

Abstract
Eukaryotic high-mobility-group-box (HMGB) proteins are nuclear factors involved in chromatin remodeling and transcription regulation. When released into the extracellular milieu, HMGB1 acts as a proinflammatory cytokine that plays a central role in the pathogenesis of several immune-mediated inflammatory diseases. We found that the Plasmodium genome encodes two genuine HMGB factors, Plasmodium HMGB1 and HMGB2, that encompass, like their human counterparts, a proinflammatory domain. Given that these proteins are released from parasitized red blood cells, we then hypothesized that Plasmodium HMGB might contribute to the pathogenesis of experimental cerebral malaria (ECM), a lethal neuroinflammatory syndrome that develops in C57BL/6 (susceptible) mice infected with Plasmodium berghei ANKA and that in many aspects resembles human cerebral malaria elicited by P. falciparum infection. The pathogenesis of experimental cerebral malaria was suppressed in C57BL/6 mice infected with P. berghei ANKA lacking the hmgb2 gene (Δhmgb2 ANKA), an effect associated with a reduction of histological brain lesions and with lower expression levels of several proinflammatory genes. The incidence of ECM in pbhmgb2-deficient mice was restored by the administration of recombinant PbHMGB2. Protection from experimental cerebral malaria in Δhmgb2 ANKA-infected mice was associated with reduced sequestration in the brain of CD4+ and CD8+ T cells, including CD8+ granzyme B+ and CD8+ IFN-γ+ cells, and, to some extent, neutrophils. This was consistent with a reduced parasite sequestration in the brain, lungs, and spleen, though to a lesser extent than in wild-type P. berghei ANKA-infected mice. In summary, Plasmodium HMGB2 acts as an alarmin that contributes to the pathogenesis of cerebral malaria.
INTRODUCTION
Malaria, the disease caused by Plasmodium infection, accounts for an estimated 660,000 deaths per year, mainly in sub-Saharan African regions (1). Among the five Plasmodium subspecies infecting humans, Plasmodium falciparum is the most prevalent in these regions, where it can cause the development of clinical symptoms ranging from asymptomatic or flu-like illness to the so-called severe forms of malaria, which include severe anemia, respiratory distress, acidosis, multiorgan failure, and, eventually, cerebral malaria (2, 3). Of these clinical outcomes, cerebral malaria (CM) is the most common, is fatal, and occurs mainly in children under the age of 5 years (for a review, see references 3 and 4). There is currently no cure for CM, and therefore, identification and functional characterization of host and parasite molecules involved in the pathogenesis of this disease are of major importance, as they could lead to the development of new therapeutic interventions.
The molecular mechanisms underlying the pathogenesis of CM remain poorly understood. Presumably, sequestration of parasitized red blood cells (pRBCs) in the brain microvasculature, in association with a systemic inflammation with production of free radicals and proinflammatory cytokines, is critical to the onset of this disease. While several host genes, encoding, e.g., cytokines, chemokines, and adhesion molecules (5–8), and the products of hemoglobin degradation (9–11) have been implicated in the pathogenesis of CM as related to the proinflammatory nature of the host immune response, the individual contributions of genes encoded by Plasmodium to the onset of CM are less clear. Recently, some Plasmodium proteins were reported to play a role in severe malaria. First, in human malaria, the expression of several specific PfEMP1 proteins promoted cytoadherence to brain endothelial cells and correlated with the severity of cerebral malaria (12, 13), suggesting that these proteins constitute a major virulence factor for cerebral malaria. Second, in rodents, the plasmepsin-4 (pm4) gene, encoding an aspartic protease involved in hemoglobin digestion, was shown to be implicated in experimental cerebral malaria (ECM) (14, 15). Finally, several studies also reported a crucial role for ICAM-1 in malaria pathogenesis, and notably, ICAM-1-deficient mice are protected from ECM (16). The combined effect of these pathological events is associated with disruption of the physical integrity of the blood-brain barrier (BBB) and the development of brain edema, leading to coma and, ultimately, death (2, 17–20). One of the characteristic features of ECM is the increased sequestration of CD4+ and CD8+ T cells, which have been reported to be associated with disease pathogenesis (21, 22). More recently, a subpopulation of CD8+ T cells expressing granzyme B (GzmB) was found to be critically involved in ECM (23) and was associated with the parasite burdens within the brains of C57BL/6 mice infected with P. berghei ANKA parasites (24). This finding underlines a direct link occurring in the brain between parasite load and CD8+ T-cell recruitment and the accumulation of products of hemoglobin oxidation in the pathogenesis of ECM. Proinflammatory cytokines, such as gamma interferon (IFN-γ), have been shown to be implicated in the pathogenesis of ECM (25, 26). The production of interleukin-6 (IL-6) in response to tumor necrosis factor alpha (TNF-α) by endothelial cells of brain capillaries was also reported for mice genetically sensitive to CM development (27). More recently, various reports showed that type I IFN governs cerebral malaria (28). High-mobility-group-box (HMGB) proteins are nuclear proteins originally shown to be loosely associated with DNA and to participate, at least to some extent, in the regulation of gene transcription (for a review, see references 29 and 30). Subsequent studies showed that the human HMGB (huHMGB) proteins are composed of two HMGB domains, box A and box B, in tandem, and that the HMGB1 isoform is actively secreted from activated innate immune cells, namely, macrophages (31), and can be released from injured cells (32) as well. In humans, HMGB1 has an important role as an extracellular soluble protein that signals tissue injury and initiates an inflammatory response. This “alarmin” protein is considered a danger signal that alerts the innate immune system to trigger a defensive and inflammatory response. Indeed, once released, extracellular HMGB1 becomes associated with a variety of endogenous ligands that are recognized by several pattern recognition receptors, including members of the Toll-like receptor (TLR) family as well as the receptor for advanced glycation end products (RAGE) (33). This explains the ability of HMGB1 to trigger inflammatory responses (34) contributing to the pathogenesis of inflammatory disorders (35). Plasma or serum HMGB1 levels were increased in patients with sepsis (36, 37), hemorrhagic shock (38), or trauma (39), and high circulating levels of HMGB1 are often associated with critical illness (40, 41). This proinflammatory effect has been mapped to a single 20-amino-acid domain of box B of the mammalian protein that alone bears the proinflammatory activity of this protein (42). Moreover, huHMGB1 can promote vascular permeability, leading to edema (43). Presumably, the combined proinflammatory and vasoactive effects of HMGB1 participate subsequently to the establishment of an amplification loop (44), which explains the involvement of the protein in the pathogenesis of a variety of immune-mediated inflammatory diseases. In malaria, the huHMGB1 serum levels evaluated in P. falciparum-infected children who developed fatal cases of CM were significantly higher than those for uncomplicated malaria cases (45, 46), suggesting that as for other immune-mediated inflammatory diseases, HMGB1 might also be involved in the pathogenesis of CM.
Considering the established role of human HMGB proteins in the pathogenesis of several inflammatory diseases, we hypothesized that Plasmodium HMGB proteins might contribute to inflammation during severe malaria. Among these disorders, CM is defined as a lethal outcome of Plasmodium infection that is frequently associated with multiple-organ failure (47).
We previously showed that several Plasmodium genes encoding transcription-associated proteins (TAPs) implicated in chromatin remodeling (48) include the genes for the HMGB isoform orthologues PfHMGB1 and PfHMGB2 (49). As for mammalian HMGB proteins, in Plasmodium there are two HMGB proteins, HMGB1 and HMGB2, which consist of only one HMGB domain and also comprise a 20-amino-acid peptide potentially bearing the proinflammatory activity. In keeping with this notion, P. falciparum HMGB proteins are released into culture medium and present in the plasmas of infected C57BL/6 mice, and a recombinant protein was shown to induce TNF-α and IL-6 production (50). Since we consider that cerebral vascular obstruction and systemic inflammation probably combine to trigger cerebral malaria, we studied the role of parasitic HMGB proteins in the onset of cerebral malaria. On the basis of these observations, we hypothesized that Plasmodium HMGB proteins might act in a proinflammatory and vasoactive manner and thus be involved in the pathogenesis of severe forms of malaria, including CM. We showed that the involvement of the hmgb2 gene in the pathogenicity of the parasite is governed by the parasite itself—P. berghei ANKA or P. berghei NK65—in addition to the mouse strain—C57BL/6 or BALB/c—highlighting the importance of the host/parasite context. Most importantly, we provide evidence showing that this is indeed the case, using an ECM model comprising the ECM-sensitive (ECM-S) mouse line C57BL/6 infected with either the highly lethal strain Plasmodium berghei ANKA or its pbhmgb2-deficient cognate, Δhmgb2 ANKA. The involvement of pbhmgb2 in ECM was also studied via supplementation experiments with the recombinant PbHMGB2 protein.
MATERIALS AND METHODS
Ethics statement.
All animal care and experiments involving mice described in the present study were approved by the Direction Départementale des Services Vétérinaires de Paris, France (permit A75-13-01), and performed in compliance with institutional guidelines and European regulations (http://ec.europa.eu/environment/chemicals/lab_animals/home_en.htm). All surgery was performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
Mice.
C57BL/6 and BALB/c mice were purchased from Janvier and Charles River Laboratories, respectively.
Parasites.
Mice were inoculated with red blood cells (RBCs) infected with either green fluorescent protein (GFP)-transgenic P. berghei ANKA (MRA-867) or Δhmgb2 ANKA parasites and, occasionally, with P. berghei NK65 (MRA-268) and its hmgb2-deficient counterpart.
Murine model of ECM.
In all experiments, RBCs infected with P. berghei ANKA or its knockout counterpart were used to infect 5- to 10-week-old C57BL/6 or BALB/c mice. Parasites were reactivated before every experiment by previous passages in ECM-S C57BL/6 mice. Depending on the experiment, C57BL/6 mice were then infected by intravenous (i.v.) or intraperitoneal (i.p.) inoculation of 105 pRBCs. Parasitemia was determined by flow cytometry and the results expressed as the percentage of pRBCs. C57BL/6 mice infected with P. berghei ANKA or Δhmgb2 ANKA supplemented with recombinant PbHMGB2 were monitored for clinical symptoms of ECM, including hemi- or paraplegia, deviation of the head, the tendency to roll over on stimulation, ataxia, and convulsions.
Protein preparation.
Escherichia coli BL21(DE3)/pLysS (Stratagene) was used to express the PbHMGB2 protein from the pEXP5-CT/TOPO (Invitrogen) construct. The pbhmgb2 open reading frame (ORF) was amplified by PCR from P. berghei ANKA cDNA by using the following oligonucleotides: pbhmgb2 forward, 5′ATGGCAACTAAAACACAAAA3′; and pbhmgb2 reverse, 5′TTATTCCTTGGTTTTTCTATATTCT3′. The stop codon was removed from the reverse primer to allow cloning into the pEXP5-CT/TOPO vector, in which a 6-His tag was added at the C terminus of the protein. Overnight cultures were used to inoculate 2YT medium containing appropriate antibiotics. Cultures were shaken at 230 rpm until the A600 reached 0.6. Protein expression was then induced for 4 h at 37°C by the addition of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). The bacteria were then harvested by centrifugation for 20 min at 4,000 rpm and stored frozen at −20°C.
PbHMGB2 was purified using a protocol adapted from a previous study (51). Bacteria were suspended in 1 ml of lysis buffer (1× phosphate-buffered saline [PBS], 10 mM β-mercaptoethanol, 0.5% Triton X-100, 10 mM imidazole) per 0.5 g of bacterial pellet, with a pinch of lysozyme. Bacteria were disrupted by sonication (Branson sonifier 450) at 10 W 10 times for 20 s each, with 20-s pauses, and centrifuged for 25 min at 10,000 rpm and 4°C. The supernatant was collected and loaded onto Ni-nitrilotriacetic acid (Ni-NTA) agarose beads (Qiagen) which were previously equilibrated in 5 ml of washing buffer 1 (1× PBS, 20 mM imidazole, 0.1% Triton X-114). Triton X-114 was added to this washing buffer to remove contaminating lipopolysaccharide (LPS). The mixture was rotated on a wheel for 2 h at 4°C. Wet beads (0.2 ml per 1 ml of lysate) were incubated for 2 h. Beads were centrifuged thereafter at 4°C and 3,000 rpm for 3 min and then washed 3 times for 10 min each with 25 volumes of washing buffer 1 and then 2 times for 10 min each with 10 volumes of washing buffer 2 (1× PBS, 30 mM imidazole) to remove the residual Triton X-114. Protein elution was achieved by using 1× PBS containing 150 mM to 200 mM imidazole. The recombinant protein concentration and purity were assessed by spectrophotometric quantitation (Bio-Rad protein assay) and 12% SDS-PAGE. The presence of residual endotoxin in the proteins was checked by using an E-toxate kit (Sigma) following the manufacturer's instructions.
Disruption of the pbhmgb2 gene.
We describe here the strategy for hmgb2 gene disruption (PBANKA_071290) that was used for the P. berghei ANKA and P. berghei NK65 parasites. Two PCR fragments flanking the HMGB2 ORF were amplified from the genomic DNA by using the oligonucleotides listed in Table S1 in the supplemental material. Amplification of the 5′-untranslated region (5′UTR) with the primer combination 5′-hmgb2-for and 5′-hmgb2-rev, including ApaI and SmaI restriction sites, respectively, resulted in a 526-bp fragment which was cloned upstream of the positive selection marker, the human dihydrofolate reductase gene (hudhfr), previously introduced into the pBC SK-vector (Stratagene) under the control of the EF1α promoter and the dhfr/ts (dihydrofolate reductase/thymidylate synthase) 3′UTR. Next, the 3′UTR region was amplified with the 3′-hmgb2-for and 3′-hmgb2-rev primers, including NotI and AscI restriction sites, respectively. This fragment was inserted downstream of the hudhfr box, resulting in the hmgb2 targeting vector pBC-5′B2hudhfr3′B2, allowing replacement of the endogenous hmgb2 locus in P. berghei ANKA upon double-crossover homologous recombination and subsequent selection with the antifolate pyrimethamine (52). P. berghei parasite transfections were performed with 5 μg ApaI/AscI-digested pBC-5′B2hudhfr3′B2 and gradient-purified P. berghei ANKA schizonts as described previously (53). Positive selection for successful integration of the targeting plasmid was carried out by providing 70 μg/ml of pyrimethamine in drinking water for a period of 8 days. Transfer of the emerging P. berghei population into naive animals confirmed pyrimethamine resistance. Genomic DNAs from selected parasite populations were genotyped by an integration-specific PCR using primers dhfr-for and hmgb2-anarev (see Table S2 in the supplemental material). Clonal hmgb2-deficient parasite populations were obtained by limiting dilution in 20 naive Swiss mice and confirmed by diagnostic PCR and Southern blotting. A probe spanning the 5′-hmgb2 portion was amplified by PCR and digoxigenin-dUTP (DIG) labeled using a DIG random prime labeling kit (Roche Applied Science). Southern blotting was performed after running 4 μg of MstI-digested genomic DNA on a 0.8% agarose gel, with passive transfer to a nitrocellulose membrane, and the DNA was revealed using an anti-DIG antibody coupled to peroxidase (Roche Applied Science).
Supplementation with recombinant proteins.
We assessed the best concentration of protein capable of restoring ECM in mice according to the data reported for recombinant HMGB1 (54). C57BL/6 mice infected with 105 Δhmgb2 ANKA pRBCs were injected by i.p. inoculation from day 4 to day 8 postinfection (p.i.), using 25 mg of PbHMGB2/kg of body weight twice a day (every 12 h). According to the LPS contamination of the protein preparations (25 ng or 50 ng per protein injection), control infected C57BL/6 mice were inoculated following the same procedure, with the protein elution buffer containing the same concentrations of LPS. Also, uninfected C57BL/6 mice were injected with recombinant PbHMGB2 alone to assess the lack of toxicity of the recombinant protein.
Evaluation of parasite burdens by cytometry and Giemsa counting.
The growth of wild-type (WT) P. berghei ANKA and derived mutant parasites was determined by flow cytometry using an Epics XL Beckman flow cytometer as described previously (53). Experiments were repeated three times, and statistical analysis was performed using the Student t test. Growth of WT P. berghei NK65 and derived mutant parasites was determined by microscopic examination of Giemsa-stained thin blood smears. Parasitemia was measured by counting 3,000 red blood cells and expressed as the percentage of total parasitized erythrocytes.
Histological analysis.
Brains from P. berghei ANKA- and Δhmgb2 ANKA-infected C57BL/6 mice were removed at specific time points of infection, fixed in 4% neutral buffered formalin for 4 days, and then dissected and embedded in paraffin. For each brain, five transversal large sections, each 2 mm thick, from the brain stem to the olfactory bulb, were removed before being embedded in five paraffin blocks. For each paraffin block, serial 5-μm sections stained with hematoxylin and eosin and Giemsa stain were studied, and this protocol correlated with a total of 20 sections for each mouse brain (modified from the protocol described for the mouse brain in stereotaxic coordinates [55]). Elementary lesions were studied according to the following criteria: hemorrhages, malaria pigment deposition, and attachment of red and white blood cells to the endothelium. A hemorrhagic focus was defined as a minimal surface of a 10- by 10-μm square, malaria pigment was observed only in infected red cells, and adhesion in murine animal models was observed only for white blood cells, in contrast to human pathology.
BBB permeability.
C57BL/6 mice infected with P. berghei ANKA were injected retro-orbitally with 0.1 ml of 2% (wt/vol) Evans blue (EB) in PBS when clinical symptoms of ECM were observed (head deviation, convulsions, ataxia, and paraplegia), usually on days 6 to 8 p.i. Δhmgb2 ANKA-infected C57BL/6 mice were injected following the same protocol at the same time, and also later on (day 12 p.i.), in cases of survival. One hour later, mice were perfused with 20 ml PBS under anesthesia (0.5 ml xylazine [Rompun], 1 ml ketamine [Imalgène 1000], quantity sufficient for 4 ml 1× PBS), and brains were harvested and photographed.
In the protein supplementation experiments, the brains of mice infected with either WT or knockout (KO) parasites, supplemented or not with recombinant HMGB2, were harvested as depicted above, weighed, and incubated in 2 ml formamide for 48 h at 37°C and in the dark. The quantification of BBB disruption was evaluated by measuring the absorbance at 620 nm, with a correction at 740 nm. The amount of Evans blue that infiltrated the brain was measured by comparison with a standard curve of Evans blue in formamide.
Preparation of total RNA and reverse transcription-quantitative PCR (RT-qPCR) transcript analysis.
At different times postinfection, total RNAs were extracted from brains, spleens, and lungs removed from C57BL/6 mice infected with either P. berghei ANKA or Δhmgb2 ANKA parasites. RNA preparation was performed between days 6 and 8 (coma stage) and at day 12 postinfection, i.e., the last day analyzed (survival). Total RNAs were extracted from the samples by mechanical grinding using TRIzol (Invitrogen) following the manufacturer's instructions. Contaminant DNA and proteins were removed following Qiagen's protocol for RNA cleanup (Qiagen RNeasy kit), and the integrity of the RNAs was controlled by using a Bioanalyzer 2100 instrument (Agilent Technologies).
The expression levels of diverse transcripts were analyzed by real-time RT-qPCR in an MX 3005P cycler (Stratagene), using SYBR green Jumpstart TaqReadyMix (Sigma) and various primer sets (see Table S2 in the supplemental material). Murine hmgb transcripts as well as transcripts of diverse pro- and anti-inflammatory cytokines and cellular adhesion molecules (cams) were monitored as follows. Reverse transcription of 1 μg total RNA was performed using a Superscript VILO cDNA synthesis kit (Invitrogen) according to the manufacturer's instructions. Five microliters of a 1/10 dilution of each cDNA was amplified with a 200 nM concentration of every primer set and 10 μl of SYBR green in a final volume of 20 μl. Amplification conditions comprised initial denaturation at 95°C for 2 min followed by 40 cycles of denaturation for 30 s at 95°C and annealing-elongation for 60 s at 60°C. The specificity of the reaction was controlled by the generation of a dissociation curve with 36 cycles of 30 s, starting at 60°C, with an increment of 1°C every cycle. The relative expression levels of transcripts were evaluated against that of uninfected C57BL/6 mice and normalized for each RNA sample with the hypoxanthyl-guanine phosphoribosyltransferase (hprt) transcript.
Preparation of brain and spleen cell suspensions.
Brains and spleens were obtained from wild-type P. berghei ANKA- or Δhmgb2 ANKA-infected C57BL/6 mice at the coma stage of CM (day 6 [d6]). Briefly, mice anesthetized with ketamine (600 mg/kg) and xylazine (20 mg/kg) were perfused with 50 ml of PBS. Each brain was then removed and homogenized in RPMI 1640 medium (BioWhittaker, Walkersville, MD) by passage through sterile meshes to obtain a single-cell suspension. To harvest leukocytes from brain tissue, Percoll (Pharmacia Biotech, Uppsala, Sweden) was added at a final concentration of 35% to the cell pellet and centrifuged at 400 × g for 20 min at 20°C. The cell pellet was washed twice and analyzed via flow cytometry.
Flow cytometric analysis of brain and spleen leukocytes.
Spleen and brain cells were stained for fluorescence-activated cell sorter (FACS) analysis according to standard protocols in cold PBS containing 2% fetal calf serum and 0.01% sodium azide (FACS buffer) with the following antibodies: allophycocyanin (APC)-labeled CD4, fluorescein isothiocyanate (FITC)-labeled anti-CD8α, FITC-labeled anti-F4/80 antibody, phycoerythrin (PE)-labeled anti-Ly6G antibody, PE-labeled anti-granzyme B antibody, and PE-labeled anti-IFN-γ antibody. All antibodies were purchased from eBioscience, San Diego, CA. RBCs were eliminated using cell lysis buffer, and cells were washed in FACS buffer. Living cells of the brain and spleen (4 × 104 and 105, respectively) were analyzed using a four-color FACSCalibur flow cytometer with ProCellQuest software (BD Biosciences, Mountain View, CA).
Enzyme-linked immunosorbent assay (ELISA) detection of PbHMGB1 and PbHMGB2 proteins in the sera of mice.
Serum samples were obtained from naive or infected C57BL/6 mice inoculated with the WT or Δhmgb2 ANKA strain. Titrations of PbHMGB1 and PbHMGB2 were performed using the protocol described by Barnay-Verdier et al. (56), with slight modifications. The majority of serum proteins other than PbHMGB1 and PbHMGB2 were removed by precipitation in the presence of 3% perchloric acid (PCA), as follows. A stock solution of 13.7% PCA was prepared by mixing a 1.26 volume of 70% PCA (the concentration of PCA sold commercially) with 8.69 volumes of H2O. The stock solution was stored in a tinted bottle sealed at room temperature. To 1 volume of serum (or other test sample) on ice, a 1/4 volume of 13.7% PCA was added and mixed well. A solution of 3% PCA led to the immediate formation of an abundant precipitate. The sample was immediately centrifuged for 5 min at 13,000 × g at 4°C. A known volume of the supernatant, which contained the PbHMGB proteins, was collected and neutralized by addition of a 1/5 volume of 1.5 M NaOH. In our experience, 200 μl of supernatant was neutralized with 17 μl of 10 M NaOH. Since the process of precipitation/neutralization led to an increase in volume of 50% due to the addition of the reagents, 1.5 μl of the neutralized supernatant was tested to measure the concentration of PbHMGB in 1 μl of serum. A volume (50 μl) diluted 1/4 was added to a 96-well plate precoated with rabbit anti-PbHMGB1 or anti-PbHMGB2 IgG antibody and incubated for 2 h at 37°C. After saturation with 100 μl of 1% bovine serum albumin (BSA), the wells were washed, and biotin-labeled rabbit anti-HMGB1 or anti-HMGB2 IgG was added at a predefined dilution of 1/250 and incubated for 2 h at 37°C. After washing, 50 μl was added to each well, followed by streptavidin-peroxidase. The reaction was revealed by adding ortho-phenylenediamine plus H2O2.
Statistical analysis.
Differences in mouse survival were evaluated by the generation of Kaplan-Meier survival plots and log rank analysis. Differences in growth rates (peripheral parasitemia) and parasite loads were analyzed by the Student t test. Kruskal-Wallis analysis was performed to compare means between the 3 or 4 groups of data for hemorrhagic focus numbering, survival curves, BBB disruption evaluation, brain- or spleen-infiltrating T-cell numbering, and transcript expression levels. Dunn's posttest was applied to analyze the effect of pbhmgb2 deletion. P values of <0.05 were considered statistically significant for each test.
Nucleotide sequence accession numbers.
The sequences of the P. berghei high-mobility-group protein gene pbhmgb2 (PBANKA_071290) and the P. berghei ANKA rRNA-encoding gene berg06_18s are available in PlasmoDB (http://plasmodb.org/plasmo/). Mouse gene sequences are available in NCBI GenBank under the following accession numbers: Mus musculus hypoxanthine guanine phosphoribosyltransferase (Hprt), NM_013556.2; M. musculus high-mobility-group protein 1 (muhmgb1), NM_010439.3; M. musculus high-mobility-group protein 2 (muhmgb2), NM_008252.3; M. musculus tumor necrosis factor alpha (tnfα), NM_013693.2; M. musculus interleukin 6 (il-6), NM_031168.1; M. musculus gamma interferon (ifnγ), NM_008337.3; M. musculus interleukin 10 (il-10), NM_010548.2; M. musculus intercellular adhesion molecule 1 (icam1), NM_010493.2; M. musculus vascular adhesion molecule 1 (vcam1), NM_011693.3; and M. musculus heme oxygenase 1 (hmox1), NM_010442.2.
RESULTS
Plasmodium HMGB proteins are released into the extracellular milieu.
To explore the systemic biological effect of HMGB2 on Plasmodium pathogenicity, our working hypothesis was based on the secretion of this “alarmin” protein from the parasite. We determined the occurrence of the P. berghei rodent parasite protein (PbHMGB2) in the sera of C57BL/6 mice infected with wild-type P. berghei ANKA via ELISA titration (Fig. 1). As expected, the protein was not found in the sera of naive noninfected mice but was present in mice infected by the WT P. berghei ANKA parasite, i.e., the rodent model of ECM (as PbHMGB1) (data not shown).
FIG 1.
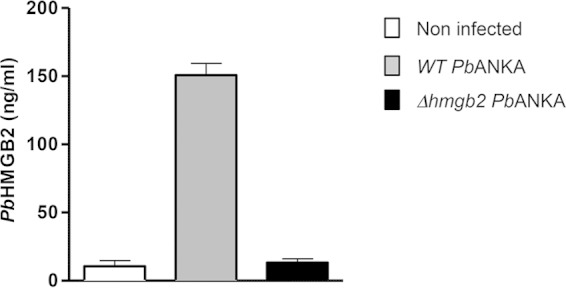
Release of the HMGB2 protein into the extracellular milieu. Release of P. berghei ANKA (PbANKA) HMGB2 into the sera of mice was analyzed by ELISA. C57BL/6 mice were inoculated with 105 erythrocytes infected with either Δhmgb2 or WT P. berghei ANKA parasites. Sera of four mice were taken at d7 postinfection and tested for their HMGB2 content by ELISA. Sera from naive mice were taken as negative controls. Results shown in the figure are expressed in nanograms per milliliter, as determined by a titration curve obtained using a range of concentrations of recombinant HMGB2.
Parasite multiplication and ECM occurrence are generated differently according to the host-parasite strain context.
We studied the implications of the HMGB2 protein in several parasite and murine strains. We used two highly pathogenic P. berghei parasites, P. berghei ANKA and P. berghei NK65, leading to the death of mice within 7 ± 1 days per ECM or 20 to 25 days per hyperparasitemia, respectively. Actually, two strains of mice, known to be susceptible to ECM (ECM-S) (C57BL/6 mice) or resistant to ECM (ECM-R) (BALB/c mice), were infected by the two WT parasites, and the effects of addition of the recombinant HMGB protein were investigated (Fig. 2A to D). The occurrence of ECM is dependent on the host-parasite combination (summarized in Table 1).
FIG 2.

Multiplication of diverse WT and hmgb2-deficient parasites according to the host mouse strain. (A and B) Survival (A) and parasitemia (B) of BALB/c mice infected with WT P. berghei ANKA and supplemented or not from days 4 to 8 p.i. (dashed line) with either recombinant PbHMGB2 (25 mg/kg) or vehicle (protein buffer plus 50 ng LPS, which takes into account the residual LPS contamination of the recombinant protein) twice a day (every 12 h). All mice were injected i.v. with 106 red blood cells infected with WT P. berghei ANKA. Differences in mortality/survival between WT-infected mice, supplemented or not with PbHMGB2, were analyzed by the log rank test. (C and D) Survival (C) and parasitemia (D) of C57BL/6 mice infected with WT P. berghei NK65 and supplemented with recombinant PbHMGB2 as described above. All mice were injected i.v. with 105 red blood cells infected with WT P. berghei NK65. For panels A to D, groups of mice contained five mice each. (E and F) Red blood cell development of P. berghei ANKA (E) and P. berghei NK65 (F), as well as their Δhmgb2 counterparts, in infected BALB/c and C57BL/6 mice, respectively. Parasitemia was measured by flow cytometry (E) and Giemsa counting (F) for sets of five mice. Values represent means ± standard deviations (SD) for one representative experiment of three. Statistical analyses of parasitemia were performed by the Student t test.
TABLE 1.
Occurrence of ECM according to host-parasite strain context
| P. berghei strain | Presence of PbHMGB2 protein | Occurrence of ECMa |
|
|---|---|---|---|
| C57BL/6 mice | BALB/c mice | ||
| WT P. berghei ANKA | − | †, 100% ECM | No ECM, 100% death by hyperparasitemia |
| + | ND | †, 65% ECM | |
| KO P. berghei ANKA | − | 65% ECM survival | No ECM, 100% death by hyperparasitemia |
| + | †, 65% ECM | ||
| WT P. berghei NK65 | − | No ECM, 100% death by hyperparasitemia | ND |
| + | No ECM, 100% death by hyperparasitemia | ND | |
| KO P. berghei NK65 | − | No ECM, no death by hyperparasitemia, 100% survival | ND |
| + | ND | ND | |
†, death of mice; ND, not determined.
In the ECM-R BALB/c mice, P. berghei ANKA parasites developed with a complete absence of neurological symptoms associated with ECM, thereafter leading to death via hyperparasitemia (Fig. 2B). However, injection of recombinant PbHMGB2 (see details in Materials and Methods) into these ECM-R mice induced the death of mice (60%) within 7 to 9 days p.i., with neurological symptoms (Fig. 2A). We verified that the protein alone was not pathogenic and therefore that ECM did not rely on the toxicity of the proteins.
In the ECM-S C57BL/6 mice, the P. berghei NK65 parasite developed in the absence of neurological symptoms (Fig. 2C, vehicle control). Supplementation with the PbHMGB2 protein did not induce ECM (Fig. 2C), and P. berghei NK65 multiplication was not affected (Fig. 2D).
We generated hmgb2-deficient P. berghei ANKA and P. berghei NK65 parasites (Δhmgb2 ANKA and Δhmgb2 NK65) as described in Fig. S1 in the supplemental material. The asexual development of the P. berghei ANKA and P. berghei NK65 strains, as well as their hmgb2-deficient derivatives, was studied in the two mouse strains. In ECM-R BALB/c mice, the multiplication of the WT was similar to that of the KO P. berghei ANKA parasite (Fig. 2E). In ECM-S C57BL/6 mice, the multiplication levels of the P. berghei NK65 and Δhmgb2 NK65 parasites appeared to be different: whereas the WT parasitemia increased for up to 15 days or more, the multiplication of Δhmgb2 NK65 parasites increased and then decreased thereafter for 15 days, when it was no longer detected in the peripheral blood (Fig. 2F). Indeed, the hmgb2 gene disruption modified the fitness of the parasite, since from similar multiplication levels at early times p.i., the hmgb2-deficient P. berghei NK65 parasites were ultimately cleared from the peripheral blood.
Finally, in the ECM-S C57BL/6 mice, P. berghei ANKA and Δhmgb2 ANKA parasites developed differently (see the next paragraph). Only P. berghei ANKA infection triggered ECM in C57BL/6 mice, in contrast to all combinations just mentioned (Table 1). We therefore decided to focus our study on the implications of HMGB2 in the occurrence of ECM by the use of C57BL/6 mice infected with P. berghei ANKA WT and hmgb2-deficient parasites.
Deletion of Plasmodium hmgb2 protects C57BL/6 mice from an ECM outcome.
As just mentioned, the rodent ECM model used in the following experiments showed the death of C57BL/6 mice within 6 to 7 days p.i. when mice were i.p. injected with 105 red blood cells infected with P. berghei ANKA (Fig. 3A). We investigated the role of the HMGB2 protein in experimental cerebral malaria via deletion of the gene from the parasite. A targeting construct was generated, i.e., pBC-5′B2hudhfr3′B2, that, after double-crossover homologous recombination, inserts the huDHFR selectable cassette in place of the Plasmodium berghei ANKA hmgb2 locus. After transfection of the linearized pBC-5′B2hudhfr3′B2 plasmid into P. berghei ANKA merozoites, recombinant parasites were selected and cloned (see Fig. S1 in the supplemental material). pbhmgb2 gene disruption in P. berghei ANKA parasites was verified in three independent clones. When the ECM-S C57BL/6 mice were infected with these three clones, we observed that the clones differed from the wild-type parasites in their lethality and occurrence of ECM (see Fig. S2). Indeed, a marked increase in survival from ECM was observed, with survival of up to 100% at d20 p.i., depending on the clone tested. We selected clone F, which we named the Δhmgb2 ANKA strain, to perform all subsequent analyses. Actually, PbHMGB2 was not detected in the sera of mice infected with Δhmgb2 ANKA, in contrast to the sera of mice infected with WT P. berghei ANKA (Fig. 1), nor was the hmgb2 transcript (see Fig. S1). Using the same procedure, no Δhmgb1 parasites were obtained after three independent transfections, suggesting that the gene is essential for parasite development.
FIG 3.

Comparative analyses of C57BL/6 mice infected with WT P. berghei ANKA and Δhmgb2 ANKA parasites. (A) Kaplan-Meier survival plot for C57BL/6 mice infected with either 105 WT P. berghei ANKA or Δhmgb2 ANKA parasites. Infected mice were monitored every day, starting on d5 p.i., for ECM symptoms. The difference in survival between WT- and Δhmgb2 ANKA-infected C57BL/6 mice was analyzed by the log rank test. This experiment was performed 3 times (n = 4, n = 7, and n = 9). (B) Parasitemia of both parasites as measured by flow cytometry. Values represent means ± SD. (C) Histological analyses of brains from WT- and Δhmgb2 ANKA-infected C57BL/6 mice. The images show representative hematoxylin- and eosin-stained sagittal sections of brains harvested at d6 and d12 p.i. from WT (d6, panels 1 to 3)- and Δhmgb2 ANKA (d6, panels 5 to 7; d12, panels 9 to 11)-infected C57BL/6 mice (n = 5 for each experimental group). The black arrows indicate the hemorrhagic foci. In each row, two incremental magnifications of the same microscopic field are presented. Evans blue dye leakage in brains of WT (d6, panel 4)- and Δhmgb2 ANKA (d6, panel 8; d12, panel 12)-infected C57BL/6 mice is presented in the far right column. (D) Microscopic counts of the numbers of hemorrhagic foci per histological brain section at d6 and d7 p.i. (n = 5 for each experimental group). Multiple comparisons of focus numbers were analyzed by the Kruskal-Wallis test (with Dunn's multiple-comparison test; P = 0.0008).
The survival and peripheral parasitemia of mice infected by either WT P. berghei ANKA or Δhmgb2 ANKA were monitored in three independent experiments. As expected, all mice infected (i.p.) with WT P. berghei ANKA died within 6 to 7 days (d6 to d7). In contrast, deletion of the hmgb2 gene was associated with a reduction of ECM incidence, with 65% ± 10.67% (mean ± standard error of the mean [SEM]) (P < 0.0001) of mice not developing ECM and succumbing 20 days after infection from hyperparasitemia, a lethal outcome of P. berghei ANKA that is unrelated to ECM (Fig. 3A). Both strains developed similar peripheral parasitemias for up to 6 days, as evaluated by the percentage of parasitized red blood cells (Fig. 3B). The Δhmgb2 ANKA parasite maintained its development until d20, leading to the death of C57BL/6 mice later by severe anemia and hyperparasitemia. To confirm that the lack of ECM onset in Δhmgb2 ANKA-infected mice was not due to an alteration of parasite growth, we compared the asexual multiplication rates of both parasites. The cloning procedure described by Janse and colleagues (53) was achieved at d6 by counting Giemsa-stained blood smears. The mean asexual multiplication rate at 24 h was calculated by assuming a total of 1.2 × 1010 erythrocytes/mouse and resulted in values of 11.94 ± 0.7 and 11.66 ± 0.2 for WT P. berghei ANKA and Δhmgb2 ANKA, respectively. The asexual multiplication of the KO parasite was not reduced compared to that of the WT parasite, underlining the identical growth rate of the knockout parasites.
Deletion of Plasmodium hmgb2 protects C57BL/6 mice from blood-brain barrier damage.
Suppression of ECM in C57BL/6 mice infected with Δhmgb2 ANKA was associated with reduced brain damage, as assessed by the extent of microvascular hemorrhages, and with reduced parasite sequestration. As early as d5 p.i., prior to ECM manifestation, no apparent brain damage was observed in Δhmgb2 ANKA-infected C57BL/6 mice, whereas minimal hemorrhages were already noticeable in 20% of mice infected with WT parasites (data not shown). P. berghei ANKA-infected C57BL/6 mice exhibited extended and severe hemorrhagic foci at d6 to d8 after infection (Fig. 3C, top row), while mice infected with the mutant parasite displayed moderate hemorrhagic foci (Fig. 3C, middle row). In an additional experiment, there was a marked decrease in the number of hemorrhagic foci per histological brain section at d6 and d7 p.i. (P < 0.05) for each time point between P. berghei ANKA and Δhmgb2 ANKA (Fig. 3D). Finally, at d12 p.i., mice surviving Δhmgb2 ANKA infection did not exhibit either hemorrhages or cerebral lesions (Fig. 3C, bottom row). These observations were confirmed by BBB disruption, as illustrated by Evans blue leakage in the brains of WT P. berghei ANKA-infected mice, whereas this was not apparent in brains of Δhmgb2 ANKA-infected mice (Fig. 3C, right column).
Exogenous PbHMGB2 protein restores Δhmgb2 ANKA virulence.
To ascertain that the increased resistance to ECM in C57BL/6 mice infected with Δhmgb2 ANKA versus WT P. berghei ANKA was due at least in part to the lack of extracellular PbHMGB2, we assessed whether recombinant PbHMGB2 was sufficient per se to reestablish ECM susceptibility in Δhmgb2 ANKA-infected mice.
Recombinant PbHMGB2 restored the incidence of ECM in Δhmgb2 ANKA-infected mice, i.e., 60% of mice died of ECM at d10, with cerebral symptoms (Fig. 4A). This experiment, which was analyzed by the Kruskal-Wallis test (P = 0.0025), was repeated 3 times with 10 mice per group. A statistical difference was observed from d10 to d20 postinfection between mice infected with the Δhmgb2 ANKA parasite in the absence or presence of the PbHMGB2 protein as analyzed by the Student t test (P = 0.0012). The pathological effect of PbHMGB2 was not associated with a modulation of peripheral parasitemia (Fig. 4B), since the parasite fitness was not affected. Moreover, a low ECM incidence was observed for vehicle-treated C57BL/6 mice infected with the Δhmgb2 ANKA strain (≤30% death), as in controls, suggesting that the low level of LPS contamination did not account for the ECM increase observed when PbHMGB2 was injected into mice. These observations were confirmed by the BBB permeability. Figure 4C shows a histogram for Evans blue evaluation of the brains of mice infected with Δhmgb2 ANKA and complemented with the recombinant protein compared to the brains of control mice infected with the hmgb2-deficient parasite alone or in the presence of vehicle. The recombinant PbHMGB2 preparations produced in E. coli contained less than 100 ng LPS per mg of recombinant protein. This residual LPS contamination was used as a vehicle control in these experiments. A statistical difference in BBB disruption was observed between the brains of Δhmgb2 ANKA- and complemented Δhmgb2 ANKA-infected mice (P = 0.0008).
FIG 4.

ECM occurrence in Δhmgb2 ANKA-infected C57BL/6 mice supplemented with recombinant PbHMGB2 protein. (A) Survival analysis of C57BL/6 mice infected i.p. with 105 Δhmgb2 ANKA pRBCs and thereafter injected from days 4 to 8 p.i. (dashed line) with either PbHMGB2 (25 mg/kg) or vehicle (protein buffer plus 50 ng LPS, which takes into account the residual LPS contamination of the recombinant protein) twice a day (every 12 h) (n = 10 for each experimental group; the experiment was done 3 times). C57BL/6 mice were also infected i.p. with 105 WT P. berghei ANKA pRBCs as an ECM positive control. Mice were monitored for ECM symptoms from d5 p.i. (B) Parasitemia was measured by flow cytometry. Values represent means ± SD for one representative experiment of three. (C) The amount of Evans blue dye (BE) that infiltrated the brain was measured as described in Materials and Methods for the three sets of mice (n = 5 for each experimental group). Multiple comparisons were analyzed by the Kruskal-Wallis test followed by Dunn's posttest for analyses of differences in survival (P = 0.0025) and BBB permeability (P = 0.0008) between all sets of mice.
Resistance of mice to development of ECM following infection with Δhmgb2 ANKA correlates with a reduction of brain-infiltrating T cells.
The development of CM is strongly associated with the activation and recruitment of T cells in the brain (21, 22). Accordingly, we examined whether such T-cell recruitment occurred when mice were infected with Δhmgb2 ANKA. Brain- and spleen-infiltrating leukocytes were isolated from naive C57BL/6 mice or mice infected with WT or Δhmgb2 ANKA 7 days after infection (Fig. 5). Compared to the uninfected controls, C57BL/6 mice infected with WT P. berghei ANKA displayed marked increases in the numbers of CD4+ T cells (7-fold) and CD8+ T cells (64-fold) in the brain (Fig. 5A and B), as expected. In addition, CD8+ GzmB+ and CD8+ IFN-γ+ T-cell sequestration (23, 24) was also increased (18- and 40-fold, respectively) compared to that in the uninfected mice. Interestingly, the recruitment of CD4+ T cells (2-fold) and CD8+ T cells (4-fold) was lower in the brains of mice infected with Δhmgb2 ANKA than in those infected with the WT P. berghei ANKA parasites, as well as the recruitment of CD8+ T cells expressing GzmB or IFN-γ (3.5-fold) (Fig. 5C and D). To verify whether the lack of the pbhmgb2 gene altered the CD4+, CD8+ GzmB+, and CD8+ IFN-γ+ T-cell sequestration in areas other than the brain, a similar analysis was performed with the spleens of naive and infected mice. All data for the spleen were consistent with those observed for the brain (Fig. 5E, F, and H), except for CD8+ GzmB+ T cells, whose numbers did not differ significantly between the two groups (Fig. 5G).
FIG 5.
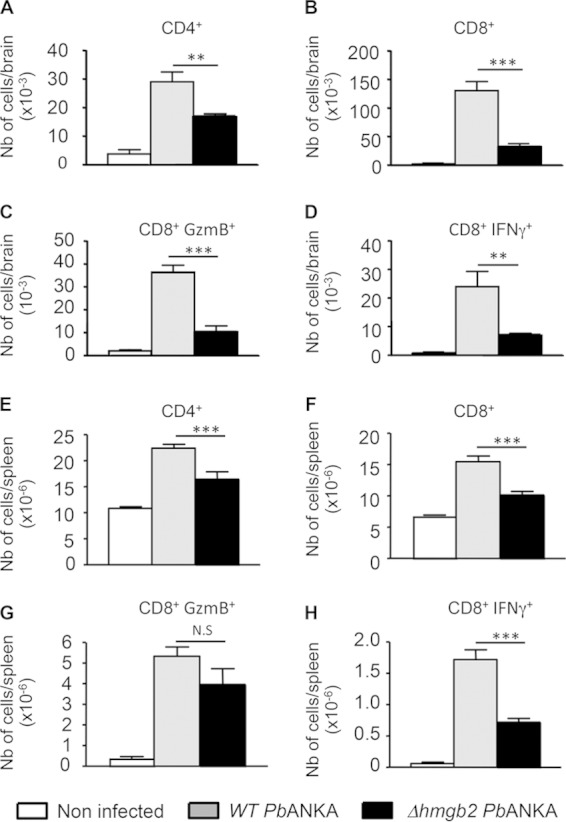
Reduced infiltration and reduced activation of CD4+ and CD8+ T cells in the brains of Δhmgb2 ANKA-infected mice. At the coma stage (day 6 postinfection), brains from WT- or Δhmgb2 ANKA-infected C57BL/6 mice, with 105 infected erythrocytes per mouse, were taken, and leukocytes associated with cerebral tissue were analyzed by FACS for the presence of the indicated leukocytes, expressed as absolute numbers per brain. Six mice per group were used. Values represent the means ± SD for one experiment of three. *, P < 0.05; **, P < 0.01; ***, P < 0.001; N.S, not significant. (A to D) CD4+, CD8+, CD8+ GzmB+, and CD8+ IFN-γ+ cells in brains. (E to H) The same cells were also analyzed in spleens. Multiple comparisons of brain-infiltrating T cells were analyzed by the Kruskal-Wallis test (with Dunn's posttest).
In search of other leukocytes that exert their effector functions during ECM, we surmised that neutrophils might represent inflammatory cells which cause the disease under particular circumstances. These cells were indeed shown to be associated with the disease pathogenesis (57). Accordingly, although the difference was not statistically significant, the percentage of Ly6G+ neutrophils in the brain was lower in Δhmgb2 ANKA-infected mice than in WT-infected mice (see Fig. S3A in the supplemental material). Analysis of sequestered F4/80+ macrophages in the brain did not show any significant difference (see Fig. S3B). Similarly, no difference could be detected in Ly6G+ neutrophils (see Fig. S3C) or F4/80+ macrophages (see Fig. S3D) within the spleens of mice from the two groups.
Do these cellular alterations that are part of the modulation of the host immune response by the HMGB2 protein correlate with parasite density (23, 24)? Parasite burdens were measured in the periphery (parasitemia) and in different tissues, including the brain, spleen, and lung (Fig. 6). We evaluated the parasite loads by RT-qPCR, measuring the relative levels of parasite 18S rRNA in C57BL/6 mice infected by both parasites, using different tissues taken at d6 to d7 p.i. Even though the peripheral parasitemia of Δhmgb2 ANKA-infected mice was comparable to that of WT P. berghei ANKA-infected mice (Fig. 6D), less parasite sequestration was noticed in mouse organs at d6 p.i. In the brain (Fig. 6A) and lung (Fig. 6B), the parasite levels of Δhmgb2 ANKA were lower (approximately 5- and 3.5-fold, respectively) than those observed for WT parasites. In the spleen (Fig. 6C), the parasite load was also lessened, but to a lesser extent (2.5-fold) than in the brain and lung.
FIG 6.

Reduced parasite biomass in the organs of Δhmgb2 ANKA-infected mice. At d6 postinfection, RNAs were extracted from the brains (A), lungs (B), and spleens (C) of WT- or Δhmgb2 ANKA-infected C57BL/6 mice (105 infected erythrocytes per mouse). Bars represent the mean ± SD parasite loads as evaluated by relative quantification of the 18S mRNA expression level, normalized via the hypoxanthyl-guanine phosphoribosyl transferase (hprt) transcript. Gray bars, WT P. berghei ANKA-infected mice; black bars, Δhmgb2 ANKA-infected mice. (D) Parasitemia was measured by counting after Giemsa staining. Values represent means ± SD. Comparisons of parasite biomass and parasitemia were analyzed by the Student t test. **, P < 0.01; ***, P < 0.001.
Deletion of Plasmodium hmgb2 decreases cytokine and adhesion protein messenger expression in brains of infected C57BL/6 mice.
The brains of infected C57BL/6 mice harvested at d6 and d12 (Fig. 3C) were analyzed to evaluate the contributions of a variety of gene transcripts to the survival of Δhmgb2 ANKA-infected C57BL/6 mice. In addition, we performed an additional transcript analysis (corresponding to Fig. 3D) by focusing our examination on d6 or d7, at the onset of ECM, and these data were included in our statistical analyses. The transcripts of the murine host, i.e., hmgb1 and hmgb2 transcripts, proinflammatory cytokine transcripts (tnfα, ifnγ, and il-6), and anti-inflammatory (il-10 and hmox-1) and adhesion (icam-1 and vcam-1) protein transcripts, were analyzed by RT-qPCR with the primer sets listed in Table S2 in the supplemental material. The levels of all transcripts (except for those of muhmgb1 and hmox-1) were increased at d6 in the brains of WT P. berghei ANKA-infected mice compared to uninfected mice, with the highest magnitude for ifnγ (Fig. 7). The levels of these transcripts were also enhanced in the brains of mice infected by Δhmgb2 ANKA, but to a lesser extent. Note that the decreased expression of the aforementioned genes was statistically significant in these mice compared to the WT parasite-infected mice. In addition, this decrease was maintained at d12 postinfection. As already mentioned, two transcripts did not follow this trend. The hmox-1 transcript, encoding a heme-degrading enzyme, showed a marked increase in Δhmgb2 ANKA-infected mice at d6, in contrast to the case in WT P. berghei ANKA-infected mice, and in turn decreased at d12. Also, the muhmgb1 transcript levels were similar in noninfected and infected mice, in contrast to the expression of the muhmgb2 transcript, which followed the common features for all other genes (markedly increased and markedly decreased in mice infected with WT P. berghei ANKA and in Δhmgb2 ANKA-infected mice, respectively).
FIG 7.

Analysis of the expression levels of cytokine, adhesion molecule, and hmgb gene transcripts in the brains of WT- and Δhmgb2 ANKA-infected C57BL/6 mice. Brains were removed from uninfected (n = 10) and WT- and Δhmgb2 ANKA-infected C57BL/6 mice at d6 (n = 14 for each group) and d12 (n = 3) p.i., and total RNAs were extracted. Bars represent the mean ± SD relative expression levels of the transcripts analyzed, normalized for each RNA sample via the hprt transcript. The expression levels of cytokine, cam, and hmgb gene transcripts were evaluated against those of naive C57BL/6 mice. White bars, noninfected mice; light gray bars, WT P. berghei ANKA-infected mice at d6; dark gray and black bars, Δhmgb2 ANKA-infected mice at d6 and d12, respectively. Multiple comparisons of expression levels were analyzed by the Kruskal-Wallis test (with Dunn's posttest). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
The molecular mechanisms underlying the pathogenesis of cerebral malaria remain poorly understood. In humans, presumably, sequestration of parasitized red blood cells in the brain microvasculature, in association with the production of inflammatory cytokines and hemoglobin oxidation, is critical for the onset of this disease. However, resources and opportunities to explore the physiopathology of this disease in humans are limited and based mainly on postmortem investigations. Although the murine model of cerebral malaria does not perfectly match the human disease, there is substantial evidence that both share common features (for a review, see references 27 and 58). Studies of the mechanisms underlying the pathophysiology of ECM revealed a number of proinflammatory proteins implicated in parasitized red blood cell sequestration and in brain pathology. Being aware that both host and parasite biological products might combine to trigger ECM, we decided to focus our study on parasite proteins. Only one pathogen-associated molecular pattern (PAMP), glycosylphosphatidylinositol (GPI), has been reported for several protozoan parasites, including Plasmodium, to elicit a deleterious host innate immune response. The P. falciparum GPI, via TLR2 and -4, signals immune cells to produce proinflammatory cytokines and induce severe malaria.
We were interested in Plasmodium HMGB proteins to assess if they might be involved in the development of ECM, since their human counterparts are involved in a number of inflammatory disorders. In the P. falciparum genome, genes encoding two orthologues of human HMGB, annotated PfHMGB1 and PfHMGB2, are present. As for the mammalian HMGB isoforms, these two proteins encompass a TNF-α-activating domain, suggesting that in addition to their involvement in chromatin remodeling (49) when released from the parasite into the extracellular milieu, they might also, like their mammalian counterparts, act as genuine inflammatory agonists. In Fig. S4A in the supplemental material, multiple-sequence alignments of different eukaryotic HMGB protein domains underline their conserved trait and the strong conservation between the Plasmodium proteins, with a higher conservation observed for box B of the mammalian proteins. In addition, alignment of the 20-amino-acid sequences of the TNF-α-activating domains (see Fig. S4B) emphasizes the greater homology encountered between the Plasmodium sequences and that of box B of the mammalian proteins (45% identity and 80% homology), reported to bear the proinflammatory activity. In keeping with this notion, P. falciparum HMGB proteins are released into culture medium (data not shown), and the P. berghei proteins are detected in the sera of C57BL/6 mice infected with P. berghei ANKA (Fig. 1). In addition, a recombinant HMGB protein induced TNF-α and IL-6 production (50). This led to the hypothesis that Plasmodium HMGB proteins might be implicated in the pathogenesis of severe forms of malaria, including cerebral malaria.
In order to study the role of HMGB in cerebral malaria, we constructed hmgb-deficient parasites (P. berghei ANKA and P. berghei NK65 derivatives) (see Fig. S1 in the supplemental material) and investigated their growth and their ability to drive ECM in C57BL/6 and BALB/c mice. Few studies have reported gene disruption at the level of erythrocytes in vivo (59–61). In mice, an absence of the hmgb1 gene is lethal, since hmgb1−/− mice are not viable and die a few hours after birth (62). The disruption of hmgb2 is not lethal, but hmgb2−/− mice displayed reduced fertility and a spermatogenesis defect (63). Along the same lines, the two P. falciparum HMGB proteins display different cellular localizations within the parasite. Whereas PfHMGB1 is essentially nuclear throughout the asexual erythrocytic cycle, PfHMGB2 is expressed predominantly in gametocytes and is present in the nucleus as well as in the cytoplasm. The two proteins also differ in their nuclear activities, with PfHMGB1 being more efficient and specific than PfHMGB2 for architectural properties (DNA binding and DNA bending), suggesting that of the two proteins, PfHMGB1 has a major role in transcription regulation and parasite development (49). This is consistent with the inability to generate Δhmgb1 P. berghei parasites, whereas several clones of Δhmgb2 parasites were obtained.
The pbhmgb2 deletion in P. berghei ANKA induced a reduction of the ECM incidence in infected ECM-S C57BL/6 mice. The pathogenic attenuation was not due to a decreased multiplication of the asexual stage, since the multiplication rate was similar to that of WT P. berghei ANKA as evaluated in the peripheral blood (Fig. 3B). Histological analysis of brain sections and EB dye leakage showed decreased brain damage already at d6 and d7 postinfection compared to WT-infected mice (Fig. 3C and D). A complete absence of hemorrhages was observed in the brains of surviving mice at d12 p.i. The injection of recombinant PbHMGB2 into Δhmgb2 ANKA-infected mice increased the incidence of ECM (up to 60%), thus reestablishing the high mortality rate observed in susceptible mice (Fig. 4). Moreover, the protein restored ECM in P. berghei ANKA-infected ECM-R BALB/c mice (Fig. 2A). In contrast, recombinant HMGB2 was not able to trigger ECM even in ECM-S mice upon infection with the P. berghei NK65 parasite (Fig. 2C), another lethal parasite that does not induce ECM. The multiplication of P. berghei NK65 parasites increased until the death of mice by hyperparasitemia, in contrast to that of the Δhmgb2 NK65 parasites, which were no longer detected in the peripheral blood after d15 p.i. (Fig. 2F). In summary, the involvement of the HMGB2 protein in the pathogenicity of the parasite is governed by the parasite as well as the mouse strain, highlighting the importance of the host/parasite context.
For this ECM model, a number of reports have proposed that sequestration of CD8+ and CD4+ T cells in the brain capillaries is a characteristic feature of the brain pathology (21, 22). Recent studies provided clear evidence that antigen-specific CD8+ T-cell-derived granzyme B (23) and CD8+ IFN-γ+ T cells (26) were critical in driving cerebral pathology. In addition, a direct causal link between parasite load and CD8+ T-cell-mediated ECM pathology was provided (24). Interestingly, the relationship between the absence of HMGB2 and the lack of ECM expression was associated with a significantly reduced recruitment of CD8+ GzmB+ and CD8+ IFN-γ+ T cells in Δhmgb2 parasite-infected mice. In addition, the recruitment of CD4+ T cells was also decreased, though to a lesser extent (Fig. 5). This suggests that the lower levels of CD4+, CD8+ GzmB+, and CD8+ IFN-γ+ T cells sequestered within the brain capillaries of mice infected with Δhmgb2 ANKA parasites could be the basis of the failure of these mice to develop ECM. It can also be inferred that HMGB2 exerts systemic effects, since it appears that sequestration of CD4+ and CD8+ T cells rather than neutrophils and macrophages also occurred in inflamed spleens.
Very recently, Renia and colleagues (26) observed that CD8+ T cells and IFN-γ drove the rapid increase in total parasite biomass and accumulation of infected RBCs in the brain and in different organs at the time when mice developed CM (24). Our data underline that in the absence of HMGB2, the decrease in the recruitment of CD8+ GzmB+ and CD8+ IFN-γ+ T cells (Fig. 5B to D) was associated with a reduced recruitment of Δhmgb2 ANKA parasites in the brain (Fig. 6A), in contrast to the similar parasitemia observed in the peripheral blood, reflecting the identical multiplication rates of the parasites (Fig. 6D). However, even though the brain was the organ where CD8+ T-cell and KO parasite sequestration was the most markedly decreased, a decrease was also observed in the lung and spleen (Fig. 5B and C), indicating that HMGB2 exerts its proinflammatory effects at a systemic level.
Compared to the brains of WT P. berghei ANKA-infected mice, the transcript levels of most proinflammatory cytokines, including tnfα and ifnγ, were decreased in the brains of Δhmgb2 ANKA-infected mice (Fig. 7). Also, the cell adhesion icam-1 transcript level was decreased, in contrast to that of vcam-1. In one study, vcam-1 was identified by microarray analysis as a gene candidate that discriminates between ECM-R and ECM-S mice in a P. berghei model (64). Our data do not support the assertion that elevated vcam-1 corresponds to ECM, as we observed slightly higher vcam-1 mRNA levels in Δhmgb2 ANKA-infected mice at d6 postinfection than in WT P. berghei ANKA-infected C57BL/6 mice, suggesting an uncertain or perhaps protective role for this adhesion molecule in our model. In concordance with our observation of a predominant role for ICAM-1 compared to VCAM-1 in malaria pathogenesis, infusion of an anti-ICAM-1 but not an anti-VCAM-1 monoclonal antibody prevented cytoadherence of infected erythrocytes in a P. yoelii model of ECM (65), in addition to in vivo evidence for the role of ICAM-1 in the sequestration of infected red blood cells in a mouse model of lethal malaria (66). In contrast, the transcript level of the cytoprotective heme oxygenase gene (hmox-1) was markedly increased at d6 in Δhmgb2 ANKA-infected C57BL/6 mice and was thus consistent with the literature (9–11). Our results showing a lower expression level of il-10 transcripts in the brains of Δhmgb2 ANKA-infected C57BL/6 mice diverge from other reports on murine CM that suggested a protective effect for IL-10 in CM (67) but are in accordance with observations in humans that showed an increase in IL-10 in CM patients compared to mild malaria patients (68). Finally, the levels of murine hmgb1 transcripts did not vary in hmgb2-deficient ANKA- and WT P. berghei ANKA-infected mice, in contrast to the murine hmgb2 transcript levels, which were highly increased in P. berghei ANKA-infected mice and remained unchanged in Δhmgb2 ANKA-infected mice.
Taken together, these data represent a breakthrough in that Plasmodium HMGB2 is ascribed a major proinflammatory cytokine-like function reminiscent of that of its mammalian HMGB counterparts, which have been shown to play key roles in several human diseases, including sepsis, lupus, rheumatoid arthritis, and cancer (69).
Our data demonstrate that deletion of a single Plasmodium gene implicated in chromatin remodeling (49) and in inflammation (50), i.e., hmgb2, contributes to reduced host lethality, an effect due to suppression of neuropathology. An infected host has two evolutionarily conserved defensive strategies that can limit its disease severity. One relies on the capacity of its immune system to reduce the pathogen load, a defensive strategy referred to as resistance to infection. The other defensive strategy acts irrespective of the pathogen load and relies instead on limiting the extent of tissue damage caused by the pathogen and/or by the immune response elicited by that pathogen. This defensive strategy is referred to as disease tolerance (reviewed in references 70 and 71). The lower virulence of hmgb2-deficient parasites than wild-type controls is not associated with modulation of the peripheral pathogen load, suggesting that PbHMGB2 can compromise disease tolerance toward blood-stage Plasmodium infection. The observation that protection from ECM in mice infected with hmgb2-deficient parasites is reversed by recombinant HMGB2 complementation suggests that it is the soluble PbHMGB2 protein that compromises the disease tolerance. This finding adds significantly to the previous observation that free heme generated as an end product of host hemoglobin oxidation also impairs disease tolerance toward Plasmodium infection (72). Whether free heme and PbHMGB2 interact functionally to trigger the development of severe forms of malaria, such as ECM, remains to be established.
Supplementary Material
ACKNOWLEDGMENTS
We thank Maurel Tefit for management of the animal house. Nathalie Bernard, Sarah Duponchel, and Nazla Bakary participated in this work as undergraduate students. We are grateful to Abiba Doukani for assistance with the genomic core facility (P3S) and to Catherine Blanc and Bénédicte Hoareau for their competent cellular contributions to flow cytometry analyses (CyPS) in Pitié-Salpêtrière, Paris. We also thank Olivier Silvie and Amélie Bigorgne for motivating discussions and Ana Ferreira in the MPS laboratory for sharing her expertise on murine models of ECM. We thank the Centre d'Elevage, de Production et d'Infection des Anophèles (CEPIA) of the Institut Pasteur, Paris, France.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.03129-14.
REFERENCES
- 1.WHO. 2014. World malaria report. WHO, Geneva, Switzerland. [Google Scholar]
- 2.Hunt NH, Golenser J, Chan-Ling T, Parekh S, Rae C, Potter S, Medana IM, Miu J, Ball HJ. 2006. Immunopathogenesis of cerebral malaria. Int J Parasitol 36:569–582. doi: 10.1016/j.ijpara.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 3.Mishra SK, Newton CR. 2009. Diagnosis and management of the neurological complications of falciparum malaria. Nat Rev Neurol 5:189–198. doi: 10.1038/nrneurol.2009.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdullah S, Adazu K, Masanja H, Diallo D, Hodgson A, Ilboudo-Sanogo E, Nhacolo A, Owusu-Agyei S, Thompson R, Smith T, Binka FN. 2007. Patterns of age-specific mortality in children in endemic areas of sub-Saharan Africa. Am J Trop Med Hyg 77:99–105. [PubMed] [Google Scholar]
- 5.Armah H, Wired EK, Dodoo AK, Adjei AA, Tettey Y, Gyasi R. 2005. Cytokines and adhesion molecules expression in the brain in human cerebral malaria. Int J Environ Res Public Health 2:123–131. doi: 10.3390/ijerph2005010123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hunt NH, Grau GE. 2003. Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol 24:491–499. doi: 10.1016/S1471-4906(03)00229-1. [DOI] [PubMed] [Google Scholar]
- 7.Brown H, Turner G, Rogerson S, Tembo M, Mwenechanya J, Molyneux M, Taylor T. 1999. Cytokine expression in the brain in human cerebral malaria. J Infect Dis 180:1742–1746. doi: 10.1086/315078. [DOI] [PubMed] [Google Scholar]
- 8.Grau GE, Piguet PF, Vassalli P, Lambert PH. 1989. Tumor-necrosis factor and other cytokines in cerebral malaria: experimental and clinical data. Immunol Rev 112:49–70. doi: 10.1111/j.1600-065X.1989.tb00552.x. [DOI] [PubMed] [Google Scholar]
- 9.Hunt NH, Stocker R. 2007. Heme moves to center stage in cerebral malaria. Nat Med 13:667–669. doi: 10.1038/nm0607-667. [DOI] [PubMed] [Google Scholar]
- 10.Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, Chora A, Rodrigues CD, Gregoire IP, Cunha-Rodrigues M, Portugal S, Soares MP, Mota MM. 2007. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med 13:703–710. doi: 10.1038/nm1586. [DOI] [PubMed] [Google Scholar]
- 11.Ferreira A, Balla J, Jeney V, Balla G, Soares MP. 2008. A central role for free heme in the pathogenesis of severe malaria: the missing link? J Mol Med 86:1097–1111. doi: 10.1007/s00109-008-0368-5. [DOI] [PubMed] [Google Scholar]
- 12.Lavstsen T, Turner L, Saguti F, Magistrado P, Rask TS, Jespersen JS, Wang CW, Berger SS, Baraka V, Marquard AM, Seguin-Orlando A, Willerslev E, Gilbert MT, Lusingu J, Theander TG. 2012. Plasmodium falciparum erythrocyte membrane protein 1 domain cassettes 8 and 13 are associated with severe malaria in children. Proc Natl Acad Sci U S A 109:E1791–E1800. doi: 10.1073/pnas.1120455109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Claessens A, Ghumra A, Gupta AP, Mok S, Bozdech Z, Rowe JA. 2011. Design of a variant surface antigen-supplemented microarray chip for whole transcriptome analysis of multiple Plasmodium falciparum cytoadherent strains, and identification of strain-transcendent rif and stevor genes. Malar J 10:180. doi: 10.1186/1475-2875-10-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spaccapelo R, Janse CJ, Caterbi S, Franke-Fayard B, Bonilla JA, Syphard LM, Di Cristina M, Dottorini T, Savarino A, Cassone A, Bistoni F, Waters AP, Dame JB, Crisanti A. 2010. Plasmepsin 4-deficient Plasmodium berghei are virulence attenuated and induce protective immunity against experimental malaria. Am J Pathol 176:205–217. doi: 10.2353/ajpath.2010.090504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spaccapelo R, Aime E, Caterbi S, Arcidiacono P, Capuccini B, Di Cristina M, Dottorini T, Rende M, Bistoni F, Crisanti A. 2011. Disruption of plasmepsin-4 and merozoites surface protein-7 genes in Plasmodium berghei induces combined virulence-attenuated phenotype. Sci Rep 1:39. doi: 10.1038/srep00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Favre N, Da Laperousaz C, Ryffel B, Weiss NA, Imhof BA, Rudin W, Lucas R, Piguet PF. 1999. Role of ICAM-1 (CD54) in the development of murine cerebral malaria. Microbes Infect 1:961–968. doi: 10.1016/S1286-4579(99)80513-9. [DOI] [PubMed] [Google Scholar]
- 17.Tripathi AK, Sullivan DJ, Stins MF. 2007. Plasmodium falciparum-infected erythrocytes decrease the integrity of human blood-brain barrier endothelial cell monolayers. J Infect Dis 195:942–950. doi: 10.1086/512083. [DOI] [PubMed] [Google Scholar]
- 18.Hunt NH, Grau GE, Engwerda C, Barnum SR, van der Heyde H, Hansen DS, Schofield L, Golenser J. 2010. Murine cerebral malaria: the whole story. Trends Parasitol 26:272–274. doi: 10.1016/j.pt.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 19.Gitau EN, Newton CR. 2005. Blood-brain barrier in falciparum malaria. Trop Med Int Health 10:285–292. doi: 10.1111/j.1365-3156.2004.01366.x. [DOI] [PubMed] [Google Scholar]
- 20.Medana IM, Turner GD. 2006. Human cerebral malaria and the blood-brain barrier. Int J Parasitol 36:555–568. doi: 10.1016/j.ijpara.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 21.Grau GE, Piguet PF, Engers HD, Louis JA, Vassalli P, Lambert PH. 1986. L3T4+ T lymphocytes play a major role in the pathogenesis of murine cerebral malaria. J Immunol 137:2348–2354. [PubMed] [Google Scholar]
- 22.Belnoue E, Kayibanda M, Vigario AM, Deschemin JC, van Rooijen N, Viguier M, Snounou G, Renia L. 2002. On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. J Immunol 169:6369–6375. doi: 10.4049/jimmunol.169.11.6369. [DOI] [PubMed] [Google Scholar]
- 23.Haque A, Best SE, Unosson K, Amante FH, de Labastida F, Anstey NM, Karupiah G, Smyth MJ, Heath WR, Engwerda CR. 2011. Granzyme B expression by CD8+ T cells is required for the development of experimental cerebral malaria. J Immunol 186:6148–6156. doi: 10.4049/jimmunol.1003955. [DOI] [PubMed] [Google Scholar]
- 24.Amante FH, Haque A, Stanley AC, Rivera FL, Randall LM, Wilson YA, Yeo G, Pieper C, Crabb BS, de Koning-Ward TF, Lundie RJ, Good MF, Pinzon-Charry A, Pearson MS, Duke MG, McManus DP, Loukas A, Hill GR, Engwerda CR. 2010. Immune-mediated mechanisms of parasite tissue sequestration during experimental cerebral malaria. J Immunol 185:3632–3642. doi: 10.4049/jimmunol.1000944. [DOI] [PubMed] [Google Scholar]
- 25.Amani V, Vigario AM, Belnoue E, Marussig M, Fonseca L, Mazier D, Renia L. 2000. Involvement of IFN-gamma receptor-medicated signaling in pathology and anti-malarial immunity induced by Plasmodium berghei infection. Eur J Immunol 30:1646–1655. doi:. [DOI] [PubMed] [Google Scholar]
- 26.Claser C, Malleret B, Gun SY, Wong AY, Chang ZW, Teo P, See PC, Howland SW, Ginhoux F, Renia L. 2011. CD8+ T cells and IFN-gamma mediate the time-dependent accumulation of infected red blood cells in deep organs during experimental cerebral malaria. PLoS One 6:e18720. doi: 10.1371/journal.pone.0018720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lou J, Lucas R, Grau GE. 2001. Pathogenesis of cerebral malaria: recent experimental data and possible applications for humans. Clin Microbiol Rev 14:810–820. doi: 10.1128/CMR.14.4.810-820.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krupka M, Seydel K, Feintuch CM, Yee K, Kim R, Lin CY, Calder RB, Petersen C, Taylor T, Daily J. 2012. Mild Plasmodium falciparum malaria following an episode of severe malaria is associated with induction of the interferon pathway in Malawian children. Infect Immun 80:1150–1155. doi: 10.1128/IAI.06008-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bianchi ME, Agresti A. 2005. HMG proteins: dynamic players in gene regulation and differentiation. Curr Opin Genet Dev 15:496–506. doi: 10.1016/j.gde.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 30.Stros M. 2010. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta 1799:101–113. doi: 10.1016/j.bbagrm.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 31.Wang H, Yang H, Czura CJ, Sama AE, Tracey KJ. 2001. HMGB1 as a late mediator of lethal systemic inflammation. Am J Respir Crit Care Med 164:1768–1773. doi: 10.1164/ajrccm.164.10.2106117. [DOI] [PubMed] [Google Scholar]
- 32.Scaffidi P, Misteli T, Bianchi ME. 2002. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 33.Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D, Morser J, Stern D, Schmidt AM. 1995. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of RAGE and amphoterin in the developing nervous system. J Biol Chem 270:25752–25761. [DOI] [PubMed] [Google Scholar]
- 34.Muller S, Scaffidi P, Degryse B, Bonaldi T, Ronfani L, Agresti A, Beltrame M, Bianchi ME. 2001. New EMBO members' review: the double life of HMGB1 chromatin protein: architectural factor and extracellular signal. EMBO J 20:4337–4340. doi: 10.1093/emboj/20.16.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erlandsson Harris H, Andersson U. 2004. The nuclear protein HMGB1 as a proinflammatory mediator. Eur J Immunol 34:1503–1512. doi: 10.1002/eji.200424916. [DOI] [PubMed] [Google Scholar]
- 36.Andersson U, Tracey KJ. 2003. HMGB1 in sepsis. Scand J Infect Dis 35:577–584. doi: 10.1080/00365540310016286. [DOI] [PubMed] [Google Scholar]
- 37.Sunden-Cullberg J, Norrby-Teglund A, Rouhiainen A, Rauvala H, Herman G, Tracey KJ, Lee ML, Andersson J, Tokics L, Treutiger CJ. 2005. Persistent elevation of high mobility group box-1 protein (HMGB1) in patients with severe sepsis and septic shock. Crit Care Med 33:564–573. doi: 10.1097/01.CCM.0000155991.88802.4D. [DOI] [PubMed] [Google Scholar]
- 38.Ombrellino M, Wang H, Ajemian MS, Talhouk A, Scher LA, Friedman SG, Tracey KJ. 1999. Increased serum concentrations of high-mobility-group protein 1 in haemorrhagic shock. Lancet 354:1446–1447. doi: 10.1016/S0140-6736(99)02658-6. [DOI] [PubMed] [Google Scholar]
- 39.Yang R, Harada T, Mollen KP, Prince JM, Levy RM, Englert JA, Gallowitsch-Puerta M, Yang L, Yang H, Tracey KJ, Harbrecht BG, Billiar TR, Fink MP. 2006. Anti-HMGB1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol Med 12:105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. 2000. HMG-1 as a mediator of acute lung inflammation. J Immunol 165:2950–2954. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]
- 41.Angus DC, Yang L, Kong L, Kellum JA, Delude RL, Tracey KJ, Weissfeld L. 2007. Circulating high-mobility group box 1 (HMGB1) concentrations are elevated in both uncomplicated pneumonia and pneumonia with severe sepsis. Crit Care Med 35:1061–1067. doi: 10.1097/01.CCM.0000259534.68873.2A. [DOI] [PubMed] [Google Scholar]
- 42.Li J, Kokkola R, Tabibzadeh S, Yang R, Ochani M, Qiang X, Harris HE, Czura CJ, Wang H, Ulloa L, Wang H, Warren HS, Moldawer LL, Fink MP, Andersson U, Tracey KJ, Yang H. 2003. Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol Med 9:37–45. [PMC free article] [PubMed] [Google Scholar]
- 43.Sappington PL, Yang R, Yang H, Tracey KJ, Delude RL, Fink MP. 2002. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology 123:790–802. doi: 10.1053/gast.2002.35391. [DOI] [PubMed] [Google Scholar]
- 44.Andersson U, Erlandsson-Harris H, Yang H, Tracey KJ. 2002. HMGB1 as a DNA-binding cytokine. J Leukoc Biol 72:1084–1091. [PubMed] [Google Scholar]
- 45.Alleva LM, Yang H, Tracey KJ, Clark IA. 2005. High mobility group box 1 (HMGB1) protein: possible amplification signal in the pathogenesis of falciparum malaria. Trans R Soc Trop Med Hyg 99:171–174. doi: 10.1016/j.trstmh.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 46.Higgins SJ, Xing K, Kim H, Kain DC, Wang F, Dhabangi A, Musoke C, Cserti-Gazdewich CM, Tracey KJ, Kain KC, Liles WC. 2013. Systemic release of high mobility group box 1 (HMGB1) protein is associated with severe and fatal Plasmodium falciparum malaria. Malar J 12:105. doi: 10.1186/1475-2875-12-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mackintosh CL, Beeson JG, Marsh K. 2004. Clinical features and pathogenesis of severe malaria. Trends Parasitol 20:597–603. doi: 10.1016/j.pt.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 48.Bischoff E, Vaquero C. 2010. In silico and biological survey of transcription-associated proteins implicated in the transcriptional machinery during the erythrocytic development of Plasmodium falciparum. BMC Genomics 11:34. doi: 10.1186/1471-2164-11-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Briquet S, Boschet C, Gissot M, Tissandie E, Sevilla E, Franetich JF, Thiery I, Hamid Z, Bourgouin C, Vaquero C. 2006. High-mobility-group box nuclear factors of Plasmodium falciparum. Eukaryot Cell 5:672–682. doi: 10.1128/EC.5.4.672-682.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar K, Singal A, Rizvi MM, Chauhan VS. 2008. High mobility group box (HMGB) proteins of Plasmodium falciparum: DNA binding proteins with pro-inflammatory activity. Parasitol Int 57:150–157. doi: 10.1016/j.parint.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 51.Reichelt P, Schwarz C, Donzeau M. 2006. Single step protocol to purify recombinant proteins with low endotoxin contents. Protein Expr Purif 46:483–488. doi: 10.1016/j.pep.2005.09.027. [DOI] [PubMed] [Google Scholar]
- 52.Menard R, Janse C. 1997. Gene targeting in malaria parasites. Methods 13:148–157. doi: 10.1006/meth.1997.0507. [DOI] [PubMed] [Google Scholar]
- 53.Janse CJ, Ramesar J, Waters AP. 2006. High-efficiency transfection and drug selection of genetically transformed blood stages of the rodent malaria parasite Plasmodium berghei. Nat Protoc 1:346–356. doi: 10.1038/nprot.2006.53. [DOI] [PubMed] [Google Scholar]
- 54.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. 1999. HMG-1 as a late mediator of endotoxin lethality in mice. Science 285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 55.Paxinos G, Watson C. 1998. The rat brain in stereotaxic coordinates, 4th ed Academic Press, San Diego, CA. [Google Scholar]
- 56.Barnay-Verdier S, Gaillard C, Messmer M, Borde C, Gibot S, Marechal V. 2011. PCA-ELISA: a sensitive method to quantify free and masked forms of HMGB1. Cytokine 55:4–7. doi: 10.1016/j.cyto.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 57.Senaldi G, Vesin C, Chang R, Grau GE, Piguet PF. 1994. Role of polymorphonuclear neutrophil leukocytes and their integrin CD11a (LFA-1) in the pathogenesis of severe murine malaria. Infect Immun 62:1144–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Souza JB, Hafalla JC, Riley EM, Couper KN. 2010. Cerebral malaria: why experimental murine models are required to understand the pathogenesis of disease. Parasitology 137:755–772. doi: 10.1017/S0031182009991715. [DOI] [PubMed] [Google Scholar]
- 59.Ting LM, Gissot M, Coppi A, Sinnis P, Kim K. 2008. Attenuated Plasmodium yoelii lacking purine nucleoside phosphorylase confer protective immunity. Nat Med 14:954–958. doi: 10.1038/nm.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aly AS, Downie MJ, Mamoun CB, Kappe SH. 2010. Subpatent infection with nucleoside transporter 1-deficient Plasmodium blood stage parasites confers sterile protection against lethal malaria in mice. Cell Microbiol 12:930–938. doi: 10.1111/j.1462-5822.2010.01441.x. [DOI] [PubMed] [Google Scholar]
- 61.Gissot M, Ting LM, Daly TM, Bergman LW, Sinnis P, Kim K. 2008. High mobility group protein HMGB2 is a critical regulator of plasmodium oocyst development. J Biol Chem 283:17030–17038. doi: 10.1074/jbc.M801637200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Calogero S, Grassi F, Aguzzi A, Voigtlander T, Ferrier P, Ferrari S, Bianchi ME. 1999. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat Genet 22:276–280. doi: 10.1038/10338. [DOI] [PubMed] [Google Scholar]
- 63.Ronfani L, Ferraguti M, Croci L, Ovitt CE, Scholer HR, Consalez GG, Bianchi ME. 2001. Reduced fertility and spermatogenesis defects in mice lacking chromosomal protein Hmgb2. Development 128:1265–1273. [DOI] [PubMed] [Google Scholar]
- 64.Delahaye NF, Coltel N, Puthier D, Flori L, Houlgatte R, Iraqi FA, Nguyen C, Grau GE, Rihet P. 2006. Gene-expression profiling discriminates between cerebral malaria (CM)-susceptible mice and CM-resistant mice. J Infect Dis 193:312–321. doi: 10.1086/498579. [DOI] [PubMed] [Google Scholar]
- 65.Kaul DK, Nagel RL, Llena JF, Shear HL. 1994. Cerebral malaria in mice: demonstration of cytoadherence of infected red blood cells and microrheologic correlates. Am J Trop Med Hyg 50:512–521. [DOI] [PubMed] [Google Scholar]
- 66.Kaul DK, Liu XD, Nagel RL, Shear HL. 1998. Microvascular hemodynamics and in vivo evidence for the role of intercellular adhesion molecule-1 in the sequestration of infected red blood cells in a mouse model of lethal malaria. Am J Trop Med Hyg 58:240–247. [DOI] [PubMed] [Google Scholar]
- 67.Kossodo S, Monso C, Juillard P, Velu T, Goldman M, Grau GE. 1997. Interleukin-10 modulates susceptibility in experimental cerebral malaria. Immunology 91:536–540. doi: 10.1046/j.1365-2567.1997.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baptista JL, Vanham G, Wery M, Van Marck E. 1997. Cytokine levels during mild and cerebral falciparum malaria in children living in a mesoendemic area. Trop Med Int Health 2:673–679. doi: 10.1046/j.1365-3156.1997.d01-355.x. [DOI] [PubMed] [Google Scholar]
- 69.Naglova H, Bucova M. 2012. HMGB1 and its physiological and pathological roles. Bratisl Lek Listy 113:163–171. [DOI] [PubMed] [Google Scholar]
- 70.Schneider DS, Ayres JS. 2008. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol 8:889–895. doi: 10.1038/nri2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Raberg L, Graham AL, Read AF. 2009. Decomposing health: tolerance and resistance to parasites in animals. Philos Trans R Soc Lond B Biol Sci 364:37–49. doi: 10.1098/rstb.2008.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seixas E, Gozzelino R, Chora A, Ferreira A, Silva G, Larsen R, Rebelo S, Penido C, Smith NR, Coutinho A, Soares MP. 2009. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc Natl Acad Sci U S A 106:15837–15842. doi: 10.1073/pnas.0903419106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.