

Abstract
Pathogenic Leptospira species cause a prevalent yet neglected zoonotic disease with mild to life-threatening complications in a variety of susceptible animals and humans. Diagnosis of leptospirosis, which primarily relies on antiquated serotyping methods, is particularly challenging due to presentation of non-specific symptoms shared by other febrile illnesses, often leading to misdiagnosis. Initiation of antimicrobial therapy during early infection to prevent more serious complications of disseminated infection is often not performed because of a lack of efficient diagnostic tests. Here we report that specific regions of leptospiral 16S ribosomal RNA molecules constitute a novel and efficient diagnostic target for PCR-based detection of pathogenic Leptospira serovars. Our diagnostic test using spiked human blood was at least 100-fold more sensitive than corresponding leptospiral DNA-based quantitative PCR assays, targeting the same 16S nucleotide sequence in the RNA and DNA molecules. The sensitivity and specificity of our RNA assay against laboratory-confirmed human leptospirosis clinical samples were 64% and 100%, respectively, which was superior then an established parallel DNA detection assay. Remarkably, we discovered that 16S transcripts remain appreciably stable ex vivo, including untreated and stored human blood samples, further highlighting their use for clinical detection of L. interrogans. Together, these studies underscore a novel utility of RNA targets, specifically 16S rRNA, for development of PCR-based modalities for diagnosis of human leptospirosis, and also may serve as paradigm for detection of additional bacterial pathogens for which early diagnosis is warranted.
Introduction
Leptospirosis is a prominent zoonotic disease caused by a diverse group of pathogenic leptospires that includes at least nine genospecies and over 200 serovars [1–7]. There are an estimated more than 800,000 cases and 50,000 deaths annually due to leptospirosis [8]. The greatest disease burden occurs in subsistence farmers [9, 10] and urban slum dwellers [2, 11–13], especially in resource-poor settings. In endemic regions, epidemics have frequently been reported following heavy rainfalls [2, 13]. Even in industrialized nations including the United States, outbreaks have been reported following sporting events [14–16], within military personnel [17–19], and in tourists [20, 21]. Additionally, there are increasing cases of the disease involving inner-city populations [7, 22], climate changes [23, 24], and expansion of urban slum populations [13, 25]. L. interrogans are transmitted from contaminated water, soil, or urine to hosts during contact with abraded skin or mucous membrane. Unlike other pathogenic spirochetes, which cause borreliosis or syphilis in humans and are unable to persist outside of a host body, leptospires can persist in aqueous environments for extended periods of time [26, 27]. The pathogen can quickly upregulate genes associated with host adaptation and virulence and can establish serious systemic infection via hematogenous dissemination to multiple internal organs, particularly the kidneys and liver [5, 28, 29]. While wild rodents serve as major natural reservoir hosts, humans and many other domesticated animals are accidental hosts in the transmission cycle of leptospirosis [1, 3, 28].
Pathogenic Leptospira spp cause a spectrum of clinical symptoms ranging from mild febrile disease to severe manifestations such as Weil’s disease and pulmonary hemorrhage syndrome, with case fatalities of >10% and >50%, respectively [1, 3, 5, 30]. Although whole cell and recombinant vaccines are shown to interfere with Leptospira infection [31–33], none of these vaccines offer complete protection. Moreover, they fail to block chronic renal colonization or urinary shedding, can elicit moderate side effects and are mostly effective against local host-adapted serovars [32, 34, 35]. Thus, given an absence of effective vaccines, prevention of disease progression is primarily reliant on timely diagnosis and antibiotic treatment. Early diagnosis of leptospirosis generally leads to effective antibiotic treatment, thereby preventing the more severe form of disseminated disease; however, there is a lack of rapid diagnostics [8]. Due to the non-specific clinical manifestations of leptospirosis, failure to diagnose the infection, or misdiagnosis, has become a significant problem in many developing countries where dengue, malaria, typhoid and other causes of acute fever are endemic [5, 36]. Diagnosis of leptospirosis still relies on classical laboratory tests including immunoassays against spirochetes or recombinant proteins, direct cultivation of bacteria grown from body fluids, or a microscopic agglutination test (MAT) using paired serum samples and Leptospira cultures [8, 37]. Although some of the immunoassays are highly sensitive, they suffer from inherent delays and variability of host immune responses as well as sequence divergence in target antigens, potentially limiting their use for early diagnosis of leptospirosis. Even the gold standard methods of direct culture and the MAT, require either weeks to grow spirochetes from body fluids or highly trained laboratory personnel and paired sera. Therefore, there is a critical need for rapid and effective diagnostics, especially for detection of early infection.
Leptospires disseminate hematogenously and spirochetemia is detectable for many days following initial exposure [3, 8, 38]. Although PCR-based diagnostic methods have been developed that can detect leptospiral DNA [8, 39–50], overall sensitivity of these assays is poor, and in general is less than 60% [8], although in some cases, higher sensitivities are reported [44, 45]. Unlike DNA targets, which usually exist as a single copy per cell, each bacterium contains hundreds to thousands of specific RNA molecules. We therefore hypothesized that an assay based on the PCR amplification of cDNA molecules representing highly and consistently transcribed Leptospira genes like16S rRNA [6, 51], which are also mostly conserved in pathogenic Leptospira [6, 52], could improve the sensitivity of Leptospira detection. In addition, detection of Leptospira transcripts in the blood would facilitate prompt and appropriate antibiotic treatment. In the current study, we report a rapid, sensitive, and specific RNA-based PCR diagnostic test for early human leptospirosis. These results could serve as a paradigm for development of novel RNA-based diagnostics of additional bacterial infections in humans, such as Lyme disease, where early diagnostics remains challenging.
Material and Methods
Ethics statement
Written informed consent was obtained from all participants prior to blood collection. The study protocol was approved prior to study initiation by the Yale Institutional Review Board (HIC#1006006956), the Ethics Committees at the Oswaldo Cruz Foundation (505.490; 16/2013) and Hospital Couto Maia (175), and the Brazilian Ministry of Health National Ethics Committee in Research (15925). Animals were treated in compliance with the Guide for the Care and Use of Laboratory Animals. All experiments involving human blood, infectious agents, and animals were performed according to the guidelines of the Institutional Biosafety Committee of the University of Maryland, and the Institutional Animal Care and Use Committee of the University of Maryland under the protocol number R-13-71.
Bacterial strains
Leptospira strains and serovars used in the study are indicated in S1 Table. Unless stated otherwise, Leptospira interrogans Fiocruz L1-130, a clinical isolate, [53] was used in most parts of the study. In some experiments, additional Leptospira serovars were also used, including isolates from 17 pathogenic and five non-pathogenic strains [54]. Spirochetes were grown in liquid Elinghausen-McCullough-Johnson-Harris (EMJH) medium [55, 56] at 29°C on a rotating platform at 100 rpm. Additional bacterial strains, such as an Escherichia coli K-12 derivative, Group A Streptococcus D471 cells [57], and Borrelia burgdorferi clone B31-A3 [58], were also used in certain experiments and grown by using the standard media and protocols.
Samples
Blood samples from uninfected Golden Syrian Hamsters (4–6 weeks old, purchased from Charles River Laboratories), a species commonly used as an animal model of Leptospirosis [29], were collected into BD Vacutainer whole blood collection tubes containing EDTA (BD Diagnostics). For human samples, twenty five patients were enrolled during active surveillance for leptospirosis at a state-run hospital, Hospital Couto Maia, in Salvador, Brazil, from June 2013 to September 2013, and blood samples were collected during early hospitalization [2]. All patients were examined for leptospirosis using previously described methods (hemoculture and MAT) [30] as well as DNA qPCR. Confirmed patients were positive for at least one of the above three tests. We determined that 22 out of 25 subjects had laboratory-confirmed leptospirosis as defined by: 1) four-fold increase in MAT titer or seroconversion (0 to ≥1:200) between paired sera [59], 2) reciprocal MAT titer of greater than 1:800 in one or more samples [59], 3) positive hemoculture, or 4) positive blood DNA PCR results. Three patients had probable leptospirosis based on the presence of a single MAT titer value of 1:100–1:400.
For samples used in the current study, about 1–2 mL of venous blood was collected directly into BD whole blood EDTA tubes. Within 5 hours of collection, 250 μL of patient blood was aliquoted from the EDTA tubes into 750 μL of TRIzol LS, thoroughly homogenized, and immediately frozen at -70°C. All patient samples were bar-coded, monitored during transport for temperature, and all cold chain data including sample receipt, processing time, and freezing time were recorded. Whole blood from 24 healthy individuals residing in non-endemic regions in the U.S. and Brazil were also collected or purchased (SeraCare Life Sciences) and processed equivalently to patient samples.
Extraction of nucleic acids and cDNA synthesis
Total RNA samples were extracted from samples stored in TRIzol (for bacterial pellets harvested from cultures grown at mid to late log phases), or TRIzol-LS (for blood samples) according to manufacturer’s (Life Technologies) instructions [60, 61]. After phase separation, RNA samples were either precipitated with isopropanol, dissolved in 20μL of RNase-free water and subjected to optional DNase1 treatment (NEB laboratories), or further purified using an RNeasy mini kit (Qiagen). For cDNA synthesis, 0.5 μg of RNA samples was reversed transcribed using VILO superscript master mix using random primers (Life technologies) according to manufacturer’s protocols. For extraction of DNA, spiked blood samples were processed using DNeasy mini kit (Qiagen) according to manufacturer’s instructions and eluted into 100 μL nuclease-free sterile water.
Primer design
The primers used for qPCR reaction were designed using NCBI Primer-BLAST primer design program based on the available L. interrogans genomic sequences. To identify primers of greatest sensitivity and specificity, we aligned the 16S sequences of 37 Leptospira serovars, including all 20 known pathogenic or non-pathogenic leptospiral species, and several other non-target bacterial species using MegAlign program (DNASTAR) (S1 Fig). We have designed specific sets of 16S forward and reverse primers expected to amplify a specific region of 16S gene that retains 1) absolute homology between all known highly pathogenic Leptospira species and serovars, 2) few base pair mismatches for intermediate pathogenic leptospires and 3) greater number of mismatches to non-pathogenic Leptospira species. The specificity of each newly designed primer was initially searched against all reference mRNA sequences using NCBI BLASTn as well as Primer-BLAST programs to rule out possible cross-reactivity with other bacteria species as well as non-targeted species including human, mice, rats, and hamsters. These primer sequences display 25% or more divergence from corresponding human or rodent genes, including at least 4–5 base pair mismatches predominantly towards the 3’ ends of the primer sequence inhibiting primer annealing to unintended targets. Similarly, primers against additional gene targets, FlaB, LipL31, LipL32, and LipL41 were also designed. All PCR primer pairs had a similar annealing temperature (60°C) and spanned nearly 200 base pairs of each of the target genes. Prior to their use in RNA measurement assays, each primer pair was tested for efficiency and non-specific amplification by melt-curve analysis using L. interrogans genomic DNA as a template.
Polymerase chain reaction
The oligonucleotide sequences for each of the primers used in specific PCR reactions are indicated in S2 Table. The relative levels of cDNA templates in each sample were assessed by quantitative PCR (qPCR) as detailed [60, 61], and whenever necessary, DNA contamination in each sample was measured using an equal volume of purified RNA as a template. All qPCR reactions were performed using the CFX96 real time PCR detection system (Bio-Rad) with the following thermal cycle conditions: 95°C for 10 min, 45 cycles of [95°C for 15 s, 60°C for 60 s], followed by a melt curve from 65°C to 95°C performed at an increment of 0.5°C per cycle. All qPCR plates included no template control wells to test for non-specific amplification or reagent contamination, and results were further tested for specificity by melt curve analysis. Detection of human or hamster β-actin or GAPDH transcripts by qPCR confirmed the integrity of cDNA samples. For detection of leptospirosis in humans, an optimized DNA qPCR analysis was also performed using nuclease (TaqMan) assay and primers that amplified a sequence of lipL32, a pathogenic Leptospira-specific gene, as detailed earlier [62, 63].
Validation of primers
L. interrogans culture was harvested at 2.9x108 leptospires per ml by centrifugation at 5000 g at room temperature (~2.9x109 total cells) and subjected to RNA isolation as detailed above. For relative assessment of each primer set, 2.5 μg RNA samples were reverse transcribed into cDNA, serially diluted to tenfold, which were used in the qPCR reactions. Standard curves and amplification efficiency of the reactions were calculated by the CFX96 instrument software, as instructed by the manufacturer. cDNA samples were also isolated from a number of pathogenic, intermediate pathogenic or non-pathogenic species and serovars. For spiking experiments, 250 μL of untreated whole human blood samples were spiked in triplicate with L. interrogans derived from at least three independent cultures at various concentrations ranging from 106–100 cells/mL. Samples were used for DNA or RNA extraction using DNeasy mini kit or TRIzol extraction procedure, followed by cleanup using RNeasy mini kit, respectively, and the RNA samples were further processed for cDNA synthesis. As controls for assessment of specificity, cDNA samples were also isolated from additional bacterial culture grown at late log phases including B. burgdorferi, E. coli, and Group A Streptococcus strains as well as from human blood and hamster liver.
RNA stability studies
Triplicate samples of 250 μl whole human blood were spiked with viable L. interrogans (100 cells/ml) derived from at least three independent cultures. Aliquots were homogenized with 750 μLTRIzol LS and stored at room temperature for 0, 4, 8, 24, 72, or 120 h before freezing at -80°C until analysis. In parallel experiments, triplicate aliquots of 250 μL human blood containing 100 leptospires per ml were stored at room temperature, 4°C, -20°C, or -80°C in the absence of any stabilization reagent, and processed together with other conditions. Samples were collected directly into 750 μL TRIzol LS after incubating at the aforementioned temperatures for 0 h, 8 h, 24 h, 7 d, and 14 d, and stored at -80°C until completion of all timepoints. RNA was extracted using the TRIzol procedure, further treated with DNase I, and finally reverse transcribed to cDNA and analyzed by qPCR using Leptospira-specific 16S primers.
Statistical analysis
Results are expressed as the mean ± standard error of the mean (SEM). The significance of the difference between the mean values of the groups was evaluated by unpaired Student t test and ANOVA.
Results
Development of an RNA-based quantitative PCR assay for detection of pathogenic leptospires
Development of a rapid and sensitive diagnostics for human leptospirosis, especially for detection of early active infection, is highly warranted. Since pathogenic leptospires are known to disseminate via blood where they are detectable for several days after infection [8], and since each bacterium may contain hundreds of copies of certain abundant transcripts, we sought to explore whether an RNA-based PCR assay would allow sensitive and specific detection of leptospires in human blood. We adopted a SYBR Green-based qRT-PCR assay, which is a highly efficient and widely used platform amongst available real-time PCR technologies [64], yet relatively simple and cost-effective. For identification of an RNA target that yields most efficient detection, we initially examined a set of characterized, abundant rRNA and mRNA gene targets: 16S rRNA, FlaB, LipL31, LipL32, and LipL41. We selected these genes not only due to their constitutive and abundant expression but also for their sequence conservation in pathogenic and intermediately pathogenic Leptospira spp, and sequence divergence or absence in non-target species. Although the above-mentioned mRNA genes are unique to pathogenic Leptospira species [6, 52], specific regions of their rRNA genes display appreciable species-specific conservation [65]. We therefore used NCBI Primer-Blast software to identify unique regions in 16S gene and created forward and reverse primers 100% identical to pathogenic L. interrogans sequences but containing several nucleotide mismatches to non-target bacterial species including non-pathogenic Leptospira (S1 Fig). These primers also lack significant similarity to mammalian species. All gene-specific primers had a similar annealing temperature and comparable amplicon sizes. The RNA samples from a highly pathogenic species, L. interrogans serovar Copenhageni strain Fiocruz L1-130 were converted into cDNA and used in a SYBR Green-based qPCR assay in the absence or presence of 10-fold excess of a control host (hamster) cDNA. While sensitivity of the assay (or the relative abundance of the transcripts) was calculated using the 2−ΔΔCT method [66], specificity of the target gene amplification was assessed using melt-curve analysis. We found that the 16S rRNA primers offered the most efficient analytical sensitivity, as evidenced by the lowest Ct values, and were nearly 1000-fold more abundant than the next efficient mRNA target, LipL32 (Fig 1A). The 16S-1 primers (Fig 1B), or other tested primers, showed PCR efficiencies between 91–99% and without detectable cross-reactivity with spiked control rodent cDNAs. As the 16S rRNA primers offered the highest sensitivity in our assay, we only used these primers in subsequent experiments (16S-1, Fig 1).
Fig 1. L. interrogans 16S rRNA primers offered highest analytical sensitivity.

A) Total RNA samples isolated from cultured spirochetes were converted into cDNA and amplification cycles (cycle threshold or Ct value) of various L. interrogans target genes are assessed in qRT-PCR assays in the presence (gray bars) or absence (black bars) of hamster cDNA. Data represent results from three independent experiments. B) 16S rRNA primers display a high PCR efficiency. L. interrogans cDNA in RNase-free water (320 ng/μL) was serially diluted to tenfold (10−1 to 10−9) and subjected to qRT-PCR assays using 16S-1 primers. Amplification cycles (left panel) were used to calculate standard curve (middle panel), which indicated detection to 10−9 dilutions with an amplification efficiency of 91.2%. A melt curve analysis (right panel) showed a melting temperature of 82°C without any non-specific amplification.
Analytical sensitivities of 16S RNA-based qPCR assays are superior to corresponding DNA-based assay in Leptospira-spiked human samples
We next assessed whether sensitivity of our 16S RNA-based assay is superior to corresponding DNA-based ones and also tested its specificity with experimentally spiked human blood samples. To accomplish this, we spiked serially diluted L. interrogans cells into 250 μl aliquots of human blood and the RNA and DNA samples were isolated using the commercial kits as detailed in the Materials and Methods. We then performed subsequent qRT-PCR and qPCR assays using corresponding templates and the same 16S primer set. Using this methodology, we found that RNA-based assays were at least 100-fold more sensitive than a DNA-based approach (Fig 2), suggesting that the difference in assay performance is not due to the target sequence rather than differences between RNA and DNA detection.
Fig 2. RNA-based detection is more sensitive than DNA.
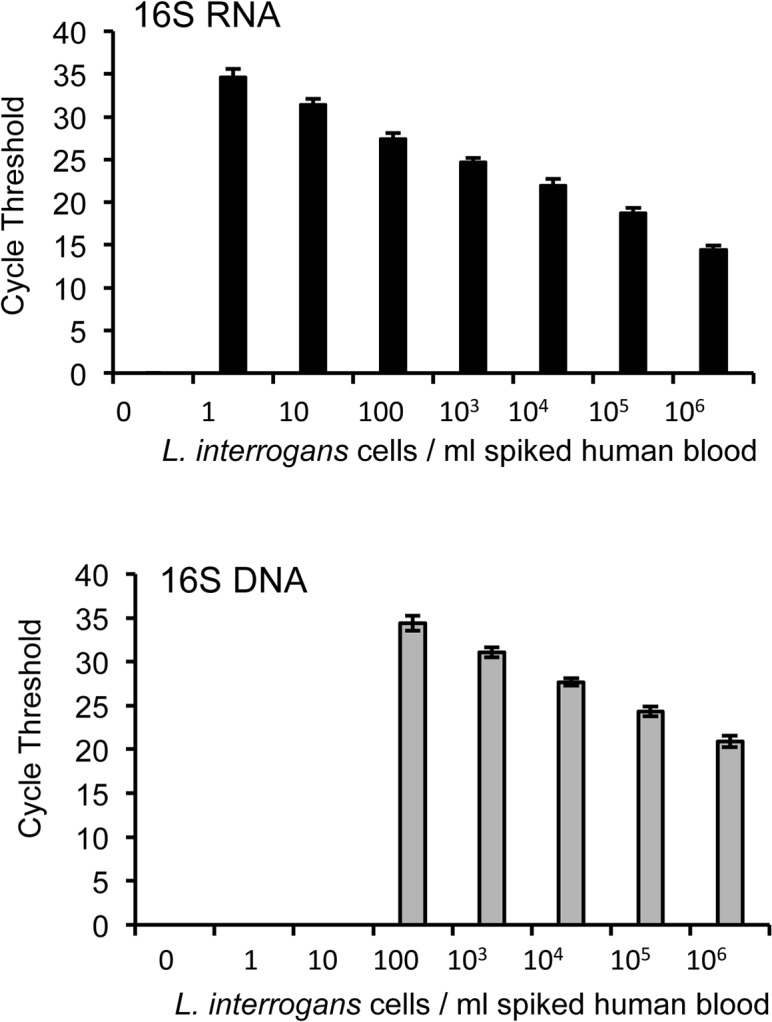
Aliquots of L. interrogans bacteria were serially diluted from 106 to 1 bacterium per milliliter of human blood and used for either RNA-based qRT-PCR (upper panel) or DNA-based qPCR (lower panel). Data represent results from three independent experiments.
Differential detection of multiple Leptospira and non-target bacterial species
We next determined the sensitivity of our 16S qRT-PCR assay in the detection of multiple pathogenic leptospiral serovars, and the specificity of the assay using non-pathogenic leptospiral species and non-target bacterial species. For this purpose, we prepared RNA samples from 17 highly or intermediately pathogenic leptospiral species and serovars as well as from five non-pathogenic species, B. burgdorferi, group A Streptococcus, E. coli or hamster and human tissues. We found that while the assay was able to detect all pathogenic species with highest sensitivity, detection of non-pathogenic leptospiral species was at least 15 Ct value (or 10,000 fold) higher (Fig 3). These results indicate a difference of several thousand folds in the concentration of target templates. In addition, weak signals at higher Ct values (39–40) were recorded for Streptococcus and Borrelia, however, this amplification was non-specific, as confirmed by the melt curve analysis. Thus, overall, we detected none of the non-target species suggesting 100% specificity of the assay (Fig 3).
Fig 3. High sensitivity and specificity of 16S rRNA qPCR assay for pathogenic and nonpathogenic leptospiral species and serovars.

Total RNA was extracted from 17 high or intermediate pathogenic, and five non-pathogenic leptospiral species, Borrelia burgdorferi, group A Streptococcus, and Escherichia coli, as well as from uninfected human blood or hamster liver, as described in the materials and methods and converted into cDNA. Equal amount (10 ng) of cDNA templates from each bacterial species were subjected to qRT-PCR assays using 16S-1 primers and amplification cycles (Ct values) were measured. Note that the sensitivity of detection is the best for all tested highly pathogenic species or serovars followed by intermediate species while non-pathogenic strains display the lowest sensitivity (an average of 15 Ct values or 10,000 fold less detectability). All tested non-target bacterial species or mammalian samples remained undetectable. Data represent results from three independent experiments.
L. interrogans 16S transcripts are remarkably stable in human blood
An ideal RNA-based diagnostic test for leptospirosis would detect stable targets, allowing for varied blood storage times and temperatures, and not require the use of toxic RNA stabilizing reagents such as TRIzol. Therefore, we compared the limit of detection of 16S transcripts in spiked human blood samples stored at room temperature in TRIzol to that of untreated spiked blood samples. We also tested how storage temperature of blood samples influenced the window of 16S detection by our assay. To accomplish our objectives, we spiked human blood with L. interrogans cells at 100 bacteria per milliliter of blood and stored aliquots at room temperature, 4°C, -20°C, or -80°C) for 1, 7 and 14 days either with or without the addition of TRIzol. A sample immediately stored in TRIzol and frozen served as the “0 hour” or baseline control for 16S RNA stability experiments. We measured the transcript levels by qRT-PCR analyses, and observed no appreciable RNA degradation in the samples stored in TRIzol at room temperature (Fig 4A) or at colder temperatures. Of note, while we recorded a significant loss of 16S RNA in samples stored at room temperature within a day, ~ 5% of the transcripts remained detectable until 7 days (Fig 4B). In contrast, we were able to detect >20% of 16S RNA transcripts from samples stored at 4°C, and >50% at -20°C and -80°C even after 14 days (Fig 4B).
Fig 4. Stability of Leptospira 16S transcripts in human blood.

A) Transcript stability in the blood treated with an RNA stabilization agent. Aliquots of human blood were spiked with leptospires (100 cells/ml), mixed with an RNase stabilization agent (TRIzol), and each aliquot was stored at room temperature for various times (0–120 hours). Following storage, levels of 16S rRNA transcripts were measured using qRT-PCR assays. Data represent results from three independent experiments. B) Transcript stability in the blood stored at various temperatures in the absence of any RNA stabilization agent. Spiked samples were prepared as described above and stored either at room temperature or at various cold temperatures (4°C, -20°C, and -80°C) up to 14 days, and transcript levels were monitored by qRT-PCR analyses. Transcript levels of “0 hour” were considered as 100%, which served as baseline controls, which displayed significant differences in transcript levels in groups marked by an asterisk (ANOVA, p<0.05). Data represent results from three independent experiments.
Assessment of clinical cases of human leptospirosis
Finally, we tested the sensitivity and specificity of our assay for the detection of Leptospira in blood samples obtained from patients, with suspected leptospirosis, as detailed in the Material and Methods section. We also compared the efficacy of our RNA assay using an established DNA-based qPCR assay. Total RNA and DNA samples were isolated from these human samples, and detection of Leptospira was examined by real-time PCR analyses, as detailed in the Materials and Methods section. Results indicated that, for 22 confirmed leptospirosis cases, Leptospira RNA was detected in 14 samples, yielding a sensitivity of 64% (Table 1). In parallel, an established qPCR assay for detection of leptospiral DNA form the corresponding human samples, as described in the Materials and Methods, identified 7 positives, thereby reflecting a significantly lower sensitivity (32%, p = 0.035) for the DNA detection assay compared to the RNA detection assay. Of note, 2 of the 3 probable clinically suspected cases were found to have detectable leptospiral RNA, whereas none of the probable cases had detectable DNA. As expected, all blood samples obtained from the 24 control individuals failed to yield a positive signal, suggesting a 100% specificity of our RNA-based assay.
Table 1. Detection of leptospiral RNA by quantitative PCR in blood samples collected from humans with suspected leptospirosis.
| Assay results | Confirmed leptospirosis (N = 22) | Probable leptospirosis (N = 3) | Healthy Subjects (N = 24) |
|---|---|---|---|
| Number (%) | |||
| RNA and DNA PCR+ | 5 (23) | 0 (0) | 0 (0) |
| RNA PCR+ alone | 9 (41) | 2 (67) | 0 (0) |
| DNA PCR+ alone | 2 (9) | 0 (0) | 0 (0) |
| Total RNA PCR+ | 14 (64) | 2 (67) | 0 (0) |
| Total DNA PCR+ | 7 (32) | 0 (0) | 0 (0) |
Discussion
As leptospirosis affects nearly a million people annually [8] and an efficient human vaccine is unavailable, development of a better diagnosis test is critical for effectively treating patients with leptospirosis. However, efficient detection of the infection is difficult to accomplish, primarily due to the fact that pathogenic leptospires not only share features of both gram-positive and gram-negative bacteria including other related spirochetes, clinical manifestation of the infection also share features of many prevalent undifferentiated febrile illnesses, such as influenza, dengue and malaria [37, 67–69]. Here, we report a use of leptospiral RNA as a diagnostic target for development of a rapid, sensitive and specific quantitative PCR assay for detection of human leptospirosis. While RNA-based diagnostic methods for detection of bacterial pathogens are uncommon, except for a few commercially-available assays for detection of certain sexually transmitted bacterial infections [70–72], to the best of our knowledge, this is the first example of an RNA-based diagnosis of human leptospirosis. Our discovery of the relative stability of 16S transcripts in untreated stored human blood samples indicate that the RNA-based assay could be widely applied for the diagnosis of leptospirosis. Additionally, our results suggest similar approaches can be employed to develop novel diagnostic tests for other bacterial diseases.
Several serological immunoassays are available to date for detection of leptospirosis [8, 37], however, diagnosis of human or animal infection is still inadequate, which is based on classical microbiological methods. The gold standard method for such diagnosis is culture or microscopic agglutination test (MAT), which are extremely slow procedures that also suffer from sensitivity, especially for diagnosis of early infection. Newer diagnostic methods that allow diagnosis of early infection at a relatively fast pace and facilitate initiation of prompt antibiotic treatment could alleviate more severe complications of disseminated infection. As spirochetemia is likely to be associated with early infection, which occurs for at least two weeks following initial infection, we hypothesized that the detection of nucleic acids with sequence specificity to pathogenic Leptospira could surrogate an active infection. Such detection in turn would yield high diagnostic efficiency if the transcripts or corresponding RNA fragments are abundant, and remain stable, in stored blood. Unlike conventional genomic DNA molecules that mostly represent a single copy per bacterial cell, or mRNA molecules, which constitute the minor population of total cellular RNA, ribosomal RNA molecules are likely to be abundant and thus offer more promising diagnostic target. In particular 16S rRNA, due to its high expression, and maintenance of species-specific sequences [65], has been widely used for taxonomic studies or as a diagnostic target to identify a particular bacterial species. Interestingly, unlike other major pathogenic spirochetes like Borrelia burgdorferi [73], L. interrogans genome houses at least two copies of 16S rRNA genes also conserved in sequenced genomes of pathogenic leptospires [6, 52]. Thus, it is perhaps not surprising that compared to even abundantly and consistently-expressed mRNA gene targets like lipL32 or flaB [51], our assay targeting 16S rRNA achieves better sensitivity that allow detection of low numbers of L. interrogans cell per milliliter of spiked human blood. This is also a notable improvement in sensitivity, compared to existing DNA-based PCRs where the limit of detection ranges from 102–103 bacteria per milliliter of blood or urine [8, 40–42]. Leptospiral 16S RNA molecules remain appreciably stable in the blood, further highlighting their potential use in the diagnosis of early infection. We do not know how 16S RNA molecules maintain notable stability in blood, however, this could be contributed by the remarkable spirochete ability to remain viable in certain aqueous environment ex vivo. In addition, existence of intact or fragmented 16S transcripts in the blood, or within phagocytic cells that enable their detection, also remains as interesting possibilities.
Although diagnosis of microbial infection based on the detection of a target RNA molecule offers multiple advantages, such as higher sensitivity and specificity, or potential indicator of early and active infection, there are inherent limitations that influence successful development of RNA PCR-based diagnostics. RNA molecules are generally less stable than other biomarkers and their cellular abundance (and thus detection) are likely to be variable, which could also be influenced by a number of additional factors. For example, efficiency of RNA detection depends on successful reverse transcription or absence of intrinsic blood factors that inhibit reverse transcription or subsequent PCR. In addition, transcript abundance may vary from bacterial cell to population levels, growth stages, or environment. Thus, absolute quantitation of microbial cells based on enumeration of RNA molecules might not be possible. Despite these challenges, due to their outstanding abundance and specificity, as also highlighted in our study, RNA targets are used in the diagnosis of a number of human infections, primarily the ones caused by viruses [74–77] and in a limited number of cases, for detection of bacterial infections, such as Mycobacteria species [78, 79], or Chlamydia trachomatis and Neisseria gonorrhoeae [70–72]. To the best of our knowledge, the current study represents the first attempt to use RNA detection to develop an improved diagnostic test for leptospirosis. Notably, 16S gene sequences were recently targeted for development of a DNA based real-time PCR assay, denoted as rt-PCR [50], which used additional primers tagged with reporter dye and quencher molecules called TaqMan probes to enhance analytical specificity, however the assay achieved overall clinical sensitivity of 34%. While our RNA assay yielded relatively superior sensitivity, we also show that use of a simpler in-house platform like SYBR Green-based qPCR assay could retain comparable specificities of TaqMan-probe based PCR. Nevertheless, we recognize that our results need to be further validated for true sensitivity and specificity values using additional studies involving larger numbers of patients, including ones from diverse epidemiological settings beyond hospitalized patients from Salvador, Brazil involved in the current study.
Although currently overall sensitivity of our RNA assay is twice as high as parallel TaqMan probe-based DNA assays and yielded statistically significant differences, it detected leptospiral RNA in 64% of the laboratory-confirmed cases. The exact reasons why our assay failed to yield positivity in a subset of laboratory-confirmed samples, including two patients where DNA assays are also positive, remained puzzling, however, could be linked to the effectiveness of antimicrobial therapy influencing spirochetemia at the time of individual sample collection, or unintended degradation of 16S RNA during less careful handling and storage conditions, amongst other unknown possibilities. On the other hand, notably, our RNA assay detected positivity in two human samples where routine laboratory diagnosis remained unconfirmed. While this could be interpreted as potential loss of specificity, it also suggests enhanced sensitivity of the RNA assay, especially in cases where pathogens are rapidly cleared before development of detectable humoral immune responses. The latter speculation of greater sensitivity of our assay is supported by its high (100%) specificity, where samples from normal humans failed to yield positivity. Notably, an earlier study also reported positivity in clinical diagnosis of Leptospira by PCR and sequencing methods in a subset of clinical samples that are serologically negative using a standard MAT panel [39]. Nevertheless, while implementation of additional care towards sample collection and storage would further enhance sensitivity of our RNA assay, use of a real-time PCR platform already has advantage over current serology-based assays in terms of rapidity, allowing quicker diagnosis of early infection and prompt antimicrobial treatment, which could prevent more severe and life-threatening complication of disseminated infection. In addition to currently-adopted SYBR Green-based qPCR assay, our RNA detection strategy is amenable to additional cheaper nucleic acid amplification methods, such as Loop Mediated Isothermal Amplification (LAMP) reaction for detection of a variety of human pathogens [80–87], including leptospirosis [88]. We also envision that future development of more efficient 16S primers or PCR platforms as well as combinatorial use of multiple gene targets could further improve the sensitivity and specificity of our assay. Therefore, our study could have far-reaching implications for development of simple, cost-effective, and rapid RNA-based PCR assays for detection of human leptospirosis as well as other bacteremic human pathogens where efficient diagnosis of early or active infection is warranted yet remains as an unmet need.
Supporting Information
Nucleotide sequences within the primer sequences of 16S rRNA genes from 37 Leptospira serovars, including all 20 known species, as well as non-target bacteria including other major pathogenic spirochetes, were derived from NCBI nucleotide sequence databases and aligned using MegAlign (DNASTAR software). Base pair mismatches between L. interrogans Fiocruz L1-130 and other species are indicated by boxes.
(PDF)
(PDF)
(PDF)
Acknowledgments
This work was supported by funding from the National Institute of Allergy and Infectious Diseases (Award Number AI114064-01 to AK, ML, and UP as well as AI052473, TW009504, and AI088752 to AK, and 1R43AI114064-01 to L2 Diagnostics (AK, ML, UP co-PIs). JL is supported by 2014 ASTMH Gorgas Memorial Institute Research Award, 2014 Global Health Equity Scholars Fellowship and Fiocruz-CNPq Ciencias Sem Fronteiras. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We sincerely thank Jin-hong Qin for her assistance with this study.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by funding from the National Institute of Allergy and Infectious Diseases (Award Number AI114064-01 to AK, ML, and UP as well as AI052473, TW009504, and AI088752 to AK, and 1R43AI114064-01 to L2 Diagnostics (AK, ML, UP co-PIs). JL is supported by 2014 ASTMH Gorgas Memorial Institute Research Award, 2014 Global Health Equity Scholars Fellowship and Fiocruz-CNPq Ciencias Sem Fronteiras. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. L2 Diagnostics provided support in the form of salaries for authors SUB and ML as well as AK and UP through subcontracts with Yale University and the University of Maryland, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bharti AR, Nally JE, Ricaldi JN, Matthias MA, Diaz MM, Lovett MA, et al. Leptospirosis: a zoonotic disease of global importance. Lancet Infect Dis. 2003;3(12):757–71. . [DOI] [PubMed] [Google Scholar]
- 2. Ko AI, Galvao Reis M, Ribeiro Dourado CM, Johnson WD Jr, Riley LW. Urban epidemic of severe leptospirosis in Brazil. Salvador Leptospirosis Study Group. Lancet. 1999;354(9181):820–5. . [DOI] [PubMed] [Google Scholar]
- 3. Ko AI, Goarant C, Picardeau M. Leptospira: the dawn of the molecular genetics era for an emerging zoonotic pathogen. Nat Rev Microbiol. 2009;7(10):736–47. doi: nrmicro2208 [pii] 10.1038/nrmicro2208 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Levett PN. Leptospirosis. Clin Microbiol Rev. 2001;14(2):296–326. 10.1128/CMR.14.2.296-326.2001 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McBride AJ, Athanazio DA, Reis MG, Ko AI. Leptospirosis. Curr Opin Infect Dis. 2005;18(5):376–86. . [DOI] [PubMed] [Google Scholar]
- 6. Ren SX, Fu G, Jiang XG, Zeng R, Miao YG, Xu H, et al. Unique physiological and pathogenic features of Leptospira interrogans revealed by whole-genome sequencing. Nature. 2003;422(6934):888–93. 10.1038/nature01597 nature01597 [pii]. . [DOI] [PubMed] [Google Scholar]
- 7. Vinetz JM, Glass GE, Flexner CE, Mueller P, Kaslow DC. Sporadic urban leptospirosis. Ann Intern Med. 1996;125(10):794–8. . [DOI] [PubMed] [Google Scholar]
- 8. Picardeau M, Bertherat E, Jancloes M, Skouloudis AN, Durski K, Hartskeerl RA. Rapid tests for diagnosis of leptospirosis: current tools and emerging technologies. Diagn Microbiol Infect Dis. 2014;78(1):1–8. 10.1016/j.diagmicrobio.2013.09.012 . [DOI] [PubMed] [Google Scholar]
- 9. Lacerda HG, Monteiro GR, Oliveira CC, Suassuna FB, Queiroz JW, Barbosa JD, et al. Leptospirosis in a subsistence farming community in Brazil. Trans R Soc Trop Med Hyg. 2008;102(12):1233–8. 10.1016/j.trstmh.2008.05.010 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sethi S, Sharma N, Kakkar N, Taneja J, Chatterjee SS, Banga SS, et al. Increasing trends of leptospirosis in northern India: a clinico-epidemiological study. PLoS Negl Trop Dis. 2010;4(1):e579 10.1371/journal.pntd.0000579 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johnson MA, Smith H, Joeph P, Gilman RH, Bautista CT, Campos KJ, et al. Environmental exposure and leptospirosis, Peru. Emerg Infect Dis. 2004;10(6):1016–22. 10.3201/eid1006.030660 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karande S, Gandhi D, Kulkarni M, Bharadwaj R, Pol S, Thakare J, et al. Concurrent outbreak of leptospirosis and dengue in Mumbai, India, 2002. J Trop Pediatr. 2005;51(3):174–81. 10.1093/tropej/fmh100 . [DOI] [PubMed] [Google Scholar]
- 13. Reis RB, Ribeiro GS, Felzemburgh RD, Santana FS, Mohr S, Melendez AX, et al. Impact of environment and social gradient on Leptospira infection in urban slums. PLoS Negl Trop Dis s. 2008;2(4):e228 10.1371/journal.pntd.0000228 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stern EJ, Galloway R, Shadomy SV, Wannemuehler K, Atrubin D, Blackmore C, et al. Outbreak of leptospirosis among Adventure Race participants in Florida, 2005. Clin Infect Dis. 2010;50(6):843–9. 10.1086/650578 . [DOI] [PubMed] [Google Scholar]
- 15. Morgan J, Bornstein SL, Karpati AM, Bruce M, Bolin CA, Austin CC, et al. Outbreak of leptospirosis among triathlon participants and community residents in Springfield, Illinois, 1998. Clin Infect Dis. 2002;34(12):1593–9. 10.1086/340615 . [DOI] [PubMed] [Google Scholar]
- 16. Jackson LA, Kaufmann AF, Adams WG, Phelps MB, Andreasen C, Langkop CW, et al. Outbreak of leptospirosis associated with swimming. Pediatr Infect Dis J. 1993;12(1):48–54. . [DOI] [PubMed] [Google Scholar]
- 17. Gale NB, Alexander AD, Evans LB, Yager RH, Matheney RG. An outbreak of leptospirosis among U. S. army troops in the Canal Zone. Am J Trop Med Hyg. 1966;15(1):64–70. . [DOI] [PubMed] [Google Scholar]
- 18. Lettieri C, Moon J, Hickey P, Gray M, Berg B, Hospenthal D. Prevalence of leptospira antibodies in U.S. Army blood bank donors in Hawaii. Mil Med. 2004;169(9):687–90. . [DOI] [PubMed] [Google Scholar]
- 19. Corwin A, Ryan A, Bloys W, Thomas R, Deniega B, Watts D. A waterborne outbreak of leptospirosis among United States military personnel in Okinawa, Japan. Int J Epidemiol. 1990;19(3):743–8. . [DOI] [PubMed] [Google Scholar]
- 20. Leshem E, Segal G, Barnea A, Yitzhaki S, Ostfeld I, Pitlik S, et al. Travel-related leptospirosis in Israel: a nationwide study. Am J Trop Med Hyg. 2010;82(3):459–63. 10.4269/ajtmh.2010.09-0239 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Crevel R, Speelman P, Gravekamp C, Terpstra WJ. Leptospirosis in travelers. Clin Infect Dis. 1994;19(1):132–4. Epub 1994/07/01. . [DOI] [PubMed] [Google Scholar]
- 22. Thiermann AB, Frank RR. Human leptospirosis in Detroit and the role of rats as chronic carriers. Int J Zoonoses. 1980;7(1):62–72. . [PubMed] [Google Scholar]
- 23. Dufour B, Moutou F, Hattenberger AM, Rodhain F. Global change: impact, management, risk approach and health measures—the case of Europe. Rev Sci Tech. 2008;27(2):529–50. . [PubMed] [Google Scholar]
- 24. Storck CH, Postic D, Lamaury I, Perez JM. Changes in epidemiology of leptospirosis in 2003–2004, a two El Nino Southern Oscillation period, Guadeloupe archipelago, French West Indies. Epidemiol Infect. 2008;136(10):1407–15. 10.1017/S0950268807000052 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lau CL, Smythe LD, Craig SB, Weinstein P. Climate change, flooding, urbanisation and leptospirosis: fuelling the fire? Trans R Soc Trop Med Hyg. 2010;104(10):631–8. 10.1016/j.trstmh.2010.07.002 . [DOI] [PubMed] [Google Scholar]
- 26. Hellstrom JS, Marshall RB. Survival of Leptospira interrogans serovar pomona in an acidic soil under simulated New Zealand field conditions. Res Vet Sci. 1978;25(1):29–33. Epub 1978/07/01. . [PubMed] [Google Scholar]
- 27. Trueba G, Zapata S, Madrid K, Cullen P, Haake D. Cell aggregation: a mechanism of pathogenic Leptospira to survive in fresh water. Int Microbiol. 2004;7(1):35–40. . [PubMed] [Google Scholar]
- 28. Adler B, de la Pena Moctezuma A. Leptospira and leptospirosis. Vet Microbiol. 2010;140(3–4):287–96. doi: S0378-1135(09)00116-3 [pii] 10.1016/j.vetmic.2009.03.012 . [DOI] [PubMed] [Google Scholar]
- 29. Coutinho ML, Matsunaga J, Wang LC, de la Pena Moctezuma A, Lewis MS, Babbitt JT, et al. Kinetics of Leptospira interrogans Infection in Hamsters after Intradermal and Subcutaneous Challenge. PLoS Negl Trop Dis. 2014;8(11):e3307 10.1371/journal.pntd.0003307 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gouveia EL, Metcalfe J, de Carvalho AL, Aires TS, Villasboas-Bisneto JC, Queirroz A, et al. Leptospirosis-associated severe pulmonary hemorrhagic syndrome, Salvador, Brazil. Emerg Infect Dis. 2008;14(3):505–8. 10.3201/eid1403.071064 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lourdault K, Wang LC, Vieira A, Matsunaga J, Melo R, Lewis MS, et al. Oral immunization with Escherichia coli expressing a lipidated form of LigA protects hamsters against challenge with Leptospira interrogans serovar Copenhageni. Infect Immun. 2014;82(2):893–902. 10.1128/IAI.01533-13 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zuerner RL, Alt DP, Palmer MV, Thacker TC, Olsen SC. A Leptospira borgpetersenii serovar Hardjo vaccine induces a Th1 response, activates NK cells, and reduces renal colonization. Clin Vaccine Immunol. 2011;18(4):684–91. 10.1128/CVI.00288-10 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cao Y, Faisal SM, Yan W, Chang YC, McDonough SP, Zhang N, et al. Evaluation of novel fusion proteins derived from extracellular matrix binding domains of LigB as vaccine candidates against leptospirosis in a hamster model. Vaccine. 2011;29(43):7379–86. 10.1016/j.vaccine.2011.07.070 . [DOI] [PubMed] [Google Scholar]
- 34. Bolin CA, Cassells JA, Zuerner RL, Trueba G. Effect of vaccination with a monovalent Leptospira interrogans serovar hardjo type hardjo-bovis vaccine on type hardjo-bovis infection of cattle. Am J Vet Res. 1991;52(10):1639–43. . [PubMed] [Google Scholar]
- 35. Koizumi N, Watanabe H. Leptospirosis vaccines: past, present, and future. J Postgrad Med. 2005;51(3):210–4. . [PubMed] [Google Scholar]
- 36. Flannery B, Costa D, Carvalho FP, Guerreiro H, Matsunaga J, Da Silva ED, et al. Evaluation of recombinant Leptospira antigen-based enzyme-linked immunosorbent assays for the serodiagnosis of leptospirosis. J Clin Microbiol. 2001;39(9):3303–10. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Musso D, La Scola B. Laboratory diagnosis of leptospirosis: a challenge. J Microbiol Immunol Infect. 2013;46(4):245–52. 10.1016/j.jmii.2013.03.001 . [DOI] [PubMed] [Google Scholar]
- 38. Boonsilp S, Thaipadungpanit J, Amornchai P, Wuthiekanun V, Chierakul W, Limmathurotsakul D, et al. Molecular detection and speciation of pathogenic Leptospira spp. in blood from patients with culture-negative leptospirosis. BMC Infect Dis. 2011;11:338 10.1186/1471-2334-11-338 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Agampodi SB, Matthias MA, Moreno AC, Vinetz JM. Utility of quantitative polymerase chain reaction in leptospirosis diagnosis: association of level of leptospiremia and clinical manifestations in Sri Lanka. Clin Infect Dis. 2012;54(9):1249–55. 10.1093/cid/cis035 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bourhy P, Bremont S, Zinini F, Giry C, Picardeau M. Comparison of real-time PCR assays for detection of pathogenic Leptospira spp. in blood and identification of variations in target sequences. J Clin Microbiol. 2011;49(6):2154–60. 10.1128/JCM.02452-10 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Slack AT, Symonds ML, Dohnt MF, Smythe LD. Identification of pathogenic Leptospira species by conventional or real-time PCR and sequencing of the DNA gyrase subunit B encoding gene. BMC Microbiol. 2006;6:95 10.1186/1471-2180-6-95 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smythe LD, Smith IL, Smith GA, Dohnt MF, Symonds ML, Barnett LJ, et al. A quantitative PCR (TaqMan) assay for pathogenic Leptospira spp. BMC Infect Dis. 2002;2:13 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Natarajaseenivasan K, Raja V, Narayanan R. Rapid diagnosis of leptospirosis in patients with different clinical manifestations by 16S rRNA gene based nested PCR. Saudi J Biol Sci. 2012;19(2):151–5. 10.1016/j.sjbs.2011.11.005 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ahmed A, Engelberts MF, Boer KR, Ahmed N, Hartskeerl RA. Development and validation of a real-time PCR for detectOon of pathogenic Leptospira species in clinical materials. PloS One. 2009;4(9):e7093 10.1371/journal.pone.0007093 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ahmed A, van der Linden H, Hartskeerl RA. Development of a recombinase polymerase amplification assay for the detection of pathogenic Leptospira . Int J Environ Res Public Health. 2014;11(5):4953–64. 10.3390/ijerph110504953 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ferreira AS, Costa P, Rocha T, Amaro A, Vieira ML, Ahmed A, et al. Direct detection and differentiation of pathogenic Leptospira species using a multi-gene targeted real time PCR approach. PloS One. 2014;9(11):e112312 Epub 2014/11/15. 10.1371/journal.pone.0112312 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goarant C, Bourhy P, D'Ortenzio E, Dartevelle S, Mauron C, Soupe-Gilbert ME, et al. Sensitivity and Specificity of a New Vertical Flow Rapid Diagnostic Test for the Serodiagnosis of Human Leptospirosis. PLoS Negl Trop Dis. 2013;7(6):e2289 10.1371/journal.pntd.0002289 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Goarant C, Laumond-Barny S, Perez J, Vernel-Pauillac F, Chanteau S, Guigon A. Outbreak of leptospirosis in New Caledonia: diagnosis issues and burden of disease. Trop Med Int Health. 2009;14(8):926–9. 10.1111/j.1365-3156.2009.02310.x . [DOI] [PubMed] [Google Scholar]
- 49. Perez J, Goarant C. Rapid Leptospira identification by direct sequencing of the diagnostic PCR products in New Caledonia. BMC Microbiol. 2010;10:325 10.1186/1471-2180-10-325 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Waggoner JJ, Balassiano I, Abeynayake J, Sahoo MK, Mohamed-Hadley A, Liu Y, et al. Sensitive Real-Time PCR Detection of Pathogenic Leptospira spp. and a Comparison of Nucleic Acid Amplification Methods for the Diagnosis of Leptospirosis. PloS One. 2014;9(11):e112356 10.1371/journal.pone.0112356 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Matsui M, Soupe ME, Becam J, Goarant C. Differential in vivo gene expression of major Leptospira proteins in resistant or susceptible animal models. Appl Environ Microbiol. 2012;78(17):6372–6. 10.1128/AEM.00911-12 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nascimento AL, Ko AI, Martins EA, Monteiro-Vitorello CB, Ho PL, Haake DA, et al. Comparative genomics of two Leptospira interrogans serovars reveals novel insights into physiology and pathogenesis. J Bacteriol. 2004;186(7):2164–72. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tucunduva de Faria M, Athanazio DA, Goncalves Ramos EA, Silva EF, Reis MG, Ko AI. Morphological alterations in the kidney of rats with natural and experimental Leptospira infection. J Comp Pathol. 2007;137(4):231–8. 10.1016/j.jcpa.2007.08.001 . [DOI] [PubMed] [Google Scholar]
- 54. Ricaldi JN, Fouts DE, Selengut JD, Harkins DM, Patra KP, Moreno A, et al. Whole genome analysis of Leptospira licerasiae provides insight into leptospiral evolution and pathogenicity. PLoS Negl Trop Dis. 2012;6(10):e1853 10.1371/journal.pntd.0001853 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ellinghausen HC Jr., McCullough WG. Nutrition of Leptospira Pomona and Growth of 13 Other Serotypes: Fractionation of Oleic Albumin Complex and a Medium of Bovine Albumin and Polysorbate 80. Am J Vet Res. 1965;26:45–51. . [PubMed] [Google Scholar]
- 56. Noubade R, Krishnamurthy GV, Murag S, Venkatesha MD, Krishnappa G. Differentiation of pathogenic and saprophytic leptospires by polymerase chain reaction. Indian J Med Microbiol. 2002;20(1):33–6. . [PubMed] [Google Scholar]
- 57. Raz A, Talay SR, Fischetti VA. Cellular aspects of the distinct M protein and SfbI anchoring pathways in Streptococcus pyogenes . Mol Microbiol. 2012;84(4):631–47. 10.1111/j.1365-2958.2012.08047.x ;. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Elias AF, Stewart PE, Grimm D, Caimano MJ, Eggers CH, Tilly K, et al. Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infect Immun. 2002;70(4):2139–50. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lessa-Aquino C, Borges Rodrigues C, Pablo J, Sasaki R, Jasinskas A, Liang L, et al. Identification of seroreactive proteins of Leptospira interrogans serovar copenhageni using a high-density protein microarray approach. PLoS Negl Trop Dis. 2013;7(10):e2499 10.1371/journal.pntd.0002499 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Promnares K, Kumar M, Shroder DY, Zhang X, Anderson JF, Pal U. Borrelia burgdorferi small lipoprotein Lp6.6 is a member of multiple protein complexes in the outer membrane and facilitates pathogen transmission from ticks to mice. Mol Microbiol. 2009;74(1):112–25. 10.1111/j.1365-2958.2009.06853.x . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yang X, Coleman AS, Anguita J, Pal U. A chromosomally encoded virulence factor protects the Lyme disease pathogen against host-adaptive immunity. PLoS Pathogens. 2009;5(3):e1000326 Epub 2009/03/07. 10.1371/journal.ppat.1000326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Reller ME, Wunder EA Jr., Miles JJ, Flom JE, Mayorga O, Woods CW, et al. Unsuspected leptospirosis is a cause of acute febrile illness in Nicaragua. PLoS Negl Trop Dis. 2014;8(7):e2941 10.1371/journal.pntd.0002941 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Stoddard RA, Gee JE, Wilkins PP, McCaustland K, Hoffmaster AR. Detection of pathogenic Leptospira spp. through TaqMan polymerase chain reaction targeting the LipL32 gene. Diagn Microbiol Infect Dis. 2009;64(3):247–55. 10.1016/j.diagmicrobio.2009.03.014 . [DOI] [PubMed] [Google Scholar]
- 64. Arikawa E, Sun Y, Wang J, Zhou Q, Ning B, Dial SL, et al. Cross-platform comparison of SYBR Green real-time PCR with TaqMan PCR, microarrays and other gene expression measurement technologies evaluated in the MicroArray Quality Control (MAQC) study. BMC Genomics. 2008;9:328 10.1186/1471-2164-9-328 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Clarridge JE 3rd. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin Microbiol Rev. 2004;17(4):840–62, table of contents. 10.1128/CMR.17.4.840-862.2004 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. . [DOI] [PubMed] [Google Scholar]
- 67. Bruce MG, Sanders EJ, Leake JA, Zaidel O, Bragg SL, Aye T, et al. Leptospirosis among patients presenting with dengue-like illness in Puerto Rico. Acta Trop. 2005;96(1):36–46. Epub 2005/08/09. 10.1016/j.actatropica.2005.07.001 . [DOI] [PubMed] [Google Scholar]
- 68. Biggs HM, Galloway RL, Bui DM, Morrissey AB, Maro VP, Crump JA. Leptospirosis and human immunodeficiency virus co-infection among febrile inpatients in northern Tanzania. Vector Borne Zoonotic Dis. 2013;13(8):572–80. 10.1089/vbz.2012.1205 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Biggs HM, Bui DM, Galloway RL, Stoddard RA, Shadomy SV, Morrissey AB, et al. Leptospirosis among hospitalized febrile patients in northern Tanzania. Am J Trop Med Hyg. 2011;85(2):275–81. 10.4269/ajtmh.2011.11-0176 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chong S, Jang D, Song X, Mahony J, Petrich A, Barriga P, et al. Specimen processing and concentration of Chlamydia trachomatis added can influence false-negative rates in the LCx assay but not in the APTIMA Combo 2 assay when testing for inhibitors. J Clin Microbiol. 2003;41(2):778–82. Epub 2003/02/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lowe P, O'Loughlin P, Evans K, White M, Bartley PB, Vohra R. Comparison of the Gen-Probe APTIMA Combo 2 assay to the AMPLICOR CT/NG assay for detection of Chlamydia trachomatis and Neisseria gonorrhoeae in urine samples from Australian men and women. J Clin Microbiol. 2006;44(7):2619–21. 10.1128/JCM.00476-06 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cheng A, Kirby JE. Evaluation of the Hologic gen-probe PANTHER, APTIMA Combo 2 assay in a tertiary care teaching hospital. Am J Clin Pathol. 2014;141(3):397–403. 10.1309/AJCPFQ25SQVZAWHZ . [DOI] [PubMed] [Google Scholar]
- 73. Bugrysheva JV, Godfrey HP, Schwartz I, Cabello FC. Patterns and regulation of ribosomal RNA transcription in Borrelia burgdorferi . BMC Microbiol. 2011;11:17 10.1186/1471-2180-11-17 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhu Z, Fan H, Qi X, Qi Y, Shi Z, Wang H, et al. Development and evaluation of a SYBR green-based real time RT-PCR assay for detection of the emerging avian influenza A (H7N9) virus. PloS One. 2013;8(11):e80028 10.1371/journal.pone.0080028 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Paulino LC, de Mello MP, Ottoboni LM. Differential gene expression in response to copper in Acidithiobacillus ferrooxidans analyzed by RNA arbitrarily primed polymerase chain reaction. Electrophoresis. 2002;23(4):520–7. . [DOI] [PubMed] [Google Scholar]
- 76. Jiang W, Yu HT, Zhao K, Zhang Y, Du H, Wang PZ, et al. Quantification of Hantaan virus with a SYBR green I-based one-step qRT-PCR assay. PloS One. 2013;8(11):e81525 10.1371/journal.pone.0081525 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Maquart M, Temmam S, Heraud JM, Leparc-Goffart I, Cetre-Sossah C, Dellagi K, et al. Development of real-time RT-PCR for the detection of low concentrations of Rift Valley fever virus. J Virol Methods. 2014;195:92–9. 10.1016/j.jviromet.2013.10.001 . [DOI] [PubMed] [Google Scholar]
- 78. Beissner M, Symank D, Phillips RO, Amoako YA, Awua-Boateng NY, Sarfo FS, et al. Detection of viable Mycobacterium ulcerans in clinical samples by a novel combined 16S rRNA reverse transcriptase/IS2404 real-time qPCR assay. PLoS Negl Trop Dis. 2012;6(8):e1756 10.1371/journal.pntd.0001756 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jiang LJ, Wu WJ, Wu H, Ryang SS, Zhou J, Wu W, et al. Rapid detection and monitoring therapeutic efficacy of Mycobacterium tuberculosis complex using a novel real-time assay. J Microbiol Biotechnol. 2012;22(9):1301–6. . [DOI] [PubMed] [Google Scholar]
- 80. Imai M, Ninomiya A, Minekawa H, Notomi T, Ishizaki T, Van Tu P, et al. Rapid diagnosis of H5N1 avian influenza virus infection by newly developed influenza H5 hemagglutinin gene-specific loop-mediated isothermal amplification method. J Virol Methods. 2007;141(2):173–80. 10.1016/j.jviromet.2006.12.004 . [DOI] [PubMed] [Google Scholar]
- 81. Jayawardena S, Cheung CY, Barr I, Chan KH, Chen H, Guan Y, et al. Loop-mediated isothermal amplification for influenza A (H5N1) virus. Emerg Infect Dis. 2007;13(6):899–901. 10.3201/eid1306.061572 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pan L, Zhang L, Wang G, Liu Q, Yu Y, Wang S, et al. Rapid, simple, and sensitive detection of Anaplasma phagocytophilum by loop-mediated isothermal amplification of the msp2 gene. J Clin Microbiol. 2011;49(12):4117–20. 10.1128/JCM.01085-11 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Curtis KA, Niedzwiedz PL, Youngpairoj AS, Rudolph DL, Owen SM. Real-Time Detection of HIV-2 by Reverse Transcription-Loop-Mediated Isothermal Amplification. J Clin Microbiol. 2014;52(7):2674–6. 10.1128/JCM.00935-14 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhang G, Brown EW, Gonzalez-Escalona N. Comparison of real-time PCR, reverse transcriptase real-time PCR, loop-mediated isothermal amplification, and the FDA conventional microbiological method for the detection of Salmonella spp. in produce. Appl Environ Microbiol. 2011;77(18):6495–501. Epub 2011/08/02. 10.1128/AEM.00520-11 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Parida M, Posadas G, Inoue S, Hasebe F, Morita K. Real-time reverse transcription loop-mediated isothermal amplification for rapid detection of West Nile virus. J Clin Microbiol. 2004;42(1):257–63. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Patel JC, Lucchi NW, Srivastava P, Lin JT, Sug-Aram R, Aruncharus S, et al. Field evaluation of a real-time fluorescence loop-mediated isothermal amplification assay, RealAmp, for the diagnosis of malaria in Thailand and India. J Infect Dis. 2014;210(8):1180–7. 10.1093/infdis/jiu252 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lucchi NW, Demas A, Narayanan J, Sumari D, Kabanywanyi A, Kachur SP, et al. Real-time fluorescence loop mediated isothermal amplification for the diagnosis of malaria. PloS One. 2010;5(10):e13733 10.1371/journal.pone.0013733 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sonthayanon P, Chierakul W, Wuthiekanun V, Thaipadungpanit J, Kalambaheti T, Boonsilp S, et al. Accuracy of loop-mediated isothermal amplification for diagnosis of human leptospirosis in Thailand. Am J Trop Med Hyg. 2011;84(4):614–20. 10.4269/ajtmh.2011.10-0473 . [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Nucleotide sequences within the primer sequences of 16S rRNA genes from 37 Leptospira serovars, including all 20 known species, as well as non-target bacteria including other major pathogenic spirochetes, were derived from NCBI nucleotide sequence databases and aligned using MegAlign (DNASTAR software). Base pair mismatches between L. interrogans Fiocruz L1-130 and other species are indicated by boxes.
(PDF)
(PDF)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.