


Abstract
Base excision repair (BER) of an oxidized base within a trinucleotide repeat (TNR) tract can lead to TNR expansions that are associated with over 40 human neurodegenerative diseases. This occurs as a result of DNA secondary structures such as hairpins formed during repair. We have previously shown that BER in a TNR hairpin loop can lead to removal of the hairpin, attenuating or preventing TNR expansions. Here, we further provide the first evidence that AP endonuclease 1 (APE1) prevented TNR expansions via its 3′-5′ exonuclease activity and stimulatory effect on DNA ligation during BER in a hairpin loop. Coordinating with flap endonuclease 1, the APE1 3′-5′ exonuclease activity cleaves the annealed upstream 3′-flap of a double-flap intermediate resulting from 5′-incision of an abasic site in the hairpin loop. Furthermore, APE1 stimulated DNA ligase I to resolve a long double-flap intermediate, thereby promoting hairpin removal and preventing TNR expansions.
INTRODUCTION
The expansion of trinucleotide repeats (TNR) is associated with over 40 human neurodegenerative diseases including Huntington's disease (CAG/CTG), Friedreich's ataxia (GAA/TTC) and myotonic dystrophy (CTG/CAG) (1,2), among others. The repeat instability results from non-B form DNA secondary structures including hairpins, triplexes, tetraplexes and sticky DNA (3) formed during DNA replication, repair, recombination and gene transcription (4–6). Recent studies have shown that the repair of oxidative DNA damage within TNRs is associated with somatic TNR instability (7–11). During the repair of oxidized base lesions such as 8-oxoguanine (8-oxoG), an abasic site is incised at its 5′-side generating a nick in the DNA backbone. If this occurs in TNRs, it allows the formation of hairpins in the DNA, which leads to repeat expansions and deletions if the hairpins are incorporated into the DNA and sustained upon completion of repair (8,11). It has been proposed that repeated cycles of oxidative DNA damage and BER lead to cumulative repeat expansions through a ‘toxic oxidation cycle,’ which can result in the onset of disease caused by TNR expansion (2,7,12).
We previously reported that the location of a DNA base lesion within a TNR repeat tract determines whether an expansion or deletion occurs, with a lesion near the 5′-end leading to expansions and a lesion in the middle leading to deletions (11). An 8-oxoG located in the hairpin loop has been found to be resistant to DNA repair (12), resulting in the accumulation of the lesion and sustainment of the hairpin loop that leads to repeat expansion. However, we have discovered that during BER in a TNR hairpin loop, the hairpin can be converted to a double-flap intermediate, containing an upstream 3′-flap and a downstream 5′-flap, which can be subsequently resolved by a 3′-5′ endo/exonuclease, Mus81/Eme1 and flap endonuclease 1 (FEN1), respectively. This subsequently results in hairpin removal and prevention or attenuation of TNR expansions (13).
AP endonuclease 1 (APE1) is a multi-functional protein abundant in human cells (14–17) that is essential in maintaining multiple cellular functions. This is demonstrated by the lethality of APE1 knockout in mice (18). The enzyme can endonucleolytically incise the 5′-end of an abasic site in duplex DNA and in single-stranded DNA (19), including a TNR hairpin loop (13,19), by hydrolyzing the phosphodiester bond during BER. In addition, APE1 can promote the recycling of DNA glycosylases by dislodging the glycosylase from an abasic site, resulting in stimulation of the activity of the enzymes such as 8-oxoG DNA glycosylase 1 (OGG1) (20,21) which efficiently initiates BER (22–24). APE1 can also stimulate the activity of FEN1 via a physical protein–protein interaction and the activity of DNA ligase I (LIG I) (25,26), as well as stimulate the activities of both the polymerase and deoxyribose phosphate (dRP) lyase activities of polymerase β (pol β) (27) by facilitating pol β binding to a gapped DNA and dRP residue (28), thereby enhancing the efficiency of BER (25,27–29). Furthermore, APE1 can physically interact with proliferating cell nuclear antigen (PCNA) (26), suggesting its involvement in BER coordination (30,31). In addition, APE1 has 3′-5′ exonuclease activity (32) that is 100-fold less efficient than its AP endonuclease activity (17). The exonuclease activity of APE1 can be regulated by poly(ADP-ribose) polymerase-1 (PARP-1), as these proteins have been shown to compete for binding to the 5′-end of a gapped intermediate during BER (33). However, the competition between PARP-1 and APE1 in binding to BER intermediates stimulates APE1 3′-5′ exonuclease activity, suggesting that APE1 is dislodged by PARP-1 from the 5′-end of a BER intermediate, allowing its binding to the 3′-end of a BER intermediate on which the exonuclease activity can act (33). It has been shown that APE1 3′-5′ exonuclease activity removes a 3′-mismatch and may provide proofreading activity for pol β during BER (34–36), indicating a crucial role of APE1 3′-5′ exonuclease in maintaining genome integrity. The APE1 exonuclease activity has also been found to be able to remove 3′-blocking groups such as L-configuration nucleoside analogs (37), suggesting its importance in trimming the ‘dirty 3′-end’ of a BER intermediate to facilitate the completion of BER. However, the role of the exonuclease activity of APE1 in sustaining genome stability remains unknown. We previously showed that the 3′-5′ flap endonuclease activity of the Mus81/Eme1 complex promotes the removal of a TNR hairpin by cleaving the 3′-flap of a double-flap intermediate generated by BER in a TNR hairpin loop (13). Yet, no BER protein that can process a 3′-5′ flap during BER in a TNR hairpin has been identified. Because APE1 exhibits 3′-5′ exonuclease activity that can process mismatches at the 3′-end of an upstream DNA strand, we hypothesized that the 3′-5′ exonuclease activity may shorten the upstream 3′-flap of a double-flap intermediate resulting from the incision of the loop region of a TNR hairpin during BER. This could subsequently promote the removal of the hairpin, thereby facilitating prevention or attenuation of TNR expansions. We tested this hypothesis by reconstituting BER on both TNR hairpins containing a base lesion in the loop region and on the double-flap intermediates that are generated during BER. We found that APE1 promoted the removal of a TNR hairpin during BER of a base lesion in the hairpin loop region. This was accomplished by the 3′-5′ exonuclease activity of the enzyme that cleaved the upstream 3′-region exonucleolytically, resolving the double-flap intermediate and preventing TNR expansions. Surprisingly, we also found that APE1 significantly stimulated the ligation activity of LIG I to specifically facilitate the completion of hairpin removal. This is the first evidence of APE1 preventing TNR expansions by facilitating hairpin removal.
MATERIALS AND METHODS
Materials
The DNA oligonucleotides which contain an 8-oxoG were synthesized by Eurofins MWG Operon (Huntsville, AL, USA), and all others were synthesized by Integrated DNA Technologies (IDT, Coralville, IA, USA). Deoxynucleoside 5′-triphosphates (dNTPs) were from Fermentas (Glen Burnie, MD, USA). Terminal deoxynucleotidyl transferase and T4 polynucleotide kinase were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Radionucleotides [γ-32P] ATP (6000 mCi/mmol) and Cordycepin 5′-triphosphate 3′-[α-32P] (5000 mCi/mmol) were purchased from Perkin Elmer Inc. (Boston, MA, USA). Micro Bio-Spin 6 chromatography columns were purchased from Bio-Rad (Hercules, CA, USA). All standard chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) and from Thermo Fisher Scientific (Waltham, MA, USA). Purified OGG1, pol β and LIG I were generous gifts from Dr Samuel H. Wilson at the National Institute of Environmental Health Sciences/National Institutes of Health. APE1 and FEN1 were expressed in Escherichia coli and purified as described below. Oligonucleotide substrates were prepared as described previously (13). Briefly, substrates containing a (CAG)7 or (CAG)14 hairpin were constructed by annealing a damaged strand containing an 8-oxoG or THF, an analog of an abasic site, in the loop-forming region of a (CAG)13 or (CAG)20 tract with a template strand containing (CTG)7 repeats at a molar ratio of 1:2. Substrates mimicking the double-flap intermediates with a 5′-sugar phosphate residue, the THF residue, were constructed by annealing an upstream primer containing a 3′-(CAG)4 or (CAG)7 flap and a downstream primer containing a 5′-(CAG)3 or (CAG)7 flap with the template strand at a molar ratio of 1:2:2. For each substrate, three CAG repeats located at both the 5′- and 3′-side of the hairpins/flaps, base paired with three CTG repeats in the template strand. Oligonucleotide sequences were described previously (13). Substrates were labeled with 32P at the 5′- or 3′-end of the damaged strand, or at the upstream primer or downstream primer, as indicated.
Protein expression and purification
FEN1 was expressed in E. Coli BL21(DE3). Two liters of lysogeny broth (LB) medium were inoculated with one colony each of the transformed BL21(DE3) cells and were incubated overnight without shaking. The cells were then incubated at 37°C at 225 rpm until OD600 reached 0.6. The protein expression was induced with 1 mM IPTG for 3.5 h and harvested by centrifugation at 2600 rpm for 45 min at 4°C. Cells were lysed with a French press cell disruptor (Glen Mills, Clifton, NJ, USA) in lysis buffer which contained 30 mM 4-(2-hydroxyethyl)-1-piperazineethane-sulfonic acid (HEPES), pH 7.5, 30 mM KCl, 1 mM dithiothreitol (DTT), 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride (PMSF) and 0.5% inositol. The cell lysates were centrifuged at 12 000 rpm for 30 min at 4°C. The supernatant was loaded onto a 10-ml Sepharose Q column operated by an AKTA Fast Protein Liquid Chromatography system (FPLC) (GE Healthcare, Piscataway, NJ, USA). The flow-through was collected and dialyzed into buffer containing 30 mM HEPES, pH 7.5, 30 mM KCl, 0.5% inositol and 1 mM PMSF, and subsequently loaded onto a 5-ml CM sepharose column (Bio-Rad), with fractions eluted using a linear gradient of KCl from 30 mM to 2 M. Peak fractions were combined and dialyzed into buffer containing 30 mM HEPES, pH 7.5, 0.5% inositol, 1.7 M (NH4)2SO4 and 1 mM PMSF. Samples were then loaded onto a 2-ml phenyl sepharose column, with fractions eluted using a linear gradient of (NH4)2SO4 from 1.7 M to 0 M. The peak fractions were combined and dialyzed into buffer containing 30 mM HEPES, pH 7.5, 30 mM KCl, 0.5% inositol and 1 mM PMSF. Samples were then loaded onto a 1-ml Mono-S column (GE Healthcare, Piscataway, NJ, USA), and eluted using a linear gradient of KCl (30 mM to 2 M). Purified FEN1 was aliquoted and frozen at −80°C until further use.
APE1 was expressed in E. Coli BL21(DE3). Two liters of LB medium were inoculated with one colony each of the transformed BL21(DE3) cells and were incubated overnight without shaking. The cells were then incubated at 37°C at 225 rpm until OD600 reached 0.6. The APE1 expression was induced by 0.5 mM IPTG for 3.5 h. Cells were harvested by centrifugation at 2500 rpm for 30 min at 4°C. The supernatant was discarded and cell pellets were lysed in lysis buffer, which contained 50 mM HEPES, pH 7.5, 30 mM NaCl, 1 mM DTT, 1 mM EDTA and 1 mM PMSF. The cell lysates were centrifuged at 12 000 rpm for 30 min at 4°C. The supernatant was loaded onto a 10-ml sepharose Q column operated by an AKTA FPLC. The flow-through was collected and dialyzed into buffer containing 50 mM HEPES, pH 7.5, 30 mM NaCl and 1 mM PMSF, and then loaded onto a 5-ml CM sepharose column (Bio-Rad), with fractions eluted using a linear gradient of NaCl (30 mM to 2 M). The peak fractions were combined and dialyzed into buffer containing 50 mM HEPES, pH 7.5, 30 mM NaCl and 1 mM PMSF. Samples were then loaded onto a 1-ml Mono-S column, and eluted using a linear gradient of NaCl from 30 mM to 2 M. Peak fractions were combined and dialyzed into buffer containing 50 mM HEPES, pH 7.5, 1.7 M (NH4)2SO4 and 1 mM PMSF. Samples were then loaded onto a 2-ml phenyl sepharose column, with fractions eluted using a linear gradient of (NH4)2SO4 ranging from 1.7 M to 0 M. Purified APE1 was aliquoted and frozen at −80°C until further use.
Reconstituted BER assay
In vitro BER of an 8-oxoG or abasic site analog, THF, in the loop region of a (CAG)7 or (CAG)14 hairpin was carried out by incubating 50 nM substrate with the indicated concentrations of OGG1, APE1, pol β, FEN1 and LIG I. Substrates containing an 8-oxoG were initially incubated with OGG1 (100 nM) and APE1 (50 nM) at 37°C for 30 min. Substrates were then subject to a phenol:chloroform extraction for removing the OGG1 and APE1. The cleaved substrates were then precipitated with ethanol and resuspended for the BER assay. Substrates containing a THF in the hairpin loop were pre-cleaved with APE1 alone and subject to the BER assay. BER of an abasic site, the THF residue, within the loop of the (CAG)7 and (CAG)14 hairpins was measured by incubating 25 nM substrates with the indicated concentrations of APE1, pol β, FEN1 and LIG I. All reactions were carried out in reaction buffer containing 50 mM Tris-HCl, pH 7.5, 50 mM KCl, 0.1 mM EDTA, 0.1 mg/ml bovine serum albumin (BSA), 0.2 mM DTT and 0.01% Nonidet P-40, with 5 mM MgCl2, 2 mM ATP and 50 μM dNTPs. The reaction mixtures were incubated at 37°C for 15 min, and terminated by addition of stopping buffer containing 95% formamide and 10 mM EDTA. Reaction mixtures were denatured at 95°C for 10 min and separated by 15 or 18% urea-denaturing polyacrylamide gel electrophoresis. Substrates and products were detected and analyzed using a Pharos FX Plus Phosphorimager from Bio-Rad.
BER enzymatic activity assay
The activities of APE1 AP endonuclease and APE1 3′-5′ exonuclease, as well as FEN1 flap cleavage activity on the (CAG)7 and (CAG)14 hairpin substrates and double-flap substrates were measured by incubating 50 or 100 nM substrates with the indicated concentrations of FEN1, APE1 and pol β in reaction buffer containing 50 mM Tris-HCl, pH 7.5, 50 mM KCl, 0.1 mM EDTA, 0.1 mg/ml BSA, 0.2 mM DTT and 0.01% Nonidet P-40, with 5 mM MgCl2 and 50 μM dNTPs. The reaction mixtures were incubated at 37°C for 15 min, and terminated by addition of stopping buffer containing 95% formamide and 10 mM EDTA. Reaction mixtures were denatured at 95°C for 10 min and separated by 15% urea-denaturing polyacrylamide gel electrophoresis. Substrates and products were detected using a Pharos FX Plus Phosphorimager (Bio-Rad Laboratory).
The effects of APE1 on LIG I activity
The effects of APE1 on LIG I activity were determined via in vitro reconstitution of BER on the double-flap substrates resulting from APE1 5′-incision of an abasic site within the loop of the (CAG)7 and (CAG)14 hairpins at low concentrations of APE1 (0.5 and 1 nM) that exhibit little3′-5′ exonuclease activity, and LIG I (1 nM). Twenty five nanomolar substrates were incubated with the indicated concentrations of FEN1, APE1, pol β and LIG I in reaction buffer containing 50 mM Tris-HCl, pH 7.5, 50 mM KCl, 0.1 mM EDTA, 0.1 mg/ml BSA, 0.2 mM DTT and 0.01% Nonidet P-40, with 5 mM MgCl2, 2 mM ATP and 50 μM dNTPs. The reaction mixtures were incubated at 37°C for 15 min, and terminated by stopping buffer containing 95% formamide and 10 mM EDTA. Reaction mixtures were denatured at 95°C for 10 min and separated by 15 or 18% urea-denaturing polyacrylamide gel electrophoresis. Results were analyzed using a Pharos FX Plus Phosphorimager (Bio-Rad Laboratory).
RESULTS
APE1 stimulates CAG repeat hairpin removal during BER of a base lesion in a hairpin loop
To determine whether APE1 can facilitate the removal of a CAG repeat hairpin during BER within the loop of the hairpin, we reconstituted BER using synthesized oligonucleotide substrates containing either a (CAG)7 or (CAG)14 hairpin with an 8-oxoG (Figure 1) or abasic site analog, tetrahydrofuran (THF) (Figure 2), in the loop region of the hairpin. We found that during BER of an 8-oxoG in the loop of the small (CAG)7 hairpin, the production of the ‘repaired unexpanded product’, the product with the same length as the template strand, was significantly increased in the presence of 50 and 100 nM APE1 (Figure 1, lanes 7–8 and lanes 10–11) compared to the amount of product resulting from BER in the absence of APE1 (Figure 1, lanes 6 and 9). During BER of an 8-oxoG in the loop of the large (CAG)14 hairpin, 50 and 100 nM APE1 resulted in the production of the repaired unexpanded product (Figure 1, lanes 18–19 and lanes 21–22), whereas little unexpanded product was generated in the absence of APE1 (Figure 1, lanes 17 and 20). APE1 alone failed to produce any repair products (Figure 1, lanes 4–5 and lanes 15–16). Consistent with the findings from BER of the 8-oxoG in the hairpin loop, 50 and 100 nM APE1 also resulted in the production or an increase in the amount of repaired unexpanded product during BER of a THF in the loop region of the (CAG)7 and (CAG)14 hairpins (Figure 2, compare lanes 7–8 with lane 6 and lanes 10–11 with lane 9, lanes 18–19 with lane 17 and lanes 21–22 with lane 20). The results indicated that APE1 promoted the removal of the (CAG)7 and (CAG)14 repeat hairpins during BER of both an 8-oxoG and abasic lesion in the loop region of the hairpin, thereby preventing TNR expansions.
Figure 1.

APE1 promotes the removal of a trinucleotide repeat hairpin during BER of 8-oxoG in a TNR hairpin loop. The effect of APE1 on hairpin removal during BER of an 8-oxoG in the loop of a small (CAG)7 hairpin (left panel) and a large (CAG)14 hairpin (right panel) was examined by reconstituting BER with the hairpin-containing substrates. Lanes 1 and 12 are markers that indicate the length of the template strand or unexpanded product. Lanes 2 and 13 are markers that indicate the length of the damage-containing strand or expanded product. Lanes 3 and 14 correspond to the substrates that were pre-incubated with 100 nM OGG1 and 50 nM APE1 and isolated after phenol-chloroform extraction. Lanes 4–5 and lanes 15–16 correspond to the reaction mixtures with OGG1/APE1 pretreated substrates and 50 and 100 nM APE1 only. Lanes 6–8 and lanes 17–19 correspond to BER reconstituted with FEN1 (1 nM) and LIG I (5 nM) in the absence and presence of APE1. Lanes 9–11 and lanes 20–22 correspond to BER reconstituted with FEN1 (1 nM), LIG I (5 nM) and pol β (10 nM) in the absence and presence of APE1. Substrates were 32P-labeled at the 5′-end of the damage-containing strand and are illustrated above each gel.
Figure 2.
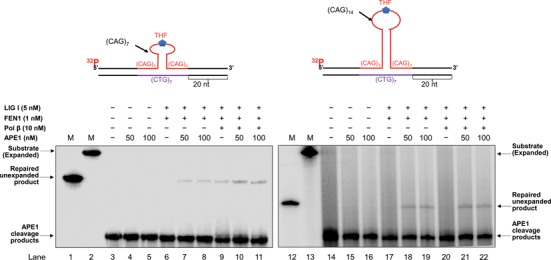
APE1 promotes the removal of a trinucleotide repeat hairpin during BER of an abasic site in a CAG repeat hairpin loop. The effect of APE1 on CAG repeat hairpin removal during BER of a THF residue, an abasic site analog, in the loop of a small (CAG)7 hairpin (left panel) and a large (CAG)14 hairpin (right panel) was examined by reconstituting BER with the substrates containing a THF in the (CAG)7 and (CAG)14 loop. Lanes 1 and 12 indicate markers that illustrate the length of the template strand or unexpanded product, corresponding to the removal of the entire hairpin. Lanes 2 and 13 correspond to markers that represent the length of the substrate only. Lanes 3 and 14 correspond to the substrates that were pre-incubated with APE1 (50 nM) and isolated after phenol-chloroform extraction. Lanes 4–5 and lanes 15–16 correspond to the reaction mixture with the APE1-pretreated substrates and 50 and 100 nM APE1 only. Lanes 6–8 and 17–19 correspond to BER reactions reconstituted with FEN1 (1 nM) and LIG I (5 nM) in the absence and presence of APE1. Lanes 9–11 and lanes 20–22 correspond to BER reaction mixture reconstituted with FEN1 (1 nM), LIG I (5 nM) and pol β (10 nM) in the absence and presence of APE1.
APE1 stimulates the processing of a double-flap intermediate during BER
Our previous studies have shown that during BER of a base lesion within the loop of a CAG repeat hairpin, the hairpin is converted to a double-flap intermediate as a result of APE1 5′-incision of an abasic site (13). We found that the double-flap intermediate was then processed by a 3′-5′ flap endonuclease, Mus81/Eme1 and FEN1 5′-flap cleavage (13). This led to cleavage of the flaps, thereby resulting in the removal of the hairpin. We further hypothesized that APE1 is also involved in the processing of a double-flap intermediate by employing its 3′-5′ exonuclease activity to process the upstream 3′-flap, thereby promoting removal of a TNR hairpin structure. To test this, we reconstituted BER with a substrate containing an upstream 3′-(CAG)4 flap and a downstream 5′-(CAG)3 flap with a THF residue, as well as with a substrate containing a (CAG)7 flap on both the upstream and downstream strands with a 5′-THF residue, in the absence and presence of APE1. These substrates simulate the double-flap intermediates produced by APE1 5′-incision of the (CAG)7 and (CAG)14 hairpins, respectively. We found that BER reconstituted with FEN1 (0.5 and 1 nM) and LIG I (5 nM) without and with pol β (5 nM) on the (CAG)3/(CAG)4 double-flap substrate, resulted in the production of a significant amount of the repaired unexpanded product (Figure 3A, lanes 6, 9 and 12). However, 50 and 100 nM APE1 did not significantly alter the production of the unexpanded repaired product with the substrate (Figure 3A, compare lanes 7–8 with lane 6, lanes 10–11 with lane 9 and lanes 13–14 with lane 12). Further characterization of the 3′-5′ exonuclease activity of APE1 showed that 50 and 100 nM APE1 exhibited efficient 3′-5′ exonuclease activity that generated a significant amount of exonucleolytic cleavage products (Figure 3A, lanes 4–5). This indicated that APE1 cleaved the upstream CAG repeats exonucleolytically. The results showed that APE1 exonuclease did not play a significant role in the removal of the short double-flaps, suggesting that FEN1 flap cleavage plays a predominant role in processing the short double-flaps. Interestingly, we found that BER with the substrate containing long double-flaps in the presence of FEN1 (1 nM) and LIG I (5 nM) only resulted in a series of repaired products that are longer than the repaired unexpanded product (Figure 3A, lanes 20 and 23), but shorter than the original hairpin-containing substrate. They were termed ‘repaired expanded product’ (Figure 3A, lanes 20–25). This occurred in the absence and presence of pol β (10 nM) (Figure 3A, lanes 20–25) indicating that the production of the expanded products was independent of pol β. However, we found that the presence of 50 and 100 nM APE1 resulted in the formation of repaired unexpanded product (Figure 3A, lanes 21–22 and lanes 24–25). APE1 alone failed to generate the product (Figure 3A, lanes 18–19), indicating that the product was specifically generated through BER. However, APE1 failed to affect the production of repaired expansion products during BER (Figure 3A, compare the amount of repaired expanded products in lanes 21–22 and lanes 24–25 with that in lanes 20 and 23). This suggests that APE1 promoted the complete resolution of the long double-flap intermediate resulting from a (CAG)14 hairpin, thereby specifically stimulating the production of repaired unexpanded product. To further determine the effect that is specifically from APE1 on the production of repaired unexpanded products during BER, we reconstituted BER with 50 and 100 nM APE1 without or with a low concentration of FEN1 (1 nM) with the double-flap substrates without a THF residue. We found that on the short (CAG)3/(CAG)4 double-flap substrate, APE1 (50 and 100 nM) along with LIG I (5 nM) was sufficient to generate repaired unexpanded product in the absence and presence of 10 nM pol β (Figure 3B, lanes 7–8 and lanes 10–11). This indicated that APE1 facilitated the processing of the short (CAG)3/(CAG)4 double-flap by cleaving the upstream strand via its 3′-5′ exonuclease activity independent of FEN1. The production of the repaired unexpanded product was significantly stimulated by the presence of 1 nM FEN1 (Figure 3B, left panel, lanes 13–14 and lanes 16–17). To further determine the specific effect of APE1 on the removal of the long (CAG)7/(CAG)7 double-flaps, we reconstituted BER with the substrate containing (CAG)7/(CAG)7 double-flaps without the 5′-THF residue in the absence and presence of FEN1 (1 nM) (Figure 3B, right panel). We found that 50 and 100 nM APE1 alone or APE1 along with LIG I (5 nM) in the absence and presence of pol β (10 nM) failed to produce any repair products (Figure 3B, lanes 21–22, lanes 24–25 and lanes 27–28). BER reconstituted with FEN1 and LIG I produced only repaired expanded products without and with pol β (10 nM) (Figure 3B, lanes 29 and 32). However, BER reconstituted with APE1, FEN1 and LIG I in the absence and presence of pol β resulted in the production of a significant amount of the repaired unexpanded product (Figure 3B, lanes 30–31 and lanes 33–34). The results indicated that APE1 significantly promoted the removal of both the short (CAG)3/(CAG)4 and long (CAG)7/(CAG)7 double-flap intermediates. Thus, we conclude that APE1 can facilitate the resolution of the double-flap intermediate formed during BER in a CAG repeat hairpin loop and stimulate the formation of the unexpanded repaired product. APE1 alone can lead to complete removal of a short double-flap intermediate through its 3′-5′ exonuclease activity (Figure 3B, lanes 7–8). However, it can only promote the processing of an intermediate with long double-flaps by cooperating with FEN1 flap cleavage (Figure 3B, lanes 30–31 and lanes 33–34).
Figure 3.
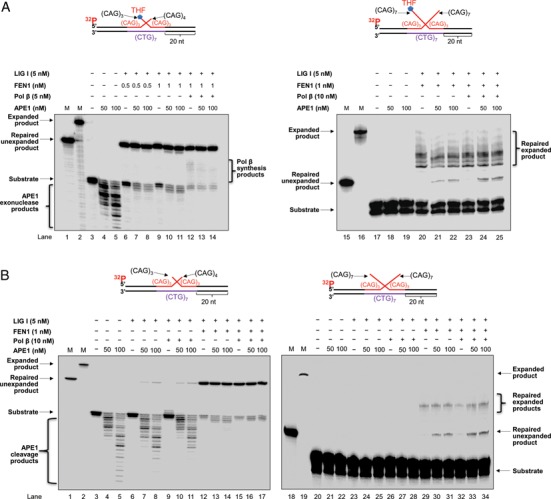
APE1 stimulates removal of a double-flap intermediate during BER. APE1 stimulation of resolution of the double-flap intermediate resulting from (CAG)7 and (CAG)14 hairpins during BER was examined by reconstituting BER with the substrates containing short (CAG)3/(CAG)4 double-flaps (left panel) or long (CAG)7/(CAG)7 double-flaps (right panel) with a THF residue (A) and without a THF residue (B) in the absence and presence of APE1. (A) Lanes 1 and 15 indicate the size markers of the template strand or unexpanded product. Lanes 2 and 16 represent the size markers of the damaged strand or expanded product. Lanes 3 and 17 represent the substrate only. Lanes 4–5 and lanes 18–19 correspond to the reaction mixture containing the substrates and 50 or 100 nM APE1. Lanes 6–11 and lanes 20–22 correspond to reactions with FEN1 (0.5 or 1 nM) and LIG I (5 nM) in the absence or presence of APE1. Lanes 12–14 and lanes 23–25 correspond to reactions with FEN1 (1 nM), LIG I (5 nM) and pol β (5 or 10 nM) in the absence and presence of APE1. (B) Lanes 1 and 18 indicate the size markers that represent the template strand or unexpanded product. Lanes 2 and 19 correspond to the size markers that illustrate the damaged strand. Lanes 3 and 20 correspond to the substrate only. Lanes 4–5 and lanes 21–22 correspond to the reaction mixture with the substrates and 50 or 100 nM APE1 only. Lanes 6–8 and lanes 23–25 correspond to reactions with the substrates and LIG I (5 nM) in the absence and presence of 50 or 100 nM APE1. Lanes 9–11 and lanes 26–28 correspond to reactions containing the substrates, pol β (10 nM) and LIG I (5 nM) in the absence and presence of APE1. Lanes 12–14 and lanes 29–31 correspond to reactions with the substrates, FEN1 (1 nM) and LIG I (5 nM) without or with APE1. Lanes 15–17 and lanes 32–34 correspond to reactions mixtures with the substrates, pol β (10 nM), FEN1 (1 nM) and LIG I (5 nM) without or with APE1.
APE1 3′-5′ exonuclease activity shortens the upstream 3′-region of a double-flap intermediate
Because our previous studies have shown that a 3′-5′ flap endonuclease, Mus81/Eme1, can facilitate the removal of a TNR hairpin by endonucleolytically cleaving a 3′-flap from the double-flap intermediate during BER (13), we further hypothesized that APE1 may promote the removal of a CAG repeat hairpin by shortening the upstream 3′-flap of a double-flap intermediate through its 3′-5′ exonuclease activity. To test this, we examined the 3′-5′ exonuclease activity of APE1 on both the (CAG)3/(CAG)4 and (CAG)7/(CAG)7 double-flap substrates without or with the THF residue, that were labeled at the 5′-end of the upstream flaps. This allowed the detection of shortened upstream flaps (Figure 4). We found that 10–100 nM APE1 exonucleolytically cleaved the upstream 3′-flap of the (CAG)3/(CAG)4 double-flap substrate, leading to the production of cleavage products that are shorter than the substrate (Figure 4, lanes 2–5 and lanes 7–10). APE1 exonucleolytic cleavage was slightly greater on the substrate lacking the THF residue (compare lanes 7–10 with lanes 2–5). The results indicated that the APE1 3′-5′ exonuclease activity efficiently processed the short 3′-upstream strand of the double-flap intermediate resulting from a small CAG repeat hairpin. To further determine if APE1 can process a long 3′-CAG repeat flap, we examined the APE1 activity on the (CAG)7/(CAG)7 double-flap substrate, without or with the THF residue. The results showed that 10–100 nM APE1 resulted in the production of a small amount of exonuclease cleavage product from the (CAG)7/(CAG)7 double-flap substrate with a THF (Figure 4 lanes 12–15). For the double-flap substrate without a THF, a small amount of APE1 exonucleolytic cleavage products was also generated (Figure 4, lanes 17–20). This indicated that APE1 3′-5′ exonuclease cleaved a long upstream 3′-flap with a low efficiency. This further suggests that the resolution of the long double-flap intermediate formed from the larger (CAG)14 hairpin during BER requires a coordinated activity between APE1 and other nucleases such as FEN1 that can resolve the double-flap intermediate through its endonucleolytic cleavage of a 5′-flap.
Figure 4.

APE1 3′–5′ exonuclease activity shortens the 3′-flap of the double-flap intermediates. APE1 3′-5′ exonuclease activity of shortening the 3′-flap of the double-flap intermediates was examined by incubating the substrates with the short (CAG)3/(CAG)4 and long (CAG)7/(CAG)7 double-flaps without or with a THF residue with increasing concentrations of APE1 ranging from 10 to 100 nM. Lanes 1, 6, 11 and 16 correspond to the substrate only. Lanes 2–5, lanes 7–10, lanes 12–15 and lanes 17–20 correspond to increasing concentrations of APE1 ranging from 10 to 100 nM. Substrates were 32P-labeled at the 5′-end of the upstream strands and are illustrated above the gels.
APE1 fails to directly stimulate FEN1 cleavage on a double-flap intermediate
APE1 has been shown to stimulate FEN1 flap cleavage during BER of an abasic lesion located in a random DNA sequence (25). To further determine if APE1 can promote the removal of a CAG repeat hairpin by facilitating FEN1 flap cleavage of the 5′-flap of a double-flap intermediate, we tested whether APE1 could stimulate FEN1 flap cleavage on the (CAG)3/(CAG)4 and (CAG)7/(CAG)7 double-flap intermediates (Figure 5). We found that in the absence and presence of 50 and 100 nM APE1, similar amounts of FEN1 cleavage products were generated by 0.5 and 1 nM FEN1 during BER of the (CAG)3/(CAG)4 and (CAG)7/(CAG)7 double-flap substrate (Figure 5, compare lanes 4–5 with lane 3, lanes 10–11 with lane 9, lanes 18–19 with lane 17 and lanes 24–25 with lane 23). We further examined if APE1 could stimulate FEN1 cleavage in the presence of pol β (10 nM). We did not observe any increase of FEN1 cleavage products in the presence of 50 and 100 nM APE1 with 10 nM pol β (Figure 5, compare lanes 7–8 with lane 6, lanes 13–14 with lane 12, lanes 21–22 with lane 20 and lanes 27–28 with lane 26). Thus, the results indicated that APE1 failed to stimulate FEN1 cleavage activity in processing the short and long double-flap intermediates during BER. We therefore concluded that the stimulation of the removal of a CAG repeat hairpin by APE1 was not the result of APE1 stimulation of FEN1 cleavage activity.
Figure 5.
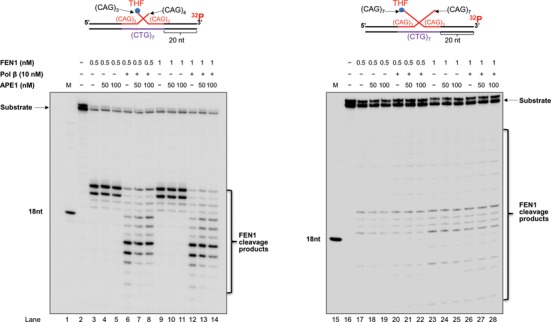
APE1 fails to stimulate FEN1 cleavage of the double-flap intermediate. The stimulatory effects of APE1 on FEN1 cleavage activity on the short (CAG)3/(CAG)4 and long (CAG)7/(CAG)7 double-flap intermediates during BER were examined by incubating 0.5 or 1 nM FEN1 with the double-flap substrates in the absence and presence of 50 or 100 nM APE1. Lanes 1 and 15 are size markers. Lanes 2 and 16 correspond to the substrate only. Lanes 3–5 and lanes 17–19 correspond to reaction mixture with FEN1 (0.5 nM) in the absence and presence of 50 or 100 nM APE1. Lanes 6–8 and lanes 20–22 correspond to reaction mixture with FEN1 (0.5 nM) and pol β (10 nM) in the absence and presence of 50 or 100 nM APE1. Lanes 9–11 and lanes 23–25 correspond to reactions with FEN1 (1 nM) in the absence and presence of 50 or 100 nM APE1. Lanes 12–14 and lanes 26–28 correspond to reactions with FEN1 (1 nM) and pol β (10 nM) in the absence and presence of 50 or 100 nM APE1. Substrates were 32P-labeled at the 3′-end of the downstream strands and are illustrated above each gel.
APE1 promotes the production of the unexpanded repair product by stimulating LIG I
A study from the Bambara group showed that APE1 can stimulate LIG I activity to promote ligation of a nick and facilitate the formation of repaired products during BER (25). Thus, it is possible that APE1 may also stimulate LIG I activity to promote the removal of a CAG repeat hairpin and facilitate the production of the repaired products during BER of a base lesion in a hairpin loop. To test this possibility, we determined the effects of APE1 on LIG I activity independent of its 3′-5′ exonuclease activity by measuring the production of the repaired products in the presence of low concentrations of APE1 (0.5 and 1 nM) that exhibited a low 3′-5′ exonuclease activity on the (CAG)3/(CAG)4 double-flap substrate (Supplementary Figure S1, lanes 3–4) and little exonucleolytic activity on the (CAG)7/(CAG)7 double-flap substrate (Supplementary Figure S1, lanes 9–10). We found that low concentrations of APE1 failed to alter the production of repaired products resulting from a short double-flap substrate (Figure 6, compare lanes 5–6 with lane 4 and lanes 8–9 with lane 7) indicating that APE1 did not significantly alter LIG I activity during removal of a small hairpin. However, the same concentrations of APE1 resulted in the production of the unexpanded product on the long (CAG)7/(CAG)7 double-flap substrate (Figure 6, compare lanes 14–15 with lane 13 and lanes 17–18 with lane 16). The results indicated that APE1 stimulated the ligation activity of LIG I, thereby promoting the production of the unexpanded repair product. Interestingly, we observed that the same concentrations of APE1 failed to stimulate the production of shortened repaired expanded products (Figure 6, lanes 14–15, lanes 17–18) suggesting that APE1 failed to stimulate the ligation of a nick by LIG I that was adjacent a hairpin structure.
Figure 6.
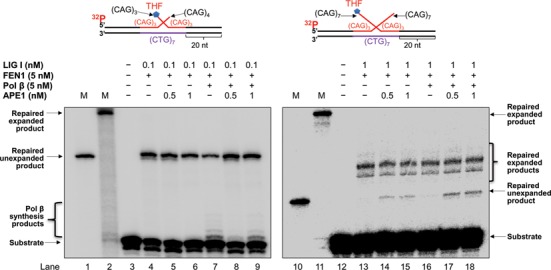
APE1 stimulates LIG I activity on a double-flap intermediate. The effects of APE1 to stimulate the ligation activity of LIG I on the short (CAG)3/(CAG)4 and long (CAG)7/(CAG)7 double-flap intermediates during BER were determined by reconstituting BER in the absence and presence of low concentrations of APE1 (0.5 and 1 nM) at which a little or no 3′-5′ exonuclease activity was observed (Supplementary Figure S1) on the short and long double-flap substrates, respectively. Lanes 1 and 10 indicate the size markers of the template strand, corresponding to complete removal of the double-flaps and full repair. Lanes 2 and 11 correspond to the markers that illustrate the length of the damaged strand containing a CAG repeat hairpin and the size of repaired expanded product. Lanes 3 and 12 correspond to the substrate only. Lanes 4–6 and lanes 13–15 correspond to reactions containing the substrates, FEN1 (5 nM) and LIG I (0.1 nM or 1 nM) in the absence and presence of 0.5 or 1 nM APE1. Lanes 7–9 and lanes 16–18 correspond to reaction mixture containing the substrates, FEN1 (5 nM), LIG I (0.1 nM or 1 nM) and pol β (5 nM) in the absence and presence of 0.5 or 1 nM APE1. Substrates were 32P-labeled at the 5′-end of the upstream strands and are illustrated above each gel.
DISCUSSION
In this study, we made the first discovery that APE1 promoted the removal of a TNR hairpin (Figures 1 and 2) by shortening the upstream 3′-flap via its 3′-5′ exonuclease activity (the left panels of Figures 3 and 4). We found that APE1 specifically facilitated the production of the repaired unexpanded product by stimulating the ligation activity of LIG I (the right panel of Figure 6). We further demonstrated that APE1 also stimulated the removal of a large (CAG)14 repeat hairpin by exonucleolytically cleaving the 3′-flap of a long (CAG)7/(CAG)7 repeat double-flap intermediate (Figure 4) as well as stimulating LIG I activity (Figure 6). This specifically facilitated the formation of the repaired unexpanded product during BER (the right panels of Figures 3A and B and 6). In addition, we showed that the stimulatory effects of APE1 on TNR hairpin removal were not the result of APE1 stimulation of FEN1, because APE1 did not show any stimulatory effects on FEN1 cleavage activity or pol β DNA synthesis (Figure 5 and Supplementary Figure S2). Our results support a model by which APE1 stimulates TNR hairpin removal to prevent TNR expansions (Figure 7), showing that upon exposure of TNR tracts to oxidative DNA damaging agents and the formation of an oxidized base lesion in the loop region of a TNR hairpin, or relocation of the lesion to the loop, a DNA glycosylase removes the damaged base leaving an abasic site. APE1 incises the 5′-side of the abasic site in the hairpin loop, converting the hairpin into a double-flap intermediate. A short double-flap intermediate converted from a small hairpin can be directly processed by APE1 via its exonucleolytic cleavage of the upstream 3′-region, shortening the flap. In coordination with FEN1 cleavage of a 5′- sugar phosphate residue, this leads to the removal of a small hairpin and prevention of repeat expansion (Figure 7, subpathway 1). A long double-flap intermediate resulting from a large hairpin is resolved by the coordination among APE1, FEN1, pol β and LIG I that results in the formation of different intermediates leading to either repeat expansion or no expansion. For the long flaps that form two adjacent small hairpins with a short 5′-flap containing a sugar phosphate, FEN1 cleaves the flap with its alternate flap cleavage activity leaving a gapped DNA. Pol β then fills in the gap leaving a nick for ligation by LIG I. The nicked intermediate with small hairpins traps a large amount of LIG I, resulting in ligation of hairpins by LIG I and the production of repaired expanded product. For this intermediate, APE1 cannot access the nick to interact with LIG I and stimulate the activity of the enzyme (Figure 7, the left side of the subpathway 2). For the long double-flap intermediate that forms a downstream long flap, FEN1 removes the flap leaving a short upstream 3′-flap. This allows the upstream flap to reanneal into an intermediate with an upstream small repeat bubble with a 3′-annealed region that is subsequently cleaved by APE1 3′-5′ exonuclease activity leaving a gapped DNA. Pol β then fills in the gap resulting in a nick, which is sealed by the limited amount of LIG I that is not bound to the nick located in between the two small hairpins. Subsequently, APE1 facilitates the binding of the limited amount of LIG I to the nick. This stimulates the ligation activity of LIG I, thereby promoting the generation of the repaired unexpanded product (Figure 7, right, subpathway 2).
Figure 7.
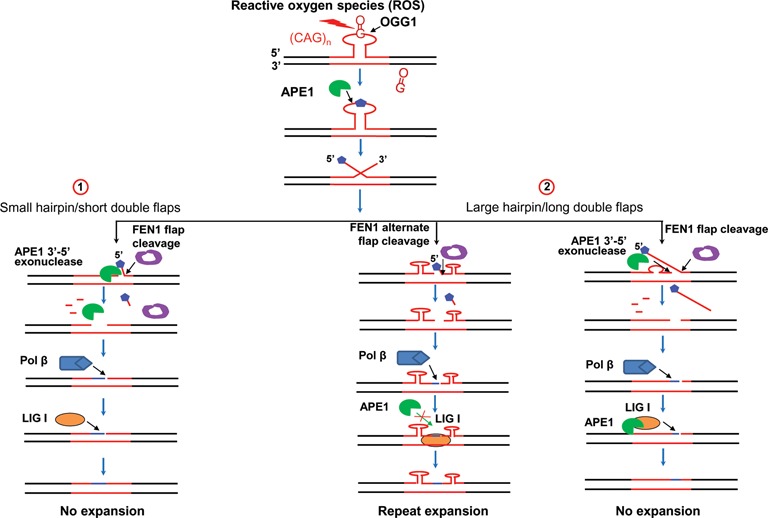
APE1 prevents trinucleotide repeat expansions during BER in a hairpin loop. During BER of a base lesion in the loop of a TNR hairpin, the damaged base is removed by a DNA glycosylase creating an abasic site. APE1 incises the 5′-side of the abasic site converting the hairpin into a double-flap intermediate. A short double-flap intermediate resulting from a small hairpin undergoes flap equilibration to allow annealing of the upstream flap to the template strand. The annealed upstream 3′-region can be processed by APE1 3′-5′ exonuclease cleavage, shortening the upstream strand to allow annealing of the downstream 5′-flap, leading to removal of a small hairpin upon FEN1 cleavage of a sugar phosphate residue, synthesis by pol β and ligation by LIG I (subpathway 1). A long double-flap intermediate formed by APE1 5′-incision of an abasic site in a large hairpin can result in different consequences upon the formation of hairpin structures by the flaps. If the double-flaps form two small hairpins adjacent to each other, FEN1 removes a sugar phosphate with a short flap through alternate flap cleavage activity, leaving a gapped DNA. Pol β then fills in the gap, leaving a nick for ligation by LIG I. This intermediate traps a large quantity of LIG I, resulting in ligation of hairpins by LIG I and repeat expansion. For this intermediate, APE1 fails to access the nick to interact with LIG I to stimulate the activity of the enzyme (left, subpathway 2). For the long double-flap intermediates that form a downstream long flap, FEN1 removes the flap leaving a short upstream 5′-flap. This allows the upstream flap to reanneal into an intermediate with an upstream small repeat bubble with a 3′-annealed region that is subsequently cleaved by APE1 3′-5′ exonuclease activity leaving gapped DNA. Pol β then fills in the gap resulting in a nick that is sealed by the limited amount of LIG I. Subsequently, APE1 facilitates the limited amount of LIG I to bind to the nick, stimulating LIG I activity, facilitating the removal of the double-flaps and generation of the unexpanded product (right, subpathway 2).
Here, for the first time, we identified a new role of APE1 in the resolution of the upstream 3′-flap formed during removal of a TNR hairpin through BER. Our findings also provide new insight into how the BER enzymes APE1, pol β, FEN1 and LIG I can cooperate to remove TNR hairpins and maintain TNR stability. Although the APE1 exonuclease activity has been shown to be able to remove 3′-mismatched bases and 3′-blocking groups (17,34,36–38), the biological function of the 3′-5′ exonuclease of APE1 remains to be elucidated. Our study demonstrates that during BER in a hairpin loop, APE1 promoted the formation of repaired unexpanded product specifically by exonucleolytically cleaving the upstream strand of a double-flap intermediate and stimulating the LIG I activity in sealing a nick. We further verified the exonucleolytic cleavage of APE1 on both the short and long double-flap substrates specifically to examine if the 3′-5′ exonuclease activity of APE1 is indeed involved in the resolution of both of the short and long double-flaps. Our results showed that APE1 exhibited efficient 3′-5′ exonuclease activity on the short double-flap substrate by predominantly cleaving two repeat units from the short upstream strand (Figure 4, lanes 2–5 and lanes 7–10), whereas it mainly cleaved one repeat unit from the long upstream flap (Figure 4, lanes 13–15 and lanes 19–20). The results indicate that APE1 3′-5′ exonucleolytic cleavage of the upstream flaps plays an important role in preventing and attenuating TNR expansion during the repair of both small- and large-hairpin structures.
Our previous study has shown that the 3′-5′ flap endonuclease Mus81/Eme1 protein complex prevents TNR expansions by cleaving the upstream 5′-flap (13). This indicates that any nuclease that can cleave the upstream 5′-flap may lead to the prevention of TNR repeat expansions during BER in a hairpin loop. Although the 3′-5′ exonuclease activity of APE1 has been well established and proposed to serve as a proof-reading enzyme for pol β during BER, its biological function remains to be elucidated. In this study, for the first time, we reveal a novel function of APE1 3′-5′ exonuclease activity and APE1 stimulation on the activity of LIG I in preventing TNR expansions via its unique coordination with other BER enzymes. Although the APE1 exonuclease activity is weaker than its AP endonuclease activity and accomplished by a relatively high concentration of the enzyme, the abundance of the enzyme in mammalian cells (105–106 molecules/cell) (16) appears to allow it to act as an efficient 3′-5′ exonuclease for sustaining TNR stability in cells.
Our results also suggested that for a double-flap intermediate, the upstream flap annealed back to the template strand creating a downstream 5′-flap. Subsequently, APE1 3′-5′ exonuclease cleaved the upstream strand creating an upstream gap into which the downstream 5′-flap reannealed to generate a nick. This is supported by the observation that APE1 exonuclease failed to make cleavage on a substrate containing an upstream 3′-flap that fails to anneal with the template strand, indicating that APE1 3′-5′ exonuclease cannot endonucleolytically cleave a single-stranded 3′-flap (36). This further indicates that the APE1 3′-5′ exonuclease activity was accomplished through the reannealing of the upstream 3′-flap to the template strand that creates a nick or gap in a DNA duplex through flap equilibration (39).
Our results showed that APE1 specifically stimulated the production of the repaired unexpanded product, but not the production of repaired expanded products via its 3′-5′ exonuclease activity (Figure 4) as well as its stimulation of the activity of LIG I (Figure 6, lanes 14–15 and lanes 17–18). This was indicated by the production of the repaired unexpanded product during BER with the long double-flap substrate in the presence of low concentrations of APE1 (Figure 6, compare lanes 14–15 with lane 13 and lanes 17–18 with lane 16) that exhibit little 3′-5′ exonuclease activity (Supplementary Figure S1). This indicates that APE1 can specifically promote the formation of repaired unexpanded product from BER of a long double-flap substrate by stimulating LIG I activity independent of its 3′-5′ exonuclease activity. The production of a large quantity of repaired expanded products suggests that the products were generated by ligation of a nick that was flanked by a small upstream and downstream CAG repeat hairpin structure that provided a limited space for LIG I alone to bind to the nick. This further prevented the binding of both LIG I and APE1 to the nick, thereby preventing any effects from APE1 on LIG I activity. Moreover, it is possible that the nick flanked by two CAG repeat hairpins may also trap LIG I, thereby resulting in depletion of LIG I that reduced the availability of LIG I for generating the unexpanded product. In this scenario, APE1 became critical for recruiting the limited amount of LIG I to bind to the nick at a duplex DNA resulting from the complete removal of the double flaps. This led to stimulation of the formation of the unexpanded product. This notion was supported by our results showing that APE1 failed to stimulate LIG I activity during BER with the short double-flap substrate that led to the production of a nick in a duplex DNA. This further suggests that there was a sufficient amount of LIG I to carry out efficient ligation on a nick in duplex DNA during BER with a short double-flap intermediate because the short double-flaps did not form hairpin structures. This was also supported by the fact that LIG I activity at a nick on duplex DNA can be stimulated by APE1 at a very low concentration of LIG I (0.06–0.1 nM) (25). Our results indicate that APE1 promoted the removal of a small TNR hairpin by employing its 3′-5′ exonuclease activity. It facilitated the removal of a large TNR hairpin by both its 3′-5′ exonuclease activity and its stimulation of LIG I activity.
FEN1 was previously shown by the Bambara group to be stimulated by APE1 through a direct interaction between the two enzymes (25). However, we failed to observe the stimulatory effect on the double-flap substrates. It is possible that with the double flap intermediate, the upstream flap may interrupt the direct interaction between APE1 and FEN1, thereby eliminating APE1 stimulatory effect on FEN1 flap cleavage. It is conceivable that APE1 can be bound to the upstream region of the double-flap intermediate, which is too far away from the base of the downstream 5′-flap of the intermediate. This would prevent the direct interaction between the enzymes and the stimulatory effect.
It should be noted that we found that the formation of unexpanded repaired product from both the short and long double-flap substrates was promoted in the presence of pol β (Figure 1, lanes 10–11 and lanes 21–22, Figure 2, lanes 10–11 and lanes 21–22, Figure 3A, lanes 24–25, Figure 3B, lanes 33–34 and Figure 6, lanes 17–18). However, the formation of the unexpanded product from both the short and long double-flap substrates was also observed in the absence of pol β (Figure 1, lanes 6–8 and lanes 18–19, Figure 2, lanes 7–8 and lanes 18–19, Figure 3A, lanes 6–11 and lanes 21–22, Figure 3B, lanes 7–8, lanes 12–14 and lanes 30–31 and Figure 6, lanes 4–6 and lanes 14–15). This indicates that pol β is not required for the production of the unexpanded product during BER. This is because for both a short and long double-flap intermediate, FEN1 cleavage of the downstream flap along with APE1 3′-5′ exonucleolytic cleavage of the upstream strand can result in the production of nicked DNA for ligation as well as gapped intermediates that need to be filled in by pol β gap-filling synthesis for generating nicked DNA for ligation and completion of repair. In addition, we found that the production of the unexpanded product was slightly stimulated by the presence of pol β with both the short and long double-flap substrates, indicating that there is no preference for pol β in the repair of a short or long double-flap intermediate during BER.
Our results showed that repair of the long double-flap substrates led to the formation of expanded products. This is because the long flaps can reanneal with the template to form an intermediate with a small hairpin in the upstream or/and the downstream strand attached with a short flap as described in our previous study (13). This further indicates that a hairpin intermediate formed in the downstream flap can be further processed by FEN1 alternate flap cleavage resulting in ligation of a small hairpin and production of shortened expanded products, thereby attenuating repeat expansion (13). However, our results also demonstrated that a significant amount of unexpanded product was generated during repair of the long double-flap intermediate, suggesting that the upstream and downstream flaps were removed completely by the cooperative cleavage activities of APE1 3′-5′ exonuclease activity and FEN1 flap cleavage through their direct cleavage of the flaps as well as their cleavage of a series of short flaps formed by a long flap in a stepwise manner.
Previous studies from the Delaney group have shown that a hairpin loop is more susceptible to the formation of oxidative DNA damage such as 8-oxoG than duplex DNA (40). Moreover, an 8-oxoG formed in the stem region of a hairpin can relocate to the loop region (41), indicating that the oxidized base lesion preferentially accumulates in a hairpin loop. Although this can further reduce OGG1 binding ability to the oxidized base lesion (12), we have demonstrated that OGG1 can efficiently remove an 8-oxoG located in the loop of a TNR hairpin within a TNR duplex tract, leaving an abasic site in the loop region. Subsequently, APE1 incises the 5′-side of the abasic site cleaving the loop and converting the hairpin into a double-flap intermediate. This results in removal of the hairpin through BER (13), demonstrating that BER in a TNR hairpin loop can lead to prevention and attenuation of TNR expansion. Employing synthetic zinc finger endonucleases, which specifically target hairpin structures of a specific sequence (CAG/CTG), the Leffak group has found that a CAG and CTG repeat hairpin structure containing about 46 repeats can be generated in cells (42). Thus, it is possible that the hairpin substrates with seven more repeats in the upper strand than the lower strand used in our study can be readily generated as a natural DNA replication and repair intermediate in cells. Here, we used the substrates containing an 8-oxoG and abasic lesion in the loop region of a TNR hairpin to mimic a damaged TNR hairpin and its repair intermediate that can be generated during BER to study the mechanism of BER in a hairpin loop. We found that BER was not significantly affected by a TNR hairpin structure.
In this study, we have identified a mechanism by which APE1 prevents TNR expansion with an in vitro biochemical approach. Since there is no in vivo system that has been established to study a mechanism that involves BER protein coordination in modulating TNR stability as yet, development of such a type of in vivo system would be helpful for further elucidating this mechanism in mammalian cells.
Our previous studies have shown that alkylated DNA damage can be induced in expanded GAA repeats associated with Friedreich's ataxia by the chemotherapeutic drug, temozolomide and BER of the alkylated DNA damage subsequently results in large GAA repeat deletions (43). This occurs as a result of the formation of a TTC loop in the template strand and a downstream GAA repeat flap. Pol β then skips over the loop, and FEN1 cleaves the GAA repeat flap allowing the removal of more GAA repeats than pol β synthesizes. This subsequently leads to repeat deletions (43). The study indicates that chemotherapeutically induced DNA damage can shorten expanded TNR tracts via BER, and thus potentially be used as novel treatment of TNR expansion-induced neurodegeneration. In this study, we further demonstrated that BER of a DNA base lesion in a hairpin loop resulted in removal of the hairpin through the coordination among APE1 3′-5′ exonuclease activity, FEN1 flap cleavage activity and LIG I activity, indicating that the removal of a TNR hairpin via the coordination among major BER enzymes during BER in a hairpin loop also serves as one of the mechanisms that shortens expanded TNR tracts. Our results suggest that multiple mechanisms underlie the shortening of expanded TNR tracts induced by oxidative or alkylating DNA damage via BER as a potential therapy for TNR expansion-related neurodegenerative diseases.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Samuel H. Wilson, Laboratory of Structural Biology, National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health (NIH) for generously providing purified BER enzymes and plasmids for expressing BER enzymes.
FUNDING
National Institutes of Health [R01 ES023569 to Y.L., R25 GM061347 to J.M.B.]. Florida International University RISE Fellowship [to J.M.B.]. Funding for open access charge: National Institutes of Health [R01 ES023569].
Conflict of interest statement. None declared.
REFERENCES
- 1.Paulson H.L., Fischbeck K.H. Trinucleotide repeats in neurogenetic disorders. Annu. Rev. Neurosci. 1996;19:79–107. doi: 10.1146/annurev.ne.19.030196.000455. [DOI] [PubMed] [Google Scholar]
- 2.McMurray C.T. Mechanisms of trinucleotide repeat instability during human development. Nat. Rev. Genet. 2010;11:786–799. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wells R.D., Dere R., Hebert M.L., Napierala M., Son L.S. Advances in mechanisms of genetic instability related to hereditary neurological diseases. Nucleic Acids Res. 2005;33:3785–3798. doi: 10.1093/nar/gki697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McMurray C.T. DNA secondary structure: a common and causative factor for expansion in human disease. Proc. Natl. Acad. Sci. U.S.A. 1999;96:1823–1825. doi: 10.1073/pnas.96.5.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wells R.D. Molecular basis of genetic instability of triplet repeats. J. Biol. Chem. 1996;271:2875–2878. doi: 10.1074/jbc.271.6.2875. [DOI] [PubMed] [Google Scholar]
- 6.Mirkin S.M. Expandable DNA repeats and human disease. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 7.Kovtun I.V., Liu Y., Bjoras M., Klungland A., Wilson S.H., McMurray C.T. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447–452. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu M., Gabison J., Liu Y. Trinucleotide repeat deletion via a unique hairpin bypass by DNA polymerase beta and alternate flap cleavage by flap endonuclease 1. Nucleic Acids Res. 2013;41:1684–1697. doi: 10.1093/nar/gks1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y., Prasad R., Beard W.A., Hou E.W., Horton J.K., McMurray C.T., Wilson S.H. Coordination between polymerase beta and FEN1 can modulate CAG repeat expansion. J. Biol. Chem. 2009;284:28352–28366. doi: 10.1074/jbc.M109.050286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y., Wilson S.H. DNA base excision repair: a mechanism of trinucleotide repeat expansion. Trends Biochem. Sci. 2012;37:162–172. doi: 10.1016/j.tibs.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lai Y., Xu M., Zhang Z., Liu Y. Instability of CTG repeats is governed by the position of a DNA base lesion through base excision repair. PLoS One. 2013;8:e56960. doi: 10.1371/journal.pone.0056960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jarem D.A., Wilson N.R., Schermerhorn K.M., Delaney S. Incidence and persistence of 8-oxo-7,8-dihydroguanine within a hairpin intermediate exacerbates a toxic oxidation cycle associated with trinucleotide repeat expansion. DNA Repair (Amst.) 2011;10:887–896. doi: 10.1016/j.dnarep.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu M., Lai Y., Torner J., Zhang Y., Zhang Z., Liu Y. Base excision repair of oxidative DNA damage coupled with removal of a CAG repeat hairpin attenuates trinucleotide repeat expansion. Nucleic Acids Res. 2014;42:3675–3691. doi: 10.1093/nar/gkt1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demple B., Herman T., Chen D.S. Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes. Proc. Natl. Acad. Sci. U.S.A. 1991;88:11450–11454. doi: 10.1073/pnas.88.24.11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demple B., Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 16.Chen D.S., Herman T., Demple B. Two distinct human DNA diesterases that hydrolyze 3′-blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Res. 1991;19:5907–5914. doi: 10.1093/nar/19.21.5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li M., Wilson D.M. 3rd. Human apurinic/apyrimidinic endonuclease 1. Antioxid. Redox Signal. 2014;20:678–707. doi: 10.1089/ars.2013.5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xanthoudakis S., Smeyne R.J., Wallace J.D., Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8919–8923. doi: 10.1073/pnas.93.17.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marenstein D.R., Wilson D.M. 3rd, Teebor G.W. Human AP endonuclease (APE1) demonstrates endonucleolytic activity against AP sites in single-stranded DNA. DNA Repair (Amst.) 2004;3:527–533. doi: 10.1016/j.dnarep.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 20.Hill J.W., Hazra T.K., Izumi T., Mitra S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001;29:430–438. doi: 10.1093/nar/29.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vidal A.E., Hickson I.D., Boiteux S., Radicella J.P. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: bypass of the AP lyase activity step. Nucleic Acids Res. 2001;29:1285–1292. doi: 10.1093/nar/29.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waters T.R., Gallinari P., Jiricny J., Swann P.F. Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J. Biol. Chem. 1999;274:67–74. doi: 10.1074/jbc.274.1.67. [DOI] [PubMed] [Google Scholar]
- 23.Parikh S.S., Mol C.D., Slupphaug G., Bharati S., Krokan H.E., Tainer J.A. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 1998;17:5214–5226. doi: 10.1093/emboj/17.17.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parikh S.S., Mol C.D., Hosfield D.J., Tainer J.A. Envisioning the molecular choreography of DNA base excision repair. Curr. Opin. Struct. Biol. 1999;9:37–47. doi: 10.1016/s0959-440x(99)80006-2. [DOI] [PubMed] [Google Scholar]
- 25.Ranalli T.A., Tom S., Bambara R.A. AP endonuclease 1 coordinates flap endonuclease 1 and DNA ligase I activity in long patch base excision repair. J. Biol. Chem. 2002;277:41715–41724. doi: 10.1074/jbc.M207207200. [DOI] [PubMed] [Google Scholar]
- 26.Dianova II, Bohr V.A., Dianov G.L. Interaction of human AP endonuclease 1 with flap endonuclease 1 and proliferating cell nuclear antigen involved in long-patch base excision repair. Biochemistry. 2001;40:12639–12644. doi: 10.1021/bi011117i. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y., Prasad R., Beard W.A., Kedar P.S., Hou E.W., Shock D.D., Wilson S.H. Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta. J. Biol. Chem. 2007;282:13532–13541. doi: 10.1074/jbc.M611295200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bennett R.A., Wilson D.M. 3rd, Wong D., Demple B. Interaction of human apurinic endonuclease and DNA polymerase beta in the base excision repair pathway. Proc. Natl. Acad. Sci. U.S.A. 1997;94:7166–7169. doi: 10.1073/pnas.94.14.7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sukhanova M.V., Khodyreva S.N., Lebedeva N.A., Prasad R., Wilson S.H., Lavrik O.I. Human base excision repair enzymes apurinic/apyrimidinic endonuclease1 (APE1), DNA polymerase beta and poly(ADP-ribose) polymerase 1: interplay between strand-displacement DNA synthesis and proofreading exonuclease activity. Nucleic Acids Res. 2005;33:1222–1229. doi: 10.1093/nar/gki266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mol C.D., Izumi T., Mitra S., Tainer J.A. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected] Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 31.Wilson S.H., Kunkel T.A. Passing the baton in base excision repair. Nat. Struct. Biol. 2000;7:176–178. doi: 10.1038/73260. [DOI] [PubMed] [Google Scholar]
- 32.Seki S., Ikeda S., Watanabe S., Hatsushika M., Tsutsui K., Akiyama K., Zhang B. A mouse DNA repair enzyme (APEX nuclease) having exonuclease and apurinic/apyrimidinic endonuclease activities: purification and characterization. Biochim. Biophys. Acta. 1991;1079:57–64. doi: 10.1016/0167-4838(91)90024-t. [DOI] [PubMed] [Google Scholar]
- 33.Cistulli C., Lavrik O.I., Prasad R., Hou E., Wilson S.H. AP endonuclease and poly(ADP-ribose) polymerase-1 interact with the same base excision repair intermediate. DNA Repair (Amst.) 2004;3:581–591. doi: 10.1016/j.dnarep.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 34.Chou K.M., Cheng Y.C. An exonucleolytic activity of human apurinic/apyrimidinic endonuclease on 3′ mispaired DNA. Nature. 2002;415:655–659. doi: 10.1038/415655a. [DOI] [PubMed] [Google Scholar]
- 35.Chou K.M., Cheng Y.C. The exonuclease activity of human apurinic/apyrimidinic endonuclease (APE1). Biochemical properties and inhibition by the natural dinucleotide Gp4G. J. Biol. Chem. 2003;278:18289–18296. doi: 10.1074/jbc.M212143200. [DOI] [PubMed] [Google Scholar]
- 36.DM 3rd Wilson. Properties of and substrate determinants for the exonuclease activity of human apurinic endonuclease Ape1. J. Mol. Biol. 2003;330:1027–1037. doi: 10.1016/s0022-2836(03)00712-5. [DOI] [PubMed] [Google Scholar]
- 37.Chou K.M., Kukhanova M., Cheng Y.C. A novel action of human apurinic/apyrimidinic endonuclease: excision of L-configuration deoxyribonucleoside analogs from the 3′ termini of DNA. J. Biol. Chem. 2000;275:31009–31015. doi: 10.1074/jbc.M004082200. [DOI] [PubMed] [Google Scholar]
- 38.Parsons J.L., Dianova II, Dianov G.L. APE1-dependent repair of DNA single-strand breaks containing 3′-end 8-oxoguanine. Nucleic Acids Res. 2005;33:2204–2209. doi: 10.1093/nar/gki518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y., Zhang H., Veeraraghavan J., Bambara R.A., Freudenreich C.H. Saccharomyces cerevisiae flap endonuclease 1 uses flap equilibration to maintain triplet repeat stability. Mol. Cell Biol. 2004;24:4049–4064. doi: 10.1128/MCB.24.9.4049-4064.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jarem D.A., Wilson N.R., Delaney S. Structure-dependent DNA damage and repair in a trinucleotide repeat sequence. Biochemistry. 2009;48:6655–6663. doi: 10.1021/bi9007403. [DOI] [PubMed] [Google Scholar]
- 41.Volle C.B., Jarem D.A., Delaney S. Trinucleotide repeat DNA alters structure to minimize the thermodynamic impact of 8-oxo-7,8-dihydroguanine. Biochemistry. 2012;51:52–62. doi: 10.1021/bi201552s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu G., Chen X., Bissler J.J., Sinden R.R., Leffak M. Replication-dependent instability at (CTG) x (CAG) repeat hairpins in human cells. Nat. Chem. Biol. 2010;6:652–659. doi: 10.1038/nchembio.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lai Y., Beaver J.M., Lorente K., Melo J., Ramjagsingh S., Agoulnik I.U., Zhang Z., Liu Y. Base excision repair of chemotherapeutically-induced alkylated DNA damage predominantly causes contractions of expanded GAA repeats associated with Friedreich's ataxia. PLoS One. 2014;9:e93464. doi: 10.1371/journal.pone.0093464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.