

Abstract
The ubiquitin-like domain-containing C-terminal domain phosphatase 1 (UBLCP1) has been implicated as a negative regulator of the proteasome, a key mediator in the ubiquitin-dependent protein degradation. Small molecule inhibitors that block UBLCP1 activity would be valuable as research tools and potential therapeutics for human diseases caused by the cellular accumulation of misfold/damaged proteins. We report a salicylic acid fragment-based library approach aimed at targeting both the phosphatase active site and its adjacent binding pocket for enhanced affinity and selectivity. Screening of the focused libraries led to the identification of the first potent and selective UBLCP1 inhibitor 13. Compound 13 exhibits an IC50 of 1.0 μM for UBLCP1 and greater than 5-fold selectivity against a large panel of protein phosphatases from several distinct families. Importantly, the inhibitor possesses efficacious cellular activity and is capable of inhibiting UBLCP1 function in cells, which in turn up-regulates nuclear proteasome activity. These studies set the groundwork for further developing compound 13 into chemical probes or potential therapeutic agents targeting the UBLCP1 phosphatase.
Keywords: Protein phosphatase, UBLCP1, Inhibitor development, Fragment-based drug discovery
1. Introduction
The proteasome is responsible for ubiquitin-dependent protein degradation, which is essential for cellular homeostasis.1,2 Proper operation of the proteasome is central to diverse pathways regulating cell cycle progression, DNA repair, transcription and inflammation. Accordingly, proteasome activity is under intricate and dynamic control whereas its deregulation is widely associated with many human diseases.3,4 A great number of proteasome interacting proteins have been identified that play crucial roles in modulating proteasome function. Among these are a group of ubiquitin-like (UBL) domain-containing proteins, including substrate shuttle proteins, E3 ubiquitin ligases, and deubiquitinating enzymes.5,6 Reversible protein phosphorylation, catalyzed by the opposing activities of protein kinases and phosphatases, represents one of the most common post-translational modifications of proteins. Not surprisingly, proteasome activity is also regulated by phosphorylation. Although many kinases have been documented to phosphorylate the proteasome,7–13 much less is known about the identity of protein phosphatases that dephosphorylate the proteasome.
Recently it was discovered that the ubiquitin-like domain-containing C-terminal domain phosphatase 1 (UBLCP1) binds the 19S regulatory particle via the UBL domain and dephosphorylates the 26S proteasome, leading to inhibition of proteasome activity.14 UBLCP1 is a member of a growing family of novel phosphatases belonging to the haloacid dehalogenase (HAD) superfamily of hydrolases.15 Other notable members of the HAD phosphatases include the C-terminal domain (CTD) phosphatases Fcp1/Scp1 that serve as key regulators of gene transcription by controlling the level of phosphorylation in the C-terminal domain of RNA polymerase II,16,17 and the Eyes absent (Eya) family of transcription factors, which possess intrinsic phosphatase activity that are vital for organ formation.18–20 Though UBLCP1 has been identified as a proteasome phosphatase, its physiological role is still poorly understood. Given the dynamic nature of protein phosphorylation, pharmacological manipulation of the UBLCP1 phosphatase activity may afford a powerful approach to investigate the mechanisms by which UBLCP1 regulates the diverse proteasome-mediated cellular processes. Moreover, since UBLCP1 knockdown enhances nuclear proteasome activity,14 inhibition of UBLCP1 should promote activation of proteasome activity, which may be beneficial for alleviating pathological conditions caused by increased misfolded/damaged proteins in the cell. We describe here a salicylic acid fragment-based library approach, which led to the identification of the first potent and selective UBLCP1 inhibitor. Importantly, the inhibitor possesses highly efficacious cellular activity and is capable of inhibiting UBLCP1 function in cells, which in turn upregulates nuclear proteasome activity.
2. Results and discussion
While all protein kinases share a common three-dimensional core structure and catalyze the phosphoryl transfer reaction with the same chemical mechanism,21 nature has evolved multiple strategies for enzyme-mediated phosphate monoester hydrolysis, with distinct families of protein phosphatases. Unlike the more established Cys-dependent protein tyrosine phosphatases (PTPs)22 and metallo Ser/Thr protein phosphatases (the PPP and PPM families),23 the HAD phosphatases employ an active site nucleophilic Asp residue for covalent catalysis.24,25 Despite the differences in structure, substrate specificity, and mechanism of catalysis, the unifying feature for all protein phosphatases is their ability to bind the phosphoryl group from various substrates. Consequently, the development of small-molecule inhibitors for all protein phosphatases faces two major challenges: (i) the active sites of protein phosphatases are positively charged, which favors negatively charged molecules with limited cell permeability, and (ii) the active sites are highly conserved among each distinct family of protein phosphatases, so it is not trivial to develop inhibitors that can selectively differentiate one phosphatase from the others in the same family.
2.1. Identification of a potent and selective UBLCP1 inhibitor from a salicylic acid-based focused library approach
We previously reported that salicylic acid derivatives can function as nonhydrolyzable phosphotyrosine (pTyr) mimetics and possess PTP inhibitory activity.26,27 We subsequently found that bicyclic benzofuran and indole-based salicylic acids show enhanced affinity for the PTPs and are sufficiently polar to bind the PTP active site, yet still capable of cell membrane penetration. 28–34 The excellent bioavailability associated with these PTP inhibitors is not surprising given the fact that salicylic acid, and various natural and synthetic benzofuran and indole derivatives are widely found in clinic drugs. Interestingly, salicylic acid derivatives have also subsequently been found as pTyr mimetics for SHP2 domain proteins.35–37 Importantly, we demonstrated that PTP inhibitor potency and selectivity can be achieved by tethering molecular fragments to an appropriate bicyclic salicylic acid core to engage both the active site and an adjacent binding pocket.28,30–33 Given the success with the PTPs, we initially set out to discover UBLCP1 inhibitors by screening the benzofuran and indole-based salicylic acid libraries already prepared in the laboratory. Library compounds were screened against UBLCP1 at 25 °C and pH 6.0 using a phosphatase activity-based assay with para-nitrophenyl phosphate (pNPP) as a substrate. Unfortunately, no compounds showed significant inhibitory activity against UBLCP1 at 3 μM compound concentration.
Examination of the crystal structure of the Drosophila UBLCP114 revealed that the active site of the enzyme may be too small to accommodate the bicyclic salicylic acid-containing substituted benzofuran and indole derivatives. Nonetheless, previous structural analyses of PTP-bicyclic salicylic acid-based inhibitor complexes indicated that the hydroxyl group and the carboxylic acid within the inhibitors serve as an effective phosphate mimetic.28,30,32,33 We reasoned that single ring salicylic acids may better suit the UBLCP1 active site, and diversity elements attached to the salicylic acid core may increase inhibitor potency and selectivity through engagement of binding pockets in the vicinity of the active site.38,39
Figure 1 depicts our salicylic acid based focused library approach for potent and selective UBLCP1 inhibitors that are capable of bridging both the active site and an adjacent peripheral site. The active site directed single ring salicylic acid cores were generated through size reduction of the bicyclic substituted benzofuran. To identify an optimal salicylic acid core for UBLCP1, we initially prepared compounds 4a–e (Scheme 1) with R3 being methoxy, thiophenyl, cyclopentyl, cyclohexyl, and phenyl group. Cores 4a–e were synthesized in four steps from the starting compound 1.31 Compound 1 was converted into 2 via a SN2 substitution reaction using methyl 2-bromohexanoate in DMSO in the presence of potassium carbonate at room temperature. Compounds 3a–e were obtained at room temperature via standard Sonogashira reaction conditions with appropriate alkyne. Hydrolysis of 3a–e in MeOH with lithium hydroxide yielded cores 4a–e, which were then purified by HPLC. To capture additional interactions with adjacent pockets surrounding the active site, we build into the salicylic acid cores a substituted acetic acid, which serves as an easily functionizable synthetic handle to introduce diverse elements through amide chemistry. Thus a structurally diverse and commercially available set of 192 amines (Fig. 2) were condensed with hexanoic acid in 4a–e to generate five focused libraries (Scheme 1) aimed at capturing additional interactions with adjacent pockets surrounding the active site. The amide libraries 5a–c were assembled in 96 well plates in the presence of HBTU, HOBt, and DIPEA in DMF. Representative wells from each plate were monitored by LC–MS, which indicated that the reactions went well affording products in 60–80% conversions.
Figure 1.
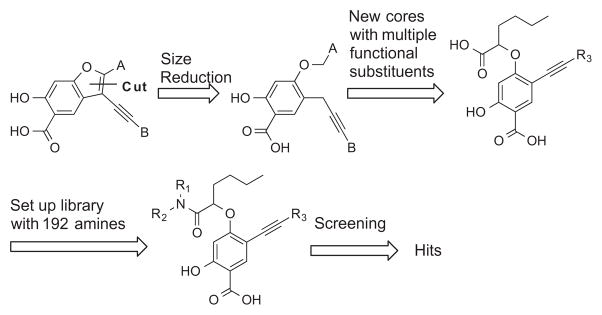
Synthesis strategy for single ring salicylic-based UBLCP1 inhibitors.
Scheme 1.

Synthesis of salicylic acid-based UBLCP1 inhibitors. Reaction conditions: (a) Methyl 2-bromohexanoate, K2CO3, DMSO, rt, 96%; (b) R3CCH, pd(pph3)2Cl2, Cul, DMF, rt, overnight, 75–89%; (c) LiOH, MeOH/H2O, reflux 90–95%; (d) R1R2NH, HBTU, HOBt, DIPEA, DMF, 70–80%.
Figure 2.

Chemical structures of the 192 amines used for library construction.
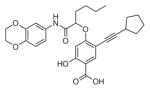
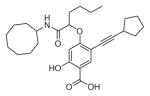
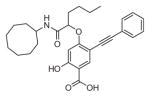
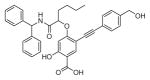
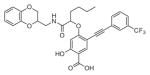
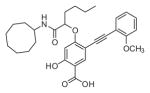
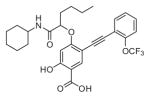
The ability of the library compounds to inhibit the UBLCP1-catalyzed hydrolysis of pNPP was assessed in situ at pH 6.0 and 25 °C. Out of five 192-member libraries, 4 compounds (6, 7, 8, and 9, Table 1) displayed measurable inhibitory activity at ~3 μM concentration. They were re-synthesized, purified, and their IC50 values for UBLCP1 were determined. As shown in Table 1, although the affinity of salicylic acid for UBLCP1 is weak (IC50 = 66 mM), compounds 6 to 9 exhibited IC50 values of 4.4, 13.5, 4.2, and 8.3 μM respectively, which are 4400–15,700 fold more potent than that of salicylic acid alone. The results indicate that functionalities attached to salicylic acid contribute significantly to UBLCP1 inhibition. Encouraged by these results, we proceeded to further optimize compound 4e with variations in the R4 substituent on the phenyl ring. For this purpose, 13 new cores were synthesized with various electron withdrawing/donating substituent’s at the ortho-, meta-, or para-position of the benzene ring. The synthesis of cores 4f–r and libraries 5f–r employed the same procedures described above (Scheme 1). High throughput screening of the 13 new libraries revealed that compounds with R4 groups at the ortho-position of the benzene ring are consistently better inhibitors than those with R4 at the meta- and para-positions. Resynthesis of the top hits 10, 11, 12, and 13 confirmed that they were genuine UBLCP1 inhibitors with IC50 values of 4.2, 5.4, 3.0, and 0.5 μM, respectively, (Table 1). Thus compound 13 appeared to be the most potent for UBLCP1, and was selected for further characterization.
Table 1.
Structures and IC50 values (μM) of top hits from libraries 5a–r at pH 6.0
| Compd# | Compound structure | IC50 |
|---|---|---|
| 6 |
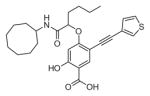
|
4.4 ± 0.2 |
| 7 |
|
13.5 ± 0.6 |
| 8 |
|
4.2 ± 0.2 |
| 9 |
|
8.3 ± 0.4 |
| 10 |
|
4.2 ± 0.3 |
| 11 |
|
5.4 ± 0.2 |
| 12 |
|
3.0 ± 0.2 |
| 13 |
|
0.5 ± 0.1 |
We then evaluated the selectivity of compound 13 at pH 7.0 and room temperature against a large panel of protein phosphatases, including the PTPs: LAR, PTPε, PTPγ, PTP1B, TC-PTP, SHP1, SHP2, STEP, PTPH1, HePTP, PEZ, VHR, VHZ, CDC14A, MKP5, and low molecular weight PTP; and Ser/Thr protein phosphatase 5, and the HAD family CTD phosphatase Scp1. IC50 determination for all phosphatases was performed under the same conditions except that the pNPP concentrations used for each enzyme corresponded to the Km value of the phosphatase under study. As shown in Table 2, compound 13 exhibits greater than 5-fold selectivity for UBLCP1 over all phosphatases examined. Of particular note, compound 13 exhibits no inhibition of HAD-like phosphatase Scp1 even at 50 μM concentration. Lineweaver–Burk plot analysis revealed that the mode of UBLCP1 inhibition by compound 13 is competitive with respect to the substrate pNPP with a Ki of 1.0 ± 0.1 μM (Fig. 3). Most importantly, compound 13 displays excellent cellular activity as shown below.
Table 2.
Selectivity of compound 13 against a large panel of protein phosphatases
| PTPs | IC50 (μM) for 13 |
|---|---|
| UBLCP1 | 1.0 ± 0.05 |
| CDC14A | 7.2 ± 0.04 |
| PTPH1 | 13.9 ± 1.3 |
| PEZ | 8.3 ± 0.5 |
| LAR | >50 |
| LMWPTP | >50 |
| PTP1B | 12.8 ± 0.7 |
| STEP | >50 |
| TC-PTP | 10.1 ± 0.4 |
| Laforin | 10.3 ± 0.4 |
| VHR | 6.0 ± 0.5 |
| MKP5 | 5.7 ± 0.5 |
| VHZ | 11.4 ± 0.93 |
| SHP1 | 8.0 ± 0.12 |
| SHP2 | 5.4 ± 0.18 |
| HePTP | 8.7 ± 0.48 |
| PTPε | >50 |
| PTPγ | >50 |
| PP5 | >50 |
| Scp1 | >50 |
Figure 3.

Lineweaver–Burk plot for SHP2 inhibition by compound 13. Compound 13 is a competitive inhibitor of SHP2 with Ki at 1.0 μM. Compound 13 concentrations were 0 (●), 0.5 (○), 1.0 (◆), and 1.5 (▽) μM, respectively.
2.2. Molecular basis of potent and specific inhibition of UBLCP1 by compound 13
To understand the molecular basis of the potency and selectivity of compound 13 toward UBLCP1, we carried out molecular modeling studies and analyzed the most likely binding mode of compound 13 with UBLCP1. Since the crystal structure of the human UBLCP1 has not been reported, we generated a three-dimensional structure of the phosphatase domain of human UBLCP1 by homology modeling40 based on the crystal structure of the Drosophila UBLCP1 (PDBID: 3SHQ).14 This homology structure was then used for molecular docking calculations in AutoDock4.2.6.41 The binding mode for compound 13 was determined by free energy comparisons and conformation cluster analyses of 800 docking runs. As shown in Figure 4A and B, the salicylic acid moiety of compound 13 binds into UBLCP1 phosphatase active site and has a number of Van der Waals contacts with nearby residues, mainly including D143, D145, S183, A184, D252, D253, I254 and N257. Moreover, the carboxyl oxygen form H-bonds with the backbone amide of A184 and ζ-amine of K230, and the hydroxyl group makes a polar interaction with the carboxylate of D252. These interactions anchor compound 13 in the active site and provide the essential driving force for binding. The trifluoromethoxy benzene group is positioned by the rigid alkyne to a positively charged surface primarily constituted by Y146, H151 and R152, and the fluorine atoms are in the right place for polar interactions with the imidazole in H151 and guanidine in R152. The other branch attached to the salicylic acid core extends from the ridge of the active site pocket to a peripheral hydrophobic cavity. The butane forms hydrophobic interactions with the aliphatic carbon atoms of I254 and R256, the amide nitrogen is hydrogen bonded to the side chain carboxyl of D253, and terminal cyclohexane hydrophobically interacts with P270 and M272. Collectively, the abundant interactions observed in the model can account for the potency of compound 13 for UBLCP1.
Figure 4.

The binding mode of compound 13 with UBLCP1, based on homology modeling and docking analysis. (A) The salicylic acid of compound 13 binds into the phosphatase active site of UBLCP1, while the two attached branches bind to the edge of the active site pocket and a peripheral hydrophobic cavity respectively. Compound 13 is shown as stick; UBLCP1 is represented in surface with electrostatic potential mapped, and the key residues constituting the interacting surface are labeled. (B) The interaction details of compound 13 with UBLCP1. Compound 13 is shown as stick (yellow carbon); residues within 5 Å distance of compound 13 are also shown in stick (gray carbon) with names labeled. H-bond and/or polar interactions are highlighted by dash lines. (C) Sequence alignment of UBLCP1 with Scp1. Residue numbers are shown at upper for UBLCP1 and bottom for Scp1. Interaction residues shown in (B) are marked with *, and the 4 conserved and 13 divergent residues are respectively highlighted in red and yellow. (D) Structure alignment of UBLCP1 (gray with black residue name) with Scp1 (green with blue residue name). The catalytic loop, the highly divergent fragment and subsequent insertion are indicated by arrows. (E) The active site pocket of Scp1 with electrostatic potential mapped, the color scale is same as that in (A). The binding conformation of compound 13 in UBLCP1 was mapped to show the much less efficient interactions with Scp1.
Since the HAD family phosphatases are unrelated to the classical PTPs and the PPP and PPM Ser/Thr protein phosphatases, it is not surprising that compound 13 exhibits selectivity for UBLCP1 over those tested protein phosphatases (Table 2). A more impressive observation is the fact that the IC50 of compound 13 for UBLCP1 is >50 fold lower than that for Scp1, which has the same DXDXT catalytic motif as UBLCP1. Through sequence and structure alignment of Scp1 with UBLCP1, we found that only 4 out of 17 interacting residues are conserved in both enzymes (Fig. 4C). Moreover, the 11-residues fragment right after the catalytic loop is highly divergent, and a 16-residues insertion forms two additional β-sheets in Scp1 (Fig. 4C and D). These structural differences cause significantly different active site pocket for Scp1. As shown in Figure 4E, the electropositive surface interacting with trifluoromethoxy benzene in UBLCP1 is changed to an electronegative and more flat surface in Scp1; the long insertion largely broadens the active pocket of Scp1; the peripheral hydrophobic cavity interacting with cyclohexane in UBLCP1 is converted to a shallow groove, all of which are predicted to diminish the interaction between compound 13 and Scp1. Together, although Scp1 has the same DXDXT catalytic motif and overall similar folds with UBLCP1, the divergent fragment and insertion right after the catalytic loop significantly alter the peripheral portion of the active pocket, make it impossible or much less effective to compound 13.
2.3. Cellular activity of compound 13
Given the potency and selectivity of 13 toward UBLCP1, we proceeded to evaluate its ability to inhibit UBLCP1-dependent activity inside the cell. UBLCP1 is a novel proteasome-associated phosphatase that negatively controls proteasome activity in the nucleus.14 Therefore, inhibition of UBLCP1 by compound 13 is expected to result in an increase of nuclear proteasome activity in cells. To test this hypothesis, we first directly measured proteasome activity from nuclear extracts of HaCaT cells (human keratinocytes) pretreated with vehicle control (DMSO) or compound 13. Cleavage of the fluorogenic peptide substrate (Suc-LLVY-AMC) by nuclear proteasomes was significantly increased by 50–60% following compound 13 treatment (Fig. 5A). This level of proteasome activation was comparable to that observed in the same cell type expressing UBLCP1 shRNA, which enhanced nuclear proteasome activity by 30–40% under the same assay conditions.14 We then determined whether this activating effect of compound 13 also holds true for folded protein substrates of the proteasome. To this end, we expressed in 293T cells a nuclear proteasome activity reporter, which is a modified version of UbG76V-GFP42 that has a nuclear localization signal (NLS) fused to its C-terminus (UbG76V-GFP-NLS). Pre-treatment of cells with compound 13 markedly accelerated the degradation of this reporter protein, again arguing for an enhancement of nuclear proteasome activity (Fig. 5B). These data suggest that compound 13 is cell-permeable and also inhibits UBLCP1 function in cells, which in turn upregulates nuclear proteasome activity.
Figure 5.
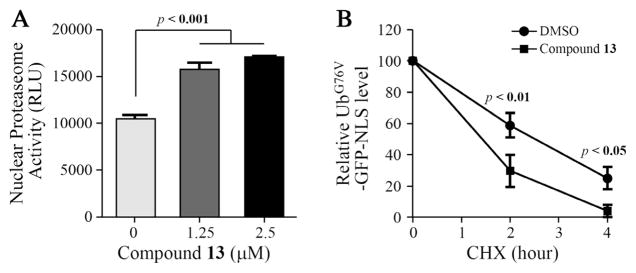
Compound 13 inhibits UBLCP1 activity in cells. (A) HaCaT cells (human keratinocytes) were treated with DMSO or the indicated concentrations of 13 for 24 h. Cells were fractionated and nuclear proteasome activity was measured by incubating with the fluorogenic peptide substrate Suc-LLVY-AMC. (B) 293T cells were transfected with the nuclear-targeted proteasome reporter, UbG76V-GFP-NLS. Cells were pre-treated with DMSO or 13 (10 μM) before cycloheximide (CHX) was added. GFP signal was measured at the indicated time points after CHX treatment.
3. Conclusion
Small molecule inhibitors are useful tools for probing the biological function and therapeutic potential of individual phosphatases, but achieving selectivity is challenging when the target phosphatase shares similar catalytic domains with other family members. The novel HAD family of protein phosphatases have attracted wide interest because of their emerging roles in transcriptional control, epigenetics, neuronal silencing and proteasome regulation. To date, only a few compounds targeting Scp143 and Eya244,45 have been reported owing to the challenges associated with protein phosphatase inhibitor development. We describe here a salicylic acid fragment-based library approach designed to target both the UBLCP1 active site and its adjacent binding pocket for enhanced affinity and selectivity. High throughput screening of the libraries led to the identification of the first potent (IC50 = 1.0 μM) and selective (>5 fold against a large panel of protein phosphatases from three distinct families) inhibitor 13 for UBLCP1. Of particular note, compound 13 exhibits greater than 50-fold selectivity for UBLCP1 against its HAD phosphatase family member Scp1, despite the similar fold of their phosphatase domains with almost identical catalytic site structure.14,24 Molecular modeling studies revealed the molecular basis of the observed potency and specificity of compound 13 for UBLCP1. More importantly, compound 13 possesses efficacious cellular activity and is capable of inhibiting UBLCP1 function in cells, which in turn up-regulates nuclear proteasome activity. Given the level of specificity and cellular activity, we expect that compound 13 will serve as a useful probe for in vivo interrogation of UBLCP1 function.
In addition to basic science application, small molecule UBLCP1 inhibitors may also have potential therapeutic applications. Although tremendous efforts have been dedicated to the development of therapeutic proteasome inhibitors, agents that activate the proteasome, which would have significant clinical utilities for treating human diseases, such as lysosomal storage disease, cancer, cystic fibrosis, Alzheimer, Parkinson’s and Huntington’s diseases46–52 that are caused by the cellular accumulation of ‘gain-of-function’ misfold/damaged protein, are scarce. Recently, small-molecule inhibitors of USP14, a proteasome-interacting deubiquitinating enzyme that antagonizes the degradation of ubiquitinated substrates, have been shown to enhance proteasome activity and facilitate the clearance of misfolded/aggregated proteins in cells.53 In fact, UBLCP1 and USP14 both bind to similar areas on the proteasome and inhibit proteasomal degradation. Therefore, it is anticipated that UBLCP1 inhibitors such as compound 13 may produce similar protective effects on cells with nuclear protein aggregates (e.g., mutant Huntingtin) and deregulated proteasome phosphorylation. Our studies set the foundation for further developing compound 13 into chemical probes or potential therapeutic agents targeting the UBLCP1 phosphatase. Given the ease and versatility of the salicylic acid fragment-based chemistry described herein, we expect that the strategy can be employed to develop cell permeable, potent and selective inhibitors for other members of the HAD family of phosphatases.
4. Experimental section
4.1. Materials
Polyethylene glycol (PEG3350) and buffers for crystallization were purchased from Hampton Research Co. p-nitrophenyl phosphate (pNPP) was purchased from Fluke Co. Dithiothreitol (DTT) was provided by Fisher (Fair Lawn, NJ).
4.2. Chemistry
1H NMR spectra were obtained on a Bruker 500 MHz and 300 MHz NMR instrument. 13C NMR spectra were obtained on a Bruker 125 MHz and 75 MHz NMR instrument. The chemical shifts were reported as δ ppm relative to TMS, using the residual solvent peak as the reference unless otherwise noted. The following abbreviations were used to express the multiplicities: s = singlet; d = doublet; t = triplet; q = quartet; m = multiplet; br = broad. High-performance liquid chromatography (HPLC) purification was carried out on a Waters Delta 600 equipped with a Sunfire Prep C18 OBD column (30 × 150 mm, 5 mM) with methanol-water (both containing 0.1% TFA) as the mobile phase (gradient: 30–100% methanol, flow 10 mL/min). All the final compounds were obtained in a highly pure form (>95%). Resolution mass spectra were obtained on an Agilent Technologies 6130 Quadrupole LC/MS. High-resolution mass spectra (HRMS) were obtained using electrospray ionization (ESI) on a time of flight (TOF) mass spectrometer at the Department of Chemistry, Indiana University as a technical service. All reactions were monitored by thin layer chromatography (TLC) carried out on Dynamic Adsorbents silica gel plates (0.25 mm thick, 60F254), visualized by using UV (254 nm). All compounds used for biological assays were purified by HPLC and are at least of 95% purity based on HPLC analytical results monitored with 254 nm wavelengths. All reagents and solvents were purchased from commercially available sources (FisherSci, Aldrich, Acros, Alfa Aesar, TCI).
4.3. Synthesis of methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4Hbenzo[ d][1,3]dioxin-7-yl)oxy)hexanoate (2)
A mixture of 7-hydroxy-6-iodo-2,2-dimethyl-4H-benzo[d][1,3]-dioxin-4-one (1) (10.00 g, 31.2 mmol), methyl 2-bromohexanoate (6.1 mL, 37.4 mmol), K2CO3 (8.64 g, 62.5 mmol), and DMSO (100 mL) was stirred at room temperature for overnight. The mixture was then diluted with water and extracted with EtOAc. The combined organic phase was dried over Na2SO4 and evaporated under reduced pressure. The product was purified by chromatography on a column of silica gel with EtOAc/Hexanes. Yield 13.43 g, 96%. 1H NMR (500 MHz, CDCl3) δ 8.32 (s, 1H), 6.22 (s, 1H), 4.72 (m, 1H), 3.78 (s, 3H), 2.05 (m, 2H), 1.72 (s, 3H), 1.71 (s, 3H), 1.58 (m, 2H), 1.40 (m, 2H), 0.95 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 170.34, 162.41, 159.31, 157.81, 140.05, 108.44, 106.72, 100.37, 78.26, 77.77, 52.51, 32.06, 27.15, 25.76, 25.57, 22.14, 13.85. Mass spectra (ESI): m/e 449 (M+H)+.
4.4. General procedures for the preparation of compounds 4a–r
A Schlenk flask was charged with methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (1 mmol), Pd(PPh3)Cl2 (0.03 mmol), iPr2NEt (4 mmol), alkyne (1.5 mmol). The DMF (4 mL) was added and the flask was evacuated and backfilled with N2 three times. After stirring 12 h at room temperature, the reaction was then diluted with water, extracted with EtOAc, washed with water and brine. The combined ethyl acetate layers were dried over anhydrous Na2SO4, filtered, concentrated to give compounds 3a–r, which was directly used in the next step without further purification. To a suspension of 3a–r (0.5 mmol) in MeOH (3 mL) was added 10% LiOH (3 mL). The obtained mixture was refluxed for 2 h, then poured into water, acidified with 3 M HCl aq and extracted with ethyl acetate. The organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by HPLC to furnish the corresponding products 4a–r. Protocol for the Prep-HPLC purification method: reverse phase HPLC was carried out on Sunfire Prep C18 OBD column (30 ×150 mm, 5 μm). Solvent A: water with 0.1% trifluoroacetic acid: Solvent B: methanol with 0.1% trifluoroacetic acid. Gradient: After 5 min at the initial condition of 90% A and 10% B, solvent B was increased to 100% within 45 min, then maintained at 100% B for 10 min. Flow rate was 50 mL/min, UV detector at 254 nm.
4.4.1. 4-((1-Carboxypentyl)oxy)-2-hydroxy-5-(3-hydroxyprop-1-yn-1-yl)benzoic acid (4a)
Compound 4a was synthesized from methyl 2-((6-iodo-2, 2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and prop-2-yn-1-ol following the general procedure for the preparation of compounds 4a–r. Yield 63.4% from compound 2. 1H NMR (300 MHz, Acetone-d6) δ 7.87 (s, 1H), 6.38 (s, 1H), 4.89 (t, J = 6.1 Hz, 1H), 4.40 (s, 2H), 2.01 (m, 2H), 1.58 (m, 2H), 1.42 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, Acetone-d6) δ 171.70, 171.63, 164.76, 164.60, 136.00, 106.16, 105.72, 101.24, 91.99, 79.67, 77.13, 50.95, 32.60, 27.70, 22.76, 14.04. Mass spectra (ESI): m/e 321 (M−H)−.
4.4.2. 4-((1-Carboxypentyl)oxy)-2-hydroxy-5-(thiophen-3-ylethynyl)benzoic acid (4b)
Compound 4b was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and 3-ethynylthiophene following the general procedure for the preparation of compounds 4a–r. Yield 69.8% from compound 2. 1H NMR (300 MHz, DMSO-d6) δ 7.84 (s, 1H), 7.76 (dd, J = 2.9, 1.2 Hz, 1H), 7.64 (dd, J = 5.0, 2.9 Hz, 1H), 7.17 (dd, J = 5.0, 1.2 Hz, 1H), 6.37 (s, 1H), 4.91 (t, J = 5.9 Hz, 1H), 1.93 (q, J = 7.1 Hz, 2H), 1.45 (m, 4H), 0.85 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ 171.72, 171.09, 163.31, 163.16, 134.59, 129.40, 129.00, 126.95, 121.77, 106.23, 104.10, 100.72, 87.14, 84.18, 76.05, 31.52, 26.69, 21.86, 13.94. Mass spectra (ESI): m/e 373 (M−H)−.
4.4.3. 4-((1-Carboxypentyl)oxy)-5-(cyclopentylethynyl)-2-hydroxybenzoic acid (4c)
Compound 4c was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and ethynylcyclopentane following the general procedure for the preparation of compounds 4a–r. Yield 82.3% from compound 2. 1H NMR (500 MHz, DMSO-d6) δ 13.30 (br, 1H), 11.60 (br, 1H), 7.67 (s, 1H), 6.27 (s, 1H), 4.82 (m, 1H), 2.86 (m, 1H), 2.09 (s, 1H), 1.97–1.87 (m, 4H), 1.72–1.67 (m, 2H), 1.65–1.42 (m, 6H), 1.38–1.32 (m, 2H), 0.89 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.15, 163.77, 162.93, 134.68, 105.51, 100.91, 97.26, 76.35, 75.48, 33.99, 33.97, 32.13, 31.12, 30.70, 27.10, 24.97, 22.35, 14.29. Mass spectra (ESI): m/e 359 (M−H)−.
4.4.4. 4-((1-Carboxypentyl)oxy)-5-(cyclohexylethynyl)-2-hydroxybenzoic acid (4d)
Compound 4d was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and ethynylcyclohexane following the general procedure for the preparation of compounds 4a–r. Yield 79.5% from compound 2. 1H NMR (300 MHz, Acetone-d6) δ 7.82 (s, 1H), 6.35 (s, 1H), 4.87 (t, J = 6.0 Hz, 1H), 2.66 (m, 1H), 1.80 (m, 4H), 1.60 (m, 6H), 1.41 (m, 6H), 0.94 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, Acetone-d6) δ 171.88, 165.00, 164.29, 135.51, 106.96, 106.09, 101.28, 97.25, 77.18, 76.28, 33.42, 33.06, 27.99, 26.69, 25.21, 23.11, 14.27. Mass spectra (ESI): m/e 373 (M−H)−.
4.4.5. 4-((1-Carboxypentyl)oxy)-2-hydroxy-5-(phenylethynyl) benzoic acid (4e)
Compound 4e was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and ethynylbenzene following the general procedure for the preparation of compounds 4a–r. Yield 86.7% from compound 2. 1H NMR (300 MHz, Acetone-d6) δ 11.50 (s, 1H), 8.00 (s, 1H), 7.54 (m, 2H), 7.40 (m, 3H), 6.44 (s, 1H), 4.97 (t, J = 5.9 Hz, 1H), 2.11 (m, 2H), 1.65 (m, 2H), 1.46 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, Acetone-d6) δ 171.82, 171.74, 165.02, 164.91, 135.81, 131.99, 129.36, 129.00, 124.54, 106.46, 105.94, 101.48, 92.63, 85.51, 77.24, 32.88, 27.88, 23.03, 14.25. Mass spectra (ESI): m/e 367 (M−H)−.
4.4.6. 4-((1-Carboxypentyl)oxy)-5-((2-chlorophenyl)ethynyl)-2-hydroxybenzoic acid (4f)
Compound 4f was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and 1-chloro-2-ethynylbenzene following the general procedure for the preparation of compounds 4a–r. Yield 65.3% from compound 2. 1H NMR (300 MHz, Acetone-d6) δ 8.04 (s, 1H), 7.64 (m, 1H), 7.52 (m, 1H), 7.38 (m, 2H), 6.45 (s, 1H), 4.91 (t, J = 6.0 Hz, 1H), 2.11 (m, 2H), 1.65 (m, 2H), 1.43 (m, 2H), 0.91 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, Acetone-d6) δ 171.80, 165.36, 164.99, 136.30, 135.72, 134.06, 130.39, 130.18, 127.83, 124.30, 106.66, 105.47, 101.47, 90.96, 89.30, 77.38, 33.01, 28.03, 23.07, 14.25. Mass spectra (ESI): m/e 401 (M−H)−.
4.4.7. 4-((1-Carboxypentyl)oxy)-5-((3-chlorophenyl)ethynyl)-2-hydroxybenzoic acid (4g)
Compound 4g was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and 1-chloro-3-ethynylbenzene following the general procedure for the preparation of compounds 4a–r. Yield 67.2% from compound 2. 1H NMR (300 MHz, Acetone-d6) δ 8.01 (s, 1H), 7.54 (m, 1H), 7.48 (m, 1H), 7.43 (m, 2H), 6.44 (s, 1H), 4.98 (t, J = 5.9 Hz, 1H), 2.11 (m, 2H), 1.66 (m, 2H), 1.46 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, Acetone-d6) δ 170.93, 170.84, 164.46, 164.16, 135.25, 133.83, 130.65, 130.26, 129.56, 128.26, 125.62, 105.74, 104.51, 100.67, 90.27, 86.21, 76.42, 32.03, 27.06, 22.23, 13.45. Mass spectra (ESI): m/e 401 (M−H)−.
4.4.8. 4-((1-Carboxypentyl)oxy)-5-((4-chlorophenyl)ethynyl)-2-hydroxybenzoic acid (4h)
Compound 4h was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and 1-chloro-4-ethynylbenzene following the general procedure for the preparation of compounds 4a–r. Yield 62.3% from compound 2. 1H NMR (300 MHz, Acetone-d6) δ 8.00 (s, 1H), 7.54 (dt, J = 8.7, 2.1 Hz, 2H), 7.44 (dt, J = 8.7, 2.1 Hz, 2H), 6.44 (s, 1H), 4.97 (t, J = 5.9 Hz, 1H), 2.10 (m, 2H), 1.64 (m, 2H), 1.45 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, Acetone-d6) δ 169.91, 163.41, 163.17, 134.20, 132.62, 131.76, 127.85, 121.57, 104.79, 103.81, 99.74, 89.66, 84.95, 75.50, 31.07, 26.12, 21.24, 12.50. Mass spectra (ESI): m/e 401 (M−H)−.
4.4.9. 4-((1-Carboxypentyl)oxy)-5-((3,4-dichlorophenyl) ethynyl)-2-hydroxybenzoic acid (4i)
Compound 4i was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and 1,2-dichloro-4-ethynylbenzene following the general procedure for the preparation of compounds 4a–r. Yield 58.6% from compound 2. 1H NMR (300 MHz, Acetone-d6) δ 8.01 (s, 1H), 7.69 (d, J = 1.9 Hz, 1H), 7.62 (dd, J = 8.3 Hz, 1H), 7.48 (dd, J = 8.3, 1.9 Hz, 1H), 6.44 (s, 1H), 4.99 (t, J = 5.9 Hz, 1H), 2.12 (m, 2H), 1.64 (m, 2H), 1.46 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, Acetone-d6) δ 171.76, 171.67, 165.45, 165.04, 136.21, 133.46, 132.90, 132.55, 131.73, 131.68, 125.01, 106.65, 105.14, 101.56, 90.23, 88.01, 77.30, 32.85, 27.93, 23.07, 14.33. Mass spectra (ESI): m/e 435 (M−H)−.
4.4.10. 4-((1-Carboxypentyl)oxy)-5-((3,5-difluorophenyl) ethynyl)-2-hydroxybenzoic acid (4j)
Compound 4j was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and 1-ethynyl-3,5-difluorobenzene following the general procedure for the preparation of compounds 4a–r. Yield 61.9% from compound 2. 1H NMR (300 MHz, Acetone-d6) δ 8.02 (s, 1H), 7.11 (m, 3H), 6.45 (s, 1H), 5.00 (t, J = 5.6 Hz, 1H), 2.11 (m, 2H), 1.64 (m, 2H), 1.46 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H). Mass spectra (ESI): m/e 403 (M−H)−.
4.4.11. 4-((1-Carboxypentyl)oxy)-2-hydroxy-5-((2-methoxyphenyl) ethynyl)benzoic acid (4k)
Compound 4k was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and 1-ethynyl-2-methoxybenzene following the general procedure for the preparation of compounds 4a–r. Yield 71.1% from compound 2. 1H NMR (300 MHz, Acetone-d6) δ 7.99 (s, 1H), 7.48 (dd, J = 7.6, 1.6 Hz, 1H), 7.36 (ddd, J = 8.4, 7.4, 1.7 Hz, 1H), 7.06 (d, J = 8.0 Hz, 1H), 6.97 (td, J = 7.5, 1.0 Hz, 1H), 6.44 (s, 1H), 4.97 (t, J = 6.0 Hz, 1H), 3.91 (s, 3H), 2.10 (m, 2H), 1.67 (m, 2H), 1.45 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, Acetone-d6) δ 171.85, 164.81, 160.87, 135.76, 133.93, 130.57, 121.17, 113.71, 111.95, 106.58, 106.45, 101.56, 89.45, 89.14, 77.33, 56.09, 32.92, 27.84, 23.01, 14.23. Mass spectra (ESI): m/e 397 (M−H)−.
4.4.12. 4-((1-Carboxypentyl)oxy)-2-hydroxy-5-((3-methoxyphenyl) ethynyl)benzoic acid (4l)
Compound 4l was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and 1-ethynyl-3-methoxybenzene following the general procedure for the preparation of compounds 4a–r. Yield 76.3% from compound 2. 1H NMR (300 MHz, Acetone-d6) δ 8.00 (s, 1H), 7.31 (t, J = 7.8 Hz, 1H), 7.11 (dt, J = 7.6, 1.3 Hz, 1H), 7.07 (m, 1H), 6.95 (m, 1H), 6.43 (s, 1H), 4.97 (t, J = 5.9 Hz, 1H), 3.83 (s, 3H), 2.10 (m, 2H), 1.66 (m, 2H), 1.46 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, Acetone-d6) δ 171.77, 164.04, 164.94, 160.55, 135.89, 130.45, 125.57, 124.42, 116.90, 115.35, 106.51, 105.91, 101.51, 92.66, 85.32, 77.28, 55.58, 32.88, 27.91, 23.05, 14.28. Mass spectra (ESI): m/e 397 (M−H)−.
4.4.13. 4-((1-Carboxypentyl)oxy)-2-hydroxy-5-((4-methoxy-phenyl)ethynyl)benzoic acid (4m)
Compound 4m was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and 1-ethynyl-4-methoxybenzene following the general procedure for the preparation of compounds 4a–r. Yield 71.9% from compound 2. 1H NMR (300 MHz, Acetone-d6) δ 7.97 (s, 1H), 7.48 (d, J = 8.0 Hz, 1H), 6.97 (d, J = 8.0 Hz, 1H), 6.43 (s, 1H), 4.96 (t, J = 5.8 Hz, 1H), 3.84 (s, 3H), 2.09 (m, 2H), 1.67 (m, 2H), 1.45 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, Acetone-d6) δ 171.83, 164.78, 164.73, 160.64, 135.52, 133.50, 116.52, 114.98, 106.43, 101.47, 92.72, 83.99, 77.27, 55.66, 32.91, 27.91, 23.05, 14.29. Mass spectra (ESI): m/e 397 (M−H)−.
4.4.14. 4-((1-Carboxypentyl)oxy)-2-hydroxy-5-((3-(trifluoromethyl) phenyl)ethynyl)benzoic acid (4n)
Compound 4n was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and 1-ethynyl-3-(trifluoromethyl)benzene following the general procedure for the preparation of compounds 4a–r. Yield 63.4% from compound 2. 1H NMR (500 MHz, DMSO-d6) δ 7.93 (s, 1H), 7.76 (m, 3H), 7.65 (t, J = 7.8 Hz, 1H), 6.39 (s, 1H), 4.95 (t, J = 5.8 Hz, 1H), 1.95 (q, J = 7.1 Hz, 2H), 1.74 (m, 4H), 0.84 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ 171.61, 171.04, 163.67, 163.51, 135.11, 134.71, 130.07, 129.71 (q, J = 31.8 Hz), 127.25, 124.07, 123.77 (q, J = 3.8 Hz), 124.07, 123.77 (q, J = 270.9 Hz), 103.35, 100.79, 90.14, 86.93, 76.05, 31.56, 26.73, 21.92, 13.82. Mass spectra (ESI): m/e 435 (M−H)−.
4.4.15. 4-((1-Carboxypentyl)oxy)-2-hydroxy-5-((2-(trifluoromethoxy) phenyl)ethynyl)benzoic acid (4o)
Compound 4o was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and 1-ethynyl-2-(trifluoromethoxy)benzene following the general procedure for the preparation of compounds 4a–r. Yield 59.2% from compound 2. 1H NMR (500 MHz, DMSO-d6) δ 7.87 (s, 1H), 7.65 (dd, J = 7.4, 1.3 Hz, 1H), 7.48 (m, 3H), 6.42 (s, 1H), 4.93 (t, J = 5.8 Hz, 1H), 1.92 (q, J = 7.2 Hz, 2H), 1.47 (m, 2H), 1.33 (m, 2H), 0.84 (t, J = 7.2 Hz, 3H). Mass spectra (ESI): m/e 451 (M−H)−.
4.4.16. 4-((1-Carboxypentyl)oxy)-2-hydroxy-5-((4-(trifluoromethoxy) phenyl)ethynyl)benzoic acid (4p)
Compound 4p was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and 1-ethynyl-4-(trifluoromethoxy)benzene following the general procedure for the preparation of compounds 4a–r. Yield 58.6% from compound 2. 1H NMR (300 MHz, Acetone-d6) δ 11.52 (s, 1H), 8.01 (s, 1H), 7.66 (dt, J = 8.9, 2.2 Hz, 2H), 7.38 (m, 2H), 6.45 (s, 1H), 4.98 (t, J = 5.9 Hz, 1H), 2.10 (m, 2H), 1.65 (m, 2H), 1.45 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, Acetoned6) δ 170.93, 170.86, 164.41, 164.13, 148.52, 148.50, 135.18, 132.97, 122.97, 121.28, 120.46 (q, J = 254.4 Hz), 105.70, 104.64, 100.69, 90.27, 85.80, 76.43, 31.99, 27.05, 22.15, 13.39. Mass spectra (ESI): m/e 451 (M−H)−.
4.4.17. 4-((1-Carboxypentyl)oxy)-2-hydroxy-5-((4-(hydroxymethyl) phenyl)ethynyl)benzoic acid (4q)
Compound 4q was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and (4-ethynylphenyl)methanol following the general procedure for the preparation of compounds 4a–r. Yield 73.7% from compound 2. 1H NMR (500 MHz, DMSO-d6) δ 13.40 (br, 1H), 11.80 (br, 1H), 7.87 (s, 1H), 7.45 (d, J = 8.2 Hz, 2H), 7.36 (d, J = 8.2 Hz, 2H), 6.38 (s, 1H), 4.93 (t, J = 5.9 Hz, 1H), 4.53 (s, 2H), 1.96 (m, 2H), 1.57 (m, 1H), 1.49 (m, 1H), 1.37 (m, 2H), 0.88 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.10, 171.50, 163.77, 163.63, 143.54, 135.04, 131.18, 127.06, 121.55, 106.71, 104.60, 101.14, 92.32, 84.99, 76.47, 62.98, 32.02, 31.13, 27.10, 22.33, 14.34. Mass spectra (ESI): m/e 397 (M−H)−.
4.4.18. 4-((1-Carboxypentyl)oxy)-2-hydroxy-5-((4-(phenoxymethyl) phenyl)ethynyl)benzoic acid (4r)
Compound 4r was synthesized from methyl 2-((6-iodo-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)hexanoate (2) and (prop-2-yn-1-yloxy)benzene following the general procedure for the preparation of compounds 4a–r. Yield 81.3% from compound 2. 1H NMR (300 MHz, Acetone-d6) δ 7.90 (s, 1H), 7.31 (m, 2H), 7.08 (m, 2H), 6.96 (m, 2H), 6.40 (s, 1H), 5.01 (s, 2H), 4.90 (t, J = 6.2 Hz, 1H), 2.01 (m, 2H), 1.56 (m, 2H), 1.40 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, Acetone-d6) δ 171.76, 165.17, 158.95, 136.49, 130.24, 121.91, 115.73, 106.47, 105.11, 101.43, 87.88, 82.65, 77.34, 57.03, 33.77, 27.94, 22.93, 14.26. Mass spectra (ESI): m/e 473 (M−H)−.
4.5. General procedure for the preparation of compounds 6–13
For resynthesis of hits in large scale, the 4-carboxymethoxy in 4-(carboxymethoxy)-2-hydroxybenzoic acid (0.1 mmol, 1.0 equiv) was activated by 1-O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluoro-phosphate (HBTU) (0.1 mmol, 1.0 equiv) in DMF for 5 min, then 1-hydroxybenzotriazole (HOBt) (0.1 mmol, 1.0 equiv) and N,N-diisopropylethylamine (0.4 mmol, 4.0 equiv) and amine (0.12 mmol, 1.2 equiv) was added. After stirring at room temperature for 2 h, the reaction liquids were directly sent to HPLC purification. Protocol for the Prep-HPLC purification method: reverse phase HPLC was carried out on Sunfire Prep C18 OBD column (30 × 150 mm, 5 μm). Solvent A: water with 0.1% trifluoroacetic acid: Solvent B: Methanol with 0.1% trifluoroacetic acid. Gradient: After 5 min at the initial condition of 90% A and 10% B, solvent B was increased to 100% within 45 min, then maintained at 100% B for 10 min. Flow rate was 50 mL/min, UV detector at 254 nm.
4.5.1. 4-((1-(Cyclooctylamino)-1-oxohexan-2-yl)oxy)-2-hydroxy-5-(thiophen-3-ylethynyl)benzoic acid (6)
4-((1-Carboxypentyl)oxy)-2-hydroxy-5-(thiophen-3-ylethynyl)-benzoic acid (4b) was reacted with cyclooctanamine according to the general procedure for the preparation of compounds 6–13 and the product was purified by reverse phase HPLC to provide compound 6. Yield 81.6%. 1H NMR (500 MHz, DMSO-d6) δ 8.01 (d, J = 8.0 Hz, 1H), 7.83 (s, 1H), 7.77 (dd, J = 2.9, 1.1 Hz, 1H), 7.64 (dd, J = 5.0, 2.9 Hz, 1H), 7.18 (dd, J = 5.0, 1.1 Hz, 1H), 6.41 (s, 1H), 4.66 (m, 1H), 3.80 (m, 1H), 1.83 (m, 2H), 1.66–1.35 (m, 19H), 0.85 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 170.98, 168.03, 163.26, 163.21, 134.56, 129.37, 129.01, 126.86, 121.68, 104.20, 101.05, 87.07, 84.22, 78.23, 48.54, 32.01, 31.19, 31.14, 26.86, 26.81, 26.62, 24.83, 23.12, 23.10, 21.82, 13.87. Mass spectra (ESI): m/e 482 (M−H)−.
4.5.2. 5-(Cyclopentylethynyl)-4-((1-((2,3-dihydrobenzo[b][1,4] dioxin-6-yl)amino)-1-oxohexan-2-yl)oxy)-2-hydroxybenzoic acid (7)
4-((1-Carboxypentyl)oxy)-5-(cyclopentylethynyl)-2-hydroxybenzoic acid (4c) was reacted with 2,3-dihydrobenzo[b][1,4]-dioxin-6-amine according to the general procedure for the preparation of compounds 6–13 and the product was purified by reverse phase HPLC to provide compound 7. Yield 88.2%. 1H NMR (500 MHz, DMSO-d6) δ 11.60 (br, 1H), 10.05 (s, 1H), 7.68 (s, 1H), 7.22 (d, J = 2.5 Hz, 1H), 6.99 (dd, J = 8.7, 2.5 Hz, 1H), 6.79 (d, J = 8.7 Hz, 1H), 6.32 (s, 1H), 4.73 (m, 1H), 4.20 (m, 5H), 2.86 (m, 1H), 1.93 (m, 4H), 1.71 (m, 2H), 1.64–1.45 (m, 7H), 1.34 (m, 3H), 0.89 (m, 3H). 13C NMR (125 MHz, DMSO-d6) δ 167.95, 163.25, 162.44, 142.98, 139.77, 134.36, 131.88, 116.85, 112.79, 108.70, 105.31, 100.73, 96.94, 78.17, 75.10, 64.18, 63.94, 33.52, 33.49, 32.31, 32.06, 30.25, 26.79, 24.52, 21.91, 13.86. Mass spectra (ESI): m/e 492 (M−H)−.
4.5.3. 4-((1-(Cyclooctylamino)-1-oxohexan-2-yl)oxy)-5-(cyclopentylethynyl)-2-hydroxybenzoic acid (8)
4-((1-Carboxypentyl)oxy)-5-(cyclopentylethynyl)-2-hydroxy-benzoic acid (4c) was reacted with cyclooctanamine according to the general procedure for the preparation of compounds 6–13 and the product was purified by reverse phase HPLC to provide compound 8. Yield 75.1%. 1H NMR (500 MHz, DMSO-d6) δ 11.50 (br, 1H), 7.96 (d, J = 8.1 Hz, 1H), 7.66 (s, 1H), 6.33 (s, 1H), 4.60 (m, 1H), 3.81 (m, 1H), 2.86 (m, 1H), 1.94 (m, 2H), 1.79 (m, 2H), 1.71–1.39 (m, 22H), 1.33 (m, 3H), 0.88 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 168.12, 163.28, 162.57, 134.24, 105.22, 100.84, 96.79, 77.96, 75.13, 48.51, 33.53, 33.51, 32.20, 31.25, 31.17, 30.24, 26.88, 26.82, 26.67, 24.87, 24.51, 23.16, 23.11, 21.89, 13.85. Mass spectra (ESI): m/e 468 (M−H)−.
4.5.4. 4-((1-(Cyclooctylamino)-1-oxohexan-2-yl)oxy)-2-hydroxy-5-(phenylethynyl)benzoic acid (9)
4-((1-Carboxypentyl)oxy)-2-hydroxy-5-(phenylethynyl)benzoic acid (4e) was reacted with cyclooctanamine according to the general procedure for the preparation of compounds 6–13 and the product was purified by reverse phase HPLC to provide compound 9. Yield 86.3%. 1H NMR (300 MHz, DMSO-d6) δ 8.08 (d, J = 8.0 Hz, 1H), 7.86 (s, 1H), 7.49 (m, 2H), 7.41 (m, 3H), 6.40 (s, 1H), 4.68 (m, 1H), 1.86 (m, 2H), 1.46 (m, 19H), 0.85 (t, J = 7.0 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ 171.54, 168.50, 163.82, 163.78, 135.12, 131.44, 129.19, 128.93, 123.29, 106.68, 104.59, 101.43, 92.15, 85.54, 78.57, 49.02, 32.57, 31.62, 31.56, 27.34, 27.29, 27.14, 25.56, 22.35, 14.38. Mass spectra (ESI): m/e 476 (M−H)−.
4.5.5. 4-((1-(Benzhydrylamino)-1-oxohexan-2-yl)oxy)-2-hydroxy-5-((4-(hydroxymethyl)phenyl)ethynyl)benzoic acid (10)
4-((1-Carboxypentyl)oxy)-2-hydroxy-5-((4-(hydroxymethyl) phenyl)ethynyl)benzoic acid (4q) was reacted with diphenyl-methanamine according to the general procedure for the preparation of compounds 6–13 and the product was purified by reverse phase HPLC to provide compound 10. Yield 89.3%. 1H NMR (500 MHz, DMSO-d6) δ 12.80 (br, 1H), 9.15 (d, J = 8.3 Hz, 1H), 7.86 (s, 1H), 7.42 (d, J = 8.2 Hz, 2H), 7.35–7.29 (m, 8H), 7.26 (m, 4H), 6.51 (s, 1H), 6.15 (d, J = 8.3 Hz, 1H), 4.91 (m, 1H), 4.52 (s, 2H), 1.91 (m, 2H), 1.34 (m, 2H), 0.84 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 171.00, 168.80, 163.34, 163.19, 143.07, 141.96, 134.62, 130.75, 128.43, 128.41, 127.30, 127.17, 127.07, 126.57, 121.02, 106.20, 104.27, 101.05, 91.88, 84.55, 77.67, 62.51, 56.01, 32.18, 26.72, 21.84, 13.89. Mass spectra (ESI): m/e 562 (M−H)−.
4.5.6. 4-((1-(((2,3-Dihydrobenzo[b][1,4]dioxin-2-yl)methyl) amino)-1-oxohexan-2-yl)oxy)-2-hydroxy-5-((3-(trifluoromethyl) phenyl)ethynyl)benzoic acid (11)
4-((1-Carboxypentyl)oxy)-2-hydroxy-5-((3-(trifluoromethyl) phenyl)ethynyl)benzoic acid (4n) was reacted with (2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methanamine according to the general procedure for the preparation of compounds 6–13 and the product was purified by reverse phase HPLC to provide compound 11. Yield 79.4%. 1H NMR (500 MHz, DMSO-d6) δ 8.47 (m, 1H), 7.93 (d, J = 7.7 Hz, 1H), 7.78 (m, 2H), 7.74 (d, J = 7.9 Hz, 1H), 6.82 (m, 4H), 6.41 (d, J = 1.4 Hz, 1H), 4.76 (m, 1H), 4.22 (m, 2H), 3.88 (m, 1H), 3.51–3.37 (m, 3H), 1.89 (m, 2H), 1.49 (m, 2H), 1.35 (m, 2H), 0.83 (m, 3H). 13C NMR (125 MHz, DMSO-d6) δ 170.84, 170.00, 169.95, 163.59, 142.74, 142.73, 142.61, 142.59, 134.97, 134.56, 129.90, 129.56 (q, J = 31.7 Hz), 127.14 (q, J = 3.5 Hz), 124.70, 123.94, 123.62 (q, J = 270.8 Hz), 121.25, 121.09, 116.93, 116.73, 106.53, 103.47, 101.01, 90.06, 86.89, 78.37, 78.27, 71.29, 71.25, 65.30, 65.25, 38.69, 38.65, 32.09, 32.06, 29.47, 26.61, 21.77, 13.66. Mass spectra (ESI): m/e 582 (M−H)−.
4.5.7. 4-((1-(Cyclooctylamino)-1-oxohexan-2-yl)oxy)-2-hydroxy-5-((2-methoxyphenyl)ethynyl)benzoic acid (12)
4-((1-Carboxypentyl)oxy)-2-hydroxy-5-((2-methoxyphenyl) ethynyl)benzoic acid (4k) was reacted with cyclooctanamine according to the general procedure for the preparation of compounds 6–13 and the product was purified by reverse phase HPLC to provide compound 12. Yield 73.1%. 1H NMR (500 MHz, DMSO-d6) δ 7.96 (d, J = 8.1 Hz, 1H), 7.82 (s, 1H), 7.42 (dd, J = 7.6, 1.7 Hz, 1H), 7.36 (m, 1H), 7.08 (d, J = 8.15 Hz, 1H), 6.97 (m, 1H), 6.45 (s, 1H), 4.70 (m, 1H), 3.85 (s, 3H), 3.80 (m, 1H), 1.83 (m, 2H), 1.58 (m, 4H), 1.45 (m, 12H), 1.33 (m, 2H), 0.86 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 171.03, 168.06, 163.18, 163.11, 159.44, 134.47, 132.78, 130.02, 120.42, 111.83, 111.38, 106.24, 104.73, 101.12, 88.50, 88.45, 78.22, 55.62, 48.53, 32.04, 31.15, 31.09, 26.85, 26.80, 26.48, 24.78, 23.11, 23.04, 21.85, 13.82. Mass spectra (ESI): m/e 506 (M−H)−.
4.5.8. 4-((1-(Cyclohexylamino)-1-oxohexan-2-yl)oxy)-2-hydroxy-5-((2-(trifluoromethoxy)phenyl)ethynyl)benzoic acid (13)
4-((1-Carboxypentyl)oxy)-2-hydroxy-5-((2-(trifluoromethoxy) phenyl)ethynyl)benzoic acid (4o) was reacted with cyclohexanamine according to the general procedure for the preparation of compounds 6–13 and the product was purified by reverse phase HPLC to provide compound 13. Yield 69.8%. 1H NMR (500 MHz, DMSO-d6) δ 7.97 (d, J = 8.1 Hz, 1H), 7.87 (s, 1H), 7.67 (dd, J = 7.6, 1.7 Hz, 1H), 7.54 (m, 1H), 7.47 (m, 2H), 6.45 (s, 1H), 4.69 (m, 1H), 3.58 (m, 1H), 1.84 (m, 2H), 1.68 (m, 4H), 1.54 (m, 1H), 1.44 (m, 2H), 1.32 (m, 2H), 1.27–1.08 (m, 5H), 0.85 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 171.37, 168.75, 164.20, 163.74, 148.32, 135.68, 133.94, 130.72, 128.32, 122.23, 120.67(q, J = 255.9 Hz), 117.92, 106.92, 103.99, 101.52, 90.83, 86.01, 78.70, 47.82, 32.61, 32.56, 32.49, 27.05, 25.56, 24.81, 22.28, 14.21. Mass spectra (ESI): m/e 532 (M−H)−. HRMS (ESI) calcd for C28H30F3NO6H+: 534.2098. Found: [M+H]+: 534.2103.
4.6. Expression and purification of UBLCP1
Overnight cultures from Escherichia coli BL21 cells transformed with the pET21a-UBL/DUBL expression vector were grown at 37 °C, with Amp (100 μg/ml). Protein expression was induced with 400 μM IPTG for 15 h at 25 °C. Cell pellets were lysed by French-Press at 0 °C in lysis buffer (50 mM Tris, pH 8.0, 300 mM NaCl, 5 mM MgCl2, 5 mM 2-mercaptoethanol, protease inhibitors and 20 mM imidazole). Cell debris was removed by high speed centrifugation and the supernatant was loaded onto a Ni-NTA column (Qiagen). The (His)6-tagged protein was eluted with 200 mM imidazole in the lysis buffer, concentrated and buffer-exchanged into 50 mM Tris, pH 8.0, 50 mM NaCl, 10 mM MgCl2 and 1 mM DTT. UBLCP1 protein was stored at −80 °C in 30% glycerol.
4.7. Kinetic analysis of UBLCP1 inhibition
UBLCP1 phosphatase activity was assayed using pNPP as a substrate at 25 °C in a pH 6.0 buffer (50 mM succinate, 100 mM NaCl, 2 mM MgCl2). The assays were performed in 96-well plates in a total reaction volume of 200 μL. The reaction was initiated by the addition of enzyme to a reaction mixture containing pNPP and the isolated. The reaction was quenched by the addition of 5 M NaOH. The nonenzymatic hydrolysis of pNPP was corrected by measuring the control without the addition of enzyme. The amount of product p-nitrophenol was determined from the absorbance at 405 nm detected by a Spectra MAX340 microplate spectrophotometer (Molecular Devices) using a molar extinction coefficient of 18,000 M−1 cm−1. The kcat and Km values were determined by fitting the data using the Enzyme Kinetics module in Sigma Plot. For compound screening, the same assay buffer was used and the assays were carried out at 25 °C. The salicylic acid based library was screened in a 96-well format at 10 μM compound concentration. The IC50 values for hit compounds were determined at 10 mM pNPP concentration.
For selectivity studies, a large panel of phosphatases including members in the PTP family, Cdc14A, PTPH1, PEZ, LAR, LMWPTP, PTP1B, STEP, TC-PTP, Laforin, VHR, MKP5, VHZ, SHP1, SHP2, HePTP, PTPε, and PTPγ; the Ser/Thr protein phosphatase family member PP5, and the HAD phosphatase family member Scp1. IC50 determination for all phosphatases was performed under the same conditions except that the pNPP concentrations used for each enzyme corresponded to the Km value of the phosphatase under study. Kinetic measurements for other phosphatases were conducted at pH 7 and 25 °C in a buffer containing 50 mM 3, 3-dimethylglutarate and 1 mM EDTA with an ionic strength of 0.15 M adjusted by addition of NaCl. The Inhibitor concentrations used for IC50 measurements cover the range from 0.2 to 5× of the IC50 value.
4.8. Molecular modeling study
The three dimensional structure of the phosphatase domain of human UBLCP1 was generated by homology modeling in MODELLER 9.14.40 The crystal structure of the Drosophila UBLCP1 (PDBID: 3SHQ) was used as a template, and the sequence alignment was performed in ClusterX2.54 During homology modeling, the default values were used for most parameters except for setting up the following parameters for thorough VTFM and MD optimizations: library_schedule = autosched.slow; max_var_iterations = 1000; md_level = refine.very_slow; repeat_optimization = 2. In total, 800 homology models were generated and the qualities were evaluated by the DOPE-HR method, the model with the lowest DOPE-HR score was used for subsequent docking study. The 3D-structure of compound 13 was generated and energy-minimized using the Chem3D program. Both compound 13 and UBLCP1 were introduced into the AutoDockTools1.4.655 for predocking processing, such as merging non-polar hydrogens, adding Gasteiger charges, setting rotatable bond for ligand, and adding solvation parameter for the receptor. The docking space was visually set around the UBLCP1 phosphatase active site, the energy grid size was set to 40 × 44 × 44 points with 0.375 Å spacing on each axis, and the energy grid maps for each atom type (i.e., A, C, HD, N, OA, SA and Mg), as well as the electrostatics and de-solvation maps were calculated using the AutoGrid4. The molecular dockings were carried out in AutoDock4.2.641 using LGALS (Lamarckian Genetic Algorithm with Local Search) algorithm. 800 separate docking runs were performed and the resulted 800 binding conformations were classified into different clusters and ranked according to the calculated binding free energy. Finally, binding mode analyses were performed in AutoDockTools1.4.6 by visual inspections and energy comparisons.
4.9. Cell culture and measurement of proteasome activity
HaCaT and HEK293T cells were cultured in Dulbecco modified Eagle medium (DMEM) with 10% fetal bovine serum and penicillin/streptomycin. HaCaT cells were plated in triplicates in a black 96-well plate (2.0 × 104 cells/well). DMSO or compound 13 was added the next day and cells were treated for 24 h. In-well fractionation of cells and measurement of nuclear proteasome activity using Suc-LLVY-AMC (Enzo Life Sciences) were done as previously described.14 To measure UbG76V-GFP-NLS turnover, the reporter was transfected into 293T cells using X-tremeGENE 9 (Roche). Seven hours later, cells were pre-treated with DMSO or 13 (10 μM) for 3 h before cycloheximide (CHX, 50 μg/ml) was added. GFP signal from cell lysates was measured at the indicated time points after CHX treatment using a Tecan Infinite M200 PRO plate reader.
Acknowledgments
This work was supported by National Institutes of Health Grants CA69202 and CA126937 to Z.Y.Z., and DK018849 to J.E.D. X.G. was supported by the Susan G. Komen postdoctoral fellowship.
Abbreviations
- UBLCP1
ubiquitin-like domain-containing C-terminal domain (CTD) phosphatase 1
- PTP
protein tyrosine phosphatase
References and notes
- 1.Finley D. Annu Rev Biochem. 2009;78:477. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schrader EK, Harstad KG, Matouschek A. Nat Chem Biol. 2009;5:815. doi: 10.1038/nchembio.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoeller D, Dikic I. Nature. 2009;458:438. doi: 10.1038/nature07960. [DOI] [PubMed] [Google Scholar]
- 4.Bingol B, Sheng M. Neuron. 2011;69:22. doi: 10.1016/j.neuron.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 5.Hartmann-Petersen R, Gordon C. Semin Cell Dev Biol. 2004;15:247. doi: 10.1016/j.semcdb.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 6.Hochstrasser M. Nature. 2009;458:422. doi: 10.1038/nature07958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castano JG, Mahillo E, Arizti P, Arribas J. Biochemistry. 1996;35:3782. doi: 10.1021/bi952540s. [DOI] [PubMed] [Google Scholar]
- 8.Satoh K, Sasajima H, Nyoumura KI, Yokosawa H, Sawada H. Biochemistry. 2001;40:314. doi: 10.1021/bi001815n. [DOI] [PubMed] [Google Scholar]
- 9.Feng Y, Longo DL, Ferris DK. Cell Growth Differ. 2001;12:29. [PubMed] [Google Scholar]
- 10.Zhang F, Hu Y, Huang P, Toleman CA, Paterson AJ, Kudlow JE. J Biol Chem. 2007;282:22460. doi: 10.1074/jbc.M702439200. [DOI] [PubMed] [Google Scholar]
- 11.Djakovic SN, Schwarz LA, Barylko B, DeMartino GN, Patrick GN. J Biol Chem. 2009;284:26655. doi: 10.1074/jbc.M109.021956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee SH, Park Y, Yoon SK, Yoon JB. J Biol Chem. 2010;285:41280. doi: 10.1074/jbc.M110.182188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Um JW, Im E, Park J, Oh Y, Min B, Lee HJ, Yoon JB, Chung KC. J Biol Chem. 2010;285:36434. doi: 10.1074/jbc.M110.133777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo X, Engel JL, Xiao J, Tagliabracci VS, Wang X, Huang L, Dixon JE. Proc Natl Acad Sci USA. 2011;108:18649. doi: 10.1073/pnas.1113170108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seifried A, Schultz J, Gohla A. FEBS J. 2013;280:549. doi: 10.1111/j.1742-4658.2012.08633.x. [DOI] [PubMed] [Google Scholar]
- 16.Archambault J, Pan G, Dahmus GK, Cartier M, Marshall N, Zhang S, Dahmus ME, Greenblatt J. J Biol Chem. 1998;273:27593. doi: 10.1074/jbc.273.42.27593. [DOI] [PubMed] [Google Scholar]
- 17.Yeo M, Lin PS, Dahmus ME, Gill GN. J Biol Chem. 2003;278:26078. doi: 10.1074/jbc.M301791200. [DOI] [PubMed] [Google Scholar]
- 18.Tootle TL, Silver SJ, Davies EL, Newman V, Latek RR, Mills IA, Selengut JD, Parlikar BE, Rebay I. Nature. 2003;426:299. doi: 10.1038/nature02097. [DOI] [PubMed] [Google Scholar]
- 19.Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, Nigam SK, Aggarwal AK, Maas R, Rose DW, Rosenfeld MG. Nature. 2003;426:247. doi: 10.1038/nature02083. [DOI] [PubMed] [Google Scholar]
- 20.Rayapureddi JP, Kattamuri C, Steinmetz BD, Frankfort BJ, Ostrin EJ, Mardon G, Hegde RS. Nature. 2003;426:295. doi: 10.1038/nature02093. [DOI] [PubMed] [Google Scholar]
- 21.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. Science. 2002;298:1912. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 22.Alonso A, Sasin J, Bottini N, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon JE, Mustelin T. Cell. 2004;117:699. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 23.Shi Y. Cell. 2009;139:468. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Kim Y, Genoud N, Gao J, Kelly JW, Pfaff SL, Gill GN, Dixon JE, Noel JP. Mol Cell. 2006;24:759. doi: 10.1016/j.molcel.2006.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang M, Liu J, Kim Y, Dixon JE, Pfaff SL, Gill GN, Noel JP, Zhang Y. Protein Sci. 2010;19:974. doi: 10.1002/pro.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sarmiento M, Wu L, Keng YF, Song L, Luo Z, Huang Z, Wu GZ, Yuan AK, Zhang ZY. J Med Chem. 2000;43:146. doi: 10.1021/jm990329z. [DOI] [PubMed] [Google Scholar]
- 27.Liang F, Huang Z, Lee SY, Liang J, Ivanov MI, Alonso A, Bliska JB, Lawrence DS, Mustelin T, Zhang ZY. J Biol Chem. 2003;278:41734. doi: 10.1074/jbc.M307152200. [DOI] [PubMed] [Google Scholar]
- 28.Yu X, Sun JP, He Y, Guo XL, Liu S, Zhou B, Hudmon A, Zhang ZY. Proc Natl Acad Sci USA. 2007;104:19767. doi: 10.1073/pnas.0706233104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou B, He Y, Zhang X, Xu J, Luo Y, Wang Y, Franzblau SG, Yang Z, Chan RJ, Liu Y, Zheng J, Zhang ZY. Proc Natl Acad Sci USA. 2010;107:4573. doi: 10.1073/pnas.0909133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, He Y, Liu S, Yu Z, Jiang ZX, Yang Z, Dong Y, Nabinger SC, Wu L, Gunawan AM, Wang L, Chan RJ, Zhang ZY. J Med Chem. 2010;53:2482. doi: 10.1021/jm901645u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He Y, Xu J, Yu ZH, Gunawan AM, Wu L, Wang L, Zhang ZY. J Med Chem. 2013;56:832. doi: 10.1021/jm301781p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He Y, Liu S, Menon A, Stanford S, Oppong E, Gunawan AM, Wu L, Wu DJ, Barrios AM, Bottini N, Cato AC, Zhang ZY. J Med Chem. 2013;56:4990. doi: 10.1021/jm400248c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeng LF, Zhang RY, Yu ZH, Liu S, Wu L, Gunawan AM, Lane BS, Mali RS, Li X, Chan RJ, Kapur R, Wells CD, Zhang ZY. J Med Chem. 2014;57:6594. doi: 10.1021/jm5006176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeng LF, Zhang RY, Bai Y, Wu L, Gunawan AM, Zhang ZY. Antioxidants Redox Signalling. 2014;20:2130. doi: 10.1089/ars.2013.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fletcher S, Singh J, Zhang X, Yue P, Page BDG, Sharmeen S, Shahani VM, Zhao W, Schimmer AD, Turkson J, Gunning PT. ChemBio Chem. 2009;10:1959. doi: 10.1002/cbic.200900172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, Yue P, Fletcher S, Zhao W, Gunning PT, Turkson J. Biochem Pharmacol. 2010;79:1398. doi: 10.1016/j.bcp.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Page BDG, Khoury H, Laister RC, Fletcher S, Vellozo M, Manzoli A, Yue P, Turkson J, Minden MD, Gunning PT. J Med Chem. 2012;55:1047. doi: 10.1021/jm200720n. [DOI] [PubMed] [Google Scholar]
- 38.Puius YA, Zhao Y, Sullivan M, Lawrence DS, Almo SC, Zhang ZY. Proc Natl Acad Sci USA. 1997;94:13420. doi: 10.1073/pnas.94.25.13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang ZY. Annu Rev Pharmacol Toxicol. 2002;42:209. doi: 10.1146/annurev.pharmtox.42.083001.144616. [DOI] [PubMed] [Google Scholar]
- 40.Sali A, Blundell TL. J Mol Biol. 1993;234:779. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 41.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J Comput Chem. 2009;30:2785. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG. Nat Biotechnol. 2000;18:538. doi: 10.1038/75406. [DOI] [PubMed] [Google Scholar]
- 43.Zhang M, Cho EJ, Burstein G, Siegel D, Zhang Y. ACS Chem Biol. 2011;6:511. doi: 10.1021/cb100357t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krueger AB, Dehdashti SJ, Southall N, Marugan JJ, Ferrer M, Li X, Ford HL, Zheng W, Zhao R. J Biomol Screening. 2013;18:85. doi: 10.1177/1087057112453936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pandey RN, Wang TS, Tadjuidje E, McDonald MG, Rettie AE, Hegde RS. PLoS ONE. 2013;8:e84582. doi: 10.1371/journal.pone.0084582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen B, Retzlaff M, Roos T, Frydman J. Cold Spring Harb Perspect Biol. 2011;3:a004374. doi: 10.1101/cshperspect.a004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sawkar AR, D’Haeze W, Kelly JW. Cell Mol Life Sci. 2006;63:1179. doi: 10.1007/s00018-005-5437-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dai C, Whitesell L, Rogers AB, Lindquist S. Cell. 2007;130:1005. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koulov AV, Lapointe P, Lu B, Razvi A, Coppinger J, Dong MQ, Matteson J, Laister R, Arrowsmith C, Yates JR, III, Balch WE. Mol Biol Cell. 2010;21:871. doi: 10.1091/mbc.E09-12-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caughey B, Lansbury PT. Annu Rev Neurosci. 2003;26:267. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 51.Cohen FE, Kelly JW. Nature. 2003;426:905. doi: 10.1038/nature02265. [DOI] [PubMed] [Google Scholar]
- 52.Morimoto RI. Genes Dev. 2008;22:1427. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, Wilson SM, King RW, Finley D. Nature. 2010;467:179. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Bioinformatics. 2007;23:2947. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 55.Sanner MF. J Mol Graphics Modell. 1999;17:57. [PubMed] [Google Scholar]