids that are part of the envelope of nitrogen-fixing Frankia vesicles [111], and menaquinone, required for electron transport.
Which bacterial group “invented” lipochitooligosaccharide (LCO) Nod factors?
The host receptors for microsymbiont signaling molecules that use the common symbiotic signaling pathway [112] evidently evolved from chitin receptors [113]. While arbuscular mycorrhizal fungi use LCOs and short-chain chitin oligomer (COs) for signaling to their plant hosts [23,31], rhizobia, with a few exceptions [33], seem to use only LCOs. It is unknown thus far which signal factors are used by Frankia strains in Clusters I and III.
Rhizobia, the microsymbionts of legumes and Parasponia, are polyphyletic. Most rhizobia belong to the α-proteobacteria, but several legumes are nodulated primarily by β-proteobacteria (Burkholderia spp., Cupriavidus spp.; [114]). It has been assumed that the nodulation genes originated in α-rhizobia and that β-rhizobia gained them through multiple lateral nod gene transfers [115], but our phylogenetic analyses now indicate that nodA and nodB are likely to have been transferred originally from actinobacteria to rhizobia; the relationships among the nodC genes are less certain.
The Dg1 protein sequences of NodA and NodC are basal to the rhizobial orthologs, and the two Dg1 NodB proteins also have basal positions in the NodB phylogeny (Fig 3). The functional role of the NodA protein in symbiosis is to transfer an acyl chain to the backbone of Nod factor precursor chito-oligosaccharides, assembled by NodC and deacetylated at the non-reducing end by NodB. This NodA-mediated acyl group transfer is key to Nod factor signaling function, rendering the LCO amphiphilic, and presumably permitting it to reach the host membrane-localized LysM receptor kinase [116]. The nodB and nodC genes are members of multigene families, coding for polysaccharide deacetylases and glycosyl transferases, respectively, with homologs occurring throughout the eubacteria. In contrast, nodA genes are not part of a ubiquitous gene family with duplications, a difference reflected in the phylogenetic trees (Figs 2, 3 and 4). Thus, the existence of a series of NodA-like acyl transferases across the phylum Actinobacteria but otherwise only in the nodulating members of the Proteobacteria supports an actinobacterial origin of nodA genes, and thus of LCO Nod factors. The detailed phylogeny suggests that the common ancestor of Dg1 and S. bottropensis specifically among the actinobacteria was the source of the rhizobial nodulation protein. The fact that α- and β-Proteobacteria lack NodA paralogs further supports the integrity of the overall topology of the tree.
Thus, the phylogenetic evidence suggests that NodA-type acyl-transferases first evolved in Actinobacteria and then laterally transferred to rhizobia. The positioning of nodA genes in the non-Frankia actinobacterial genomes tentatively suggests a link with cell wall biosynthesis, and it is plausible that an acyltransferase is involved in the biosynthesis of actinobacterial cell walls as many of them (though not those of Frankia sp.) contain mycolic acids which are linked to the peptidoglycan [117]. An origin of nodA (or nodABC) outside the Proteobacteria would be analogous to the situation of nodIJ, which evolved in β-Proteobacteria and were subsequently acquired by α-Proteobacteria via lateral gene transfer [80]. Interestingly, also in Dg1 the nodB2C operon was found to be linked to a nodIJ-like operon. However, phylogenetic analysis of NodI and NodJ homologs (S6 Fig) showed that the linkage between nodBC and nodIJ type genes is unlikely to be based on lateral gene transfer from Actinobacteria, but seems to have evolved independently in Actinobacteria and β-Proteobacteria (and later in α-Proteobacteria [80]).
The relatively low GC content of the nod genes in Dg1 relative to the whole genome, and similar patterns in the corresponding rhizobial genes, suggest a comparatively rapid evolution of these genes, related to the functional specialization of symbiosis. It is conceivable that multiple lateral gene transfer between the two phyla could explain the relatively low GC content of the nod genes relative to genomic GC content. However, this is not a scenario that is plausible based on any of the strongly-supported lines of phylogenetic evidence.
No close homologs of the canonical nod genes could be identified in the sequenced genomes of any Frankia strain of Cluster I or Cluster III, as can be seen in the phylogenetic trees. Thus in Frankia, the nod gene orthologs seem to be exclusive to Cluster II. Nod gene expression in nodules was observed not only in Dg1 in symbiosis with D. glomerata as described here; expression has also been detected in Ceanothus velutinus nodules collected in California (T. Persson, A.M. Berry and K. Pawlowski, unpublished observations). It is all the more striking that this strain—or group of strains—that has one of the smallest genomes and the largest number of pseudogenes of Frankia strains analyzed so far, and the most reduced number of gene clusters for the synthesis of secondary metabolites (S4 Table), nevertheless retains canonical nod genes. Assuming that the Dg1 nod genes expressed in symbiosis catalyze the production of LCOs for signaling to host plants, and given the well-demonstrated basal position of Cluster II genes relative to other Frankia, shown both here and in studies of a wide range of genes (e.g., [10]), it seems most likely that the common ancestor of all the symbiotic Frankia strains contained the canonical nod genes which were transmitted to the rhizobia, but subsequently lost in the progenitor of Frankia Clusters I and III. At any rate, the fact that Dg1 cannot be cultured so far has prevented any isolation of LCO Nod factors formed by this strain, as nodules proved to be too complex a source to isolate LCOs present at very low concentrations.
Conclusions
In summary, the results of the analysis of the first genome of a Cluster II Frankia strain, Dg1, supports the hypothesis that symbioses between Cluster II Frankia strains and actinorhizal Cucurbitales are likely to represent the oldest actinorhizal symbioses. Comparative analysis of Frankia genomes revealed more than 800 unique genes in Dg1. Among those are rhizobial-type canonical nod genes, which are expressed in symbiosis and, based on detailed phylogenetic analysis, are likely to have originated in Actinobacteria. Transcriptome analysis supported the hypothesis that Cluster II strains in nodules export an assimilated form of nitrogen, rather than ammonium, most likely arginine. Analyses of D. glomerata nodule occupancy via 454 OTU sequencing showed that (a) more than one strain was found in a nodule lobe, and (b) seemingly the same, or at least very similar strains were present in a Californian inoculant and in an inoculant originating in Pakistan, though two different strains were dominating in both inoculants. Transcriptome analysis underlined the low genetic diversity between the genomes of these different strains.
Supporting Information
The cumulative distribution function (CDF) for the rpkb data is illustrated as black circular data points. The CDF generated from the fit of a negative binomial function to the empirical data is illustrated as a solid red line.
(DOCX)
The core genome between the four sequenced Frankia strains (ACN14a, CcI3, EAN1pec, CcI3, and Dg1) was calculated using EDGAR (http://edgar.cebitec.uni-bielefeld.de; [59]).
(DOCX)
(A) Fraction of the genome consisting of IS elements in different Frankia strains. Detailed analysis of IS elements in two representatives of the basal non-symbiotic strains (CN3, EuI1c), in four strains of cluster I (two Alnus-infective strains, ACN14a and QA3, and two Casuarina-infective strains, CcI3 and BMG5.12), one representative of cluster II (Dg1) and four representatives of cluster III (two Elaeagnus-infective strains, EUN1f and EAN1pec, and one Discaria-infective strain, BCU110501) showed that three genomes among those analysed—those of CcI3, Dg1 and EAN1pec—show an increase in relative amounts of IS elements, and that among these three, the Dg1 genome contains the highest relative amount of IS elements. (B) Distribution of size of IS elements in Frankia strains.
(DOCX)
Acidothermus cellulolyticus 11B (Acido), Stackebrandtia nassauensis DSM 44728 (Stack), Geodermatophilus obscurus DSM 43160 (Go), Nakamurella multipartita DSM 44233 (Naka), and Thermobifida fusca YX (Thermo) were used as outgroups. Forty housekeeping genes were analyzed. Each gene sequence was identified in Candidatus Frankia datiscae Dg1. After identification, the gene was used in a Blast search as the query. The corresponding Blast was restricted to Frankia alni ACN14a, Frankia sp. Ccl3, Frankia sp. EaN1pec, A. cellulolyticus 11B, G. obscurus DSM 43160, S. nassauensis DSM 44728, N. multipartita DSM 44233 and T. fusca YX. All alignments were created using MUSCLE (multiple sequence comparison by log- expectation; Edgar 2004) at the EMBL-EBI website. Maximum parsimony analyses were performed using the software package PAUP* version 4.0b10 (Swofford 1999). All characters were weighted equally and gaps in the alignment were treated as missing. A heuristic search strategy with 10 random replicates, TBR branch-swapping and the MULTREES optimization was used. MAXTREES parameter was set to 10,000. Support for branches was evaluated using bootstrap analysis (Felsenstein 1985) and random sequence addition for 100 replicates, using the same parameters.
(DOCX)
Identical amino acids in highly conserved positions are highlighted in blue, identical amino acids in less conserved positions are highlighted in grey. Results are depicted in the order NodA, NodB, NodC, NodI, NodJ.
(DOCX)
All sequences from Dg1 are given in red. Sequences from β-proteobacteria where the rhizobial nodIJ genes evolved are given in green, sequences from α-proteobacteria are given in turquoise. Names of actinobacterial NltI/NltJ sequences the genes of which are part of a nodBCnltIJ operon are indicated in blue. The sequences from Streptomyces bottropensis are given in purple. All sequences used for the phylogenetic analysis are given in S6 Table.
(PDF)
(XLSX)
(ZIP)
One inoculant goes back to a D. glomerata plant from California (UCD), the other one to a Coriaria nepalensis plant from Pakistan (SU).
(XLSX)
The analysis of the genome sequences with regard to biochemical pathways in Dg1 was performed using Pathway tools [47], MAGE, IMG/ER and based on Udwary et al. [67].
(XLSX)
Palindromic Repeats were analyzed using the palindrome tool from EMBOSS (http://bips.u-strasbg.fr/EMBOSS/) with no mismatches and the following parameters: 1. Repeat units between 8 and 11 bases with up to a 3 base gap. 2. Repeat units between 12 and 19 bases with up to a 7 base gap. 3. Repeat units between 20 and 90 bases with up to a 20 base gap. 4. Repeat units less than 12 bases must occur at least 10 times in the genome. 5. Repeat units less than 20 bases must occur twice in the genome. Tandem repeats were analyzed with the MUMmer 3.13 package (http://www.tigr.org/software/mummer/) with the following parameters: Minimum match length = 20 bases. 2. It is assumed that one copy of a tandem repeat in a genome is not very significant unless it is long. Therefore, a genome-wide screen for the repeat used was added. The total number of bases incorporated into repeats for a particular repeat unit must total 50 or more bases.
(DOCX)
(XLSX)
(DOCX)
Acknowledgments
This research was funded by grants from the Swedish Research Councils Formas (229-2005-679) and VR (2007-17840-52674-16) to KP; by CA Experiment Station project PLS-2173-H to AMB; by a grant from the French ANR (BugsInaCell ANR-13-BSV7-0013-03) to PN; and UC Mexus-Conacyt grant to AMB and AMH. KB was supported by UCDavis Plant Sciences Graduate Fellowship; AO’B was supported by NSF GRFP DGE-1148897; EGW was supported by NSF EF-0949453 to MTF. Thanks go to Y-Y Guo for technical assistance and to A Jumpponen, Kansas State University, for guidance in 454 sequencing. We appreciate the sponsorship of the Joint Genome Institute for the Genome Jamboree of the Frankia_symbiont of Datisca glomerata genome, Walnut Creek, CA on July 27–28, 2009, especially the guidance of N. Karpides, N. Ivanova, and A. Copeland.
Data Availability
All relevant data are within the paper and its Supporting Information files. All sequences files are available from GenBank or JGI IMG, and all accession numbers are listed within the paper.
Funding Statement
This work was supported by Swedish Research Council Formas (229-2005-679) to KP; Swedish Research Council VR (2007-17840-52674-16) to KP; CA Experiment Station project PLS-2173-H to AMB; UC Davis Plant Sciences Graduate Fellowship to KB; UC Mexus-Conacyt 05-24 to AMB and AMH; NSF GRFP DGE-1148897 to A'OB; NSF EF-0949453 to MTF; Sesam ANR-10-BLAN-1708 to PN; and BugsInACell ANR-13-BSV7-0013-03 to PN. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Benson DR, Silvester WB (1993) Biology of Frankia strains, actinomycete symbionts of actinorhizal plants. Microbiol Rev 57: 293–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Soltis DE, Soltis PS, Morgan DR, Swensen SM, Mullin BC, Dowd JM, et al. (1995) Chloroplast gene sequence data suggest a single origin of the predisposition for symbiotic nitrogen fixation in angiosperms. Proc Natl Acad Sci USA 92: 2647–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Doyle JJ (2011) Phylogenetic perspectives on the origins of nodulation. Mol Plant Microbe Interact 24: 1289–1295. 10.1094/MPMI-05-11-0114 [DOI] [PubMed] [Google Scholar]
- 4. Werner GDA, Cornwell WK, Sprent JI, Kattge J, Kiers TE (2014) A single evolutionary innovation drives the deep evolution of symbiotic N2-fixation in angiosperms. Nature Commun 5: 4087 10.1038/ncomms5087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zanne AE, Tank DC, Cornwell WK, Eastman JM, Smith SA, FitzJohn RG, et al. (2014) Three keys to the radiation of angiosperms into freezing environments. Nature 506: 89–92. 10.1038/nature12872 [DOI] [PubMed] [Google Scholar]
- 6. Normand P, Chapelon C (1997) Direct characterization of Frankia and of close phyletic neighbors from an Alnus viridis rhizosphere. Physiol Plant 99: 722–731. [Google Scholar]
- 7. Normand P, Orso S, Cournoyer B, Jeannin P, Chapelon C, Dawson J, et al. (1996) Molecular phylogeny of the genus Frankia and related genera and emendation of family Frankiaceae. Int J Syst Bacteriol 46: 1–9. [DOI] [PubMed] [Google Scholar]
- 8. Ghodhbane-Gtari F, Nouioui I, Chair M, Boudabous A, Gtari M (2010) 16S-23S rRNA intergenic spacer region variability in the genus Frankia . Microb Ecol 60: 487–495. 10.1007/s00248-010-9641-6 [DOI] [PubMed] [Google Scholar]
- 9. Nouioui I, Ghodhbane-Gtari F, Beauchemin NJ, Tisa LS, Gtari M (2011) Phylogeny of members of the Frankia genus based on gyrB, nifH and glnII sequences. Antonie Van Leeuwenhoek 100: 579–587. 10.1007/s10482-011-9613-y [DOI] [PubMed] [Google Scholar]
- 10. Sen A, Daubin V, Abrouk D, Gifford I, Berry AM, Normand P (2014) Phylogeny of the class Actinobacteria revisited in the light of complete genomes. The orders ‘Frankiales’ and Micrococcales should be split into coherent entities: proposal of Frankiales ord. nov., Geodermatophilales ord. nov., Acidothermales ord. nov. and Nakamurellales ord. nov. Int J Syst Evol Microbiol 64: 3821–332. 10.1099/ijs.0.063966-0 [DOI] [PubMed] [Google Scholar]
- 11. Normand P, Lapierre P, Tisa LS, Gogarten JP, Alloisio N, Bagnarol E, et al. (2007) Genome characteristics of facultatively symbiotic Frankia sp. strains reflect host range and host plant biogeography. Genome Res 17: 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tisa LS, Beauchemin N, Gtari M, Sen A, Wall LG (2013) What stories can the Frankia genomes start to tell us? J Biosci 38: 719–726 [DOI] [PubMed] [Google Scholar]
- 13. Nouioui I, Beauchemin N, Cantor MN, Chen A, Detter JC, Furnholm T, et al. (2013) Draft genome sequence of Frankia sp. strain BMG5.12, a nitrogen-fixing actinobacterium isolated from Tunisian soils. Genome Announc 1: e00468–13. 10.1128/genomeA.00468-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sen A, Beauchemin N, Bruce D, Chain P, Chen A, Walston Davenport K, et al. (2013) Draft genome sequence of Frankia sp. strain QA3, a nitrogen-fixing actinobacterium isolated from the root nodule of Alnus nitida . Genome Announc 1: e001031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wall LG, Beauchemin N, Cantor MN, Chaia E, Chen A, Detter JC, et al. (2013) Draft genome Sequence of Frankia sp. strain BCU110501, a nitrogen-fixing actinobacterium isolated from nodules of Discaria trinevis . Genome Announc 1: e00503–13. 10.1128/genomeA.00503-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vanden Heuvel BD, Benson DR, Bortiri E, Potter D (2004) Low genetic diversity among Frankia spp. strains nodulating sympatric populations of actinorhizal species of Rosaceae, Ceanothus (Rhamnaceae) and Datisca glomerata (Datiscaceae) west of the Sierra Nevada (California). Can J Microbiol 50: 989–1000. [DOI] [PubMed] [Google Scholar]
- 17. Lawrence D, Schoenike R, Quispel A, Bond G (1967) The role of Dryas drummondii in vegetation development following ice recession at Glacier Bay, Alaska, with special reference to its nitrogen fixation by root nodules. J Ecol 55: 793–813. [Google Scholar]
- 18. Bond G (1976) The results of the IBP survey of root nodule formation in non-leguminous angiosperms In Symbiotic Nitrogen Fixation in Plants. Edited by Nutman P.. London: Cambridge University Press, pp. 443–474. [Google Scholar]
- 19. Liston A, Rieseberg LH, Elias T (1989) Morphological statis and molecular divergence in the intercontinal disjunct genus Datisca (Datiscaceae). Aliso 12: 525–542. [Google Scholar]
- 20. Yokoyama J, Suzuki M, Iwatsuki K, Hasebe M (2000) Molecular phylogeny of Coriaria, with special emphasis on the disjunct distribution. Mol Phylogenet Evol 14: 11–19. [DOI] [PubMed] [Google Scholar]
- 21. Persson T, Benson DR, Normand P, Vanden Heuvel B, Pujic P, Chertkov O, et al. (2011) The genome of Candidatus Frankia datiscae Dg1, the uncultured microsymbiont from nitrogen-fixing root nodules of the dicot Datisca glomerata . J Bacteriol 193: 7017–7018. 10.1128/JB.06208-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kistner C, Parniske M (2002) Evolution of signal transduction in intracellular symbiosis. Trends Plant Sci 7: 511–518. [DOI] [PubMed] [Google Scholar]
- 23. Maillet F, Poinsot V, André O, Puech-Pagès V, Haouy A, Gueunier M, et al. (2011) Fungal lipochitooligosaccharide symbiotic signals in arbuscular mycorrhiza. Nature 469: 58–63. 10.1038/nature09622 [DOI] [PubMed] [Google Scholar]
- 24. Op den Camp R, Streng A, De Mita S, Cao Q, Polone E, Liu W, et al. (2010). LysM-type mycorrhizal receptor recruited for Rhizobium symbiosis in nonlegume Parasponia . Science 331: 909–912. 10.1126/science.1198181 [DOI] [PubMed] [Google Scholar]
- 25. Oldroyd GE (2013) Speak, friend, and enter: signalling systems that promote beneficial symbiotic associations in plants. Nat Rev Microbiol 11: 252–263. 10.1038/nrmicro2990 [DOI] [PubMed] [Google Scholar]
- 26. Markmann K, Giczey G, Parniske M (2008) Functional adaptation of a plant receptor-kinase paved the way for the evolution of intracellular root symbioses with bacteria. PLoS Biol 6: e68 10.1371/journal.pbio.0060068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gherbi H, Markmann K, Svistoonoff S, Estevan J, Autran D, Giczey G, et al. (2008) SymRK defines a common genetic basis for plant root endosymbioses with arbuscular mycorrhiza fungi, rhizobia, and Frankia bacteria. Proc Natl Acad Sci USA 105: 4928–4932. 10.1073/pnas.0710618105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Svistoonoff S, Benabdoun FM, Nambiar-Veetil M, Imanishi L, Vaissayre V, Cesari S, et al. (2013) The independent acquisition of plant root nitrogen-fixing symbiosis in Fabids recruited the same genetic pathway for nodule organogenesis. PLoS One 8: e64515 10.1371/journal.pone.0064515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hocher V, Alloisio N, Auguy F, Fournier P, Doumas P, et al. (2011) Transcriptomics of actinorhizal symbioses reveals homologs of the whole common symbiotic signaling cascade. Plant Physiol 156: 700–711. 10.1104/pp.111.174151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Demina IV, Persson T, Santos P, Plaszczyca M, Pawlowski K (2013) Comparison of the nodule vs. root transcriptome of the actinorhizal plant Datisca glomerata: actinorhizal nodules contain a specific class of defensins. PLoS One 8: e72442 10.1371/journal.pone.0072442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Genre A, Chabaud M, Balzergue C, Puech-Pagès V, Novero M, Rey T, et al. (2013) Short-chain chitin oligomers from arbuscular mycorrhizal fungi trigger nuclear Ca2+ spiking in Medicago truncatula roots and their production is enhanced by strigolactone. New Phytol 198: 190–202. 10.1111/nph.12146 [DOI] [PubMed] [Google Scholar]
- 32. Cérémonie H, Debelle F, Fernandez MP (1999) Structural and functional comparison of Frankia root hair deforming factor and rhizobia Nod factor. Can J Bot 77: 1293–1301. [Google Scholar]
- 33. Giraud E, Moulin L, Vallenet D, Barbe V, Cytryn E, Avarre JC, et al. (2007) Legumes symbioses: Absence of nod genes in photosynthetic bradyrhizobia. Science 316: 1307–1312 [DOI] [PubMed] [Google Scholar]
- 34. Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Swofford DL (1999) PAUP 4.0: Phylogenetic analysis using parsimony (and other methods) Sunderland, MA, Sinauer Associates, Inc. [Google Scholar]
- 36. Felsenstein J (1985) Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39: 783–791. [DOI] [PubMed] [Google Scholar]
- 37. Benson AD, Cavanaugh M, Clark KM, Karsch-Mizrachi I, Lipman DJ, et al. (2013) GenBank. Nucl Acids Res 41: D36–D42. 10.1093/nar/gks1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Abascal F, Zardoya R, Posada D (2005) ProtTest: Selection of best-fit models of protein evolution. Bioinformatics 21: 2104–2105. [DOI] [PubMed] [Google Scholar]
- 39. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst Biol 59:307–321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- 40. Felsenstein J (2005) Using the quantitative genetic threshold model for inferences between and within species. Philos Trans R Soc Lond B Biol Sci 360: 1427–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. (2011) Integrative Genomics Viewer. Nature Biotechnol 29: 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. R Development Core Team (2008) R: A language and environment for statistical computing R Foundation for Statistical Computing, Vienna, Austria: ISBN 3-900051-07-0, URL http://www.R-project.org. [Google Scholar]
- 45.Delignette-Muller ML, Pouillot R, Denis JB, Dutang C (2013) fitdistrplus: help to fit of a parametric distribution to non-censored or censored data. Version 1.0–2
- 46. Caspi R, Altman T, Dale JM, Dreher K, Fulcher CA, Gilham F, et al. (2010) The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucl Acids Res 38: D473–D479. 10.1093/nar/gkp875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Karp PD, Paley SM, Krummenacker M, Latendresse M, Dale JM, Lee TJ, et al. (2010) Pathway Tools version 13.0: Integrated software for pathway/genome informatics and systems biology. Brief Bioinform 11: 40–79. 10.1093/bib/bbp043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT–PCR. Nucl Acids Res 29: e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.ABI PRISM 7700 Sequence Detection System. User Bulletin #2 (2007) Applied Biosystems. P/N 4303859B, Stock No. 777802–002.
- 50. Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, et al. (2011) antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucl Acids Res 39: W339–W346. 10.1093/nar/gkr466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Blom J, Albaum SP, Doppmeier D, Pühler A, Vorhölter FJ, Zakrzewski M, et al. (2009) EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinformat 10:154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S, et al. (2006) MaGe: a microbial genome annotation system supported by synteny results. Nucl Acids Res 34: 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Price AL, Jones NC, Pevzner PA (2005) De novo identification of repeat families in large genomes. Bioinformatics 21: i351–i358. [DOI] [PubMed] [Google Scholar]
- 54.Vigil-Stenman V, Larsson J, Nylander JAA, Bergman B (2014) Local hopping mobile DNA implicated in pseudogene formation and reductive evolution in an obligate cyanobacteria-plant symbiosis. BMC Genomics, in press. [DOI] [PMC free article] [PubMed]
- 55. Siguier P, Pérochon J, Lestrade L, Mahillon J, Chandler M (2006) ISfinder: the reference centre for bacterial insertion sequences. Nucl Acids Res 34: D32–D36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mirza MS, Hameed S, Akkermans ADL (1994) Genetic diversity of Datisca-compatible Frankia strains determined by sequence analysis of PCR-amplified 16S rRNA gene. Appl Environ Microbiol 60: 2371–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Reeder J, Knight R (2010) Rapid denoising of pyrosequencing amplicon data: exploiting the rank-abundance distribution. Nature Methods 7: 668–669. 10.1038/nmeth0910-668b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman DF, et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- 60. Katoh K, Standley DM (2014) MAFFT: iterative refinement and additional methods. Methods Mol Biol 1079: 131–146. 10.1007/978-1-62703-646-7_8 [DOI] [PubMed] [Google Scholar]
- 61. Swofford DL (2003) PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sinauer Associates, Sunderland, Massachusetts. [Google Scholar]
- 62. Downie JA, Knight CD, Johnston AWB, Rossen L (1985) Identification of genes and gene products involved in the nodulation of peas by Rhizobium leguminosarum . Mol Gen Genet 198: 255–262. [Google Scholar]
- 63. Khan SR, Gaines J, Roop RM 2nd, Farrand SK (2008) Broad-host-range expression vectors with tightly regulated promoters and their use to examine the influence of TraR and TraM expression on Ti plasmid quorum sensing. Appl Environ Microbiol 74: 5053–5062. 10.1128/AEM.01098-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Garg B, Dogra RC, Sharma PK (1999) High-efficiency transformation of Rhizobium leguminosarum by electroporation. Appl Environ Microbiol 65: 2802–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fåhraeus G (1957) The infection of clover root hairs by nodule bacteria studied by simple glass technique. J Gen Microbiol 16: 374–381. [DOI] [PubMed] [Google Scholar]
- 66. Beringer J (1974) R factor transfer in Rhizobium leguminosarum . J Gen Microbiol 84: 188–198. [DOI] [PubMed] [Google Scholar]
- 67. Udwary DW, Gontang EA, Jones AC, Jones CS, Schultz AW, Winter JM, et al. (2011) Significant natural product biosynthetic potential of actinorhizal symbionts of the genus Frankia, as revealed by comparative genomic and proteomic analyses. Appl Environ Microbiol 77: 3617–3625. 10.1128/AEM.00038-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pfeifer F (2012) Distribution, formation and regulation of gas vesicles. Nat Rev Microbiol 10: 705–715. 10.1038/nrmicro2834 [DOI] [PubMed] [Google Scholar]
- 69. Siguier P, Gourbeyre E, Chandler M (2014) Bacterial insertion sequences: their genomic impact and diversity. FEMS Microbiol Rev 38: 865–891. 10.1111/1574-6976.12067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mahillon J, Chandler M (1998) Insertion sequences. Microbiol Mol Biol Rev 62:725–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Moran NA, Plague GR (2004) Genomic changes following host restriction in bacteria. Curr Opin Genet Dev 14: 627–633. [DOI] [PubMed] [Google Scholar]
- 72. Bickhart DM, Gogarten JP, Lapierre P, Tisa LS, Normand P, Benson DR (2009) Insertion sequence content reflects genome plasticity in strains of the root nodule actinobacterium Frankia . BMC Genom 10: 468 10.1186/1471-2164-10-468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Moran NA (2003) Tracing the evolution of gene loss in obligate bacterial symbionts. Curr Opin Microbiol 6: 512–518. [DOI] [PubMed] [Google Scholar]
- 74. Ran L, Larsson J, Vigil-Stenman T, Nylander JA, Ininbergs K, Zheng WW, et al. (2010) Genome erosion in a nitrogen-fixing vertically transmitted endosymbiotic multicellular cyanobacterium. PLoS One 5: e11486 10.1371/journal.pone.0011486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ivanova N, Sikorski J, Jando M, Munk C, Lapidus A, Glavina Del Rio T, et al. (2010) Complete genome sequence of Geodermatophilus obscurus type strain (G-20). Stand Genomic Sci 2: 158–167. 10.4056/sigs.711311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Barabote RD, Xie G, Leu DH, Normand P, Necsulea A, Daubin V, et al. (2009) Complete genome of the cellulolytic thermophile Acidothermus cellulolyticus 11B provides insights into its ecophysiological and evolutionary adaptations. Genome Res 19: 1033–1043. 10.1101/gr.084848.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Munk C, Lapidus A, Copeland A, Jando M, Mayilraj S, Glavina Del Rio T, et al. (2009) Complete genome sequence of Stackebrandtia nassauensis type strain (LLR-40K-21). Stand Genomic Sci 1: 234–241. 10.4056/sigs.37633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tice H, Mayilraj S, Sims D, Lapidus A, Nolan M, Lucas S, et al. (2010) Complete genome sequence of Nakamurella multipartita type strain (Y-104). Stand Genomic Sci 2: 168–175. 10.4056/sigs.721316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lee KB, De Backer P, Aono T, Liu CT, Suzuki S, Suzuki T (2008) The genome of the versatile nitrogen fixer Azorhizobium caulinodans ORS571. BMC Genomics 9: 271 10.1186/1471-2164-9-271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Aoki S, Ito M, Iwasaki W (2013) From β- to α-proteobacteria: the origin and evolution of rhizobial nodulation genes nodIJ . Mol Biol Evol 30: 2494–2508. 10.1093/molbev/mst153 [DOI] [PubMed] [Google Scholar]
- 81. Husník F, Chrudimský T, Hypša V (2011) Multiple origins of endosymbiosis within the Enterobacteriaceae (γ-Proteobacteria): convergence of complex phylogenetic approaches. BMC Biol 9: 87 10.1186/1741-7007-9-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Young JPW, Johnston AW, Thomson NR, Ghayoui ZF, Hull KH, et al. (2006) The genome of Rhizobium leguminosarum has recognizable core and accessory components. Genome Biol 7: R34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Daubin V, Perrière G (2003) G+C3 structuring along the genome: a common feature in prokaryotes. Mol Biol Evol 20: 471–483 [DOI] [PubMed] [Google Scholar]
- 84. Alloisio N, Queiroux C, Fournier P, Pujic P, Normand P, Vallenet D, Médigue C, Yamaura M, Kakoi K, Kucho K (2010) The Frankia alni symbiotic transcriptome. Mol Plant Microbe Interact 23: 593–607. 10.1094/MPMI-23-5-0593 [DOI] [PubMed] [Google Scholar]
- 85. Pawlowski K, Demchenko KN (2012) The diversity of actinorhizal symbiosis. Protoplasma 249: 967–979. [DOI] [PubMed] [Google Scholar]
- 86. Hruska K, Kaevska M (2012) Mycobacteria in water, soil, plants and air: a review. Veterinarni Medicina 57: 623–679. [Google Scholar]
- 87. Peterson SB, Dunn AK, Klimowicz AK, Handelsman J (2006) Peptidoglycan from Bacillus cereus mediates commensalism with rhizosphere bacteria from the Cytophaga-Flavobacterium group. Appl Env Microbiol 72: 5421–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Labeda DP (2010) Multilocus sequence analysis of phytopathogenic species of the genus Streptomyces . Int J Syst Evol Microbiol 61: 2525–2531. 10.1099/ijs.0.028514-0 [DOI] [PubMed] [Google Scholar]
- 89. Chelius MK, Triplett EW (2000). Dyadobacter fermentans gen. nov., sp. nov., a novel Gram-negative bacterium isolated from surface-sterilized Zea mays stems. Int J Syst Evol Microbiol 50: 751–758. [DOI] [PubMed] [Google Scholar]
- 90. Edulamudi P, Masilamani AJA, Divi VRSG, Konada VM (2011) Novel root nodule bacteria belonging to the genus Caulobacter . Lett Appl Microbiol 53: 587–591. 10.1111/j.1472-765X.2011.03151.x [DOI] [PubMed] [Google Scholar]
- 91. Takeuchi M, Hamana K, Hiraishi A (2001) Proposal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium and Sphingopyxis, on the basis of phylogenetic and chemotaxonomic analyses. Int J Syst Evol Microbiol 51: 1405–1417. [DOI] [PubMed] [Google Scholar]
- 92. Madhaiyan M, Poonguzhali S, Kwon SW, Sa TM (2009) Methylophilus rhizosphaerae sp. nov., a restricted facultative methylotroph isolated from rice rhizosphere soil. Int J Syst Evol Microbiol 59: 2904–2908. 10.1099/ijs.0.009811-0 [DOI] [PubMed] [Google Scholar]
- 93. Moran NA (2006) Symbiosis. Curr Biol. 16: R866–R871. [DOI] [PubMed] [Google Scholar]
- 94. Gray MW (1999) Evolution of organellar genomes. Curr Opin Genet Dev 9: 678–687. [DOI] [PubMed] [Google Scholar]
- 95. Redecker D, Kodner R, Graham LE. (2000a) Glomalean fungi from the Ordovician. Science 289: 1920–1921. [DOI] [PubMed] [Google Scholar]
- 96. Sędzielewska KA, Fuchs J, Temsch EM, Baronian K, Watzke R, Kunze G (2011) Estimation of the Glomus intraradices nuclear DNA content. New Phytol 192: 794–797. 10.1111/j.1469-8137.2011.03937.x [DOI] [PubMed] [Google Scholar]
- 97. Nouioui I, Sbissi I, Ghodhbane-Gtari F, Benbrahim KF, Normand P, Gtari M (2013) First report on the occurrence of the uncultivated cluster 2 Frankia microsymbionts in soil outside the native actinorhizal host range area. J Biosci 38: 695–698. [DOI] [PubMed] [Google Scholar]
- 98. Burleigh SH, Dawson JO (1995) Spores of Frankia strain HFPCcI3 nodulate Casuarina equisetifolia after passage through the digestiv tracts of captive parakeets (Melopsittacus undulatus). Can J Bot 73: 1527–1530. [Google Scholar]
- 99. Huss-Danell K, Uliassi D, Renberg I (1997) River and lake sediments as sources of infective Frankia (Alnus). Plant Soil 197: 35–39. [Google Scholar]
- 100. Clawson ML, Caru M, Benson DR (1998) Diversity of Frankia strains in root nodules of plants from the families Elaeagnaceae and Rhamnaceae. Appl Environ Microbiol. 64: 3539–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ghodhbane-Gtari F, Beauchemin N, Bruce D, Chain P, Chen A, Walston Davenport K, et al. (2013) Draft genome sequence of Frankia sp. strain CN3, an atypical, noninfective (Nod-) ineffective (Fix-) isolate from Coriaria nepalensis . Genome Announc 1: e0008513 10.1128/genomeA.00085-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mansour SR, Oshone R, Hurst SG 4th, Morris K, Thomas WK, Tisa LS (2014) Draft genome sequence of Frankia sp. strain CcI6, a salt-tolerant nitrogen-fixing actinobacterium isolated from the root nodule of Casuarina cunninghamiana . Genome Announc 2: e01205–13. 10.1128/genomeA.01205-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Clark LC, Seipke RF, Prieto P, Willemse J, van Wezel GP, Hutchings MI, et al. (2013) Mammalian cell entry genes in Streptomyces may provide clues to the evolution of bacterial virulence. Sci Rep. 3: 1109 10.1038/srep01109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mirza MS, Akkermans WM, Akkermans ADL (1994) PCR-amplified 16S ribosomal RNA sequence analysis to confirm nodulation of Datisca cannabina by the endophyte of Coriaria nepalensis Wall. Plant Soil 160: 147–152. [Google Scholar]
- 105. Schlaman HR, Horvath B, Vijgenboom E, Okker RJ, Lugtenberg BJ (1991) Suppression of nodulation gene expression in bacteroids of Rhizobium leguminosarum biovar viciae . J Bacteriol 173: 4277–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Limpens E, Mirabella R, Fedorova E, Franken C, Franssen H, Bisseling T, et al. (2005) Formation of organelle-like N2-fixing symbiosomes in legume root nodules is controlled by DMI2. Proc Natl Acad Sci USA 102: 10375–10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Berry AM, Murphy TM, Okubara PA, Jacobsen KR, Swensen SM, Pawlowski K (2004) Novel expression pattern of cytosolic glutamine synthetase in nitrogen-fixing root nodules of the actinorhizal host, Datisca glomerata . Plant Physiol 135: 1849–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Berry AM, Mendoza-Herrera A, Guo Y-Y, Hayashi J, Persson T, Barabote R, Demchenko K, Zhang SX, Pawlowski K (2011) New perspectives on nodule nitrogen assimilation in actinorhizal symbioses. Funct Plant Biol 38: 645–652. [DOI] [PubMed] [Google Scholar]
- 109. Prell J, White JP, Bourdes A, Bunnewell S, Bongaerts RJ, Poole PS (2009) Legumes regulate Rhizobium bacteroid development and persistence by the supply of branched-chain amino acids. Proc Natl Acad Sci USA 106: 12477–12482. 10.1073/pnas.0903653106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Chen WM, Prell J, James EK, Sheu DS, Sheu SY (2012) Biosynthesis of branched-chain amino acids is essential for effective symbioses between β-rhizobia and Mimosa pudica . Microbiology 158(Pt7): 1758–1766. 10.1099/mic.0.058370-0 [DOI] [PubMed] [Google Scholar]
- 111. Berry AM, Harriott OT, Moreau RA, Osman SF, Benson DR, Jones AD (1993) Hopanoid lipids compose the Frankia vesicle envelope, presumptive barrier of oxygen diffusion to nitrogenase. Proc Natl Acad Sci USA 90: 6091–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Parniske M (2008) Arbuscular mycorrhiza: the mother of plant root endosymbioses. Nat Rev Microbiol 6:763–775. 10.1038/nrmicro1987 [DOI] [PubMed] [Google Scholar]
- 113. Zhang XC, Cannon SB, Stacey G (2009) Evolutionary genomics of LysM genes in land plants. BMC Evol Biol 9: 183 10.1186/1471-2148-9-183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Chen WM, de Faria SM, Straliotto R, Pitard RM, Simões-Araùjo JL, Chou JH, et al. (2005) Proof that Burkholderia strains form effective symbioses with legumes: a study of novel Mimosa-nodulating strains from South America. Appl Environ Microbiol 71: 7461–7471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bontemps C, Elliott GN, Simon MF, Dos Reis Júnior FB, Gross E, et al. (2010) Burkholderia species are ancient symbionts of legumes. Mol Ecol 19: 44–52. 10.1111/j.1365-294X.2009.04458.x [DOI] [PubMed] [Google Scholar]
- 116. Goedhart J, Röhrig H, Hink MA, van Hoek A, Visser AJ, Bisseling T, et al. (1999) Nod factors integrate spontaneously in biomembranes and transfer rapidly between membranes and to root hairs, but transbilayer flip-flop does not occur. Biochem 38: 10898–10907. [DOI] [PubMed] [Google Scholar]
- 117. Embley TM, Stackebrandt E (1994) The molecular phylogeny and systematics of the actinomycetes. Annu Rev Microbiol 48: 257–289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The cumulative distribution function (CDF) for the rpkb data is illustrated as black circular data points. The CDF generated from the fit of a negative binomial function to the empirical data is illustrated as a solid red line.
(DOCX)
The core genome between the four sequenced Frankia strains (ACN14a, CcI3, EAN1pec, CcI3, and Dg1) was calculated using EDGAR (http://edgar.cebitec.uni-bielefeld.de; [59]).
(DOCX)
(A) Fraction of the genome consisting of IS elements in different Frankia strains. Detailed analysis of IS elements in two representatives of the basal non-symbiotic strains (CN3, EuI1c), in four strains of cluster I (two Alnus-infective strains, ACN14a and QA3, and two Casuarina-infective strains, CcI3 and BMG5.12), one representative of cluster II (Dg1) and four representatives of cluster III (two Elaeagnus-infective strains, EUN1f and EAN1pec, and one Discaria-infective strain, BCU110501) showed that three genomes among those analysed—those of CcI3, Dg1 and EAN1pec—show an increase in relative amounts of IS elements, and that among these three, the Dg1 genome contains the highest relative amount of IS elements. (B) Distribution of size of IS elements in Frankia strains.
(DOCX)
Acidothermus cellulolyticus 11B (Acido), Stackebrandtia nassauensis DSM 44728 (Stack), Geodermatophilus obscurus DSM 43160 (Go), Nakamurella multipartita DSM 44233 (Naka), and Thermobifida fusca YX (Thermo) were used as outgroups. Forty housekeeping genes were analyzed. Each gene sequence was identified in Candidatus Frankia datiscae Dg1. After identification, the gene was used in a Blast search as the query. The corresponding Blast was restricted to Frankia alni ACN14a, Frankia sp. Ccl3, Frankia sp. EaN1pec, A. cellulolyticus 11B, G. obscurus DSM 43160, S. nassauensis DSM 44728, N. multipartita DSM 44233 and T. fusca YX. All alignments were created using MUSCLE (multiple sequence comparison by log- expectation; Edgar 2004) at the EMBL-EBI website. Maximum parsimony analyses were performed using the software package PAUP* version 4.0b10 (Swofford 1999). All characters were weighted equally and gaps in the alignment were treated as missing. A heuristic search strategy with 10 random replicates, TBR branch-swapping and the MULTREES optimization was used. MAXTREES parameter was set to 10,000. Support for branches was evaluated using bootstrap analysis (Felsenstein 1985) and random sequence addition for 100 replicates, using the same parameters.
(DOCX)
Identical amino acids in highly conserved positions are highlighted in blue, identical amino acids in less conserved positions are highlighted in grey. Results are depicted in the order NodA, NodB, NodC, NodI, NodJ.
(DOCX)
All sequences from Dg1 are given in red. Sequences from β-proteobacteria where the rhizobial nodIJ genes evolved are given in green, sequences from α-proteobacteria are given in turquoise. Names of actinobacterial NltI/NltJ sequences the genes of which are part of a nodBCnltIJ operon are indicated in blue. The sequences from Streptomyces bottropensis are given in purple. All sequences used for the phylogenetic analysis are given in S6 Table.
(PDF)
(XLSX)
(ZIP)
One inoculant goes back to a D. glomerata plant from California (UCD), the other one to a Coriaria nepalensis plant from Pakistan (SU).
(XLSX)
The analysis of the genome sequences with regard to biochemical pathways in Dg1 was performed using Pathway tools [47], MAGE, IMG/ER and based on Udwary et al. [67].
(XLSX)
Palindromic Repeats were analyzed using the palindrome tool from EMBOSS (http://bips.u-strasbg.fr/EMBOSS/) with no mismatches and the following parameters: 1. Repeat units between 8 and 11 bases with up to a 3 base gap. 2. Repeat units between 12 and 19 bases with up to a 7 base gap. 3. Repeat units between 20 and 90 bases with up to a 20 base gap. 4. Repeat units less than 12 bases must occur at least 10 times in the genome. 5. Repeat units less than 20 bases must occur twice in the genome. Tandem repeats were analyzed with the MUMmer 3.13 package (http://www.tigr.org/software/mummer/) with the following parameters: Minimum match length = 20 bases. 2. It is assumed that one copy of a tandem repeat in a genome is not very significant unless it is long. Therefore, a genome-wide screen for the repeat used was added. The total number of bases incorporated into repeats for a particular repeat unit must total 50 or more bases.
(DOCX)
(XLSX)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files. All sequences files are available from GenBank or JGI IMG, and all accession numbers are listed within the paper.