

Abstract
Anemia is a major source of morbidity and mortality worldwide. Here we review recent insights into how red blood cells (RBCs) are produced, the pathogenic mechanisms underlying various forms of anemia, and novel therapies derived from these findings. It is likely that these new insights, mainly arising from basic scientific studies, will contribute immensely to understanding frequently debilitating forms of anemia and the ability to treat affected patients. Major worldwide diseases that may stand to benefit from the new advances include the hemoglobinopathies (β-thalassemia and sickle cell disease), rare genetic disorders of red blood cell production, and anemias associated with chronic kidney disease, inflammation, and cancer. Promising new treatment approaches include drugs that target recently defined pathways in red blood cell production, iron metabolism, and fetal globin gene expression, as well as gene therapies using improved viral vectors and newly developed genome editing technologies.
Introduction
Anemia, defined as a decreased quantity of circulating red blood cells (RBCs), otherwise known as erythrocytes, is a major source of morbidity and mortality worldwide. Recent epidemiological studies suggest that one third of the world’s population is affected, with considerable resultant morbidity and mortality, particularly in children.1 Much of this burden is due to nutritional, infectious, and systemic etiologies, including iron deficiency, malaria, schistosomiasis, and chronic kidney disease.1 Genetic disorders of hemoglobin (Hb), including sickle cell disease (SCD) and thalassemia, are also common causes of clinically significant anemia, particularly among children.2
Recent studies have produced important insights into the molecular mechanisms of normal RBC homeostasis and how these processes go awry in human disease. These efforts have generated novel therapeutic approaches and several new drugs that are now being examined in clinical trials. Here we review these studies, emphasizing how investigation of the basic biology of anemia can generate new findings of clinical relevance.
Normal erythropoiesis and the pathophysiology of common anemias
Healthy human adults produce approximately 2 million RBCs each second through a coordinated process that begins with multipotential hematopoietic stem and progenitor cells (HSPCs).3,4 One model for this developmental pathway, termed erythropoiesis, is shown in Figure 1. Recent studies in mice suggest alternate routes for HSPC differentiation whereby erythroid and megakaryocytic (platelet producing cells) lineage commitment occurs earlier in the hematopoietic hierarchy than previously realized.5,6 Other new studies in mice suggest that multipotential and lineage-committed progenitors, rather than hematopoietic stem cells (HSCs), may bear the burden for most long-term output of differentiated blood cells under steady state conditions.7 Whether this is true in humans remains to be established, but these findings suggest that native erythropoiesis may not occur through one series of fixed differentiation steps, but rather, through multiple pathways that are regulated at the levels of lineage commitment and differentiation. This complexity and deviation from standard models of differentiation is important to bear in mind when considering the pathophysiology of various forms of anemia and emerging therapies.
Figure 1.

A model for hematopoiesis.
The long term hematopoietic stem cell (LT-HSC) gives rise to short term hematopoietic stem cells (ST-HSCs) that then give rise to the multipotent common myeloid progenitor (CMP) and common lymphoid progenitor (CLP). The CMP then gives rise to megakaryocyte-erythroid progenitors (MEPs) and granulocyte-macrophage progenitors (GMPs). The maturation of lineage-committed erythroid progenitors is shown on the left side. The earliest progenitor, burst forming unit erythroid (BFU-E), gives rise to the colony forming unit erythroid (CFU-E). These two progenitors are identified by colony assays. The CFU-E differentiates into morphologically distinct precursors that undergo progressive maturation: proerythroblasts (ProE), basophilic erythroblasts (BasoE), polychromatic erythroblasts (PolyE), and orthochromatic erythroblasts (OrthoE). The latter enucleate to produce reticulocytes (Retic) that are released into the circulation and mature further into red blood cells (RBCs).). EPO promotes the survival and proliferation of multiple erythroid progenitors and precursors from late BFU-E to basophilic erythroblast stages.113
The maturation of lineage-committed erythroid precursors is characterized by reduced proliferative capacity, characteristic morphological changes, and massive accumulation of Hb, the major blood oxygen transporter (Figure 1). Since Hb is an iron-containing protein, erythropoiesis is linked to and dependent upon physiological pathways that mediate iron absorption and utilization. Numerous cytokines stimulate erythropoiesis, the most clinically important one being erythropoietin (EPO). Evidence suggests that inhibitory cytokines also regulate RBC production in vivo.8–10
To maintain appropriate blood Hb levels, homeostatic mechanisms sense tissue oxygenation and adjust RBC production accordingly. In healthy individuals, steady state erythropoietic output balances RBC turnover. Anemia ensues when homeostatic mechanisms go awry and is caused by three basic problems affecting RBCs: reduced production, increased destruction (hemolysis), or loss (hemorrhage). The associated defects can be intrinsic to RBCs or their precursors, or extrinsic. Several prevalent causes of anemia worldwide including dietary iron deficiency, malaria, and other infectious diseases are not discussed further in this review.1 Rather, we focus on common and rare forms of anemia for which therapeutic approaches are emerging through exciting new discoveries in erythroid biology.
RBC-intrinsic disorders
Most forms of anemia within this class are monogenic disorders caused by inherited mutations. Salient examples are discussed here, with greater details and more comprehensive summaries provided by numerous textbooks.11 Diamond-Blackfan anemia (DBA) is a rare inherited anemia characterized by developmental anomalies and decreased RBC precursors (“hypoplastic anemia”). Most DBA patients have autosomal dominant mutations in one of at least 12 ribosomal protein (RP) genes.12 How haploinsufficiency of different RP genes causes anemia is unknown, although one common mechanism may be reduced translation of GATA1 mRNA, which encodes an essential erythroid transcription factor.13,14 Most RBC-intrinsic forms of anemia associated with hemolysis are caused by mutations that inhibit the production or function of three major classes of proteins: glycolytic or hexose monophosophate shunt enzymes (for example, glucose-6-phosphate dehydrogenase deficiency),15 RBC membrane components (for example, hereditary spherocytosis),16 and Hb (for example, sickle cell disease (SCD) and the thalassemia syndromes).2 Among these, SCD and β–thalassemia are particularly common and debilitating, affecting millions of individuals worldwide.1,2 Recent paradigm-shifting advances into the pathophysiology of these Hb disorders, mechanisms of globin gene expression, and tools for genetic manipulation have elucidated exciting new therapeutic approaches, thereby attracting considerable attention from both academia and industry.
Pathophysiology of the major hemoglobinopathies
The adult form of hemoglobin (HbA) is a heterotetramer consisting of two globin protein subunits, α- and β-globin, each encoded by separate genes. β-thalassemia is caused by mutations that inhibit the production of β-globin chains.17 Consequently, free α-globin molecules form insoluble aggregates that damage and destroy erythroid precursors in a process termed ineffective erythropoiesis. In this way, β-thalassemia resembles other diseases associated with unstable proteins, including α1-antitrypsin deficiency, Wilson disease, and some neurodegenerative disorders.18
Free α-globin interacts with various protein quality control pathways. For example, excess α-globin chains are eliminated by ubiquitin mediated proteolysis and autophagy.18 A recent study suggests that excess unstable α-globin peptide chains bind and sequester the molecular chaperone HSP70 in the cytoplasm, which in turn destabilizes GATA1, altering gene expression and contributing to ineffective erythropoiesis.19 In β-thalassemia, ineffective erythropoiesis drives EPO production, which stimulates ineffective erythropoiesis further in a vicious cycle. Consequently, massive expansion of delayed and dying erythroid precursors in blood forming tissues cause bone defects, organomegaly, and metabolic abnormalities.17 In addition, free α-globin within mature circulating RBCs induces their hemolysis. Thus, β–thalassemia causes both reduced production and accelerated destruction of RBCs.
In SCD, a single amino acid substitution in the β-globin protein induces deoxygenated HbA tetramers to form rigid polymers within RBCs, causing their deformation and the classic sickle-shaped morphology.20 Intrinsic alterations in rheology, as well as secondary effects on endothelium, inflammatory cells, and plasma proteins, promote the adherence of sickled RBCs to each other and to the inner lining of small blood vessels, causing vaso-occlusion and tissue hypoxia. This clinical consequences of this pathophysiology, termed vaso-occlusive crisis (VOC), include pain crisis, respiratory insufficiency (“acute chest syndrome”), and strokes.21
In SCD and other hemolytic disorders, circulating free Hb released by damaged RBCs degrades the vasodilator nitric oxide (NO).22,23 In addition, Hb releases free heme, which activates neutrophils through multiple mechanisms.22 The resultant inflammatory and vasoconstricting processes are believed to promote disease pathophysiology and may represent viable therapeutic targets. For example, haptoglobin and hemopexin scavenge circulating free Hb and heme, respectively.22
Severe forms of SCD and β-thalassemia are treated with regular or intermittent RBC transfusions and supportive care.17,20,24 In many regions of the world, RBC donor availability and high risk for infectious disease transmission limits blood transfusion therapy. Successful stem cell transplantation is curative for both disorders, but also limited by toxicities and donor availability.25 Thus, new treatments for these major Hb disorders are desperately needed.
Anemia of chronic kidney disease (CKD)
EPO binds the EPO receptor (EpoR) on hematopoietic progenitors to initiate a series of signaling events that promote erythroid differentiation, survival, and proliferation (Figure 2).3,26 The kidney is the major physiological source of EPO.27,28 Consequently, chronic kidney disease (CKD) is associated with EPO deficiency and anemia caused by decreased RBC production. In addition, CKD is a proinflammatory state associated with the production of numerous cytokines that damage or destroy erythroid precursors directly, or reduce iron availability for erythropoiesis.27,28 Hundreds of millions of people worldwide are affected by CKD, many of whom are EPO-deficient.29 Numerous review articles provide guidelines for the use of EPO in CKD and other anemic patients, including its benefits and potential toxicities (http://kdigo.org/home/guidelines/anemia-in-ckd/).24 Overall, the availability of recombinant EPO and its derivatives has had an enormous positive impact on many patients with CKD. However, many are EPO-refractory,27 requiring enormous doses or alternative therapies, which are discussed below.
Figure 2.

Stimulatory and inhibitory pathways regulating erythropoiesis. EPO engages EpoR on erythroid precursors to activate a signaling cascade that begins with JAK2, thereby facilitating survival and proliferation. EPO mRNA is normally secreted from a subset of adult kidney cells and its expression is regulated by hypoxia inducible transcription factor (HIF) family members. Hypoxia stabilizes HIF proteins by inhibiting the activity of prolyl hydroxylase enzymes (PHD) that stimulate HIF degradation. Pharmacological prolyl hydroxylase inhibitors (PHI) stabilize HIF to stimulate EPO production, not only in the kidney as shown, but also in the liver of anephric patients (not shown). Growth differentiation factor 11 (GDF11) may negatively regulate erythropoiesis by engaging transforming growth factor β receptors and is blocked by activin receptor traps, including Sotatercept and ACE-536. The physiological source of GDF11 is currently unknown.
Anemia of inflammation
Inflammation-associated suppression of RBC production contributes substantially to the worldwide burden of anemia.30,31 This anemia of inflammation occurs simultaneously with chronic infectious diseases, autoimmune or rheumatologic disorders, and other systemic illnesses. The pathophysiology derives largely from the actions of proinflammatory cytokines, which stimulate hepcidin production to inhibit iron absorption and utilization (see below), block the differentiation of erythroid precursors/progenitors directly, and inhibit their responsiveness to EPO.30,32,33
Anemia of cancer
Approximately 40% of cancer patients have symptomatic anemia, which can limit the quality of life and be a major cause of morbidity.34 Causes of cancer-associated anemia include chemotherapy-induced myelosuppression and the production of inhibitory cytokines.9 Myelodysplastic syndrome, a common disorder in older individuals, is associated with hematopoietic cell-intrinsic defects in RBC production.35 This anemia sometimes improves with the administration of EPO and/or chemotherapeutic agents. However, responses to these treatments are usually transient, eventually necessitating RBC transfusions.
Stimulating Erythropoiesis
Modulating EPO receptor signaling
EPO and its derivative long-acting analogs are used widely to support erythropoiesis in patients with CKD, cancer, and other forms of anemia associated with insufficient endogenous production of this cytokine.27 Alternative approaches to stimulate the Epo receptor (EpoR) theoretically may have additional clinical benefits when compared with EPO including oral bioavailability and/or additional erythropoietic activities.36 The long acting pegylated peptide Peginasatide, which bears no structural resemblance to EPO, activates EpoR signaling potently to stimulate erythropoiesis.36 This agent was used briefly to treat the anemia of CKD and to treat a rare form of anemia caused by endogenous production of EPO-neutralizing antibodies.37,38 However, Peginasatide was removed from the market after it was found to cause an unacceptably high rate of anaphylaxis.39
EPO is synthesized mainly by a group of poorly defined kidney cells that are capable of sensing hypoxia (Figure 2). However, other tissues, predominantly the liver, provide a potential alternative endogenous source that can be stimulated pharmacologically. Transcription of the EPO gene is activated by low oxygen tension through the hypoxia inducible family of transcription factors (HIFs).40 Under normoxic conditions, prolyl hydroxylase enzymes hydroxylate HIFs, which targets them for proteasomal degradation. Prolyl hydroxylase inhibitors (PHIs) have thus emerged as a promising approach to stimulate HIF activity and thereby increase endogenous EPO production (Figure 2).27 Clinical trials of orally administered PHIs in patients with CKD and other EPO-dependent anemias are underway and early results are promising.28 However, HIFs regulate the expression of many genes besides EPO, with both positive and negative implications for treating anemia.40 For example, HIFs may alter iron distribution to increase its bioavailability for erythropoiesis during chronic inflammatory states, such as CKD and cancer. On the other hand, HIFs stimulate new blood vessel formation, which is associated with tumor growth. Moreover, some PHIs are associated with clinical adverse effects, including liver necrosis.27 Use of tissue-specific and isoform-selective PHIs are being investigated and may reduce some toxicities.28 Overall, PHIs represent promising erythropoietic agents with potential widespread applications.
Suppressing GDF11
While it has been known for some time that erythroid precursors are subject to negative regulation of maturation and/or survival by cytokines, such pathways have not been exploited therapeutically.9,10 Recently, a ligand trap for the activin receptor type IIA (Sotatercept) was generated to treat osteoporosis. Development of this product was based on preclinical studies implicating a role for activin signaling in bone homeostasis.41,42 To the surprise of clinical investigators, Sotatercept also induced dramatic increases in Hb and RBC counts in healthy human subjects.42 Sotatercept, and a modified version (ACE-536), appear to stimulate erythropoiesis by blocking the activity of the bone morphogenetic protein family member GDF11 (Figure 2).43,44 These agents act independent of EPO and in preclinical models stimulate erythropoiesis in several EPO-refractory anemic states including β-thalassemia and myelodysplastic syndrome.43,44 Further studies are needed to better understand the role of GDF11 in normal erythropoiesis and various anemias.8 For example, while erythroid precursors themselves may be a source of GDF11, the most important physiologic sources of this cytokine are unknown. In addition, the stages of erythropoiesis targeted by GDF11, the precise receptors through which it acts, and the mechanisms through which it blocks erythroid maturation remain to be defined. Recent work in mice suggests that GDF11 may inhibit aging of various tissues including the heart, skeletal muscle, and brain.45–48 Whether inhibiting GDF11 through receptor traps or other means will accelerate tissue aging must be investigated in clinical studies. Regardless, this approach holds promise for treating anemia, particularly in patients with EPO-refractory states and clinical trials are underway (http://clinicaltrials.gov/show/NCT01736683, http://clinicaltrials.gov/show/NCT01464164, http://clinicaltrials.gov/show/NCT01999582, https://ash.confex.com/ash/2014/webprogram/Paper73847.html).
Other strategies
Ongoing research has revealed additional pathways that may stimulate erythropoiesis. Recent work has shown that polymeric human IgA1 can stimulate erythropoiesis by interacting with transferrin receptor 1 to augment EpoR signaling.49 During the anemia of inflammation, hepcidin-induced iron restriction inhibits the activity of aconitases, iron-dependent enzymes that convert citrate to isocitrate as part of the tricarboxylic acid cycle. Isocitrate is a precursor for the production of ATP, reducing equivalents and heme, and may also suppress inflammatory signaling. In a rodent model for the anemia of chronic disease, administration of isocitrate bypassed aconitase deficiency to relieve some erythropoietic defects.50 These interesting findings may illustrate new approaches for treating various forms of anemia, although further work is required.
Fetal Hemoglobin Induction for β-hemoglobinopathies
Targeting relevant transcription factors
Strategies to replace mutated or deficient adult β-globin are of potential benefit for SCD and β-thalassemia, respectively. Fetal Hb (HbF) is composed of two β-like γ-globin chains and two α-globin chains. HbF is the major form of Hb expressed during gestation and supports oxygen transport in the fetus. Around birth, γ-globin production from two genes (HBG1 and HBG2) declines, while transcription of the nearby HBB gene encoding adult type β-globin increases gradually (Figure 3a). This “hemoglobin switch” is typically completed around 6 months of age, explaining why patients with β-thalassemia and SCD begin to manifest major symptoms in late infancy.51 Rare individuals carry mutations in regulatory regions of the β-globin locus that inhibit the γ-to-β switch, resulting in increased HbF throughout life, a benign condition known as hereditary persistence of fetal hemoglobin (HPFH).52 Remarkably, individuals with concomitant HPFH and β-thalassemia or SCD mutations exhibit reduced disease severity and in some cases are entirely asymptomatic.51,53,54 These clinical observations indicate that preventing or reversing the γ-to-β–globin switch through drugs or genetic manipulation could be a powerful approach for treating SCD and β-thalassemia. Individuals with isolated HPFH are asymptomatic, demonstrating that high levels of HbF have no adverse consequences.
Figure 3.
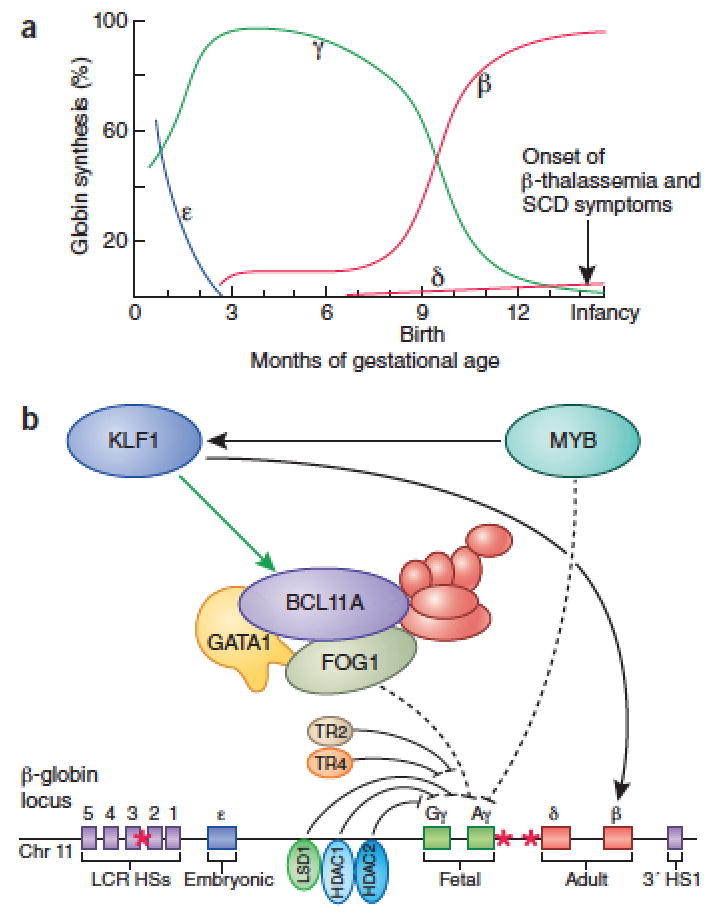
Regulation of fetal hemoglobin (HbF) production. (a) Timing of β-like globin subunit switching during human ontogeny. The embryonic, fetal, and adult stages are shown in blue, green, and red, respectively. (b) Regulators of hemoglobin switching including BCL11A, KLF1, MYB, TR2, TR4, LSD1, HDAC1, and HDAC2 as well as their modes of proposed regulation are depicted here. Some factors, including the BCL11A complex repress γ-globin through indirect mechanisms of action and are therefore shown with dotted lines. BCL11A binding sites are indicated with an asterix. Corepressor complexes that associated with BCL11A and other regulators are depicted in red.
More than 30 years ago, hydroxyurea (HU) was found to induce HbF production and alleviate symptoms and organ damage in SCD patients, through mechanisms that remain incompletely understood.55 Recent guidelines from the National Institutes of Health recommend that routine HU treatment be considered for all SCD patients beginning at age six months and regardless of symptoms (http://www.nhlbi.nih.gov/health-pro/guidelines/sickle-cell-disease-guidelines). However, HU causes only modest HbF gene expression induction and is not a panacea for SCD. Thus, there has been a long-standing search for better HbF-inducing agents.53
New approaches to promote HbF production have emerged from recent studies investigating the mechanisms of hemoglobin switching. Genome-wide association (GWAS) and functional follow up studies of HbF levels identified the multi-zinc finger transcription factor, BCL11A, to be a key repressor of γ-globin expression and physiological mediator of the γ-to-β switch (Figure 3).56 Indeed, some HPFH mutations within the β-globin gene cluster may promote γ–globin gene transcription by preventing BCL11A binding to its target regions.52,57 Subsequent work in murine models for SCD demonstrated that suppression of Bcl11a by RNA interference or gene ablation raised HbF to therapeutic levels with minimal effects on other erythroid genes, thereby reducing clinical complications.56,58,59 These studies identify BCL11A as a key therapeutic target for HbF induction. However, approaches to suppress BCL11A in patients are challenging for two important reasons. First, transcription factors are notoriously difficult to inhibit with small molecules.53 Second, BCL11A has essential functions in non-erythroid cells, including lymphocytes and neurons.53 Repression of BCL11A through RNA interference or gene editing in HSCs may circumvent some of these problems (see Box on Gene Editing strategies).60 Recent work has identified an erythroid-specific BCL11A enhancer and cis-elements within the human β-globin locus that are bound by the BCL11A protein.52,57 These DNA regions may be useful targets for gene editing approaches to raise HbF by inhibiting BCL11A or its activity specifically at the β-globin locus. In addition, BCL11A has been shown to function within large protein complexes that include epigenetic modifier enzymes (discussed below), which may represent more tractable (albeit less specific) drug targets.53,56,61
Box. Gene Editing Strategies.
With the advent of novel gene editing approaches using zinc fingers, transcription activator-like effector nucleases (TALENs), homing endonucleases, and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated proteins (Cas), the potential for endogenous gene correction has become a reality.114,115 Each of these technological advances works in a slightly different manner to create precisely engineered DNA breaks that promote insertion/deletion (indel) mutations via non-homologous end joining DNA repair pathways or that are faithfully repaired by homologous recombination with exogenous DNA templates.115 Pioneering work has demonstrated that it is possible to modify endogenous genes in primary human HSPCs, including inactivation of the HIV co-receptor CCR5 that may be able to treat acquired immune deficiency syndrome and severe combined immunodeficiency defect due resultant production of functional T lymphocytes.116–118 Current challenges for gene editing of HSPCs include optimizing the delivery of gene editing tools, improving the efficiency of introducing targeted modifications of interest, and avoiding the creation of potentially harmful off-target mutations.
Additional molecular targets for HbF induction also exist (Figure 3b).53,62 For example common DNA polymorphisms and rare mutations that reduce expression of the MYB gene in erythroid progenitors are associated with elevated HbF.63–66 MYB is a transcription factor with pleiotropic roles in hematopoiesis, raising concern that its manipulation may be toxic. However, studies in mice show that partial Myb suppression can inhibit leukemogenesis while sparing normal hematopoiesis, identifying a potential therapeutic range for HbF induction.67 Rare mutations in the gene encoding the erythroid transcription factor KLF1 have been identified in individuals with elevated HbF, although the extent of induction varies and some KLF1 mutations also cause anemia.53,68–70 The nuclear hormone receptors TR2 and TR4 have also been suggested to regulate γ-globin transcription and overexpression of these factors increased HbF levels and ameliorated clinical symptoms in a mouse model of SCD.62,71 As with BCL11A, these transcription factors pose challenges as pharmacological targets.
Targeting epigenetic enzymes
Rather than targeting transcription factors themselves, it may be more feasible to inhibit their associated enzymes that regulate gene expression by modifying histones and other nuclear proteins. The histone deacetylases (HDACs) are suggested to regulate HbF expression through modification of chromatin and these nuclear proteins physically associate with many of the transcription factors that regulate HbF.51 Chemical and RNA interference screens showed that inhibiting HDAC1 and HDAC2 induces HbF in primary human erythroblasts.72 In addition, inhibitors of LSD1/KDM1A, a flavin-dependent monoamine oxidase that demethylates lysines 4 and 9 in histone H3, including the anti-depressant tranylcypromine, were shown to induce HbF production in primary human cells and animal models.73 However, other studies show that LSD1/KDM1A inhibition can also impair normal erythropoiesis, raising concerns about the viability of this therapeutic target.61 More generally, because epigenetic enzymes are expressed ubiquitously and have multifaceted functions, their inhibition long-term may be toxic, particularly in hemoglobinopathy patients where therapies are most effective when initiated early in childhood and continued lifelong. Of note, the potential of small molecule inhibitors of HDACs and LSD1/KDM1A as anti-cancer agents is being studied in numerous clinical trials. HbF quantification, which is simple, inexpensive, and does not require extra blood sampling, should be incorporated into these studies. While the results might be confounded by intercurrent malignancy, medications, and other factors, such studies could provide valuable information for assessing the utility of these drugs and their associated molecular pathways for HbF induction in humans.
Other Strategies to Treat Hemoglobinopathies
Treatment of ineffective erythropoiesis in β-thalassemia
In β-thalassemia, ineffective erythropoiesis combined with elevated EPO causes pathological expansion of immature, damaged erythroid precursors. Thus, chemical inhibition of JAK2, which mediates the proliferative effects of EPO, reduces splenomegaly and improves anemia in β-thalassemic mice.74 Further studies to demonstrate the safety and efficacy of JAK2 inhibition in patients with β-thalassemia are required.75 As noted earlier, inhibition of GDF11 signaling via ligand traps such as Sotatercept or ACE-536 may reduce ineffective erythropoiesis and raise RBC production in β-thalassemia.43 Finally, new therapies aimed at manipulating protein quality control mechanisms to enhance degradation of free α-globin or reduce its interaction with HSP70 may be useful for treating β-thalassemia.19
Reducing inflammation and sickle cell formation in SCD
Numerous studies seek to better define and inhibit inflammation in SCD. For example, blockade of the adhesion receptors P- and E-selectins, may reduce VOC.76,77 Other anti-inflammatory agents, such as intravenous immunoglobulin and those that block CXCR2 have shown promise in model systems.78,79 Additional approaches to reduce inflammation are ongoing.21 A subset of inflammatory cells known as iNKT cells may contribute substantially to the ischemic-reperfusion injury thought to underlie VOC.80 SCD mice and patients have increased numbers of iNKT cells and blockade of these cells appears to be safe and potentially effective for preventing VOC and its associated complications.81
Only deoxygenated HbS is capable of sickling. This supports the use of inhaled oxygen as a powerful short-term therapy for VOC, although long term and excessive administration of oxygen to SCD patients can suppress EPO production.82 A potentially more effective long-term approach is to use molecules that bind HbS, increase its affinity for oxygen, and thereby prevent polymerization and sickling.83,84 5-hydroxymethyl-2-furfural is an example of one such molecule. More potent and orally-bioavailable derivatives are under clinical investigation. (https://www.clinicaltrials.gov/ct2/show/NCT02285088, https://www.clinicaltrials.gov/ct2/show/NCT01987908).83
Manipulating iron absorption and distribution
Recent insights into the physiological regulation of iron homeostasis are relevant to understanding and treating many forms of anemia (Figure 4).85–88 The membrane transporter ferroportin stimulates uptake of iron by intestinal epithelial cells and egress of iron from macrophage. Hepcidin, a small peptide secreted by the liver, binds ferroportin and stimulates its degradation, causing reduced intestinal absorption of iron and its accumulation within the reticuloendothelial system.
Figure 4.
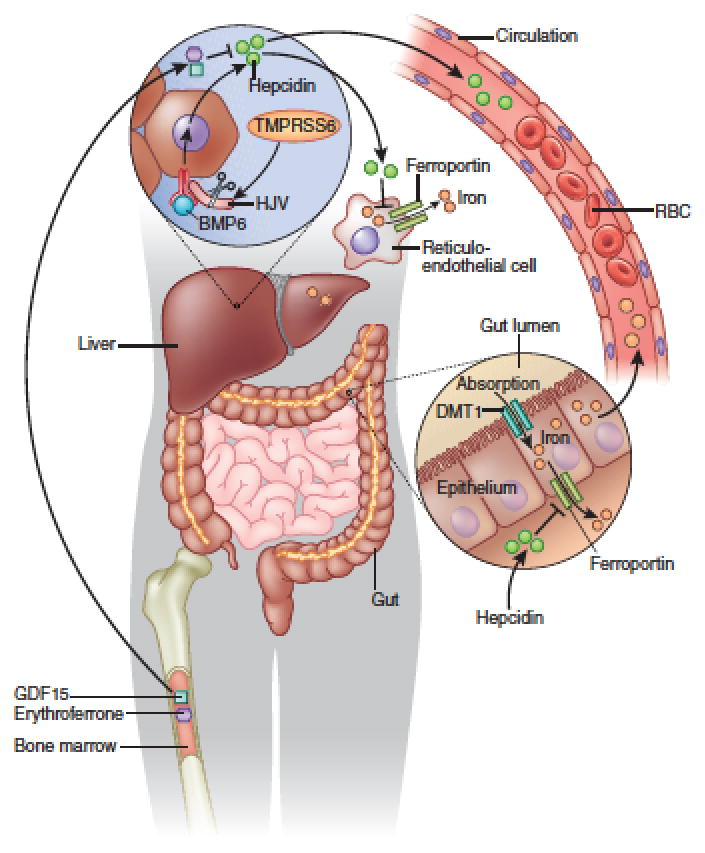
Regulation of iron homeostasis. Hepcidin gene transcription in hepatocytes is regulated by BMP6, BMP6 receptors, the co-receptor hemojuvelin (HJV) and inflammatory cytokines such as interleukin 6 (not shown). TMPRSS6 cleaves HJV to inhibit hepcidin production. Secreted hepcidin binds ferroportin and stimulates its degradation, resulting in decreased intestinal iron absorption and iron accumulation in reticuloendothelial cells. During ineffective erythropoiesis in β-thalassemia and other disorders, accumulated bone marrow erythroid precursors release erythroferrone and GDF15, which suppress liver hepcidin production, causing inappropriate iron absorption and release of iron from reticuloendothelial cells.
Inflammatory cytokines, such as interleukin 6, stimulate hepcidin production, which contributes to the anemia of chronic disease and inflammation by reducing iron availability for erythropoiesis. Prolyl hydroxylase inhibitors, discussed earlier, may alleviate anemia not only by enhancing endogenous EPO production, but also by reducing hepcidin levels to stimulate the uptake of dietary iron and its release from reticulendothelial stores.
Iron overload is a major toxicity in β-thalassemia and to a lesser extent SCD and other conditions requiring regular transfusions for two main reasons. First, RBC transfusion therapy increases the iron burden and no physiological mechanisms exist for its elimination.85,88 Second, accumulation of erythroid precursors, particularly with β-thalassemia-associated ineffective erythropoiesis, release erythroferrone and GDF15, which are believed to act on the liver to suppress hepcidin production (Figure 4). Consequently, dietary iron absorption increases and iron storage in macrophages is suppressed. Through both mechanisms, toxic free iron accumulates in the blood and various tissues including liver, pancreas, and heart. Several iron chelators are available to treat iron overload.85 However, these agents are somewhat non-specific, have toxicities, and may not be fully effective, prompting searches for additional therapies.
TMPRSS6 is a serine protease that cleaves hemojuvelin (HJV), a co-receptor for BMP6. Therefore, suppression of TMPRSS6 stimulates hepcidin production, which may be exploited therapeutically to reduce iron overload in hemoglobinopathy patients.89,90 Direct administration of hepcidin would produce similar beneficial effects, but this peptide is short lived in vivo.88 Thus, efforts are underway to produce hepcidin-like molecules with increased stability. One such group of molecules, termed minihepcidins, reduce iron overload in animal models.91,92 Ongoing studies of iron metabolism will likely identify additional approaches for treating iron overload (or deficiency). For example, further insights into the mechanisms of erythroid erythroferrone synthesis and action should identify new strategies to modulate hepcidin, which would be of particular benefit to patients with ineffective erythropoiesis, including those with β-thalassemia. Normalizing the body’s iron burden also appears to reduce ineffective erythropoiesis, at least in β-thalassemic mice models, suggesting an added benefit to inhibiting iron uptake.93,94
Manipulating the Genome to Treat Congenital Anemias
Congenital forms of anemia are prevalent worldwide, particularly the β-Hb disorders.1,2 Since β-thalassemia and SCD are well characterized disorders caused by monogenic mutations, they are amenable to cure by gene replacement therapy (Figure 5). In this approach, HSPCs are removed from the patient, modified to express a β-like globin protein in erythroid precursors and then reinfused.95 In principle, the modified HSCs will reconstitute the hematopoietic system, thereby producing normal, gene-corrected RBCs. The major challenges to this approach are requirements for genetic correction of a large proportion of HSCs, high-level engraftment of the corrected cells after transplantation, and sufficient production of the restored β-like globin protein.
Figure 5.

Gene therapy or editing to treat congenital forms of anemia. Potential corrective approaches for β thalassemia or sickle cell disease are shown. These approaches can also be used to correct various other severe anemias caused by monogenic mutations.
The earliest trials of hematopoietic cell gene therapy focused on immune disorders, such as severe combined immunodeficiency caused by defective interleukin-2 receptor γ chain.95 This disorder is tractable for gene therapy because its amelioration requires relatively modest expression levels of the deficient protein and gene-corrected lymphocytes have a strong survival advantage in vivo. In contrast, normal β–globin expression must be restored to massive levels in erythroid precursors for phenotypic correction of SCD or β-thalassemia.
Painstaking work over many years has led to the development of HIV1-based lentiviral vectors that can infect human HSCs and support high-level, erythroid specific expression of β-like globins.96,97 Most vectors encode a mutant version of β-globin that prevents sickle Hb polymerization or native γ-globin, which also has this property.95 Based on this work, a clinical trial was initiated for β-thalassemia and one treated patient became transfusion independent.98 However, a predominant hematopoietic clone was detected with aberrant expression of the HMGA2 gene near the site of lentiviral integration.98 This finding was concerning, given the leukemogenic effects of vector-induced activation of oncogenes noted in other gene therapy trials.95,99,100 New generations of vectors are now being designed to reduce the tendency of integrated viral vector to activate nearby genes and also to increase further the levels of β-like globin expression. Early clinical trial results in additional β-thalassemia patients treated with these improved vectors are promising (https://ash.confex.com/ash/2014/webprogram/Paper67341.html).
Innovative new strategies for gene therapy of SCD and β-thalassemia may circumvent the requirements for high-level expression of vector-encoded β-like globin. For example, suppression of BCL11A expression by short hairpin RNAs (shRNAs) induce γ-globin expression in adult human erythroblasts.56,60 This represents a potential gene therapy strategy for β-hemoglobinopathies, either alone or in combination with β-like globin gene replacement, although it may be important to suppress BCL11A selectively, since the protein is required for lymphopoiesis.53 Recently, a new strategy for stimulating HbF production in adult erythroblasts was demonstrated by expressing zinc fingers fused to protein dimerization domains, which bind specific DNA sequences and each other to promote physical interaction (“looping”) between γ-globin and an upstream enhancer termed the locus control region.101 Further work is necessary to ensure that this approach does not interfere with normal erythropoiesis or globin production in vivo.
The potential for using gene therapy to treat erythroid disorders extends beyond the β-hemoglobinopathies. Other monogenic erythroid diseases, particularly those where allogenic hematopoietic transplantation is used for treatment, may be ideal targets (Figure 5). For example, gene therapy has been investigated for DBA, but this would require separate vectors for each different form of the disease, which is caused by mutations in one of at least 12 RP genes.102 It was recently shown that DBA can also be caused by mutations in GATA1.14 Moreover, GATA1 mRNA translation was impaired selectively in erythroblasts of DBA patients with RP gene mutations and enforced expression of GATA1 in these cells partly rescued their defective erythropoiesis.13 Thus, viral vector encoded GATA1 may represent a more versatile gene therapy approach for multiple forms of DBA, both GATA1- and RP gene-mutated. Other erythroid diseases, such as congenital erythropoietic porphyria, where toxic porphyrin metabolites produced by erythroid precursors cause multisystemic illness, and red cell enzyme disorders, including severe forms of glucose 6-phosphate dehydrogenase and pyruvate kinase deficiency, also show promise for gene therapy correction.103–106
Until recently, all hematopoietic gene therapy approaches relied on the introduction of exogenous genetic material, mainly via viral vectors. Recent developments in the field of genome editing offer exciting new approaches (See Box on Gene Editing strategies). Both β-thalassemia and SCD mutations have been corrected by gene editing in pluripotent stem cells.107,108 It may also be possible to treat these disorders by using a similar approach to disrupt BCL11A expression in erythroid cells, thereby derepressing γ-globin synthesis. It is likely that our ability to modify resident genes within HSPCs will soon be refined to the point where gene editing can be used safely and efficiently to treat numerous blood disorders, including those that affect RBC production and/or survival.
Conclusions and Future Directions
Recent scientific discoveries have increased our understanding of erythropoiesis and associated disorders, elucidating exciting new therapeutic opportunities. These include the identification of GDF11 as a negative regulator of erythropoiesis and discoveries of numerous enzymes, cytokines, and hormones that regulate iron metabolism. In addition, technological advances, such as refined lentiviral vectors and gene editing approaches raise the exciting possibility that genetic defects causing anemia can be alleviated or repaired precisely for long-lasting disease correction.
However, significant challenges remain. Further improvements in stimulating erythropoiesis pharmacologically rely on better understanding the roles of positively and negatively acting cytokines on this process and the potential toxic effects of these molecules on bystander tissues. Manipulating newly identified transcriptional regulators of HbF production, such as BCL11A and histone-modifying enzymes, remains challenging and the full-spectrum of side effects from targeting these molecules is unknown.53 Importantly, much of our knowledge regarding human hematopoiesis, including erythroid development is based on murine models.4,109 While these approaches are valuable, fundamental differences between mouse and human erythropoiesis are becoming more apparent.110–112 Thus, new approaches for studying human hematopoiesis are essential for developing new therapeutics.4,109
How to translate and prioritize major technological and scientific advances into meaningful global therapies must be considered carefully. Technologically sophisticated, expensive approaches such as gene therapy or biologic drug infusions are not feasible for large numbers of patients who live in developing countries where SCD and β-thalassemia are common.1,2 Thus, balanced perspectives and approaches are required. Continued efforts must be applied to advance the best-available therapies into widespread use, such as hydroxyurea for SCD, and to develop improved oral agents that can be manufactured inexpensively. Moreover, even basic interventions such as regular transfusions and iron chelation can have a considerable impact on patients with anemia when used appropriately and regularly. Thus, efforts to improve the treatment of these disorders must balance the application of advanced technologies with current needs to deliver optimal therapies to the greatest numbers of patients. Close collaboration between basic scientists, clinical investigators, and front-line clinicians is required to achieve these goals. Because anemia contributes considerably to the worldwide burden of disease, it is likely that our rapidly advancing ability to treat these disorders will have a profound impact upon the lives of affected individuals and their families.
Acknowledgments
We apologize for being unable to cite numerous papers and studies in this field due to space limitations. We are grateful to Narla Mohandas, Art Nienhuis, and David Nathan for comments and suggestions on this review. V.G.S received support from grants from the National Institutes of Health (R01 DK103794, R21 HL120791, U01 HL117720), a March of Dimes Basil O’Connor Scholar Award, and an award from the Diamond-Blackfan Anemia Foundation. M.J.W. received support from the National Institutes of Health (R01 DK0923128, R01 DK61692, R01 HL088554). We are grateful to the many patients with anemia and their families, who have advanced the field by participating in clinical studies and advocating research.
References
- 1.Kassebaum NJ, et al. A systematic analysis of global anemia burden from 1990 to 2010. Blood. 2014;123:615–624. doi: 10.1182/blood-2013-06-508325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115:4331–4336. doi: 10.1182/blood-2010-01-251348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palis J. Primitive and definitive erythropoiesis in mammals. Front Physiol. 2014;5:3. doi: 10.3389/fphys.2014.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10:120–136. doi: 10.1016/j.stem.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto R, et al. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell. 2013;154:1112–1126. doi: 10.1016/j.cell.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Sanjuan-Pla A, et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature. 2013;502:232–236. doi: 10.1038/nature12495. [DOI] [PubMed] [Google Scholar]
- 7.Sun J, et al. Clonal dynamics of native haematopoiesis. Nature. 2014 doi: 10.1038/nature13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paulson RF. Targeting a new regulator of erythropoiesis to alleviate anemia. Nat Med. 2014;20:334–335. doi: 10.1038/nm.3524. [DOI] [PubMed] [Google Scholar]
- 9.Spivak JL. The anaemia of cancer: death by a thousand cuts. Nat Rev Cancer. 2005;5:543–555. doi: 10.1038/nrc1648. [DOI] [PubMed] [Google Scholar]
- 10.Dai CH, Price JO, Brunner T, Krantz SB. Fas ligand is present in human erythroid colony-forming cells and interacts with Fas induced by interferon gamma to produce erythroid cell apoptosis. Blood. 1998;91:1235–1242. [PubMed] [Google Scholar]
- 11.Orkin SH, Nathan DG. Nathan and Oski’s hematology of infancy and childhood. Saunders/Elsevier; Philadelphia: 2009. [Google Scholar]
- 12.Landowski M, et al. Novel deletion of RPL15 identified by array-comparative genomic hybridization in Diamond-Blackfan anemia. Hum Genet. 2013;132:1265–1274. doi: 10.1007/s00439-013-1326-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ludwig LS, et al. Altered translation of GATA1 in Diamond-Blackfan anemia. Nat Med. 2014;20:748–753. doi: 10.1038/nm.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sankaran VG, et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J Clin Invest. 2012;122:2439–2443. doi: 10.1172/JCI63597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Wijk R, van Solinge WW. The energy-less red blood cell is lost: erythrocyte enzyme abnormalities of glycolysis. Blood. 2005;106:4034–4042. doi: 10.1182/blood-2005-04-1622. [DOI] [PubMed] [Google Scholar]
- 16.Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372:1411–1426. doi: 10.1016/S0140-6736(08)61588-3. [DOI] [PubMed] [Google Scholar]
- 17.Sankaran VG, Nathan DG. Thalassemia: an overview of 50 years of clinical research. Hematol Oncol Clin North Am. 2010;24:1005–1020. doi: 10.1016/j.hoc.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 18.Khandros E, Thom CS, D’Souza J, Weiss MJ. Integrated protein quality-control pathways regulate free alpha-globin in murine beta-thalassemia. Blood. 2012;119:5265–5275. doi: 10.1182/blood-2011-12-397729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arlet JB, et al. HSP70 sequestration by free alpha-globin promotes ineffective erythropoiesis in beta-thalassaemia. Nature. 2014;514:242–246. doi: 10.1038/nature13614. [DOI] [PubMed] [Google Scholar]
- 20.Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376:2018–2031. doi: 10.1016/S0140-6736(10)61029-X. [DOI] [PubMed] [Google Scholar]
- 21.Manwani D, Frenette PS. Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Hematology Am Soc Hematol Educ Program. 2013;2013:362–369. doi: 10.1182/asheducation-2013.1.362. [DOI] [PubMed] [Google Scholar]
- 22.Gladwin MT, Kanias T, Kim-Shapiro DB. Hemolysis and cell-free hemoglobin drive an intrinsic mechanism for human disease. J Clin Invest. 2012;122:1205–1208. doi: 10.1172/JCI62972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gladwin MT, Ofori-Acquah SF. Erythroid DAMPs drive inflammation in SCD. Blood. 2014;123:3689–3690. doi: 10.1182/blood-2014-03-563874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chou ST. Transfusion therapy for sickle cell disease: a balancing act. Hematology Am Soc Hematol Educ Program. 2013;2013:439–446. doi: 10.1182/asheducation-2013.1.439. [DOI] [PubMed] [Google Scholar]
- 25.Locatelli F, et al. Outcome of patients with hemoglobinopathies given either cord blood or bone marrow transplantation from an HLA-identical sibling. Blood. 2013;122:1072–1078. doi: 10.1182/blood-2013-03-489112. [DOI] [PubMed] [Google Scholar]
- 26.Jelkmann W. Physiology and pharmacology of erythropoietin. Transfus Med Hemother. 2013;40:302–309. doi: 10.1159/000356193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macdougall IC. New anemia therapies: translating novel strategies from bench to bedside. Am J Kidney Dis. 2012;59:444–451. doi: 10.1053/j.ajkd.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 28.Malyszko J. New renal anemia drugs: is there really anything new on the horizon? Expert Opin Emerg Drugs. 2014;19:1–4. doi: 10.1517/14728214.2014.872239. [DOI] [PubMed] [Google Scholar]
- 29.Jha V, et al. Chronic kidney disease: global dimension and perspectives. Lancet. 2013;382:260–272. doi: 10.1016/S0140-6736(13)60687-X. [DOI] [PubMed] [Google Scholar]
- 30.Witmer CM. Hematologic manifestations of systemic disease (including iron deficiency, anemia of inflammation and DIC) Pediatr Clin North Am. 2013;60:1337–1348. doi: 10.1016/j.pcl.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 31.Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med. 2005;352:1011–1023. doi: 10.1056/NEJMra041809. [DOI] [PubMed] [Google Scholar]
- 32.Nemeth E, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113:1271–1276. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang CQ, Udupa KB, Lipschitz DA. Interferon-gamma exerts its negative regulatory effect primarily on the earliest stages of murine erythroid progenitor cell development. J Cell Physiol. 1995;162:134–138. doi: 10.1002/jcp.1041620116. [DOI] [PubMed] [Google Scholar]
- 34.Birgegard G, et al. Cancer-related anemia: pathogenesis, prevalence and treatment. Oncology. 2005;68 (Suppl 1):3–11. doi: 10.1159/000083128. [DOI] [PubMed] [Google Scholar]
- 35.Bejar R, Steensma DP. Recent developments in myelodysplastic syndromes. Blood. 2014;124:2793–2803. doi: 10.1182/blood-2014-04-522136. [DOI] [PubMed] [Google Scholar]
- 36.Wrighton NC, et al. Small peptides as potent mimetics of the protein hormone erythropoietin. Science. 1996;273:458–464. doi: 10.1126/science.273.5274.458. [DOI] [PubMed] [Google Scholar]
- 37.Macdougall IC, et al. Peginesatide for anemia in patients with chronic kidney disease not receiving dialysis. N Engl J Med. 2013;368:320–332. doi: 10.1056/NEJMoa1203166. [DOI] [PubMed] [Google Scholar]
- 38.Fishbane S, et al. Peginesatide in patients with anemia undergoing hemodialysis. N Engl J Med. 2013;368:307–319. doi: 10.1056/NEJMoa1203165. [DOI] [PubMed] [Google Scholar]
- 39.Bennett CL, Jacob S, Hymes J, Usvyat LA, Maddux FW. Anaphylaxis and hypotension after administration of peginesatide. N Engl J Med. 2014;370:2055–2056. doi: 10.1056/NEJMc1400883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 41.Raje N, Vallet S. Sotatercept, a soluble activin receptor type 2A IgG-Fc fusion protein for the treatment of anemia and bone loss. Curr Opin Mol Ther. 2010;12:586–597. [PubMed] [Google Scholar]
- 42.Sherman ML, et al. Multiple-dose, safety, pharmacokinetic, and pharmacodynamic study of sotatercept (ActRIIA-IgG1), a novel erythropoietic agent, in healthy postmenopausal women. J Clin Pharmacol. 2013;53:1121–1130. doi: 10.1002/jcph.160. [DOI] [PubMed] [Google Scholar]
- 43.Dussiot M, et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in beta-thalassemia. Nat Med. 2014;20:398–407. doi: 10.1038/nm.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suragani RN, et al. Transforming growth factor-beta superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. 2014;20:408–414. doi: 10.1038/nm.3512. [DOI] [PubMed] [Google Scholar]
- 45.Loffredo FS, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. 2013;153:828–839. doi: 10.1016/j.cell.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sinha M, et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. 2014;344:649–652. doi: 10.1126/science.1251152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katsimpardi L, et al. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science. 2014;344:630–634. doi: 10.1126/science.1251141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Villeda SA, et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat Med. 2014;20:659–663. doi: 10.1038/nm.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coulon S, et al. Polymeric IgA1 controls erythroblast proliferation and accelerates erythropoiesis recovery in anemia. Nat Med. 2011;17:1456–1465. doi: 10.1038/nm.2462. [DOI] [PubMed] [Google Scholar]
- 50.Richardson CL, et al. Isocitrate ameliorates anemia by suppressing the erythroid iron restriction response. J Clin Invest. 2013;123:3614–3623. doi: 10.1172/JCI68487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sankaran VG, Orkin SH. The switch from fetal to adult hemoglobin. Cold Spring Harb Perspect Med. 2013;3:a011643. doi: 10.1101/cshperspect.a011643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sankaran VG, et al. A functional element necessary for fetal hemoglobin silencing. N Engl J Med. 2011;365:807–814. doi: 10.1056/NEJMoa1103070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sankaran VG. Targeted therapeutic strategies for fetal hemoglobin induction. Hematology Am Soc Hematol Educ Program. 2011;2011:459–465. doi: 10.1182/asheducation-2011.1.459. [DOI] [PubMed] [Google Scholar]
- 54.Musallam KM, et al. Fetal hemoglobin levels and morbidity in untransfused patients with beta-thalassemia intermedia. Blood. 2012;119:364–367. doi: 10.1182/blood-2011-09-382408. [DOI] [PubMed] [Google Scholar]
- 55.Platt OS. Hydroxyurea for the treatment of sickle cell anemia. N Engl J Med. 2008;358:1362–1369. doi: 10.1056/NEJMct0708272. [DOI] [PubMed] [Google Scholar]
- 56.Sankaran VG, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839–1842. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- 57.Bauer DE, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342:253–257. doi: 10.1126/science.1242088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu J, et al. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science. 2011;334:993–996. doi: 10.1126/science.1211053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sankaran VG, et al. Developmental and species-divergent globin switching are driven by BCL11A. Nature. 2009;460:1093–1097. doi: 10.1038/nature08243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wilber A, et al. Therapeutic levels of fetal hemoglobin in erythroid progeny of beta-thalassemic CD34+ cells after lentiviral vector-mediated gene transfer. Blood. 2011;117:2817–2826. doi: 10.1182/blood-2010-08-300723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu J, et al. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc Natl Acad Sci U S A. 2013;110:6518–6523. doi: 10.1073/pnas.1303976110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Suzuki M, Yamamoto M, Engel JD. Fetal globin gene repressors as drug targets for molecular therapies to treat the beta-globinopathies. Mol Cell Biol. 2014;34:3560–3569. doi: 10.1128/MCB.00714-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sankaran VG, et al. MicroRNA-15a and -16-1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proc Natl Acad Sci U S A. 2011;108:1519–1524. doi: 10.1073/pnas.1018384108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Galarneau G, et al. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet. 2010;42:1049–1051. doi: 10.1038/ng.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sankaran VG, et al. Rare complete loss of function provides insight into a pleiotropic genome-wide association study locus. Blood. 2013;122:3845–3847. doi: 10.1182/blood-2013-09-528315. [DOI] [PubMed] [Google Scholar]
- 66.Stadhouders R, et al. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. J Clin Invest. 2014;124:1699–1710. doi: 10.1172/JCI71520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zuber J, et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011;25:1628–1640. doi: 10.1101/gad.17269211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Borg J, et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet. 2010;42:801–805. doi: 10.1038/ng.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Giardine B, et al. Systematic documentation and analysis of human genetic variation in hemoglobinopathies using the microattribution approach. Nat Genet. 2011;43:295–301. doi: 10.1038/ng.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arnaud L, et al. A dominant mutation in the gene encoding the erythroid transcription factor KLF1 causes a congenital dyserythropoietic anemia. Am J Hum Genet. 2010;87:721–727. doi: 10.1016/j.ajhg.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Campbell AD, et al. Forced TR2/TR4 expression in sickle cell disease mice confers enhanced fetal hemoglobin synthesis and alleviated disease phenotypes. Proc Natl Acad Sci U S A. 2011;108:18808–18813. doi: 10.1073/pnas.1104964108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bradner JE, et al. Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proc Natl Acad Sci U S A. 2010;107:12617–12622. doi: 10.1073/pnas.1006774107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi L, Cui S, Engel JD, Tanabe O. Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nat Med. 2013;19:291–294. doi: 10.1038/nm.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Libani IV, et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in beta-thalassemia. Blood. 2008;112:875–885. doi: 10.1182/blood-2007-12-126938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Breda L, Rivella S. Modulators of erythropoiesis: emerging therapies for hemoglobinopathies and disorders of red cell production. Hematol Oncol Clin North Am. 2014;28:375–386. doi: 10.1016/j.hoc.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gutsaeva DR, et al. Inhibition of cell adhesion by anti-P-selectin aptamer: a new potential therapeutic agent for sickle cell disease. Blood. 2011;117:727–735. doi: 10.1182/blood-2010-05-285718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chang J, et al. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood. 2010;116:1779–1786. doi: 10.1182/blood-2009-12-260513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chang J, Shi PA, Chiang EY, Frenette PS. Intravenous immunoglobulins reverse acute vaso-occlusive crises in sickle cell mice through rapid inhibition of neutrophil adhesion. Blood. 2008;111:915–923. doi: 10.1182/blood-2007-04-084061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jang JE, Hod EA, Spitalnik SL, Frenette PS. CXCL1 and its receptor, CXCR2, mediate murine sickle cell vaso-occlusion during hemolytic transfusion reactions. J Clin Invest. 2011;121:1397–1401. doi: 10.1172/JCI45336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wallace KL, et al. NKT cells mediate pulmonary inflammation and dysfunction in murine sickle cell disease through production of IFN-gamma and CXCR3 chemokines. Blood. 2009;114:667–676. doi: 10.1182/blood-2009-02-205492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Field JJ, et al. Sickle cell vaso-occlusion causes activation of iNKT cells that is decreased by the adenosine A2A receptor agonist regadenoson. Blood. 2013;121:3329–3334. doi: 10.1182/blood-2012-11-465963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Embury SH, Garcia JF, Mohandas N, Pennathur-Das R, Clark MR. Effects of oxygen inhalation on endogenous erythropoietin kinetics, erythropoiesis, and properties of blood cells in sickle-cell anemia. N Engl J Med. 1984;311:291–295. doi: 10.1056/NEJM198408023110504. [DOI] [PubMed] [Google Scholar]
- 83.Abdulmalik O, et al. 5-hydroxymethyl-2-furfural modifies intracellular sickle haemoglobin and inhibits sickling of red blood cells. Br J Haematol. 2005;128:552–561. doi: 10.1111/j.1365-2141.2004.05332.x. [DOI] [PubMed] [Google Scholar]
- 84.Safo MK, Kato GJ. Therapeutic strategies to alter the oxygen affinity of sickle hemoglobin. Hematol Oncol Clin North Am. 2014;28:217–231. doi: 10.1016/j.hoc.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Finberg KE. Striking the target in iron overload disorders. J Clin Invest. 2013;123:1424–1427. doi: 10.1172/JCI68889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tanno T, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13:1096–1101. doi: 10.1038/nm1629. [DOI] [PubMed] [Google Scholar]
- 87.Kautz L, et al. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46:678–684. doi: 10.1038/ng.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ganz T. Systemic iron homeostasis. Physiol Rev. 2013;93:1721–1741. doi: 10.1152/physrev.00008.2013. [DOI] [PubMed] [Google Scholar]
- 89.Schmidt PJ, et al. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(−/−) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood. 2013;121:1200–1208. doi: 10.1182/blood-2012-09-453977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guo S, et al. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J Clin Invest. 2013;123:1531–1541. doi: 10.1172/JCI66969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Preza GC, et al. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J Clin Invest. 2011;121:4880–4888. doi: 10.1172/JCI57693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ramos E, et al. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood. 2012;120:3829–3836. doi: 10.1182/blood-2012-07-440743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gardenghi S, et al. Hepcidin as a therapeutic tool to limit iron overload and improve anemia in beta-thalassemic mice. J Clin Invest. 2010;120:4466–4477. doi: 10.1172/JCI41717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li H, et al. Transferrin therapy ameliorates disease in beta-thalassemic mice. Nat Med. 2010;16:177–182. doi: 10.1038/nm.2073. [DOI] [PubMed] [Google Scholar]
- 95.Kohn DB, Pai SY, Sadelain M. Gene therapy through autologous transplantation of gene-modified hematopoietic stem cells. Biol Blood Marrow Transplant. 2013;19:S64–69. doi: 10.1016/j.bbmt.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 96.May C, et al. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature. 2000;406:82–86. doi: 10.1038/35017565. [DOI] [PubMed] [Google Scholar]
- 97.Pawliuk R, et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science. 2001;294:2368–2371. doi: 10.1126/science.1065806. [DOI] [PubMed] [Google Scholar]
- 98.Cavazzana-Calvo M, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hacein-Bey-Abina S, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stein S, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 101.Deng W, et al. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell. 2014;158:849–860. doi: 10.1016/j.cell.2014.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Flygare J, Olsson K, Richter J, Karlsson S. Gene therapy of Diamond Blackfan anemia CD34(+) cells leads to improved erythroid development and engraftment following transplantation. Exp Hematol. 2008;36:1428–1435. doi: 10.1016/j.exphem.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 103.Bedel A, et al. Metabolic correction of congenital erythropoietic porphyria with iPSCs free of reprogramming factors. Am J Hum Genet. 2012;91:109–121. doi: 10.1016/j.ajhg.2012.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Robert-Richard E, et al. Effective gene therapy of mice with congenital erythropoietic porphyria is facilitated by a survival advantage of corrected erythroid cells. Am J Hum Genet. 2008;82:113–124. doi: 10.1016/j.ajhg.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rovira A, et al. Stable in vivo expression of glucose-6-phosphate dehydrogenase (G6PD) and rescue of G6PD deficiency in stem cells by gene transfer. Blood. 2000;96:4111–4117. [PubMed] [Google Scholar]
- 106.Meza NW, et al. Rescue of pyruvate kinase deficiency in mice by gene therapy using the human isoenzyme. Mol Ther. 2009;17:2000–2009. doi: 10.1038/mt.2009.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xie F, et al. Seamless gene correction of beta-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014;24:1526–1533. doi: 10.1101/gr.173427.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sun N, Zhao H. Seamless correction of the sickle cell disease mutation of the HBB gene in human induced pluripotent stem cells using TALENs. Biotechnol Bioeng. 2014;111:1048–1053. doi: 10.1002/bit.25018. [DOI] [PubMed] [Google Scholar]
- 109.Sankaran VG, Orkin SH. Genome-wide association studies of hematologic phenotypes: a window into human hematopoiesis. Curr Opin Genet Dev. 2013;23:339–344. doi: 10.1016/j.gde.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ulirsch JC, et al. Altered chromatin occupancy of master regulators underlies evolutionary divergence in the transcriptional landscape of erythroid differentiation. PLoS Genet. 2014;10:e1004890. doi: 10.1371/journal.pgen.1004890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pishesha N, et al. Transcriptional divergence and conservation of human and mouse erythropoiesis. Proc Natl Acad Sci U S A. 2014;111:4103–4108. doi: 10.1073/pnas.1401598111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.An X, et al. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood. 2014;123:3466–3477. doi: 10.1182/blood-2014-01-548305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bunn HF. Erythropoietin. Cold Spring Harb Perspect Med. 2013;3:a011619. doi: 10.1101/cshperspect.a011619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gupta RM, Musunuru K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J Clin Invest. 2014;124:4154–4161. doi: 10.1172/JCI72992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Holt N, et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28:839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lombardo A, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 118.Genovese P, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]