

Abstract
Dosage compensation in Drosophila increases the transcription of genes on the single X chromosome in males to equal that of both X chromosomes in females. Site-specific histone acetylation by the male-specific lethal (MSL) complex is thought to play a fundamental role in the increased transcriptional output of the male X. Nucleation and sequence-independent spreading of the complex to active genes serves as a model for understanding the targeting and function of epigenetic chromatin-modifying complexes. Interestingly, two noncoding RNAs are key for MSL assembly and spreading to active genes along the length of the X chromosome.
In Drosophila, gene transcription on the X chromosome in males doubles to match that of both X chromosomes in females. This involves site-specific histone acetylation by the MSL complex, which requires two noncoding RNAs.
OVERVIEW
In recent years, the long-standing dogma that cellular differentiation and development require the coordinate regulation of different sets of genes in time and space has led to the search for regulatory signals that would affect the activity of groups of functionally related genes. An example of coordinate regulation had been described in Drosophila long before these investigations were initiated. This involved a group of genes whose activity was regulated in unison, yet were not related by function. Rather, they shared a common location in the genetic material—the X chromosome. The purpose of the regulation was to ensure that females with two X chromosomes and males with only one X would have equal levels of gene products, in other words, to compensate for differences in the doses of X-linked genes between the sexes, commonly referred to as “dosage compensation.” In studying this level of regulation, the question “How are groups of unrelated genes coordinately regulated?” became “What are the mechanisms that can regulate the activity of a whole chromosome?”
The study of dosage compensation in Drosophila, a mechanism that enhances the transcription of most of the genes on the single X chromosome in males, reveals the involvement of site-specific histone acetylation, X-specific noncoding RNAs (called roX1 and roX2), and chromosome-wide targeting of an evolutionarily conserved chromatin-modifying machine (called the MSL complex, for male-specific lethal complex).
1. THE PHENOMENON OF DOSAGE COMPENSATION WAS DISCOVERED IN DROSOPHILA
The karyotypes (i.e., ensemble of chromosomes) of many organisms include a pair of sex chromosomes. In Drosophila, females have two sex chromosomes called the X chromosomes that are identical in shape and genetic content; both X chromosomes are active in all somatic cells. Males have one X and a Y chromosome that differs from the X in morphology and genetic information that it contains. On the sex chromosomes there are genes that are responsible for sex determination and sexual differentiation. The Y chromosome is male specific, but the X chromosome carries many genes involved in basic cellular housekeeping functions or developmental pathways. Females with two X chromosomes have twice the number of these genes; males with a single X have only one dose. Yet, the level of the products of most of these genes is the same in the two sexes. In the early 1930s, this paradox was first noticed in Drosophila by H.J. Muller while he was studying the eye pigment level of individuals carrying partial loss-of-function X-linked mutations (Muller 1932). Muller reasoned that there must be a regulatory mechanism that helps flies to compensate for the difference in dosage of X-linked genes in males and females by equalizing the level of X-linked gene products between the two sexes. He called this hypothetical regulatory mechanism “dosage compensation” (Fig. 1).
Figure 1.

Diagrammatic representation of the results that led H.J. Muller to formulate the hypothesis of dosage compensation. The mutant allele of the X-linked white gene (wa) is a hypomorph and allows partial eye-pigment synthesis; its presence on the X chromosomes is indicated. The level of pigmentation is directly proportional to the dosage of the wa allele within each sex; yet, males with one dose and females with two doses have comparable amounts of pigment because of dosage compensation.
Following its discovery in Drosophila, the phenomenon of dosage compensation was observed in additional species. We now know that in organisms belonging to distantly related groups—from round worms to mammals—transcriptional regulation leading to equal products of X-linked genes in males and females has been achieved in different ways: by decreasing the level of transcription of the two doses of X-linked genes in hermaphrodites relative to males (Caenorhabditis elegans) or by hypertranscribing the X chromosome in both males and females and then shutting down one of the two X chromosomes throughout most of its length in the somatic cells of females (mammals). The mechanisms underlying dosage compensation in these forms are described in Strome et al. (2014) and Brockdorff and Turner (2014).
The first evidence that dosage compensation in Drosophila is achieved by regulating the transcription of X-linked genes was obtained more than 30 years after Muller’s seminal observations, by A.S. Mukherjee and W. Beermann (Mukherjee and Beermann 1965). Using transcription autoradiography of the giant polytenic chromosomes of larval salivary glands, a molecular technique that represented the state of the art at that time, these investigators observed that the level of [3H]uridine incorporation by the single X in males and both Xs in females was equivalent. It appeared, therefore, that the rate of RNA synthesis by the single X chromosome in males was approximately twice the rate of each of the two Xs in females. The next experimental breakthrough consisted of the genetic identification by J. Belote and J. Lucchesi of four genes: msl1, msl2, msl3, and mle, with loss-of-function mutations that appeared inconsequential in females but lethal in males; notably, the mutant males showed approximately half of the normal level of [3H]uridine incorporation by their X chromosome (Belote and Lucchesi 1980a,b). Furthermore, the X chromosome had lost its normal paler and somewhat puffed appearance that had been interpreted as an indication of an enhanced level of transcriptional activity in relation to each of the two X chromosomes in females. These results suggested that the equalization of X-linked gene products was achieved by doubling, on average, the transcriptional activity of the X chromosome in males rather than by halving the transcriptional activity of each X in females.
An alternate hypothesis was proposed based on an “inverse dosage effect,” in which the activity of all chromosomes is set by general transcriptional regulators (reviewed in Birchler et al. 2011). In males, because of the absence of one X chromosome, a greater concentration of these regulators would be available than in females, driving the activity of all chromosomes to higher levels. For appropriate compensation to occur, the products of the msl loci would sequester some of these regulators away from the autosomes in males, thus leaving only the X chromosome with increased expression. In this model, msl gene mutations result in elevation of the expression of autosomal genes rather than a reduction of X-linked gene expression. However, a number of experimental results are incompatible with the inverse hypothesis (Arkhipova et al. 1997; Hamada et al. 2005; Straub et al. 2005; Deng et al. 2011). Particularly compelling is the recent observation that ectopic MSL complex on autosomes leads to a localized increase in transcription and suppression of phenotypes caused by haplo-insufficient mutants in the same region (Park et al. 2010).
Among the four genes introduced above, two were newly discovered (male-specific lethal 1, msl1; and male-specific lethal 2, msl2), whereas the other two (maleless, mle; and male-specific lethal 3, msl3) had been previously identified by other investigators in natural populations (specific references to this early phase of the study of dosage compensation can be found in Lucchesi and Manning 1987). For ease of reference, all of the gene products identified to date, on the basis of the male-specific lethal phenotype of their loss-of-function mutations, are called the MSLs. The next phase in the study of dosage compensation was initiated with the cloning of mle and the three msl genes, and the discovery and cloning of the mof histone acetyltransferase gene. By cytoimmunofluorescence, the five gene products were found to associate in an identical pattern at numerous sites along the polytene X chromosome in males (reviewed in Gelbart and Kuroda 2009). This observation and the interdependence of the different gene products for X-chromosome binding suggested that they form a complex. It is crucial for viability that the complex is present in males (XY) and absent in females (XX); therefore, the first step in dosage compensation is to establish this sex-specificity.
2. REGULATORS OF DOSAGE COMPENSATION
2.1. Regulation of Dosage Compensation Starts with Counting the Number of X Chromosomes
Each embryo needs to count its X chromosomes to make the critical decision whether or not to implement dosage compensation. An incorrect decision, such as failure to up-regulate the single male X or aberrant up-regulation of both female XXs, results in lethality. In Drosophila, the X-counting process is coordinated with the sex determination decision (reviewed in Cline and Meyer 1996). Phenotypic sex is determined by the number of X chromosomes per nucleus, such that XX embryos are females and XY embryos are male. The Y chromosome is required for male fertility, but unlike in mammals, it plays no role in phenotypic sex. Formally it is the X:autosome ratio that controls both sex and dosage compensation, as the X counting mechanism is sensitive to the number of sets of autosomes. This becomes apparent in 2X:3A triploids, which have an intermediate X:A ratio between XY:2A males and XX:2A females. 2X:3A triploids differentiate as intersexes with a mixture of both male and female cells.
The X:A ratio controls both sex determination and dosage compensation by regulating a critical binary switch gene, Sex lethal (Sxl). Sxl encodes a female-specific RNA-binding protein that regulates splicing and translation of key messenger RNAs (mRNAs) in the sex determination and dosage compensation pathways respectively (Fig. 2). The Sex lethal gene resides on the X chromosome and is positively regulated by transcription factors encoded by the X, such that embryos with two X chromosomes are able to initiate Sxl expression from an early, regulated promoter, Pe, whereas embryos with a single X per nucleus fail to express Sxl from Pe. This initial transient difference in activation of Sxl in early embryos is stabilized by an autoregulatory loop in which SXL protein positively regulates splicing of its own mRNA from a maintenance promoter that is expressed constitutively. SXL initiates differentiation in the female mode by regulating the splicing of the transformer (tra) gene in a sex-specific manner. In turn, this gene product (together with the product of another gene, transformer2 (tra2), present in both sexes) directs the splicing of the doublesex (dsx) primary transcript to yield a regulatory protein that acts to repress genes required for male development, thus achieving female sexual differentiation. In male embryos, an alternate mode of splicing of the dsx transcripts occurs by default and leads to a product that represses genes required for female development, resulting in male sexual differentiation.
Figure 2.

Diagram of the control of sex determination and dosage compensation. If the X/A ratio is equal to 1, a regulatory cascade leads to female sexual development. In females, the presence of the Sxl gene product prevents the translation of the msl2 message and the assembly of the MSL complex. If the X/A ratio is only 0.5, absence of the cascade leads by default to male sexual development and to the formation of the MSL complex.
2.2. The SXL Protein Prevents Formation of the MSL Complex in Females
The key target of SXL in the dosage compensation pathway is msl2 mRNA (Bashaw and Baker 1997; Kelley et al. 1997). SXL binding sites are located in both the 5′ and 3′ untranslated regions (UTRs) of msl2 mRNA. SXL is normally present only in females, in which it represses translation of the msl2 mRNA through association with its UTRs (see Fig. 2). If SXL is absent in females, dosage compensation is aberrantly turned on and these females die. Conversely, if SXL is ectopically expressed in males, dosage compensation is turned off and males die. Ectopic expression of MSL2 in females is sufficient to assemble MSL complexes on both female X chromosomes, indicating that all other MSL components are either turned on or stabilized by expression of MSL2.
In summary, dosage compensation must respond to the number of X chromosomes in the nucleus, and these are counted early in embryonic development. Females repress MSL2 translation, preventing inappropriate dosage compensation when two X chromosomes are present. In the absence of SXL-mediated repression, males express MSL2 protein and this leads to the assembly of a functional MSL complex. What are the components of the complex and how do they function together to regulate dosage compensation?
3. ASSEMBLY OF THE CHROMATIN-REMODELING COMPLEX RESPONSIBLE FOR COMPENSATION
The MSL complex, which is essential for male viability, consists of five known protein subunits and two noncoding RNAs (ncRNAs). The unifying function of the individual components of the MSL complex appears to be the targeting of histone H4K16 acetylation and additional chromatin-modifying activities to active X-chromosomal genes (Fig. 3). Association of MSL1 and MSL2 is essential for binding of the MSL complex to chromatin because these are the only subunits that associate with the X chromosome in the absence of the other MSL subunits (Copps et al. 1998; Gu et al. 1998). Whether or not there is a direct interaction with DNA is not known, as neither of these proteins contains an identifiable DNA-binding domain. Two MSL2 subunits interact with an MSL1 dimer and may initiate the assembly of the MSL complex (Hallacli et al. 2012). The direct interaction of MSL2 with MSL1 maps near the RING finger of MSL2 (a C3HC4 zinc-binding domain) and to an amino-terminal coiled coil domain in MSL1 (Scott et al. 2000). RING fingers are also associated with E3 ubiquitin ligase activity in many proteins, and both human and fly MSL2 show ubiquitin ligase activity directed toward histone H2B in vitro, dependent on interaction with MSL1 (Wu et al. 2011). MSL2 also ubiquitinates itself and other members of the MSL complex components, including MSL1, MSL3, and MOF, but not MLE in vitro (Villa et al. 2012). Current experiments are directed toward understanding the physiological role for this activity in vivo.
Figure 3.

The various components of the MSL complex. Known or proposed functions of MSL components include the acetylation of histone H4K16 by MOF and the ubiquitination of H2BK34 by MSL2; MLE has ATPase and RNA/DNA helicase activity, and JIL-1 phosphorylates histone H3. The male-specific complex promotes enrichment of the general factors JIL-1 and topoisomerase II to the male X chromosome.
In addition to its essential role in chromatin binding, MSL1 forms a scaffold for interaction with MSL3 and MOF via adjacent conserved carboxy-terminal domains (Morales et al. 2004; Kadlec et al. 2011). In the absence of interaction with MSL2, MSL1 is destabilized, thus leading to failure of MSL complex formation (Chang and Kuroda 1998).
MSL3 belongs to a group of proteins that may have coevolved with the chromodomain-bearing histone acetyltransferases (HATs; Pannuti and Lucchesi 2000; see Marmorstein and Zhou 2014 for more on HAT structure and function). Yeast Eaf3, a member of the MRG15/MSL3 protein family, interacts via the chromodomain with methylated H3K36 and recruits the histone deacetylase complex Rpd3S to protect active genes from spurious transcription initiation within the coding regions (Carrozza et al. 2005; Joshi and Struhl 2005; Keogh et al. 2005). A similar interaction between the MSL3 chromodomain and active chromatin marks such as H3K36me3 may help the MSL complex to locate target genes (Fig. 3) (Larschan et al. 2007; Bell et al. 2008; Sural et al. 2008). Of particular interest is the existence of a human complex related to the MSL complex that includes human homologs of MOF, MSL1, MSL2, and MSL3 (Smith et al. 2005; Taipale et al. 2005). This complex, which specifically acetylates histone H4 at lysine 16 and is responsible for the majority of this histone isoform in human cells, can include one of three different versions of a Drosophila MSL3 homolog encoded by two different genes (Smith et al. 2005).
Dosage compensation in Drosophila provided an early and compelling argument for a link between site-specific histone modifications and the regulation of gene expression. In 1992, Turner and colleagues made the seminal discovery that antibodies recognizing site-specific acetylation of histone H4 revealed distinct patterns on the Drosophila polytene chromosomes, with acetylation at position 16 (H4K16ac) showing strong enrichment on the male X chromosome (Turner et al. 1992). This enrichment requires the function of the msl genes (Bone et al. 1994), and the causal link between dosage compensation and chromatin modification was firmly established by the discovery that mof (males absent on the first) encodes a founding member of a new family of HATs (Hilfiker et al. 1997).
MOF is a member of the MYST subfamily of HATs. This subfamily, characterized by the presence of a chromodomain, can be further subdivided into enzymes that specifically acetylate lysine 16 in vivo (MOF and human MOF; Smith et al. 2005) and those such as Esa1p (essential SAS-related acetyl transferase 1) in yeast that acetylate all four terminal lysines of H4. Another MYST family member, SAS2, specifically acetylates lysine 16 in yeast, but lacks a chromodomain. Because the mof gene resides on the X chromosome, the discovery and characterization of mutants as male-specific lethals required a special genetic scheme to determine that an X-linked mutation lethal in males was viable in homozygous condition in females (Hilfiker et al. 1997).
mof encodes the MSL activity that is best implicated in gene regulation; therefore, one of the principal roles of the rest of the complex may be to localize MOF to its targets on the X chromosome. MOF recruitment is particularly important as MOF also participates in the nonspecific lethal (NSL) or MBD-R2 complex in both sexes (Raja et al. 2010; Feller et al. 2012; Lam et al. 2012). The NSL complex is found at 5′ ends of most active genes, coincident with MSL complex-independent H4K16 acetylation, and is essential for viability in both sexes. Because females are viable in the absence of MOF (albeit with low fertility), it is currently not clear how central MOF is to NSL function (Hilfiker et al. 1997; Gelbart et al. 2009).
MLE shows RNA/DNA helicase, adenosine triphosphatase (ATPase), and single-stranded RNA/single-stranded DNA binding activities in vitro (Lee et al. 1997), foreshadowing a potential role for RNA in MSL function. Mutants that retain the ATPase function but lack the helicase activity can still enhance transcription, but fail to support spreading of the complex along the X chromosome (Morra et al. 2008). Orthologs of MLE, which include human RNA helicase A (RHA), belong to the DEXH RNA helicase subfamily and are characterized by an additional domain implicated in double-stranded RNA binding (Pannuti and Lucchesi 2000). RHA is an abundant, essential protein in mammals, involved in numerous biological processes (Lee et al. 1998). It is likely that MLE performs its function in dosage compensation by interacting with or altering RNA structure, in particular the roX RNAs (see Sec. 4).
In addition to male-specific factors, it is likely that general factors involved in chromatin organization and transcription in both sexes also participate in dosage compensation. JIL-1, a tandem kinase, is found along all chromosomes in both males and females, but is more highly concentrated on the male X chromosome. This enrichment is dependent on the MSL complex. JIL-1 mediates histone H3 phosphorylation at Serine 10 and maintains open chromatin structure in transcriptionally active regions of the genome (Wang et al. 2001). However, despite its localization on active genes, definition of a direct role in transcription remains elusive (Ivaldi et al. 2007; Cai et al. 2008; Regnard et al. 2011).
In summary, the MSL proteins and JIL1 kinase have the ability to modify nucleosomes specifically on the male X chromosome. A central question is how these chromatin-modifying activities become targeted to a single chromosome. ncRNAs, DNA sequences, and active chromatin marks have all been implicated in the targeting process.
4. NONCODING roX RNAs FACILITATE ASSEMBLY AND TARGETING OF THE MSL COMPLEX ON THE X CHROMOSOME
One of the most intriguing and mysterious aspects of dosage compensation in both mammals and Drosophila is the role of ncRNAs in targeting compensation to the correct chromosome (reviewed in Gelbart and Kuroda 2009; also see Brockdorff and Turner 2014). Two ncRNAs, called RNA on X (roX), are dissimilar in size and sequence, and yet function redundantly to target the MSL complex to the male X chromosome in Drosophila (Meller and Rattner 2002). Traditional mutant screens usually do not reveal the existence of genes that encode products with redundant functions such as the roX RNAs. Rather, roX RNAs were discovered by serendipity as male-specific RNAs in the adult brain (Amrein and Axel 1997; Meller et al. 1997). On closer examination, both RNAs displayed a lack of significant open reading frames and colocalization with the MSL complex along the length of the X. roX RNA function was not revealed until an X-chromosome mutant for both roX1 and roX2 was isolated. Most double mutant males die, with severely mislocalized MSL complexes, whereas single mutant males have no known phenotype (Meller and Rattner 2002). This is surprising in view of the fact that the two roX RNAs are very different in size (3.7 kb vs. 0.5–1.4 kb) and share little sequence similarity. A likely explanation for the apparent flexibility and sequence divergence of the roX RNAs comes from the discovery of a degenerate sequence, termed the roX box, which is found in multiple copies in each RNA and can participate in conserved secondary structures (Fig. 4) (Park et al. 2007; Kelley et al. 2008). An RNA, comprised primarily of tandem copies of these secondary structures, is capable of stimulating the H4K16 acetylation activity of the MSL complex in vivo (Park et al. 2007).
Figure 4.

Evolutionarily conserved regions of roX RNAs. (A) Gene structure of roX1 and roX2 RNA showing location of stem-loop region (SL) and GUUNUACG roX boxes. (B) Stem-loop structures of the RNAs. (Modified from Maenner et al. 2013, © Elsevier.)
roX RNAs are recovered after coimmunoprecipitation of MSL proteins, demonstrating physical association of the RNAs with the complex (Meller et al. 2000; Smith et al. 2000). Partial purification of the complex suggests the presence of a tight core consisting of MSL1, MSL2, MSL3, and MOF proteins, with roX RNA and the MLE helicase lost except under very low salt concentrations (Smith et al. 2000). The minimal protein core complex lacking roX RNAs can still specifically acetylate histone H4 on lysine 16 within nucleosomes in vitro (Morales et al. 2004), and overexpression of MSL proteins can partially overcome the lack of roX RNAs, suggesting that the proteins possess all of the essential functions of dosage compensation but require the RNAs to stimulate assembly and spreading (Oh et al. 2003). Assembly of the complex is initiated when the secondary structure of the roX RNAs is modified, allowing the binding of MSL2 and providing the core for the full recruitment of the other MSL subunits (Ilik et al. 2013; Maenner et al. 2013).
5. HIGH-RESOLUTION ANALYSIS OF MSL BINDING ON THE X CHROMOSOME
Genomic analyses of MSL-binding sites have provided valuable insights into both the targeting principles and mechanism of action of the MSL complex. Chromatin immunoprecipitation (ChIP), enriching for MSL-associated DNA fragments, has been coupled to microarray or high-throughput DNA sequencing (ChIP-chip or ChIP-seq technology). Results show strong enrichment on the X chromosome, consistent with the previous cytological analyses. With the greatly improved resolution of ChIP, it was discovered that the MSL complex is enriched over the bodies of active X-linked genes, rather than binding primarily at the promoter or upstream intergenic regions as would be expected of a typical transcription factor (Fig. 5A,B) (Alekseyenko et al. 2006; Gilfillan et al. 2006). In fact, MSL binding is biased toward the 3′ ends of active genes, suggesting that it might act downstream from initiation, potentially at the level of elongation or the recycling of RNA polymerase II back to the promoter for reinitiation (Smith et al. 2001).
Figure 5.

Localization of the MSL complex. (A) High-resolution ChIP-chip analysis of MSL3 binding and H3K36me3 along 180-kb sections of chromosomes X and 2L in males, illustrated on a logarithmic scale. MSL3 was tagged (MSL-TAP) to purify it using the tandem affinity purification (TAP) technique during ChIP analysis. Concomitant H3K36me3 ChIP-chip analysis shows that the MSL complex colocalizes with H3K36me3 on the middle and 3′ ends of transcribed genes in SL2 cells. Genes on the top line represent those expressed from left to right and those shown below are genes expressed from right to left. Rectangles represent exons, connected by lines that represent introns. Red genes are expressed, whereas black genes are not expressed. (B) Comparison of gene expression state and MSL binding. Genes were divided into quantiles by increasing Affymetrix expression values and graphed to show the percent of genes in each quantile that were clearly bound by MSL complex in ChIP-chip analysis. (C–F) Maximal autosomal spreading is achieved when a roX transgene is the only source of roX RNA in the cell. (C) Chromosomes from a male with a wild-type (WT) X; the presence of an autosomal roX transgene is indicated by the arrow, showing a narrow MSL band (red). X, X chromosome; A, autosome. (D) A male with only one active X-linked roX gene. The MSL complex spreads slightly more from the autosomal roX transgene than in wild type, but binding is reduced on the X chromosome. (E,F) Extensive spreading of the MSL complex in two roX transgenic male lines, in which the X chromosome has both roX genes deleted. (A, Adapted, with permission, from Alekseyenko et al. 2006, © Cold Spring Harbor Laboratory Press; and, with permission, from Larschan et al. 2007, © Elsevier; B, adapted, with permission, from Alekseyenko et al. 2006; C–F, adapted from Park et al. 2002, © AAAS.)
The full binding pattern does not reveal a simple solution to the targeting question. How might this pattern arise solely on the X chromosome? One can envisage two very general models for regulating a whole chromosome. A single site or a very limited number of sites might control the chromosome in cis, as is the case in mammalian X inactivation via the region called the X inactivation center (see Brockdorff and Turner 2014). This mechanism requires either compartmentalization of the complex to a specific place in the nucleus or regulation over very long distances through the spreading of factors from the central control region to the rest of the chromosome. On the other extreme, a chromosome could have unique identifying sequences at every regulated gene along its entire length. In this case, any segment of the chromosome could be regulated autonomously. The targeting model for the MSL complex appears to share characteristics of both of these possibilities, with a set of initiation sites potentially dispersing the complex in cis to its full set of targets along the chromosome.
roX RNAs are normally encoded by the X chromosome; the roX1 gene is near the tip and the roX2 gene is around the middle of the euchromatic part of the X. Like the Xist gene in mammals, the roX genes may reside on the X to target MSL complex assembly to this chromosome. When roX genes are moved to the autosomes as transgenes, they potently attract MSL proteins to their novel insertion sites (Fig. 5C–F), where the complex appears to spread in cis, variably into flanking sequences (Kelley et al. 1999). Under specific genetic conditions (e.g., when there are no competing endogenous roX genes on the X chromosome), extensive spreading from autosomal roX transgenes is consistently seen (Fig. 5E,F) (Park et al. 2002). This extensive spreading is augmented by overexpression of MSL1 and MSL2, the key limiting MSL proteins, and diminished by overexpression of roX RNA from competing transgenes, suggesting that successful cotranscriptional assembly of MSL complexes may drive local spreading (Oh et al. 2003). The ability of roX genes to direct MSL complexes to the wrong chromosome is one of the most intriguing aspects of the dosage compensation mechanism. However, in all cases in which roX genes direct spreading in cis on autosomes, they also provide roX RNA in trans to cover the X chromosome (Meller and Rattner 2002). Therefore, it is clear the X chromosome has additional targeting signals beyond the two known roX genes.
In the absence of either MSL1 or MSL2, none of the remaining MSL proteins or roX RNAs appears to retain specific recognition for the X chromosome. However, in the absence of MLE, MSL3, or MOF, partial MSL complexes bind a subset of approximately 35–70 sites by cytological mapping, including the two roX genes (Fig. 6A). Termed high-affinity sites (HASs) or chromatin entry sites (CESs), these were postulated to be nucleation sites that might enable the MSL complex to access the X chromosome. Again, a genomic approach has provided key insights into how targeting might occur. Results from ChIP of MSL2 in msl3 mutant embryos, or MSL1 or MSL2 in suboptimal crosslinking conditions, identified a set of 130–150 candidate entry sites by their high affinity (Alekseyenko et al. 2008; Straub et al. 2008). These sites included the previously characterized entry sites in the roX1 and roX2 genes (Park et al. 2003). Motif searches yielded a 21-bp GA-rich (or TC-rich) common sequence motif, named the MSL recognition element or MRE (Fig. 6B). In functional assays, CES sequences attract the MSL complex when moved to autosomes. Conversely, MRE mutations abolish MSL recruitment, whereas scrambling the surrounding sequences has no effect (Alekseyenko et al. 2008). Therefore, MREs sprinkled along the length of the X are likely to play a key role in MSL recognition of the X chromosome. Recently, a zinc finger protein, CLAMP (chromatin-linked adaptor for MSL proteins), was found to bind directly to the MREs, with particular affinity for those MREs present within the CES sites; in the absence of CLAMP, the MSL complex was depleted along the entire X chromosome, suggesting that it functions in recruiting the complex to its initial binding sites (Larschan et al. 2012; Soruco et al. 2013).
Figure 6.

Targeting of the MSL complex. (A) The MSL complex is found associated with numerous sites along the X chromosome in males (upper panel); a mutant or incomplete complex containing at least MSL1 + MSL2 is found at fewer sites called CESs or HASs. (B) A GA-rich motif is a common feature in MSL CESs. The motif logo is shown with two examples of CESs presented below. This motif occurs once in CES11D1 and three times in CES5C2. The GA-rich core is highlighted in red. (A, Modified from Gu et al. 2000; B, reprinted, with permission, from Alekseyenko et al. 2008, © Elsevier.)
The identification of a functional DNA sequence element is a significant step toward understanding how the MSL complex initiates binding to the X chromosome. However, the MRE is less than twofold enriched on the X, and like the sites for most sequence-specific binding factors, there is an excess of nonused sites that match the consensus motif. Although it is enriched on the X chromosome, the CLAMP protein is present at the 5′ end of active genes on all chromosomes in both sexes. Therefore, there must be additional features, such as the local chromatin context (Alekseyenko et al. 2012), that help distinguish the MREs on the X that are used in vivo.
6. TRANSITION FROM INITIATION SITES TO TARGET GENES
If entry sites enable sequence-specific binding of the MSL complex to the X chromosome, how does the complex reach the majority of its target sites (i.e., transcriptionally active genes on the X chromosome)? Experiments to date support a model in which initial recruitment of the MSL complex to CESs generates high local concentrations that drive spreading to nearby sites of lower affinity. Recent evidence, based on high-shear ChIP-seq, suggests that MSL2 and MLE contact the HASs directly and provide a platform for the indirect association of the other subunits and roX RNA at these sites (Straub et al. 2013). Movement of the complex to active genes is facilitated by the activity of MOF and MLE (Gu et al. 2000; Morra et al. 2008) and is stabilized by the binding of MSL3 to H3K36me3 (Larschan et al. 2007).
It is not known whether spreading is a linear process, whereby the complex scans along chromatin and is only stabilized at active genes, or whether spreading is discontinuous, sampling chromatin in three-dimensional space. In either case, the majority of the MSL complex is targeted within 10 kb of a CES (Sural et al. 2008). Interestingly, the MSL-dependent H4K16ac mark is more widely distributed along the X chromosome than documented MSL binding, suggesting the transient interaction of the complex with a larger fraction of the X than that observed to have stable binding (Fig. 7) (Gelbart et al. 2009; Conrad et al. 2012; Straub et al. 2013).
Figure 7.

Correlation of H4K16 acetylation and MSL complex binding on the male X chromosomes. The distribution of H4K16ac on the male X chromosome is broader than MSL complex; active genes that lack stable MSL binding are nonetheless associated with H4K16ac. See Figure 5 for explanation of gene representation. (Adapted from Gelbart et al. 2009.)
The mechanism for recognizing active genes may involve additional sequence elements, features of transcription, or both. Ectopic MSL binding appears at regulated transgenes inserted on the X only upon induction of expression (Fig. 8) (Sass et al. 2003). The localization of the MSL complex to active genes on autosomes when ectopically recruited by a roX transgene suggests that the critical targeting features are not restricted to X-linked genes (Kelley et al. 1999). One clue suggesting the identity of one of these features came from the observation that the distribution of MSL complex along genes is strongly coincident with the pattern of H3K36me3, a histone modification associated with active genes on all chromosomes (Fig. 5A). This interaction is the hallmark of coding regions and is less pronounced in long intronic sequences in which H3K36me3 is less abundant (Straub et al. 2013). SET2, the enzyme responsible for H3K36 trimethylation, is attracted to gene bodies by the elongating form of RNA polymerase II. In the absence of SET2 activity, the MSL complex shows diminished binding to target genes on the X chromosome, suggesting a role for H3K36me3 in spreading (Larschan et al. 2007). The fact that binding is decreased, but not eliminated, in a set2 mutant suggests that transcriptionally active genes are recognized by additional, partially redundant mechanisms. For example, roX RNAs participate in spreading by an unknown mechanism.
Figure 8.

The MSL complex targets activated genes. (A) A construct containing a promoter under the control of the trans-activator GAL4 has been inserted at a site on the X indicated by the yellow arrowhead. This region is normally devoid of the MSL complex in larval salivary gland chromosomes. (B) When GAL4 is introduced, it binds to the construct (red), activates it, and recruits the MSL complex (blue). (Adapted, with permission, from Sass et al. 2003, © National Academy of Sciences.)
An 80-kb segment of autosomal DNA inserted on the X is capable of MSL recruitment to its active genes with no evidence for skipping, suggesting that there is not an obligate requirement for an X-specific sequence within active genes (Gorchakov et al. 2009). However, deletion mapping of X-linked genes inserted on autosomes has revealed specific gene segments that function in conjunction with transcription to attract the MSL complex (Kind and Akhtar 2007). As yet, not enough of these examples have been described to deduce common sequence characteristics that can be tested experimentally by mutagenesis. In stable translocation stocks, spreading of MSL complexes from the X into contiguous autosomal sequences is not evident (Fagegaltier and Baker 2004; Oh et al. 2004). Therefore, even if spreading of MSL complexes is a major mechanism for covering the X chromosome, there is very likely an additional characteristic of the X that causes MSL complex to strongly favor X over autosomal binding. Possibilities include a distinct overall sequence composition, including enrichment for simple repeats that can be detected by principal component analysis (Stenberg and Larsson, 2011), and the distinct three-dimensional organization of each chromosome into its own “territory” within the nucleus (see Sec. 9).
How can these various observations be accommodated into an integrated scheme for X-chromosome targeting of the MSL complex? A model that is the best fit for existing data is depicted in Figure 9. In this model, MSL complexes assemble at CESs, in particular, the sites of roX RNA transcription, and subsequently access flanking and distant sites on the X based on their X-linkage and transcriptional activity.
Figure 9.
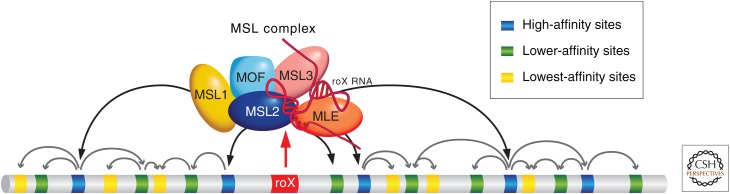
Model for the targeting of the MSL complex to the X chromosome. The dosage compensation complex in Drosophila is proposed to implement at least three targeting principles: (i) interaction with the sites of noncoding roX RNA synthesis, (ii) chromatin context–dependent binding to degenerate DNA sequences of varying affinities, and (iii) DNA sequence–independent movement from initiation sites to chromatin marks signaling gene expression (reviewed in Gelbart and Kuroda 2009). This movement has been characterized as “spreading” based on its apparent restriction in cis to the chromosome of origin, but the underlying molecular mechanism remains to be understood.
7. CHROMATIN MODIFICATIONS ASSOCIATED WITH DOSAGE COMPENSATION
A key modification that is correlated with the association of the MSL complex with the X chromosome in males is the presence of a high level of histone H4 acetylated at lysine 16 (Turner et al. 1992; Bone et al. 1994). This chromatin mark occurs throughout active transcriptional units with a bias toward the middle and the 3′ end (Fig. 7) (Kind et al. 2008; Gelbart et al. 2009). In yeast, this particular covalent modification of histone H4 plays a key role in maintaining the boundary between silent and active chromatin; loss of function of Sas2, the HAT responsible for H4K16ac, allows the spreading of telomeric heterochromatin into adjacent subtelomeric chromatin (Suka et al. 2002; for more detail, see Grunstein and Gasser 2013). Structural studies have indicated that a key internucleosomal interaction may occur between an acidic patch of the histone H2A-H2B dimer on one nucleosome and a positively charged segment of the histone H4 tail (residues 16–26) extending from a neighboring nucleosome (Luger et al. 1997; Schalch et al. 2005). When lysine 16 is acetylated, its positive charge becomes neutral suggesting that weakening a repressive internucleosomal structure could play a key role in dosage compensation. This contention was supported by the demonstration that reconstituted nucleosomal arrays acetylated at lysine 16 of histone H4 cannot achieve the level of salt-induced condensation of nonacetylated arrays (Shogren-Knaak et al. 2006; Robinson et al. 2008) and that this acetylation also weakens the self-association of reconstituted single nucleosome particles, reflecting the specific role of H4K16 in nucleosome–nucleosome stacking (Liu et al. 2011). Using molecular force spectroscopy, the acetylation of H4K16 was observed to weaken nucleosome packing in reconstituted chromatin fibers and to result in a more disordered architecture (Dunlap et al. 2012). Whether, in vivo, it is the formation of the 30-nm fiber (intramolecular compaction) or the higher-order 100- to 400-nm fibers (intermolecular compaction) that are affected by the presence of H4K16ac is not known (Shogren-Knaak et al. 2006). In either case, the presence of H4K16ac renders the chromatin of dosage compensated genes more accessible to factors or complexes. This is evidenced by the significantly greater accessibility of the compensated male X chromosome to an extrinsic DNA-binding protein (e.g., the bacterial DNA methyltransferase). The elevated accessibility of this protein follows the distribution of H4K16ac along the X (Bell et al. 2010). Given the long-standing correlation between active chromatin and early DNA replication (Hiratani and Gilbert 2009; Schubeler et al. 2002), it is not surprising that another feature of the compensated X chromosome is that it initiates replication earlier in S phase than the rest of the genome (Lakhotia and Mukherjee 1970; Bell et al. 2010).
Another finding is that the X chromosome is more susceptible to mechanical shearing than the autosomes in both males and females, indicating that it has a more open chromatin structure. In fact, the histone marks associated with active gene transcription (H3K4me2 and H3S10ph), as well as those specifically enriched on the dosage compensated X (H4K16ac) in males, are also slightly enriched on the X chromosomes of females. These findings suggest that the evolution of the distinct chromatin structure responsible for dosage compensation in males has affected the female X (Zhang and Oliver 2010).
In addition to the effect of H4K16 acetylation, there is mounting evidence that dosage compensation involves changes in the torsional stress of X-linked genes. Reducing the level of supercoiling factor, a protein known to associate at the 5′ end of active genes (Ogasawara et al. 2007), preferentially affects male viability because of a sex-specific decrease in the transcription of X-linked genes (Furuhashi et al. 2006). Compensated chromatin is topologically different from noncompensated chromatin. The difference requires the function of topoisomerase II, which associates with the MSL complex and is recruited to compensated genes in excess of the amount present on autosomal genes with similar transcription levels (Cugusi et al. 2013).
The rate of histone variant H3.3 incorporation into the X chromosome in male cells is enhanced in relation to the autosomes (Mito et al. 2005). This is to be expected, as the replication-independent nucleosome deposition of H3.3 occurs in transcriptionally active regions of chromatin and involves the replacement of histone H3 with the variant H3.3 (see Henikoff and Smith 2014). However, contrary to the expectation that this increased level of H3.3 on the X may contribute to the mechanism of dosage compensation is the observation that the absence of the two genes that encode H3.3, although causing sterility in both sexes, has no effect on viability of mutant flies (Hodl and Basler 2009).
8. A MODEL FOR THE MECHANISM OF COMPENSATION
Gene expression can be regulated at multiple steps, particularly during transcription initiation, release from pausing, or elongation. A number of considerations have suggested that the transcriptional enhancement of X-linked genes responsible for dosage compensation occurs at the elongation step of transcription rather than at initiation. Foremost was the observation that the high level of H4K16 acetylation mediated by the MSL complex occurs throughout the length of transcriptional units with a bias that favors their 3′ ends rather than the promoter regions (Smith et al. 2001). Furthermore, genes with “weak” promoters and genes with “strong” promoters coexist on the X chromosome; in males, the activity of both types of genes is enhanced approximately twofold by the dosage compensation mechanism.
Recently, supporting evidence for the hypothesis that the MSL complex enhances transcription during elongation was obtained using GRO-seq (global run-on sequencing; Core et al. 2008). The experiments, measuring the relative density of RNA polymerase in male nuclei, found an increase in density specifically on X-linked gene bodies compared to autosomal genes, which was no longer evident after RNAi depletion of the MSL complex (Larschan et al. 2011). The nature of the mechanism underlying this experimental observation is not yet fully understood. In particular, it is not known whether increased density of RNA polymerase in gene bodies reflects a corresponding increase in initiation not evident in the GRO-seq assay, improved processivity (i.e., a decrease in premature termination), or both. However, the localization of the MSL complex and increased H4K16 acetylation is consistent with a direct effect on elongation. As modeled in vitro, the H4K16 acetylation of nucleosomes, widely spread along dosage compensated transcriptional units, might diminish internucleosomal interactions in vivo, thereby facilitating nucleosome eviction and RNAPII progression. MSL2 is an E3 ubiquitin ligase that ubiquitinates histone H2B in vitro; its human ortholog is an E3 ubiquitin ligase that ubiquitinates histone H2B and facilitates the methylation of H3K4 and H3K79 (Wu et al. 2011). Ubiquitinated H2B and methylated H3K79 represent histone modifications that are important for transcription elongation (Minsky et al. 2008). A similar function for MSL2 in Drosophila would bolster the role of the MSL complex in facilitating transcription elongation.
Clearly, an enhanced rate of elongation of RNAPII alone is not sufficient to generate the increase in steady-state level of X-linked gene transcripts necessary for dosage compensation. One possibility is that transcription along the length of genes is not always successful, and that the processivity of RNA polymerase II can be improved to increase final RNA production. If the dosage compensation mechanism is, in fact, based on enhancing the rate of elongation, then it is clear that to achieve an increase in the steady-state level of X-linked gene transcripts, there must occur a concomitant increase in the frequency of recruitment of polymerase or release from pausing. Given the tight relationship between these two phases of transcription, distinguishing the primary mechanism for up-regulation may require additional experimentation and perhaps technical advances not yet realized.
9. DOSAGE COMPENSATION AND NUCLEAR ORGANIZATION
During interphase, chromosomes are seen to occupy individual territories rather than an intermingling of unraveled chromatin strands (Cremer and Cremer 2010; see Dekker and Misteli 2014). This organization is particularly evident in cells of Drosophila males in which the X chromosome can be identified by the presence of the MSL complex (Strukov et al. 2011). Within this compartment, the chromatin modifications that underlie the mechanism of dosage compensation appear to induce a particular higher-order topography to the X chromosome. Throughout development, X-linked sites that are separated by approximately a dozen megabases are located much closer in male than in female nuclei (Fig. 10) (Grimaud and Becker 2009). This difference is dependent on the presence of the MSL1-MSL2 chromatin-recognition component of the complex and is not affected by the absence of the other three MSL proteins. Because, in the absence of any one of the latter, the partial complex that includes MSL1-MSL2 is found only at HASs, the proximity of X-linked loci in male cells must be mediated by their clustering (Grimaud and Becker 2009).
Figure 10.

Male-specific conformation of the dosage-compensated X chromosome. A pair of high-affinity chromosomal sites (roX2 and usp) were visualized by two-color FISH (fluorescence in situ hybridization) in female or male embryos. DNA was stained with DAPI (blue) and the X-chromosome territory (magenta) was painted with an antibody against MSL2 in male nuclei (there is no MSL2 in female nuclei). A merge of the channels reveals the proximity of the HASs and their residence relative to the MSL2 territory in male nuclei, clearly summarized in the cartoon on the right. The schematic diagram showing part of the X chromosome below indicates the distances separating the different HASs. (Modified, with permission, from Grimaud and Becker 2009, © Cold Spring Harbor Laboratory Press.)
It is interesting to note that the MSL proteins copurify with the nuclear pore complex proteins Nup153 and Megator (Mtor). Regions of the genome at the nuclear periphery that are proximal to the nuclear pore complex contain groups of active genes, suggesting this compartment may have a regulatory effect on transcription (Vaquerizas et al. 2010). Depletion of the Nup153 and Mtor nucleoporins leads to the loss of dosage compensation (Mendjan et al. 2006), although this analysis could be complicated by general viability issues (Grimaud and Becker 2009).
10. INFLUENCE OF GLOBAL CHROMATIN FACTORS ON THE MALE X CHROMOSOME
The male polytene X chromosome shows special sensitivity to changes in the dosage or activity of several chromatin regulators that are thought to be general, non-X-chromosome-specific factors. For example, a functional interaction between ISWI-bearing complexes and the MSL complex was brought to light by the observation that loss-of-function mutations of Iswi (imitation switch) lead to a global structural defect on the X chromosome in salivary gland preparations of males (Deuring et al. 2000). ISWI is an ATPase found in four chromatin-remodeling complexes of Drosophila: NURF (nucleosome remodeling factor), ACF (ATP-dependent chromatin assembly and remodeling factor), CHRAC (chromatin accessibility complex; Hargreaves and Crabtree 2011), and RSF (remodeling and spacing factor; Hanai et al. 2008). In vivo, ACF and CHRAC behave as assembly factors, promoting the formation of chromatin, particularly repressive chromatin states. Nevertheless, high-resolution mapping of nucleosomes in Iswi mutant males and females indicates that the chromosome condensation defects that they show, especially of the X chromosome in males, are not correlated with global ISWI-dependent nucleosome spacing changes (Sala et al. 2011). A more convincing explanation is provided by the observation that ISWI facilitates the association of linker histone H1 and that a reduction of the H1 leads to a chromosomal phenotype similar to the one described above for ISWI loss of function (Siriaco et al. 2009). Somewhat surprisingly, loss of function of the RSF complex does not appear to alter chromosome structure (Hanai et al. 2008).
Early functional studies of the NURF complex indicated that it might be involved in enhancing transcription in some cases or repressing it in others (Badenhorst et al. 2002). Mutants in nurf301 show the abnormally decondensed male X seen in Iswi mutants (Badenhorst et al. 2002). NURF also has a specific effect on roX1 and roX2 transcription; wild-type NURF negatively regulates these genes in females and reduces the level of transcription of roX2 in males by approximately one half (Bai et al. 2007).
The X-chromosome defect visible in Iswi and nurf mutant male salivary glands does not occur when the MSL complex is inactive (i.e., in the absence of H4K16ac). In contrast, the absence of either roX gene reduces the abnormal puffing of the X in the region of the roX mutation. This observation highlights the localized nature of opposing activities that are responsible for normal chromatin organization (Bai et al. 2007).
The down-regulation of certain structural components of heterochromatin, such as Su(var)3-7 (suppressor of variegation 3-7) and HP1 (heterochromatin protein 1) leads to a polytene X-chromosome phenotype that is similar to the one resulting from ISWI knockdown (Delattre et al. 2004; Spierer et al. 2005). In these cases as well, the bloated appearance requires the presence of an active MSL complex.
In wild-type males, the distribution of heterochromatin factors along the X chromosome may be regulated by the action of JIL-1, a histone H3 serine 10 kinase. JIL-1 localizes to active gene bodies with a 3′ bias, is approximately twofold more abundant on the X chromosome than the autosomes in males (Jin et al. 1999, 2000) and is necessary for proper dosage compensation of eye pigmentation (Lerach et al. 2005). Loss-of-function alleles result in global changes in the morphology of polytene chromosomes in both males and females, whereas the male X is once again shorter, fatter, and without any evidence of banding (Deng et al. 2005). JIL-1 loss-of-function alleles allow the spreading of H3 dimethylated at lysine 9 (H3K9me2), the modification that attracts HP1 to inactive chromatin, suggesting that JIL-1 normally marks and preserves the limits of euchromatic domains (Ebert et al. 2004; Zhang et al. 2006).
11. HOW DID DOSAGE COMPENSATION EVOLVE?
In Drosophila, the evolution of the regulatory mechanism of dosage compensation has been correlated to the evolution of heteromorphic sex chromosomes represented by an X chromosome that is similar to the major autosomes in the density of transcribing genes per unit length and a Y that is largely heterochromatic. The sex chromosomes are thought to have originated from the occurrence of an autosomal mutation that determined one of two mating types. In Drosophila this mating type became the heterogametic male sex. Over time, the autosome bearing the initial mutation degenerated by the concomitant reduction of recombination and the random occurrence of deleterious mutations that were retained if they were linked to an occasional beneficial mutation. In many organisms, the difference in levels of expression of genes present in two doses in the homogametic sex and in a single dose in the heterogametic sex appears to be tolerated and specific adjustments regulate the transcription of critical individual genes in the two sexes. In flies, mammals, and worms, however, the degeneration of the genetic content of the incipient Y provided the selective pressure necessary for the evolution of a chromosome-wide compensatory mechanism that increases the expression of the alleles present on its homolog, the X chromosome (Charlesworth 1978; Lucchesi 1978). Evidence for the occurrence of this evolutionary phenomenon can be found in several Drosophila species that have undergone whole chromosome arm fusions (reviewed in Charlesworth 1978). The wealth of classical cytological information, as well as the complete genomic sequencing of numerous Drosophila species, provides an invaluable window into the evolution of sex chromosomes and the concomitant mechanism of dosage compensation.
12. OUTLOOK
Dosage compensation is an epigenetic regulatory mechanism that presents several unique characteristics: It is a paramount example of the control of the expression of individual genes at the level of an entire chromosome, and it is mediated by a multiprotein complex (MSL) that includes not only enzymatic subunits with known chromatin-modifying functions, but also two long ncRNAs that are required for its assembly. Very substantial progress has been accomplished in the past few years in elucidating various aspects of the chromatin modifications effected by the MSL complex and in identifying some of the parameters that regulate its targeting. Further understanding of the targeting process will be facilitated by the molecular analysis of evolving sex chromosomes in different Drosophila species with fully sequenced genomes (see www.FlyBase.org). An understanding of the function of the roX RNAs in spreading of the MSL complex will make use of new RNA, rather than protein-based ChIP mapping techniques (Chu et al. 2011; Simon et al. 2011). The role of histone modifications, namely H4K16 acetylation and H2B ubiquitination, will be assessed by replacing canonical histones with multiple copies of modified histone transgenes (Gunesdogan et al. 2010). Topological characteristics of dosage compensated chromatin units will be determined by biophysical experiments (Kruithof et al. 2009; Allahverdi et al. 2011). An important insight into the function of the MSL complex will be to determine its status during the cell cycle and how it manages to survive through DNA replication and mitosis (Lavender et al. 1994; Strukov et al. 2011). In summary, a full understanding of all of these aspects of the regulatory mechanism will contribute key information regarding the organization of chromatin into transcriptional domains and the role that chromosomal organization and nuclear topology play in gene regulation (the topic of Dekker and Misteli 2014); it will also provide important insights into molecular mechanisms that fine-tune the heritable expression of genes within precise ranges.
Footnotes
Editors: C. David Allis, Marie-Laure Caparros, Thomas Jenuwein, and Danny Reinberg
Additional Perspectives on Epigenetics available at www.cshperspectives.org
REFERENCES
*Reference is also in this collection.
- Alekseyenko AA, Larschan E, Lai WR, Park PJ, Kuroda MI. 2006. High-resolution ChIP-chip analysis reveals that the Drosophila MSL complex selectively identifies active genes on the male X chromosome. Genes Dev 20: 848–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, Peng S, Larschan E, Gorchakov AA, Lee OK, Kharchenko P, McGrath SD, Wang CI, Mardis ER, Park PJ, et al. 2008. A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell 134: 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, Ho JWK, Peng S, Gelbart M, Tolstorukov MY, Plachetka A, Kharchenko PV, Jung YL, Gorchakov AA, Larschan E, et al. 2012. Sequence-specific targeting of dosage compensation in Drosophila favors an active chromatin context. PLoS Genet 8: e1002646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allahverdi A, Yang R, Korolev N, Fan Y, Davey CA, Liu CF, Nordenskiold L. 2011. The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Res 39: 1680–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrein H, Axel R. 1997. Genes expressed in neurons of adult male Drosophila. Cell 88: 459–469. [DOI] [PubMed] [Google Scholar]
- Arkhipova I, Li J, Meselson M. 1997. On the mode of gene-dosage compensation in Drosophila. Genetics 145: 729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badenhorst P, Voas M, Rebay I, Wu C. 2002. Biological functions of the ISWI chromatin remodeling complex NURF. Genes Dev 16: 3186–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Larschan E, Kwon SY, Badenhorst P, Kuroda MI. 2007. Regional control of chromatin organization by noncoding roX RNAs and the NURF remodeling complex in Drosophila melanogaster. Genetics 176: 1491–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashaw GJ, Baker BS. 1997. The regulation of the Drosophila msl-2 gene reveals a function for Sex-lethal in translational control. Cell 89: 789–798. [DOI] [PubMed] [Google Scholar]
- Bell O, Conrad T, Kind J, Wirbelauer C, Akhtar A, Schubeler D. 2008. Transcription-coupled methylation of histone H3 at lysine 36 regulates dosage compensation by enhancing recruitment of the MSL complex in Drosophila melanogaster. Mol Cell Biol 28: 3401–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell O, Schwaiger M, Oakeley EJ, Lienert F, Beisel C, Stadler MB, Schubeler D. 2010. Accessibility of the Drosophila genome discriminates PcG repression, H4K16 acetylation and replication timing. Nat Struct Mol Biol 17: 894–900. [DOI] [PubMed] [Google Scholar]
- Belote JM, Lucchesi JC. 1980a. Control of X chromosome transcription by the maleless gene in Drosophila. Nature 285: 573–575. [DOI] [PubMed] [Google Scholar]
- Belote JM, Lucchesi JC. 1980b. Male-specific lethal mutations of Drosophila melanogaster. Genetics 96: 165–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler J, Sun L, Fernandez H, Donohue R, Xie W, Sanyal A. 2011. Re-evaluation of the function of the male specific lethal complex in Drosophila. J Genet Genomics 38: 327–332. [DOI] [PubMed] [Google Scholar]
- Bone JR, Lavender J, Richman R, Palmer MJ, Turner BM, Kuroda MI. 1994. Acetylated histone H4 on the male X chromosome is associated with dosage compensation in Drosophila. Genes Dev 8: 96–104. [DOI] [PubMed] [Google Scholar]
- *.Brockdorff N, Turner BM. 2014. Dosage compensation in mammals. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a019406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Bao X, Deng H, Jin Y, Girton J, Johansen J, Johansen KM. 2008. RNA polymerase II–mediated transcription at active loci does not require histone H3S10 phosphorylation in Drosophila. Development 135: 2917–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, et al. 2005. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123: 581–592. [DOI] [PubMed] [Google Scholar]
- Chang KA, Kuroda MI. 1998. Modulation of MSL1 abundance in female Drosophila contributes to the sex specificity of dosage compensation. Genetics 150: 699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. 1978. Model for evolution of Y chromosomes and dosage compensation. Proc Natl Acad Sci 75: 5618–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. 2011. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell 44: 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline TW, Meyer BJ. 1996. Vive la difference: Males vs females in flies vs worms. Annu Rev Genet 30: 637–702. [DOI] [PubMed] [Google Scholar]
- Conrad T, Cavalli FM, Holz H, Hallacli E, Kind J, Ilik I, Vaquerizas JM, Luscombe NM, Akhtar A. 2012. The MOF chromobarrel domain controls genome-wide H4K16 acetylation and spreading of the MSL complex. Dev Cell 22: 610–624. [DOI] [PubMed] [Google Scholar]
- Copps K, Richman R, Lyman LM, Chang KA, Rampersad-Ammons J, Kuroda MI. 1998. Complex formation by the Drosophila MSL proteins: Role of the MSL2 RING finger in protein complex assembly. EMBO J 17: 5409–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. 2008. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322: 1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer T, Cremer M. 2010. Chromosome territories. Cold Spring Harb Perspect Biol 2: a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cugusi S, Ramos E, Ling H, Yokoyama R, Luk KM, Lucchesi JC. 2013. Topoisomerase II plays a role in dosage compensation in Drosophila. Transcription 4 10.4161/trns.26185. [DOI] [PubMed] [Google Scholar]
- *.Dekker J, Misteli T. 2014. Long-range chromatin interactions. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a019356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delattre M, Spierer A, Jaquet Y, Spierer P. 2004. Increased expression of Drosophila Su(var)3-7 triggers Su(var)3-9-dependent heterochromatin formation. J Cell Sci 117: 6239–6247. [DOI] [PubMed] [Google Scholar]
- Deng H, Zhang W, Bao X, Martin JN, Girton J, Johansen J, Johansen KM. 2005. The JIL-1 kinase regulates the structure of Drosophila polytene chromosomes. Chromosoma 114: 173–182. [DOI] [PubMed] [Google Scholar]
- Deng X, Hiatt JB, Nguyen DK, Ercan S, Sturgill D, Hillier LW, Schlesinger F, Davis CA, Reinke VJ, Gingeras TR, et al. 2011. Evidence for compensatory upregulation of expressed X-linked genes in mammals, Caenorhabditis elegans and Drosophila melanogaster. Nat Genet 43: 1179–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuring R, Fanti L, Armstrong JA, Sarte M, Papoulas O, Prestel M, Daubresse G, Verardo M, Moseley SL, Berloco M, et al. 2000. The ISWI chromatin-remodeling protein is required for gene expression and the maintenance of higher order chromatin structure in vivo. Mol Cell 5: 355–365. [DOI] [PubMed] [Google Scholar]
- Dunlap D, Yokoyama R, Ling H, Sun HY, McGill K, Cugusi S, Lucchesi JC. 2012. Distinct contributions of MSL complex subunits to the transcriptional enhancement responsible for dosage compensation in Drosophila. Nucleic Acids Res 40: 11281–11291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert A, Schotta G, Lein S, Kubicek S, Krauss V, Jenuwein T, Reuter G. 2004. Su(var) genes regulate the balance between euchromatin and heterochromatin in Drosophila. Genes Dev 18: 2973–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagegaltier D, Baker BS. 2004. X chromosome sites autonomously recruit the dosage compensation complex in Drosophila males. PLoS Biol 2: e341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller C, Prestel M, Hartmann H, Straub T, Söding J, Becker PB. 2012. The MOF-containing NSL complex associates globally with housekeeping genes, but activates only a defined subset. Nucleic Acids Res 40: 1509–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi H, Nakajima M, Hirose S. 2006. DNA supercoiling factor contributes to dosage compensation in Drosophila. Development 133: 4475–4483. [DOI] [PubMed] [Google Scholar]
- Gelbart ME, Kuroda MI. 2009. Drosophila dosage compensation: A complex voyage to the X chromosome. Development 136: 1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelbart ME, Larschan E, Peng S, Park PJ, Kuroda MI. 2009. Drosophila MSL complex globally acetylates H4K16 on the male X chromosome for dosage compensation. Nat Struct Mol Biol 16: 825–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilfillan GD, Straub T, de Wit E, Greil F, Lamm R, van Steensel B, Becker PB. 2006. Chromosome-wide gene-specific targeting of the Drosophila dosage compensation complex. Genes Dev 20: 858–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorchakov AA, Alekseyenko AA, Kharchenko P, Park PJ, Kuroda MI. 2009. Long-range spreading of dosage compensation in Drosophila captures transcribed autosomal genes inserted on X. Genes Dev 23: 2266–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaud C, Becker PB. 2009. The dosage compensation complex shapes the conformation of the X chromosome in Drosophila. Genes Dev 23: 2490–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Grunstein M, Gasser SM. 2013. Epigenetics in Saccharomyces cerevisiae. Cold Spring Harb Perspect Biol 5: a017491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Szauter P, Lucchesi JC. 1998. Targeting of MOF, a putative histone acetyl transferase, to the X chromosome of Drosophila melanogaster. Dev Genet 22: 56–64. [DOI] [PubMed] [Google Scholar]
- Gu W, Wei X, Pannuti A, Lucchesi JC. 2000. Targeting the chromatin-remodeling MSL complex of Drosophila to its sites of action on the X chromosome requires both acetyl transferase and ATPase activities. EMBO J 19: 5202–5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunesdogan U, Jackle H, Herzig A. 2010. A genetic system to assess in vivo the functions of histones and histone modifications in higher eukaryotes. EMBO Rep 11: 772–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallacli E, Lipp M, Georgiev P, Spielman C, Cusack S, Akhtar A, Kadlec J. 2012. Msl1-mediated dimerization of the dosage compensation complex is essential for male X-chromosome regulation in Drosophila. Mol Cell 48: 587–600. [DOI] [PubMed] [Google Scholar]
- Hamada FN, Park PJ, Gordadze PR, Kuroda MI. 2005. Global regulation of X chromosomal genes by the MSL complex in Drosophila melanogaster. Genes Dev 19: 2289–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanai K, Furuhashi H, Yamamoto T, Akasaka K, Hirose S. 2008. RSF governs silent chromatin formation via histone H2Av replacement. PLoS Genet 4: e1000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves DC, Crabtree GR. 2011. ATP-dependent chromatin remodeling: Genetics, genomics and mechanisms. Cell Res 21: 396–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Henikoff S, Smith MM. 2014. Histone variants and epigenetics. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker A, Hilfiker-Kleiner D, Pannuti A, Lucchesi JC. 1997. mof, a putative acetyl transferase gene related to the Tip60 and MOZ human genes and to the SAS genes of yeast, is required for dosage compensation in Drosophila. EMBO J 16: 2054–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I, Gilbert DM. 2009. Replication timing as an epigenetic mark. Epigenetics 4: 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodl M, Basler K. 2009. Transcription in the absence of histone H3.3. Curr Biol 19: 1221–1226. [DOI] [PubMed] [Google Scholar]
- Ilik IA, Quinn JJ, Georgiev P, Tavares-Cadete F, Maticzka D, Toscano S, Wan Y, Spitale RC, Luscombe N, Backofen R, et al. 2013. Tandem stem-loops in roX RNAs act together to mediate X chromosome dosage compensation in Drosophila. Mol Cell 51: 156–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivaldi MS, Karam CS, Corces VG. 2007. Phosphorylation of histone H3 at Ser10 facilitates RNA polymerase II release from promoter-proximal pausing in Drosophila. Genes Dev 21: 2818–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Wang Y, Walker DL, Dong H, Conley C, Johansen J, Johansen KM. 1999. JIL-1: A novel chromosomal tandem kinase implicated in transcriptional regulation in Drosophila. Mol Cell 4: 129–135. [DOI] [PubMed] [Google Scholar]
- Jin Y, Wang Y, Johansen J, Johansen KM. 2000. JIL-1, a chromosomal kinase implicated in regulation of chromatin structure, associates with the male specific lethal (MSL) dosage compensation complex. J Cell Biol 149: 1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AA, Struhl K. 2005. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol Cell 20: 971–978. [DOI] [PubMed] [Google Scholar]
- Kadlec J, Hallacli E, Lipp M, Holz H, Sanchez-Weatherby J, Cusack S, Akhtar A. 2011. Structural basis for MOF and MSL3 recruitment into the dosage compensation complex by MSL1. Nat Struct Mol Biol 18: 142–149. [DOI] [PubMed] [Google Scholar]
- Kelley RL, Wang J, Bell L, Kuroda MI. 1997. Sex lethal controls dosage compensation in Drosophila by a non-splicing mechanism. Nature 387: 195–199. [DOI] [PubMed] [Google Scholar]
- Kelley RL, Meller VH, Gordadze PR, Roman G, Davis RL, Kuroda MI. 1999. Epigenetic spreading of the Drosophila dosage compensation complex from roX RNA genes into flanking chromatin. Cell 98: 513–522. [DOI] [PubMed] [Google Scholar]
- Kelley RL, Lee OK, Shim YK. 2008. Transcription rate of noncoding roX1 RNA controls local spreading of the Drosophila MSL chromatin remodeling complex. Mech Dev 125: 1009–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, Schuldiner M, Chin K, Punna T, Thompson NJ, et al. 2005. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell 123: 593–605. [DOI] [PubMed] [Google Scholar]
- Kind J, Akhtar A. 2007. Cotranscriptional recruitment of the dosage compensation complex to X-linked target genes. Genes Dev 21: 2030–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J, Vaquerizas JM, Gebhardt P, Gentzel M, Luscombe NM, Bertone P, Akhtar A. 2008. Genome-wide analysis reveals MOF as a key regulator of dosage compensation and gene expression in Drosophila. Cell 133: 813–828. [DOI] [PubMed] [Google Scholar]
- Kruithof M, Chien FT, Routh A, Logie C, Rhodes D, van Noort J. 2009. Single-molecule force spectroscopy reveals a highly compliant helical folding for the 30-nm chromatin fiber. Nat Struct Mol Biol 16: 534–540. [DOI] [PubMed] [Google Scholar]
- Lakhotia SC, Mukherjee AS. 1970. Chromosomal basis of dosage compensation in Drosophila. 3. Early completion of replication by the polytene X-chromosome in male: Further evidence and its implications. J Cell Biol 47: 18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam KC, Mühlpfordt F, Vaquerizas JM, Raja SJ, Holz H, Luscombe NM, Manke T, Akhtar A. 2012. The NSL complex regulates housekeeping genes in Drosophila. PLoS Genet 8: e1002736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larschan E, Alekseyenko AA, Gortchakov AA, Peng S, Li B, Yang P, Workman JL, Park PJ, Kuroda MI. 2007. MSL complex is attracted to genes marked by H3K36 trimethylation using a sequence-independent mechanism. Mol Cell 28: 121–133. [DOI] [PubMed] [Google Scholar]
- Larschan E, Bishop EP, Kharchenko PV, Core LJ, Lis JT, Park PJ, Kuroda MI. 2011. X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature 471: 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larschan E, Soruco MM, Lee OK, Peng S, Bishop E, Chery J, Goebel K, Feng J, Park PJ, Kuroda MI. 2012. Identification of chromatin-associated regulators of MSL complex targeting in Drosophila dosage compensation. PLoS Genet 8: e1002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavender JS, Birley AJ, Palmer MJ, Kuroda MI, Turner BM. 1994. Histone H4 acetylated at lysine 16 and proteins of the Drosophila dosage compensation pathway co-localize on the male X chromosome through mitosis. Chromosome Res 2: 398–404. [DOI] [PubMed] [Google Scholar]
- Lee CG, Chang KA, Kuroda MI, Hurwitz J. 1997. The NTPase/helicase activities of Drosophila maleless, an essential factor in dosage compensation. EMBO J 16: 2671–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CG, da Costa Soares V, Newberger C, Manova K, Lacy E, Hurwitz J. 1998. RNA helicase A is essential for normal gastrulation. Proc Natl Acad Sci 95: 13709–13713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerach S, Zhang W, Deng H, Bao X, Girton J, Johansen J, Johansen KM. 2005. JIL-1 kinase, a member of the male-specific lethal (MSL) complex, is necessary for proper dosage compensation of eye pigmentation in Drosophila. Genesis 43: 213–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lu C, Yang Y, Fan Y, Yang R, Liu CF, Korolev N, Nordenskiold L. 2011. Influence of histone tails and h4 tail acetylations on nucleosome-nucleosome interactions. J Mol Biol 414: 749–764. [DOI] [PubMed] [Google Scholar]
- Lucchesi JC. 1978. Gene dosage compensation and the evolution of sex chromosomes. Science 202: 711–716. [DOI] [PubMed] [Google Scholar]
- Lucchesi JC, Manning JE. 1987. Gene dosage compensation in Drosophila melanogaster. Adv Genet 24: 371–429. [DOI] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389: 251–260. [DOI] [PubMed] [Google Scholar]
- Maenner S, Müller M, Fröhlich J, Langer D, Becker PB. 2013. ATP-dependent roX RNA remodeling by the helicase maleless enables specific association of MSL proteins. Mol Cell 51: 174–184. [DOI] [PubMed] [Google Scholar]
- *.Marmorstein R, Zhou M-M. 2014. Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb Perspect Biol 6: a018762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller VH, Rattner BP. 2002. The roX genes encode redundant male-specific lethal transcripts required for targeting of the MSL complex. EMBO J 21: 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller VH, Wu KH, Roman G, Kuroda MI, Davis RL. 1997. roX1 RNA paints the X chromosome of male Drosophila and is regulated by the dosage compensation system. Cell 88: 445–457. [DOI] [PubMed] [Google Scholar]
- Meller VH, Gordadze PR, Park Y, Chu X, Stuckenholz C, Kelley RL, Kuroda MI. 2000. Ordered assembly of roX RNAs into MSL complexes on the dosage-compensated X chromosome in Drosophila. Curr Biol 10: 136–143. [DOI] [PubMed] [Google Scholar]
- Mendjan S, Taipale M, Kind J, Holz H, Gebhardt P, Schelder M, Vermeulen M, Buscaino A, Duncan K, Mueller J, et al. 2006. Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila. Mol Cell 21: 811–823. [DOI] [PubMed] [Google Scholar]
- Minsky N, Shema E, Field Y, Schuster M, Segal E, Oren M. 2008. Monoubiquitinated H2B is associated with the transcribed region of highly expressed genes in human cells. Nat Cell Biol 10: 483–488. [DOI] [PubMed] [Google Scholar]
- Mito Y, Henikoff JG, Henikoff S. 2005. Genome-scale profiling of histone H3.3 replacement patterns. Nat Genet 37: 1090–1097. [DOI] [PubMed] [Google Scholar]
- Morales V, Straub T, Neumann MF, Mengus G, Akhtar A, Becker PB. 2004. Functional integration of the histone acetyltransferase MOF into the dosage compensation complex. EMBO J 23: 2258–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra R, Smith ER, Yokoyama R, Lucchesi JC. 2008. The MLE subunit of the Drosophila MSL complex uses its ATPase activity for dosage compensation and its helicase activity for targeting. Mol Cell Biol 28: 958–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee AS, Beermann W. 1965. Synthesis of ribonucleic acid by the X-chromosomes of Drosophila melanogaster and the problem of dosage compensation. Nature 207: 785–786. [DOI] [PubMed] [Google Scholar]
- Muller HJ. 1932. Further studies on the Nature and causes of gene mutations. Proc 6th Int Congr Genet 1: 213–255. [Google Scholar]
- Ogasawara Y, Furuhashi H, Hirose S. 2007. DNA supercoiling factor positively regulates expression of the homeotic gene Abdominal-B in Drosophila melanogaster. Genes Cells 12: 1347–1355. [DOI] [PubMed] [Google Scholar]
- Oh H, Park Y, Kuroda MI. 2003. Local spreading of MSL complexes from roX genes on the Drosophila X chromosome. Genes Dev 17: 1334–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H, Bone JR, Kuroda MI. 2004. Multiple classes of MSL binding sites target dosage compensation to the X chromosome of Drosophila. Curr Biol 14: 481–487. [DOI] [PubMed] [Google Scholar]
- Pannuti A, Lucchesi JC. 2000. Recycling to remodel: Evolution of dosage-compensation complexes. Curr Opin Genet Dev 10: 644–650. [DOI] [PubMed] [Google Scholar]
- Park Y, Kelley RL, Oh H, Kuroda MI, Meller VH. 2002. Extent of chromatin spreading determined by roX RNA recruitment of MSL proteins. Science 298: 1620–1623. [DOI] [PubMed] [Google Scholar]
- Park Y, Mengus G, Bai X, Kageyama Y, Meller VH, Becker PB, Kuroda MI. 2003. Sequence-specific targeting of Drosophila roX genes by the MSL dosage compensation complex. Mol Cell 11: 977–986. [DOI] [PubMed] [Google Scholar]
- Park SW, Kang Y, Sypula JG, Choi J, Oh H, Park Y. 2007. An evolutionarily conserved domain of roX2 RNA is sufficient for induction of H4-Lys16 acetylation on the Drosophila X chromosome. Genetics 177: 1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SW, Oh H, Lin YR, Park Y. 2010. MSL cis-spreading from roX gene up-regulates the neighboring genes. Biochem Biophys Res Commun 399: 227–231. [DOI] [PubMed] [Google Scholar]
- Raja SJ, Charapitsa I, Conrad T, Vaquerizas JM, Gebhardt P, Holz H, Kadlec J, Fraterman S, Luscombe NM, Akhtar A. 2010. The nonspecific lethal complex is a transcriptional regulator in Drosophila. Mol Cell 38: 827–841. [DOI] [PubMed] [Google Scholar]
- Regnard C, Straub T, Mitterweger A, Dahlsveen IK, Fabian V, Becker PB. 2011. Global analysis of the relationship between JIL-1 kinase and transcription. PLoS Genet 7: e1001327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PJ, An W, Routh A, Martino F, Chapman L, Roeder RG, Rhodes D. 2008. 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J Mol Biol 381: 816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala A, Toto M, Pinello L, Gabriele A, Di Benedetto V, Ingrassia AM, Lo Bosco G, Di Gesu V, Giancarlo R, Corona DF. 2011. Genome-wide characterization of chromatin binding and nucleosome spacing activity of the nucleosome remodelling ATPase ISWI. EMBO J 30: 1766–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sass GL, Pannuti A, Lucchesi JC. 2003. Male-specific lethal complex of Drosophila targets activated regions of the X chromosome for chromatin remodeling. Proc Natl Acad Sci 100: 8287–8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalch T, Duda S, Sargent DF, Richmond TJ. 2005. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature 436: 138–141. [DOI] [PubMed] [Google Scholar]
- Schubeler D, Scalzo D, Kooperberg C, van Steensel B, Delrow J, Groudine M. 2002. Genome-wide DNA replication profile for Drosophila melanogaster: A link between transcription and replication timing. Nat Genet 32: 438–442. [DOI] [PubMed] [Google Scholar]
- Scott MJ, Pan LL, Cleland SB, Knox AL, Heinrich J. 2000. MSL1 plays a central role in assembly of the MSL complex, essential for dosage compensation in Drosophila. EMBO J 19: 144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. 2006. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311: 844–847. [DOI] [PubMed] [Google Scholar]
- Simon MD, Wang CI, Kharchenko PV, West JA, Chapman BA, Alekseyenko AA, Borowsky ML, Kuroda MI, Kingston RE. 2011. The genomic binding sites of a noncoding RNA. Proc Natl Acad Sci 108: 20497–20502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siriaco G, Deuring R, Chioda M, Becker PB, Tamkun JW. 2009. Drosophila ISWI regulates the association of histone H1 with interphase chromosomes in vivo. Genetics 182, 661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ER, Pannuti A, Gu W, Steurnagel A, Cook RG, Allis CD, Lucchesi JC. 2000. The Drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol Cell Biol 20: 312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ER, Allis CD, Lucchesi JC. 2001. Linking global histone acetylation to the transcription enhancement of X-chromosomal genes in Drosophila males. J Biol Chem 276: 31483–31486. [DOI] [PubMed] [Google Scholar]
- Smith ER, Cayrou C, Huang R, Lane WS, Cote J, Lucchesi JC. 2005. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol Cell Biol 25: 9175–9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soruco MM, Chery J, Bishop EP, Siggers T, Tolstorukov MY, Leydon AR, Sugden AU, Goebel K, Feng J, Xia P, et al. 2013. The CLAMP protein links the MSL complex to the X chromosome during Drosophila dosage compensation. Genes Dev 27: 1551–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spierer A, Seum C, Delattre M, Spierer P. 2005. Loss of the modifiers of variegation Su(var)3-7 or HP1 impacts male X polytene chromosome morphology and dosage compensation. J Cell Sci 118: 5047–5057. [DOI] [PubMed] [Google Scholar]
- Stenberg P, Larsson J. 2011. Buffering and the evolution of chromosome-wide gene regulation. Chromosoma 120: 213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub T, Gilfillan GD, Maier VK, Becker PB. 2005. The Drosophila MSL complex activates the transcription of target genes. Genes Dev 19: 2284–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub T, Grimaud C, Gilfillan GD, Mitterweger A, Becker PB. 2008. The chromosomal high-affinity binding sites for the Drosophila dosage compensation complex. PLoS Genet 4: e1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub T, Zabel A, Gilfillan GD, Feller C, Becker PB. 2013. Different chromatin interfaces of the Drosophila dosage compensation complex revealed by high-shear ChIP-seq. Genome Res 23: 473–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Strome S, Kelly WG, Ercan S, Lieb JD. 2014. Regulation of the X chromosomes in Caenorhabditis elegans. Cold Spring Harb Perspect Biol 6: a018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strukov YG, Sural TH, Kuroda MI, Sedat JW. 2011. Evidence of activity-specific, radial organization of mitotic chromosomes in Drosophila. PLoS Biol 9: e1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suka N, Luo K, Grunstein M. 2002. Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine16 and spreading of heterochromatin. Nat Genet 32: 378–383. [DOI] [PubMed] [Google Scholar]
- Sural TH, Peng S, Li B, Workman JL, Park PJ, Kuroda MI. 2008. The MSL3 chromodomain directs a key targeting step for dosage compensation of the Drosophila melanogaster X chromosome. Nat Struct Mol Biol 15: 1318–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M, Rea S, Richter K, Vilar A, Lichter P, Imhof A, Akhtar A. 2005. hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol Cell Biol 25: 6798–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BM, Birley AJ, Lavender J. 1992. Histone H4 isoforms acetylated at specific lysine residues define individual chromosomes and chromatin domains in Drosophila polytene nuclei. Cell 69: 375–384. [DOI] [PubMed] [Google Scholar]
- Vaquerizas JM, Suyama R, Kind J, Miura K, Luscombe NM, Akhtar A. 2010. Nuclear pore proteins nup153 and megator define transcriptionally active regions in the Drosophila genome. PLoS Genet 6: e1000846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa R, Forné I, Müller M, Imhof A, Straub T, Becker PB. 2012. MSL2 combines sensor and effector functions in homeostatic control of the Drosophila dosage compensation machinery. Mol Cell 48: 647–654. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang W, Jin Y, Johansen J, Johansen KM. 2001. The JIL-1 tandem kinase mediates histone H3 phosphorylation and is required for maintenance of chromatin structure in Drosophila. Cell 105: 433–443. [DOI] [PubMed] [Google Scholar]
- Wu L, Zee BM, Wang Y, Garcia BA, Dou Y. 2011. The RING finger protein MSL2 in the MOF complex is an E3 ubiquitin ligase for H2B K34 and is involved in crosstalk with H3 K4 and K79 methylation. Mol Cell 43: 132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Oliver B. 2010. An evolutionary consequence of dosage compensation on Drosophila melanogaster female X-chromatin structure? BMC Genomics 11: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Deng H, Bao X, Lerach S, Girton J, Johansen J, Johansen KM. 2006. The JIL-1 histone H3S10 kinase regulates dimethyl H3K9 modifications and heterochromatic spreading in Drosophila. Development 133: 229–235. [DOI] [PubMed] [Google Scholar]
WWW RESOURCES
- www.FlyBase.org A database of Drosophila genes and genomes.