

ABSTRACT
Inositol pyrophosphates (PP-IPs) comprising inositol, phosphate, and pyrophosphate (PP) are essential for multiple functions in eukaryotes. Their role in fungal pathogens has never been addressed. Cryptococcus neoformans is a model pathogenic fungus causing life-threatening meningoencephalitis. We investigate the cryptococcal kinases responsible for the production of PP-IPs (IP7/IP8) and the hierarchy of PP-IP importance in pathogenicity. Using gene deletion and inositol polyphosphate profiling, we identified Kcs1 as the major IP6 kinase (producing IP7) and Asp1 as an IP7 kinase (producing IP8). We show that Kcs1-derived IP7 is the most crucial PP-IP for cryptococcal drug susceptibility and the production of virulence determinants. In particular, Kcs1 kinase activity is essential for cryptococcal infection of mouse lungs, as reduced fungal burdens were observed in the absence of Kcs1 or when Kcs1 was catalytically inactive. Transcriptome and carbon source utilization analysis suggested that compromised growth of the KCS1 deletion strain (Δkcs1 mutant) in the low-glucose environment of the host lung is due to its inability to utilize alternative carbon sources. Despite this metabolic defect, the Δkcs1 mutant established persistent, low-level asymptomatic pulmonary infection but failed to elicit a strong immune response in vivo and in vitro and was not readily phagocytosed by primary or immortalized monocytes. Reduced recognition of the Δkcs1 cells by monocytes correlated with reduced exposure of mannoproteins on the Δkcs1 mutant cell surface. We conclude that IP7 is essential for fungal metabolic adaptation to the host environment, immune recognition, and pathogenicity.
IMPORTANCE
Cryptococcus neoformans is responsible for 1 million cases of AIDS-associated meningitis and ~600,000 deaths annually. Understanding cellular pathways responsible for pathogenicity might have an impact on new drug development. We characterized the inositol polyphosphate kinases Kcs1 and Asp1, which are predicted to catalyze the production of inositol pyrophosphates containing one or two diphosphate moieties (PP-IPs). Using gene deletion analysis and inositol polyphosphate profiling, we confirmed that Kcs1 and Asp1 are major IP6 and IP7 kinases, respectively. Kcs1-derived IP7, but not Asp1-derived IP8, is crucial for pathogenicity. Global expression profiling and carbon source utilization testing suggest that IP7-deficient cryptococci cannot adapt their metabolism to allow growth in the glucose-poor environment of the host lung, and consequently, fungal burdens are significantly reduced. Persistent asymptomatic Δkcs1 mutant infection correlated with decreased mannoprotein exposure on the Δkcs1 mutant surface and reduced phagocytosis. We conclude that IP7 is crucial for the metabolic adaptation of C. neoformans to the host environment and for pathogenicity.
INTRODUCTION
Cryptococcus neoformans var. grubii is a basidiomycetous opportunistic human fungal pathogen of global significance that commonly infects immunocompromised hosts, including those with AIDS. Cryptococcus neoformans is responsible for ~1 million cases of HIV-associated cryptococcal meningitis and ~600,000 deaths per year worldwide, predominantly in sub-Saharan Africa (1). Cryptococcal lung infection within an immunocompromised host can result from the inhalation of infectious propagules or the reactivation of a latent infection (2). Dissemination from the lungs to the central nervous system (CNS) occurs via the blood or lymphatic system (2). Survival of Cryptococcus neoformans within the adverse environment of the host depends on the coordinated functions of multiple stress-related signaling pathways that regulate the expression of a suite of virulence traits and remodeling of the fungal cell wall (3).
Cryptococcal virulence determinants include the ability to grow at host physiological temperature (37°C) and the production of melanin and polysaccharide capsule (for a review, see references 4, to ,7). Melanin protects cells from damage caused by reactive oxygen species, which macrophages produce as part of their antimicrobial defense (8). It is deposited in the cell wall via the catalytic activity of the cell wall-associated enzyme laccase (9). The capsule surrounds the cell wall and is comprised of glucuronoxylomannan (GXM) (90%), galactoxylomannan (GalXM), and highly mannosylated and immunogenic mannoproteins (1%) (10, 11). It protects cryptococci within the host by having a suppressive effect on the host proinflammatory response, by inhibiting the antigen-presenting capability of monocytes, and by rendering cryptococcal cells resistant to phagocytosis by shielding cell wall glucans and mannoproteins from recognition by macrophages and dendritic cells (7, 12, 13). Many mannoproteins are tethered to the cell wall by a glycosylphosphatidylinositol (GPI) anchor (14) and are localized predominantly in the innermost layer of the capsule. However, they can become detached from the anchor and secreted through the capsular layer (15–19). Several of these mannoproteins are potential vaccine candidates against cryptococcal infection (14, 20–22).
The cryptococcal enzyme phospholipase C1 (Plc1) is a component of a stress-related signaling pathway governing the production of a suite of virulence traits, which are compromised when the Plc1-encoding gene is deleted (23). We demonstrated that recombinant Plc1 hydrolyzes the membrane lipid, phosphatidylinositol-bisphosphate (PIP2) to produce inositol 1,4,5-trisphosphate (IP3) (23) and that IP3 is a substrate for cryptococcal Arg1, an inositol polyphosphate kinase (IPK) (24). As with the Δplc1 mutant, multiple cryptococcal virulence traits were compromised in the Δarg1 mutant (24), suggesting that the absence of more complex inositol polyphosphates (IPs) and/or inositol pyrophosphates (PP-IPs) downstream of Arg1 contributes to the reduced virulence observed for both mutants.
PP-IPs, of which IP7 and IP8 are the most physiologically relevant, differ from IPs in that they possess a diphosphate on one or more carbons of the inositol ring. They regulate diverse cellular functions in eukaryotes (25–32). In the nonpathogenic yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe, different IP7 isoforms are essential for growth, vacuole biogenesis, salt tolerance, endocytic trafficking, the function of the cortical actin cytoskeleton, surface adhesion, and dimorphic switching in S. pombe (26, 33–37). PP-IPs affect cellular function at both the transcriptional and the posttranslational level via mechanisms that remain to be fully understood. These molecules modulate protein function directly through binding or indirectly by phosphorylating prephosphorylated proteins via a nonenzymatic reaction (38, 39). IP7 isoforms can also compete with membrane PI(3,4,5)P3 for binding to pleckstrin homology domain-containing proteins, affecting the association of these proteins with the plasma membrane, as observed in Dictyostelium discoideum (25). Recently, Worley et al. (40) showed that PP-IPs act in parallel with the TORC1 pathway to control the activity of the class I histone deacetylase Rpd3L and elicit global transcriptional changes in response to different types of stress (40). PP-IPs are therefore central regulators of eukaryotic cellular function, and targeting their synthesis holds promise for the development of new antifungal therapies. However, the kinases directly responsible for the synthesis of PP-IPs in fungal pathogens have not been determined, and the direct role of PP-IPs in regulating fungal pathogenicity remains unclear.
In the present study, we identified two PP-IP-producing kinases, Kcs1 and Asp1 (homolog of S. cerevisiae Vip1) in C. neoformans, characterized their enzymatic activities, and investigated the role of their products in pathogenicity and cellular function. Since IP7 was found to be the most crucial PP-IP for cryptococcal pathogenicity, we further investigated its role in cellular function using gene expression and carbon source utilization studies.
RESULTS
Kcs1 is an IP6 kinase in C. neoformans.
We identified a putative IP6 kinase-encoding gene (CNAG_02897/KCS1) in C. neoformans var. grubii (strain H99) using Kcs1 from S. cerevisiae in a homology search. The protein encoded by KCS1 is predicted to be 1,249 amino acids long and to contain an IPK domain at the C terminus. C. neoformans Kcs1 (CnKcs1) has 53% similarity to S. cerevisiae Kcs1 (ScKcs1) at the amino acid level. While the long N-terminal sequences are highly varied among fungal Kcs1 homologs, the C-terminal IPK domains are more conserved: the CnKcs1 and ScKcs1 IPK domains share 50% similarity. Phylogenetic analysis of the fungal Kcs1 homologs using full Kcs1 sequences or IPK domains demonstrates that CnKcs1 is clustered with other Kcs1 homologs of Basidiomycetes (Fig. 1A).
FIG 1 .

Kcs1 is an IP6 kinase in C. neoformans. (A) Phylogenetic analysis of Kcs1 homologs in fungi. Full-length C. neoformans Kcs1 protein (indicated by the arrow) was used to generate the phylogenetic tree. Cg, Candida glabrata XP_445605; Sc, Saccharomyces cerevisiae Q12494; Af, Aspergillus fumigatus XP_753330; An, Aspergillus nidulans XP_664484; Ci, Coccidioides immitis EAS27460.2; Um, Ustilago maydis XP_757030.1; Cc, Coprinopsis cinerea XP_001836888; Pc, Phanerochaete carnosa EKM59468; Ta, Trichosporon asahii EKD02283; Cn, Cryptococcus neoformans AFR93794. Numbers represent bootstrap values. (B) Analysis of the inositol polyphosphate profile of the WT, Δkcs1, and Δarg1 strains. The inset represents IP profiles of the reconstituted Δkcs1 strains: the Δkcs1::KCS1 strain (expressing native Kcs1) and the Δkcs1::KCS1AAA strain (expressing kinase-inactive Kcs1). Intracellular IPs were labeled with [3H]myo-inositol, extracted, and analyzed by anion-exchange HPLC. The elution time for different IP species was determined using standards. The position of each IP species is indicated.
Next, we assessed the role of Kcs1 in cryptococcal IP metabolism by creating a KCS1 deletion strain (Δkcs1 mutant) and comparing its IP profile with that of the wild type (WT) and Δarg1 mutant (see Text S1 and Fig. S1A and B in the supplemental material; Fig. 1B). Intracellular IPs in the WT and mutant strains were radiolabeled by adding [3H]myo-inositol to the culture medium, extracted from the cells and fractionated using anion-exchange high-performance liquid chromatography (HPLC). As in S. cerevisiae and mammalian cells, the most abundant IP species in the WT was IP6. Accumulation of IP3 in the C. neoformans Δarg1 mutant was consistent with the findings of our previous study, where raised levels of IP3 were detected using a radiometric IP3 binding assay (24). The Δarg1 mutant was also deficient in more highly phosphorylated IP species, including the predicted products of Arg1 (IP4 and IP5) and IP6 to IP8 (IP6–8). In the C. neoformans Δkcs1 mutant, the amount of IP3–6 was similar to that in the WT. However, the more highly phosphorylated PP-IP5 (IP7) and PP2-IP4 (IP8) were not detected, consistent with Kcs1 functioning as the major IP6 kinase in C. neoformans. These results also establish that Arg1 and Kcs1 are components of the same metabolic pathway.
The mammalian IP6 kinase IP6K1 has been reported to perform physiological functions that are independent of its IP6 kinase activity (41). Furthermore, CnKcs1 and ScKcs1 contain an extended N terminus, which may have additional physiological functions. To distinguish IP6 kinase-dependent and -independent effects, we created the Δkcs1::KCS1AAA strain, in which the sequence encoding conserved amino acids in the Kcs1 catalytic domain (1032 SLL1034) was altered to encode AAA, resulting in the expression of kinase-inactive Kcs1 (Text S1; Fig. S1A. This same substitution abrogated Kcs1 catalytic activity in S. cerevisiae (34). The Δkcs1::KCS1AAA strain was created by ectopic insertion of the mutagenized KCS1 gene with its native promoter and terminator into the genome of the Δkcs1 mutant. A control strain (the Δkcs1::KCS1 strain) was similarly created by inserting the native KCS1 genomic fragment ectopically into the genome of the Δkcs1 mutant. Integration of KCS1AAA and KCS1 into a single genomic location in each strain was confirmed by Southern blotting (Fig. S1C). The levels of expression of ectopically integrated KCS1 and KCS1AAA in the respective reconstituted strains were comparable to the expression of native KCS1 in the WT (Fig. S1D). Like the Δkcs1 mutant, the Δkcs1::KCS1AAA strain did not produce IP7–8 (Fig. 1B, inset). The IP profile of the Δkcs1::KCS1 strain was similar to that of the WT (Fig. 1B, inset). The Δkcs1 and Δkcs1::KCS1AAA strains were subsequently used to identify phenotypes that were specifically associated with the absence of Kcs1 catalytic activity.
Loss of IP6 kinase activity leads to growth and cell wall integrity defects.
The ability of C. neoformans to colonize host tissue is dependent on its ability to grow at mammalian physiological temperatures. Since PP-IP products generated by ScKcs1 play a role in the response to heat stress, the growth of the WT, Δkcs1, and Δkcs1::KCS1AAA strains was assessed at 30°C, 37°C, and 39°C using a drop dilution test. Like that of the ScΔkcs1 mutant, the growth of the Δkcs1 and Δkcs1::KCS1AAA strains at all three temperatures was reduced compared to that of the WT and Δkcs1::KCS1 strain (Fig. 2).
FIG 2 .
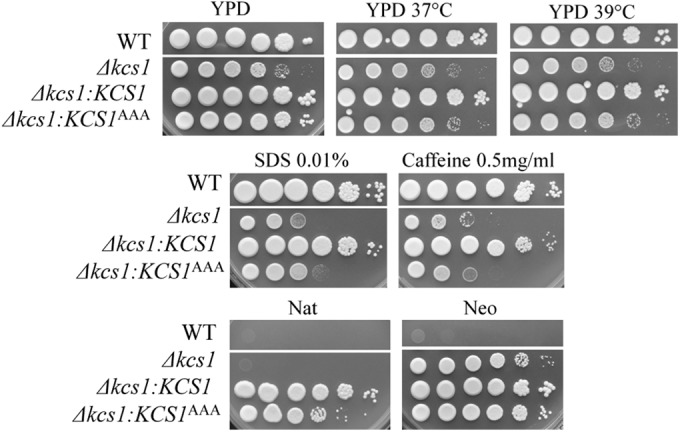
Spot dilution assays demonstrating a cell wall defect in the Δkcs1 and kinase-inactive Δkcs1::KCS1AAA strains but not in the kinase-active Δkcs1::KCS1 strain. The strains were serially diluted 10-fold (106 to 10 cells/spot from left to right) and spotted onto the test plates indicated. The dual nourseothricin (Nat) and neomycin (Neo) resistance of the Δkcs1::KCS1 and Δkcs1::KCS1AAA strains confirms that integration of the KCS1 construct (providing Nat resistance) occurred in the Δkcs1 mutant strain background (Neo resistance).
We also investigated the integrity of the cell wall in the Δkcs1 and Δkcs1::KCS1AAA mutants and their ability to produce mating filaments. Cell wall integrity was assessed by comparing the extent of growth in the presence of cell wall-perturbing agents. Figure 2 demonstrates that the growth of the Δkcs1 and Δkcs1::KCS1AAA strains was similarly reduced in the presence of SDS and caffeine, compared with that of the WT and Δkcs1::KCS1 strains, consistent with a cell wall integrity defect. The Δkcs1 and Δkcs1::KCS1AAA (mating type α) strains also produced fewer mating filaments following a unilateral cross with KN99 (mating type a), while filament production was restored to WT levels in the Δkcs1::KCS1 strain (Fig. S2). Since the cell wall integrity and mating defects persisted in the Δkcs1::KCS1AAA strain (Fig. 2; Fig. S2), it can be concluded that the PP-IPs produced by Kcs1 are essential for cell wall integrity and mating.
The Δkcs1 mutant is hypersusceptible to antifungal agents.
To investigate whether disruption of PP-IP synthesis alters the susceptibility of C. neoformans to antifungal drugs, the extent of growth of the WT and the Δkcs1 mutant was compared in the presence of amphotericin B, flucytosine, echinocandins, and a range of azoles (Table S1). The Δkcs1 mutant was 8 times more susceptible than the WT to posaconazole, voriconazole, itraconazole, and fluconazole and twice as susceptible to flucytosine and amphotericin B but remained resistant to the echinocandins, caspofungin, micafungin, and anidulafungin. The Δarg1 mutant demonstrated a similar drug susceptibility profile (Table S1 and Text S1).
The Δkcs1 mutant has defective melanin production and an increased capsule size.
Next, we assessed the ability of the Kcs1-deficient strains to produce melanin. Melanin protects cryptococci from a variety of stresses, including macrophage-derived oxidative stress and UV light. The Δkcs1 and Δkcs1::KCS1AAA strains produced less melanin than the WT when grown on minimal medium (MM) without glucose and supplemented with l-3,4-dihydroxyphenylalanine (l-DOPA) (Fig. 3A). The extent of melanization in the Δkcs1::KCS1 strain was similar to that of the WT. Since the cell wall-associated enzyme laccase 1 catalyzes melanin synthesis in C. neoformans, allowing it to be deposited in the cell wall, the extracellular laccase activity associated with intact fungal cells was also measured. The results in Fig. 3B demonstrate that the reduced melanization of the Δkcs1 and Δkcs1::KCS1AAA strains (Fig. 3A) coincided with reduced extracellular laccase activity as measured by oxidation of the laccase substrate 2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid) (ABTS). Once again, laccase activity was restored to WT levels in the Δkcs1::KCS1 strain. To determine whether reduced laccase activity in the Kcs1-deficient strains is due to the absence of LAC1 gene induction under melanin-inducing conditions, we measured LAC1 expression in the WT, Δkcs1, Δkcs1::KCS1, and Δkcs1::KCS1AAA strains following 5 h of growth in MM without glucose. Figure 3C demonstrates that the LAC1 expression profile in all strains is similar to that observed for laccase activity and melanin production. These data indicate that Kcs1-derived PP-IPs are essential for induction of LAC1.
FIG 3 .

Melanization, extracellular laccase activity, and LAC1 gene expression are reduced in the Δkcs1 and kinase-dead Δkcs1::KCS1AAA strains. (A) Cumulative melanization in the WT, Δkcs1, Δkcs1::KCS1AAA, and Δkcs1::KCS1 strains was compared by spotting similar numbers of cells (5 µl; OD600 = 3) onto MM plates lacking glucose (laccase inducing) and containing the laccase substrate l-DOPA, and incubating them for 3 days at 30°C. (B) Extracellular laccase activity was quantified spectrophotometrically by measuring the oxidation of the laccase substrate ABTS (ABTSox) by fungal cells following 4 h of growth in MM broth without glucose at 30°C. (C) LAC1 gene expression was quantified in cultures grown for 5 h in MM broth without glucose by real-time PCR. ACT1 was used for normalization of expression. The differences between different strains in panels B and C are statistically significant (P < 0.05), as determined by Student’s t test.
Capsule production was also assessed following the growth of fungal strains on Sabouraud (SAB) agar. Notably, colonies formed by the Kcs1-deficient strains had a mucoid (glossy) appearance, in contrast to the dull surface of the WT (Fig. 4A). The capsules produced by the Δkcs1 and Δkcs1::KCS1AAA strains were on average larger than those produced by the WT (Fig. 4B and C). Colony appearance and capsule size were partially restored in the Δkcs1::KCS1 strain.
FIG 4 .

Kcs1 deficiency is associated with a mucoid (glossy) colony appearance (A) and enlarged capsules (B and C). (A) Fungal strains were grown on Sabouraud (SAB) agar plates at ambient temperature for 3 days. (B) The capsules of the SAB agar-grown cells were visualized by negative staining with India ink. (C) Capsule width was measured in at least 30 cells. The increases in capsule sizes observed for the Δkcs1 and Δkcs1::KCS1AAA strains, relative to the WT and Δkcs1::KCS1 strains, respectively, are statistically significant (*, P < 0.05), as determined using a Tukey-Kramer multiple-comparison test. Error bars indicate standard errors.
Phenotypic defects in the Δkcs1 mutant are caused by the absence of IP7, not IP8.
Kcs1 is essential for the production of the PP-IPs IP7 and IP8, as neither species was detected in the Δkcs1 and Δkcs1::KCS1AAA strains (Fig. 1B). To determine the relative contribution of IP8 to the Kcs1-dependent traits, we identified a homolog of the IP6–7 kinase of S. cerevisiae (ScVip1) in C. neoformans, designated Asp1. Asp1 protein (encoded by CNAG_02161) is predicted to be 1,117 amino acids long and is 33% identical and 24% similar to ScVip1. Like ScVip1 and Asp1 in Schizosaccharomyces pombe, Asp1 of C. neoformans contains an N-terminal ATP grasp domain responsible for the kinase activity and a C-terminal histidine acid phosphatase domain of unknown function. C. neoformans ASP1 was deleted as described for KCS1, creating the Δasp1 mutant. IP7 accumulated and PP2-IP4 (IP8) was not detected in the Δasp1 mutant (Fig. 5), consistent with C. neoformans Asp1 functioning as an IP7 kinase, as reported for S. cerevisiae Vip1 (42, 43). A reduction in IP5 and IP6 was also noted in the Δasp1 strain (Fig. 5). The growth of the Δasp1 mutant was assessed under the same conditions as those for the Δksc1 mutant, including 37°C, in the presence of cell wall-perturbing agents, and on mating agar (Fig. S3). The Δasp1 strain was also assessed for the production of melanin and capsule. Under all growth conditions, the Δasp1 strain exhibited a phenotype similar to that of the WT, indicating that Kcs1-derived PP-IP5 (IP7), rather than Asp1-derived PP2-IP4 (IP8), is the most crucial PP-IP for cryptococcal cellular function.
FIG 5 .
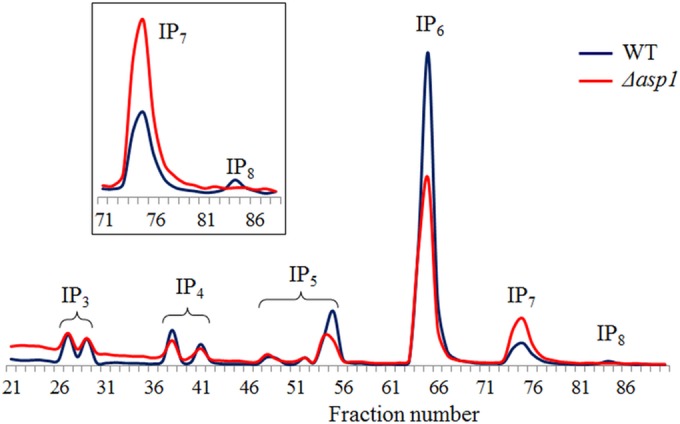
Inositol polyphosphate profile of the Δasp1 mutant demonstrating that Asp1 is an IP7 kinase. The intracellular IPs in the WT and the Δasp1 mutant were labeled with [3H]myo-inositol, extracted, and analyzed by anion-exchange HPLC. The elution times for the different IP species indicated were determined using standards. Several IP3, IP4, and IP5 isoforms are discernible in this trace. The inset represents a magnified section of the main graph and demonstrates the accumulation of IP7 and the reduction of IP8 in the Δasp1 strain.
The Δkcs1 mutant is less pathogenic than the WT in animal models.
The pathogenicity of the Δkcs1 mutant was compared to that of the WT in an invertebrate model, the greater wax moth (Galleria mellonella), and a mouse inhalation model. Larvae of Galleria mellonella were infected by inoculation of fungi into the lower prolegs and incubated at 30°C. The average survival of Δkcs1 mutant-infected larvae was 14 days, compared with 5 days for WT-infected larvae (Fig. 6A). Mice were inoculated via the nares. The median survival time of mice infected with the WT was 14 days. However, no mice succumbed to infection with the Δkcs1 mutant for up to 50 days postinoculation (Fig. 6B). Virulence was almost restored by the introduction of KCS1 into the Δkcs1 mutant, with the median survival time of Δkcs1::KCS1 strain-infected mice being 18 days. Despite the avirulent phenotype, Δkcs1 cells were detected in mouse lungs (3 × 103 to 90 × 103 CFU per total lung tissue) over the 50-day postinfection period (Fig. 6C). The Δkcs1 mutant infection burden was ~8-fold lower than that of the WT at 3 days postinfection. However, the difference between the WT and the Δkcs1 mutant pulmonary burdens increased to ~140-fold at the onset of the inflammatory response 7 days postinfection (see Fig. 7) and to ~286-fold 14 days postinfection. These increases were due primarily to the more rapid increase in WT pulmonary burden than in Δkcs1 mutant burden over this time. Despite the persistence of Δkcs1 mutant in lungs up to 50 days postinfection, no colonization of the brain was observed during this time (Fig. 6D). This was in contrast to what occurred with mice infected with the WT, which developed a CNS infection by day 14 (the median survival time of WT-infected mice).
FIG 6 .

Ksc1 is essential for pathogenicity in Galleria mellonella and mice. (A) G. mellonella larvae were inoculated with the WT or the Δkcs1 mutant (106 yeast cells/10 µl). Two groups of larvae were also mock infected with water, or not at all, to serve as controls. The larvae were incubated at 30°C for 10 days, their health was monitored, and their deaths were recorded. (B to D) Anesthetized mice were inoculated intranasally with 5 × 105 CFU/20 µl of the indicated strains and euthanized after exhibiting debilitating symptoms of infection for the survival study (B) or at 3, 7, and 14 days postinfection for organ burden analysis (C and D). (A and B) Δkcs1 mutant-infected animals not showing clinical symptoms of illness were culled at days 15 and 50, and censorship of these infection groups is indicated by the cross. The Kaplan-Meier log rank test was used to determine differences in survival, with Δkcs1 mutant-infected larvae and mice surviving longer than WT-infected animals and the reconstituted strain in the case of the mouse study (*, P < 0.05). The difference in survival of mice infected with the WT and Δkcs1::KCS1 strain was insignificant (P = 0.161). Lungs (C) and brain (D) were removed and homogenized for organ burden analysis (CFU per organ) using quantitative culture.
FIG 7 .
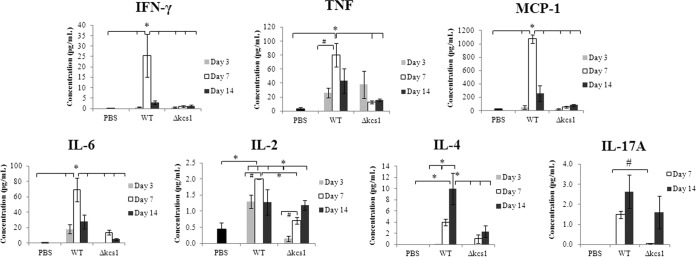
Cytokine profile in mock (PBS)-, WT-, and Δkcs1 mutant-infected mouse lung. Lungs of control and C. neoformans-infected mice (3 mice/strain/time point) were homogenized, and the concentrations of the indicated cytokines in the supernatant were measured using a cytometric bead array. The IL-10 responses on all days and the IL-17A response at day 3 for both the WT- and the Δkcs1 mutant-infected mice were below the level of sensitivity (not shown). *, P < 0.05 using a one-way analysis of variance (ANOVA) and a Tukey-Kramer multiple-comparison test; #, P < 0.05 using a two-tailed Student t test.
Periodic acid-Schiff (PAS)-stained sections of WT- and Δkcs1 mutant-infected lungs at day 7 and day 14 are shown in Fig. S4 in the supplemental material. A dense WT infection was observed on both days within the alveolar spaces and bronchi, with evidence of extensive infiltration of monocytes and polymorphs. In contrast, but consistent with organ burden analysis, Δkcs1 mutant infection was sparse and lung tissue was healthier than in WT-infected lung, as indicated by large areas of well-aerated alveoli and minimal evidence of inflammation. Despite the death of Δkcs1 mutant cells over the infection time course, budding cells were observed in the infected lung tissue.
The Δasp1 mutant was also tested for virulence in a mouse inhalation model, and the progression of disease in Δasp1 mutant-infected animals was found to be similar to that of WT-infected animals. This was expected, as no phenotypic differences were observed between the WT and the Δasp1 strain (Fig. S3 and S5).
Δkcs1 mutant infection elicits a weak host immune response.
The persistent, asymptomatic Δkcs1 mutant infection observed in the mouse model (Fig. 6C) prompted us to compare the immune responses elicited by the WT and the Δkcs1 mutant. The cytokine profile in infected lung tissue at 3, 7, and 14 days postinoculation was determined by analyzing the supernatants of homogenized lung extracts prepared from WT-, Δkcs1 mutant-, and mock-infected animals using a cytokine bead array (CBA) and flow cytometry (Fig. 7).
WT infection elicited a strong proinflammatory response in the lung (gamma interferon [IFN-γ] and tumor necrosis factor [TNF]), which peaked at day 7 postinfection; the day 7 response for IFN-γ and TNF-α was 36- and 3-fold higher than the day 3 response, respectively, and ~688- and 27-fold higher than the response observed in mock-infected animals (Fig. 7). TNF is required for neutrophil recruitment into pulmonary tissue during a cryptococcal infection (44), and IFN-γ maintains monocyte-derived macrophages in an activated state conducive to fungal killing and clearance. The strong proinflammatory response in WT-infected mice was accompanied by a strong monocyte chemoattractant protein-1 (MCP-1) response, which also peaked at day 7. MCP-1 is a crucial mediator of monocyte and T cell recruitment to the sites of infection. At day 7, the MCP-1 response was 22-fold higher than the day 3 response and 40-fold higher than the response observed in mock-infected animals. Despite the decrease in MCP-1 levels at day 14, significant numbers of monocytes were present at this time point in WT-infected lung (Fig. S4). In contrast to WT, no increase in the level of any of these three cytokines was observed in Δkcs1 mutant-infected lung at 7 days, despite evidence of persistent Δkcs1 mutant infection. Interleukin 6 (IL-6) production also peaked in WT-infected lung at day 7, with the response being ~4-fold higher than at day 3 and ~84-fold higher than the response observed in mock-infected animals. In contrast to the proinflammatory cytokines and MCP-1, IL-6 production peaked at day 7 in Δkcs1 mutant-infected lungs and was ~36-fold higher than the day 3 response and ~17-fold higher than the response observed in mock-infected animals.
IL-2 levels increased in Δkcs1 mutant-infected lungs over the time course but particularly from day 3 to day 7. However, the day 3 level was lower than the mock-infected control and WT values, while the day 7 level was similar to that of the mock-infected control but lower than the WT value. Interestingly, similar IL-2 responses were obtained for WT- and Δkcs1 mutant-infected lungs at day 14 despite the significantly lower Δkcs1 mutant fungal burden. In contrast to that of the other cytokines, IL-4 and IL17-A production was higher at day 14 than at day 7 in WT-infected lungs. However, the IL-4 response observed for Δkcs1 mutant-infected lungs did not change significantly between days 7 and 14 but was still higher than the response observed in mock-infected lungs (Fig. 7).
For both the WT and Δkcs1 strains, IL-17A levels at day 3 were below the level of detection and are not shown. At day 7, the IL-17A level in Δkcs1 mutant-infected lung was lower than that in WT-infected lung, while at day 14, a similar IL-17A response occurred in WT- and Δkcs1 mutant-infected lungs, despite the significantly lower Δkcs1 mutant burden.
The phagocytosis of the Δkcs1 mutant by monocytes and subsequent monocyte activation are compromised.
Monocytes and macrophages are the first line of defense against cryptococcal infection. Since we observed an altered surface phenotype in the Kcs1-deficient mutants (Fig. 4A), which may affect cryptococcal recognition by monocytes, we compared the efficiencies of phagocytosis of the WT and Δkcs1 mutant in vitro, using the THP-1 monocytic cell line. Fungal cells were labeled with fluorescein isothiocyanate (FITC) and opsonized with human serum (containing complement and antifungal antibodies), and their uptake was measured by flow cytometry. Figure 8A demonstrates that after 4 h of coincubation, ~40% of THP-1 cells were associated with WT C. neoformans. However, only ~10% of THP-1 cells were associated with Δkcs1 cells. Figure S6A shows a representative scatterplot used for quantification of adhesion/uptake. The extent of THP-1 cell association with the WT and Δkcs1 mutant was also measured when IgG antibodies purified from human blood were used as the opsonizing agent. The results in Fig. 8A show that only ~20% of THP-1 cells were associated with WT C. neoformans, indicating that the combination of complement and antibody is a more potent opsonin of WT cryptococci than antibody alone. As with the results observed for serum opsonization, the number of THP-1 cells associated with IgG-opsonized Δkcs1 cells was reduced. Notably, the difference in THP-1 cell association between the WT and mutant was even greater when IgG was used as an opsonin than when serum was used (9-fold versus 4-fold). The extent of THP-1 cell association with the WT and Δkcs1 strains was also measured when anticapsular antibodies were used as the opsonizing agent (Fig. 8A). However, in this case, the extents of THP-1 association with the WT and Δkcs1 mutant were similar.
FIG 8 .

Reduced phagocytosis of Δkcs1 cells by monocytes correlates with reduced monocyte activation and reduced binding of concanavalin A. (A) Live FITC-labeled WT and Δkcs1 cells were opsonized with 100% serum, purified human IgG, or anti-GXM antibodies and cocultured with activated THP-1 monocytes for 4 h, and the extent of adhesion/uptake was measured by flow cytometry. (B) As in panel A, except that cells were heat killed and opsonized with 10% human serum. (C) As in panel B, except that CD14-positive monocytes within a PBMC preparation were used instead of THP-1 cells. (D) The expression of chemokine- and interleukin-encoding genes in activated THP-1 cells following coculture with serum-opsonized WT and Δkcs1 cells was quantified using an RT2 profiler PCR array designed for analysis of the antifungal response. The expression data were normalized to the expression of the housekeeping genes indicated and then calculated as the fold change from the expression of activated THP-1 cells that had not been coincubated with C. neoformans. (E) Each culture was incubated with ConA-FITC and the fluorescence quantified using flow cytometry. The bar graph represents the average fluorescence of WT and Δkcs1 cells. Except for those in panel D, all results represent the means and standard deviations of results from biological triplicates. *, P < 0.005 using an unpaired, two-tailed t test.
To rule out the possibility that Δkcs1 cells are taken up less by THP-1 monocytes due to their lower rate of proliferation, we compared the phagocytosis of heat-killed WT and Δkcs1 mutant. As with live cryptococci, the phagocytosis of dead Δkcs1 cells was reduced (Fig. 8B). The extents of adhesion/uptake of heat-killed cryptococcal cells were also compared in primary human CD14-positive monocytes in a peripheral blood mononuclear cell (PBMC) preparation. Monocytes were distinguished from T and B cells by the binding of fluorescent anti-CD14. As with THP-1 cells (Fig. 8A and B), monocyte uptake of heat-killed, serum-opsonized Δkcs1 cells by CD14-positive monocytes was reduced (Fig. 8C). Microscopy of the cocultures also revealed reduced association of the Δkcs1 mutant with THP-1 cells and PBMCs (Fig. S6B), consistent with the flow cytometry analysis of fungal adhesion/uptake.
To determine whether reduced phagocytosis of Δkcs1 cells by monocytes coincided with reduced monocyte activation, we compared levels of THP-1 activation in the presence of live WT or Δkcs1 cells after 4 h of coincubation, using an RT2 profiler PCR array. The results in Fig. 8D indicate that the expression of proinflammatory cytokine- and interleukin-encoding genes is significantly higher in THP-1 cells coincubated with WT than with Δkcs1 cells, consistent with the stronger cytokine induction profile observed in the WT-infected mouse lung (Fig. 7). For example, the induction of the gene encoding the monocyte chemoattractant protein (Mcp1/Ccl2) was >9-fold higher in THP-1 cells coincubated with the WT than in cells coincubated with the Δkcs1 mutant. Thus, the reduced rate of Δkcs1 cell phagocytosis correlated with reduced monocyte activation.
Mannoprotein exposure at the Δkcs1 cell surface is reduced.
The similar rates of phagocytosis of anti-GXM-opsonized WT and Δkcs1 cells suggest that the antibody binds similarly to the surfaces of both strains despite their microscopic (larger capsule size) and macroscopic (mucoid colony morphology) differences. The difference in phagocytosis of WT and Δkcs1 cells observed when serum or human IgG were used for opsonization suggests that the respective opsonins bind to surface epitopes other than the sugar moiety binding targets of anti-GXM antibodies. Furthermore, the data suggest that the targets of serum and human IgG opsonins are present in greater amounts on WT than on Δkcs1 cells. The capsule contains mannoproteins, in addition to polysaccharides, and human serum contains antibodies against fungal mannoproteins that can initiate classical pathway activation upon binding to macrophages (45). To investigate mannoprotein exposure at the surfaces of WT and Δkcs1 cells, the extents of concanavalin A (ConA) binding were compared. ConA is a lectin that reacts with surface mannose sugars found on many microbes and is available as an FITC conjugate so that binding can be assessed by flow cytometry. The results in Fig. 8E show that ConA-FITC binding to the Δkcs1 cell surface is decreased in comparison with binding to WT cells, suggesting that mannosylated proteins are not displayed normally on the surface of the mutant cells.
To determine whether the reduced presence of mannoproteins on the Δkcs1 cell surface correlates with reduced expression of genes encoding mannoproteins, we performed transcriptome sequencing (RNA-seq) of the WT and Δkcs1 strains. We focused on genes encoding proteins with a signal peptide which targets them to the endoplasmic reticulum (ER)/Golgi complex and then to the cell periphery, excluding transmembranal proteins and proteins likely to be mitochondrial, peroxisomal, or ER resident. The expression of 22 extracellular proteins was down-regulated in the mutant, with 17 predicted to be glycosylated and 4 containing a GPI anchor (Table 1). Taken together, the data indicate that Kcs1-derived IP7 affects the fungal surface composite at the transcriptional level, influencing the extent of opsonization and phagocytosis and the ensuing immune response.
TABLE 1 .
The expression of genes encoding extracellular proteins is lower in Δkcs1 cells than in WT cells
| Identifier | Description | Relative expression ofa: |
Presence of: |
||||
|---|---|---|---|---|---|---|---|
| WT cells (fpkm) | Δkcs1 cells (fpkm) |
WT/Δkcs1 (fold change) |
O-linked glyco- sylation |
N-linked glyco- sylation |
GPI | ||
| CNAG_06291 | Polysaccharide deacetylase Fpd1 (D25) | 410.5 | 28.88 | 14.2 | |||
| CNAG_03465 | Laccase | 99.52 | 9.259 | 10.7 | + | + | |
| CNAG_06346 | Lipoprotein A | 180.3 | 20.23 | 8.91 | |||
| CNAG_02225 | Glucan 1,3-β-glucosidase | 335.3 | 53.46 | 6.27 | |||
| CNAG_01758 | Polygalacturonase | 67.41 | 12.8 | 5.27 | + | ||
| CNAG_06260 | α-Glucosidase | 42.12 | 8.581 | 4.91 | |||
| CNAG_04944 | Glycosyl hydrolase family 88 | 176.5 | 34.34 | 5.14 | + | ||
| CNAG_04735 | Elastinolytic metalloproteinase | 517.4 | 111.7 | 4.63 | + | + | |
| CNAG_06213 | Pepsin, aspartic protease | 180.8 | 38.86 | 4.65 | + | + | |
| CNAG_06501 | 1,3-β-Glucanosyltransferase | 187 | 46.21 | 4.05 | + | + | + |
| CNAG_04380 | Aspartic peptidase | 14.6 | 3.913 | 3.73 | + | + | |
| CNAG_00488 | Phospholipase B1-like lipase | 26.23 | 5.983 | 4.38 | + | + | |
| CNAG_01040 | Carboxypeptidase D | 11.72 | 3.38 | 3.47 | + | ||
| CNAG_07638 | PLC-like phosphodiesterase | 206.5 | 70.22 | 2.94 | + | + | + |
| CNAG_00581 | Aspartic peptidase | 1652 | 607.3 | 2.72 | + | ||
| CNAG_06251 | Ser/Thr protein phosphatase | 5.526 | 2.085 | 2.65 | + | + | |
| CNAG_07600 | β-Glucosidase | 112.5 | 40.76 | 2.76 | + | ||
| CNAG_04625 | Serine-type endopeptidase | 339.1 | 139.7 | 2.43 | + | ||
| CNAG_05471 | α-Glucosidase | 15.81 | 6.75 | 2.34 | + | + | |
| CNAG_05973 | Serine carboxypeptidase | 15.17 | 6.594 | 2.3 | + | ||
| CNAG_05411 | Endoglucanase | 80.12 | 37.08 | 2.16 | + | + | |
| CNAG_07865 | Ferro-O2-oxidoreductase | 44.08 | 21.82 | 2.02 | + | ||
Normalized fpkm values (fragments per kilobase of exon per million reads mapped) were generated by the Galaxy-based Cuffdiff tool and used as a measure of gene expression.
Transcriptome analysis identifies metabolic changes in the Δkcs1 mutant.
The gene expression profiles of the WT and Δkcs1 strains obtained by RNA-seq were further analyzed to identify changes in gene expression that could be responsible for the altered phenotype and virulence profile of the Δkcs1 mutant. Initial functional classification of differentially expressed genes was made using gene ontology (GO) term assignment generated by the Joint Genome Institute (JGI) and then refined further based on published literature and by performing a homology search. We found that the absence of IP7 resulted in an 11-fold reduction in the expression of the LAC1 gene involved in melanization, consistent with the results in Fig. 3. However, the most significant changes were observed in metabolic function. Tables 2 and S3 demonstrate that genes encoding proteins involved in glycolysis are more highly expressed in the Δkcs1 mutant than in the WT. Genes involved in mRNA translation and ribosomal biosynthesis are also more highly expressed in the Δkcs1 mutant. However, expression of genes encoding numerous sugar transporters and enzymes that comprise metabolic pathways involved in the utilization of alternative carbon sources (citric acid and glyoxylate cycles, gluconeogenesis, and fatty acid β-oxidation) is reduced in the Δkcs1 mutant. These metabolic differences suggest that WT, but not Δkcs1, cells actively express genes involved in the utilization of carbon sources other than glucose, including lactate and fatty acids, which require mitochondrial function.
TABLE 2 .
Summary of the RNA-seq data analysis
| Functional group | No. of genes upregulated in the Δkcs1 mutant | No. of genes downregulated in the Δkcs1 mutant | Total no. of genes in the functional group | Significance of enrichment (P value) |
|---|---|---|---|---|
| Glycolysis and pyruvate decarboxylation | 4 | 6 | 0.0055 | |
| Translation/ribosomal biogenesis | 91 | 160 | 5.32e–37 | |
| Citric acid cycle | 5 | 16 | 0.0273 | |
| Fatty acid β-oxidation and peroxisomal organization | 18 | 24 | 4.58e–13 | |
| Glyoxylate cycle and gluconeogenesis | 3 | 5 | 0.0120 | |
| Sugar transporters | 23 | 51 | 9.06e–10 | |
| GO term-assigned genes | 545 | 417 | 3689 |
The environment of the host lung is low in glucose (46), and enzymes involved in the use of alternative carbon sources are essential for cryptococcal pathogenicity (47–49). We therefore compared the extent of growth of both strains on glucose and three nonfermentable carbon sources, lactate, glycerol, and oleic acid, each present as the sole carbon source. We also used minimal medium (MM), which is more physiological than yeast extract-peptone-dextrose (YPD) (Fig. 2) . In MM supplemented with glucose Δkcs1 mutant grew similar to the WT strain (Fig. 9A). Glycerol is incorporated into the glycolytic/gluconeogenesis pathway following conversion to dihydroacetone phosphate by mitochondrial flavin adenine dinucleotide (FAD)-dependent glycerol-3-phosphate dehydrogenase. Lactate is converted to pyruvate, which is channeled into the citric acid cycle and gluconeogenesis. Oleic acid undergoes β-oxidation in mitochondria and/or peroxisomes to generate acetyl coenzyme A (acetyl-CoA), which can be used for energy generation via the citric acid cycle. The results in Fig. 9A demonstrate that in contrast to that on glucose, the growth of Δkcs1 cells on the alternative carbon sources was severely compromised.
FIG 9 .
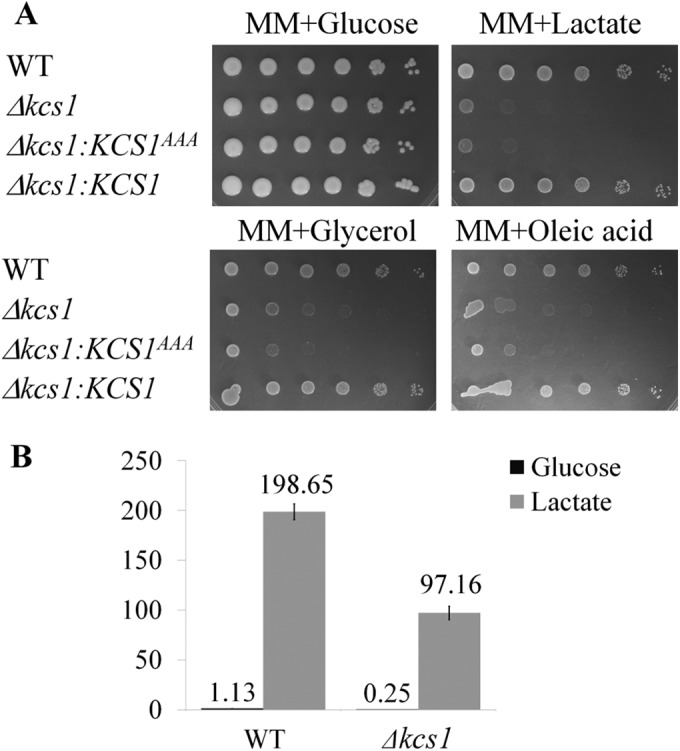
The Δkcs1 mutant is defective in its ability to utilize nonfermentable carbon sources. (A) Drop dilution assay. The strains were serially diluted 10-fold (106 to 10 cells/spot from left to right) and spotted onto the test plates as indicated. (B) To compare the levels of expression of phosphoenolpyruvate carboxykinase PCK1 in WT and Δkcs1 cells, the strains were grown on YPD, washed, and incubated in MM plus 1% glucose or MM plus 1% sodium lactate broth for 4 h. PCK1 expression was quantified by qPCR using the housekeeping gene ACT1 for normalization. The differences between WT and Δkcs1 cells grown in the same medium are statistically significant (P < 0.05), as determined by Student’s t test. Error bars indicate standard deviations.
Using quantitative PCR (qPCR) and minimal medium containing glucose or lactate, we compared the levels of expression of a gene encoding a key enzyme in gluconeogenesis, phosphoenolpyruvate carboxykinase (PCK1; CNAG_04217) (Fig. 9B). Pck1 catalyzes an irreversible step of gluconeogenesis, and its expression was 20-fold lower in Δkcs1 cells grown overnight in YPD than in WT cells under the same conditions (Table S3). In minimal medium supplemented with glucose, PCK1 expression was 4.5-fold lower in Δkcs1 than in WT cells (Fig. 9B), consistent with the global expression data. In the absence of glucose (minimal medium with lactate), PCK1 was induced in both strains, but its expression was 2-fold lower in the Δkcs1 mutant, indicating that gluconeogenesis is not fully activated in the absence of IP7. This result confirms that the mutant is compromised in its ability to metabolize alternative carbon sources and that the defect is caused, at least in part, by inadequate transcriptional regulation of metabolic enzymes.
DISCUSSION
Delineation of the IP biosynthesis pathway in C. neoformans.
Using gene deletion analysis and HPLC-based radiometric IP profiling, we have confirmed that Arg1 is an IP3 kinase. Furthermore, we have demonstrated that the role of Arg1 is to provide IP precursors for the PP-IP-producing kinase Kcs1, which, like Arg1, is essential for cryptococcal virulence. We determined that Kcs1 is the major IP6 kinase in C. neoformans, since it is essential for the synthesis of PP-IP5 (IP7) and PP2-IP4 (IP8). In particular, neither the Δkcs1 mutant nor the kinase-inactive mutant produced detectable amounts of IP7 (or IP8). We also showed that Asp1 functions primarily as an IP7 kinase, converting Kcs1-generated IP7 to IP8. In S. cerevisiae, IP7 is also produced by an Asp1 homolog, Vip1 (42). However, our IP profiling revealed that Asp1 does not contribute significantly to IP7 production in C. neoformans. Based on the IP profile obtained for the Δarg1, Δkcs1, and Δasp1 mutants, we propose a model of the IP biosynthesis pathway in C. neoformans depicted in Fig. 10. The presence of a putative IP5 kinase (Ipk1) in the middle of the pathway can be inferred from its homology to Ipk1 in S. cerevisiae; its role in C. neoformans is currently being investigated in our laboratory.
FIG 10 .
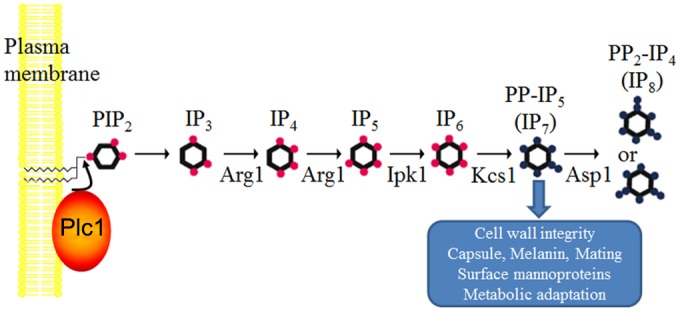
Schematic representation of the inositol polyphosphate biosynthesis pathway in C. neoformans. Phospholipase C1 (Plc1) hydrolyzes PIP2 to produce IP3. Arg1 is a dual-specificity kinase converting IP3 to IP4 and IP4 to IP5. Ipk1 is the proposed IP5 kinase, providing IP6 as a substrate for Kcs1. Kcs1 is the major IP6 kinase generating PP-IP5 (IP7), and Asp1 is an IP7 kinase generating PP2-IP4 (IP8). The physiological roles of Kcs1-derived IP7 are indicated.
The IP biosynthetic pathway as a drug target.
Two observations support investigation of the fungal PP-IP biosynthetic pathway as a potential antifungal drug target: (i) the absence of redundancy in the fungal pathway compared with the mammalian pathway and (ii) low sequence similarity between fungal and mammalian enzymes. For example, while Arg1 in C. neoformans is the only enzyme able to convert IP3 to IP4, three IP3 kinase isoforms and the inositol polyphosphate multikinase (IPMK) catalyze this reaction in humans. Arg1 differs structurally and functionally from mammalian IP3 kinases, implying potential selectivity of inhibition (50, 51). Arg1 and IPMK are only 19% identical, while Kcs1 is even less similar (13% identity) to its mammalian equivalent, inositol hexakisphosphate kinase 1 isoform 1. The hypersusceptibility of Δarg1 and Δkcs1 cells to antifungal agents (Table S1) also suggests that inhibitors of fungal Kcs1 or Arg1 may act synergistically with these marketed antifungal drugs to provide a better treatment outcome.
Kcs1-derived IP7, but not Asp1-derived IP8, is required for optimal growth and the production of virulence traits.
We observed delayed growth of the CnΔkcs1 mutant under standard growth conditions and a cell wall integrity defect but no high-temperature growth defect. In a cross with the KN99 MATa strain, the Δkcs1 mutant, but not the Δasp1 mutant, produced stunted mating filaments. Defective Δkcs1 cell walls may affect retention of the sexual agglutinins and adhesins, which are required for efficient cell contact prior to filamentation (52), as well as capsule attachment. A mucoid phenotype, typical of the Δkcs1 mutant, is likely to be associated with increased capsule size. Such a correlation was also observed in C. neoformans following phenotypic switching (53) and in the allergen 1 (ALL1) deletion mutant (54). During glucose deprivation, the Δkcs1 strain, but not the Δasp1 strain, fails to produce extracellular laccase and, consequently, melanin. We established that Kcs1-derived IP7 affects melanization at the transcriptional level, as the gene encoding the major melanin-producing enzyme in C. neoformans, laccase 1 (LAC1), was not induced in Δkcs1 cells. Transcriptional profiling data supported this finding.
IP7-dependent metabolic adaptation affects growth and pathogenicity.
The inability of the Δkcs1 mutant to achieve high infection burdens in mouse lung is most likely multifactorial, due to its weakened cell walls, inability to produce melanin, and reduced growth rate. High-throughput analysis of gene expression in the WT and Δkcs1 mutant suggested that the Δkcs1 mutant has a reduced ability to utilize carbon sources other than glucose and that this metabolic dysfunction may be a potential reason for its reduced ability to colonize glucose-deficient mouse lungs (46, 55). It has been demonstrated that WT cryptococci harvested during pulmonary infection exhibit a gene expression profile consistent with a starvation response (47, 56, 57). Metabolic dysfunction due to the absence of IP7, as suggested by our gene expression data, was supported by our observation of the severely reduced growth of the Δkcs1 mutant on alternative carbon sources, such as oleic acid, glycerol, and lactate (Fig. 9A). Utilization of these carbon sources requires functional mitochondria and, in the case of oleic acid, peroxisomes (58). A similar metabolic imbalance was reported for the Δkcs1 mutant of S. cerevisiae (59), whose cells did not grow on ethanol or glycerol as a sole carbon source.
Characterization of cryptococcal mutants defective in key metabolic enzymes established unequivocally the importance of β-oxidation and gluconeogenesis for the survival of C. neoformans in the nutrient-poor environment of the host and for pathogenicity. For example, a mutant in the peroxisomal β-oxidation pathway (Δmfe1 mutant) failed to grow on fatty acids as a carbon source and displayed hypovirulence in mice and reduced dissemination to the brain (49). Consistent with this report and with our observation that Δkcs1 mutant growth was reduced on oleic acid as the sole carbon source, MFE1 expression in Δkcs1 cells was 21-fold lower than in WT cells (Table S3). Similarly, a gluconeogenesis-defective cryptococcal mutant deficient in phosphoenolpyruvate carboxykinase Pck1(Δpck1 mutant) failed to grow on lactate as a sole carbon source and was also markedly less virulent in mice (48). Figure 9B and Table S3 demonstrate that the expression of PCK1 was lower in Δkcs1 cells than in the WT; the difference was 20-fold when the WT and Δkcs1 cells were grown in YPD and 2-fold when the strains were grown in minimal medium with lactate as a sole carbon source. Taken together, our findings suggest that the reduced pathogenicity of the Δkcs1 mutant may be due to a failure to adjust its metabolic response to a nutrient-poor environment, and this failure is caused, at least in part, by compromised regulation of gene expression. Such metabolic adaptation is essential for successful host infection.
Persistence of Δkcs1 mutant infection correlates with reduced surface mannoprotein.
Despite the reduced pathogenicity and metabolic defects observed in the Δkcs1 mutant, a low level of apparently asymptomatic infection persisted in vivo. Our results suggest that this persistence of Δkcs1 mutant infection in lung is due primarily to the reduction in the display of surface mannoproteins in the Δkcs1 strain, as evidenced by decreased ConA binding and reduced expression of genes encoding immunogenic mannoproteins, leading to reduced recognition by phagocytes. It is likely that the majority of the opsonic anticryptococcal antibodies in serum target mannoproteins (45, 60). Thus, the reduced mannoprotein content on the surfaces of Δkcs1 cells is expected to result in reduced opsonization by antibodies against mannoprotein and less efficient recognition of the fungal cells by phagocytic Fc receptors (45). Reduced phagocytosis of Δkcs1 cells indeed coincided with evidence of less phagocyte activation. The levels of phagocytosis of heat-killed WT and Δkcs1 cells were also compared to rule out the possibility that the reduction in phagocytosis of Δkcs1 cells by THP-1 monocytes was due to the lower proliferation rate of Δkcs1 cells. Again, the phagocytosis of killed Δkcs1 cells was reduced relative to that of WT cells (Fig. 8B). This reduction was even greater than that observed for live cells (~16-fold versus ~4-fold), possibly because a different concentration of serum was used for opsonization of the killed cells, as indicated in the legend of Fig. 8.
Persistent Δkcs1 strain lung infection may also be due to impaired sampling of mannoproteins by resident macrophages and dendritic cells for subsequent presentation to T cells, once again as a result of diminished mannoprotein production in Δkcs1 cells. Notably, the secreted polysaccharide deacetylase homolog Fpd1/D21 (CNAG_06291), which stimulates T-cell activation in mice and therefore contributes to the adaptive immune response to WT C. neoformans (61, 62), was down-regulated in Δkcs1 cells (Table 1). Transcription of many genes encoding proteases, including aspartic proteases (CNAG_06213, CNAG_04380, and CNAG_00581), carboxypeptidases (CNAG_01040 and CNAG_05973), serine-type endopeptidase (CNAG_04625), and elastinolytic metalloproteinase (CNAG_04735), was also reduced. Fungal proteases are known to elicit inflammatory responses via activation of protease-activated receptors (PARs), leading to allergic inflammation (63, 64). Thus, reduced secretion of proteases by the Δkcs1 mutant potentially contributes to the weak proinflammatory cytokine response and impaired infiltration of inflammatory neutrophils and monocytes observed in Δkcs1 mutant-infected lung (Fig. 7 and S4).
The persistence of Δkcs1 strain infection also correlated with a reduced recognition of Δkcs1 cells by the host immune system, as indicated by the lung cytokine profile (Fig. 7). First, in contrast to what occurred in WT-infected lung, no peak at day 7 was observed for the TNF, IFN-γ, and MCP-1 responses in Δkcs1 mutant-infected lung. This is consistent with a greater reduction in the proinflammatory response to Δkcs1 cells at day 7 when fungal burden is taken into account. Interestingly, however, similar IL-2 and IL-17A responses were obtained in WT- and Δkcs1-infected lungs at day 14 despite the significantly lower Δkcs1 mutant burden. Taken together, the phagocytosis, lung cytokine, and histological data suggest that the persistence of a low-grade, asymptomatic Δkcs1 mutant infection is attributable to reduced recruitment of monocytes to sites of Δkcs1 mutant infection and reduced uptake of the Δkcs1 mutant by resident lung macrophages and infiltrating monocytes.
In summary, we have demonstrated for the first time that the IP biosynthetic pathway is essential for cryptococcal pathogenicity in a mammalian infection model. We have shown that Kcs1 is the major IP6 kinase in C. neoformans producing IP7, while Asp1 is an IP7 kinase essential for the production of IP8. We have established that, of the two PP-IPs, IP7 exerts the greatest influence on cryptococcal pathogenicity. Our findings indicate that IP7 affects cryptococcal cellular functions, at least in part, via gene expression and that successful host infection requires IP7 to activate starvation-related metabolic pathways.
MATERIALS AND METHODS
Strains and media.
Wild-type C. neoformans var. grubii strain H99 (serotype A, MATa) was used in this study. The ARG1 (CNAG_06500) deletion mutant (Δarg1 mutant) was described previously (24). The KN99 (MATα) strain was a kind gift from Joseph Heitman’s laboratory (Duke University, Durham, NC, USA) (65). C. neoformans strains were maintained on SAB agar plates (4% dextrose, 1% peptone, 2% agar, pH 5.6), and incubated in YNB (6.7 g/liter yeast nitrogen base, 0.5% glucose), YPD (1% yeast extract, 2% peptone, and 2% dextrose), or MM (minimal medium; 15 mM glucose, 10 mM MgSO4, 29.4 mM KH2PO4, 13 mM glycine, 3 µM thiamine) broth in preparation for the experiments. Mating was induced on 5% V8 juice (pH 5), 2% agar plates. MM without glucose was used for laccase induction in broth culture and for the preparation of broth and plates with different carbon sources (1% oleic acid, sodium lactate, or glycerol). Minimal medium plates supplemented with 1 mM l-DOPA (l-3,4-dihydroxyphenylalanine) were used to assess melanization.
Quantification of extracellular laccase activity.
The protocol was adapted from reference 66. C. neoformans strains were grown overnight in YPD at 30°C. Cells were pelleted, washed with water, resuspended in MM without glucose at an optical density at 600 nm (OD600) of 1, and incubated for 4 h at 30°C. To measure cell-associated laccase activity, 200 µl of the culture was removed and the cells were pelleted by centrifugation. The pellet was resuspended in 1 ml of a 3 mM concentration of a chromogenic laccase substrate, ABTS [2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid)], and incubated for 30 min at 30°C until a light blue color was produced, indicative of ABTS oxidation. The cells were pelleted, and the OD of the supernatant was measured at 436 nm. Laccase-specific activity was expressed as micromoles of ABTS substrate oxidized/minute/milliliter of culture at an OD600 of 1. The concentration of oxidized ABTS was calculated using the molar extinction coefficient (ε = 29.3 mM−1 cm−1).
[3H]inositol labeling.
[3H]inositol labeling of cryptococcal cells was performed according to the protocol of reference 67, with several modifications. Briefly, fungal strains from overnight YPD culture were diluted in 5 ml fresh YPD to an OD600 of 0.01 (WT versus the Δkcs1 and Δarg1 mutants) or to an OD600 of 0.05 (WT versus the Δkcs1::KCS1, Δkcs1::KCS1AAA, and Δasp1 strains), supplemented with 10 µCi/ml [3H]myo-inositol (PerkinElmer), and incubated until the OD of the cultures reached at least 6 (16 to 76 h). The cells were pelleted, washed twice with 1 ml of fresh YPD, and snap-frozen in liquid nitrogen. To extract inositol polyphosphates, the cells were resuspended in extraction buffer (1 M HClO4, 3 mM EDTA, 0.1 mg/ml IP6) and homogenized in the presence of glass beads using a bead beater (four 30-s cycles with 1-min rests in between cycles). After the debris was pelleted, supernatants were neutralized using neutralization buffer (1 M K2CO3, 3 mM EDTA) and incubated on ice for 2 h. HPLC inositol phosphate analysis was performed as previously described (67).
Murine inhalation model of cryptococcosis.
All procedures were approved by the Sydney West Local Health District Animal Ethics Committee, Department of Animal Care. Survival and organ burden/cytokine analyses were conducted using 7-week-old female BALB/c mice obtained from the Animal Resource Centre, Floreat Park, Western Australia. For both sets of analyses, mice were anesthetized using isoflurane (in oxygen) delivered via an isoflurane vaporizer attached to a Stinger small-animal anesthetic machine (Advanced Anaesthesia Specialists). Groups of 8 to 10 mice were then inoculated intranasally with the C. neoformans WT or IPK mutant strain (5 × 105 yeast cells in 20 µl phosphate-buffered saline [PBS]) and observed daily for signs of ill health. The number of viable yeast cells inoculated into the nares was later confirmed by quantitative culture.
For the survival study, mice which had lost 20% of their preinfection weight or which showed debilitating clinical signs prior to losing this weight, including hunching, respiratory distress, excessive fur ruffling, or sluggish/unsteady movement, were euthanized by CO2 inhalation followed by cervical dislocation. Δkcs1 mutant-infected mice were sacrificed 50 days postinoculation. Differences in survival were determined with SPSS version 21 statistical software, using the Kaplan-Meier method (log rank test), where a P value of <0.05 was considered statistically significant.
For calculating lung and brain infection burdens and determining lung cytokine profiles, 3 from a group of 9 mice infected with a particular strain (the WT or Δkcs1 mutant) were sacrificed 3, 7, and 14 days postinoculation. For lung cytokine analysis, 3 mock-infected mice sacrificed on days 3, 7, and 14, respectively, served as controls. For determining infection burdens, lungs and brains were harvested from infected mice, weighed, and homogenized in a total of 1 ml sterile PBS. One hundred microliters of each homogenate was removed and serially diluted 10-fold in sterile PBS, and 100 µl of each dilution was plated onto SAB agar. The plates were incubated for 2 to 3 days at 30°C, and the number of CFU was determined. The remainder of each lung homogenate (900 µl) and 900 µl of lung homogenate obtained from each of the control mice were centrifuged at 10,000 × g to pellet tissue debris, and the supernatants were collected and frozen for cytokine analysis.
Cytokine profiling in infected lung supernatant.
Cytokine concentrations in lung homogenates collected postinfection were determined using mouse Th1/Th2/Th17 cytokine bead arrays (CBA; Becton, Dickinson Biosciences). MCP-1 was measured in a separate assay using the mouse MCP-1 flex set (bead B7) (BD Biosciences). CBA kits were used according to the manufacturer’s instructions, data were collected using an LSRII flow cytometer, and results were analyzed using FCAP software version 3.0 (BD Biosciences).
Virulence in Galleria mellonella.
C. neoformans WT and Δkcs1 mutant cells were grown overnight in YPD broth, pelleted by centrifugation, and resuspended in water at a concentration of 108 cells/ml. G. mellonella larvae (10 per strain) were inoculated with 10 µl of cell suspension (106 yeast cells) by injection into the hemocoel via the lower prolegs. The viability of each inoculum was assessed by performing serial 10-fold dilutions, plating the lowest dilutions on SAB agar plates, and counting the CFU after a 3-day incubation at 30°C. Inoculated larvae were monitored daily for 10 days after daily removal of their cocoon. Larvae were considered dead when they no longer responded to touch (68).
Phagocytosis experiments.
THP-1 cells were maintained in RPMI 1640-10% fetal bovine serum (FBS) medium. IFN-γ (10 ng/ml) was added 24 h prior to the phagocytosis experiment to activate the monocytes. Cryptococcal cells were grown overnight in YNB broth, pelleted by centrifugation, and resuspended in staining solution (0.5 mg/ml FITC in PBS) for 30 min at 30°C. To heat kill fungal cells in some of the experiments, YNB cultures were centrifuged and the cell pellets were incubated at 56°C for 30 min prior to FITC staining. Following FITC staining, fungal cell pellets were washed three times with PBS and opsonized for 30 min at 30°C with human serum (10 to 100%, as indicated in the figures) or human IgG (Sigma-Aldrich I4506) or anti-GXM antibody 18B7 (a kind gift from Arturo Casadevall) in PBS (69). Opsonized fungal cells were pelleted and mixed with THP-1 monocytes in a ratio of 3:1 or 5:1 in THP-1 growth medium. Fungal and mammalian cells were coincubated for 3 to 5 h at 37°C in 5% CO2. To quantify the adhesion/uptake of the fungal cells by THP-1, the cultures were mixed by pipetting, diluted with PBS, and analyzed by flow cytometry and microscopy. Reverse transcription-PCR (RT-PCR) was used to assess the THP-1 activation response to WT and Δkcs1 cells after 4 h of coincubation, using an RT2 profiler PCR array designed for analysis of the antifungal immune response (SABiosciences). PCR was performed using a QuantStudio6 Flex real-time PCR system (Life Technologies). The expression data were normalized to the expression of the housekeeping GAPDH, ACTB, B2M, HPRT1, and RPLPO genes and then calculated as the fold change from expression in activated THP-1 cells that had not been coincubated with cryptococci.
Peripheral blood monocytes (PBMCs) were isolated from fresh human blood using a Ficoll-Paque gradient. Up to 20 ml of blood was layered on top of 15 ml of Ficoll-Paque and centrifuged at 400 × g for 30 min. The interphase containing monocytes and T and B lymphocytes was collected in a fresh tube and washed twice with PBS before resuspension of the final pellet in RPMI 1640-10% FBS medium. PBMCs were visualized with crystal violet stain and counted using a hemocytometer. PBMCs (4 × 106) were mixed with 4 × 106 heat-killed, FITC-labeled, 10%-serum-opsonized cryptococci in 100 µl of RPMI 1640-10% FBS medium and coincubated for 2 h at 37°C in 5% CO2. The cell pellets were washed once with PBS. Nonspecific Fc receptor-mediated antibody binding was blocked before cells were stained with antigen-presenting cell (APC)-H7-conjugated anti-CD14 antibody (monocyte marker) (BD Biosciences; catalog number 560180). Adhesion/uptake of cryptococci by the CD14-positive cell population was quantified by flow cytometry and visualized by microscopy (70, 71; see Text S1 in the supplemental material).
Mannoprotein staining with ConA-FITC.
Cryptococcal cells were grown overnight in YNB, washed with PBS, and resuspended in 100 µl PBS supplemented with 0.5% gelatin at an OD600 of 0.5. Each suspension was incubated for 30 min at room temperature. Ten microliters of ConA-FITC (Sigma; C7642) from a 2.5-mg/ml stock solution in water was added to each culture sample, followed by incubation for 30 min at room temperature. The extent of ConA-FITC binding was assessed using flow cytometry.
High-throughput gene expression analysis.
Fungal strains were grown in YPD broth for 22 h. RNA was isolated using TRIzol and further purified using a Qiagen RNA isolation kit. Library preparation and sequencing were carried out by the Ramaciotti Centre for Genomics (UNSW, Sydney, Australia) using a TruSeq sample preparation kit and the Illumina HiSeq2000 sequencing platform to generate 100-bp paired-end reads. The resulting data were analyzed using the Galaxy platform (https://usegalaxy.org/). The analysis included data from three biological replicates for each strain; a ≥2-fold difference in gene expression between WT and Δkcs1 cells was considered to be significant. The presence of a signal peptide and N- and O-linked glycosylation were predicted using CBS prediction tools (http://www.cbs.dtu.dk/services/). The presence of a GPI anchor was predicted using the Big-PI fungal predictor (http://mendel.imp.ac.at/sat/gpi/fungi_server.html). Initial functional classification of cryptococcal genes was performed using GO term assignment from the Joint Genome Institute (JGI) database (http://genome.jgi.doe.gov/cgi-bin/ToGo?species=Cryne_ H99_1) and further improved based on the literature and by performing a homology search. A hypergeometric test was used to assess whether Kcs1-dependent genes were overrepresented in specific functional groups.
SUPPLEMENTAL MATERIAL
Supplemental methods. Download
Verification of KCS1 and ASP1 deletions and KCS1 reconstitution in the Δkcs1 mutant. (A) Diagram of the KCS1 and ASP1 deletion strategy and the reconstitution of the Δkcs1 mutant with native and mutagenized KCS1. Primer names and their binding positions are shown, and their direction of amplification (-s for forward and -a for reverse) are indicated by arrows. Diagonal lines represent adaptors introduced to perform overlap PCR, and “ots” indicates “outside” the region of integration of the deletion construct. ORF, open reading frame. (B) PCR verification of the Δkcs1 and Δasp1 mutants (Δ), and KCS1 reconstitution of the Δkcs1 mutant (rec). The wild type (WT) was used as a control. PCRs were performed using the indicated primer pairs and genomic DNA as a template. ActP, actin promoter; Ttrp, tryptophan terminator. (C) The expression of ectopically integrated KCS1 and KCS1AAA in the Δkcs1::KCS1 and Δkcs1::KCS1AAA strains, respectively, is comparable to the expression of the native KCS1 gene (WT sample). As expected, KCS1 expression was not detected in the deletion mutant (Δkcs1). Expression was quantified by real-time PCR. The actin-encoding gene ACT1 was used for normalization. (D) Southern blot analysis with digoxigenin-labeled Neo and Nat probes to confirm integration of the KCS1 deletion and reconstitution constructs into a single genomic location in the target strain. Download
Mating filament formation is compromised in the Δkcs1 and Δkcs1::KCS1AAA strains. The indicated strains (MATα) were crossed with KN99 (MATa) on V8 agar plates and incubated for 10 days to assess mating filaments macroscopically and by light microscopy. Both the Δkcs1 and Δkcs1::KCS1AAA strains exhibited reduced filament formation, while in the Δkcs1::KCS1 strain, it was restored to that of the WT. Download
(A) Phenotypic characterization of the Δasp1 strain. (B) Overnight cultures of the WT and the Δasp1 strain were counted and serially diluted 10-fold to give 106 to 10 cells/5 µl (from left to right). Dilutions were spotted onto the test plates indicated. Melanization of the Δasp1 strain in panel B was tested on minimal medium (MM) agar containing the laccase substrate l-DOPA. (C) The WT and the Δasp1 strain were grown in MM broth to induce capsule production. (D) Mating filament production by the WT and the Δasp1 strain (MATα strains) was tested by performing a unilateral cross with WT strain KN99 (MATa) on V8 mating agar. Following strain mixing, the plates were incubated for 10 days and observed under a light microscope to assess the formation of mating filaments. Download
Histology of WT- and Δkcs1 mutant-infected lung. Lungs were removed postinfection, fixed, sectioned, and stained with periodic acid-Schiff (PAS) stain. Fungal cell bodies are dark pink and surrounded by a white halo, which may be capsule or alveolar space (white arrows). The day 7 Δkcs1 ×60 magnification image, where a budding Δkcs1 cell is observed (black arrow), represents the enclosed area demarcated by the square in the day 7 Δkcs1 ×10 magnification image. Areas of inflammation are indicated by black broken arrows. Download
The virulence of the Δasp1 strain in mice is similar to that of the WT. Anesthetized mice were inoculated intranasally with 5 × 105 CFU/20 µl of the indicated strains and euthanized after showing debilitating symptoms of infection. The Kaplan-Meier log rank test was used to establish that there was no significant difference (P = 0.587) in survival between WT- and Δasp1 strain-infected mice (the median percentages of survival of WT- and Δasp1 strain-infected mice were 14% ± 2.1% and 15% ± 1.6%, respectively). Cum, cumulative. Download
The absence of Kcs1 affects the association and uptake of cryptococcal cells by mammalian phagocytes. (A) Representative scatter plots used to quantify the extent of adhesion/uptake of the FITC-labeled fungal cells by THP-1 monocytes. Green fluorescence (FITC-A) is plotted against forward scatter (FSC-A). Populations demarcated by the black, purple, and red gates represent nonfluorescent THP-1 cells, free fluorescent fungal cells, and fluorescent fungal cells associated with THP-1 cells, respectively. (B) Reduced association and uptake of Δkcs1 mutant by THP1 cells and monocytes within a PBMC preparation following a 4-h coculture, as visualized by microscopy. Arrows indicate fungal cells, and arrowheads indicate mammalian cells. Download
Δkcs1 and Δarg1 cells exhibit increased susceptibility to antifungals. MICs were determined by assessing the growth of the WT and mutant strains in the presence of serially diluted antifungal compounds. AND, anidulafungin; AMB, amphotericin B; MF, micafungin; CAS, caspofungin; FC, flucytosine; PZ, posaconazole; VOR, voriconazole; IZ, itraconazole; FZ, fluconazole.
Primers used in this study. Uppercase nucleotides in the oligonucleotide sequences are complementary to the template, while lowercase nucleotides indicate added adaptor sequences.
RNA-seq analysis of the gene expression in the WT and Δkcs1 mutant. The data are based on the analysis of triplicate samples. FPKM values (fragments per kilobase of exon per million reads mapped) as a normalized measure of gene expression were generated by the Galaxy-based Cuffdiff tool. The difference in gene expression between Δkcs1 mutant and WT samples was considered significant if log2(Δkcs1 mutant/WT) was at least 1 or not more than −1, and the Q value (false-discovery rate at which the test result may be called significant) was <0.05.
ACKNOWLEDGMENTS
This work was supported by funding from a National Health and Medical Research Council of Australia project grant (APP1058779) and by Bioplatforms Australia through the Commonwealth Government National Collaborative Research Infrastructure Strategy (RNA-seq). A.S. is supported by the Medical Research Council (MRC core support to the MRC/UCL Laboratory for the Molecular Cell Biology University Unit [grant MC_U122680443]), and T.C.S. is a Sydney Medical School Foundation Fellow.
We thank Ana Traven for helpful suggestions and Robert Lev for generous help with the RNA-seq data analysis. We also thank Virginia James and the Histology Department at WMIMR for performing mouse lung histology, Christabel F. Wilson for help with the animal infection studies, the WMI genomics facility for assistance with qPCR, the WMI flow cytometry and microscopy facilities for assistance with phagocytosis studies, and the sequencing team at the Ramaciotti Centre for Genomics (UNSW, Sydney, Australia).
Footnotes
Citation Lev S, Li C, Desmarini D, Saiardi A, Fewings NL, Schibeci SD, Sharma R, Sorrell TC, Djordjevic JT. 2015. Fungal Inositol pyrophosphate IP7 is crucial for metabolic adaptation to the host environment and pathogenicity. mBio 6(3):e00531-15. doi:10.1128/mBio.00531-15.
REFERENCES
- 1.Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM. 2009. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23:525–530. doi: 10.1097/QAD.0b013e328322ffac. [DOI] [PubMed] [Google Scholar]
- 2.Hull CM, Heitman J. 2002. Genetics of Cryptococcus neoformans. Annu Rev Genet 36:557–615. doi: 10.1146/annurev.genet.36.052402.152652. [DOI] [PubMed] [Google Scholar]
- 3.Kozubowski L, Lee SC, Heitman J. 2009. Signalling pathways in the pathogenesis of Cryptococcus. Cell Microbiol 11:370–380. doi: 10.1111/j.1462-5822.2008.01273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eisenman HC, Casadevall A. 2012. Synthesis and assembly of fungal melanin. Appl Microbiol Biotechnol 93:931–940. doi: 10.1007/s00253-011-3777-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perfect JR. 2006. Cryptococcus neoformans: the yeast that likes it hot. FEMS Yeast Res 6:463–468. doi: 10.1111/j.1567-1364.2006.00051.x. [DOI] [PubMed] [Google Scholar]
- 6.Doering TL. 2009. How sweet it is! Cell wall biogenesis and polysaccharide capsule formation in Cryptococcus neoformans. Annu Rev Microbiol 63:223–247. doi: 10.1146/annurev.micro.62.081307.162753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Meara TR, Alspaugh JA. 2012. The Cryptococcus neoformans capsule: a sword and a shield. Clin Microbiol Rev 25:387–408. doi: 10.1128/CMR.00001-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Aisen P, Casadevall A. 1995. Cryptococcus neoformans melanin and virulence: mechanism of action. Infect Immun 63:3131–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waterman SR, Hacham M, Panepinto J, Hu G, Shin S, Williamson PR. 2007. Cell wall targeting of laccase of Cryptococcus neoformans during infection of mice. Infect Immun 75:714–722. doi: 10.1128/IAI.01351-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bose I, Reese AJ, Ory JJ, Janbon G, Doering TL. 2003. A yeast under cover: the capsule of Cryptococcus neoformans. Eukaryot Cell 2:655–663. doi: 10.1128/EC.2.4.655-663.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaragoza O, Rodrigues ML, De Jesus M, Frases S, Dadachova E, Casadevall A. 2009. The capsule of the fungal pathogen Cryptococcus neoformans. Adv Appl Microbiol 68:133–216. doi: 10.1016/S0065-2164(09)01204-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kozel TR. 1995. Virulence factors of Cryptococcus neoformans. Trends Microbiol 3:295–299. doi: 10.1016/S0966-842X(00)88957-X. [DOI] [PubMed] [Google Scholar]
- 13.García-Rodas R, Zaragoza O. 2012. Catch me if you can: phagocytosis and killing avoidance by Cryptococcus neoformans. FEMS Immunol Med Microbiol 64:147–161. doi: 10.1111/j.1574-695X.2011.00871.x. [DOI] [PubMed] [Google Scholar]
- 14.Levitz SM, Specht CA. 2006. The molecular basis for the immunogenicity of Cryptococcus neoformans mannoproteins. FEMS Yeast Res 6:513–524. doi: 10.1111/j.1567-1364.2006.00071.x. [DOI] [PubMed] [Google Scholar]
- 15.Djordjevic JT, Del Poeta M, Sorrell TC, Turner KM, Wright LC. 2005. Secretion of cryptococcal phospholipase B1 (PLB1) is regulated by a glycosylphosphatidylinositol (GPI) anchor. Biochem J 389:803–812. doi: 10.1042/BJ20050063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siafakas AR, Sorrell TC, Wright LC, Wilson C, Larsen M, Boadle R, Williamson PR, Djordjevic JT. 2007. Cell wall-linked cryptococcal phospholipase B1 is a source of secreted enzyme and a determinant of cell wall integrity. J Biol Chem 282:37508–37514. doi: 10.1074/jbc.M707913200. [DOI] [PubMed] [Google Scholar]
- 17.Eigenheer RA, Jin Lee Y, Blumwald E, Phinney BS, Gelli A. 2007. Extracellular glycosylphosphatidylinositol-anchored mannoproteins and proteases of Cryptococcus neoformans. FEMS Yeast Res 7:499–510. doi: 10.1111/j.1567-1364.2006.00198.x. [DOI] [PubMed] [Google Scholar]
- 18.Biondo C, Mancuso G, Midiri A, Bombaci M, Messina L, Beninati C, Teti G. 2006. Identification of major proteins secreted by Cryptococcus neoformans. FEMS Yeast Res 6:645–651. doi: 10.1111/j.1567-1364.2006.00043.x. [DOI] [PubMed] [Google Scholar]
- 19.Biondo C, Messina L, Bombaci M, Mancuso G, Midiri A, Beninati C, Cusumano V, Gerace E, Papasergi S, Teti G. 2005. Characterization of two novel cryptococcal mannoproteins recognized by immune sera. Infect Immun 73:7348–7355. doi: 10.1128/IAI.73.11.7348-7355.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pietrella D, Cherniak R, Strappini C, Perito S, Mosci P, Bistoni F, Vecchiarelli A. 2001. Role of mannoprotein in induction and regulation of immunity to Cryptococcus neoformans. Infect Immun 69:2808–2814. doi: 10.1128/IAI.69.5.2808-2814.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pietrella D, Corbucci C, Perito S, Bistoni G, Vecchiarelli A. 2005. Mannoproteins from Cryptococcus neoformans promote dendritic cell maturation and activation. Infect Immun 73:820–827. doi: 10.1128/IAI.73.2.820-827.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang C, Nong SH, Mansour MK, Specht CA, Levitz SM. 2002. Purification and characterization of a second immunoreactive mannoprotein from Cryptococcus neoformans that stimulates T-cell responses. Infect Immun 70:5485–5493. doi: 10.1128/IAI.70.10.5485-5493.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chayakulkeeree M, Sorrell TC, Siafakas AR, Wilson CF, Pantarat N, Gerik KJ, Boadle R, Djordjevic JT. 2008. Role and mechanism of phosphatidylinositol-specific phospholipase C in survival and virulence of Cryptococcus neoformans. Mol Microbiol 69:809–826. doi: 10.1111/j.1365-2958.2008.06310.x. [DOI] [PubMed] [Google Scholar]
- 24.Lev S, Desmarini D, Li C, Chayakulkeeree M, Traven A, Sorrell TC, Djordjevic JT. 2013. Phospholipase C of Cryptococcus neoformans regulates homeostasis and virulence by providing inositol trisphosphate as a substrate for Arg1 kinase. Infect Immun 81:1245–1255. doi: 10.1128/IAI.01421-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo HR, Huang YE, Chen JC, Saiardi A, Iijima M, Ye K, Huang Y, Nagata E, Devreotes P, Snyder SH. 2003. Inositol pyrophosphates mediate chemotaxis in Dictyostelium via pleckstrin homology domain-PtdIns(3,4,5)P3 interactions. Cell 114:559–572. doi: 10.1016/S0092-8674(03)00640-8. [DOI] [PubMed] [Google Scholar]
- 26.Saiardi A, Sciambi C, McCaffery JM, Wendland B, Snyder SH. 2002. Inositol pyrophosphates regulate endocytic trafficking. Proc Natl Acad Sci U S A 99:14206–14211. doi: 10.1073/pnas.212527899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.York SJ, Armbruster BN, Greenwell P, Petes TD, York JD. 2005. Inositol diphosphate signaling regulates telomere length. J Biol Chem 280:4264–4269. doi: 10.1074/jbc.M412070200. [DOI] [PubMed] [Google Scholar]
- 28.Saiardi A, Resnick AC, Snowman AM, Wendland B, Snyder SH. 2005. Inositol pyrophosphates regulate cell death and telomere length through phosphoinositide 3-kinase-related protein kinases. Proc Natl Acad Sci U S A 102:1911–1914. doi: 10.1073/pnas.0409322102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagata E, Luo HR, Saiardi A, Bae BI, Suzuki N, Snyder SH. 2005. Inositol hexakisphosphate kinase-2, a physiologic mediator of cell death. J Biol Chem 280:1634–1640. doi: 10.1074/jbc.M409416200. [DOI] [PubMed] [Google Scholar]
- 30.Bhandari R, Juluri KR, Resnick AC, Snyder SH. 2008. Gene deletion of inositol hexakisphosphate kinase 1 reveals inositol pyrophosphate regulation of insulin secretion, growth, and spermiogenesis. Proc Natl Acad Sci U S A 105:2349–2353. doi: 10.1073/pnas.0712227105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Illies C, Gromada J, Fiume R, Leibiger B, Yu J, Juhl K, Yang SN, Barma DK, Falck JR, Saiardi A, Barker CJ, Berggren PO. 2007. Requirement of inositol pyrophosphates for full exocytotic capacity in pancreatic beta cells. Science 318:1299–1302. doi: 10.1126/science.1146824. [DOI] [PubMed] [Google Scholar]
- 32.Azevedo C, Burton A, Ruiz-Mateos E, Marsh M, Saiardi A. 2009. Inositol pyrophosphate mediated pyrophosphorylation of AP3B1 regulates HIV-1 gag release. Proc Natl Acad Sci U S A 106:21161–21166. doi: 10.1073/pnas.0909176106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saiardi A, Caffrey JJ, Snyder SH, Shears SB. 2000. The inositol hexakisphosphate kinase family. Catalytic flexibility and function in yeast vacuole biogenesis. J Biol Chem 275:24686–24692. doi: 10.1074/jbc.M002750200. [DOI] [PubMed] [Google Scholar]
- 34.Dubois E, Scherens B, Vierendeels F, Ho MM, Messenguy F, Shears SB. 2002. In Saccharomyces cerevisiae, the inositol polyphosphate kinase activity of Kcs1p is required for resistance to salt stress, cell wall integrity, and vacuolar morphogenesis. J Biol Chem 277:23755–23763. doi: 10.1074/jbc.M202206200. [DOI] [PubMed] [Google Scholar]
- 35.Padmanabhan U, Dollins DE, Fridy PC, York JD, Downes CP. 2009. Characterization of a selective inhibitor of inositol hexakisphosphate kinases: use in defining biological roles and metabolic relationships of inositol pyrophosphates. J Biol Chem 284:10571–10582. doi: 10.1074/jbc.M900752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pöhlmann J, Fleig U. 2010. Asp1, a conserved 1/3 inositol polyphosphate kinase, regulates the dimorphic switch in Schizosaccharomyces pombe. Mol Cell Biol 30:4535–4547. doi: 10.1128/MCB.00472-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feoktistova A, McCollum D, Ohi R, Gould KL. 1999. Identification and characterization of Schizosaccharomyces pombe asp1(+), a gene that interacts with mutations in the Arp2/3 complex and actin. Genetics 152:895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhandari R, Saiardi A, Ahmadibeni Y, Snowman AM, Resnick AC, Kristiansen TZ, Molina H, Pandey A, Werner JK Jr, Juluri KR, Xu Y, Prestwich GD, Parang K, Snyder SH. 2007. Protein pyrophosphorylation by inositol pyrophosphates is a posttranslational event. Proc Natl Acad Sci U S A 104:15305–15310. doi: 10.1073/pnas.0707338104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saiardi A, Bhandari R, Resnick AC, Snowman AM, Snyder SH. 2004. Phosphorylation of proteins by inositol pyrophosphates. Science 306:2101–2105. doi: 10.1126/science.1103344. [DOI] [PubMed] [Google Scholar]
- 40.Worley J, Luo X, Capaldi AP. 2013. Inositol pyrophosphates regulate cell growth and the environmental stress response by activating the HDAC Rpd3L. Cell Rep 3:1476–1482. doi: 10.1016/j.celrep.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chakraborty A, Latapy C, Xu J, Snyder SH, Beaulieu JM. 2014. Inositol hexakisphosphate kinase-1 regulates behavioral responses via GSK3 signaling pathways. Mol Psychiatry 19:284–293. doi: 10.1038/mp.2013.21. [DOI] [PubMed] [Google Scholar]
- 42.Mulugu S, Bai W, Fridy PC, Bastidas RJ, Otto JC, Dollins DE, Haystead TA, Ribeiro AA, York JD. 2007. A conserved family of enzymes that phosphorylate inositol hexakisphosphate. Science 316:106–109. doi: 10.1126/science.1139099. [DOI] [PubMed] [Google Scholar]
- 43.Onnebo S, Saiardi A. 2009. Inositol pyrophosphates modulate hydrogen peroxide signalling. Biochem J 423:109–118. doi: 10.1042/BJ20090241. [DOI] [PubMed] [Google Scholar]
- 44.Herring AC, Lee J, McDonald RA, Toews GB, Huffnagle GB. 2002. Induction of interleukin-12 and gamma interferon requires tumor necrosis factor alpha for protective T1-cell-mediated immunity to pulmonary Cryptococcus neoformans infection. Infect Immun 70:2959–2964. doi: 10.1128/IAI.70.6.2959-2964.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levitz SM. 2010. Innate recognition of fungal cell walls. PLoS Pathog 6:e1000758. doi: 10.1371/journal.ppat.1000758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garnett JP, Baker EH, Baines DL. 2012. Sweet talk: insights into the nature and importance of glucose transport in lung epithelium. Eur Respir J 40:1269–1276. doi: 10.1183/09031936.00052612. [DOI] [PubMed] [Google Scholar]
- 47.Hu G, Cheng PY, Sham A, Perfect JR, Kronstad JW. 2008. Metabolic adaptation in Cryptococcus neoformans during early murine pulmonary infection. Mol Microbiol 69:1456–1475. doi: 10.1111/j.1365-2958.2008.06374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Panepinto J, Liu L, Ramos J, Zhu X, Valyi-Nagy T, Eksi S, Fu J, Jaffe HA, Wickes B, Williamson PR. 2005. The DEAD-box RNA helicase Vad1 regulates multiple virulence-associated genes in Cryptococcus neoformans. J Clin Invest 115:632–641. doi: 10.1172/JCI23048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kretschmer M, Wang J, Kronstad JW. 2012. Peroxisomal and mitochondrial beta-oxidation pathways influence the virulence of the pathogenic fungus Cryptococcus neoformans. Eukaryot Cell 11:1042–1054. doi: 10.1128/EC.00128-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bosch D, Saiardi A. 2012. Arginine transcriptional response does not require inositol phosphate synthesis. J Biol Chem 287:38347–38355. doi: 10.1074/jbc.M112.384255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Resnick AC, Snowman AM, Kang BN, Hurt KJ, Snyder SH, Saiardi A. 2005. Inositol polyphosphate multikinase is a nuclear PI3-kinase with transcriptional regulatory activity. Proc Natl Acad Sci U S A 102:12783–12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lipke PN, Kurjan J. 1992. Sexual agglutination in budding yeasts: structure, function, and regulation of adhesion glycoproteins. Microbiol Rev 56:180–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guerrero A, Jain N, Goldman DL, Fries BC. 2006. Phenotypic switching in Cryptococcus neoformans. Microbiology 152:3–9. doi: 10.1099/mic.0.28451-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jain N, Li L, Hsueh YP, Guerrero A, Heitman J, Goldman DL, Fries BC. 2009. Loss of AllerGen 1 confers a hypervirulent phenotype that resembles mucoid switch variants of Cryptococcus neoformans. Infect Immun 77:128–140. doi: 10.1128/IAI.01079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saumon G, Martet G, Loiseau P. 1996. Glucose transport and equilibrium across alveolar-airway barrier of rat. Am J Physiol 270:L183–L190. [DOI] [PubMed] [Google Scholar]
- 56.Fan W, Kraus PR, Boily MJ, Heitman J. 2005. Cryptococcus neoformans gene expression during murine macrophage infection. Eukaryot Cell 4:1420–1433. doi: 10.1128/EC.4.8.1420-1433.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Derengowski LDS, Paes HC, Albuquerque P, Tavares AH, Fernandes L, Silva-Pereira I, Casadevall A. 2013. The transcriptional response of Cryptococcus neoformans to ingestion by Acanthamoeba castellanii and macrophages provides insights into the evolutionary adaptation to the mammalian host. Eukaryot Cell 12:761–774. doi: 10.1128/EC.00073-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turcotte B, Liang XB, Robert F, Soontorngun N. 2010. Transcriptional regulation of nonfermentable carbon utilization in budding yeast. FEMS Yeast Res 10:2–13. doi: 10.1111/j.1567-1364.2009.00555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Szijgyarto Z, Garedew A, Azevedo C, Saiardi A. 2011. Influence of inositol pyrophosphates on cellular energy dynamics. Science 334:802–805. doi: 10.1126/science.1211908. [DOI] [PubMed] [Google Scholar]
- 60.Zhang MX, Lupan DM, Kozel TR. 1997. Mannan-specific immunoglobulin G antibodies in normal human serum mediate classical pathway initiation of C3 binding to Candida albicans. Infect Immun 65:3822–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Biondo C, Beninati C, Delfino D, Oggioni M, Mancuso G, Midiri A, Bombaci M, Tomaselli G, Teti G. 2002. Identification and cloning of a cryptococcal deacetylase that produces protective immune responses. Infect Immun 70:2383–2391. doi: 10.1128/IAI.70.5.2383-2391.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Biondo C, Beninati C, Bombaci M, Messina L, Mancuso G, Midiri A, Galbo R, Teti G. 2003. Induction of T helper type 1 responses by a polysaccharide deacetylase from Cryptococcus neoformans. Infect Immun 71:5412–5417. doi: 10.1128/IAI.71.9.5412-5417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boitano S, Flynn AN, Sherwood CL, Schulz SM, Hoffman J, Gruzinova I, Daines MO. 2011. Alternaria alternata serine proteases induce lung inflammation and airway epithelial cell activation via PAR2. Am J Physiol Lung Cell Mol Physiol 300:L605–L614. doi: 10.1152/ajplung.00359.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yike I. 2011. Fungal proteases and their pathophysiological effects. Mycopathologia 171:299–323. doi: 10.1007/s11046-010-9386-2. [DOI] [PubMed] [Google Scholar]
- 65.Nielsen K, Cox GM, Wang P, Toffaletti DL, Perfect JR, Heitman J. 2003. Sexual cycle of Cryptococcus neoformans var. grubii and virulence of congenic a and alpha isolates. Infect Immun 71:4831–4841. doi: 10.1128/IAI.71.9.4831-4841.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heung LJ, Luberto C, Plowden A, Hannun YA, Del Poeta M. 2004. The sphingolipid pathway regulates Pkc1 through the formation of diacylglycerol in Cryptococcus neoformans. J Biol Chem 279:21144–21153. doi: 10.1074/jbc.M312995200. [DOI] [PubMed] [Google Scholar]
- 67.Azevedo C, Saiardi A. 2006. Extraction and analysis of soluble inositol polyphosphates from yeast. Nat Protoc 1:2416–2422. doi: 10.1038/nprot.2006.337. [DOI] [PubMed] [Google Scholar]
- 68.Mylonakis E, Moreno R, El Khoury JB, Idnurm A, Heitman J, Calderwood SB, Ausubel FM, Diener A. 2005. Galleria mellonella as a model system to study Cryptococcus neoformans pathogenesis. Infect Immun 73:3842–3850. doi: 10.1128/IAI.73.7.3842-3850.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Casadevall A. 1995. Antibody immunity and invasive fungal infections. Infect Immun 63:4211–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fraser JA, Subaran RL, Nichols CB, Heitman J. 2003. Recapitulation of the sexual cycle of the primary fungal pathogen Cryptococcus neoformans var. gattii: implications for an outbreak on Vancouver Island, Canada. Eukaryot Cell 2:1036–1045. doi: 10.1128/EC.2.5.1036-1045.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Toffaletti DL, Rude TH, Johnston SA, Durack DT, Perfect JR. 1993. Gene transfer in Cryptococcus neoformans by use of biolistic delivery of DNA. J Bacteriol 175:1405–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental methods. Download
Verification of KCS1 and ASP1 deletions and KCS1 reconstitution in the Δkcs1 mutant. (A) Diagram of the KCS1 and ASP1 deletion strategy and the reconstitution of the Δkcs1 mutant with native and mutagenized KCS1. Primer names and their binding positions are shown, and their direction of amplification (-s for forward and -a for reverse) are indicated by arrows. Diagonal lines represent adaptors introduced to perform overlap PCR, and “ots” indicates “outside” the region of integration of the deletion construct. ORF, open reading frame. (B) PCR verification of the Δkcs1 and Δasp1 mutants (Δ), and KCS1 reconstitution of the Δkcs1 mutant (rec). The wild type (WT) was used as a control. PCRs were performed using the indicated primer pairs and genomic DNA as a template. ActP, actin promoter; Ttrp, tryptophan terminator. (C) The expression of ectopically integrated KCS1 and KCS1AAA in the Δkcs1::KCS1 and Δkcs1::KCS1AAA strains, respectively, is comparable to the expression of the native KCS1 gene (WT sample). As expected, KCS1 expression was not detected in the deletion mutant (Δkcs1). Expression was quantified by real-time PCR. The actin-encoding gene ACT1 was used for normalization. (D) Southern blot analysis with digoxigenin-labeled Neo and Nat probes to confirm integration of the KCS1 deletion and reconstitution constructs into a single genomic location in the target strain. Download
Mating filament formation is compromised in the Δkcs1 and Δkcs1::KCS1AAA strains. The indicated strains (MATα) were crossed with KN99 (MATa) on V8 agar plates and incubated for 10 days to assess mating filaments macroscopically and by light microscopy. Both the Δkcs1 and Δkcs1::KCS1AAA strains exhibited reduced filament formation, while in the Δkcs1::KCS1 strain, it was restored to that of the WT. Download
(A) Phenotypic characterization of the Δasp1 strain. (B) Overnight cultures of the WT and the Δasp1 strain were counted and serially diluted 10-fold to give 106 to 10 cells/5 µl (from left to right). Dilutions were spotted onto the test plates indicated. Melanization of the Δasp1 strain in panel B was tested on minimal medium (MM) agar containing the laccase substrate l-DOPA. (C) The WT and the Δasp1 strain were grown in MM broth to induce capsule production. (D) Mating filament production by the WT and the Δasp1 strain (MATα strains) was tested by performing a unilateral cross with WT strain KN99 (MATa) on V8 mating agar. Following strain mixing, the plates were incubated for 10 days and observed under a light microscope to assess the formation of mating filaments. Download
Histology of WT- and Δkcs1 mutant-infected lung. Lungs were removed postinfection, fixed, sectioned, and stained with periodic acid-Schiff (PAS) stain. Fungal cell bodies are dark pink and surrounded by a white halo, which may be capsule or alveolar space (white arrows). The day 7 Δkcs1 ×60 magnification image, where a budding Δkcs1 cell is observed (black arrow), represents the enclosed area demarcated by the square in the day 7 Δkcs1 ×10 magnification image. Areas of inflammation are indicated by black broken arrows. Download
The virulence of the Δasp1 strain in mice is similar to that of the WT. Anesthetized mice were inoculated intranasally with 5 × 105 CFU/20 µl of the indicated strains and euthanized after showing debilitating symptoms of infection. The Kaplan-Meier log rank test was used to establish that there was no significant difference (P = 0.587) in survival between WT- and Δasp1 strain-infected mice (the median percentages of survival of WT- and Δasp1 strain-infected mice were 14% ± 2.1% and 15% ± 1.6%, respectively). Cum, cumulative. Download
The absence of Kcs1 affects the association and uptake of cryptococcal cells by mammalian phagocytes. (A) Representative scatter plots used to quantify the extent of adhesion/uptake of the FITC-labeled fungal cells by THP-1 monocytes. Green fluorescence (FITC-A) is plotted against forward scatter (FSC-A). Populations demarcated by the black, purple, and red gates represent nonfluorescent THP-1 cells, free fluorescent fungal cells, and fluorescent fungal cells associated with THP-1 cells, respectively. (B) Reduced association and uptake of Δkcs1 mutant by THP1 cells and monocytes within a PBMC preparation following a 4-h coculture, as visualized by microscopy. Arrows indicate fungal cells, and arrowheads indicate mammalian cells. Download
Δkcs1 and Δarg1 cells exhibit increased susceptibility to antifungals. MICs were determined by assessing the growth of the WT and mutant strains in the presence of serially diluted antifungal compounds. AND, anidulafungin; AMB, amphotericin B; MF, micafungin; CAS, caspofungin; FC, flucytosine; PZ, posaconazole; VOR, voriconazole; IZ, itraconazole; FZ, fluconazole.
Primers used in this study. Uppercase nucleotides in the oligonucleotide sequences are complementary to the template, while lowercase nucleotides indicate added adaptor sequences.
RNA-seq analysis of the gene expression in the WT and Δkcs1 mutant. The data are based on the analysis of triplicate samples. FPKM values (fragments per kilobase of exon per million reads mapped) as a normalized measure of gene expression were generated by the Galaxy-based Cuffdiff tool. The difference in gene expression between Δkcs1 mutant and WT samples was considered significant if log2(Δkcs1 mutant/WT) was at least 1 or not more than −1, and the Q value (false-discovery rate at which the test result may be called significant) was <0.05.