

Abstract
Upon obesity, adipose tissue is excessively expanded and characterized by pathologic processes like hypoxia, fibrosis, and inflammation. Death ligands belonging to the TNF superfamily such as TNF-α are important contributors to these derangements and exert a pronounced influence on the metabolic and cellular homeostasis of adipose tissue. Here, we sought to identify the effect of the death ligand TNF-related apoptosis-inducing ligand (TRAIL) on the adipose tissue precursor cell pool and therefore investigated its influence on preadipocyte proliferation. Treatment of human preadipocytes with TRAIL resulted in a time- and dose-dependent increase in proliferation (EC50 3.4 ng/ml) comparable to IGF-1. Although no apoptosis was observed, TRAIL triggered a rapid cleavage of caspase-8 and -3. Neither inhibition of caspase activity by zVAD.fmk (20 µM) nor ablation of caspase-8 expression by lentivirus-delivered small hairpin RNA (shRNA) abolished the proliferative response. TRAIL triggered a delayed and sustained activation of ERK1/2, leaving Akt, p38, JNK, and NF-κB unaffected. Importantly, inhibition of ERK1/2 activation by PD0325901 (300 nM) or AZD6244 (5 or 10 µM) completely abolished the proliferative response. We thus reveal a hitherto unknown function of TRAIL in regulating adipose tissue homeostasis by promoting the proliferation of tissue-resident precursor cells.—Funcke, J.-B., Zoller, V., Abd El Hay, M., Debatin, K.-M., Wabitsch, M., Fischer-Posovszky, P. TNF-related apoptosis-inducing ligand promotes human preadipocyte proliferation via ERK1/2 activation.
Keywords: adipose tissue homeostasis, adipocyte progenitor, noncanonical signaling, death ligand
With the growing epidemic of obesity, white adipose tissue has come to the center of metabolic research. It has long been regarded a passive organ for energy storage, but is nowadays well recognized as an active endocrine organ (1). Adipose tissue releases more than 100 different factors to the circulation (1). These hormones, growth factors, inflammatory cytokines, and metabolites exert their functions either locally in an auto-/paracrine manner or systemically and thereby regulate whole-body energy balance and metabolism (1).
Adipose tissue displays an exceptionally high plasticity (2, 3). It can rapidly expand or shrink in response to changes in the energy state of the body (2, 3). In obesity, it is excessively enlarged and characterized by pathologic alterations such as hypoxia, fibrosis, and inflammation involving the infiltration and accumulation of macrophages (2, 3). These local alterations play a causal role in the development of obesity-associated comorbidities, especially insulin resistance and type 2 diabetes mellitus, but also hepatic steatosis and cardiovascular diseases (4).
TRAIL belongs to the TNF superfamily (5). In the canonical pathway, TRAIL binding to its cognate receptors at the cell surface (TRAIL-R1 and TRAIL-R2) leads to formation of a primary complex also called death-inducing signaling complex, consisting of receptor oligomers, the adaptor molecule Fas-associated death domain (FADD), the initiator caspases 8 or 10, and the regulatory protein cellular FLICE-like inhibitory protein (cFLIP) (5). The oligomerization of initiator caspases in the complex triggers their activation by self-cleavage, allowing further downstream events and induction of apoptosis to occur (5–7). In a noncanonical pathway, FADD, cFLIP, as well as caspase-8 and -10 can be released from the primary complex and contribute to the formation of a cytosolic complex, which additionally encompasses the adaptor protein TNF receptor type 1-associated death domain (TRADD), receptor-interacting protein kinase 1 (RIPK1), and TNF receptor-associated factor 2 (TRAF2) (8–10). This secondary complex can activate diverse cellular signaling cascades including the Akt, mitogen-activated protein kinase (MAPK, i.e., p38, JNK, ERK1/2), and NF-κB pathways (10). However, the biological significance of this kinase cascade activation is still elusive.
TRAIL has gained fame as an anticancer agent. It is a promising candidate for the treatment of malignancies because it selectively induces apoptosis in cancer cells, but not in nonmalignant cells (11). Its safety and efficacy are currently tested in several phase I/II studies (11).
TRAIL and its receptors are expressed in many tissues, including adipose tissue (12). We started studying the function of TRAIL in adipose tissue because the circulating TRAIL level was shown to correlate positively with the body mass index (BMI) and serum lipid levels (13). Interestingly, the expression of TRAIL receptors in adipose tissue is up-regulated with increasing BMI in humans as well as in mouse models of obesity (14), suggesting a functional role of these receptors in adipose tissue biology as already demonstrated for TNF-R1 and Fas (15, 16). Indeed, we recently demonstrated that TRAIL can regulate adipocyte metabolism by caspase-dependent cleavage of peroxisome proliferator-activated receptor γ (PPARγ) (14).
White adipose tissue is composed of different cell types, mainly lipid-filled adipocytes, which make up for most of tissue mass and are responsible for energy storage and metabolism. Other cell types include progenitor cells at different stages of determination or differentiation (e.g., mesenchymal stem cells and preadipocytes), endothelial cells, nerve cells, and immune cells (2, 3). The presence of progenitor cells capable of differentiating into mature adipocytes is of crucial importance for the expandability of adipose tissue (2, 3). Therefore, the aim of this study was to elucidate the impact of TRAIL on preadipocyte function, specifically preadipocyte proliferation.
MATERIALS AND METHODS
Materials
Recombinant human TRAIL was obtained from R&D Systems (Wiesbaden-Nordenstadt, Germany). Fully human agonistic monoclonal TRAIL-R1 (mapatumumab) and TRAIL-R2 (lexatumumab) antibodies were kind gifts of Human Genome Sciences (11). Recombinant human FasL was obtained from Enzo Life Sciences (Lörrach, Germany) and recombinant human TNF-α from Biochrom (Berlin, Germany). zVAD.fmk was obtained from Bachem (Bubendorf, Switzerland), PD0325901 and AZD6244 from Selleckchem (Munich, Germany).
Experimental subjects and human primary stromal-vascular cell isolation
Subcutaneous adipose tissue samples were obtained from 4 female patients undergoing plastic surgery. The mean age of the patients was 31.8 ± 5.7 years, the mean BMI was 30.0 ± 8.3 kg/m2. All procedures were performed in accordance with the Declaration of Helsinki guidelines and approved by the ethics committee of Ulm University. Written informed consent was obtained from all patients in advance. Stromal-vascular cells were isolated from adipose tissue samples by established protocols (17).
Cell culture
Simpson-Golabi-Behmel syndrome (SGBS) preadipocytes (18) and human primary stromal-vascular cells from adipose tissue were maintained in their undifferentiated state in growth medium consisting of DMEM/F-12 (1:1) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin (Life Technologies, Darmstadt, Germany), 17 µM d-pantothenic acid, and 33 µM biotin (Sigma-Aldrich, Munich, Germany).
Microscopic cell counting
SGBS preadipocytes were seeded into 12 well plates (7000 cells/well). For each treatment, triplicate wells were prepared. On the next day, the cells were given fresh growth medium with or without TRAIL (1, 10, or 100 ng/ml). At 0, 24, 48, and 72 hours after stimulation the number of adherent cells was determined by directly counting 3 random microscopic fields (1 mm2 area each) per well using a net micrometer.
[3H]-thymidine incorporation
SGBS preadipocytes (5000 cells/well) and human primary stromal-vascular cells (10,000–20,000 cells/well) were seeded into 24 well plates. For each treatment, at least triplicate wells were prepared. On the next day, the cells were given fresh growth medium supplied with the indicated substances. After 48 hours, 2.5 µCi of methyl-[3H]-thymidine (Hartmann Analytic, Braunschweig, Germany) were added. After 16 hours, the cells were washed with Dulbecco’s phosphate-buffered saline (DPBS), trypsinized, and harvested using a Filtermat Harvester (Inotech Biosystems, Brandon, FL, USA). Solid scintillation counting of β- decay events was performed on a MicroBeta TriLux 1450 (Perkin Elmer, Rodgau, Germany).
Carboxyfluorescein diacetate succinimidyl ester fluorescence staining
SGBS cells were trypsinized, centrifuged, and resuspended in basal medium consisting of DMEM/F-12 (1:1) supplemented with100 U/ml penicillin, 100 µg/ml streptomycin (Life Technologies), 17 µM d-pantothenic acid, and 33 µM biotin (Sigma-Aldrich). 5/6-carboxyfluorescein diacetate N-succinimidyl ester (Affymetrix eBioscience, Frankfurt am Main, Germany) was added to a final concentration of 4.5 µM, and the cells were incubated in the dark for 15 minutes at 37°C. The cells were washed 3 times with basal medium and seeded into 6 well plates (30,000 cell/well) in growth medium. For each treatment, at least triplicate wells were prepared. On the next day, the cells were given fresh basal medium or growth medium supplied with the indicated substances. After 72 hours, the cells were trypsinized and analyzed on a FACSCalibur flow cytometer (BD, Heidelberg, Germany).
Hoechst 33342 fluorescence staining
SGBS cells were seeded into 4 well chamber slides (BD; 10,000 cells/well). On the next day, the cells were given fresh growth medium supplied with the indicated substances. After 72 hours, the cells were washed twice with DPBS, fixed with formaldehyde, washed with DPBS and incubated with 500 ng/ml Hoechst 33342 (Sigma-Aldrich) in the dark for 15 minutes at room temperature. The cells were washed with DPBS, mounted with fluorescence mounting medium (DAKO, Hamburg, Germany) and analyzed on an AX-70 fluorescence microscope (Olympus, Hamburg, Germany).
Nicoletti assay
SGBS preadipocytes were seeded into 12 well plates (7000 cells/well). For each treatment, at least triplicate wells were prepared. Cell death by apoptosis was quantified as described by Nicoletti et al. (19, 20). Specific apoptosis was calculated by the following formula: 100% × [experimental apoptosis (%) − spontaneous apoptosis in growth medium (%)] / [100% − spontaneous apoptosis in growth medium (%)].
Whole protein extraction and Western blot analysis
The cells were washed once with ice cold DPBS, detached using a cell scraper, and lysed for 20 minutes at 4°C in whole protein lysis buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 10% glycerol) freshly supplemented with 1 mM DTT, 1X cOmplete proteinase inhibitors, and 1X phosSTOP phosphatase inhibitors (Roche, Mannheim, Germany). Lysates were cleared by high-speed centrifugation at 4°C.
Protein concentration was determined using the Pierce BCA protein assay kit (Thermo Scientific, Munich, Germany). SDS-PAGE and Western blotting were carried out using NuPAGE and iBlot products obtained from Life Technologies. Briefly, 20–30 µg of sample protein were separated on NuPAGE 4–12% Bis-Tris gradient gels in 1X NuPAGE MES SDS running buffer and transferred onto iBlot nitrocellulose membranes. The following primary antibodies were used: rabbit anti-phospho-Akt (S473), rabbit anti-Akt, mouse anti-phospho-ERK1/2 (T202/Y204), rabbit anti-phospho-JNK1/2 (T183/Y185), rabbit anti-phospho-p38 (T180/Y182), mouse anti-p38, mouse anti-phospho-IκBα (S32/S36), rabbit anti-IκBα, and rabbit anti-caspase-3 (Cell Signaling Technology), mouse anti-β-actin, and rabbit anti-ERK1/2 (Sigma-Aldrich), mouse anti-caspase-8 (Enzo Life Sciences), mouse anti-α-tubulin (Calbiochem/EMD Millipore, Darmstadt, Germany), mouse anti-JNK (BD). Horseradish peroxidase-conjugated secondary antibodies were obtained from Santa Cruz Biotechnology (Heidelberg, Germany). Enhanced chemiluminescence was carried out using Amersham ECL Western blotting detection reagent (GE Healthcare, Munich, Germany).
Lentiviral-mediated knockdown of caspase-8
Lentiviral-mediated knockdown of caspase-8 was carried out using the BLOCK-iT inducible H1 Lentiviral RNAi kit, the HEK293FT cell line, Lipofectamine 2000, and ViraPower lentiviral packaging mix obtained from Life Technologies.
Two independent shRNAs targeting caspase-8, shC8.1 (start 411, 5′-GAA CAA CTG GAC AGT GAA GA-3′) and shC8.2 (start 683, 5′-GGT CAT GCT CTA TCA GAT TT-3′), as well as a sequence with no corresponding counterpart in the human genome (hyper random sequence), shHRS (5′-GAT CAT GTA GAT ACG CTC A-3′), were first cloned into pENTR/H1/TO and then transferred into pLenti4/BLOCK-iT-DEST by targeted recombination. Lentiviral particles were produced in HEK293FT cells using the ViraPower lentiviral packaging mix according to the manufacturer’s recommendations. SGBS preadipocytes were transduced with lentiviral supernatants supplemented with 8 µg/ml Sequabrene (Sigma-Aldrich) at a multiplicity of infection of 1–1.5. Stably transduced cells were selected with Zeocin (Life Technologies). Caspase-8 knockdown was controlled by Western blot.
NF-κB p65 immunofluorescence staining
SGBS cells were seeded into 4 well chamber slides (BD; 10,000 cells/well). After 72 hours, the cells were given fresh growth medium supplied with the indicated substances. At 30 minutes and 2 hours after stimulation the cells were washed twice with DPBS, fixed with formaldehyde, washed twice with DPBS and permeabilized with Triton X-100. The cells were washed twice with DPBS, incubated in blocking buffer (DPBS supplemented with 10% fetal bovine serum) for 1 hour at room temperature. The cells were stained overnight shaking at 4°C with rabbit anti-NF-κB p65 primary antibody (Santa Cruz Biotechnology) diluted 1:100 in blocking buffer. The cells were washed 4 times with blocking buffer and stained for 1 hour in the dark at room temperature with goat anti-rabbit IgG-FITC secondary antibody (Santa Cruz Biotechnology) diluted 1:200 in blocking buffer. The cells were washed 4 times with blocking buffer, mounted with fluorescence mounting medium (DAKO) and analyzed on an AX-70 fluorescence microscope (Olympus).
Nuclear protein extraction and NF-κB electrophoretic mobility shift assay
Nuclear protein extraction was performed as described by Andrews and Faller (21) with modifications by Kasperczyk et al. (22). In brief, the cells were washed once with ice cold DPBS, detached using a cell scraper, and incubated for 15 minutes at 4°C in low salt buffer (10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid-potassium hydroxide (HEPES-KOH) pH 7.9, 1.5 mM MgCl2, 10 mM KCl) freshly supplemented with 500 µM DTT, 200 µM PMSF, 1 mM Na3VO4, and 1.33X protease inhibitor (Sigma-Aldrich). Nonidet P-40 substitute (0.6%; Sigma-Aldrich) was added, the cells were vortexed, and the cytosolic extract was separated by centrifugation for 5 minutes at 2500 rpm, 4°C. The cell pellet was incubated for 20 minutes shaking at 4°C in high salt buffer (20 mM HEPES-KOH pH 7.9, 1.5 mM MgCl2, 420 mM NaCl, 200 µM EDTA, 25% glycerol) freshly supplemented with 500 µM DTT, 200 µM PMSF, 1 mM Na3VO4, and 1.33X protease inhibitor (Sigma-Aldrich). Remaining cellular debris were removed by centrifugation for 10 minutes at 13,000 rpm, 4°C. The nuclear extract was collected, snap-frozen in liquid nitrogen, and stored at −80°C until use. Before use, the protein concentration of the nuclear extracts was determined using the Pierce BCA protein assay kit obtained from Thermo Scientific.
NF-κB sense probe (5′-AGT TGA GGG GAC TTT CCC AGG C-3′) was labeled with γ-[32P]-ATP (Perkin Elmer) using T4 polynucleotide kinase (MBI Fermentas, St. Leon-Rot, Germany). After labeling, a 2-fold molar excess of unlabeled NF-κB antisense probe (5′-GCC TGG GAA AGT CCC CTC AAC T-3′) was added and annealed by short heating to 90°C and slow cooling overnight. The resulting double-stranded labeled NF-κB probe was purified on Micro Bio-Spin P30 columns (BioRad, Munich, Germany).
Binding reactions were prepared on ice in binding buffer (10 mM Tris-HCl pH 7.5, 1 mM MgCl2, 50 mM NaCl, 500 µM EDTA, 4% glycerol, 500 µM DTT) with 5 µg nuclear extract protein, 1 µg poly(dI:dC) (Sigma-Aldrich), and 10,000-12,000 cpm of labeled NF-κB probe and incubated for 30 minutes at room temperature. The complexes formed were separated on a nondenaturing 6% polyacrylamide gel with 0.3X TBE as the running buffer. The polyacrylamide gel was then dried onto Whatman paper (GE Healthcare), and the radioactive signal was detected using Amersham Hyperfilm ECL films (GE Healthcare).
Statistical analysis
Statistical analysis was carried out using GraphPad Prism software version 6.01 (GraphPad Software, La Jolla, CA, USA). A 1-way ANOVA was used to analyze the data displayed in Figs. 1B, 1E, 1F, 2B, 3A, 3B, 4C, 4E, 6C, and 6D. A 2-way ANOVA was used to analyze the data displayed in Fig. 1A. Where either a 1-way or 2-way ANOVA were employed, a Bonferroni post-test comparing the indicated conditions was performed. A 2-tailed, paired Student's t test was used to analyze the data displayed in Fig. 1D. In all cases, the significance level (α) was set to be 0.05.
Figure 1.
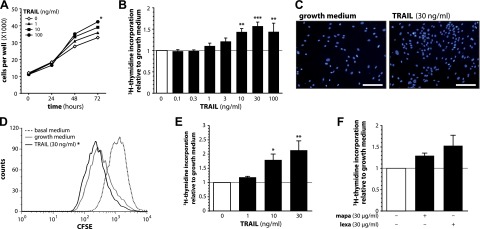
TRAIL stimulates human preadipocyte proliferation. A–C) SGBS preadipocytes were stimulated with growth medium alone or with increasing doses of TRAIL. A) At the indicated time points, the number of adherent cells was counted using a net micrometer. Data are expressed as means ± sem of 3 independent experiments performed. *P ≤ 0.05 for TRAIL 100 ng/ml compared with growth medium alone at 72 hours. B) After 72 hours, [3H]-thymidine incorporation was measured. Data are expressed as means ± sem of at least 3 independent experiments performed. **P ≤ 0.01; ***P ≤ 0.001 compared with growth medium alone. C) After 72 hours, Hoechst 33342 fluorescence staining was performed. One representative of 3 independent experiments performed is shown. Scale bars, 200 µm. D) SGBS preadipocytes were stained with CFSE and stimulated with basal medium, growth medium alone or with TRAIL. After 72 hours, the remaining CFSE fluorescence was measured. A representative histogram of 1 of 3 independent experiments performed is shown. The loss of CFSE mean fluorescence intensity in comparison to cells stimulated with basal medium was calculated. *P ≤ 0.05 for TRAIL 30 ng/ml compared with growth medium alone. E) Human primary stromal-vascular cells were isolated from adipose tissue samples of 4 patients and stimulated with growth medium alone or with increasing doses of TRAIL. After 72 hours, [3H]-thymidine incorporation was measured. Data are expressed as means ± sem. *P ≤ 0.05; **P ≤ 0.01 compared with growth medium alone. F) SGBS preadipocytes were stimulated with growth medium alone or human agonistic antibodies specific for either TRAIL-R1 (mapatumumab, mapa) or TRAIL-R2 (lexatumumab, lexa). After 72 hours, [3H]-thymidine incorporation was measured. Data are expressed as means ± sem of 3 independent experiments performed.
Figure 2.
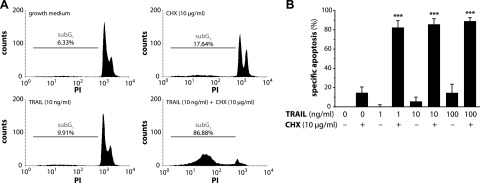
TRAIL does not induce human preadipocyte apoptosis. A, B) SGBS preadipocytes were stimulated with growth medium alone or with increasing doses of TRAIL in the absence or presence of cycloheximide (CHX). After 48 hours, apoptotic cell death was measured by flow cytometry. A) Representative histograms of 1 of 3 independent experiments performed are shown. B) Specific apoptosis was calculated and analyzed statistically. Data are expressed as means ± sem of 3 independent experiments performed. ***P ≤ 0.001 compared with growth medium alone.
Figure 3.
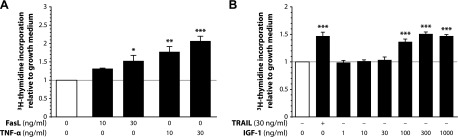
Death ligands stimulate human preadipocyte proliferation. A) SGBS preadipocytes were stimulated with growth medium alone or with increasing doses of either FasL or TNF-α. After 72 hours, [3H]-thymidine incorporation was measured. Data are expressed as means ± sem of 3 independent experiments performed. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001 compared with growth medium alone. B) SGBS preadipocytes were stimulated with growth medium alone or with either TRAIL or increasing doses of IGF-1. After 72 hours, [3H]-thymidine incorporation was measured. Data are expressed as means ± sem of at least 3 independent experiments performed. ***P ≤ 0.001 compared with growth medium alone.
Figure 4.
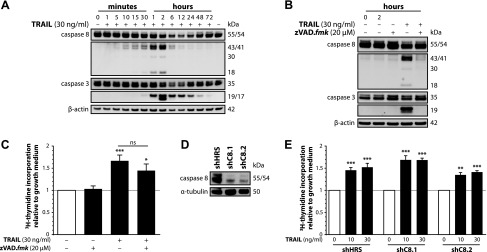
TRAIL triggers caspase activation, but caspases are not involved in the induction of proliferation. A) SGBS preadipocytes were stimulated with growth medium alone or with TRAIL. At the indicated time points, proteins were isolated and subjected to Western blot analysis to assess the processing of caspase-8 and -3. β-actin was used as a loading control. One representative of 3 independent experiments performed is shown. B, C) SGBS preadipocytes were stimulated with growth medium alone or with TRAIL and/or zVAD.fmk. B) At the indicated time points, proteins were isolated and subjected to Western blot analysis to assess the inhibition of caspase-8 and -3 processing by zVAD.fmk. β-actin was used as a loading control. One representative of 3 independent experiments performed is shown. C) After 72 hours, [3H]-thymidine incorporation was measured. Data are expressed as means ± sem of six independent experiments performed. *P ≤ 0.05; ***P ≤ 0.001 compared with growth medium alone (directly above the bars). ns, not significant comparing the indicated treatments (above the connecting line). D, E) A stable, lentiviral-mediated knockdown of caspase-8 in SGBS preadipocytes was performed. Two shRNAs targeting caspase-8 (shC8.1 and shC8.2) were used. A hyper random sequence (shHRS) was used as a control. Subsequent experiments were performed with cells from bulk cultures. D) Western blot analysis to assess the extent of caspase-8 knockdown by shRNA expression. α-tubulin was used as a loading control. E) Stably modified SGBS preadipocytes were stimulated with growth medium alone or with increasing doses of TRAIL. After 72 hours, [3H]-thymidine incorporation was measured. Data are expressed as means ± sem of 3 independent experiments performed. **P ≤ 0.01; ***P ≤ 0.001 compared with growth medium alone.
Figure 6.
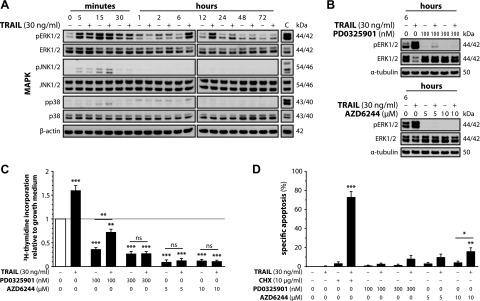
TRAIL-mediated activation of ERK1/2 is responsible for the induction of proliferation. A) SGBS preadipocytes were stimulated with growth medium alone or with TRAIL. At the indicated time points, proteins were isolated and subjected to Western blot analysis to assess the activation state of the ERK1/2, p38, and JNK pathways. β-actin was used as a loading control. “C” indicates a positive control. One representative of 3 independent experiments performed is shown. B, C) SGBS preadipocytes were stimulated with growth medium alone or with TRAIL in the absence or presence of different doses of PD0325901 or AZD6244. B) At the indicated time points, proteins were isolated and subjected to Western blot analysis to assess the inhibition of ERK1/2 activation by PD0325901 or AZD6244. α-tubulin was used as a loading control. One representative of 3 independent experiments performed is shown. C) After 72 hours, [3H]-thymidine incorporation was measured. Data are expressed as means ± sem of 3 independent experiments performed. **P ≤ 0.01; ***P ≤ 0.001 compared with growth medium alone (directly above the bars) or comparing the indicated treatments (above the connecting line). D) SGBS preadipocytes were stimulated with growth medium alone or with TRAIL in the absence or presence of cycloheximide (CHX) or different doses of PD0325901 or AZD6244. After 48 hours, apoptotic cell death was measured by flow cytometry and specific apoptosis was calculated. Data are expressed as means ± sem of 3 independent experiments performed. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001 compared with growth medium alone (directly above the bars) or comparing the indicated treatments (above the connecting line).
RESULTS
To study the impact of TRAIL on preadipocyte proliferation, we took advantage of the human SGBS cell strain. These cells are neither transformed nor immortalized and behave functionally like human primary stromal-vascular cells isolated from adipose tissue (18, 23). They are characterized by a high capacity for adipogenic differentiation and yield adipocytes that closely mimic ex vivo differentiated human primary adipocytes (18, 23). Therefore, the SGBS cell strain represents a useful tool to study both preadipocyte and adipocyte biology.
TRAIL induces human preadipocyte proliferation
SGBS preadipocytes were treated with increasing doses of human recombinant TRAIL and their proliferative behavior was studied. Utilizing microscopic cell counting, a time- and dose-dependent increase in the number of cells adherent to the cell culture dish was observed upon treatment with TRAIL (Fig. 1A). As estimated by [3H]-thymidine incorporation, already at concentrations as low as 1 or 3 ng/ml, TRAIL induced a slight increase in proliferation (Fig. 1B). A maximal increase in proliferation by ∼57% was detected at 30 ng/ml TRAIL. The mitogenic effect of this death ligand was further confirmed by a nuclear staining, which clearly indicated higher cell numbers in TRAIL-treated cultures (Fig. 1C), and by flow cytometry of CFSE-labeled and TRAIL-treated cells (Fig. 1D). TRAIL-induced proliferation was dependent on TRAIL binding to its cognate receptors as addition of a human recombinant TRAIL-R2/Fc construct completely abolished its proliferative effect (Supplemental Fig. 1A). Importantly, TRAIL exerted a comparable or even stronger mitogenic function in human primary stromal-vascular cells isolated from white adipose tissue samples of subjects undergoing plastic surgery (Fig. 1E). Moreover, treatment of SGBS preadipocytes with agonistic antibodies specific for either TRAIL-R1 (mapatumumab) or TRAIL-R2 (lexatumumab) resulted in a proliferative response comparable to treatment with the recombinant protein (Fig. 1F). Lexatumumab seemed more efficient than mapatumumab, which may reflect the higher expression levels of TRAIL-R2 compared with TRAIL-R1 in human preadipocytes (14, 24).
Of note, no significant induction of apoptosis was observed under the chosen experimental conditions (Fig. 2A, B). After 48 hours, less than 6% of cells had undergone apoptosis when treated with 10 ng/ml TRAIL (Fig. 2B). Only upon coincubation with the protein biosynthesis inhibitor cycloheximide was a robust sensitization for cell death, increasing apoptosis to over 82%, detected (Fig. 2A, B). This confirms earlier studies of our group (14, 20, 24) and is also in line with the observation that TRAIL induces apoptosis favorably in cancer cells, sparing nonmalignant cells (11).
Death ligands (TRAIL, FasL, and TNF-α) induce human preadipocyte proliferation in an extent comparable to IGF-1
We then wanted to know whether the proliferative effect of TRAIL also extends to other death ligands of the TNF-α family. Interestingly, both Fas ligand (FasL) and TNF-α stimulated proliferation in SGBS preadipocytes with FasL being comparable to TRAIL, but TNFα even being more efficient (maximal increases by ∼57% for TRAIL, ∼53% for FasL, and ∼107% for TNF-α at 30 ng/ml, respectively) (Fig. 3A). These data suggest that death ligands in general are important and potent mitogenic factors in preadipocytes. This is further supported by the finding that IGF-1, a well-known mitogen in many cell types (25, 26), exerted a proliferative response in a comparable range to that of the death ligands used in this study (maximal increase by ∼51% at 300 ng/ml IGF-1) (Fig. 3B).
TRAIL triggers caspase activation, though caspase activity and presence are not required for its proliferative effect
In canonical signaling, the TRAIL-induced cleavage and activation of caspases constitutes a central event (5). From earlier studies of our group, we knew that TRAIL stimulation triggers caspase cleavage in mature adipocytes without inducing apoptosis (14). Therefore, we studied the occurrence of caspase activation in SGBS preadipocytes under the experimental conditions used in this study. Strikingly, in response to 30 ng/ml TRAIL, a rapid cleavage of caspase-8 was observed with the intermediate p43/41 fragment detectable after only 5–10 minutes and the active p18 fragment detectable after 30 minutes (Fig. 4A). Furthermore, cleavage of caspase-3 to the active p17 fragment was evident after 30 minutes and peaked after 2 hours (Fig. 4A). No difference was observed between control and TRAIL-treated cells concerning the activation state of the caspases 10, 6, 2, and 1 (data not shown). To probe the role of caspase activity in the proliferative effect of TRAIL, the pan-caspase inhibitor zVAD.fmk was used. A concentration of 20 µM was sufficient to completely block the activation of caspase-8 and -3 after 2 hours (Fig. 4B). However, the inhibition of caspase activity had no significant impact on the proliferative effect of TRAIL (increase by ∼67% for TRAIL alone compared with ∼45% for TRAIL with zVAD.fmk) (Fig. 4C). To corroborate this finding, caspase-8-deficient SGBS preadipocytes were generated using a lentiviral-mediated shRNA approach. Two different constructs targeting caspase-8 (shC8.1 and shC8.2) were designed, both of them causing a knockdown of caspase-8 protein expression by ∼80% compared with cells expressing a hyper-random sequence (shHRS) without any known target, which was used as negative control (Fig. 4D). Both the control cells and the caspase-8 knockdown cells displayed a robust proliferative response upon TRAIL treatment, and no significant difference was detectable between the 3 used cell types (Fig. 4E). This set of data demonstrates that caspases are not involved in mediating the mitogenic effect of TRAIL on human preadipocytes.
TRAIL does not alter the activation state of the Akt and NF-κB cascades
By noncanonical signaling, TRAIL can activate diverse cellular cascades including the Akt, NF-κB, and mitogen-activated protein kinase (MAPK, i.e., p38, JNK, and ERK1/2) pathways (10). To study the activation of these kinases, SGBS preadipocytes were stimulated with 30 ng/ml TRAIL for several time points and Western blot analysis was carried out (Fig. 5A). In control as well as TRAIL-treated cells, a phosphorylation of Akt was detectable from 5 minutes to 2 hours with no difference between treatments. As a surrogate of NF-κB activation, the abundance of phosphorylated and nonphosphorylated IκBα was analyzed. Although a positive control (macrophage-conditioned medium) induced a phosphorylation and subsequent degradation of IκBα, we detected only weak and inconclusive phosphorylation signals upon treatment with TRAIL (Fig. 5A). We therefore decided to apply additional, more specific methods to study NF-κB activation. Immunofluorescence of the NF-κB subunit p65 revealed a clear translocation of p65 to the nucleus upon treatment with 30 ng/ml TNF-α, whereas upon treatment with 30 ng/ml TRAIL p65 remained sequestered in the cytoplasm (Fig. 5B). Likewise, in EMSA, a treatment with 30 ng/ml TNF-α robustly triggered NF-κB transcriptional activity in contrast to a treatment with 30 ng/ml TRAIL, which did not evoke any signal (Fig. 5C). This set of data demonstrates that neither Akt nor NF-κB play a causal role in mediating the effect of TRAIL on human preadipocyte proliferation.
Figure 5.
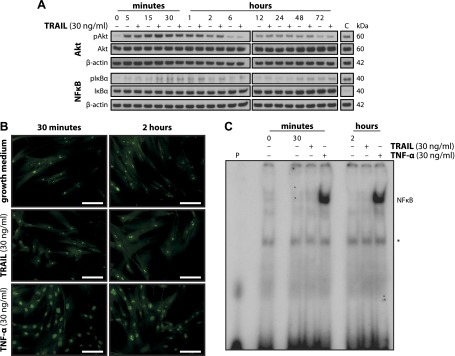
TRAIL does not alter Akt or NF-κB signaling. A) SGBS preadipocytes were stimulated with growth medium alone or with TRAIL. At the indicated time points, proteins were isolated and subjected to Western blot analysis to assess the activation state of the Akt and NF-κB pathways. β-actin was used as a loading control. “C” indicates a positive control. One representative of 3 independent experiments performed is shown. B, C) SGBS preadipocytes were stimulated with growth medium alone or with either TRAIL or TNF-α. B) At the indicated time points, NF-κB p65 immunofluorescence staining was performed. One representative of 3 independent experiments performed is shown. Scale bars, 100 µm. C) At the indicated time points, nuclear proteins were isolated and subjected to EMSA analysis to assess the transcriptional activity of NF-κB. “P” indicates a control for the probe alone. Asterisk indicates nonspecific signals. One representative of 3 independent experiments performed is shown.
TRAIL does not lead to phosphorylation of p38 and JNK, but activates the ERK1/2 cascade, which proves indispensable for the induction of proliferation
We then continued to investigate the activation of MAPKs in response to TRAIL treatment. Western blot analysis revealed hardly any activation of p38 and a weak phosphorylation of JNK after 5 to 15 minutes, which was comparable between control and TRAIL-treated cells (Fig. 6A). Also for ERK1/2, a similar initial wave of comparable activation was observed under both conditions, which is most likely caused by the media change at the beginning of the experiment. Importantly though, we detected a strong, delayed, and sustained phosphorylation of ERK1/2 after 6 hours, which persisted up to 24 hours. The activation of the ERK1/2 cascade upon TRAIL treatment was completely blocked using a TRAIL-R2/Fc construct (Supplemental Fig. 1B). To define the role of the observed ERK1/2 activation for the mitogenic effect of TRAIL, we decided to block the ERK1/2 pathway. To do so, 2 small molecule inhibitors of MEK1/2, the specific upstream kinases of ERK1/2, PD0325901, and AZD6244 (selumetinib), were chosen. For both these inhibitors, clinical studies exploring their anticancer potential are currently in progress (clinicaltrials.gov). In SGBS preadipocytes, not only the basal but also the TRAIL-induced activation of ERK1/2 was completely abolished by both inhibitors at concentrations of 300 nM for PD0325901 as well as 5 and 10 µM for AZD6244 (Fig. 6B). Moreover, both PD0325901 and AZD6244 robustly inhibited the basal proliferation and also the TRAIL-induced proliferation of SGBS preadipocytes (Fig. 6C). Of note, a significant increase in [3H]-thymidine incorporation in response to TRAIL stimulation was observed at 100 nM PD0325901, a concentration where the TRAIL-induced phosphorylation of ERK1/2 was not fully blocked by the inhibitor. Importantly, no significant increase in apoptosis was observed upon treatment with TRAIL in presence of 100 and 300 nM PD0325901 as well as 5 µM AZD6244 (Fig. 6D). This set of experiments underlines the general importance of the ERK1/2 signaling pathway for human preadipocyte proliferation and suggests that ERK1/2 mediates the proliferative effect of TRAIL.
DISCUSSION
We identified TRAIL as a potent mitogen in human preadipocytes. We initially started to explore the effect of TRAIL on proliferation when we observed an increase in the viability of SGBS preadipocytes upon treatment with TRAIL in a study, which primarily focused on the apoptotic function of the death ligand in human preadipocytes and adipocytes (24). In the present study, TRAIL was found to evoke a pronounced increase in proliferation in both the SGBS cell strain as well as in human primary stromal-vascular cells isolated from white adipose tissue. A proliferative effect was also observed for FasL and TNF-α, suggesting that death ligands in general are important and potent mitogenic factors in human preadipocytes. FasL and TNF-α were identified as proliferative factors in human mesenchymal stem cells and adipocyte precursor cells, respectively, earlier (27–29). In contrast, this to the best of our knowledge is the first study to demonstrate that TRAIL can increase preadipocyte numbers by stimulating their proliferation. The relevance of this finding is underlined by the observation that the proliferative effect of TRAIL was comparable to IGF-1. The latter is a well-known mitogen in many types of malignant and nonmalignant cells (25, 26) and according to Hausman et al. constitutes the most important trigger of proliferation in human preadipocytes (30).
After the discovery of TRAIL as an apoptosis-inducing member of the TNF superfamily in 1995/96 (31, 32), the TRAIL/TRAIL-R system rapidly became an attractive target for anticancer treatment strategies, because it seemed that TRAIL selectively only induced cell death in transformed cells (11). However, almost 20 years of research succeeding its initial discovery revealed that the initial view of TRAIL as a mere cell death-inducing agent was far too simplistic (10).
In the clinical trials conducted so far, the vast majority of cancers were found to be resistant to cell death induction by targeting the TRAIL/TRAIL receptor system (11). Quite on the contrary, cancer cells not succumbing to cell death frequently show a proliferative, prosurvival, proinflammatory, promigratory, and proinvasive outcome upon TRAIL treatment due to activation of noncanonical signaling pathways (10). Besides that, the occurrence of both apoptotic and nonapoptotic effects of TRAIL has been discovered in benign cell types (e.g., in the immune, hematopoietic, and vascular systems) (33–35). In the vascular system, TRAIL was identified as a mitogenic factor in human primary vascular smooth muscle cells and vascular endothelial cells (36–39).
Our group recently added human adipocytes to the list of nonmalignant cell types regulated by TRAIL in a manner not involving cell death induction (14). In adipocytes, TRAIL impacts key metabolic functions by a rapid activation of caspases resulting in the cleavage and inactivation of PPARγ (14). In the present study, caspase-8 and caspase-3 cleavage was also observed in preadipocytes. However, neither the inhibition of caspase activity with a pan-caspase inhibitor nor the lentiviral-mediated knockdown of caspase-8 expression had any significant impact on TRAIL-induced proliferation. This clearly demonstrates that the mitogenic effect of TRAIL is independent of caspase activity and presence.
TRAIL did not significantly alter the activation pattern of Akt, NF-κB, and the MAPKs p38 and JNK. However, we detected a delayed and sustained activation of ERK1/2 upon treatment with TRAIL. TRAIL-stimulated proliferation via ERK1/2 activation was reported in malignant small cell lung cancer (40) and glioma cells (41), pseudomalignant rheumatoid arthritis synovial fibroblasts (42, 43), as well as nonmalignant vascular smooth muscle and vascular endothelial cells (36, 37). In these studies, ERK1/2 activation typically ensued within 5–15 minutes and lasted for ∼30 minutes (36, 37, 40–43). We also observed an early wave of ERK1/2 phosphorylation within 5 minutes to 2 hours, which was seen upon both control and TRAIL treatment, and is most likely attributable to the addition of fresh serum-supplemented medium at the beginning of the experiment. The fact that a strong ERK1/2 phosphorylation upon TRAIL treatment was detected after 6 hours that lasted for up to 24 hours raised the question whether this effect is secondary and caused by TRAIL-induced changes in protein production and/or secretion resulting in auto-/paracrine stimulation. However, experiments performed with brefeldin A to block protein secretion did not reveal any alterations in the phosphorylation of ERK1/2 by TRAIL (data not shown) ruling out this possibility.
The ERK1/2 pathway is a central regulator of growth and proliferation in many cell types. Likewise, blocking ERK1/2 phosphorylation by inhibition of the specific upstream kinases MEK1/2 using the small molecule inhibitors PD0325901 and AZD6244 (selumetinib) strongly reduced the basal, serum-induced proliferation of human preadipocytes. Importantly, blocking the ERK1/2 pathway did not sensitize the cells for TRAIL-induced apoptosis. This highlights that ERK1/2 activity is of importance for the proliferation but not the survival of human preadipocytes and is supported by previous findings from our group, demonstrating the Akt pathway to constitute the main anti-apoptotic signaling route in human preadipocytes (20). Most importantly, though, blocking EKR1/2 phosphorylation completely abrogated TRAIL-induced proliferation demonstrating that it is the activation of ERK1/2 that mediates the proliferative effect of TRAIL.
We also compared the TRAIL-induced intracellular signaling cascades to FasL and TNF-α stimulated signaling. The 3 ligands displayed distinct activation patterns of signaling cascades in human preadipocytes (Supplemental Fig. 2). This is interesting since the TRAIL/TRAIL-R, FasL/Fas, and TNF-α/TNF-R1 systems do not only share structural features but also possess comparable signaling capacities (5, 10).
Our study has several important implications. We propose that TRAIL represents an important regulator of adipose tissue homeostasis. The most important determinants of adipose tissue mass are the volume and number of present adipocytes. We have shown earlier that TRAIL inhibits the insulin-stimulated glucose uptake and de novo lipogenesis in human adipocytes thereby regulating adipocyte volume (14). Moreover, we have found that TRAIL interferes with the adipogenic differentiation of human preadipocytes thus also regulating adipocyte numbers (44). The current study elucidates that TRAIL stimulates preadipocyte proliferation and might thereby increase the number of adipose tissue resident precursor cells. Adipose tissue is a very dynamic organ. Its capability to expand contributes considerably to metabolic flexibility and thereby systemically to metabolic health (2, 3). Healthy adipose tissue expansion is defined by the controlled recruitment of precursor cells for adipogenic differentiation along with the establishment of appropriate vascularization (2, 3).
We suggest that TRAIL exerts differential effects in adipose tissue in the lean and the obese state. It is rather probable that TRAIL expression is spatially restricted within adipose tissue in the lean state. The stem and progenitor cell compartment of adipose tissue mostly resides in close association with resident vascular, especially microvascular structures (45). The cells potentially contributing to the local expression of TRAIL in these structures are vascular smooth muscle cells and vascular endothelial cells. Although the expression of TRAIL by vascular smooth muscle cells is well documented (35), its expression by vascular endothelial cells is still a subject of debate (46, 47). Strikingly, SGBS preadipocytes as well as human primary stromal-vascular cells express TRAIL (data not shown), thus also implying that the stem and progenitor cells in adipose tissue add to the local presence of TRAIL at sites of their accumulation. By regulating proliferation and differentiation, the local expression of TRAIL in vascular and microvascular structures might contribute decisively to the maintenance of the adipose stem and progenitor cell compartment. Moreover, TRAIL might sustain the structure of the adipose tissue stem cell niche by regulating proliferation, survival, and migration of the involved vascular smooth muscle and endothelial cells (36–39). Thus, in the lean state, TRAIL likely contributes to the preservation of adipose tissue function primarily by affecting the adipose tissue stem cell niche. Detailed studies focusing on the expression of TRAIL by the different types of cells found in adipose tissue will be necessary to explore this theory.
By contrast, it is well conceivable that TRAIL expression is spatially unrestricted within adipose tissue in the obese state. Upon excessive pathologic expansion, adipose tissue gets infiltrated by different types of immune cells, especially classically activated proinflammatory M1 macrophages, which are known to express high levels of TRAIL (48). In agreement with this, TRAIL mRNA levels are increased in adipose tissue of obese mice (14). Under these conditions, high levels of TRAIL within adipose tissue might not only impact the pool of resident precursor cells, but also exert additional functions, such as influencing the metabolic properties of adipocytes (14).
Interestingly, the serum levels of TRAIL correlate positively with numerous obesity-related parameters such as the BMI, waist circumference, total body fat, serum triglyceride, and cholesterol levels (13, 49, 50). Based on our in vitro data, which showed that TRAIL inhibited insulin-stimulated metabolic processes in human adipocytes (14), one might conclude that elevated TRAIL levels lead not only to adipose tissue but also to systemic insulin resistance. However, recent data from animal experiments gave contrary results (51, 52). The intraperitoneal application of recombinant TRAIL to mice decreased the severity of adiposity, glucose intolerance, and systemic inflammation upon high-fat feeding (52). Similarly, a systemic lack of TRAIL increased the severity of the glucose intolerance and systemic inflammation in TRAIL knockout mice on a high-fat diet (51). In both cases, the observed beneficial effects of circulating TRAIL might primarily be explained by its immunosuppressive and immunoregulatory functions that counteract inflammation (33). Supporting the latter notion, the immunosuppressive and immunoregulatory functions of TRAIL, especially its effects on T cells but likely also on macrophages, also protect from the development of type 1 diabetes mellitus in several mouse models of this disease (53–55). The distinct impact of a local versus a systemic elevation of TRAIL levels, however, will have to be elucidated in cell type-specific knockout mice.
Taken together, the present study identified TRAIL as a potent mitogen in human preadipocytes. TRAIL stimulated a delayed and sustained phosphorylation of ERK1/2, which was responsible for mediating the proliferative response. An increase in preadipocyte proliferation might contribute to the maintenance of the adipose tissue-resident precursor pool, thereby supporting the formation of new adipocytes and healthy expansion of adipose tissue. In conclusion, manipulating the activity of the TRAIL/TRAIL-R system either in an adipose tissue-selective or systemic manner might thus provide a novel approach to restore adipose tissue function in pathophysiological states.
Supplementary Material
Acknowledgments
The authors thank Alexandra Killian for her outstanding technical assistance and Mike Andrew Westhoff for helpful discussions throughout this project. J.B.F. and V.Z. were funded by the International Graduate School in Molecular Medicine Ulm (IGradU).
Glossary
- BMI
body mass index
- cFLIP
cellular FLICE-like inhibitory protein
- DPBS
Dulbecco’s phosphate-buffered saline
- FADD
Fas-associated death domain
- PPARγ
peroxisome proliferator-activated receptor γ
- SGBS
Simpson-Golabi-Behmel syndrome
- shRNA
small hairpin RNA
- TRAIL
TNF-related apoptosis-inducing ligand
- TRAIL-R1/2
TNF-related apoptosis-inducing ligand receptor 1/2
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Fischer-Posovszky P., Wabitsch M., Hochberg Z. (2007) Endocrinology of adipose tissue—an update. Horm. Metab. Res. 39, 314–321 [DOI] [PubMed] [Google Scholar]
- 2.Sun K., Kusminski C. M., Scherer P. E. (2011) Adipose tissue remodeling and obesity. J. Clin. Invest. 121, 2094–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosen E. D., Spiegelman B. M. (2014) What we talk about when we talk about fat. Cell 156, 20–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samocha-Bonet D., Dixit V. D., Kahn C. R., Leibel R. L., Lin X., Nieuwdorp M., Pietilainen K. H., Rabasa-Lhoret R., Roden M., Scherer P. E., Klein S., Ravussin E. (2014) Metabolically healthy and unhealthy obese—the 2013 Stock Conference report. Obes. Rev. 15, 697–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashkenazi A., Salvesen G. (2014) Regulated cell death: signaling and mechanisms. Annu. Rev. Cell Dev. Biol. 30, 337–356 [DOI] [PubMed] [Google Scholar]
- 6.Dickens L. S., Boyd R. S., Jukes-Jones R., Hughes M. A., Robinson G. L., Fairall L., Schwabe J. W., Cain K., Macfarlane M. (2012) A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol. Cell 47, 291–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schleich K., Warnken U., Fricker N., Ozturk S., Richter P., Kammerer K., Schnolzer M., Krammer P. H., Lavrik I. N. (2012) Stoichiometry of the CD95 death-inducing signaling complex: experimental and modeling evidence for a death effector domain chain model. Mol. Cell 47, 306–319 [DOI] [PubMed] [Google Scholar]
- 8.Varfolomeev E., Maecker H., Sharp D., Lawrence D., Renz M., Vucic D., Ashkenazi A. (2005) Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J. Biol. Chem. 280, 40599–40608 [DOI] [PubMed] [Google Scholar]
- 9.Jin Z., El-Deiry W. S. (2006) Distinct signaling pathways in TRAIL- versus tumor necrosis factor-induced apoptosis. Mol. Cell. Biol. 26, 8136–8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Azijli K., Weyhenmeyer B., Peters G. J., de Jong S., Kruyt F. A. (2013) Non-canonical kinase signaling by the death ligand TRAIL in cancer cells: discord in the death receptor family. Cell Death Differ. 20, 858–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lemke J., von Karstedt S., Zinngrebe J., Walczak H. (2014) Getting TRAIL back on track for cancer therapy. Cell Death Differ. 21, 1350–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spierings D. C., de Vries E. G., Vellenga E., van den Heuvel F. A., Koornstra J. J., Wesseling J., Hollema H., de Jong S. (2004) Tissue distribution of the death ligand TRAIL and its receptors. J. Histochem. Cytochem. 52, 821–831 [DOI] [PubMed] [Google Scholar]
- 13.Choi J. W., Song J. S., Pai S. H. (2004) Associations of serum TRAIL concentrations, anthropometric variables, and serum lipid parameters in healthy adults. Ann. Clin. Lab. Sci. 34, 400–404 [PubMed] [Google Scholar]
- 14.Keuper M., Wernstedt Asterholm I., Scherer P. E., Westhoff M. A., Moller P., Debatin K. M., Strauss G., Wabitsch M., Fischer-Posovszky P. (2013) TRAIL (TNF-related apoptosis-inducing ligand) regulates adipocyte metabolism by caspase-mediated cleavage of PPARgamma. Cell Death Dis. 4, e474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hotamisligil G. S., Shargill N. S., Spiegelman B. M. (1993) Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259, 87–91 [DOI] [PubMed] [Google Scholar]
- 16.Wueest S., Rapold R. A., Schumann D. M., Rytka J. M., Schildknecht A., Nov O., Chervonsky A. V., Rudich A., Schoenle E. J., Donath M. Y., Konrad D. (2010) Deletion of Fas in adipocytes relieves adipose tissue inflammation and hepatic manifestations of obesity in mice. J. Clin. Invest. 120, 191–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hauner H., Skurk T., Wabitsch M. (2001) Cultures of human adipose precursor cells. Methods Mol. Biol. 155, 239–247 [DOI] [PubMed] [Google Scholar]
- 18.Wabitsch M., Brenner R. E., Melzner I., Braun M., Moller P., Heinze E., Debatin K. M., Hauner H. (2001) Characterization of a human preadipocyte cell strain with high capacity for adipose differentiation. Int. J. Obes. Relat. Metab. Disord. 25, 8–15 [DOI] [PubMed] [Google Scholar]
- 19.Nicoletti I., Migliorati G., Pagliacci M. C., Grignani F., Riccardi C. (1991) A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 139, 271–279 [DOI] [PubMed] [Google Scholar]
- 20.Fischer-Posovszky P., Tornqvist H., Debatin K. M., Wabitsch M. (2004) Inhibition of death-receptor mediated apoptosis in human adipocytes by the insulin-like growth factor I (IGF-I)/IGF-I receptor autocrine circuit. Endocrinology 145, 1849–1859 [DOI] [PubMed] [Google Scholar]
- 21.Andrews N. C., Faller D. V. (1991) A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 19, 2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kasperczyk H., La Ferla-Bruhl K., Westhoff M. A., Behrend L., Zwacka R. M., Debatin K. M., Fulda S. (2005) Betulinic acid as new activator of NF-kappaB: molecular mechanisms and implications for cancer therapy. Oncogene 24, 6945–6956 [DOI] [PubMed] [Google Scholar]
- 23.Fischer-Posovszky P., Newell F. S., Wabitsch M., Tornqvist H. E. (2008) Human SGBS cells—a unique tool for studies of human fat cell biology. Obes. Facts 1, 184–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mader I., Wabitsch M., Debatin K. M., Fischer-Posovszky P., and Fulda S. (2010) Identification of a novel proapoptotic function of resveratrol in fat cells: SIRT1-independent sensitization to TRAIL-induced apoptosis. FASEB J. 24, 1997–2009 [DOI] [PubMed] [Google Scholar]
- 25.Jones J. I., Clemmons D. R. (1995) Insulin-like growth factors and their binding proteins: biological actions. Endocr. Rev. 16, 3–34 [DOI] [PubMed] [Google Scholar]
- 26.Gallagher E. J., LeRoith D. (2010) The proliferating role of insulin and insulin-like growth factors in cancer. Trends in endocrinology and metabolism. Trends Endocrinol. Metab. 21, 610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Isakson P., Hammarstedt A., Gustafson B., Smith U. (2009) Impaired preadipocyte differentiation in human abdominal obesity: role of Wnt, tumor necrosis factor-alpha, and inflammation. Diabetes 58, 1550–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rippo M. R., Babini L., Prattichizzo F., Graciotti L., Fulgenzi G., Tomassoni Ardori F., Olivieri F., Borghetti G., Cinti S., Poloni A., Fazioli F., Procopio A. D. (2013) Low FasL levels promote proliferation of human bone marrow-derived mesenchymal stem cells, higher levels inhibit their differentiation into adipocytes. Cell Death Dis. 4, e594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kras K. M., Hausman D. B., Martin R. J. (2000) Tumor necrosis factor-alpha stimulates cell proliferation in adipose tissue-derived stromal-vascular cell culture: promotion of adipose tissue expansion by paracrine growth factors. Obes. Res. 8, 186–193 [DOI] [PubMed] [Google Scholar]
- 30.Hausman D. B., DiGirolamo M., Bartness T. J., Hausman G. J., Martin R. J. (2001) The biology of white adipocyte proliferation. Obes. Rev. 2, 239–254 [DOI] [PubMed] [Google Scholar]
- 31.Wiley S. R., Schooley K., Smolak P. J., Din W. S., Huang C. P., Nicholl J. K., Sutherland G. R., Smith T. D., Rauch C., Smith C. A., et al. (1995) Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 3, 673–682 [DOI] [PubMed] [Google Scholar]
- 32.Pitti R. M., Marsters S. A., Ruppert S., Donahue C. J., Moore A., Ashkenazi A. (1996) Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 271, 12687–12690 [DOI] [PubMed] [Google Scholar]
- 33.Falschlehner C., Schaefer U., Walczak H. (2009) Following TRAIL's path in the immune system. Immunology 127, 145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zauli G., Secchiero P. (2006) The role of the TRAIL/TRAIL receptors system in hematopoiesis and endothelial cell biology. Cytokine Growth Factor Rev. 17, 245–257 [DOI] [PubMed] [Google Scholar]
- 35.Kavurma M. M., Bennett M. R. (2008) Expression, regulation and function of trail in atherosclerosis. Biochem. Pharmacol. 75, 1441–1450 [DOI] [PubMed] [Google Scholar]
- 36.Secchiero P., Gonelli A., Carnevale E., Milani D., Pandolfi A., Zella D., Zauli G. (2003) TRAIL promotes the survival and proliferation of primary human vascular endothelial cells by activating the Akt and ERK pathways. Circulation 107, 2250–2256 [DOI] [PubMed] [Google Scholar]
- 37.Secchiero P., Zerbinati C., Rimondi E., Corallini F., Milani D., Grill V., Forti G., Capitani S., Zauli G. (2004) TRAIL promotes the survival, migration and proliferation of vascular smooth muscle cells. Cell. Mol. Life Sci. 61, 1965–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kavurma M. M., Schoppet M., Bobryshev Y. V., Khachigian L. M., Bennett M. R. (2008) TRAIL stimulates proliferation of vascular smooth muscle cells via activation of NF-kappaB and induction of insulin-like growth factor-1 receptor. J. Biol. Chem. 283, 7754–7762 [DOI] [PubMed] [Google Scholar]
- 39.Chan J., Prado-Lourenco L., Khachigian L. M., Bennett M. R., Di Bartolo B. A., Kavurma M. M. (2010) TRAIL promotes VSMC proliferation and neointima formation in a FGF-2-, Sp1 phosphorylation-, and NFkappaB-dependent manner. Circ. Res. 106, 1061–1071 [DOI] [PubMed] [Google Scholar]
- 40.Belyanskaya L. L., Ziogas A., Hopkins-Donaldson S., Kurtz S., Simon H. U., Stahel R., Zangemeister-Wittke U. (2008) TRAIL-induced survival and proliferation of SCLC cells is mediated by ERK and dependent on TRAIL-R2/DR5 expression in the absence of caspase-8. Lung Cancer 60, 355–365 [DOI] [PubMed] [Google Scholar]
- 41.Vilimanovich U., Bumbasirevic V. (2008) TRAIL induces proliferation of human glioma cells by c-FLIPL-mediated activation of ERK1/2. Cell. Mol. Life Sci. 65, 814–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morel J., Audo R., Hahne M., Combe B. (2005) Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces rheumatoid arthritis synovial fibroblast proliferation through mitogen-activated protein kinases and phosphatidylinositol 3-kinase/Akt. J. Biol. Chem. 280, 15709–15718 [DOI] [PubMed] [Google Scholar]
- 43.Audo R., Combe B., Coulet B., Morel J., Hahne M. (2009) The pleiotropic effect of TRAIL on tumor-like synovial fibroblasts from rheumatoid arthritis patients is mediated by caspases. Cell Death Differ. 16, 1227–1237 [DOI] [PubMed] [Google Scholar]
- 44.Keuper M., Asterholm I., Scherer P., Wolf A. M., Knippschild U., Wabitsch M., Fischer-Posovszky P. (2011) TRAIL (TNF related apoptosis inducing ligand) affects adipogenic differentiation and adipocyte metabolism. Horm. Res. Paediatr. 76, 20–4821912153 [Google Scholar]
- 45.Baer P. C. (2014) Adipose-derived mesenchymal stromal/stem cells: An update on their phenotype in vivo and in vitro. World J. Stem Cells 6, 256-265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Secchiero P., Zauli G. (2008) The puzzling role of TRAIL in endothelial cell biology. Arterioscler. Thromb. Vasc. Biol. 28, e4; author reply e5–e6 [DOI] [PubMed] [Google Scholar]
- 47.O’Brien L. A., Richardson M. A., Berg D. T., Gerlitz B., Gupta A., Grinnell B. W. (2008) The puzzling role of TRAIL in endothelial cell biology. Arterioscler. Thromb. Vasc. Biol. 28, e5–e6 [DOI] [PubMed] [Google Scholar]
- 48.Martinez F. O., Gordon S., Locati M., Mantovani A. (2006) Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J. Immunol. 177, 7303–7311 [DOI] [PubMed] [Google Scholar]
- 49.Ashley D. T., O'Sullivan E. P., Davenport C., Devlin N., Crowley R. K., McCaffrey N., Moyna N. M., Smith D., O'Gorman D. J. (2011) Similar to adiponectin, serum levels of osteoprotegerin are associated with obesity in healthy subjects. Metabolism 60, 994–1000 [DOI] [PubMed] [Google Scholar]
- 50.Brombo G., Volpato S., Secchiero P., Passaro A., Bosi C., Zuliani G., Zauli G. (2013) Association of soluble Tumor necrosis factor-Related Apoptosis-Inducing Ligand (TRAIL) with central adiposity and low-density lipoprotein cholesterol. PLoS ONE 8, e58225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Di Bartolo B. A., Chan J., Bennett M. R., Cartland S., Bao S., Tuch B. E., Kavurma M. M. (2011) TNF-related apoptosis-inducing ligand (TRAIL) protects against diabetes and atherosclerosis in Apoe (-)/(-) mice. Diabetologia 54, 3157–3167 [DOI] [PubMed] [Google Scholar]
- 52.Bernardi S., Zauli G., Tikellis C., Candido R., Fabris B., Secchiero P., Cooper M. E., Thomas M. C. (2012) TNF-related apoptosis-inducing ligand significantly attenuates metabolic abnormalities in high-fat-fed mice reducing adiposity and systemic inflammation. Clin. Sci. 123, 547–555 [DOI] [PubMed] [Google Scholar]
- 53.Lamhamedi-Cherradi S. E., Zheng S. J., Maguschak K. A., Peschon J., Chen Y. H. (2003) Defective thymocyte apoptosis and accelerated autoimmune diseases in TRAIL−/− mice. Nat. Immunol. 4, 255–260 [DOI] [PubMed] [Google Scholar]
- 54.Mi Q. S., Ly D., Lamhamedi-Cherradi S. E., Salojin K. V., Zhou L., Grattan M., Meagher C., Zucker P., Chen Y. H., Nagle J., Taub D., Delovitch T. L. (2003) Blockade of tumor necrosis factor-related apoptosis-inducing ligand exacerbates type 1 diabetes in NOD mice. Diabetes 52, 1967–1975 [DOI] [PubMed] [Google Scholar]
- 55.Zauli G., Toffoli B., di Iasio M. G., Celeghini C., Fabris B., Secchiero P. (2010) Treatment with recombinant tumor necrosis factor-related apoptosis-inducing ligand alleviates the severity of streptozotocin-induced diabetes. Diabetes 59, 1261–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.