

ABSTRACT
Vaginal inflammation (vaginitis) is the most common disease caused by the human-pathogenic fungus Candida albicans. Secretory aspartyl proteinases (Sap) are major virulence traits of C. albicans that have been suggested to play a role in vaginitis. To dissect the mechanisms by which Sap play this role, Sap2, a dominantly expressed member of the Sap family and a putative constituent of an anti-Candida vaccine, was used. Injection of full-length Sap2 into the mouse vagina caused local neutrophil influx and accumulation of the inflammasome-dependent interleukin-1β (IL-1β) but not of inflammasome-independent tumor necrosis factor alpha. Sap2 could be replaced by other Sap, while no inflammation was induced by the vaccine antigen, the N-terminal-truncated, enzymatically inactive tSap2. Anti-Sap2 antibodies, in particular Fab from a human combinatorial antibody library, inhibited or abolished the inflammatory response, provided the antibodies were able, like the Sap inhibitor Pepstatin A, to inhibit Sap enzyme activity. The same antibodies and Pepstatin A also inhibited neutrophil influx and cytokine production stimulated by C. albicans intravaginal injection, and a mutant strain lacking SAP1, SAP2, and SAP3 was unable to cause vaginal inflammation. Sap2 induced expression of activated caspase-1 in murine and human vaginal epithelial cells. Caspase-1 inhibition downregulated IL-1β and IL-18 production by vaginal epithelial cells, and blockade of the IL-1β receptor strongly reduced neutrophil influx. Overall, the data suggest that some Sap, particularly Sap2, are proinflammatory proteins in vivo and can mediate the inflammasome-dependent, acute inflammatory response of vaginal epithelial cells to C. albicans. These findings support the notion that vaccine-induced or passively administered anti-Sap antibodies could contribute to control vaginitis.
IMPORTANCE
Candidal vaginitis is an acute inflammatory disease that affects many women of fertile age, with no definitive cure and, in its recurrent forms, causing true devastation of quality of life. Unraveling the fungal factors causing inflammation is important to be able to devise novel tools to fight the disease. In an experimental murine model, we have discovered that aspartyl proteinases, particularly Sap2, may cause the same inflammatory signs of vaginitis caused by the fungus and that anti-Sap antibodies and the protease inhibitor Pepstatin A almost equally inhibit Sap- and C. albicans-induced inflammation. Sap-induced vaginitis is an early event during vaginal infection, is uncoupled from fungal growth, and requires Sap and caspase-1 enzymatic activities to occur, suggesting that Sap or products of Sap activity activate an inflammasome sensor of epithelial cells. Our data support the notion that anti-Sap antibodies could help control the essence of candidal vaginitis, i.e., the inflammatory response.
INTRODUCTION
Secretory aspartyl proteinases (Sap) of Candida albicans are considered to play important roles in the pathogenicity of this fungus. They constitute a family of at least 10 members with subfamilies, for instance, Sap1-3 and Sap4-6, each characterized by close homology and physiological relevance. Various lines of evidence have suggested that Sap expression enables the fungus to adhere and/or invade and damage host tissues and, perhaps more importantly, cause deviations in, if not exacerbate, host immunity (1–3). Although SAP gene expression studies and experimental systemic C. albicans infections with SAP-deleted mutants have generated some contradictory findings (4, 5), the pathological significance of Sap production by this fungus in mucosal infections rests upon rather consistent experimental and clinical evidence (6–8). In particular, Sap2 is a dominant member of the Sap family that is secreted at high levels in the vaginas of experimentally infected rats and in women affected by vulvovaginal candidiasis (VVC) (7, 8). Antibodies (Abs) against Sap2 are protective against experimental vaginal infection in estrogen-treated rats (9, 10). A virosomal preparation of a recombinant, enzymatically inactive Sap2 has been shown to confer protection in the above model and has been proposed as a vaccine against recurrent vaginal candidiasis (11). However, little is known about the mechanisms whereby Sap2 and other Sap contribute to the pathogenesis of vaginal candidiasis, and the mechanisms enabling anti-Sap immune responses to control vaginal candidiasis in experimental models are unclear.
Recent investigations by our group have shown that Sap2 (and other members of the Sap family) can induce an inflammatory response by human monocytes, macrophages, and dendritic cells in vitro, likely through activation of NLRP3 inflammasome and induction of different caspases (12, 13). However, it is unknown whether Sap exert proinflammatory activity in vivo and whether neutralization of this activity is relevant for protection against vaginal candidiasis, a disease characterized by typical signs and symptoms of acute inflammation (14–17).
To elucidate the mechanisms by which Sap cause vaginal inflammation in vivo, we used two recombinant forms of Sap2, one with an N-terminal-truncated, enzymatically inactive protein (tSap2, i.e., the antigenic constituent of the virosomal vaccine) and the other a full-length protein with preserved enzymatic activity (Sap2). Both proteins were directly injected into the vaginal cavity of mice under conditions that are predisposing to C. albicans infection (estrogen treatment) and at concentrations in the range of those found both in the vagina of experimentally infected rats and in the vaginal cavity of naturally infected women (8, 10). Other Sap were also tested and/or used as controls. We observed the mice for two classical signs of inflammation and inflammasome activation: polymorphonuclear cells (PMN, neutrophil) influx and the presence of cytokines, particularly interleukin-1β (IL-1β), in the vaginal fluid of mice. We also asked whether Sap2 and other Sap could be directly involved in the vaginal inflammation caused by C. albicans in our mouse model. We observed that the vaccine antigen tSap2 was devoid of proinflammatory activity in the mouse vagina and that anti-tSap2 Abs and Pepstatin A, a protease inhibitor known to inhibit Sap, were able to markedly reduce or abolish the inflammatory activity of the full-length Sap2 as well as the vaginal inflammation caused by C. albicans.
RESULTS
Proinflammatory activity of Sap2, but not tSap2, in the mouse vaginal cavity.
In previous studies, we found relatively high concentrations of Sap2 in the vaginal fluid of rats experimentally infected with C. albicans and in women with acute, recurrent vaginitis (in some subjects, concentrations even higher than 500 ng/ml in vaginal fluid were found) (7–9). Moreover, anti-Sap2 Abs conferred protection against experimental vaginal candidiasis, leading to the proposal of a Sap2-based anticandidal vaccine (3, 11). For these reasons and the availability of both a full-length, enzymatically very active recombinant Sap2 and a truncated, enzymatically inactive tSap2 as an appropriate control (see below; see also Fig. S1A in the supplemental material), we tested active and tSap2 versions for their capacities to induce vaginal inflammation (vaginitis) in mice. Following dose-finding experiments (data not shown), doses of 0.5 µg Sap2 and tSap2 were found to be optimal. Sap2 or tSap2 was directly injected into the mouse vaginal cavity, and vaginal fluid was harvested 24 h later and examined for numbers of GR-1-positive cells (PMN) and IL-1β concentrations. As a comparator and nonspecific marker of inflammation, lipopolysaccharide (LPS; 50 µg) was injected into the vaginal cavity of other mice (positive control), whereas mice injected with saline only served as negative controls. Figure 1 shows the cumulative data of all mice tested. Despite the expected variability, the graphs show that both Sap2 and LPS, but not tSap2, were capable of inducing a marked influx of PMN and IL-1β production in the mouse vagina compared to saline-injected mice. Based on these results, tSap2 was further used as a suitable negative control, whenever appropriate. In addition, we assayed for the presence of tumor necrosis factor alpha (TNF-α) upon intravaginal injection of Sap2 or LPS. Elevated levels of this cytokine were found following LPS stimulation, but it was not detected upon Sap2 stimulation (see Fig. S1B). Of note, the most active Sap2 concentration used in the above-described experiments fell in the range of Sap2 levels found in rats experimentally infected with C. albicans and those in clinical samples from women with candidal vaginitis (7, 8).
FIG 1 .
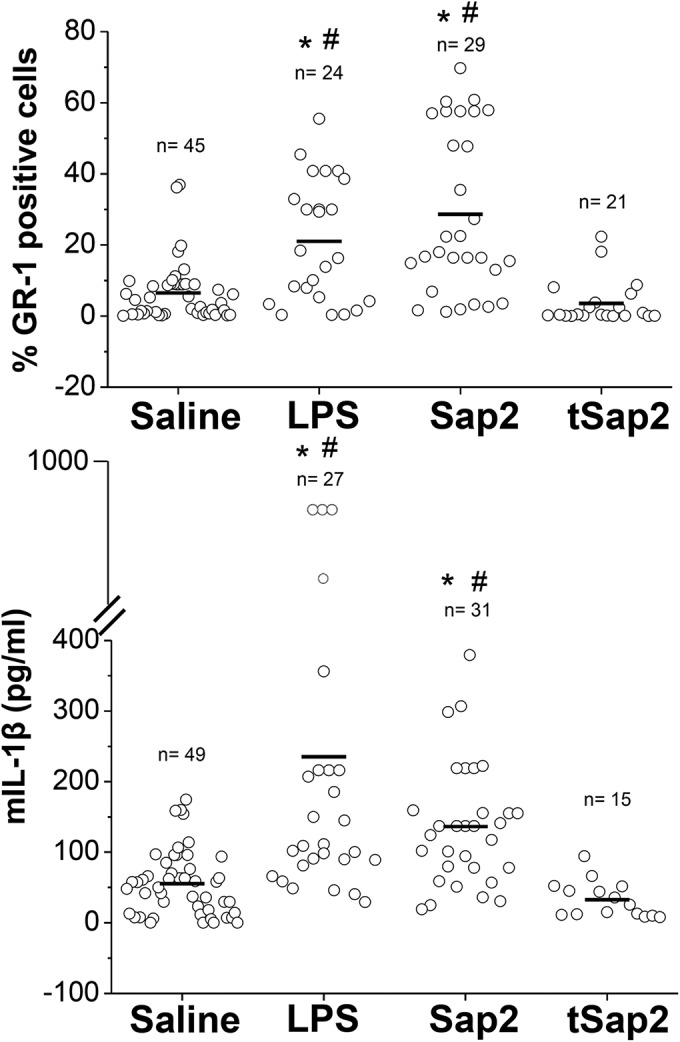
Proinflammatory activity of Sap2 in the mouse vaginal cavity. Dot plot graphs of the percentage of GR-1-positive cells (PMN) (upper panel) or IL-1β production (lower panel) from vaginal washes of mice after 24 h of intravaginal injection of saline, LPS (50 µg/10 µl/mouse), or Sap2 or tSap2 (both at 0.5 µg/10 µl/mouse). *, P < 0.01 for LPS- or Sap2-treated versus saline-treated mice; #, differences between LPS- and Sap2-treated animals were not statistically significant.
We also tested other recombinant Sap, in dose-response experiments (data not shown), to examine their capacity to induce vaginal inflammation. As also expected from Sap protein homology (1) and published data on monocyte inflammatory responses in vitro (12, 18), other Sap, in particular Sap1, Sap3, and Sap6, were shown to activate a similar response, but none of these proteinases caused a stimulation which was superior to the inflammation induced by Sap2 at doses compatible with those found in vivo (7, 8) (see Fig. S2 in the supplemental material).
Specificity of Sap2-induced vaginal inflammation and its inhibition by Sap2 binders.
Anti-Sap neutralizing Abs, induced by a virosomal, tSap2-based vaccine, have been suggested to be responsible for a protective response in a rat vaginitis model (6, 11). Thus, we wondered whether anti-Sap2 Abs, raised in rabbits immunized with the above vaccine, could directly inhibit Sap2-induced vaginal inflammation in mice. We also controlled for specificity of this serum by testing its effect on LPS-induced inflammation, and we used nonimmune serum as a negative control.
As shown in Fig. 2A and B, nonimmune and immune rabbit sera were unable to cause significant PMN influx or IL-1β production over the saline background in nontreated mice. In contrast, the immune serum caused significant inhibition of Sap2-stimulated PMN influx and IL-1β production, with no significant effect on stimulation by LPS. Since the serum may contain nonspecific Sap2 inhibitors that could be enriched in immune serum, more specific evidence was sought via the use of monoclonal Abs (MAbs). The Fab format was selected to avoid any potential interference due to Ab binding to Fc receptors of cells present in or recruited into the vaginal cavity. Two of these latter Abs were selected, one capable of binding Sap2 and inhibiting its enzymatic activity (HuCal I) and another one equally capable of Sap2 binding but unable to inhibit its enzymatic activity (HuCal nI). As shown in Fig. 2C and D, neither compound showed proinflammatory activity in itself; however, HuCal I nearly abolished Sap2-induced PMN influx (Fig. 2C) and significantly inhibited IL-1β production (Fig. 2D). On the contrary, HuCal nI caused a nonsignificant reduction of PMN influx and minor effects on IL-1β production.
FIG 2 .

Effect of immune serum, HuCal Abs, and Pepstatin A on Sap2 proinflammatory activity. Vaginal washes of mice treated for 24 h with saline, LPS (50 µg/10 µl/mouse), or Sap2 (0.5 µg/10 µl/mouse) in the presence or absence of nonimmune serum, immune serum, noninhibitory or inhibitory (HuCal nI or HuCal I) MAb, or Pepstatin A were analyzed for the percentage of GR-1-positive cells (A and C) or IL-1β production (B and D). Data are expressed as means ± SEM. *, P < 0.05 LPS- or Sap2-treated versus saline-treated mice; #, P < 0.05 for immune serum, HuCal I, or Pepstatin A plus Sap2 treatment, versus Sap2 treatment.
Both tSap2’s inability to cause inflammation and the marked difference between HuCal I and HuCal nI Fab suggested a critical role exerted by Sap2 enzymatic activity in the inflammation. Thus, we tested the effect of Pepstatin A, a well-known inhibitor of aspartyl proteinase activity. Data in Fig. 2C and D show that Pepstatin A had roughly the same inhibitory effects as the HuCal I Fab in decreasing PMN influx and cytokine production compared to the saline background. Neither HuCal I nor Pepstatin A inhibited LPS-induced inflammation (data not shown). Together with the data of the previous section, the results of these experiments suggest that proinflammatory Sap2 activity at the vaginal level is specific and requires functional enzymatic activity to cause inflammation. These data also suggest that the inability of some Sap (see Fig. S2 in the supplemental material) to cause vaginal inflammation could also be due to their low level of enzymatic activity or different specificities in the vaginal environment.
Sap and vaginal inflammation caused by C. albicans.
C. albicans cells, particularly in their hyphal morphological form, cause inflammation through inflammasome activation (19, 20), and this inflammatory response is currently considered the main pathogenic mechanism of experimental vaginal candidiasis in mice (19, 20). Given that Sap2 is largely secreted during vaginal infections, in both some experimental models and women (21), we first asked whether and to what extent the fungal cells caused inflammation in our experimental model, and also whether the Sap2 inhibitors shown in the previous section could also inhibit C. albicans-induced vaginal inflammation. To this aim, estrogenized mice were treated with anti-Sap2 immune serum or Pepstatin A or enzyme-inhibitory HuCal I and then challenged intravaginally with live C. albicans cells. The saline, nonimmune serum, and HuCal nI groups served as controls. Twenty-four hours after challenge, all mice were examined for neutrophils and IL-1β content in the vaginal fluid. As shown in Fig. 3A and B, the fungal cells (strain CA-6) induced marked PMN influx (Fig. 3A) and high IL-1β production (Fig. 3B), and these responses were nearly abolished (PMN) or strongly inhibited (the cytokine) by treatment with the anti-Sap2 immune serum or HuCal I or Pepstatin A. Some reduction of both PMN influx and IL-1β was also measured with nonimmune serum or HuCal nI, but this lowering did not reach statistical significance. We also examined whether the blockade of the IL-1β receptor was affected by C. albicans-induced neutrophil influx. As shown in Table 1, the blockade of IL-1β receptor, while obviously not affecting production of IL-1β, inhibited in a dose-dependent manner the PMN influx in the mouse vagina, strengthening the functional role of inflammasome-dependent cytokine production in our model.
FIG 3 .

Effects of immune serum, HuCal I, and Pepstatin A on vaginal inflammation induced by C. albicans (A and B) and on PMN influx in mice challenged with Δsap and control strains (C). Vaginal washes of mice treated for 24 h with saline or CA-6 (2 × 107 yeast cells/10 µl/mouse) in the presence or absence of nonimmune serum, immune serum, HuCal nI, HuCal I, or Pepstatin A were analyzed for the percentage of GR-1-positive cells (A) or IL-1β production (B). (C) PMN influx (percentage of GR-1 positive cells) of vaginal washes of mice challenged with CA-6, CAI4, or Δsap strains (2 × 107 yeast cells/10 µl/mouse). In all panels, data are expressed as means ± SEM. *, P < 0.05 for CA-6 or CAI4- or Δsap-treated versus saline-treated mice; #, P < 0.05 for immune serum, HuCal I, or Pepstatin A plus CA-6 treatment versus CA-6 treated mice (for bar graphs including these treatment groups); #, P < 0.05 for Δsap1 Δsap2 Δsap3 (Δsap1-3)-treated mice versus CAI4-treated mice (where these treatments groups are indicated in bar graphs).
Table 1 .
Effects of anakinra on CA-6-induced vaginal inflammation
| Endpoint | Treatmenta |
||||
|---|---|---|---|---|---|
| Saline | CA-6 | CA-6 + Anak at 1 µM | CA-6 + Anak at 10 µM | CA-6 + Anak at 100 µM | |
| % GR-1-positive cells | 5.61 ± 3.11 | 35.85 ± 6.83 | 59.42 ± 3.66 | 13.38 ± 3.77 | 6.63 ± 1.68 |
| IL-1β (pg/ml) | 24.36 ± 8.99 | 257.70 ± 91.30 | 176.9 ± 27.89 | 167.0 ± 37.29 | 170.66 ± 32.60 |
Vaginal washes from groups of six mice after 24 h of CA-6 challenge (2 × 107 yeast cells /10 µl/mouse) in the presence or absence of the indicated concentration of anakinra were analyzed for the percentage of GR-1-positive cells or IL-1β production. Saline-treated mice were used as controls. Data are expressed as means ± SEM.
Since (i) the inflammatory potential of Sap2 appeared to be shared, at least in part, by its close homologs Sap1 and Sap3 (see Fig. S2 in the supplemental material); (ii) both the immune serum and the HuCal I Ab also recognized Sap1 and Sap3 in addition to Sap2 (11) (data not shown), we next examined whether mutant strains of C. albicans with deletion of one or all three of the SAP1, SAP2, and SAP3 genes could retain the capacity to induce vaginal inflammation. In these experiments, the strain CAI4 (SC5314 genetic background), from which the SAP-deleted mutants were derived, was used as a positive control. Figure 3C shows that the deletion of single SAP genes did not abolish the inflammatory properties of the fungus and caused a slight, statistically not significant, reduction of neutrophil influx into the vaginal cavity compared to the parental strain. Only the deletion of all three genes caused complete loss of PMN influx, with values similar to those of the negative, saline-only injected control. The same data were obtained with the production of IL-1β (15.4 ± 7.8, 153.3 ± 22.9, and 20.9 ± 8.3 pg/ml of vaginal fluid in saline controls or CAI4 or triple mutant treated mice, respectively) (means ± standard errors of the means [SEM]). Overall, these findings suggest that the proinflammatory potential of C. albicans in the vagina can be mediated, though to different magnitudes, by each of the three proteins of the Sap1-3 subfamily. However, deletion of a single gene, including SAP2, is compensated by the expression of the other two genes. On the other hand, the lack of inflammatory activity by the triple mutant also suggests that most of the inflammatory potential of the fungus is due, under our experimental conditions, to the expression and activity of the SAP1, SAP2, and SAP3 genes. This conclusion is also in agreement with the observations reported above that IL-1β production and neutrophil influx in C. albicans-challenged mice are strongly inhibited by Abs recognizing the three Sap (Fig. 3A and B).
It is known that vaginal inflammation is, in the mouse model, largely uncoupled to the burden of intravaginal fungal growth (20, 22). Thus, we investigated whether this uncoupling was also verified in our CD1 mouse model, which is known to be relatively resistant to vaginal infection but not, as shown above, to vaginal inflammation caused by C. albicans. In these experiments, the capacities of Pepstatin A and enzyme-inhibitory Abs to inhibit C. albicans vaginal colonization, as reported in the rat model (9, 10), were tested by CFU enumeration and real-time imaging in vivo technology. These experiments requested obvious protocol variations (no vaginal wash at 24 h postchallenge and the first CFU measurement was on day +2, when counts are more stable and representative than on day 1) and the use of a luciferase-expressing mutant strain of the fungus (already shown to cause vaginal inflammation, similar to the strain used in the above-described experiments) (23). Saline, nonimmune serum, and noninhibitory HuCal nI, as well as fluconazole (FLZ), served as negative- and positive-control treatments, respectively. Fungal colonization was assayed on days 2 and 5 postchallenge. As shown in Fig. 4A to E, none of the anti-Sap2 reagents caused a significant reduction of the even-moderate intravaginal fungus burden, independent from the quantification protocol. Fluconazole exerted a moderate inhibitory effect that was detectable on day +2 at a statistically significant level only with the more sensitive bioluminescence imaging technique (24) and was detectable with both techniques on day +5. Determinations of intravaginal fungal loads on day +2 after challenge were also performed with the SAP-deleted mutants. We found that the fungal burden was moderate (around 4 log CFU/g of vaginal tissue) and did not significantly differ between the parental CAI4 strain and each single SAP deletion mutant (data not shown).
FIG 4 .

Effects of immune serum, HuCal I, and Pepstatin A on vaginal colonization by C. albicans. Anesthetized mice under the pseudoestrus condition were treated intravaginally 30 min before and again 30 min after challenge with gLUC59 (2 × 107 yeast cells/10 µl/mouse), with rabbit nonimmune or immune serum and anti-tSap2, HuCal nI or HuCal I, Pepstatin A, or FLZ. At 2 and 5 days postchallenge, mice were treated intravaginally with 10 µl of coelenterazine (0.5 mg/ml) and imaged with the IVIS-200TM imaging system under anesthesia with 2.5% isofluorane (A). Quantification of total photon emission from ROI was evaluated, and the statistical significance was determined (B and D). #, P < 0.05 for FLZ-treated versus saline-treated mice. Other groups of mice were treated as described above, and at 2 and 5 days postchallenge the fungal burdens of vaginas were evaluated based on CFU measurements, and the statistical significance was determined (C and E). Data are expressed as means ± SEM. #, P < 0.05 for FLZ-treated versus saline-treated mice.
Some mechanistic clues about Sap2-induced inflammation at the vaginal level.
Since (i) intracellular pattern recognition receptors (PRRs), including inflammasomes, play key roles in inflammation and anti-Candida defense (22, 25), and (ii) Sap2 (and other Sap) is capable of activating NLRP3 inflammasome in human monocytes and other immune cells (12), we examined whether the vaginal inflammation caused by Sap2 could also be attributed to inflammasome activation. To this end, we analyzed the cells of the vaginal exudate for the expression of activated caspase-1 and IL-1β by using flow cytometry. As shown in Fig. 5A, B, and C, the vaginal fluid harvested 24 h after Sap2 injection contained numerous epithelial cells (expressing the CD326 marker) that were positive for both active caspase-1 and IL-1β proteins. In addition, both caspase-1 and IL-1β expression levels in these cells were abolished by treatment with the caspase-1 inhibitor (IC-1) (Fig. 5B and C). This inhibitor did also almost completely block the production of another inflammasome-relevant cytokine, IL-18, in the vaginal fluid of Sap2-injected mice (Fig. 5F).
FIG 5 .

Sap2-induced inflammasome activation in vaginal epithelial cells. Vaginal washes of mice treated for 24 h with saline, LPS (50 µg/10 µl/mouse), or Sap2 (0.5 µg/10 µl/mouse) in the presence or absence of IC-1 were analyzed for the percentage of double-positive CD326+/caspase-1+ (A and B) and CD326+/IL-1β+ (A and C) cells, or for IL-18 production (F). Data are expressed as means ± SEM. *, P < 0.05 for LPS- or Sap2-treated versus saline treated mice and for IC-1 plus LPS- or Sap2-treated mice versus LPS- or Sap2-treated mice. For the gating strategy (A) of flow cytometry analysis, vaginal cells were gated on CD326-positive epithelial cells (based on side light scatter and CD326 staining; R1), caspase-1-positive cells (based on side light scatter and caspase-1 staining; R2), and IL-1β-positive cells (based on side light scatter and IL-1β staining; R3). Hence, the logical “AND” operator was used: for R4 results from the intersection between R1 and R2 and for R5 from the intersection between R1 and R3. For Western blotting, the A-431 cell line was untreated (NS) or treated for 2 h with LPS (10 µg/ml) plus ATP (5 mM) or Sap2 (20 µg/ml) in the presence or absence of IC-1 or Pepstatin A. After incubation, cell lysates were analyzed by Western blotting. Membranes were incubated with Abs to caspase-1 and actin. Caspase-1 results were normalized against those for actin (D). Culture supernatants were collected and tested for IL-1β production (E). Data are expressed as means ± SEM. *, P < 0.05 for LPS plus ATP or Sap2 treatment groups versus NS; #, P < 0.05 for IC-1 plus LPS plus ATP or Sap2 or for Pepstatin A plus Sap2-treated versus LPS- or Sap2-treated.
The activation of caspase-1 and the induction of IL-1β production in the human vaginal epithelial cell line A-431 treated with LPS plus ATP or Sap2 was also assayed by Western blotting (Fig. 5D). Both treatments induced caspase-1, and for both treatments IC-1 caused inhibition of caspase-1 cleavage (more manifest, however, with Sap2 than LPS under our conditions). Treatment of cells with Pepstatin A almost completely abolished caspase-1 activation (Fig. 5D) and IL-1β production (Fig. 5E) by Sap2-stimulated vaginal epithelial cells. Together with the lack of TNF-α induction (see above and Fig. 1B), these findings are indicative of inflammasome activation in Sap2-stimulated vaginal epithelial cells, although they do not specify which specific inflammasome complex (NLRP1 or -3 or NLRC4) is activated. However, it appears clear and in agreement with the markers of vaginal inflammation shown in previous sections, that Sap2 requires enzymatic activity for this effect to occur.
DISCUSSION
Secretory aspartyl proteinases (Sap) have long been considered key virulence traits of C. albicans, with rather strong experimental and clinical evidence for a major role in vaginal candidiasis (1, 3). However, the mechanisms by which this family of enzymes is involved in vaginal disease have remained unclear. Sap are active enzymes with a wide range of substrate specificities (26). Since some of these substrates (e.g., complement, histatins, and E-cadherin, and also Abs) play critical roles in both innate and adaptive immune responses, Sap expression is thought to enable the fungus to evade host immunity by enzymatic proteolysis of one or more of the above factors (1, 3). Concurrently, studies in well-established animal models and reconstituted human vaginal epithelia have provided indirect clues for a role of some members of the Sap family in facilitating fungus adherence and penetration into epithelial tissues (27–30). Evidence gathered with the use of anti-Sap Abs supports this proadherence role, although Sap are not consistently expressed on the C. albicans cell surface (10). It is relevant that anti-Sap Abs are not or are only erratically found either in serum or vaginal fluid of colonized or even infected subjects. To our knowledge, there has been no report on Abs capable of neutralizing Sap enzymatic activity in any animal or human organs or secretions. This is of particular importance for arguing for or against the assumption that most, if not all, of Sap pathogenic effects are mediated by their enzymatic activities.
In this study, we particularly focused on Sap2 as a potential proinflammatory factor in vaginal candidiasis. Sap2 is a member of the Sap family of C. albicans that is dominantly expressed under various in vitro and in vivo conditions, including experimental and clinical candidiasis (1, 31, 32). The high expression levels of this protein in women affected by vaginal candidiasis, as well as the capacity of anti-Sap2 Abs to confer some protection in animals intravaginally challenged with this fungus, have constituted the rationale for proposing a Sap2-based virosomal vaccine (11). This vaccine was proved to be safe and immunogenic in a phase 1 clinical trial in Switzerland and is currently under consideration for a combined anti-Candida vaccine by a U.S. company (Pevion, NovaDigm). For progress in this area, it will be important to understand how Sap2 and other Sap, particularly those closely related to and serologically cross-reactive with Sap2, participate in the pathogenesis of the disease and also which of the immune responses to the vaccine is critical for protection. The literature reported above, although of interest, does not address the immunological aspects of Sap-related pathogenicity mechanisms and the mechanisms of anti-Sap-mediated protection. In particular, it is unclear which of the anti-Sap immune responses is effective for controlling vaginal infection and whether this control is exerted through inhibition of C. albicans growth or direct inhibition of vaginal inflammation by the fungus, or both.
Although the capacity of some members of the Sap family to cause inflammation has long been suspected, and proinflammatory cytokine production upon Sap stimulation has been reported from studies in in vitro systems (33), only recently has there been a rather convincing demonstration that Sap2 and other Sap can induce a classical inflammatory cascade mediated by NLRP3 inflammasome activation in cultured human monocytes and other hematopoietic cells (12).
Based on the rationale above and the aforementioned in vitro evidence, here we have studied the ability of Sap proteinases to cause vaginal inflammation in vivo, focusing on Sap2 as a dominant member of the family and a proposed protective vaccine antigen. To this end, we used two recombinant Sap2 preparations: the virosomal vaccine antigen, a truncated version lacking the first N-terminal 76 amino acids, which is enzymatically inactive (tSap2) (34), and a full-length, enzymatically active protein (Sap2). These two proteins were directly injected into the mouse vaginal cavity, and classical signs of local inflammation such as the influx of PMN and the presence of a dominant proinflammatory cytokine, such as IL-1β, were monitored 24 h after Sap inoculation. By this approach, we obtained the following original findings in our vaginitis model: (i) Sap2, but not the vaccine antigen tSap2, is capable of inducing inflammation in the vaginal cavity that is substantially equivalent, in our model, to the inflammation induced by early colonization with C. albicans; (ii) anti-Sap2 Abs (cross-reactive with Sap1 and Sap3), as well as the Sap inhibitor Pepstatin A, can neutralize Sap2-induced vaginal inflammation; (iii) the same Abs and Pepstatin A equally inhibited C. albicans-induced inflammation, provided that, for both findings ii and iii, Abs, akin to Pepstatin A, were able to inhibit Sap enzymatic activity. We also show here for the first time that human-engineered anti-Sap2 Abs, in the Fab format, can inhibit Sap enzyme activity and inflammation and, by this mechanism, inhibit C. albicans-induced vaginal inflammation. The apparent similar effects of anti-Sap2 immune serum, Pepstatin A, and inhibitory HuCal I in dampening C. albicans-induced vaginal inflammation also suggest that sera from suitably immunized animals may indeed contain Sap enzyme-inhibitory Abs.
Both Sap2-induced inflammatory signs, i.e., neutrophil influx and cytokine production in the vaginal cavity, were obviously variable in our model system. This may have been related not only to the intrinsic variability of mouse responses but also to our own approach of using unprotected Sap2 administration. We selected this unconventional modality of Sap2 intravaginal administration to avoid vaginal stimulation or irritation inevitably caused by exogenous materials added to Sap2 preparations, such as protein-coating materials, gels, or preservants. Nonetheless, the evidence for inflammatory processes occurring in the vaginal cavity following Sap2 administration was consistent and reproducible when measured both as PMN recruitment and cytokine production. This proinflammatory activity did not appear to be mediated by occasional pyrogenic contaminants, as our Sap2 preparations were highly purified and essentially devoid of endotoxin. Importantly, the Sap2-specific binders capable of inhibiting both Sap2- and C. albicans-induced inflammation were unable to affect LPS-induced inflammation. This is also supported by the observation that only those Abs capable of inhibiting Sap enzymatic activity inhibited vaginal inflammation.
Our proposal that enzyme activity is critical for Sap2-induced proinflammatory activity in the vaginal environment is clearly supported by the inability of tSap2, which is devoid of enzymatic activity, to cause vaginal inflammation, as well as by the potent inhibition of inflammation caused by Pepstatin A, a well-known inhibitor of aspartyl proteinase and Sap enzyme activity (35). This would appear to make a rather remarkable and intriguing difference from Sap-induced inflammatory responses of human monocytes (and other immune cells) that do not require Sap enzyme activity (12). However, the different nature and functions of hemopoietic and vaginal epithelial cells should be considered. Monocytes, macrophages, and dendritic cells are capable of quickly internalizing particles or molecules by phagocytosis or other pathways, while vaginal epithelial cells are coated by mucus and a thick keratin-like material normally induced on the vaginal mucosa by estrogen treatment (36). It is possible that enzymatic activity is only required to hydrolyze the above-described material and allow Sap penetration by clathrin-dependent mechanisms, as has been suggested to occur in human monocytes (12). Notably, keratin and keratin-like proteins are exquisitely sensitive to Sap hydrolysis (6). It is also possible that vaginal inflammation is caused by Sap entering the epithelium and therein degrading an intracellular protein, or simply the result of a Sap-mediated degradation of one or more components of vaginal tissue or the vaginal microbiota. Inflammation could be contributed to, or enhanced by, cytokines or other mediators released not only by epithelial cells, but also from PMNs and other inflammatory cells recruited into the vaginal cavity. In fact, Sap2 is a PMN chemoattractant (37, 38). Studies by Yano and collaborators and Peters and collaborators (19, 20) have identified the calcium binding proteins associated with the alarmin response as the epithelial factors responsible for the vaginal influx of PMN. In theory, Sap2 enzymatic activity could favor the release of these proinflammatory mediators by the epithelial cells.
Importantly, our initial data on mechanisms of Sap2-induced vaginitis call into play a similar activation of an inflammasome sensor to that shown in previous studies with hematopoietic cells (12, 18). Caspase-1 is expressed in murine vaginal epithelial cells and a human vaginal epithelial cell line, with the consequent cleavage of pro-IL-1β into the secreted cytokine. Inhibition of caspase-1, therefore, causes the reduction of both IL-1β and another inflammasome-relevant cytokine, IL-18. The role of IL-1β and inflammasome activation for neutrophil recruitment into the vaginal cavity is further emphasized by the inhibition of PMN influx via the blockade of the IL-1β receptor. Nonetheless, the full elucidation of the mechanistic differences between hematopoietic versus epithelial inflammasome activation in response to Sap2 awaits further comparative studies, which will include the use of mice deficient in various inflammasome components.
A particularly important result of our study is that the early (24 h) vaginal inflammation caused by C. albicans is inhibited by the same Abs and Pepstatin A that inhibit Sap2-induced inflammation. Neither anti-Sap2 immune serum nor HuCal I MAbs react with the fungus itself and hence cannot inhibit adherence to or entry into the vaginal epithelium. Therefore, the block of inflammation cannot be explained by a reduced direct fungal contact. Coupled with the observation that Pepstatin A is also capable of strongly inhibiting PMN influx and IL-1β production during vaginal colonization by C. albicans, the data overall suggest that vaginal inflammation caused by this fungus in our mouse model is, at least in part, mediated by Sap2 and/or the closely related, antigenically cross-reactive proteinases Sap1 and Sap3. The observation that the triple SAP1 SAP2 SAP3 deletion mutant was unable to cause vaginal inflammation would suggest that, under our experimental conditions, vaginal inflammation caused by C. albicans is predominantly due to members of the Sap1-3 subfamily. However, these data should be interpreted within the context and limitations of our experimental model, which include a relatively early stage of fungal establishment in the mouse vagina (24 h) and the use of animals (CD1 mice) which are known to be relatively resistant to C. albicans vaginal infection (but not, as shown here, to vaginal inflammation). Therefore, we cannot exclude that mechanisms independent of Sap1-3 could cause C. albicans-mediated vaginal inflammation. Other Sap are produced by the fungus during the infection (1), and some of them can even prevail on Sap2 expression in other models of vaginal infection (39). They could add to or replace Sap2, given that Sap enzymatic activity appears to be the essential factor for inflammation to occur. In fact, Sap6 was also capable of inducing vaginal inflammation (see Fig. S2 in the supplemental material).
No PRR is known for Sap, but it has been shown that these proteins may enter the cell through clathrin-dependent pinocytosis, hence acting as intracellular danger signals (12). Some authors have suggested that inflammasome activation with its corollary of IL-1β production is critical to induce a potent Th17 response, with IL-17A and -F production in turn essential for control of infection. Nonetheless, contrasting data have been reported on this topic (23, 40, 41). In this context, it was logical to ask whether inhibition of Sap2-induced inflammation by immune serum or Pepstatin A had any effect on C. albicans vaginal growth and colonization. Our experiments showed that inhibition of Sap2-induced vaginal inflammation had no consistent effect on vaginal colonization, at least on its early, critical establishment stages, and when the burden of fungal cells, like in our CD1 mouse model, is not elevated (Fig. 4). Importantly, uncoupling of inflammation and fungal growth has been reported in murine experimental models (42), but not in other models of vaginal infection where anti-Sap Abs and Pepstatin A have been shown to limit C. albicans intravaginal growth (9). Nonetheless, pioneering work by Fidel’s research group with experimental infections in women suggests that the “inflammatory” mouse model may mimic inflammatory signs of human candidal vaginitis (43). Overall, our data demonstrate that Sap, in particular Sap2, or proteolytic products of Sap activity are proinflammatory molecules which can be recognized as danger signals by intracytoplasmic receptors and hence induce a potent inflammatory cascade in vivo. In women affected by vaginal candidiasis, inflammatory signs are dominant, and the findings illustrated in this study, linked to those of previous studies by ourselves and others, point to a dominant role of Sap as direct or indirect inducers of inflammation during C. albicans infection. The demonstration that C. albicans-induced inflammation may be inhibited by anti-Sap Abs corroborates the notion that a vaccine based on tSap2 antigen, hence inducing anti-Sap Abs, could be efficacious in limiting or controlling vaginal inflammation. This anti-inflammatory activity could usefully add to other protection-relevant properties, such as the capacity of inhibiting adherence and neutralizing other virulence properties expressed by Sap in vitro and in other models of candidal vaginitis (3). It should be noted that Abs inhibiting Sap2 enzyme activity have been found in women during the phase 1 clinical trial of the virosomal tSap2 vaccine (unpublished data).
MATERIALS AND METHODS
Ethics statement.
All animal experiments were performed in agreement with the EU Directive 2010/2063, the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes, and National Law 116/92. The protocol was approved by Perugia University Ethics Committee for animal care and use (Comitato Universitario di Bioetica; permit no. 149/2009-B). All the animals were housed in the animal facility of the University of Perugia (authorization 34/2003A).
Sap.
Recombinant full-length Sap2 was expressed in Escherichia coli BL21(pLysS) as a zymogen without the secretion header (39). Inclusion bodies were dissolved in 8 M urea and renatured by dialysis with 10 mM Tris (pH 8.0). Sap2 was autocatalytically activated by incubation at 37°C for 30 min in 50 mM sodium citrate (pH 3.2) and buffer exchange over a G25 Sephadex column (GE Healthcare) into 10 mM methylpiperazin (pH 5.2). Highly purified active Sap2 was eluted at 30 ms/cm (pH 5.2) from Q Sepharose FF (GE Healthcare) and was stored frozen. The in vitro proteolytic activity of the resulting protein was comparable to that of Pichia pastoris-expressed Sap2 when assessed as described by Wagener et al. (44): it was Pepstatin A sensitive, >90% inhibited by HuCal 10826 (see below) at a stereochemical HuCal:Sap2 ratio of 2:1 when preincubated for 30 min at neutral pH on ice, and it was insensitive to HuCal 10818 at a ratio of 4:1 under identical conditions.
An E. coli recombinant truncated Sap2 (tSap2) preparation was produced and purified as described by Sandini et al. (34). While tSap2 was enzymatically inactive (Fig. 1A), the anti-tSap2 Abs used throughout this study efficiently recognized and bound to Sap2, and some of them inhibited Sap2 enzymatic activity in vitro (see below). Where Sap2 has been indicated in our text (with or without the t prefix), it means that the indicated serum or MAb recognized with apparently similar reactivities both Sap2 and tSap2. Both Sap2 and tSap2 were essentially endotoxin free (<0.05 EU/ml), as determined in a Limulus amebocyte gel assay (45). Recombinant Sap1, Sap3, Sap5, Sap6, Sap9, and Sap10 were expressed in Pichia pastoris as described previously. Proteins were purified from P. pastoris culture supernatants via anion-exchange chromatography, followed by desalting by passage through a Sephadex G25 column, using 0.1 M citrate buffer (pH 4.5; rSap1, rSap3, and rSap6) or pH 5.5 (rSap9 and rSap10). rSap5 was purified via ultrafiltration (46, 47).
Human combinatorial Ab library (HuCal Abs 10818 and 10826).
The two recombinant HuCal Abs were produced by AbD-Serotec (Germany) after screening a human combinatorial phage-display library against tSap2. The HuCal Abs used in the experiment were bivalent Fab mini-Abs containing a heavy chain C-terminal dHLX dimerization domain followed by Myc and His6 tags. Both Abs bind specifically to tSap2 protein, both in native and denatured form. However, one of them (HuCal 10826; here, HuCal I) inhibited Sap2 enzymatic activity when assayed at a stereochemical HuCal I:Sap2 ratio of 2:1, whereas HuCal 10818 (referred to here as HuCal nl) bound to Sap2 but did not inhibit the enzymatic activity under the same condition. Neither HuCal I nor HuCal nI bound to C. albicans.
Rabbit sera.
A rabbit immune serum against tSap2 was generated by ProSci Inc. (CA, USA) in New Zealand white rabbits. The standard protocol included repeated immunization with 0.1 mg tSap2, adjuvanted with complete Freund’s adjuvant (CFA) for priming and incomplete Freund’s adjuvant (IFA) for the subsequent 3 boosts, respectively. Blood from the terminal bleeds from three immunized animals was pooled to yield the immune serum. The nonimmune serum was derived from three control rabbits that received PBS and adjuvant. The immune serum recognized with apparently similar efficiencies both the immunized antigen (tSap2) and Sap2 antigen in a standard ELISA. No serum binding to C. albicans in yeast or hyphal form was detected by immunofluorescence.
Candida albicans strains and culture.
The origin and characteristics of the highly virulent C. albicans strain used in this study (CA-6) have been previously described (48). C. albicans 1398 carrying the ACT1p-gLUC59 fusion (gLUC59) was also used, as previously described (49). Δsap1, Δsap2, and Δsap3 single deletion mutants (Δsap::hisG/Δsap::hisG::URA3::hisG) and a Δsap1 Δsap2 Δsap3 triple deletion mutant (Δsap1::hisG/Δsap1::hisG Δsap2::hisG/Δsap2::hisG Δsap3::hisG/ Δsap3::hisG and the integration of the plasmid CIp10) were used and compared to the CIp10-containing parental strain CAI4 (50–52). The cultures were maintained by serial passages on Sabouraud agar plus chloramphenicol (50 µg/ml) (both from Sigma-Aldrich). The yeast cells were harvested by suspending a single colony in saline, washing twice, and counting in a hemocytometer, and counts were adjusted to the desired concentration.
Mice.
Female CD1 mice, obtained from Harlan Nossan Laboratories (Milan, Italy), were purchased at 4 to 5 weeks of age. Mice were allowed to rest for 1 week before the experiment; by that time the animals were roughly 5 to 6 weeks old. Mice were used under specific-pathogen-free conditions that included testing sentinels for unwanted infections; according to the Federation of European, Laboratory Animal Science Association standards, no infections were detected.
Infection and treatment.
Mice were maintained under a pseudoestrus condition by subcutaneous (s.c.) injection of 0.2 mg of estradiol valerate in 100 µl of sesame oil (both from Sigma-Aldrich) 2 days prior to infection. Mice, anesthetized with 2.5 to 3.5 (vol/vol) isoflurane gas, were injected into the vaginal lumen close to the cervix, 30 min before and again 30 min after treatment with LPS (50 µg/10 µl/mouse) or Sap2, tSap2, or another Sap (all at 0.5 µg/10 µl/mouse), with rabbit nonimmune or immune serum anti-tSap2 (both diluted 1:500 in saline, with injection volumes of 10 µl/mouse), nonenzyme inhibitory HuCal nI, HuCal I strongly inhibiting Sap2 enzyme activity (both at 0.36 ng/10 µl/mouse), Pepstatin A (1 µg/10 µl/mouse; Sigma-Aldrich), or a selective caspase-1 inhibitor, Ac-YVAD-CMK (IC-1; 250 µM/10 µl/mouse; Dba Italia srl) (53). In selected experiments anesthetized mice were injected into the vaginal lumen, 30 min before and again 30 min after challenge with C. albicans (CA-6) or gLUC59 (both at 2 × 107 yeast cells/10 µl/mouse), with rabbit nonimmune or immune serum anti-tSap2 (both at a dilution of 1:500 in saline/10 µl/mouse), HuCal nI or HuCal I (both at 0.36 ng/10 µl/mouse), Pepstatin A (1 µg/10 µl/mouse), FLZ (60 µg/10 µl/mouse), or an antagonist of IL1beta receptor, anakinra (1, 10, or 100 µM). To favor vaginal contact and adsorption of the treatments, mice were held head down for 1 min following inoculation.
Monitoring of vaginal C. albicans burden.
At days 2 and 5 postinfection, mice were treated with 10 µl of coelenterazine (0.5 mg/ml in methanol-H2O [1:10]; SynChem, OHM) in the vaginal lumen. Afterward, mice were imaged with the IVIS-200TM imaging system (Xenogen Inc.) under anesthesia with 2.5% isoflurane. Total photon emission from vaginal areas within the images (region of interest [ROI]) of each mouse was quantified using the Living ImageR software package.
At days 2 and 5 postinfection, the fungal burdens of vaginas were also evaluated by plating serial dilutions of organ homogenates onto yeast extract-peptone-dextrose agar plus chloramphenicol (50 µg/ml) (both from Sigma-Aldrich) and counting the CFU.
Vaginal washes.
Twenty-four hours postinfection, the vaginal lumens were thoroughly washed with 150 µl of saline, given in three separate 50 µl volumes. The washes were centrifuged, and the supernatants were collected and tested for cytokine production. The total cellular fraction was used for the flow cytometry analysis.
Flow cytometry analysis.
Total cellular fractions obtained from vaginal washes were fixed with 1.5% formalin, washed, allowed to react with a fluorescein isothiocyanate-conjugated MAb to mouse Ly-6G (GR-1; 0.05 µg/test; rat IgG2bκ; eBioscience, Inc., San Diego, CA) for 20 min at room temperature (RT) in the dark. After incubation, cells were washed twice with fluorescence buffer (FB), resuspended in 0.5 ml of FB, and then analyzed by flow cytometry using a FACSCalibur system (Becton, Dickinson). Data are expressed as the percentage of GR-1-positive cells. In selected experiments, cells were fixed with 1.5% formalin for 10 min at RT, washed, and incubated with an allophycocyanin-labeled MAb to mouse CD326 (0.05 µg/test; rat IgG2bκ; eBioscience) for 20 min at RT in the dark. After incubation, cells were washed twice with FB, permeabilized with absolute methanol (500 µl/106 cells) for 10 min on ice, and incubated with purified Abs to mouse cleaved caspase-1 or IL-1β (both goat secondary Abs, at a dilution 1:50; Santa Cruz Biotechnology, Santa Cruz, CA) for 20 min at RT followed by phycoerythrin-labeled conjugated affinity-purified secondary Ab (dilution of 1:100; Chemicon Inc., Temecula, CA) (54). Briefly, for the gating strategy of flow cytometry analysis, cells were gated on CD326-positive epithelial cells (based on side light scatter and CD326 staining; R1), cleaved caspase-1-positive cells (based on side light scatter and cleaved caspase-1 staining; R2), and cleaved IL-1β-positive cells (based on side light scatter and cleaved IL-1β staining; R3). Hence, the logical “AND” operator, which indicates whether both operands are true, was used for R4 results from the intersection between R1 and R2, and for R5 from the intersection between R1 and R3. Autofluorescence was assessed by using untreated cells. Data are expressed as the percentage of positive cells. Control staining of cells with irrelevant Abs was used to obtain background fluorescence values.
Cytokine production.
The supernatants from vaginal washes were collected and tested for IL-1β, IL-18, and TNF-α levels by specific ELISAs (all from eBioscience). Cytokine titers were calculated relative to standard curves.
A-431 vaginal cell line.
The human A-431 vaginal epithelial cell line, obtained from ATCC (LGC Standards, Milan, Italy) were grown in Dulbecco’s modified Eagle’s medium plus 10% fetal calf serum as previously described (55, 56). For stimulation experiments, A-431 cells were grown in 6-well dishes, preincubated in serum-free medium for 2 h, and stimulated for 2 h at 37°C plus 5% CO2 with LPS (10 µg/ml) and ATP (5 mM) or Sap2 (20 µg/ml) in the presence or absence of IC-1 (25 µM) (12) or Pepstatin A (1 µg/ml).
Western blot analysis.
After stimulation, A-431 cells were recovered by adding trypsin-EDTA (BioWhittaker), washing, and lysing with mammalian protein extraction reagent in the presence of protease and phosphatase inhibitors (all obtained from Pierce, Rockford, IL). Protein concentrations were determined with a bicinchoninic acid protein assay reagent kit (Pierce). The lysates (60 µg) were separated by sodium dodecyl sulfate–10% PAGE and transferred to a nitrocellulose membrane (Pierce) for 1 h at 100 V in a blotting system (Bio-Rad, Hercules, CA). The membranes were incubated overnight with polyclonal Abs to human cleaved caspase-1 (goat) and cleaved IL-1β (goat) (both diluted 1:200; Santa Cruz Biotechnology) in blocking buffer. Detection was achieved with the appropriate secondary Ab coupled to horseradish peroxidase, followed by use of a chemilucent trial kit (Chemicon Inc.). Immunoblotting with rabbit polyclonal Ab to actin (dilution of 1:200; Santa Cruz Biotechnology) was used as an internal loading control. Immunoreactive bands were visualized and quantified by using a Chemidoc instrument (Bio-Rad). In particular, quantitative analysis of the region of interest was performed using the Fiji Is Just ImageJ program.
Determination of Sap2 and tSap2 activities.
Bovine serum albumin (BSA; 1% [wt/vol]) was incubated in citrate buffer (0.1 M, pH 3.2, at 37°C) for 15 min in the presence or absence of Sap2 or tSap2 (both at 100 µg/ml) with or without Pepstatin A (100 µg/ml). After incubation, BSA hydrolysis, performed by SDS-PAGE, was observed in the presence of Sap2 but not tSap2 (see Fig. S1A in the supplemental material).
Statistical analysis.
The in vivo results illustrated in bar graphs in the figures are reported as means ± SEM from 3 to 5 different mice in 3 to 4 separate experiments. For data reported as dot plot graphs, the numbers of analyzed mice for each treatment are indicated in the figure. Quantitative variables were tested for normal distribution and compared by means of Student’s two-tailed t test. A two-tailed test of significance at a P level of <0.05 was used to determine statistical significance.
SUPPLEMENTAL MATERIAL
Sap2 activity and effect of Sap2 on TNF-α production. Bovine serum albumin (BSA; 1% [wt/vol]) was incubated in citrate buffer (0.1 M, pH 3.2, at 37°C) for 15 min in the presence or absence of Sap2 or tSap2 (both at 100 µg/ml) with or without Pepstatin A. After incubation, BSA hydrolysis was performed via SDS-PAGE (A). The fold change of TNF-α production from vaginal washes of mice after 24 h of saline, LPS (50 µg/10 µl/mouse), or Sap2 (0.5 µg/10 µl/mouse) treatment is shown. SEM (always below 10%) are not shown. Download
Effects of different Sap on PMN recruitment. The percentages of GR-1-positive cells from vaginal washes of mice after 24 h of saline, LPS (50 µg/10 µl/mouse), Sap1, Sap2, Sap3, Sap5, Sap6, Sap9, or Sap10 treatment (all at 0.5 µg/10 µl/mouse) are shown. *, P < 0.05 for LPS- or Sap-treated mice versus saline-treated mice. Download
ACKNOWLEDGMENTS
The work described in this paper was supported in part by a grant from Fondazione Cassa di Risparmio, Umbria Region (2012.01.28.021), and the Gilead Fellowships Program, 2013, Catholic University, Rome, Italy.
Footnotes
Citation Pericolini E, Gabrielli E, Amacker M, Kasper L, Roselletti E, Luciano E, Sabbatini S, Kaeser M, Moser C, Hube B, Vecchiarelli A, Cassone A. 2015. Secretory aspartyl proteinases cause vaginitis and can mediate vaginitis caused by Candida albicans in mice. mBio 6(3):e00724-15. doi:10.1128/mBio.00724-15.
ADDENDUM
While our manuscript was being revised in preparation for submission, a paper appeared in mBio that described a role for NLRP3 inflammasome and hyphae-associated aspartyl proteinases in murine vaginal candidiasis [see V. M. Bruno, A. C. Shetty, J. Yano, P. L. Fidel, Jr., M. C. Noverr, and B. M. Peters, mBio 6(2):e00182-15, 2015, http://dx.doi.org/10.1128/mBio.00182-15].
REFERENCES
- 1.Naglik JR, Challacombe SJ, Hube B. 2003. Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol Mol Biol Rev 67:400–428, table of contents. doi: 10.1128/MMBR.67.3.400-428.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Bernardis F, Sullivan PA, Cassone A. 2001. Aspartyl proteinases of Candida albicans and their role in pathogenicity. Med Mycol 39:303–313. doi: 10.1080/mmy.39.4.303.313. [DOI] [PubMed] [Google Scholar]
- 3.Cassone A. 2014. Vulvovaginal Candida albicans infections: pathogenesis, immunity and vaccine prospects. BJOG 122:785–795. doi: 10.1111/1471-0528.12996. [DOI] [PubMed] [Google Scholar]
- 4.Lermann U, Morschhäuser J. 2008. Secreted aspartic proteases are not required for invasion of reconstituted human epithelia by Candida albicans. Microbiology 154:3281–3295. doi: 10.1099/mic.0.2008/022525-0. [DOI] [PubMed] [Google Scholar]
- 5.Correia A, Lermann U, Teixeira L, Cerca F, Botelho S, da Costa RM, Sampaio P, Gärtner F, Morschhäuser J, Vilanova M, Pais C. 2010. Limited role of secreted aspartyl proteinases Sap1 to Sap6 in Candida albicans virulence and host immune response in murine hematogenously disseminated candidiasis. Infect Immun 78:4839–4849. doi: 10.1128/IAI.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cassone A, Cauda R. 2012. Candida and candidiasis in HIV-infected patients: where commensalism, opportunistic behavior and frank pathogenicity lose their borders. AIDS 26:1457–1472. doi: 10.1097/QAD.0b013e3283536ba8. [DOI] [PubMed] [Google Scholar]
- 7.Cassone A, De Bernardis F, Mondello F, Ceddia T, Agatensi L. 1987. Evidence for a correlation between proteinase secretion and vulvovaginal candidosis. J Infect Dis 156:777–783. doi: 10.1093/infdis/156.5.777. [DOI] [PubMed] [Google Scholar]
- 8.De Bernardis F, Agatensi L, Ross IK, Emerson GW, Lorenzini R, Sullivan PA, Cassone A. 1990. Evidence for a role for secreted aspartate proteinase of Candida albicans in vulvovaginal candidiasis. J Infect Dis 161:1276–1283. doi: 10.1093/infdis/161.6.1276. [DOI] [PubMed] [Google Scholar]
- 9.De Bernardis F, Boccanera M, Adriani D, Spreghini E, Santoni G, Cassone A. 1997. Protective role of antimannan and anti-aspartyl proteinase antibodies in an experimental model of Candida albicans vaginitis in rats. Infect Immun 65:3399–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Bernardis F, Liu H, O’Mahony R, La Valle R, Bartollino S, Sandini S, Grant S, Brewis N, Tomlinson I, Basset RC, Holton J, Roitt IM, Cassone A. 2007. Human domain antibodies against virulence traits of Candida albicans inhibit fungus adherence to vaginal epithelium and protect against experimental vaginal candidiasis. J Infect Dis 195:149–157. doi: 10.1086/509891. [DOI] [PubMed] [Google Scholar]
- 11.De Bernardis F, Amacker M, Arancia S, Sandini S, Gremion C, Zurbriggen R, Moser C, Cassone A. 2012. A virosomal vaccine against candidal vaginitis: immunogenicity, efficacy and safety profile in animal models. Vaccine 30:4490–4498. doi: 10.1016/j.vaccine.2012.04.069. [DOI] [PubMed] [Google Scholar]
- 12.Pietrella D, Pandey N, Gabrielli E, Pericolini E, Perito S, Kasper L, Bistoni F, Cassone A, Hube B, Vecchiarelli A. 2013. Secreted aspartic proteases of Candida albicans activate the NLRP3 inflammasome. Eur J Immunol 43:679–692. doi: 10.1002/eji.201242691. [DOI] [PubMed] [Google Scholar]
- 13.Gabrielli E, Pericolini E, Luciano E, Sabbatini S, Roselletti E, Perito S, Kasper L, Hube B, Vecchiarelli A. 2015. Induction of caspase-11 by aspartyl proteinases of Candida albicans and implication in promoting inflammatory response. Infect Immun 83:1940–1948. doi: 10.1128/IAI.02895-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eckert LO. 2006. Clinical practice. Acute vulvovaginitis. N Engl J Med 355:1244–1252. doi: 10.1056/NEJMcp053720. [DOI] [PubMed] [Google Scholar]
- 15.Sobel JD. 1992. Pathogenesis and treatment of recurrent vulvovaginal candidiasis. Clin Infect Dis 14(Suppl 1):S148–S153. doi: 10.1093/clinids/14.Supplement_1.S148. [DOI] [PubMed] [Google Scholar]
- 16.Peters BM, Yano J, Noverr MC, Fidel PL Jr. 2014. Candida vaginitis: when opportunism knocks, the host responds. PLoS Pathog 10:e1003965. doi: 10.1371/journal.ppat.1003965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cassone A, Casadevall A. 2012. Recent progress in vaccines against fungal diseases. Curr Opin Microbiol 15:427–433. doi: 10.1016/j.mib.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pietrella D, Rachini A, Pandey N, Schild L, Netea M, Bistoni F, Hube B, Vecchiarelli A. 2010. The inflammatory response induced by aspartic proteases of Candida albicans is independent of proteolytic activity. Infect Immun 78:4754–4762. doi: 10.1128/IAI.00789-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yano J, Palmer GE, Eberle KE, Peters BM, Vogl T, McKenzie AN, Fidel PL Jr. 2014. Vaginal epithelial cell-derived S100 alarmins induced by Candida albicans via pattern recognition receptor interactions are sufficient but not necessary for the acute neutrophil response during experimental vaginal candidiasis. Infect Immun 82:783–792. doi: 10.1128/IAI.00861-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peters BM, Palmer GE, Nash AK, Lilly EA, Fidel PL Jr, Noverr MC. 2014. Fungal morphogenetic pathways are required for the hallmark inflammatory response during Candida albicans vaginitis. Infect Immun 82:532–543. doi: 10.1128/IAI.01417-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cassone A, De Bernardis F, Santoni G. 2007. Anticandidal immunity and vaginitis: novel opportunities for immune intervention. Infect Immun 75:4675–4686. doi: 10.1128/IAI.00083-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, Fitzgerald KA. 2009. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 5:487–497. doi: 10.1016/j.chom.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pietrella D, Rachini A, Pines M, Pandey N, Mosci P, Bistoni F, d’Enfert C, Vecchiarelli A. 2011. Th17 cells and IL-17 in protective immunity to vaginal candidiasis. PLoS One 6:e22770. doi: 10.1371/journal.pone.0022770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gabrielli E, Roselletti E, Luciano E, Sabbatini S, Mosci P, Pericolini E. 2015. Comparison between bioluminescence imaging technique and CFU count for the study of oropharyngeal candidiasis in mice. Cytometry A 87:428–436. doi: 10.1002/cyto.a.22666. [DOI] [PubMed] [Google Scholar]
- 25.Tomalka J, Ganesan S, Azodi E, Patel K, Majmudar P, Hall BA, Fitzgerald KA, Hise AG. 2011. A novel role for the NLRC4 inflammasome in mucosal defenses against the fungal pathogen Candida albicans. PLoS Pathog 7:e1002379. doi: 10.1371/journal.ppat.1002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aoki W, Kitahara N, Miura N, Morisaka H, Yamamoto Y, Kuroda K, Ueda M. 2011. Comprehensive characterization of secreted aspartic proteases encoded by a virulence gene family in Candida albicans. J Biochem 150:431–438. doi: 10.1093/jb/mvr073. [DOI] [PubMed] [Google Scholar]
- 27.Schaller M, Bein M, Korting HC, Baur S, Hamm G, Monod M, Beinhauer S, Hube B. 2003. The secreted aspartyl proteinases Sap1 and Sap2 cause tissue damage in an in vitro model of vaginal candidiasis based on reconstituted human vaginal epithelium. Infect Immun 71:3227–3234. doi: 10.1128/IAI.71.6.3227-3234.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Consolaro ME, Gasparetto A, Svidzinski TI, Peralta RM. 2006. Effect of Pepstatin A on the virulence factors of Candida albicans strains isolated from vaginal environment of patients in three different clinical conditions. Mycopathologia 162:75–82. doi: 10.1007/s11046-006-0026-9. [DOI] [PubMed] [Google Scholar]
- 29.Gropp K, Schild L, Schindler S, Hube B, Zipfel PF, Skerka C. 2009. The yeast Candida albicans evades human complement attack by secretion of aspartic proteases. Mol Immunol 47:465–475. doi: 10.1016/j.molimm.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 30.Villar CC, Kashleva H, Nobile CJ, Mitchell AP, Dongari-Bagtzoglou A. 2007. Mucosal tissue invasion by Candida albicans is associated with E-cadherin degradation, mediated by transcription factor Rim101p and protease Sap5p. Infect Immun 75:2126–2135. doi: 10.1128/IAI.00054-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schaller M, Schäfer W, Korting HC, Hube B. 1998. Differential expression of secreted aspartyl proteinases in a model of human oral candidosis and in patient samples from the oral cavity. Mol Microbiol 29:605–615. doi: 10.1046/j.1365-2958.1998.00957.x. [DOI] [PubMed] [Google Scholar]
- 32.Lian CH, Liu WD. 2007. Differential expression of Candida albicans secreted aspartyl proteinase in human vulvovaginal candidiasis. Mycoses 50:383–390. doi: 10.1111/j.1439-0507.2007.01384.x. [DOI] [PubMed] [Google Scholar]
- 33.Schaller M, Korting HC, Borelli C, Hamm G, Hube B. 2005. Candida albicans-secreted aspartic proteinases modify the epithelial cytokine response in an in vitro model of vaginal candidiasis. Infect Immun 73:2758–2765. doi: 10.1128/IAI.73.5.2758-2765.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sandini S, La Valle R, Deaglio S, Malavasi F, Cassone A, De Bernardis F. 2011. A highly immunogenic recombinant and truncated protein of the secreted aspartic proteases family (rSap2t) of Candida albicans as a mucosal anticandidal vaccine. FEMS Immunol Med Microbiol 62:215–224. doi: 10.1111/j.1574-695X.2011.00802.x. [DOI] [PubMed] [Google Scholar]
- 35.Pichová I, Pavlícková L, Dostál J, Dolejsí E, Hrusková-Heidingsfeldová O, Weber J, Ruml T, Soucek M. 2001. Secreted aspartic proteases of Candida albicans, Candida tropicalis, Candida parapsilosis and Candida lusitaniae. Inhibition with peptidomimetic inhibitors. Eur J Biochem 268:2669–2677. doi: 10.1046/j.1432-1327.2001.02152.x. [DOI] [PubMed] [Google Scholar]
- 36.Mosci P, Pietrella D, Ricci G, Pandey N, Monari C, Pericolini E, Gabrielli E, Perito S, Bistoni F, Vecchiarelli A. 2013. Mouse strain-dependent differences in estrogen sensitivity during vaginal candidiasis. Mycopathologia 175:1–11. doi: 10.1007/s11046-012-9589-9. [DOI] [PubMed] [Google Scholar]
- 37.El Messaoudi K, Thiry L, Van Tieghem N, Liesnard C, Englert Y, Moguilevsky N, Bollen A. 1999. HIV-1 infectivity and host range modification by cathepsin D present in human vaginal secretions. AIDS 13:333–339. doi: 10.1097/00002030-199902250-00005. [DOI] [PubMed] [Google Scholar]
- 38.Ran Y, Iwabuchi K, Yamazaki M, Tsuboi R, Ogawa H. 2013. Secreted aspartic proteinase from Candida albicans acts as a chemoattractant for peripheral neutrophils. J Dermatol Sci 72:191–193. doi: 10.1016/j.jdermsci.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 39.Koelsch G, Tang J, Loy JA, Monod M, Jackson K, Foundling SI, Lin X. 2000. Enzymic characteristics of secreted aspartic proteases of Candida albicans. Biochim Biophys Acta 1480:117–131. doi: 10.1016/S0167-4838(00)00068-6. [DOI] [PubMed] [Google Scholar]
- 40.Hernández-Santos N, Gaffen SL. 2012. Th17 cells in immunity to Candida albicans. Cell Host Microbe 11:425–435. doi: 10.1016/j.chom.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaffen SL, Jain R, Garg AV, Cua DJ. 2014. The IL-23–IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 14:585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Black CA, Eyers FM, Russell A, Dunkley ML, Clancy RL, Beagley KW. 1999. Increased severity of Candida vaginitis in BALB/c nu/nu mice versus the parent strain is not abrogated by adoptive transfer of T cell enriched lymphocytes. J Reprod Immunol 45:1–18. doi: 10.1016/S0165-0378(99)00017-0. [DOI] [PubMed] [Google Scholar]
- 43.Fidel PL Jr., Barousse M, Espinosa T, Ficarra M, Sturtevant J, Martin DH, Quayle AJ, Dunlap K. 2004. An intravaginal live Candida challenge in humans leads to new hypotheses for the immunopathogenesis of vulvovaginal candidiasis. Infect Immun 72:2939–2946. doi: 10.1128/IAI.72.5.2939-2946.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wagener J, Schneider JJ, Baxmann S, Kalbacher H, Borelli C, Nuding S, Küchler R, Wehkamp J, Kaeser MD, Mailänder-Sanchez D, Braunsdorf C, Hube B, Schild L, Forssmann WG, Korting HC, Liepke C, Schaller M. 2013. A peptide derived from the highly conserved protein GAPDH is involved in tissue protection by different antifungal strategies and epithelial immunomodulation. J Invest Dermatol 133:144–153. doi: 10.1038/jid.2012.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gabrielli E, Pericolini E, Cenci E, Monari C, Magliani W, Ciociola T, Conti S, Gatti R, Bistoni F, Polonelli L, Vecchiarelli A. 2012. Antibody constant region peptides can display immunomodulatory activity through activation of the Dectin-1 signalling pathway. PLoS One 7:e43972. doi: 10.1371/journal.pone.0043972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borg-von Zepelin M, Beggah S, Boggian K, Sanglard D, Monod M. 1998. The expression of the secreted aspartyl proteinases Sap4 to Sap6 from Candida albicans in murine macrophages. Mol Microbiol 28:543–554. doi: 10.1046/j.1365-2958.1998.00815.x. [DOI] [PubMed] [Google Scholar]
- 47.Albrecht A, Felk A, Pichova I, Naglik JR, Schaller M, de Groot P, Maccallum D, Odds FC, Schäfer W, Klis F, Monod M, Hube B. 2006. Glycosylphosphatidylinositol-anchored proteases of Candida albicans target proteins necessary for both cellular processes and host-pathogen interactions. J Biol Chem 281:688–694. doi: 10.1074/jbc.M509297200. [DOI] [PubMed] [Google Scholar]
- 48.Romani L, Mencacci A, Cenci E, Spaccapelo R, Mosci P, Puccetti P, Bistoni F. 1993. CD4+ subset expression in murine candidiasis. Th responses correlate directly with genetically determined susceptibility or vaccine-induced resistance. J Immunol 150:925–931. [PubMed] [Google Scholar]
- 49.Enjalbert B, Rachini A, Vediyappan G, Pietrella D, Spaccapelo R, Vecchiarelli A, Brown AJ, d’Enfert C. 2009. A multifunctional, synthetic Gaussia princeps luciferase reporter for live imaging of Candida albicans infections. Infect Immun 77:4847–4858. doi: 10.1128/IAI.00223-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hube B, Sanglard D, Odds FC, Hess D, Monod M, Schäfer W, Brown AJ, Gow NA. 1997. Disruption of each of the secreted aspartyl proteinase genes SAP1, SAP2 and SAP3 of Candida albicans attenuates virulence. Infect Immun 65:3529–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murad AM, Lee PR, Broadbent ID, Barelle CJ, Brown AJ. 2000. CIp10, an efficient and convenient integrating vector for Candida albicans. Yeast 16:325–327. doi: 10.1002/1097-0061(20000315)16:4<325::AID-YEA538>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 52.Fonzi WA, Irwin MY. 1993. Isogenic strain construction and gene mapping in Candida albicans. Genetics 134:717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schierle GS, Hansson O, Leist M, Nicotera P, Widner H, Brundin P. 1999. Caspase inhibition reduces apoptosis and increases survival of nigral transplants. Nat Med 5:97–100. doi: 10.1038/4785. [DOI] [PubMed] [Google Scholar]
- 54.Pericolini E, Gabrielli E, Bistoni G, Cenci E, Perito S, Chow SK, Riuzzi F, Donato R, Casadevall A, Vecchiarelli A. 2010. Role of CD45 signaling pathway in galactoxylomannan-induced T cell damage. PLoS One 5:e12720. doi: 10.1371/journal.pone.0012720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, Parks WP. 1973. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst 51:1417–1423. [DOI] [PubMed] [Google Scholar]
- 56.Graness A, Hanke S, Boehmer FD, Presek P, Liebmann C. 2000. Protein-tyrosine-phosphatase-mediated epidermal growth factor (EGF) receptor transinactivation and EGF receptor-independent stimulation of mitogen-activated protein kinase by bradykinin in A431 cells. Biochem J 347:441–447. doi: 10.1042/0264-6021:3470441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sap2 activity and effect of Sap2 on TNF-α production. Bovine serum albumin (BSA; 1% [wt/vol]) was incubated in citrate buffer (0.1 M, pH 3.2, at 37°C) for 15 min in the presence or absence of Sap2 or tSap2 (both at 100 µg/ml) with or without Pepstatin A. After incubation, BSA hydrolysis was performed via SDS-PAGE (A). The fold change of TNF-α production from vaginal washes of mice after 24 h of saline, LPS (50 µg/10 µl/mouse), or Sap2 (0.5 µg/10 µl/mouse) treatment is shown. SEM (always below 10%) are not shown. Download
Effects of different Sap on PMN recruitment. The percentages of GR-1-positive cells from vaginal washes of mice after 24 h of saline, LPS (50 µg/10 µl/mouse), Sap1, Sap2, Sap3, Sap5, Sap6, Sap9, or Sap10 treatment (all at 0.5 µg/10 µl/mouse) are shown. *, P < 0.05 for LPS- or Sap-treated mice versus saline-treated mice. Download