

Abstract
Antibodies are key reagents to investigate cellular processes. The development of recombinant antibodies and binders derived from natural protein scaffolds has expanded traditional applications, such as immunofluorescence, binding arrays, and immunoprecipitation. In addition, their small size and high stability in ectopic environments have enabled their use in all areas of cell research, including structural biology, advanced microscopy, and intracellular expression. Understanding these novel reagents as genetic modules that can be integrated into cellular pathways opens up a broad experimental spectrum to monitor and manipulate cellular processes.
Deciphering the inner workings of the cell requires specific molecular probes to measure the spatial and temporal dynamics of cellular structures. One class of probes that has been pivotal to modern cell biology is antibodies. Their ability to bind specifically to antigens such as proteins and even their posttranslational modifications (PTMs) has been exploited extensively to interrogate cellular function. For example, fluorescently labeled antibodies have been crucial staining reagents in molecular imaging techniques to reveal information on subcellular localization, abundance, and molecular interactions of biological antigens of interest. In addition, antibodies are essential to a vast range of biochemical analyses, including classic diagnostic techniques, such as Western blots and ELISA, but also to systems biology methods, such as mass spectrometry and ChIP-Seq (chromatin immunoprecipitation sequencing; Kidder et al., 2011), in which antibodies mediate the initial purification of the biological specimen.
Although conventional full-length antibodies are still the most widely used binding reagents for biochemistry and cell biology applications, their complex structural organization and their tedious manufacturing procedures have urged the development of new, alternative binding reagents. Such binders are either recombinantly generated immunoglobulin derivatives or synthetically designed from very different protein scaffolds. As such, recombinant binders are made to complement antibody-based fields of application or even enable completely new and innovative experiments. Of particular interest are binders that can be robustly expressed in living cells, a feature that is exclusive to small and stable binding molecules and cannot be performed easily with full-length antibodies, as a result of crucial inter- and intramolecular disulphide bridges that do not form in the cytoplasm. Thus, researchers have found a plethora of new applications in which binders have been combined with enzymatic or structural functionalities in living systems.
The development of in vitro screening techniques has been a decisive step for the rise and generation of recombinant binding reagents. These methods include classic phage display but also bacterial and yeast display as well as ribosomal and mRNA display. With such in vitro display techniques at hand, directed evolution strategies and genetic manipulation of binder sequences allow targeted engineering of key features, such as specificity, valence, affinity, and stability, enable derivatization toward smaller and more stable binding entities, and facilitate expression in heterologous hosts. Yet, it is the virtually limitless combinability of binder-mediated target recognition with any other chemical or biological function (viruses, translocation peptides, enzymes, structural proteins, dyes, toxins, and therapeutic agents) that opens up a whole universe of biotechnological innovations. Here, we provide a short overview of the most important recombinant binder formats and how they are generated followed by a few examples of how these valuable reagents can promote innovation and enable new discoveries in various fields of cell research. An in-depth discussion of molecular and structural aspects of some of these formats can be found in recent specialized reviews (Muyldermans, 2013; Plückthun, 2015).
In vitro binder selection with display techniques
Several polypeptide display techniques are available to identify antigen-specific binders in vitro. Although these methods use different biological vehicles, they share common features, including the capacity to screen large gene libraries, physical coupling of the encoding DNA sequence with its respective protein, and the possibility to increase binding specificity and affinity by repetitive mutagenesis and selection cycles (panning).
Phage display
Phage display, the most common display technique, involves the display of a recombinant binder library on the surface of bacteriophages upon genetic fusion with a viral coat protein. Individual phages comprise a defined binder on the surface and the respective gene within a phagemid inside the phage particle. Challenging this phage library with an immobilized antigen allows for in vitro selection of specific binders that can be amplified and identified by reinfection of Escherichia coli. Most widely used are M13 filamentous phages.
Bacterial and yeast display
Both techniques rely on fusing the gene of the binder library to respective surface proteins. In contrast to phages, bacteria and yeast can be screened via flow cytometry. Displaying binders on yeast is additionally advantageous because the expression is mediated by a eukaryotic machinery, which has been demonstrated to be more effective in comparison to phage display (Bowley et al., 2007).
Ribosome and mRNA display
Ribosome display involves the in vitro transcription and translation of a DNA library. A genetic trick allows immobilization of the translated, correctly folded protein on the ribosome in a noncovalent complex with the respective mRNA, thereby providing the necessary genotype–phenotype link. The variable pool of binder–ribosome–mRNA complexes is then incubated with immobilized antigen, and specific binders are amplified by reverse transcription.
mRNA display differs from ribosome display in that the in vitro translated polypeptide is covalently linked to its cognate mRNA via puromycin, a tRNA mimetic. In contrast to other display techniques, ribosome and mRNA display are independent of E. coli transformation and the accompanying potential loss of library diversity. In addition, the reverse transcription and PCR amplification steps in between successive selection cycles introduce sporadic mutations that mimic an affinity maturation step.
Development of different recombinant binders
Immunoglobulin derivatives
Naturally produced immunoglobulins (IgG, IgM, IgA, IgD, and IgE) areuniversal weapons against pathogenic threats. The predominant isotype in nature is IgG, a 150-kD multichain/multidomain protein. IgG consists of two heavy chains and two light chains with variable domains (VH and VL; Fig. 1 A). The binding occurs via six (three in VH, three in VL) complementarity determining regions (CDRs). Derivatization of IgG allows the generation of functional Fab (∼50 kD), scFv (∼25 kD), and single, variable domain VH or VL fragments (Fig. 1 A). In Fab and scFv formats, the antigen binding surface is established via specific association of the VH and the VL domain, mediated by hydrophobic framework residues (Chothia et al., 1985). The necessity of noncovalent interdomain interactions for functional domain assembly impairs the thermodynamic stability of Fabs and scFvs (Willuda et al., 1999; Wörn and Plückthun, 2001). This problem is aggravated with isolated VH and VL domains, as these hydrophobic residues are exposed at the surface and thus decrease protein solubility. Therefore, functional applications of single domain antibodies derived from IgGs often require dedicated protein engineering (Tanha et al., 2006).
Figure 1.

Comparison of immunoglobulin derivatives and nonimmunoglobulin binder formats. (A) Immunoglobulins and their derivatives. (left) A conventional IgG molecule is comprised of two heavy and two light chains. Heavy chains comprise three constant domains (CH1–3, dark blue) and one variable domain (VH, dark green); light chains comprise one constant (CL, light blue) and one variable domain (VL, light green). The naturally functional antigen binding unit is formed by noncovalent association of the VH and the VL domain. This association is mediated by hydrophobic framework regions, indicated in pink. IgG can be derivatized to Fab, scFv, and single domain VH or VL binders. (right) Heavy chain antibodies (hcAb) are found in Camelidae, lack the light chain and the CH1 domain, and comprise a single, antigen binding domain, the VHH domain. (B) Nonimmunoglobulin binders are based on natural and designed protein scaffolds. Shown are fibronectin-derived Adnectins/monobodies that are characterized by an Ig-like β-sandwich structure, anticalins that are based on the lipocalin fold, affibodies that derive from protein A and comprise three α helices, and DARPins, which are designer proteins composed of ankyrin repeats. The randomized residues that mediate the respective ligand binding are marked in red. Protein Data Bank accession numbers are given in parentheses. N, N terminus; C, C terminus.
As a result of these biochemical limitations of IgGs and their derivatives, the discovery of naturally evolved heavy chain antibodies (hcAbs) derived from camels (Hamers-Casterman et al., 1993) has raised great interest in the recombinant antibody field (Fig. 1 A). The functional antigen-binding unit of hcAbs is reduced to only one single variable domain (VHH domain; nanobody). VHHs have a size of ∼13–14 kD and have evolved biochemical features that are favorable for a wide range of biotechnological applications. These properties include the substitution of hydrophobic with hydrophilic residues in framework regions (Muyldermans et al., 1994; Vu et al., 1997; Harmsen et al., 2000), increasing the overall stability and solubility of these single domain antibodies and allowing robust, heterologous expression in bacterial hosts and functional expression in eukaryotic cells (Rothbauer et al., 2006). Moreover, the mode of binding tends to differ from conventional antibodies. Conventional IgG paratopes often form cavities, grooves, or flat surfaces to bind small chemical groups, peptides, and planar epitopes on large folded proteins, respectively (Sundberg and Mariuzza, 2002). In contrast, the prolate shape of a VHH exposes a convex paratope that is well suited to bind cavities or cryptic epitopes, which are likely unavailable for bulkier IgG paratopes as was exemplified for the blocking of the active site of lysozyme and the recognition of elusive structures of pathogenic trypanosomes (Nguyen et al., 2000; Stijlemans et al., 2004; De Genst et al., 2006).
Nonimmunoglobulin binders
The specific needs of the various fields of application as well as patenting issues motivated the development of several alternative binder formats that are based on defined nonimmunoglobulin protein folds. Here, we describe a few that are well established and suited for cellular research (Fig. 1 B).
There are different strategies to engineer completely new classes of antibody mimetics. On the one hand, researchers have used natural protein folds as universal scaffolds for the generation of recombinant binding reagents. For example, the 10-kD fibronectin protein fold serves as a template for the bioengineered Adnectins/monobodies (Fig. 1 B). They are structured similarly to immunoglobulin domains with seven β sheets and three CDR-like loops located on the top of the barrel-like fold (Koide et al., 1998). Genetic randomization of the CDR-like loops allows the generation of libraries from which to retrieve binders with desired specificities after in vitro display, as was for example demonstrated for a monobody directed against the Abl SH2 domain (Wojcik et al., 2010). In contrast to variable Ig domains, the fibronectin structure does not depend on intramolecular disulphide bridges, thus facilitating functional applications in reducing environments such as the cytoplasm of living cells (Gross et al., 2013).
Another example of a synthetic binding reagent is anticalins, which derive from the 20-kD lipocalin fold (Beste et al., 1999). Lipocalins are eight-stranded β-barrel structures that mediate binding and transport of small molecules in their endogenous environment. Upon randomization of the four natural binding loops, the synthetic variant may be designed to specifically recognize antigens of interest with high affinities. The goblet-like binding pocket significantly differs from antibody paratopes (Fig. 1 B). Thus, the anticalin format is considered especially useful to generate binders against small molecules (Korndörfer et al., 2003) and protruding conformational epitopes (Eggenstein et al., 2014).
Another class of affinity proteins has been designed based on the immunoglobulin binding protein A from Staphylococcus aureus. These so-termed affibodies comprise three α helices without disulphide bonds and have a molecular mass of ∼6.5 kD (Fig. 1 B). Random mutagenesis of defined residues within the two binding helices allows screening for target-specific binding reagents for biotechnological use (Nord et al., 1997).
In terms of size and binding mode, all aforementioned binder formats are more or less restricted to the fold of their natural template. On the other hand, engineering repeat proteins as recombinant antibody mimetics allows more modular design strategies. The basic principle of repeat binders lies within the consecutive arrangement of multiple repeat units (Fig. 1 B). The overall length of the repeat protein may be adjusted with respect to specific target properties. A prominent example for repeat protein binders are designed ankyrin repeat proteins (DARPins). DARPins are based on the ankyrin fold, which naturally mediates protein–protein interactions (PPIs) and has been observed in diverse protein families (Li et al., 2006; Al-Khodor et al., 2010). The structural subunit of a DARPin consists of a β turn followed by a pair of antiparallel α helices and a loop, typically comprising 33 amino acids. Randomization of defined helix residues enables selection of high-affinity binders (Binz et al., 2003; Forrer et al., 2003; Kohl et al., 2003). In contrast to barrel-like binder folds with antigen-reactive loop structures, DARPins form slightly concave binding surfaces that favor large, conformational epitopes. As a result of the complete absence of disulphide bridges, DARPins are well suited as potential intracellular binders (Parizek et al., 2012) and can be expressed in large amounts in bacteria.
In addition to these established formats, a plethora of novel protein scaffolds has been developed and includes recombinant binders such as avimers (Silverman et al., 2005), affilins (Ebersbach et al., 2007), fynomers (Grabulovski et al., 2007), affitins (Mouratou et al., 2007), knottins (Smith et al., 1998), armadillo repeat proteins (Parmeggiani et al., 2008), and the very recently published adhirons, which can be stably produced in large amounts (Tiede et al., 2014). As a complement to conventional antibodies, man-made recombinant binders, based on immunoglobulin or nonimmunoglobulin folds, open up new possibilities for the life sciences. The available formats have common and unique properties (Table 1) that can be used to choose the best format for a given application. For all formats, however, the success in retrieving a potent binder with a desired target specificity largely depends on the library size as well as the techniques and conditions used for their screening.
Table 1.
Recombinant binders and their molecular characteristics
| Format | Size | Template | Structure (disulphides?) | Binding mode | Characteristic features |
|---|---|---|---|---|---|
| kD | |||||
| Ig derivative | |||||
| Fab | ∼50 | IgG | Four Ig domains, β-sandwich (yes) | Via six CDR loops, VH/VL interface | Nonrecombinant generation by papain digest of IgG is possible |
| scFv | ∼25 | IgG | Two Ig domains , β-sandwich(yes) | Via six CDR loops, VH/VL interface | A synthetic link stabilizes the noncovalent interaction between VH and VL domains |
| VH | ∼13 | IgG | β-sandwich (yes) | Three VH CDR loops | Single domain antibody; exposed hydrophobic stretch |
| VL | ∼13 | IgG | β-sandwich (yes) | Three VL CDR loops | Single domain antibody; exposed hydrophobic stretch |
| VHH | ∼13 | hcAb | β-sandwich (yes) | Three VHH CDR loops | Small, stable Ig antigen binding unit; recognition of cryptic epitopes |
| V-NAR | ∼9 | Ig-NAR | β-sandwich (yes) | Three V-NAR CDR loops | Small, stable Ig antigen binding unit; recognition of cryptic epitopes |
| Non-Ig binders | |||||
| Monobodies | ∼10 | Fibronectin | β-sandwich (no) | Three CDR-like loops | Ig-like structure lacking disulphides |
| Anticalins | ∼20 | Lipocalin | Goblet-like β-barrel (yes) | Four CDR-like loops | Binding pocket for small molecules and protruding epitopes |
| Affibodies | ∼6.5 | Protein A | Three α-helices (no) | helix-mediated | Bacterial origin, smallest recombinant binder format |
| DARPins | ∼18 | Ankyrin repeat | Helix-turn-helix (no) | planar, flexible surface | Repetitive, modular design |
Applications of recombinant binders in molecular and cellular biology
Cellular proteomics
The analysis of proteomes relies on the availability of high-quality binding reagents. They are necessary for the molecular analysis of specific target proteins and their spatiotemporal cellular abundance using standard detection methods such as microscopy and Western blot analysis. Furthermore, binder-mediated affinity purification in combination with systems biology techniques has contributed to a comprehensive understanding of cellular interactomes in development and disease. Although such experiments were historically performed with conventional antibodies, the use of recombinant binding reagents for proteomic application becomes increasingly popular, and international consortia have been installed with the long-term goal to cover the entire human proteome with renewable binders (Taussig et al., 2007; Colwill and Gräslund, 2011).
Biochemical and proteomic analyses of crude samples include affinity-based purification or depletion of specific components using chromatography columns that, however, require large amounts of affinity material. Thus, expensive monoclonal and polyclonal antibodies are less attractive to purify or deplete endogenous proteins, whereas inexpensive immobilized-metal affinity chromatography (IMAC) systems, such as the Ni-NTA (nitrilotriacetic acid)/His-tag system, are widely used but also restricted to artificially tagged proteins. Consequently, recombinant binders that can be produced at low costs in high quantities offer valuable alternatives for immunoaffinity chromatography of biological samples (Blank et al., 2002; Grönwall et al., 2007).
The rise of comprehensive proteomic analyses started with the advent of protein microarrays enabling quantification of cellular protein components (Fig. 2 A). However, array analyses of complex protein samples require highly sensitive and specific detection reagents to measure low-abundant proteins (Fig. 2 A). In terms of recombinant probes, high-affinity scFvs have proven especially useful for protein arrays (Wingren et al., 2007) and allowed for biomarker profiling of cancers (Ingvarsson et al., 2008; Carlsson et al., 2010) and autoimmune diseases (Carlsson et al., 2011). However, the high cost per array is still the limiting factor in protein array technologies.
Figure 2.

Applications of recombinant binders in biochemistry and structural biology. (A) Use of recombinant binders for purification and detection in vitro. Fields of applications involve classic capture assays such as ELISA, high-throughput screening (HTS) techniques and immunoprecipitation (IP) as well as proteomic and array technologies. (B) Recombinant binders for assisted crystallography. Shown are three different structures and binder formats (Nanobody, Affibody, and DARPin) that have been used for the elucidation of challenging structures. Protein Data Bank accession numbers are given in parentheses.
State-of-the-art proteomics nowadays mostly relies on the use of mass spectrometry to analyze complex protein mixtures (Walther and Mann, 2010). Affinity tools are used in mass spectrometry–based proteomics to purify or enrich defined targets and subsequently identify PTMs and interacting factors (Fig. 2 A). To bypass the time-consuming generation of specific binders, proteins are often tagged with established foreign epitopes. The use of GFP as tag in cellular proteomics allows the experimental link between live cell microscopy and proteomic analysis (Cristea et al., 2005) and a high-affinity GFP-binding nanobody—termed GFP binding protein (GBP)—enables highly efficient one-step purification of GFP fusion proteins to study PPIs and protein–DNA interactions (Rothbauer et al., 2008; Trinkle-Mulcahy et al., 2008; Frauer and Leonhardt, 2009; Pichler et al., 2012). Similarly, an RFP-specific nanobody was genetically immobilized to biogenic, magnetic nanoparticles produced by magnetotactic bacteria (Pollithy et al., 2011) for efficient magnetic bead pull-down of RFP fusion proteins.
Recent efforts demonstrated that proteomics may be used to identify and generate recombinant binders by combining next generation DNA sequencing with mass spectrometry analysis of immune repertoires (Fridy et al., 2014). For this purpose, nanobody cDNA libraries from an immunized llama were sequenced and matched with the respective antigen-specific hcAb peptide sequences determined by mass spectrometry. This approach yielded high-affinity GFP- and mCherry-specific nanobodies, which complement the available toolbox of fluorescent protein (FP)–specific nanobodies (Rothbauer et al., 2006; Kirchhofer et al., 2010) and DARPins (Brauchle et al., 2014). Advances in the analysis of single-molecule complexes with DNA-barcoded proteins (Gu et al., 2014) might pave the way toward massive parallel screening of antibody libraries against antigen libraries and thus significantly boost the throughput during binder development.
Structural biology
High-resolution structural information, preferably provided by x-ray crystallography, is essential for a thorough understanding of complex biological systems. Unfortunately, high-quality diffracting crystals of intrinsic disordered or flexible proteins, transient protein assemblies, multiprotein complexes, and membrane proteins are difficult to produce even when highly purified recombinant proteins are available. Nevertheless, the crystallization of such recalcitrant proteins is now possible as a result of the recognition of the beneficial effect of auxiliary affinity reagents. The early successes with Fab (Kovari et al., 1995) or Fv (Ostermeier et al., 1995) as so-called crystallization chaperones have been extended and repeated on a large scale with nanobodies, monobodies (FN3 domains), DARPins, and to a lesser extent, also anticalins and affibodies (Fig. 2 B).
The efficacy of these affinity reagents to assist the crystallization process originates from their shared favorable properties: facile identification and large-scale production of stable and soluble target binders. The DARPins, monobodies, anticalins, and affibodies have an additional advantage over nanobodies, Fabs, and scFvs because they lack disulphide bridges, allowing standard cytoplasmic expression to produce functional chaperones. Apart from these shared critical factors to initiate crystallography screens, the chaperones may promote crystal formation by a variety of possible mechanisms that will improve the surface area capable to form crystal contacts (Korotkov et al., 2009; Uysal et al., 2009; Derewenda, 2010) and lower the entropic cost of lattice formation. In particular, recombinant binders might facilitate crystals by: (a) increasing the total amount of structured polypeptide (Loris et al., 2003) or by organizing intrinsically disordered proteins (Skrabana et al., 2010; Serrière et al., 2011; Abskharon et al., 2014); (b) reducing the inherent conformational heterogeneity within the sample after selection and fixation of one native conformer in the ensemble (Zhou et al., 2001; Uysal et al., 2009; Rasmussen et al., 2011a,b; Kruse et al., 2013) or by minimizing local flexibility (Chaikuad et al., 2014); (c) shielding highly charged and flexible regions such as in the Polo-like kinase 1 crystal structure (Bandeiras et al., 2008); (d) masking problematic peptide patches with unwanted self-polymerizing propensity (Hoyer et al., 2008; Domanska et al., 2011; Baranova et al., 2012); and (e) introducing or by extending a hydrophobic and rigid surface area available to form crystal contacts (Sennhauser et al., 2007; Bandeiras et al., 2008; Schönfeld et al., 2009; Veesler et al., 2009; Rasmussen et al., 2011a; Koide et al., 2012; Krishnamurthy and Gouaux, 2012).
Especially, β-rich chaperones (nanobodies and monobodies) have a pronounced tendency to form intermolecular self-assemblies via an exposed edge of their β sheet (Tereshko et al., 2008). However, DARPins were shown to be involved in crystal packing as well (Veesler et al., 2009).
The availability of these diverse types of chaperones permits the generation of binders against all possible topologies. It is well established that the convex binding surface of nanobodies is better suited to interact with concave surfaces on the antigen, whereas DARPins use the opposite mode of interaction. The different interactions of a DARPin and a nanobody with the same receptor binding protein of P2 bacteriophage illustrate this nicely (Spinelli et al., 2006; Veesler et al., 2009). Interestingly, the monobody recombinant binder can be developed to associate with its target in a loop-focused fashion (like nanobodies) or with the combination of one solvent exposed loop and the side of the domain (Koide et al., 2012). These different libraries (“loops-only” or “side and loop”) are expected to generate binders recognizing different epitope architectures. Engineered anticalins, with a goblet-like shaped target-recognizing structure, favor protruding epitopes as was shown in the structure of the complex with human fibronectin ED-B (Extra Domain-B; Gebauer et al., 2013). Finally, affibodies seem to prefer a flat interacting surface (Hoyer et al., 2008). Therefore, all these chaperones should be considered complementary to each other, rather that competitive, to tackle the most difficult proteins to crystallize and to improve the diffraction quality. Moreover, different crystals of the same target protein complexed with alternative chaperones or with the same type of chaperone associated at different epitopes are instrumental to identify various alternative conformations of macromolecules, which might be linked to mechanism of action (Pecqueur et al., 2012; Chaikuad et al., 2014) or to degenerate binding with alternative partners.
Apart from the availability of various libraries of recombinant binders, also multiple dedicated selection protocols have been developed to retrieve the best possible candidate chaperone for protein crystallization (Kim et al., 2011; Paduch et al., 2013; Pardon et al., 2014). With this toolbox (libraries of different recombinant binders and dedicated selection methods) at hand, several crystallization chaperones have been identified, and an impressive and expanding list of high-resolution structures of intact proteins that were otherwise difficult to crystallize has been collected.
Affinity-based biosensors, inhibitors, and imaging tools
Fluorescent biosensors are essential analytical tools to study molecular target structures at the cellular level. Although conventional antibodies are traditionally used to stain antigens in fixed cells, providing endpoint information only, FP-based biosensors are widely used for the spatiotemporal analysis of target structures in living cells. The genetic tagging of a protein of interest with a fluorescent reporter enables the dynamic visualization of molecular features in living cells. Such studies include the subcellular localization, conformational changes, and PPI that can be measured e.g., by distance-dependent Förster resonance energy transfer (FRET). However, the fusion with FPs requires genetic manipulation and may compromise the biological function of the protein of interest (Hosein et al., 2003). Moreover, recombinant expression rarely reflects endogenous expression levels, and nonprotein targets cannot be studied with this strategy. For these applications, recombinant affinity binders offer new options to engineer novel intracellular biosensors.
Tracing and tracking
A straightforward application for dynamic intracellular tracking of endogenous target structures consists of the genetic fusion of specific binders with FPs (Fig. 3 A). For these applications, any recombinant binders can be used, as long as they are functionally expressed in living cells. The reported reagents cover diverse aspects of cell biology, including the visualization of cytoskeletal components (Rothbauer et al., 2006; Riedl et al., 2008, 2010), the DNA replication machinery (Burgess et al., 2012), and viral infections (Jones et al., 2010; Helma et al., 2012) as well as reporting on apoptotic progression (Zolghadr et al., 2012) and ubiquitin signaling (Sims et al., 2012). However, it is important to note that live-cell visualization of endogenous structures and their respective cell biology with affinity entities requires careful monitoring of potential interference upon antigen binding, such as effects on stability or localization of the target. To prevent that genetic fusion of a nanobody impairs its affinity and specificity, fusion partners should be preferentially added at the C terminus of nanobodies, which is the natural connection site for the constant domain and thus distal from the antigen binding site. Moreover, such visualization inherently depends on antigen-specific subcellular patternings (e.g., focal or filamentous structures), because the fluorescence signal is constitutively derived from the biosensor itself and thus does not report on the antigen abundance per se. One approach to quantify endogenous factors with recombinant binders is based on solvatochromatic fluorescent dyes whose fluorescence increases upon antigen binding (Toutchkine et al., 2003; Nalbant et al., 2004). Site-specific attachment of these dyes to a Src-specific monobody for example allowed the quantitation of Src dynamics in living cells (Gulyani et al., 2011). So far, however, this approach requires in vitro dye conjugation and intracellular application via microinjection, which limits its utility in cell biology. In general, recombinant SNAP-/Halo-/CLIP-tag techniques (Keppler et al., 2003; Gautier et al., 2008; Los et al., 2008) rely on self-labeling protein tags that covalently react with chemically modified and fluorescently labeled substrates. This approach offers new experimental options for site-specific dye attachment in living cells and have been applied to label binders such as DARPins and scFvs (Kampmeier et al., 2009; Hussain et al., 2011; Gu et al., 2013).
Figure 3.

Recombinant binders as intracellular biosensors, effectors, and tools for nanoscopy. (A) Categories of affinity-based live-cell biosensors and effectors. (top left) Fluorescently tagged tracers are used to monitor antigen-specific localization patterns. (top right) Conformation-specific and PTM-specific binders are recruited to respective sites of action. (bottom left) Binders that block biologically active target sites or modulate target function upon binding. (bottom right) An F3H assay to investigate PPIs in living cells. A GFP-specific nanobody is anchored to defined subcellular structures such as the artificially introduced LacO array visible as a spot in the nucleus or the endogenous nuclear lamina or centrosomes. In all of these cases, RFP-preys colocalize with GFP-bait fusions in the presence of an interaction. (B) Recombinant binder tools for nanoscopy. Superresolution techniques such as 3D-structured illumination microscopy require repetitive imaging of a single cell, which leads to severe bleaching of FPs. Nanobodies can thus be used to stabilize or enhance FPs and enable high quality image acquisition conditions. Importantly, their small size reduces the linkage error, as the distance between the fluorescent label and the actual specimen is minimized. Bar, 10 µm.
Conformation and PTM sensors
As discussed in the structural biology section, affinity reagents can detect and stabilize specific conformational states of proteins. Consequently, reframing such binders as intracellular biosensors potentially enables the spatiotemporal analysis of specific conformational changes and their biological implications in living cells (Fig. 3 A). Such application was shown for a nanobody that binds the activated conformation of the β2-adrenoceptor (Rasmussen et al., 2011a). Fusing this binder with GFP reports on the subcellular localization of activated β2-adrenoceptor and lead to the identification of endosomal membranes as initiation sites of acute G protein–coupled receptor signaling, which has previously been considered to occur exclusively from the plasma membrane (Irannejad et al., 2013).
Similarly, a DARPin that recognizes the activated, phosphorylated conformation of extracellular signal–regulated kinase (ERK; Kummer et al., 2012) was conjugated with a solvatochromatic merocyanine to quantitatively report on active pERK localization in living cells in absence and presence of an inhibitor of the upstream regulatory kinase MEK1/2 (Kummer et al., 2013). Interestingly, this DARPin recognizes a conformational change within the activation loop that is activation-dependent and thus indirectly reports on the primary modification, the ERK phosphorylation. Like conventional antibodies, recombinant binders can be generated against PTMs and nonprotein epitopes. Thus, live-cell application of recombinant binders paves the way toward dynamic analysis of PTMs as was shown with a GFP-tagged scFv that specifically recognizes histone acetylation (H3K9ac) and was used for dynamic live-cell and live-animal monitoring of epigenetic chromatin modulation (Sato et al., 2013).
PPIs
Systematic probing of PPIs is often performed in vitro or in yeast. In living cells, PPIs can be tested with FRET, which is technically demanding, or with protein fragment complementation assays, which do not report in real time and are irreversible. In contrast, the recently developed fluorescent-3-hybrid (F3H) assay is based on recombinant immunorecruitment and allows dynamic and reversible monitoring of PPIs in living mammalian cells with a simple optical readout (Fig. 3 A; Herce et al., 2013). GFP fusions with proteins of interest (bait) were recruited to discrete subcellular structures such as artificial LacO DNA arrays by fusing GBP to the Lac repressor. In addition, naturally occurring major satellite repeats, the nuclear periphery, and cytoplasmic structures such as centrosomes can be used as anchor points. Enrichment of red fluorescent prey proteins at the respective anchor sites then indicates specific interaction. The nanobody-based F3H assay is also suited for drug discovery, as small molecule compounds or peptides can be identified that prevent or disrupt PPIs. With this F3H assay, e.g., the disruption of the p53–HDM2 interaction by potential cancer drugs such as Nutlin-3 was monitored in living cells, providing direct information on dose response, kinetics, and bioavailability (Herce et al., 2013).
Nanoscopy
In recent years, several novel super-resolution microscopy technologies revolutionized the field of fluorescence microscopy and enabled the analysis of cellular structures at subdiffraction resolution (Schermelleh et al., 2010). However, the higher resolution also imposes new requirements on detection reagents (Fig. 3 B). As a result of the size of primary and secondary antibodies, the attached fluorophore is positioned at a distance from the actual antigen, leading to so-called linkage errors. Thus, using the smallest possible immunofluorescence binding reagents is of utmost importance to unleash the full potential of super-resolution microscopy. In a first exemplary study, the GFP-specific nanobody was used for structured illumination microscopy to reveal the molecular machinery that effects the intercellular abscission during cell division (Guizetti et al., 2011). This gain in resolution was then systematically demonstrated arguing for the use of directly labeled nanobodies for advanced nanoscopy (Ries et al., 2012). Similarly, nanobodies have been used for photothermal single-molecule tracking in living cells with functionalized gold nanoparticles (Leduc et al., 2013).
Target modulation and validation
The molecular interaction between binder and antigen potentially alters or inhibits the biological function of the target structure (Fig. 3 A). In combination with intracellular expression, recombinant binders may thus be used for targeted modulation of antigen activity. The conceptual realization of such binder-mediated modulation was demonstrated by targeting the active site of the potato starch branching enzyme A in plant cells (Jobling et al., 2003). In a related approach, estrogen receptor–specific monobodies were used to discriminate ligand-induced conformational changes of estrogen receptor in yeast (Koide et al., 2002). Finally, nanobodies were used to modulate protein conformation and thereby either enhance or minimize fluorescence properties of FPs (Kirchhofer et al., 2010).
Novel recombinant binder formats have been developed as innovative biotherapeutics that bind and block defined disease-related antigens. Many of these potential target antigens, however, reside on the inside of cells, and thus, therapeutic applications depend on efficient techniques for delivery of proteins into affected cells. For research purposes, however, the ectopic expression of inhibitory binders in cells is a promising workaround for the functional validation of potential drug targets and does not require the physical target ablation by knockout or knockdown. For such purposes, DARPins were used to selectively inhibit JNK isoforms in human cells (Parizek et al., 2012), a class of enzymes that is critically involved in stress-induced signaling and is discussed as a potential drug target for various indications (Cui et al., 2007). Furthermore, a fibronectin-derived monobody was designed to block an intramolecular interaction of the fusion oncoprotein Bcr-Abl in primary chronic myeloid leukemia cells, revealing a novel interface for therapeutic intervention in chronic myeloid leukemia and demonstrating the general use of recombinant intracellular binders against elusive drug targets (Grebien et al., 2011; Sha et al., 2013).
Toward synthetic biology
Synthetic library design
Immunoglobulin-based binders derived from natural postimmunization repertoires reliably deliver high-quality recombinant affinity reagents. However, the natural antibody modularity also allows the rational design of entirely synthetic antibody libraries using advanced molecular biology techniques. Over the last two decades, such synthetic antibody technologies have constantly evolved and are now arguably as good as or even better than natural libraries (Adams and Sidhu, 2014). Design strategies for such synthetic libraries are based on different concepts and include the recreation of natural CDR diversity or, as a next step, the rational implementation of well-known structure–function parameters to create recombinant antibody libraries with predefined, favorable biophysical properties regarding both antigen recognition and overall stability (Hoet et al., 2005; Tiller et al., 2013). Lacking a natural mechanism of diversity, nonimmunoglobulin binder libraries have to be synthetically generated and diversified with continuous improvements in design and performance (Seeger et al., 2013).
Recombinant binders in synthetic biology
Synthetic biology not only helps in the generation of recombinant binders but also utilizes them to control gene transcription, protein turnover, or reroute signaling cascades (Lienert et al., 2014). Central claims of the still young field of synthetic biology involve the rational redesign of genetic building blocks to engineer novel cellular functions for a broad range of purposes and applications. In consequence, the conception of antibody derivatives and synthetic binders as cellular affinity modules that can be combined with any other gene adds to the molecular toolbox for synthetic biology. As a result of the widespread use of GFP fusion proteins in all areas of cell biology, the GFP-binding nanobody has often been used for initial proof-of-concept experiments that, however, demonstrate the potential of recombinant binders for future applications in synthetic biology. Thus, a nanobody was combined with the Drosophila melanogaster ubiquitin pathway and used for specific degradation of GFP fusion proteins in vivo (Caussinus et al., 2012). For this purpose, the WD40 domain of an F-box protein that naturally mediates the interaction with the protein to be degraded was replaced with GBP, allowing targeted proteasomal knockdown of GFP fusion proteins (Fig. 4 A). In principle, this approach can be adapted with any binder-mediated specificity to subtly control target protein abundance and thus specifically modulate cellular functions in vivo.
Figure 4.
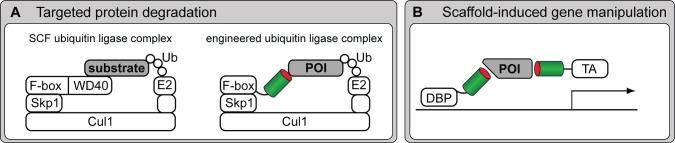
Recombinant binders as modular entities in conceptual, cell-based assay design. (A) A targeted protein knockout via an engineered proteasomal degradation device. The natural WD40 domain that mediates the interaction with a substrate to be degraded via the Skp1-Cul1-F-box-protein (SCF) ubiquitin (Ub) ligase complex is substituted with a recombinant binder, allowing targeted ablation of a protein of interest (POI; Caussinus et al., 2012). (B) A scaffold-induced system to manipulate gene activity. Two different binders, recognizing distinct epitopes of a scaffold protein of interest are fused to a DNA-binding protein (DBP) and a transcriptional activator (TA) enabling protein of interest–dependent gene activation (Tang et al., 2013).
Likewise, the F3H assay that was described in the biosensors section for monitoring PPIs in living cells is based on a synthetic binding device and demonstrates the concept of intracellular immunorecruitment. It can be used to target practically any biochemical activity to a defined site of action. This was shown with an engineered Plo1 kinase, in which the natural targeting sequence had been replaced by GBP, thereby enriching enzymatic activity at intracellular sites of GFP localization (Grallert et al., 2013). Moreover, this GBP-anchoring system can be combined with proteins recognizing particular DNA sequences within the genome to manipulate the localization of chromosomal segments and chromatin domains in living cells. With this approach, topological effects can be studied as recently demonstrated by rescuing the ability of an MECP2 Rett syndrome mutant protein to bind and remodel heterochromatin (Casas-Delucchi et al., 2012). The combination of this strategy with the controlled nuclear import of the anchor allowed additional temporal control.
In a further study, engineered scaffold proteins have been used as tools for the artificial control of cellular signaling events (Good et al., 2011). In a related approach, GFP was recently used as a scaffold protein to induce gene activation in a cell type–specific manner and to demonstrate that nanobodies can be used as dimerization-mediating binding entities (Fig. 4 B; Tang et al., 2013). The GFP/GBP system was also used in the mouse brain for molecular profiling of defined projective neurons (Ekstrand et al., 2014).
These studies illustrate the plethora of novel experimental designs enabled by intracellular binders. In general, the small size and intracellular stability of these recombinant binders allow the exchange of natural binding modules and thereby redirect enzymatic activities and redesign cellular structures and signaling pathways.
Conclusions and perspectives
Some of the pioneering applications described in this review were initially demonstrated with only few recombinant binders. This was in part caused by the limited availability of well-characterized binders against a broad range of antigens. The extremely laborious procedures of library screening to generate specific and reliable binders still remain the major bottlenecks and represent a particular challenge to most cell biology laboratories. Thus, recent efforts in developing new binder formats and automatized screening, together with the establishment of publically available binder resources (Taussig et al., 2007), ideally with sequence information (Bradbury and Plückthun, 2015) will hopefully soon lead to a situation in which suitable binders for a specific target and a given application are just a click away.
Although conventional antibodies are still dominating in classic antibody realms, such as proteomics, Western blot, and immunofluorescence techniques, recombinant formats enable completely novel types of applications in structural biology, living cells, and synthetic biology. The possibility to genetically combine their target affinity with biological executor functions will be of interest not only for cell biology but also for future therapies. Here, the concomitant development of cellular delivery strategies will be crucial to enable direct uptake of functional binders in living cells. In addition, with gene therapy finally realizing its decades-old promises, recombinant binders will increasingly become important as shown for innovations such as chimeric antigen receptors to fight cancers (Kochenderfer et al., 2010). Once binders are understood as recombinant genetic tools that can be custom tailored and combined with any existing functionality, the possibilities seem unlimited, for research as well as therapy.
Acknowledgments
This work was supported by the Center for Nanoscience, Nanosystems Initiative Munich, and the Deutsche Forschungsgemeinschaft (SPP 1623 CA 198/8-1 and HE 721/13-1). S. Muyldermans received support from AFFINOMICS (Protein Binders for Characterization of Human Proteome Function: Generation, Validation, Application), Seventh Framework program (contract number 241481).
The authors are co-inventors on several patents in the field of recombinant binder technologies.
Footnotes
Abbreviations used in this paper:
- DARPin
- designed ankyrin repeat protein
- ERK
- extracellular signal–regulated kinase
- F3H
- fluorescent-3-hybrid
- FP
- fluorescent protein
- GBP
- GFP-binding protein
- hcAb
- heavy chain antibody
- PPI
- protein–protein interaction
- PTM
- posttranslational modification
References
- Abskharon R.N., Giachin G., Wohlkonig A., Soror S.H., Pardon E., Legname G., and Steyaert J.. 2014. Probing the N-terminal β-sheet conversion in the crystal structure of the human prion protein bound to a nanobody. J. Am. Chem. Soc. 136:937–944. 10.1021/ja407527p [DOI] [PubMed] [Google Scholar]
- Adams J.J., and Sidhu S.S.. 2014. Synthetic antibody technologies. Curr. Opin. Struct. Biol. 24:1–9. 10.1016/j.sbi.2013.11.003 [DOI] [PubMed] [Google Scholar]
- Al-Khodor S., Price C.T., Kalia A., and Abu Kwaik Y.. 2010. Functional diversity of ankyrin repeats in microbial proteins. Trends Microbiol. 18:132–139. 10.1016/j.tim.2009.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandeiras T.M., Hillig R.C., Matias P.M., Eberspaecher U., Fanghänel J., Thomaz M., Miranda S., Crusius K., Pütter V., Amstutz P., et al. 2008. Structure of wild-type Plk-1 kinase domain in complex with a selective DARPin. Acta Crystallogr. D Biol. Crystallogr. 64:339–353. 10.1107/S0907444907068217 [DOI] [PubMed] [Google Scholar]
- Baranova E., Fronzes R., Garcia-Pino A., Van Gerven N., Papapostolou D., Péhau-Arnaudet G., Pardon E., Steyaert J., Howorka S., and Remaut H.. 2012. SbsB structure and lattice reconstruction unveil Ca2+ triggered S-layer assembly. Nature. 487:119–122. [DOI] [PubMed] [Google Scholar]
- Beste G., Schmidt F.S., Stibora T., and Skerra A.. 1999. Small antibody-like proteins with prescribed ligand specificities derived from the lipocalin fold. Proc. Natl. Acad. Sci. USA. 96:1898–1903. 10.1073/pnas.96.5.1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binz H.K., Stumpp M.T., Forrer P., Amstutz P., and Plückthun A.. 2003. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 332:489–503. 10.1016/S0022-2836(03)00896-9 [DOI] [PubMed] [Google Scholar]
- Blank K., Lindner P., Diefenbach B., and Plückthun A.. 2002. Self-immobilizing recombinant antibody fragments for immunoaffinity chromatography: generic, parallel, and scalable protein purification. Protein Expr. Purif. 24:313–322. 10.1006/prep.2001.1575 [DOI] [PubMed] [Google Scholar]
- Bowley D.R., Labrijn A.F., Zwick M.B., and Burton D.R.. 2007. Antigen selection from an HIV-1 immune antibody library displayed on yeast yields many novel antibodies compared to selection from the same library displayed on phage. Protein Eng. Des. Sel. 20:81–90. 10.1093/protein/gzl057 [DOI] [PubMed] [Google Scholar]
- Bradbury A., and Plückthun A.. 2015. Reproducibility: Standardize antibodies used in research. Nature. 518:27–29. 10.1038/518027a [DOI] [PubMed] [Google Scholar]
- Brauchle M., Hansen S., Caussinus E., Lenard A., Ochoa-Espinosa A., Scholz O., Sprecher S.G., Plückthun A., and Affolter M.. 2014. Protein interference applications in cellular and developmental biology using DARPins that recognize GFP and mCherry. Biol. Open. 3:1252–1261. 10.1242/bio.201410041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess A., Lorca T., and Castro A.. 2012. Quantitative live imaging of endogenous DNA replication in mammalian cells. PLoS ONE. 7:e45726 10.1371/journal.pone.0045726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A., Persson O., Ingvarsson J., Widegren B., Salford L., Borrebaeck C.A., and Wingren C.. 2010. Plasma proteome profiling reveals biomarker patterns associated with prognosis and therapy selection in glioblastoma multiforme patients. Proteomics Clin. Appl. 4:591–602. 10.1002/prca.200900173 [DOI] [PubMed] [Google Scholar]
- Carlsson A., Wuttge D.M., Ingvarsson J., Bengtsson A.A., Sturfelt G., Borrebaeck C.A., and Wingren C.. 2011. Serum protein profiling of systemic lupus erythematosus and systemic sclerosis using recombinant antibody microarrays. Mol. Cell. Proteomics. 10:M110.005033 10.1074/mcp.M110.005033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas-Delucchi C.S., Becker A., Bolius J.J., and Cardoso M.C.. 2012. Targeted manipulation of heterochromatin rescues MeCP2 Rett mutants and re-establishes higher order chromatin organization. Nucleic Acids Res. 40:e176 10.1093/nar/gks784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caussinus E., Kanca O., and Affolter M.. 2012. Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat. Struct. Mol. Biol. 19:117–121. 10.1038/nsmb.2180 [DOI] [PubMed] [Google Scholar]
- Chaikuad A., Keates T., Vincke C., Kaufholz M., Zenn M., Zimmermann B., Gutiérrez C., Zhang R.G., Hatzos-Skintges C., Joachimiak A., et al. 2014. Structure of cyclin G-associated kinase (GAK) trapped in different conformations using nanobodies. Biochem. J. 459:59–69. 10.1042/BJ20131399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chothia C., Novotný J., Bruccoleri R., and Karplus M.. 1985. Domain association in immunoglobulin molecules. The packing of variable domains. J. Mol. Biol. 186:651–663. 10.1016/0022-2836(85)90137-8 [DOI] [PubMed] [Google Scholar]
- Colwill K., Renewable Protein Binder Working Group and Gräslund S.. 2011. A roadmap to generate renewable protein binders to the human proteome. Nat. Methods. 8:551–558. 10.1038/nmeth.1607 [DOI] [PubMed] [Google Scholar]
- Cristea I.M., Williams R., Chait B.T., and Rout M.P.. 2005. Fluorescent proteins as proteomic probes. Mol. Cell. Proteomics. 4:1933–1941. 10.1074/mcp.M500227-MCP200 [DOI] [PubMed] [Google Scholar]
- Cui J., Zhang M., Zhang Y.Q., and Xu Z.H.. 2007. JNK pathway: diseases and therapeutic potential. Acta Pharmacol. Sin. 28:601–608. 10.1111/j.1745-7254.2007.00579.x [DOI] [PubMed] [Google Scholar]
- De Genst E., Silence K., Decanniere K., Conrath K., Loris R., Kinne J., Muyldermans S., and Wyns L.. 2006. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. USA. 103:4586–4591. 10.1073/pnas.0505379103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derewenda Z.S. 2010. Application of protein engineering to enhance crystallizability and improve crystal properties. Acta Crystallogr. D Biol. Crystallogr. 66:604–615. 10.1107/S090744491000644X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domanska K., Vanderhaegen S., Srinivasan V., Pardon E., Dupeux F., Marquez J.A., Giorgetti S., Stoppini M., Wyns L., Bellotti V., and Steyaert J.. 2011. Atomic structure of a nanobody-trapped domain-swapped dimer of an amyloidogenic β2-microglobulin variant. Proc. Natl. Acad. Sci. USA. 108:1314–1319. 10.1073/pnas.1008560108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersbach H., Fiedler E., Scheuermann T., Fiedler M., Stubbs M.T., Reimann C., Proetzel G., Rudolph R., and Fiedler U.. 2007. Affilin-novel binding molecules based on human γ-B-crystallin, an all β-sheet protein. J. Mol. Biol. 372:172–185. 10.1016/j.jmb.2007.06.045 [DOI] [PubMed] [Google Scholar]
- Eggenstein E., Eichinger A., Kim H.J., and Skerra A.. 2014. Structure-guided engineering of Anticalins with improved binding behavior and biochemical characteristics for application in radio-immuno imaging and/or therapy. J. Struct. Biol. 185:203–214. 10.1016/j.jsb.2013.03.009 [DOI] [PubMed] [Google Scholar]
- Ekstrand M.I., Nectow A.R., Knight Z.A., Latcha K.N., Pomeranz L.E., and Friedman J.M.. 2014. Molecular profiling of neurons based on connectivity. Cell. 157:1230–1242. 10.1016/j.cell.2014.03.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrer P., Stumpp M.T., Binz H.K., and Plückthun A.. 2003. A novel strategy to design binding molecules harnessing the modular nature of repeat proteins. FEBS Lett. 539:2–6. 10.1016/S0014-5793(03)00177-7 [DOI] [PubMed] [Google Scholar]
- Frauer C., and Leonhardt H.. 2009. A versatile non-radioactive assay for DNA methyltransferase activity and DNA binding. Nucleic Acids Res. 37:e22 10.1093/nar/gkn1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridy P.C., Li Y., Keegan S., Thompson M.K., Nudelman I., Scheid J.F., Oeffinger M., Nussenzweig M.C., Fenyö D., Chait B.T., and Rout M.P.. 2014. A robust pipeline for rapid production of versatile nanobody repertoires. Nat. Methods. 11:1253–1260. 10.1038/nmeth.3170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier A., Juillerat A., Heinis C., Corrêa I.R. Jr., Kindermann M., Beaufils F., and Johnsson K.. 2008. An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 15:128–136. 10.1016/j.chembiol.2008.01.007 [DOI] [PubMed] [Google Scholar]
- Gebauer M., Schiefner A., Matschiner G., and Skerra A.. 2013. Combinatorial design of an Anticalin directed against the extra-domain b for the specific targeting of oncofetal fibronectin. J. Mol. Biol. 425:780–802. 10.1016/j.jmb.2012.12.004 [DOI] [PubMed] [Google Scholar]
- Good M.C., Zalatan J.G., and Lim W.A.. 2011. Scaffold proteins: hubs for controlling the flow of cellular information. Science. 332:680–686. 10.1126/science.1198701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabulovski D., Kaspar M., and Neri D.. 2007. A novel, non-immunogenic Fyn SH3-derived binding protein with tumor vascular targeting properties. J. Biol. Chem. 282:3196–3204. 10.1074/jbc.M609211200 [DOI] [PubMed] [Google Scholar]
- Grallert A., Patel A., Tallada V.A., Chan K.Y., Bagley S., Krapp A., Simanis V., and Hagan I.M.. 2013. Centrosomal MPF triggers the mitotic and morphogenetic switches of fission yeast. Nat. Cell Biol. 15:88–95. 10.1038/ncb2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grebien F., Hantschel O., Wojcik J., Kaupe I., Kovacic B., Wyrzucki A.M., Gish G.D., Cerny-Reiterer S., Koide A., Beug H., et al. 2011. Targeting the SH2-kinase interface in Bcr-Abl inhibits leukemogenesis. Cell. 147:306–319. 10.1016/j.cell.2011.08.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grönwall C., Sjöberg A., Ramström M., Höidén-Guthenberg I., Hober S., Jonasson P., and Ståhl S.. 2007. Affibody-mediated transferrin depletion for proteomics applications. Biotechnol. J. 2:1389–1398. 10.1002/biot.200700053 [DOI] [PubMed] [Google Scholar]
- Gross G.G., Junge J.A., Mora R.J., Kwon H.B., Olson C.A., Takahashi T.T., Liman E.R., Ellis-Davies G.C., McGee A.W., Sabatini B.L., et al. 2013. Recombinant probes for visualizing endogenous synaptic proteins in living neurons. Neuron. 78:971–985. 10.1016/j.neuron.2013.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G.J., Friedman M., Jost C., Johnsson K., Kamali-Moghaddam M., Plückthun A., Landegren U., and Söderberg O.. 2013. Protein tag-mediated conjugation of oligonucleotides to recombinant affinity binders for proximity ligation. New Biotechnol. 30:144–152. 10.1016/j.nbt.2012.05.005 [DOI] [PubMed] [Google Scholar]
- Gu L., Li C., Aach J., Hill D.E., Vidal M., and Church G.M.. 2014. Multiplex single-molecule interaction profiling of DNA-barcoded proteins. Nature. 515:554–557. 10.1038/nature13761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guizetti J., Schermelleh L., Mäntler J., Maar S., Poser I., Leonhardt H., Müller-Reichert T., and Gerlich D.W.. 2011. Cortical constriction during abscission involves helices of ESCRT-III-dependent filaments. Science. 331:1616–1620. 10.1126/science.1201847 [DOI] [PubMed] [Google Scholar]
- Gulyani A., Vitriol E., Allen R., Wu J., Gremyachinskiy D., Lewis S., Dewar B., Graves L.M., Kay B.K., Kuhlman B., et al. 2011. A biosensor generated via high-throughput screening quantifies cell edge Src dynamics. Nat. Chem. Biol. 7:437–444. 10.1038/nchembio.585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamers-Casterman C., Atarhouch T., Muyldermans S., Robinson G., Hamers C., Songa E.B., Bendahman N., and Hamers R.. 1993. Naturally occurring antibodies devoid of light chains. Nature. 363:446–448. 10.1038/363446a0 [DOI] [PubMed] [Google Scholar]
- Harmsen M.M., Ruuls R.C., Nijman I.J., Niewold T.A., Frenken L.G., and de Geus B.. 2000. Llama heavy-chain V regions consist of at least four distinct subfamilies revealing novel sequence features. Mol. Immunol. 37:579–590. 10.1016/S0161-5890(00)00081-X [DOI] [PubMed] [Google Scholar]
- Helma J., Schmidthals K., Lux V., Nüske S., Scholz A.M., Kräusslich H.G., Rothbauer U., and Leonhardt H.. 2012. Direct and dynamic detection of HIV-1 in living cells. PLoS ONE. 7:e50026 10.1371/journal.pone.0050026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herce H.D., Deng W., Helma J., Leonhardt H., and Cardoso M.C.. 2013. Visualization and targeted disruption of protein interactions in living cells. Nat. Commun. 4:2660 10.1038/ncomms3660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoet R.M., Cohen E.H., Kent R.B., Rookey K., Schoonbroodt S., Hogan S., Rem L., Frans N., Daukandt M., Pieters H., et al. 2005. Generation of high-affinity human antibodies by combining donor-derived and synthetic complementarity-determining-region diversity. Nat. Biotechnol. 23:344–348. 10.1038/nbt1067 [DOI] [PubMed] [Google Scholar]
- Hosein R.E., Williams S.A., Haye K., and Gavin R.H.. 2003. Expression of GFP-actin leads to failure of nuclear elongation and cytokinesis in Tetrahymena thermophila. J. Eukaryot. Microbiol. 50:403–408. 10.1111/j.1550-7408.2003.tb00261.x [DOI] [PubMed] [Google Scholar]
- Hoyer W., Grönwall C., Jonsson A., Ståhl S., and Härd T.. 2008. Stabilization of a β-hairpin in monomeric Alzheimer’s amyloid-β peptide inhibits amyloid formation. Proc. Natl. Acad. Sci. USA. 105:5099–5104. 10.1073/pnas.0711731105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain A.F., Kampmeier F., von Felbert V., Merk H.F., Tur M.K., and Barth S.. 2011. SNAP-tag technology mediates site specific conjugation of antibody fragments with a photosensitizer and improves target specific phototoxicity in tumor cells. Bioconjug. Chem. 22:2487–2495. 10.1021/bc200304k [DOI] [PubMed] [Google Scholar]
- Ingvarsson J., Wingren C., Carlsson A., Ellmark P., Wahren B., Engström G., Harmenberg U., Krogh M., Peterson C., and Borrebaeck C.A.. 2008. Detection of pancreatic cancer using antibody microarray-based serum protein profiling. Proteomics. 8:2211–2219. 10.1002/pmic.200701167 [DOI] [PubMed] [Google Scholar]
- Irannejad R., Tomshine J.C., Tomshine J.R., Chevalier M., Mahoney J.P., Steyaert J., Rasmussen S.G., Sunahara R.K., El-Samad H., Huang B., and von Zastrow M.. 2013. Conformational biosensors reveal GPCR signalling from endosomes. Nature. 495:534–538. 10.1038/nature12000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobling S.A., Jarman C., Teh M.M., Holmberg N., Blake C., and Verhoeyen M.E.. 2003. Immunomodulation of enzyme function in plants by single-domain antibody fragments. Nat. Biotechnol. 21:77–80. 10.1038/nbt772 [DOI] [PubMed] [Google Scholar]
- Jones C.T., Catanese M.T., Law L.M.J., Khetani S.R., Syder A.J., Ploss A., Oh T.S., Schoggins J.W., MacDonald M.R., Bhatia S.N., and Rice C.M.. 2010. Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system. Nat. Biotechnol. 28:167–171. 10.1038/nbt.1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampmeier F., Ribbert M., Nachreiner T., Dembski S., Beaufils F., Brecht A., and Barth S.. 2009. Site-specific, covalent labeling of recombinant antibody fragments via fusion to an engineered version of 6-O-alkylguanine DNA alkyltransferase. Bioconjug. Chem. 20:1010–1015. 10.1021/bc9000257 [DOI] [PubMed] [Google Scholar]
- Keppler A., Gendreizig S., Gronemeyer T., Pick H., Vogel H., and Johnsson K.. 2003. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 21:86–89. 10.1038/nbt765 [DOI] [PubMed] [Google Scholar]
- Kidder B.L., Hu G., and Zhao K.. 2011. ChIP-Seq: technical considerations for obtaining high-quality data. Nat. Immunol. 12:918–922. 10.1038/ni.2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Stroud R.M., and Craik C.S.. 2011. Rapid identification of recombinant Fabs that bind to membrane proteins. Methods. 55:303–309. 10.1016/j.ymeth.2011.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhofer A., Helma J., Schmidthals K., Frauer C., Cui S., Karcher A., Pellis M., Muyldermans S., Casas-Delucchi C.S., Cardoso M.C., et al. 2010. Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol. 17:133–138. 10.1038/nsmb.1727 [DOI] [PubMed] [Google Scholar]
- Kochenderfer J.N., Wilson W.H., Janik J.E., Dudley M.E., Stetler-Stevenson M., Feldman S.A., Maric I., Raffeld M., Nathan D.A., Lanier B.J., et al. 2010. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 116:4099–4102. 10.1182/blood-2010-04-281931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl A., Binz H.K., Forrer P., Stumpp M.T., Plückthun A., and Grütter M.G.. 2003. Designed to be stable: crystal structure of a consensus ankyrin repeat protein. Proc. Natl. Acad. Sci. USA. 100:1700–1705. 10.1073/pnas.0337680100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide A., Bailey C.W., Huang X., and Koide S.. 1998. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 284:1141–1151. 10.1006/jmbi.1998.2238 [DOI] [PubMed] [Google Scholar]
- Koide A., Abbatiello S., Rothgery L., and Koide S.. 2002. Probing protein conformational changes in living cells by using designer binding proteins: application to the estrogen receptor. Proc. Natl. Acad. Sci. USA. 99:1253–1258. 10.1073/pnas.032665299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide A., Wojcik J., Gilbreth R.N., Hoey R.J., and Koide S.. 2012. Teaching an old scaffold new tricks: monobodies constructed using alternative surfaces of the FN3 scaffold. J. Mol. Biol. 415:393–405. 10.1016/j.jmb.2011.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korndörfer I.P., Beste G., and Skerra A.. 2003. Crystallographic analysis of an “anticalin” with tailored specificity for fluorescein reveals high structural plasticity of the lipocalin loop region. Proteins. 53:121–129. 10.1002/prot.10497 [DOI] [PubMed] [Google Scholar]
- Korotkov K.V., Pardon E., Steyaert J., and Hol W.G.. 2009. Crystal structure of the N-terminal domain of the secretin GspD from ETEC determined with the assistance of a nanobody. Structure. 17:255–265. 10.1016/j.str.2008.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovari L.C., Momany C., and Rossmann M.G.. 1995. The use of antibody fragments for crystallization and structure determinations. Structure. 3:1291–1293. 10.1016/S0969-2126(01)00266-0 [DOI] [PubMed] [Google Scholar]
- Krishnamurthy H., and Gouaux E.. 2012. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature. 481:469–474. 10.1038/nature10737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse A.C., Ring A.M., Manglik A., Hu J., Hu K., Eitel K., Hübner H., Pardon E., Valant C., Sexton P.M., et al. 2013. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 504:101–106. 10.1038/nature12735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer L., Parizek P., Rube P., Millgramm B., Prinz A., Mittl P.R., Kaufholz M., Zimmermann B., Herberg F.W., and Plückthun A.. 2012. Structural and functional analysis of phosphorylation-specific binders of the kinase ERK from designed ankyrin repeat protein libraries. Proc. Natl. Acad. Sci. USA. 109:E2248–E2257. 10.1073/pnas.1205399109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer L., Hsu C.W., Dagliyan O., MacNevin C., Kaufholz M., Zimmermann B., Dokholyan N.V., Hahn K.M., and Plückthun A.. 2013. Knowledge-based design of a biosensor to quantify localized ERK activation in living cells. Chem. Biol. 20:847–856. 10.1016/j.chembiol.2013.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc C., Si S., Gautier J., Soto-Ribeiro M., Wehrle-Haller B., Gautreau A., Giannone G., Cognet L., and Lounis B.. 2013. A highly specific gold nanoprobe for live-cell single-molecule imaging. Nano Lett. 13:1489–1494. [DOI] [PubMed] [Google Scholar]
- Li J., Mahajan A., and Tsai M.D.. 2006. Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry. 45:15168–15178. 10.1021/bi062188q [DOI] [PubMed] [Google Scholar]
- Lienert F., Lohmueller J.J., Garg A., and Silver P.A.. 2014. Synthetic biology in mammalian cells: next generation research tools and therapeutics. Nat. Rev. Mol. Cell Biol. 15:95–107. 10.1038/nrm3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loris R., Marianovsky I., Lah J., Laeremans T., Engelberg-Kulka H., Glaser G., Muyldermans S., and Wyns L.. 2003. Crystal structure of the intrinsically flexible addiction antidote MazE. J. Biol. Chem. 278:28252–28257. 10.1074/jbc.M302336200 [DOI] [PubMed] [Google Scholar]
- Los G.V., Encell L.P., McDougall M.G., Hartzell D.D., Karassina N., Zimprich C., Wood M.G., Learish R., Ohana R.F., Urh M., et al. 2008. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 3:373–382. 10.1021/cb800025k [DOI] [PubMed] [Google Scholar]
- Mouratou B., Schaeffer F., Guilvout I., Tello-Manigne D., Pugsley A.P., Alzari P.M., and Pecorari F.. 2007. Remodeling a DNA-binding protein as a specific in vivo inhibitor of bacterial secretin PulD. Proc. Natl. Acad. Sci. USA. 104:17983–17988. 10.1073/pnas.0702963104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyldermans S. 2013. Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 82:775–797. 10.1146/annurev-biochem-063011-092449 [DOI] [PubMed] [Google Scholar]
- Muyldermans S., Atarhouch T., Saldanha J., Barbosa J.A., and Hamers R.. 1994. Sequence and structure of VH domain from naturally occurring camel heavy chain immunoglobulins lacking light chains. Protein Eng. 7:1129–1135. 10.1093/protein/7.9.1129 [DOI] [PubMed] [Google Scholar]
- Nalbant P., Hodgson L., Kraynov V., Toutchkine A., and Hahn K.M.. 2004. Activation of endogenous Cdc42 visualized in living cells. Science. 305:1615–1619. 10.1126/science.1100367 [DOI] [PubMed] [Google Scholar]
- Nguyen V.K., Hamers R., Wyns L., and Muyldermans S.. 2000. Camel heavy-chain antibodies: diverse germline V(H)H and specific mechanisms enlarge the antigen-binding repertoire. EMBO J. 19:921–930. 10.1093/emboj/19.5.921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord K., Gunneriusson E., Ringdahl J., Ståhl S., Uhlén M., and Nygren P.A.. 1997. Binding proteins selected from combinatorial libraries of an α-helical bacterial receptor domain. Nat. Biotechnol. 15:772–777. 10.1038/nbt0897-772 [DOI] [PubMed] [Google Scholar]
- Ostermeier C., Iwata S., Ludwig B., and Michel H.. 1995. Fv fragment-mediated crystallization of the membrane protein bacterial cytochrome c oxidase. Nat. Struct. Biol. 2:842–846. 10.1038/nsb1095-842 [DOI] [PubMed] [Google Scholar]
- Paduch M., Koide A., Uysal S., Rizk S.S., Koide S., and Kossiakoff A.A.. 2013. Generating conformation-specific synthetic antibodies to trap proteins in selected functional states. Methods. 60:3–14. 10.1016/j.ymeth.2012.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardon E., Laeremans T., Triest S., Rasmussen S.G., Wohlkönig A., Ruf A., Muyldermans S., Hol W.G., Kobilka B.K., and Steyaert J.. 2014. A general protocol for the generation of Nanobodies for structural biology. Nat. Protoc. 9:674–693. 10.1038/nprot.2014.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parizek P., Kummer L., Rube P., Prinz A., Herberg F.W., and Plückthun A.. 2012. Designed ankyrin repeat proteins (DARPins) as novel isoform-specific intracellular inhibitors of c-Jun N-terminal kinases. ACS Chem. Biol. 7:1356–1366. 10.1021/cb3001167 [DOI] [PubMed] [Google Scholar]
- Parmeggiani F., Pellarin R., Larsen A.P., Varadamsetty G., Stumpp M.T., Zerbe O., Caflisch A., and Plückthun A.. 2008. Designed armadillo repeat proteins as general peptide-binding scaffolds: consensus design and computational optimization of the hydrophobic core. J. Mol. Biol. 376:1282–1304. 10.1016/j.jmb.2007.12.014 [DOI] [PubMed] [Google Scholar]
- Pecqueur L., Duellberg C., Dreier B., Jiang Q., Wang C., Plückthun A., Surrey T., Gigant B., and Knossow M.. 2012. A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proc. Natl. Acad. Sci. USA. 109:12011–12016. 10.1073/pnas.1204129109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichler G., Leonhardt H., and Rothbauer U.. 2012. Fluorescent protein specific Nanotraps to study protein-protein interactions and histone-tail peptide binding. Methods Mol. Biol. 911:475–483. [DOI] [PubMed] [Google Scholar]
- Plückthun A. 2015. Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 55:489–511. 10.1146/annurev-pharmtox-010611-134654 [DOI] [PubMed] [Google Scholar]
- Pollithy A., Romer T., Lang C., Muller F.D., Helma J., Leonhardt H., Rothbauer U., and Schuler D.. 2011. Magnetosome expression of functional camelid antibody fragments (nanobodies) in Magnetospirillum gryphiswaldense. Appl. Environ. Microbiol. 77:6165–6171. 10.1128/AEM.05282-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S.G., Choi H.J., Fung J.J., Pardon E., Casarosa P., Chae P.S., Devree B.T., Rosenbaum D.M., Thian F.S., Kobilka T.S., et al. 2011a Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature. 469:175–180. 10.1038/nature09648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S.G., DeVree B.T., Zou Y., Kruse A.C., Chung K.Y., Kobilka T.S., Thian F.S., Chae P.S., Pardon E., Calinski D., et al. 2011b Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 477:549–555. 10.1038/nature10361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J., Crevenna A.H., Kessenbrock K., Yu J.H., Neukirchen D., Bista M., Bradke F., Jenne D., Holak T.A., Werb Z., et al. 2008. Lifeact: a versatile marker to visualize F-actin. Nat. Methods. 5:605–607. 10.1038/nmeth.1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J., Flynn K.C., Raducanu A., Gärtner F., Beck G., Bösl M., Bradke F., Massberg S., Aszodi A., Sixt M., and Wedlich-Söldner R.. 2010. Lifeact mice for studying F-actin dynamics. Nat. Methods. 7:168–169. 10.1038/nmeth0310-168 [DOI] [PubMed] [Google Scholar]
- Ries J., Kaplan C., Platonova E., Eghlidi H., and Ewers H.. 2012. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat. Methods. 9:582–584. 10.1038/nmeth.1991 [DOI] [PubMed] [Google Scholar]
- Rothbauer U., Zolghadr K., Tillib S., Nowak D., Schermelleh L., Gahl A., Backmann N., Conrath K., Muyldermans S., Cardoso M.C., and Leonhardt H.. 2006. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods. 3:887–889. 10.1038/nmeth953 [DOI] [PubMed] [Google Scholar]
- Rothbauer U., Zolghadr K., Muyldermans S., Schepers A., Cardoso M.C., and Leonhardt H.. 2008. A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol. Cell. Proteomics. 7:282–289. 10.1074/mcp.M700342-MCP200 [DOI] [PubMed] [Google Scholar]
- Sato Y., Mukai M., Ueda J., Muraki M., Stasevich T.J., Horikoshi N., Kujirai T., Kita H., Kimura T., Hira S., et al. 2013. Genetically encoded system to track histone modification in vivo. Sci Rep. 3:2436 10.1038/srep02436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schermelleh L., Heintzmann R., and Leonhardt H.. 2010. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 190:165–175. 10.1083/jcb.201002018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönfeld D., Matschiner G., Chatwell L., Trentmann S., Gille H., Hülsmeyer M., Brown N., Kaye P.M., Schlehuber S., Hohlbaum A.M., and Skerra A.. 2009. An engineered lipocalin specific for CTLA-4 reveals a combining site with structural and conformational features similar to antibodies. Proc. Natl. Acad. Sci. USA. 106:8198–8203. 10.1073/pnas.0813399106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger M.A., Zbinden R., Flütsch A., Gutte P.G., Engeler S., Roschitzki-Voser H., and Grütter M.G.. 2013. Design, construction, and characterization of a second-generation DARP in library with reduced hydrophobicity. Protein Sci. 22:1239–1257. 10.1002/pro.2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sennhauser G., Amstutz P., Briand C., Storchenegger O., and Grütter M.G.. 2007. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol. 5:e7 10.1371/journal.pbio.0050007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrière J., Dugua J.M., Bossus M., Verrier B., Haser R., Gouet P., and Guillon C.. 2011. Fab’-induced folding of antigenic N-terminal peptides from intrinsically disordered HIV-1 Tat revealed by X-ray crystallography. J. Mol. Biol. 405:33–42. 10.1016/j.jmb.2010.10.033 [DOI] [PubMed] [Google Scholar]
- Sha F., Gencer E.B., Georgeon S., Koide A., Yasui N., Koide S., and Hantschel O.. 2013. Dissection of the BCR-ABL signaling network using highly specific monobody inhibitors to the SHP2 SH2 domains. Proc. Natl. Acad. Sci. USA. 110:14924–14929. 10.1073/pnas.1303640110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman J., Liu Q., Bakker A., To W., Duguay A., Alba B.M., Smith R., Rivas A., Li P., Le H., et al. 2005. Multivalent avimer proteins evolved by exon shuffling of a family of human receptor domains. Nat. Biotechnol. 23:1556–1561. 10.1038/nbt1166 [DOI] [PubMed] [Google Scholar]
- Sims J.J., Scavone F., Cooper E.M., Kane L.A., Youle R.J., Boeke J.D., and Cohen R.E.. 2012. Polyubiquitin-sensor proteins reveal localization and linkage-type dependence of cellular ubiquitin signaling. Nat. Methods. 9:303–309. 10.1038/nmeth.1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrabana R., Dvorsky R., Sevcik J., and Novak M.. 2010. Monoclonal antibody MN423 as a stable mold facilitates structure determination of disordered tau protein. J. Struct. Biol. 171:74–81. 10.1016/j.jsb.2010.02.016 [DOI] [PubMed] [Google Scholar]
- Smith G.P., Patel S.U., Windass J.D., Thornton J.M., Winter G., and Griffiths A.D.. 1998. Small binding proteins selected from a combinatorial repertoire of knottins displayed on phage. J. Mol. Biol. 277:317–332. 10.1006/jmbi.1997.1621 [DOI] [PubMed] [Google Scholar]
- Spinelli S., Desmyter A., Verrips C.T., de Haard H.J., Moineau S., and Cambillau C.. 2006. Lactococcal bacteriophage p2 receptor-binding protein structure suggests a common ancestor gene with bacterial and mammalian viruses. Nat. Struct. Mol. Biol. 13:85–89. 10.1038/nsmb1029 [DOI] [PubMed] [Google Scholar]
- Stijlemans B., Conrath K., Cortez-Retamozo V., Van Xong H., Wyns L., Senter P., Revets H., De Baetselier P., Muyldermans S., and Magez S.. 2004. Efficient targeting of conserved cryptic epitopes of infectious agents by single domain antibodies. African trypanosomes as paradigm. J. Biol. Chem. 279:1256–1261. 10.1074/jbc.M307341200 [DOI] [PubMed] [Google Scholar]
- Sundberg E.J., and Mariuzza R.A.. 2002. Molecular recognition in antibody-antigen complexes. Adv. Protein Chem. 61:119–160. [DOI] [PubMed] [Google Scholar]
- Tang J.C., Szikra T., Kozorovitskiy Y., Teixiera M., Sabatini B.L., Roska B., and Cepko C.L.. 2013. A nanobody-based system using fluorescent proteins as scaffolds for cell-specific gene manipulation. Cell. 154:928–939. 10.1016/j.cell.2013.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanha J., Nguyen T.D., Ng A., Ryan S., Ni F., and Mackenzie R.. 2006. Improving solubility and refolding efficiency of human V(H)s by a novel mutational approach. Protein Eng. Des. Sel. 19:503–509. 10.1093/protein/gzl037 [DOI] [PubMed] [Google Scholar]
- Taussig M.J., Stoevesandt O., Borrebaeck C.A., Bradbury A.R., Cahill D., Cambillau C., de Daruvar A., Dübel S., Eichler J., Frank R., et al. 2007. ProteomeBinders: planning a European resource of affinity reagents for analysis of the human proteome. Nat. Methods. 4:13–17. 10.1038/nmeth0107-13 [DOI] [PubMed] [Google Scholar]
- Tereshko V., Uysal S., Koide A., Margalef K., Koide S., and Kossiakoff A.A.. 2008. Toward chaperone-assisted crystallography: protein engineering enhancement of crystal packing and X-ray phasing capabilities of a camelid single-domain antibody (VHH) scaffold. Protein Sci. 17:1175–1187. 10.1110/ps.034892.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiede C., Tang A.A., Deacon S.E., Mandal U., Nettleship J.E., Owen R.L., George S.E., Harrison D.J., Owens R.J., Tomlinson D.C., and McPherson M.J.. 2014. Adhiron: a stable and versatile peptide display scaffold for molecular recognition applications. Protein Eng. Des. Sel. 27:145–155. 10.1093/protein/gzu007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiller T., Schuster I., Deppe D., Siegers K., Strohner R., Herrmann T., Berenguer M., Poujol D., Stehle J., Stark Y., et al. 2013. A fully synthetic human Fab antibody library based on fixed VH/VL framework pairings with favorable biophysical properties. MAbs. 5:445–470. 10.4161/mabs.24218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toutchkine A., Kraynov V., and Hahn K.. 2003. Solvent-sensitive dyes to report protein conformational changes in living cells. J. Am. Chem. Soc. 125:4132–4145. 10.1021/ja0290882 [DOI] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L., Boulon S., Lam Y.W., Urcia R., Boisvert F.M., Vandermoere F., Morrice N.A., Swift S., Rothbauer U., Leonhardt H., and Lamond A.. 2008. Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J. Cell Biol. 183:223–239. 10.1083/jcb.200805092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uysal S., Vásquez V., Tereshko V., Esaki K., Fellouse F.A., Sidhu S.S., Koide S., Perozo E., and Kossiakoff A.. 2009. Crystal structure of full-length KcsA in its closed conformation. Proc. Natl. Acad. Sci. USA. 106:6644–6649. 10.1073/pnas.0810663106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veesler D., Dreier B., Blangy S., Lichière J., Tremblay D., Moineau S., Spinelli S., Tegoni M., Plückthun A., Campanacci V., and Cambillau C.. 2009. Crystal structure and function of a DARPin neutralizing inhibitor of lactococcal phage TP901-1: comparison of DARPin and camelid VHH binding mode. J. Biol. Chem. 284:30718–30726. 10.1074/jbc.M109.037812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu K.B., Ghahroudi M.A., Wyns L., and Muyldermans S.. 1997. Comparison of llama VH sequences from conventional and heavy chain antibodies. Mol. Immunol. 34:1121–1131. 10.1016/S0161-5890(97)00146-6 [DOI] [PubMed] [Google Scholar]
- Walther T.C., and Mann M.. 2010. Mass spectrometry-based proteomics in cell biology. J. Cell Biol. 190:491–500. 10.1083/jcb.201004052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willuda J., Honegger A., Waibel R., Schubiger P.A., Stahel R., Zangemeister-Wittke U., and Plückthun A.. 1999. High thermal stability is essential for tumor targeting of antibody fragments: engineering of a humanized anti-epithelial glycoprotein-2 (epithelial cell adhesion molecule) single-chain Fv fragment. Cancer Res. 59:5758–5767. [PubMed] [Google Scholar]
- Wingren C., Ingvarsson J., Dexlin L., Szul D., and Borrebaeck C.A.. 2007. Design of recombinant antibody microarrays for complex proteome analysis: choice of sample labeling-tag and solid support. Proteomics. 7:3055–3065. 10.1002/pmic.200700025 [DOI] [PubMed] [Google Scholar]
- Wojcik J., Hantschel O., Grebien F., Kaupe I., Bennett K.L., Barkinge J., Jones R.B., Koide A., Superti-Furga G., and Koide S.. 2010. A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nat. Struct. Mol. Biol. 17:519–527. 10.1038/nsmb.1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wörn A., and Plückthun A.. 2001. Stability engineering of antibody single-chain Fv fragments. J. Mol. Biol. 305:989–1010. 10.1006/jmbi.2000.4265 [DOI] [PubMed] [Google Scholar]
- Zhou Y., Morais-Cabral J.H., Kaufman A., and MacKinnon R.. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 414:43–48. 10.1038/35102009 [DOI] [PubMed] [Google Scholar]
- Zolghadr K., Gregor J., Leonhardt H., and Rothbauer U.. 2012. Case study on live cell apoptosis-assay using lamin-chromobody cell-lines for high-content analysis. Methods Mol. Biol. 911:569–575. [DOI] [PubMed] [Google Scholar]