

Abstract
Drosophila melanogaster as a cosmopolitan species has successfully adapted to a wide range of different environments. Variation in temperature is one important environmental factor that influences the distribution of species in nature. In particular for insects, which are mostly ectotherms, ambient temperature plays a major role in their ability to colonize new habitats. Chromatin-based gene regulation is known to be sensitive to temperature. Ambient temperature leads to changes in the activation of genes regulated in this manner. One such regulatory system is the Polycomb group (PcG) whose target genes are more expressed at lower temperatures than at higher ones. Therefore, a greater range in ambient temperature in temperate environments may lead to greater variability (plasticity) in the expression of these genes. This might have detrimental effects, such that positive selection acts to lower the degree of the expression plasticity. We provide evidence for this process in a genomic region that harbors two PcG-regulated genes, polyhomeotic proximal (ph-p) and CG3835. We found a signature of positive selection in this gene region in European populations of D. melanogaster and investigated the region by means of reporter gene assays. The target of selection is located in the intergenic fragment between the two genes. It overlaps with the promoters of both genes and an experimentally validated Polycomb response element (PRE). This fragment harbors five sequence variants that are highly differentiated between European and African populations. The African alleles confer a temperature-induced plasticity in gene expression, which is typical for PcG-mediated gene regulation, whereas thermosensitivity is reduced for the European alleles.
Keywords: positive selection, gene regulation, environmental sensitivity, polycomb group
DROSOPHILA melanogaster is a species that has colonized all continents on Earth. Now a cosmopolitan species with a worldwide distribution, it started its global spread from its sub-Saharan ancestral range relatively recently. Its origin is thought to be in southern-central Africa from which it first expanded through Africa and finally reached the Eurasian continent on the order of 10,000 years ago (David and Capy 1988; Lachaise and Silvain 2004; Li and Stephan 2006; Stephan and Li 2007; Pool et al. 2012). This settlement was accompanied by a severe population size bottleneck and involved a significant loss of genetic variation (Li and Stephan 2006; Stephan and Li 2007; Pool et al. 2012). The colonization of Europe and Asia from its original source population appears to be even more recent since European and Asian populations share a most recent common ancestor (MRCA) ∼5000 years ago (Laurent et al. 2011).
For insects, which are mostly ectotherms, differences in temperature are one of the most important environmental variables that influence the distribution of species in nature (Clarke 1996). In the temperate climate of Europe, the range of possible temperatures is probably one of the major challenges D. melanogaster was confronted with during colonization.
Chromatin-based gene regulation is known to be sensitive to temperature (Fauvarque and Dura 1993; Gibert et al. 2011). In the case of the Polycomb group (PcG)-mediated gene regulation, it is known that genes under the control of this group of proteins have a higher transcriptional output when flies are reared or held at lower temperatures than at higher ones (Fauvarque and Dura 1993; Chan et al. 1994; Zink and Paro 1995; Bantignies et al. 2003; Gibert et al. 2011). The PcG and another group of proteins, the Trithorax group (TrxG), act antagonistically to epigenetically maintain repressed and activated transcription states, respectively. They act through cis-regulatory DNA elements called Polycomb response elements (PREs), which recruit the proteins of the two groups to their target genes. PREs regulate their target genes in combination with other regulatory DNA sequences (i.e., enhancers) in a cell- or tissue-specific manner. This interplay modifies the expression of PcG-regulated genes in such a way that enhancers initially determine the level of transcriptional output, which is subsequently epigenetically maintained by PREs (Schwartz et al. 2010; Kassis and Brown 2013; Steffen and Ringrose 2014).
Several recent studies (Harr et al. 2002; Levine and Begun 2008; Gibert et al. 2011) explored the question of whether the temperature-induced expression plasticity of PcG-regulated genes may have been detrimental to D. melanogaster while settling in temperate environments. These studies suggest that adaptation included selection acting to buffer this thermosensitive process in temperate populations.
In this study, we provide evidence for selection acting in cis to buffer the temperature-induced expression plasticity of PcG regulation in populations adapted to temperate environments. We carried out population genetic analyses to show that a DNA sequence region between the two PcG-regulated genes polyhomeotic proximal (ph-p) and CG3835 has been the target of a selective sweep in European populations of D. melanogaster. Furthermore, using transgenic reporter gene assays, we demonstrate that sequence variation in this 5-kb selected fragment mediates differences in gene expression between European and African sequence variants. Temperature-sensitive expression is observed in the case of the African alleles but not in the European ones. These results are consistent with positive selection favoring cis-regulatory polymorphisms that led to decreased thermosensitivity of gene expression in temperate populations.
Materials and Methods
Fly lines and sequence data
Assembled full genome sequences were taken from the Drosophila Population Genomics Project (DPGP) (http://www.dpgp.org), including those from 133 sub-Saharan African lines [among them the Zambian population sample (Siavonga) of 27 lines] and a French population sample (Lyon, 8 lines). We additionally analyzed a Dutch population from Leiden consisting of 10 lines and two Malaysian samples from Kuala Lumpur and Kota Kinabalu consisting of 7 and 16 lines, respectively. Full genomes for the Dutch and Malaysian samples were assembled following the approach of Pool et al. (2012) and are available at http://evol.bio.lmu.de/downloads. Nucleotides with known admixture or identity-by-descent according to Pool et al. (2012) were replaced with missing value labels in the analysis. The same was done for sites exhibiting heterozygosity since heterozygotes are not expected in genome data from haploid embryos (Pool et al. 2012). Additionally, 12 lines of the aforementioned Dutch population and 12 lines of one from Zimbabwe (Lake Kariba) were fully sequenced between positions 2,030,513 and 2,059,036 on the X chromosome (FlyBase release 5), applying the Sanger method (Sanger et al. 1977). This method was also used to sequence fragments containing the polymorphisms of interest in population samples from Siavonga, Zambia (10 lines); Munich, Germany (12 lines); Umea, Sweden (14 lines); and Kuala Lumpur, Malaysia (11 lines). All Sanger sequences were deposited in GenBank (accession nos. KR024038–KR024162).
Population genetic analysis
To analyze DNA sequence polymorphisms in a 73-kb region around the ph locus, sequences of the Zambia, Dutch, and French population samples generated by the DPGP were used. Nucleotide diversity was estimated in terms of π (Tajima 1989) and divergence was calculated against a D. simulans sequence (Hu et al. 2013). The composite-likelihood ratio (CLR) test of positive selection was performed, applying the software SweeD (Pavlidis et al. 2013). It computes the CLR between a selective sweep model and a neutral model based on the background genomic patterns of polymorphism (Kim and Stephan 2002). We ran the program on the complete X chromosome and calculated the significance threshold (95th quantile) by generating neutral coalescent simulations, using the demographic model of Laurent et al. (2011). To improve the power of the test statistic, the European sample was extended by adding the French population sample (Pavlidis et al. 2010, 2013) and two additional site classes of the site-frequency spectrum (SFS) consisting of sites that are monomorphic in the European sample and polymorphic in the Zambian one (Nielsen et al. 2005). Polarization was done against D. simulans (Hu et al. 2013).
Outlier analyses were performed using BayeScan version 2.1 (Foll and Gaggiotti 2008), a Bayesian method based on a logistic regression model that separates locus-specific effects of selection from population-specific effects of demography. FST coefficients (Beaumont and Balding 2004) are estimated and decomposed into a population-specific component (β) and a locus-specific one (α). Departure from neutrality at a given SNP locus is assumed when α is significantly different from zero. Positive values of α suggest positive directional selection, whereas negative α-values indicate balancing selection. BayeScan runs were carried out using default parameters for a 300-kb genomic window around the ph locus with sequences from seven European and African populations. These included samples from The Netherlands (10 lines), France (8 lines), Cameroon (10 lines), Gabon (9 lines), Ethiopia (8 lines), Rwanda (27 lines), and Zambia (27 lines).
Expression analysis in whole adult flies
Gene expression was analyzed in whole adult flies from the aforementioned population samples from The Netherlands and Zimbabwe. Flies were reared on a standard cornmeal–molasses medium at ∼28° and 18° with a 14/10-hr light/dark cycle. Expression was measured in 11 fly lines per population. For each line, RNA was extracted from five males and five females (aged 4–6 days). RNA extraction including DNase I digestion was performed using the MasterPure RNA Purification Kit (Epicentre, Madison, WI; http://www.epibio.com). RNA purity was assessed via the ratio of absorbances at 260 and 280 nm (A260/A280 > 1.8). It was then reverse transcribed into complementary DNA (cDNA), using random primers and SuperScript III Reverse Transcriptase (Invitrogen, Carlsbad, CA; http://www.lifetechnologies.com). RT-qPCR reactions were run with iQ SYBR Green Supermix (Bio-Rad, Hercules, CA; http://www.bio-rad.com) on a CFX96 real-time PCR cycler (Bio-Rad). Primers for target and reference genes were designed, applying the QuantPrime software (Arvidsson et al. 2008). Per fly line, two biological replicates were run in duplicates. No template controls (NTCs) were included to control contamination and primer specificity was confirmed by melting-curve analysis. Relative expression was calculated using the qBase relative quantification framework (Hellemans et al. 2007). Both reference genes (RpL32 and RpS20) were stably expressed across samples. This was assessed by calculating the coefficient of variation and the M stability parameter according to Hellesmans et al. (2007). Log-transformed normalized relative quantities were subjected to a paired t-test to test for statistically significant expression differences.
Reporter gene assays
The genomic region between ph-p and CG3835 reaching from 2,030,598 to 2,035,598 on the X chromosome in FlyBase release 5 (Pierre et al. 2014) was PCR amplified from one Dutch and one Zimbabwean strain (NL01 and ZK186, respectively), using the primers 5′-GCCACAGTCACAGCACTAAGT-3′ and 3′-CCTTTCATCCATAAGTCAGTG-5′. The PCR products were cloned directly into the “pCR4Blunt-TOPO” vector (Invitrogen). The insert was then excised as a HindIII/NotI fragment and cloned into the “placZ-2attB” integration vector (Bischof et al. 2007). The identity and orientation of the cloned fragments were confirmed by restriction analysis and sequencing. Integration vector DNA was purified with the QIAprep Spin Miniprep Kit (QIAGEN, Hilden, Germany; http://www.qiagen.com) and used for microinjection of early-stage embryos of the ΦX86Fb strain (attP site at cyological band 86Fb). This strain includes a stable source of ΦC31 integrase on the X chromosome. The integration site used was selected by the criteria of no binding of PcG/TrxG proteins and no occurrence of their specific histone marks within a window of ±5 kb around the site. Following microinjection, viable flies were crossed to a “white−” strain to remove the integrase and establish stable lines. Resulting offspring were screened for red eye color as a marker of successful transformants.
Reporter gene expression was measured in brains and midguts of third instar larvae via RT-qPCR. Flies containing one copy of the inserted construct were grown on a standard cornmeal–molasses medium at 28° and 17° with a 14/10-hr light/dark cycle. Five females and five males were allowed to mate and oviposit for 3 and 7 days at 28° and 17°, respectively. Tissue of the resulting progeny was dissected and immediately stored in RNAlater (QIAGEN). RNA extraction and RT-qPCR were performed as described above. Primer sequences for the lacZ reporter gene were taken from Zhang et al. (2013). For normalization the two aforementioned reference genes (RpL32 and RpS20) were used. Three biological replicates per construct in the particular tissue at the particular rearing temperature were run in triplicates. Negative controls included NTCs and no reverse-transcription controls (NRTs) to exclude contamination. Furthermore, negative controls also consisted of midgut and brain dissections of larvae reared at the two different temperatures of an “empty” ΦX86Fb strain without any integrated constructs. Both reference genes were stably expressed across samples. Log-transformed normalized relative quantities were calculated as described above and subjected to a Welch two-sample t-test to test for statistically significant expression differences between the different rearing temperatures and transgenic constructs. False discovery rate (FDR) was controlled using the multiple-testing correction method of Benjamini and Hochberg (1995).
Results
DNA sequence polymorphism in the ph region
Full-genome data provided by the Drosophila Population Genomics Project were used to analyze a 73-kb genomic region of intermediate recombination rates (Fiston-Lavier et al. 2010) that is located on the X chromosome between positions 1,990,000 and 2,063,000 (FlyBase release 5). In an African population sample from Siavonga, Zambia, a reduction of variation in the region is observed (Figure 1A) that overlaps with the valley of low polymorphism detected in previous studies in a Zimbabwean population sample from Lake Kariba (Beisswanger et al. 2006; Beisswanger and Stephan 2008). In these previous studies, evidence was presented that this reduction of polymorphism originated most likely from the action of positive directional selection in the recent past causing a selective sweep in the ancestral species range. As was shown before (Beisswanger et al. 2006), the Dutch population sample from Leiden harbors an even more pronounced valley of low polymorphism that spans >60 kb (Figure 1A).
Figure 1.

Evidence of positive selection in the Drosophila polyhomeotic (ph) genomic region. (A) Nucleotide variability (π) in European population samples from The Netherlands (blue line) and an African sample from Zambia (black line), as well as divergence (K) to D. simulans (dashed line), is shown in a sliding window across the region (window size = 3000 bp, step size = 1000 bp). (B) Composite-likelihood-ratio (CLR) analysis of a selective sweep using the site-frequency-based test statistic of SweeD (Pavlidis et al. 2013) for a European sample consisting of the Dutch and the French lines. The significance threshold of the test statistic is given by the dashed line. (C) BayeScan FST coefficients (Foll and Gaggiotti 2008) averaged over seven population samples from Europe and Africa (see Materials and Methods). BayeScan results based on a 300-kb window surrounding the ph locus (of which 73 kb are shown here) reveal a total of six outlier SNPs (FDR = 0.05) within the selective sweep region that was identified by the SweeD test. One of these is already segregating in the African samples, in contrast to the other five outlier SNPs that are located in the ph-p/CG3835 intergenic region. The latter are highlighted in red and by gray lines across panels. (D) The positions of the genes contained within the region. The arrowheads indicate the direction of transcription. The hatched box corresponds to the portion used for reporter gene analysis.
Likelihood analysis of selective sweeps in the European population
The 73-kb region shown in Figure 1A was submitted to a composite-likelihood-ratio test that is based on the site-frequency spectrum used by SweeD (Pavlidis et al. 2013). Since a larger sample size may lead to more accurate results in distinguishing selective sweeps from demographic events and inferring the genomic position of sweeps (Pavlidis et al. 2010, 2013), the French population sample from DPGP (Pool et al. 2012) was added to the Dutch sample to obtain a larger European data set. SweeD was run on the complete X chromosome and the CLR profile of the region of interest is shown in Figure 1B. The SweeD test provided a likelihood profile that is much broader than the valley of reduced variation in Africa (Beisswanger et al. 2006; Beisswanger and Stephan 2008) and spans almost the entire region of very low polymorphism found in the European sample (Figure 1, A and B).
Genetic differentiation between the European and African population samples in the ph region
Because a large fraction of the region of low variation in Europe contains no or very few SNPs, the CLR test cannot be used to identify the targets of selection. Instead, following Wilches et al. (2014), we utilized genetic differentiation between African and European populations to obtain model-based FST coefficients for each SNP (Foll and Gaggiotti 2008; Riebler et al. 2008). BayeScan analyses (Foll and Gaggiotti 2008) were run on an X-chromosomal window of 300 kb surrounding the ph locus between positions 1,900,000 and 2,200,000 (FlyBase release 5). SNP data from seven European and African populations were considered, which included samples from The Netherlands, France, Cameroon, Ethiopia, Gabon, Rwanda, and Zambia. Including all seven population samples with a total of 11,894 SNPs, BayeScan yielded 22 significant outlier SNPs (FDR = 0.05) with positive α-values, suggesting that these SNPs are targets of positive directional selection (Supporting Information, Table S2).
Six of these 22 outliers are located in the region of significant CLR values. While one of those 6 is already segregating in the African samples, the other 5 are monomorphic in the population samples from Africa. The former is also identified as an outlier SNP (FDR = 0.07), when the European samples are excluded from the BayeScan analysis (position on X chromosome, 2,039,998; see Table S2). These results suggest that the differentiation of this SNP started in Africa, whereas the differentiation of the other 5 SNPs occurred outside the ancestral species range. The 5 SNPs are located in the intergenic region between ph-p and CG3835 (Figure 1). Except for 1 of the 5, in which case no outgroup sequence was available, derived sequence variants are observed for all lines of the two European samples and ancestral variants for all lines of the five African samples. Thus, the 5 SNPs mark two distinct haplotype groups, a derived one and an ancestral one. Complementing the DPGP data with Sanger sequencing of lines from populations of Europe, Africa, and Asia, it is observed that the derived haplotype group is in very high frequency in Europe (44 of 46 lines) and the ancestral one is in very high frequency in Africa (143 of 144 lines). Interestingly, in the two Asian population samples from Malaysia a third haplotype group is quite abundant (15 of 32 lines) (Figure 2). This group is a recombinant between the derived and the ancestral haplotypes where the second and third sequence variants are identical with those of the derived group and the rest with those of the ancestral one. In addition to the recombinant haplotype group, 8 lines of the derived group and 9 lines of the ancestral one make up the Southeast Asian samples (Figure 2). Thus, in contrast to the European population samples, in which the derived variants of these 5 SNPs are near fixation, the derived alleles occur in intermediate frequencies in the Asian samples.
Figure 2.

Geographical distribution of haplotype groups in the intergenic region between ph-p and CG3835. The derived, ancestral, and recombinant haplotype groups are shown in white, black, and gray, respectively. The area of the pie chart is proportional to the size of the population sample of a given region.
Taken together, our observations suggest that the ph region was hit not only by a selective event causing a sweep in the ancestral African region, but also by another sweep that may have occurred outside the ancestral range, leading to the high frequency of the derived haplotype in Europe.
Sanger sequencing of the European sweep region
Accurate detection of short insertions and deletions is still difficult using next-generation sequencing. Therefore, to exclude insertion/deletion polymorphisms as possible targets of selection in Europe, we additionally sequenced the region of interest in population samples from Europe and Africa, using the Sanger method (see Materials and Methods). Since the target of selection in the European samples appears to be located in the upstream half of the valley of reduced variation, these 30 kb were fully sequenced in the Dutch population sample from Leiden and a sample from the ancestral range of D. melanogaster from Lake Kariba, Zimbabwe (Pool et al. 2012). Sanger sequencing supports the results of the full-genome data set in that the highest genetic differentiation is observed in the intergenic region of ph-p and CG3835 and no highly differentiated insertions/deletions were found between the European and African samples.
Expression analysis and reporter gene assays
The intergenic region between ph-p and CG3835 contains a PRE and the promoters of the two genes. This PRE was experimentally validated by using reporter gene assays and different PcG mutant backgrounds (Fauvarque and Dura 1993). The fragment exhibiting PRE activity as demonstrated by Fauvarque and Dura (1993) spans nearly the whole intergenic region and overlaps with the promoters of both genes. Since PREs function in an orientation-independent fashion (Busturia et al. 1997; Americo et al. 2002; Kozma et al. 2008), which was shown in particular for this specific PRE (Fauvarque and Dura 1993), and ph-p and CG3835 are known PcG target genes as seen by chromatin immunoprecipitation (ChIP) experiments (Schuettengruber et al. 2009; Schwartz et al. 2010), it is likely that both genes are under the control of the PRE residing in the region between them. Thermosensitivity in expression as often found for genes regulated by PcG proteins was observed for ph-p in its natural genetic environment. In a rather crude experiment using whole adult flies reared at different temperatures, we observed a significantly higher expression when temperature was lower. However, this effect was significant only for the Zimbabwean population sample, not for the Dutch one (Figure 3).
Figure 3.
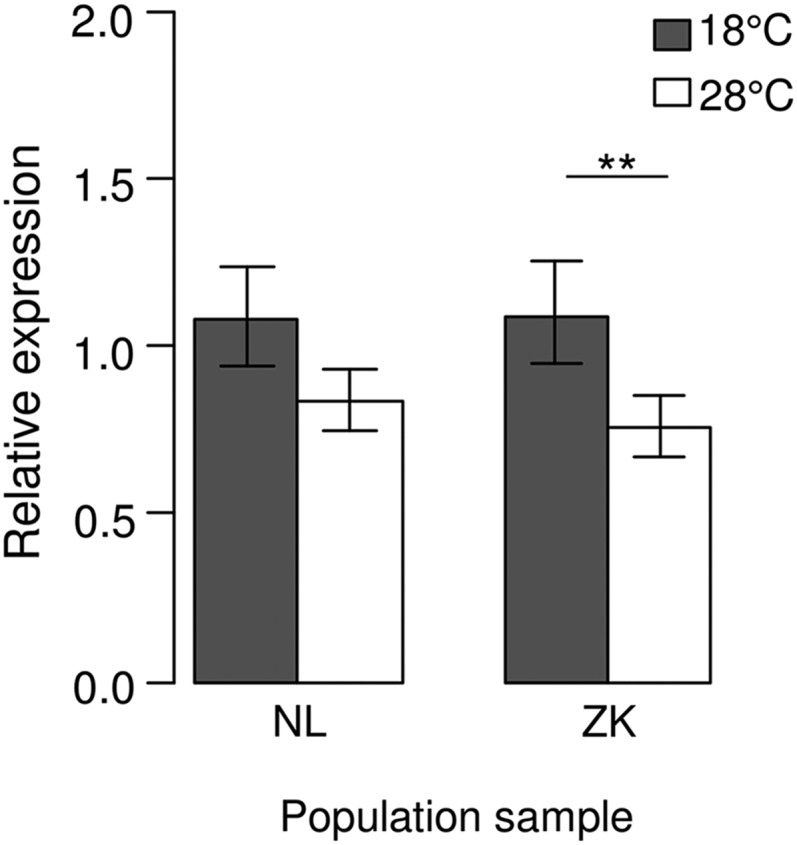
Expression of ph-p in population samples from The Netherlands (NL) and Zimbabwe (ZK). mRNA abundance in adult flies reared at 18° (shaded bars) and 28° (open bars) was measured via RT-qPCR. Error bars represent the 95% confidence interval. **P < 0.01.
To further test whether the five SNPs that define the derived and ancestral haplotype groups have an effect on gene expression, four reporter gene constructs were created in which the 5-kb intergenic region from either a Dutch (derived sequence variants) or a Zimbabwean (ancestral sequence variants) strain was fused to the Escherichia coli lacZ gene. The lacZ reporter gene was driven by either the ph-p promoter or the one of CG3835 (Figure 4). Reporter gene constructs were inserted into a common genetic background using the site-specific ΦC31 integration system (Bischof et al. 2007), allowing the comparison of the expression of the different constructs at the same genomic position in an otherwise identical genetic background. It was also checked that the selected integration site was not located in a PcG-regulated genomic region (see Materials and Methods).
Figure 4.
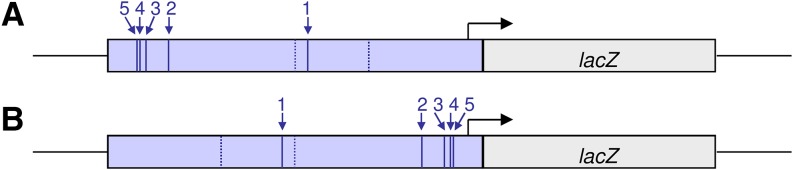
Schematic representation of the reporter gene constructs used. (A and B) ph-p promoter (A) and CG3835 promoter (B) are driving the E. coli lacZ gene. Blue lines give the relative location of the sequence variants in the 5-kb intergenic region containing the PRE and the promoters of ph-p and CG3835. The solid lines and numbers correspond to the five highly differentiated candidate SNPs that mark the different haplotype groups. Dashed lines indicate the other two sequence variants that differ between the African and European strains from which the intergenic region incorporated in the constructs originated.
To explore whether a temperature-sensitive pattern can be observed in the regulation of the two PcG target genes, flies were reared at 17° and 28° and messenger RNA (mRNA) expression levels of the lacZ reporter were quantified in the different transgenic lines via RT-qPCR. Since ph-p is highly expressed in the brain and CG3835 in the midgut of third instar larvae (Chintapalli et al. 2007), these tissues were dissected for the expression analysis. To measure lacZ mRNA levels, when expression of both genes is low, RT-qPCRs were also run for lines in which the reporter gene is controlled by the ph-p promoter on samples from the larval midgut and in those with lacZ driven by the CG3835 promoter on samples from the larval brain.
As expected from the endogenous expression, when the ph-p promoter was driving the reporter gene, a higher lacZ expression was observed in the brain than in the midgut, and vice versa in constructs with lacZ under the control of the CG3835 promoter (Table S1). For all tissues and treatments, lacZ expression due to the ph-p promoter was higher than expression due to the CG3835 promoter (Figure 5). Constructs carrying the ancestral sequence variants exhibited a temperature-sensitive expression pattern in the midgut while no such temperature-dependent expression difference was detected in the brain and for those constructs with the derived sequence variants (Figure 5). In the case of the ancestral sequence variants, midgut expression was approximately twofold higher when larvae were reared at 17° than at 28°. This difference due to temperature in lacZ expression was highly significant for the ph-p promoter, whereas no significance was reached for the promoter of CG3835 (Figure 5, C and D, and Table S1). For the constructs with the derived sequence variants and lacZ under the control of the ph-p promoter, a significantly higher reporter gene expression at 28°, compared to that of constructs with the ancestral variants, led to a buffering of the thermosensitivity (Figure 5, C and D).
Figure 5.

Reporter gene expression in larval tissues at two different rearing temperatures. mRNA abundance was measured via RT-qPCR in brains (A and B) and midguts (C and D) from third instar larvae reared at either 17° (shaded bars) or 28° (open bars). In A and C the ph-p promoter is driving lacZ expression whereas in B and D lacZ is under the control of the CG3835 promoter. The intergenic region containing the PRE and the promoters was taken from either a European strain from The Netherlands (NL) or an African one from Zimbabwe (ZK); that is, constructs were carrying either the derived or the ancestral sequence variants, respectively. Error bars represent the 95% confidence interval. *P < 0.05, **P < 0.01 (FDR = 0.05).
Therefore, we may conclude that nucleotide differences between the European and African sequences in the intergenic region have led to differences in gene expression. However, which of these differences confer the observed expression differences is currently unknown. In addition to the five candidate SNPs, there are two other sites in the 5-kb insert that differ between the two fly strains (NL01 and ZK186) from which the fragment was taken (Figure 4). One of these two sites is upstream of the first candidate SNP and the other one is downstream of this SNP (Figure 4B). The latter harbors a derived variant in the African line that is rare in Africa (6 of 139 lines) and not found in Europe, whereas the former one is also highly differentiated between Africa and Europe with the derived variant in high frequency in Europe (42 of 42 lines) and rare in Africa (7 of 138 lines). The seven sites that differ between the African and European reporter gene constructs are all candidates responsible for the observed differences in lacZ expression (Figure 4 and Figure 5). However, only the highly differentiated SNPs are expected to be causative if selection is responsible for the observed expression differences. Each of these SNPs has the potential to insert or delete a transcription factor binding site (TFBS) motif or change its binding affinity (Hauenschild et al. 2008), located either in the PRE or in any other regulatory element in the ph-p/CG3835 intergenic region. Interestingly, for the fifth of the candidate SNPs (Figure 4), the derived variant creates the Grh consensus sequence experimentally identified by Blastyák et al. (2006) and a Dsp1 consensus sequence that was demonstrated to be important in PcG recruitment (Déjardin et al. 2005). The derived state of the aforementioned additional highly differentiated SNP upstream of the first candidate SNP leads to the insertion of a motif, a GTGT sequence, which was shown to be functional in PRE activity in a number of studies (Kassis and Brown 2013).
Discussion
As was shown before (Beisswanger et al. 2006; Beisswanger and Stephan 2008), the genomic region around the ph locus exhibits a strong reduction in nucleotide polymorphism in D. melanogaster populations from Africa and Europe. Thus, the data suggest positive directional selection acting at this locus, leading to a selective sweep. In the previous studies, however, the question remained whether the sweep in the European population is independent of the African one or a result of a trans-population sweep that arose in Africa before the colonization of Europe (Beisswanger et al. 2006; Beisswanger and Stephan 2008). The much more pronounced reduction in nucleotide diversity in Europe could just be a product of the severe population size bottleneck D. melanogaster underwent during its migration out of Africa, and this bottleneck could also be the cause for the very high differentiation of the genomic region between ph-p and CG3835. However, since Asian and European populations share a MRCA after the out-of-Africa bottleneck (Laurent et al. 2011), and populations from Asia show a high genetic diversity in the aforementioned intergenic region, it is unlikely that the bottleneck is responsible for the high frequency of the derived sequence variants found in Europe.
The genes ph-p and CG3835 flanking this highly differentiated region are known PcG target genes (Schuettengruber et al. 2009; Schwartz et al. 2010) and harbor an experimentally validated PRE between each other (Fauvarque and Dura 1993). PcG-regulated genes are temperature sensitive in their expression; i.e., their transcriptional output is higher when flies are reared or held at lower temperatures than at higher ones (Fauvarque and Dura 1993; Chan et al. 1994; Zink and Paro 1995; Bantignies et al. 2003; Gibert et al. 2011). This phenomenon prompted the hypothesis that if cold temperatures disrupt PcG regulation, then adaptation to temperate environments should include the buffering of this expression plasticity (Levine and Begun 2008). Natural selection would then act to stabilize the transcriptional output, leading to a lower degree of gene expression plasticity in response to varying temperatures. As a consequence, thermosensitivity of PcG target gene expression would be reduced by limiting the influence of the environment. A genome-wide expression analysis indeed identified more genes with expression plasticity due to rearing temperature in tropical compared to temperate populations (Levine et al. 2011). Our study also supports the reduced thermosensitivity of PcG target gene expression in temperate populations. The data suggest temperature sensitivity of PcG target gene expression in African populations that was selected against in populations from Europe to stabilize the transcriptional output across temperatures. At the locus under study, this was observed for the expression of ph-p in the natural genetic background and for larval midguts, using reporter gene assays. The reporter gene analysis linked the SNPs that were detected as likely targets of positive selection in Europe to the European stabilized gene expression. Rearing temperature had no effect on gene expression in larval brains. Possible explanations could be that brain expression is under a greater selective pressure against gene expression variability, and so the expression level is already less environmentally sensitive in the African populations, or that there is no thermosensitivity of expression in the larval brain. In addition, for the ph-p promoter-driven expression, the data indicate that higher transcriptional output at lower temperatures is not in itself detrimental. The greater variability in expression due to a higher degree of variation in temperature in temperate climates, however, seems to have been disadvantageous and needed to be reduced by the action of selection during the colonization of Europe.
More recent studies focused only on selection stabilizing the temperature-sensitive transcriptional output by directly acting on the proteins of the PcG system in populations from temperate environments (Harr et al. 2002; Levine and Begun 2008; Gibert et al. 2011). In this study, we present evidence for selection acting on cis-regulatory sequences to reduce the temperature sensitivity of PcG-regulated gene expression.
Since PRE function is highly dependent on the genomic location, one drawback of our study may be that reporter gene assays were only done at one integration site in the genome (Kassis and Brown 2013; Steffen and Ringrose 2014). This position effect is mainly due to regulatory elements in the vicinity of the integration site that can have an influence on the function of the transgenic PRE. However, it is likely that redoing the study at an additional integration site would yield similar results to those reported here. One reason for this is that studies observing this position effect mainly looked at smaller PRE sequences (Kassis and Brown 2013). Our inserted fragment is ∼5 kb in length and likely contains other regulatory sequences (i.e., enhancers) in addition to the two promoters and the PRE. Furthermore, we could reproduce the endogenous expression pattern of the different tissues in the transgenic lines with a higher expression of ph-p in larval brains than in midguts and vice versa for CG3835.
Here, we report a cis-regulatory change mediating a decreased thermosensitivity of PcG regulation at a specific locus. The question arises of whether selection against temperature-sensitive expression variability in temperate populations is a global phenomenon, i.e., PcG target genes in general exhibit such a buffering, or whether it is specific for the locus examined in this study. The former is supported by other studies that have shown greater expression plasticity in tropical populations than in temperate ones (Levine et al. 2011) and given evidence for spatially varying selection targeting proteins of the Polycomb group (Harr et al. 2002; Levine and Begun 2008). It would then also be of interest to which amount either of both, cis-regulatory and trans-regulatory changes, contributes to the reduced thermosensitivity in temperate populations and whether one can observe other PcG target genes with cis-regulatory changes.
The buffering of the temperature-induced expression plasticity due to the derived sequence variants is likely to be explained by changes in TFBS motifs. There are two possibilities for how this could have happened. First, changes in TFBS motifs occurred in enhancer sequences, altering the strength of the enhancer, resulting in a change in the transcriptional output that is then maintained by the associated PRE. Second, the PRE could have been directly targeted by selection and TFBSs of PcG proteins and associated factors could have been modified, leading, e.g., to changes in PcG recruitment and therefore to differences in the expression level that is maintained by the PRE (Schwartz et al. 2010; Steffen and Ringrose 2014). For enhancers as well as for PREs, it is well documented that small changes in their sequences can have large effects on the expressed phenotype and both cis-regulatory elements are known to evolve rapidly (Hauenschild et al. 2008). Therefore, it seems likely that a change in sequence of one of them (or both) is responsible for the expression differences described in this study. To find the causative sequence variant(s) and the associated TFBS(s), further experimental studies are needed.
Supplementary Material
Acknowledgments
We acknowledge S. Lange and A. Steincke for providing technical assistance. We thank Charles Langley (University of California, Davis) for providing us with the assembled genomes for the Dutch and Malaysian populations. We are also very grateful for constructive comments from the two reviewers and David Begun. This work was supported by the Deutsche Forschungsgemeinschaft Research Unit grants STE 325/12-1 and 12-2.
Footnotes
Communicating editor: D. J. Begun
Supporting information is available online at www.genetics.org/lookup/suppl/doi:10.1534/genetics.115.177030/-/DC1.
Literature Cited
- Americo J., Whiteley M., Brown J. L., Fujioka M., Jaynes J. B., et al. , 2002. A complex array of DNA-binding proteins required for pairing-sensitive silencing by a polycomb group response element from the Drosophila engrailed gene. Genetics 160: 1561–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvidsson S., Kwasniewski M., Riaño-Pachón D. M., Mueller-Roeber B., 2008. QuantPrime–a flexible tool for reliable high-throughput primer design for quantitative PCR. BMC Bioinformatics 9: 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantignies F., Grimaud C., Lavrov S., Gabut M., Cavalli G., 2003. Inheritance of Polycomb-dependent chromosomal interactions in Drosophila. Genes Dev. 17: 2406–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont M. A., Balding D. J., 2004. Identifying adaptive genetic divergence among populations from genome scans. Mol. Ecol. 13: 969–980. [DOI] [PubMed] [Google Scholar]
- Beisswanger S., Stephan W., 2008. Evidence that strong positive selection drives neofunctionalization in the tandemly duplicated polyhomeotic genes in Drosophila. Proc. Natl. Acad. Sci. USA 105: 5447–5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisswanger S., Stephan W., De Lorenzo D., 2006. Evidence for a selective sweep in the wapl region of Drosophila melanogaster. Genetics 172: 265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y., 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 57: 289–300. [Google Scholar]
- Bischof J., Maeda R. K., Hediger M., Karch F., Basler K., 2007. An optimized transgenesis system for Drosophila using germ-line-specific φC31 integrases. Proc. Natl. Acad. Sci. USA 104: 3312–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blastyák A., Mishra R. K., Karch F., Gyurkovics H., 2006. Efficient and specific targeting of Polycomb group proteins requires cooperative interaction between Grainyhead and Pleiohomeotic. Mol. Cell. Biol. 26: 1434–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busturia A., Wightman C. D., Sakonju S., 1997. A silencer is required for maintenance of transcriptional repression throughout Drosophila development. Development 124: 4343–4350. [DOI] [PubMed] [Google Scholar]
- Chan C. S., Rastelli L., Pirrotta V., 1994. A Polycomb response element in the Ubx gene that determines an epigenetically inherited state of repression. EMBO J. 13: 2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintapalli V. R., Wang J., Dow J. A., 2007. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat. Genet. 39: 715–720. [DOI] [PubMed] [Google Scholar]
- Clarke A., 1996. The influence of climate chance of the distribution and evolution of organisms, pp. 377–407 in Animals and Temperature: Phenotypic and Evolutionary Adaptation, edited by Johnston I. A., Bennett A. F. Cambridge University Press, Cambridge, UK. [Google Scholar]
- David J. R., Capy P., 1988. Genetic variation of Drosophila melanogaster natural populations. Trends Genet. 4: 106–111. [DOI] [PubMed] [Google Scholar]
- Déjardin J., Rappailles A., Cuvier O., Grimaud C., Decoville M., et al. , 2005. Recruitment of Drosophila Polycomb group proteins to chromatin by DSP1. Nature 434: 533–538. [DOI] [PubMed] [Google Scholar]
- Fauvarque M. O., Dura J. M., 1993. Polyhomeotic regulatory sequences induce developmental regulator-dependent variegation and targeted P-element insertions in Drosophila. Genes Dev. 7: 1508–1520. [DOI] [PubMed] [Google Scholar]
- Fiston-Lavier A.-S., Singh N. D., Lipatov M., Petrov D. A., 2010. Drosophila melanogaster recombination rate calculator. Gene 463: 18–20. [DOI] [PubMed] [Google Scholar]
- Foll M., Gaggiotti O., 2008. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180: 977–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibert J.-M., Karch F., Schlötterer C., 2011. Segregating variation in the polycomb group gene cramped alters the effect of temperature on multiple traits. PLoS Genet. 7: e1001280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr B., Kauer M., Schlötterer C., 2002. Hitchhiking mapping: a population-based fine-mapping strategy for adaptive mutations in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 99: 12949–12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauenschild A., Ringrose L., Altmutter C., Paro R., Rehmsmeier M., 2008. Evolutionary plasticity of polycomb/trithorax response elements in Drosophila species. PLoS Biol. 6: e261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellemans J., Mortier G., De Paepe A., Speleman F., Vandesompele J., 2007. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 8: R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T. T., Eisen M. B., Thornton K. R., Andolfatto P., 2013. A second-generation assembly of the Drosophila simulans genome provides new insights into patterns of lineage-specific divergence. Genome Res. 23: 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassis, J. A., and J. L. Brown, 2013 Polycomb group response elements in Drosophila and vertebrates, pp. 83–118 in Advances in Genetics, edited by J. C. Dunlap, S. F. Goodwin, and T. Friedmann. Academic Press, New York/London/San Diego. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Stephan W., 2002. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics 160: 765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma G., Bender W., Sipos L., 2008. Replacement of a Drosophila Polycomb response element core, and in situ analysis of its DNA motifs. Mol. Genet. Genomics 279: 595–603. [DOI] [PubMed] [Google Scholar]
- Lachaise D., Silvain J.-F., 2004. How two Afrotropical endemics made two cosmopolitan human commensals: the Drosophila melanogaster-D. simulans palaeogeographic riddle. Genetica 120: 17–39. [DOI] [PubMed] [Google Scholar]
- Laurent S. J., Werzner A., Excoffier L., Stephan W., 2011. Approximate Bayesian analysis of Drosophila melanogaster polymorphism data reveals a recent colonization of Southeast Asia. Mol. Biol. Evol. 28: 2041–2051. [DOI] [PubMed] [Google Scholar]
- Levine M. T., Begun D. J., 2008. Evidence of spatially varying selection acting on four chromatin-remodeling loci in Drosophila melanogaster. Genetics 179: 475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M. T., Eckert M. L., Begun D. J., 2011. Whole-genome expression plasticity across tropical and temperate Drosophila melanogaster populations from Eastern Australia. Mol. Biol. Evol. 28: 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Stephan W., 2006. Inferring the demographic history and rate of adaptive substitution in Drosophila. PLoS Genet. 2: e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R., Williamson S., Kim Y., Hubisz M. J., Clark A. G., et al. , 2005. Genomic scans for selective sweeps using SNP data. Genome Res. 15: 1566–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P., Jensen J. D., Stephan W., 2010. Searching for footprints of positive selection in whole-genome SNP data from nonequilibrium populations. Genetics 185: 907–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P., Živković D., Stamatakis A., Alachiotis N., 2013. SweeD: likelihood-based detection of selective sweeps in thousands of genomes. Mol. Biol. Evol. 30: 2224–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre S. E. S., Ponting L., Stefancsik R., McQuilton P., The FlyBase Consortium , 2014. FlyBase 102—advanced approaches to interrogating FlyBase. Nucleic Acids Res. 42: D780–D788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pool J. E., Corbett-Detig R. B., Sugino R. P., Stevens K. A., Cardeno C. M., et al. , 2012. Population genomics of sub-Saharan Drosophila melanogaster: African diversity and non-African admixture. PLoS Genet. 8: e1003080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riebler A., Held L., Stephan W., 2008. Bayesian variable selection for detecting adaptive genomic differences among populations. Genetics 178: 1817–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., Coulson A. R., 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74: 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B., Ganapathi M., Leblanc B., Portoso M., Jaschek R., et al. , 2009. Functional anatomy of polycomb and trithorax chromatin landscapes in Drosophila embryos. PLoS Biol. 7: e1000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz Y. B., Kahn T. G., Stenberg P., Ohno K., Bourgon R., et al. , 2010. Alternative epigenetic chromatin states of polycomb target genes. PLoS Genet. 6: e1000805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen P. A., Ringrose L., 2014. What are memories made of? How Polycomb and Trithorax proteins mediate epigenetic memory. Nat. Rev. Mol. Cell Biol. 15: 340–356. [DOI] [PubMed] [Google Scholar]
- Stephan W., Li H., 2007. The recent demographic and adaptive history of Drosophila melanogaster. Heredity 98: 65–68. [DOI] [PubMed] [Google Scholar]
- Tajima F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilches, R., S. Voigt, P. Duchén, S. Laurent, and W. Stephan, 2014 Fine-mapping and selective sweep analysis of QTL for cold tolerance in Drosophila melanogaster. G3 4: 1635–1645. [DOI] [PMC free article] [PubMed]
- Zhang Y., Arcia S., Perez B., Fernandez-Funez P., Rincon-Limas D. E., 2013. p∆ TubHA4C, a new versatile vector for constitutive expression in Drosophila. Mol. Biol. Rep. 40: 5407–5415. [DOI] [PubMed] [Google Scholar]
- Zink D., Paro R., 1995. Drosophila Polycomb-group regulated chromatin inhibits the accessibility of a trans-activator to its target DNA. EMBO J. 14: 5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.