

Abstract
Schizophrenia is a severe mental illness with neuropathology in many regions, including the striatum. The typical symptoms of this disease are psychosis (such as hallucinations and delusions), cognitive impairments and the deficit syndrome. Not all patients respond to treatment and in those who do, only psychotic symptoms are improved. Imaging studies support a biological distinction between treatment response and resistance, but postmortem examinations of this issue are rare. The present study tests the hypotheses that abnormalities in mitochondria, the energy producing organelles in the cell, may correlate with treatment response. Postmortem striatal tissue was obtained from the Maryland Brain Collection. The density of mitochondria (in various neuropil compartments) and the number of mitochondria per synapse (all types of synapses combined) were tallied using electron microscopy and stereology in striatum from schizophrenia subjects (rated treatment responsive or not) and normal controls. The number of mitochondria per synapse was significantly different among groups for both the caudate nucleus (p<0.025) and putamen (p<0.002). Compared to controls, treatment responsive schizophrenia subjects had a 37-43% decrease in the number of mitochondria per synapse in the caudate nucleus and putamen. In the putamen, treatment responsive subjects also had decreases in this measure compared to treatment resistant subjects (34%). Our results provide further support for a biological distinction between treatment response and treatment resistance in schizophrenia. Since treatment responders have fewer mitochondria per synapse than controls, while the treatment resistant subjects have similar results to that of controls, fewer mitochondria per synapse may be related to treatment response.
Keywords: synapses, postmortem, antipsychotic drugs, treatment resistance
Introduction
Schizophrenia (SZ) is a severe mental illness affecting approximately 1% of the population. The illness has varied symptoms including hallucinations, delusions, cognitive deficits, disorganization and negativity (DSM-IV). There appears to be several risk factors for the disease, the etiology is unclear, and neuropathological findings are often nonreplicated (Lewis and Lieberman, 2000; Harrison and Weinberger, 2005). Pathologies associated with SZ are found throughout the brain and are often subtle (Harrison, 1999; Powers, 1999; Harrison and Weinberger, 2005). Among the many anomalies in SZ are those related to mitochondrial function (Ben-Shachar, 2002; Ben-Shachar and Laifenfeld, 2004; Karry et al., 2004). Evidence of mitochondrial dysfunction in SZ includes genetic (Marchbanks et al., 2003; Kvajo et al., 2008), metabolic (Prabakaran et al., 2004), enzymatic (Prince et al., 1999, 2000; Maurer et al., 2001) and anatomic abnormalities (Kung and Roberts, 1999; Kolomeets and Uranova, 2009).
Mitochondria are responsible for essential cellular functions including producing 95% of cellular energy using the electron transport chain (Wong-Riley, 1989), buffering intracellular calcium (Gunter et al., 1994; Duchen et al., 2008), producing reactive oxygen species (Chang and Reynolds 2006), regulating apoptosis (Susin et al., 1999) and modulating synaptic activity (Li et al., 2004; Miller and Sheetz, 2004). The production of ATP and calcium buffering are essential in maintaining synaptic strength and abnormalities in these processes could lead to decreased metabolism and defective synaptic activity (Ben-Shachar and Laifenfeld, 2004; Chang and Reynolds, 2006; Duchen et al., 2008).
The focus of the present study is the striatum because it is a region of convergence of glutamate and dopamine, two transmitters implicated in SZ (Carlsson and Lindqvist, 1963; Carlsson, 1978; Krystal et al., 1994; Goff and Coyle, 2001). Investigation of the striatum in SZ has shown neurochemical alterations (Carlsson, 1978; Laruelle et al., 1996, 1999), metabolic abnormalities (Wiesel et al., 1987; Resnick et al., 1988; Buchsbaum et al., 1992; Shihabuddin et al., 1998) and ultrastructural anomalies (Roberts et al., 1996; 2005a,b, 2008, 2009; Uranova et al., 1996, 2001). Several mitochondrial electron transport enzymes are affected in SZ. For example, there are reports of decreases in complex I and III activity, protein and/or mRNA levels (Prince et al., 1999; Maurer et al., 2001; Ben-Shachar and Karry, 2008), and differential changes in complex IV in the caudate nucleus and putamen (Cavelier et al., 1995; Prince et al., 1999).
Our previous studies have found an increase in synaptic number in the caudate nucleus and putamen striosomes in postmortem SZ subjects (Roberts et al., 2005a,b). In a later study we examined the relationship of striatal dopamine terminals and treatment response and found more labeled terminals in treatment responders than controls or treatment resistant subjects (Roberts et al., 2009). These results were consistent with numerous neuroimaging studies, which have shown a relationship between pathophysiology and the degree of treatment response in SZ (Beerpoot et al., 1996; Sheitman and Lieberman, 1998; Arango et al., 2003; Altamura et al., 2005). In the present study we sought to determine if there was a link in the striatum between treatment response and mitochondria. Therefore we examined mitochondrial number in postmortem striatum from SZ subjects subdivided based on treatment response/resistance as defined by Kane et al. (1988) and revised by Conley and Kelly (2000, 2001). This study has been previously presented in preliminary form (Somerville et al., 2003).
Materials and Methods
Subjects
Postmortem human striatum was obtained with family permission from the Maryland Brain Collection. The tissue was collected within 8 hours of death from adult subjects with SZ (n=13) and normal controls (NCs) (n=8). The NCs had no history of brain disease and were matched to SZ subjects for age, gender, postmortem interval and race, when possible. These protocols were approved by the Institutional Review Boards at the University of Maryland and the University of Alabama. Table I shows group means for demographics, tissue and psychiatric information.
Table I.
Demographic and Diagnostic Chart
| Controls (n=8) | SZ: Treatment Responders (n=8) | SZ: Resistant (n=3) | df (t or F)p value | |
|---|---|---|---|---|
| Age in years | 43±17 | 52±11 | 43±10 | 16 (1.020) p<0.383 |
| Race | 3AA, 5C | 4AA, 4C | 3C | 16 (1.881) p<0.185 |
| Gender | 5M, 3F | 5M, 3F | 2M, 1F | 16 (0.008) p<0.992 |
| PMI in hours | 5.4±1.6 | 4.8±1.5 | 6.7±1.5 | 16 (1.849) p<0.189 |
| pH | 7.03±0.29 | 6.97±0.26 (n=6) | 7.1±0.2 | 12 (0.264) p<0.773 |
| DSM-IV diagnosis | NA | 4CUT, 3 P, 1unk | 2CUT, 1P | 8 (0.253) p<0.807 |
| Antipsychotic drugs | NA | 6 typ, 2 atyp | 3 atyp | 9 (4.583) p<0.003) |
| Fluphenazine ~ | NA | 9.50±7.78 (n=3) | 8.00±0 (n=1) | 1 (−.0157) p<0.901 |
| Age of onset years | NA | 24.0±5.6 (n=4) | 21.5±2.1 | 4 (−0.582) p<0.592 |
| Length of illness years | NA | 22.5±12.8 (n=4) | 28.0±1.4 | 4 (0.573) p<0.597 |
The mean ± SD is shown for NCs, and SZs divided by treatment response. Antipsychotic drug status reflects the last six months before death: typ, typical; atyp, atypical. Degrees of freedom for the ANOVA are shown for within groups. Fluphenazine equivalents, age of onset, and length of illness were known only in some cases. Abbreviations: NC: normal control. SZ: subject with schizophrenia. PMI: postmortem interval. AA: African American. C: Caucasian. M: male. F: female. DSM-IV: diagnostic statistical manual subtype. CUT: chronic undifferentiated type. P: paranoid. unk: unknown. NA, not applicable.
Diagnoses
Drug therapy, duration of illness and other details were obtained from autopsy reports, psychiatric records, and family interviews using the Scheduled Clinical Interview for the DSM IV (SCID) (Spitzer et al., 1992). The diagnoses were made by two research psychiatrists according to the DSM-IV criteria using the Diagnostic Evaluation After Death (DEAD) (Salzman et al., 1983). Subjects and results were divided by treatment response into three categories: 1) normal controls, 2) treatment responsive and 3) treatment resistant. Two off drug subjects were included in the study in a separate group for the purpose of comparing fluphenazine equivalents to the number of mitochondria per synapse. The diagnoses of treatment resistance or responsiveness is not a standard feature diagnosed in postmortem collections, however we have previously done so and methods are described in detail in Roberts et al., (2009). Briefly, treatment resistance was diagnosed based on the criteria described by Conley and Kelly (Conley and Kelly 2000, 2001), which is a modification of the Kane criteria (Kane et al., 1988). These criteria are: 1) presence of a drug-refractory condition, defined as at least two treatment periods of adequate length and dosage with no clinical improvement; 2) persistence of the illness for at least five years with no periods of good social or occupational function; and 3) the presence of persistent positive psychotic symptoms (e.g., hallucinations, delusions) throughout the person's life. If all criteria were present, a diagnosis of treatment resistance was made.
Tissue Processing
Coronal blocks from the anterior caudate nucleus and putamen were immersed in a cold solution of 4% paraformaldehyde and 1% glutaraldehyde in 0.1M phosphate buffer (PB), pH=7.4 at 4 °C. Six series of sections, cut on a vibratome at 40μm, were collected in cold PB. One series of samples was flat embedded using standard techniques (Perez-Costas et al., 2007). Briefly, the sections were rinsed (3×10 minutes) in PB, placed in 1% osmium tetroxide (1 hour), dehydrated in ascending concentrations of alcohol, stained with uranyl acetate (2 hours), further dehydrated in ascending concentrations of alcohol, mixtures of propylene oxide and resin. Finally, the tissue was embedded in resin on glass slides and heated at 60 °C for 72 hours. Samples were excised from ventral and dorsal striatal regions from several of the embedded sections per case, mounted on beem capsules, then serially thin-sectioned on an ultramicrotome at a thickness of 90nm. The average length of each ribbon was 15 serial sections.
Although a short postmortem interval is the most crucial measure of tissue integrity for electron microscopy, we performed pH measurements as a marker of general tissue integrity. Previous studies have shown that the pH of the cerebellum is an acceptable and reliable measure of overall brain pH (Stan et al., 2006). To determine pH, a sample piece of cerebellum (a 1.5 - 2.0 cm block) was dissected from the frozen brain with a Stryker autopsy bone saw. After the sample was thawed out, it was homogenized with an Omni PCR Tissue Homogenizing Kit, and the pH was measured according to published techniques (Harrison et al., 1995; Johnson et al., 1996; Johnston et al., 1997).
Data Collection and Analysis
Regions were selected for photography and the disector stereology technique (Sterio, 1984) was used to tally the number of mitochondria. Synaptic density was obtained from the same cases and micrographs by the same methods and has been reported previously (Roberts et al., 2005a). In each section, 3-4 photomicrographs (at a magnification of 10,000×) were taken that formed a montage. These montages were printed (final viewing magnification was approximately 25,000×), and a counting box (with an area of approximately 100μm2) was drawn in each. To obtain volume measurements, the area of the counting box was multiplied by the thickness of the disector height (90nm) and the number of disectors as described in Geinisman et al., (1996) and in Perez-Costas et al., (2007). Mitochondria were tallied only in the neuropil and glial processes, but not in the somata. At least 2 blocks were analyzed per region, per case, yielding at least 100-200 mitochondria analyzed per region per case. The analysis of synaptic and mitochondrial number was performed with the experimenter blind to the case identity.
The criteria for identifying mitochondria were the presence of distinctive cristae and a double membrane. Neuropil structures were identified as axon terminals (presence of three or more synaptic vesicles), dendrites (postsynaptic to a synapse or having an attached spine), myelinated axons (presence of a myelin sheath), or astroglial processes (presence of fibrils and watery cytoplasm). Some neuronal processes had no distinguishing features and were unidentifiable; the mitochondria in these structures were included in total neuropil counts but excluded from the subcellular analyses. Synapses were identified by the presence of three or more synaptic vesicles at the synapse in the presynaptic terminal, parallel pre-and post-synaptic membranes with a discernable synaptic cleft, and a postsynaptic density. For this analysis all synapses were counted but the number of mitochondria per synapse was not differentiated by synaptic subtype. Both the number of mitochondria in axon terminals and the number of mitochondria per axon terminal forming a synapse (hereafter called mitochondria per synapse) were measured because the number of mitochondria in axon terminals does not take into account whether the terminal is forming a synapse or whether the total number of axon terminals varies.
Statistics
Group means and standard deviations (SD) were obtained for each group. Analysis of variance (ANOVA) followed by LSD post-hoc tests were used for between group comparisons. Diagnostic characteristics were compared between treatment responders vs. treatment resistant SZ using a Students independent t-test. Since there was a significant difference in the type of antipsychotic drugs used between the treatment responsive SZs and the treatment resistant SZs, we performed a linear regression and correlation analysis between fluphenazine equivalents and mitochondria per synapse. For this analysis we included the off drug SZ subjects. A Pearson bivariate correlation was used with a 2-tailed significance level. Mitochondrial density is reported as the number ± SD of mitochondria per 100 μm3 or the number ±SD per synapse.
Results
The electron micrographs demonstrate that the integrity of the tissue was equivalent between normal controls and cases with SZ in both the caudate nucleus (Figures 1-2) and the putamen (Figure 3). Mitochondria are evident in the neuropil in various structures including dendrites, axon terminals, and myelinated axons (Figures 1-3). Axon terminals contain variable numbers of mitochondria, usually ranging from zero to three or four.
Figure 1.

Electron micrographs of human postmortem tissue from the caudate nucleus of A) a normal control and B) a treatment responsive subject with schizophrenia. These micrographs were selected to show mitochondria (m) in various subcellular locations, not to depict average density of profiles. Abbreviations: s, spine; at, axon terminal; den, dendrite; ma, myelinated axon; solid arrows, synapses; dotted arrow, spine emerging from dendrite. Scale bars= 0.5μm.
Figure 2.

Electron micrographs of human postmortem tissue from the caudate nucleus of A) a normal control, B) a treatment resistant subject with schizophrenia, and C) a treatment responsive subject with schizophrenia. Abbreviations: m, mitochondria; spine; at, axon terminal; den, dendrite; solid arrows, synapses. Scale bars= 0.5μm.
Figure 3.

Electron micrographs of human postmortem tissue from the putamen of A) a normal control and B) a treatment responsive subject with schizophrenia. Abbreviations: m, mitochondria; s, spine; at, axon terminal; den, dendrite; ma, myelinated axon; solid arrows, synapses; dotted arrow, spine emerging from dendrite. Scale bars= 0.5μm.
Table I shows group means and statistical analyses of demographic and diagnostic characteristics. The three groups did not differ significantly from each other in terms of group composition of race, gender, pH, postmortem interval or age. Moreover, neither SZ subgroup differed from each other in terms of the distribution of DSM-IV subtypes, age of onset or length of illness. The composition of the two SZ groups was different in terms of antipsychotic drug (APD) treatment. The treatment resistant subjects were all on atypical APDs, while the treatment responsive subjects were on either typical or atypical APDs. Therefore we tested to see if there was a correlation between fluphenazine equivalents and the mitochondrial measures in which we found significant differences (mitochondria per synapse in the caudate nucleus and putamen). For this analysis we included two off-drug cases to increase the number of cases in which fluphenazine levels were known. We found no correlations, suggesting that the group difference in APDs did not affect the results. Individual values are shown in Table II.
Table II.
The number of mitochondria per synapse is shown individually for controls, and schizophrenic subjects divided into responsive, resistant or off drug cases.
| Mitochondria per Synapse Caudate Nucleus | Mitochondria per Synapse Putamen | ||||
|---|---|---|---|---|---|
| NC | Responsive | Resistant | NC | Responsive | Resistant |
| 0.99 | 0.71 T | 1.58 A | 0.97 | 0.98 T | 1.07 |
| 1.16 | 1.01 T (4) | 0.94 A | 1.32 (4) | 1.25 T (4) | 1.62 |
| 1.92 | 1.18 T | 1.67 A (8) | 1.59 | 0.96 T | 1.30 (8) |
| 2.14 | 0.99 T | 1.93 | 0.79 T | ||
| 1.70 | 1.01 T (15) | Off drug | 1.77 (15) | * | Off drug |
| 1. 77 | 1.29 T | 1.14 (0) | 1.45 | 0.62 T | 1.26 (0) |
| 0.88 | 0.74 A | 1.57 (0) | 1.60 | 1.03 A | 1.15 (0) |
| 2.15 | 1.07 A | 1.58 | * | ||
The two off drug cases were included in this analysis to provide examples of SZ subjects with fluphenazine equivalentsequal to zero. Both subjects were African American females (ages= 33,58) with pHs of 6.8 and PMIs of 3 hours. Numbers in ( ) denote the fluphenazine levels. APD status is listed as (T) for typical or (A) for atypical.
indicates that samples of the putamen were not available.
The density of mitochondria throughout the neuropil, in dendrites and in axon terminals was not significantly different among the three groups in either the caudate nucleus or the putamen (Figure 4). However, the number of mitochondria per synapse showed significant differences in both regions (Figure 5). In the caudate nucleus, there were significant differences in mitochondria per synapse across groups (ANOVA, p<0.025). The treatment responsive group had fewer mitochondria per synapse than did the controls (a 37% decrease, p<0.008), and a non significant decrease compared to the treatment resistant group (a 28% decrease). A regression analysis and Pearson correlation within the SZ group between fluphenazine equivalents and mitochondria per synapse showed no significant correlation (n=6, Pearson correlation= −0.247, R=0.247, R2 =0.061, and p<0.637). In the putamen, significant differences in the number of mitochondria per synapse were observed across the groups (ANOVA, p<0.002). Treatment responsive SZs had significantly fewer mitochondria per synapse verses controls (43% decrease, p<0.001) and SZs that were treatment resistant (34% decrease, p<0.031). A regression analysis and Pearson correlation within the SZ group between fluphenazine equivalents and mitochondria per synapse showed no significant correlation (n=4, Pearson correlation= 0.046, R=0.046, R2 = 0.002, p<0.954).
Figure 4.
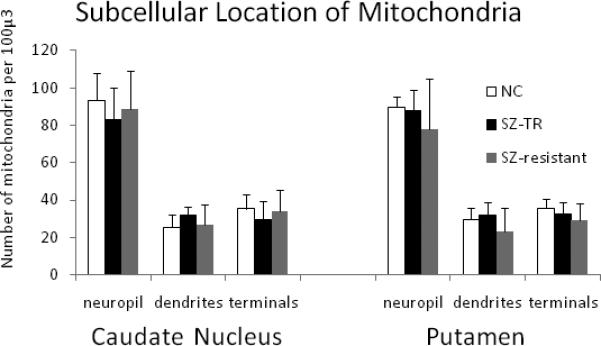
Graphs showing location of mitochondria in the caudate nucleus and putamen. The number of mitochondria is shown for the neuropil, in dendrites and in axon terminals. ANOVA did not show any significant group differences for any of the measures.
Figure 5.
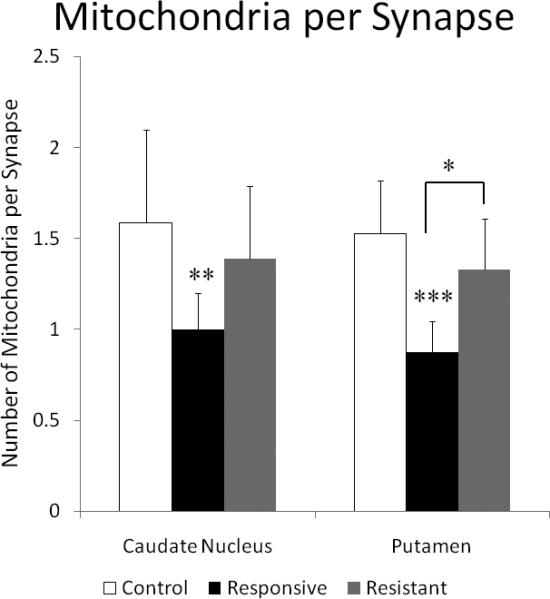
Graph comparing the number of mitochondria per synapse in the caudate nucleus and putamen. ANOVA showed significant group differences for both the caudate nucleus (p<0.025) and the putamen (p<0.002). In the caudate nucleus, treatment responsive SZ subjects had fewer mitochondria per synapse than that of the NCs. In the putamen, there are significantly fewer mitochondria per synapse in treatment responsive SZ subjects compared to both NCs and treatment resistant SZ subjects. Asterisks indicate results of LSD post-hoc t-tests:* p< 0.05, ** p<0.01, ***, p<0.001 and are placed over the group when comparing with NCs and over the bar comparing treatment responsive to treatment resistant SZ subjects.
Discussion
The main results of the present study show fewer numbers of mitochondria per synapse in treatment responsive SZs vs. NCs in both the caudate nucleus and the putamen. In addition, treatment responsive SZs had significantly fewer mitochondria per synapse than that of the treatment resistant subjects in the putamen. Our results are consistent with in vivo studies suggesting a biological basis to treatment response and resistance. The observation that the treatment responders have fewer mitochondria per synapse compared to treatment resistant SZs suggests that fewer mitochondria per synapse may be important for treatment response.
There are limitations of postmortem studies in SZ, which need to be addressed. Reliability of postmortem diagnoses in SZ research is a worry, but the use of structured instruments diminishes this caveat, as we have discussed previously (Roberts et al., 2009). The use of antipsychotic drugs (APDs) imposes certain confounds such as results being altered, caused or normalized by medication. Some APDs cause a small enlargement of the striatum (Chakos et al., 1994; Gur et al., 1998; Shihabuddin et al., 1998; Gunduz et al., 2002), which could confound density measurements. However, imaging studies have shown that the size of the caudate nucleus is similar in good and poor responders (Buchsbaum et al., 2003). And, the number of mitochondria per synapse is a measurement in which striatal size variation should have no impact. Another potential concern was that the proportion of cases on typical vs. atypical APDs was significantly different between the treatment responsive and resistant SZ groups. Importantly, with the exception of clozapine, first- and second generation antipsychotic drugs alleviate positive symptoms to the same extent (Lieberman et al., 2005; McEvoy, 2006; Kane et al., 2008). Also, there were no correlations between fluphenazine equivalents and the number of mitochondria per synapse (Table II), though the number of individuals where this information was known was small. Therefore, even though the SZ subgroups were composed of different numbers of subjects on typical versus atypical APDs, the possibility that the difference in results between the groups is related to medication seems unlikely.
Numerous studies indicate abnormalities of mitochondria in SZ (see Introduction), yet there are few electron microscopy studies with which to compare the current results. Uranova et al. (1996, 2001, 2007) reported decreased numbers of mitochondria in cell bodies of astrocytes in the caudate nucleus and in neurons in the prefrontal cortex. Their studies are not comparable to the present study as they reported on glial cell bodies and we examined neuronal processes. A previous study from our laboratory that did not examine mitochondria per synapse or treatment response showed lower numbers of mitochondria in striatal neuropil of SZs compared to NCs (Kung and Roberts, 1999). Technical considerations probably account for differing results since we used simple profile counts in our first paper (Kung and Roberts, 1999) and stereology in the present paper. Notably, stereology is considered superior to two dimensional counting techniques (Sterio, 1984; Geinisman et al., 1996). Moreover, both the amounts of mitochondrial DNA (Cavelier et al. (1995) and estimates of mitochondrial mass (Maurer et al., 2001) are similar between SZ and NCs, supporting the results of the present study.
The reason for treatment resistance is poorly understood but appears to have a biological basis (Beerpoot et al., 1996; Sheitman and Lieberman, 1998; Altamura et al., 2005). A relationship between pathophysiology in SZ and the degree of treatment response has been shown in several neuroimaging studies (Rodriguez et al., 1997; Stall et al., 2001; Arango et al., 2003). MRI studies have shown that treatment resistant SZ subjects have greater cortical atrophy in certain regions (Mitelman et al., 2005), smaller putamen volumes (Mitelman et al., 2009) and larger cerebral ventricles than do treatment responsive SZs (Stern et al., 1993; Bilder et al. 1994; Staal et al., 2001). SPECT shows differential values for cerebral perfusion, an index of neuronal activity (Gemmell et al., 1990; Turkington et al., 1993), in treatment responsive vs. resistant SZs (Rodriguez et al., 1997). APD naïve SZ subjects who eventually respond to treatment have elevated dopamine release compared to those subjects who did not eventually respond (Laruelle et al., 1999; Abi-Dargham et al., 2000). Importantly, treatment resistance does not occur because of a failure of D2 receptor blockade by APDs as these subjects show a 95% blockade of striatal D2 receptors following typical APD treatment (Coppens et al., 1991). Neurobiological differences between treatment response and treatment resistance in SZ are rarely studied at the microscopic level in postmortem tissue, but provide a strategy for trying to link psychosis with particular neuropathologies (Roberts et al., 2009). Our results provide support for a biological distinction between treatment responsive and treatment resistant SZ.
Surprisingly, treatment resistant SZ subjects presented normal numbers of mitochondria while the treatment responsive subjects showed the anomaly. This suggests that fewer mitochondria per synapse may be related to treatment response. Treatment responders may have fewer mitochondria per synapse and somehow this feature permits response to APD, or the reduction of mitochondrial number may be a possible mechanism of action of APDs. Our previous studies have shown an increase in synaptic density in the caudate nucleus and in the putamen patches of schizophrenia subjects (Roberts et al., 2005a,b). If too many synapses in the striatum play a role in the symptoms of schizophrenia, perhaps fewer mitochondria per synapse may be a compensatory mechanism to normalize overall synaptic activity, which may facilitate treatment response. Fewer mitochondria per synapse in treatment responsive schizophrenia may indicate a loss or decrease in number of mitochondria from one or more sources of axon terminals. In future studies the identity of the terminals, the types of neurons, and the striosomal compartments can be determined with immunohistochemistry to determine which populations of inputs and neurons are involved.
Although there is abundant evidence for mitochondrial malfunction in SZ, the role of mitochondria in the symptoms of SZ is still unclear. In the caudate nucleus of SZs, COX activity (complex IV) is decreased by 30-60% (Cavelier et al., 1995; Prince et al., 1999), suggesting diminished mitochondrial function. Psychostimulants, which cause psychotic symptoms in normal subjects and exacerbate psychosis in SZs (Tomiyama, 1990; Jentsch and Roth, 1999; Lahti et al., 1995, 2001), reversibly decrease COX staining in rats (Prince et al., 1997). Imaging studies have shown that SZ subjects off medication have decreased striatal metabolism, which improves when symptoms are alleviated be APDs (Buchsbaum et al., 1992). Taken together these studies imply that fewer or less functional mitochondria are correlated with psychosis. However, other findings suggest that an increase in mitochondrial number and /or function is linked to psychosis. Ben-Shachar (2002) showed using platelet analysis that SZs exhibiting psychotic symptoms have an increase in the level of complex I activity while SZs with residual symptoms had a decrease in activity of this enzyme. Still other studies do not support a direct link between mitochondrial abnormalities and psychosis (Maurer and Moller, 1997) but show a relationship with decreased cognition (Prince et al 2000). Clearly further studies are necessary to elucidate the relationship between SZ symptoms and the role of mitochondria.
Mitochondria change location in response to cellular energy demands and the stage of their own life cycle (Mjaatvedt and Wong-Riley, 1988; Isaacs et al., 1992; Ligon and Steward, 2000; Miller and Sheetz, 2004). It is unclear if the decrease in number of mitochondria per synapse, seen in the present paper, is a reflection of failure of the mitochondria to move from the cell bodies of origin or overall decreased function and return to the soma. In the same cases as those in the present paper we have shown an increase in the density of synapses in the caudate nucleus and putamen patches (Roberts et al 2005a,b). Synapses need energy to form and to function properly (Wong-Riley, 1989). The decrease in density of mitochondria in terminals forming synapses may be an adaptive response to normalize overactive neurotransmission. Future studies will address if the number of mitochondria is decreased in particular populations of synapses. Based on our data, mitochondrial density is differentially affected in SZ according to treatment response. Understanding the role that mitochondria might play in SZ could lead to better comprehension of the mechanisms of APDs to alleviate psychotic symptoms and alter brain metabolism, and what goes awry in treatment resistance.
Acknowledgements
The authors wish to acknowledge the Maryland Brain Collection for their support and cooperation and Ms. Rosie Ricks for help preparing the manuscript. This research was supported in part by MH60744 to RCR and MH073461 predoctoral fellowship to SS.
References
- Abi-Dargham A, Gil R, Krystal J, Baldwin RM, Seibyl JP, Bowers M, van Dyck CH, Charney DS, Innis RB, Laruelle M. Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am J Psychiatry. 1998;155(6):761–767. doi: 10.1176/ajp.155.6.761. [DOI] [PubMed] [Google Scholar]
- Altamura AC, Bassetti R, Cattaneo E, Vismara S. Some biological correlates of drug resistance in schizophrenia: a multidimensional approach. World J Biol Psychiatry. Suppl. 2005;2:23–30. doi: 10.1080/15622970510030027. [DOI] [PubMed] [Google Scholar]
- Arango C, Breier A, McMahon R, Carpenter WT, Jr., Buchanan RW. The relationship of clozapine and haloperidol treatment response to prefrontal, hippocampal, and caudate brain volumes. Am J Psychiatry. 2003;160:1421–1427. doi: 10.1176/appi.ajp.160.8.1421. [DOI] [PubMed] [Google Scholar]
- Beerpoot LJ, Lipska BK, Weinberger DR. Neurobiology of treatment-resistant schizophrenia: new insights and new models. Eur Neuropsychopharmacol. 1996;6:S27–34. doi: 10.1016/0924-977x(96)00008-9. [DOI] [PubMed] [Google Scholar]
- Ben-Shachar D. Mitochondrial dysfunction in schizophrenia: a possible linkage to dopamine. J Neurochem. 2002;83:1241–1251. doi: 10.1046/j.1471-4159.2002.01263.x. [DOI] [PubMed] [Google Scholar]
- Ben-Shachar D, Laifenfeld D. Mitochondria, Synaptic Plasticity, and Schizophrenia. Int Review Neurobiol. 2004;59:273–296. doi: 10.1016/S0074-7742(04)59011-6. [DOI] [PubMed] [Google Scholar]
- Bilder RM, Wu H, Chakos MH, Bogerts B, Pollack S, Aronowitz J, Ashtari M, Degreef G, Kane JM, Lieberman JA. Cerebral morphometry and clozapine treatment in schizophrenia. J Clin Psychiatry. 1994;55:53–56. [PubMed] [Google Scholar]
- Buchsbaum MS, Potkin SG, Siegel BV, Jr., Lohr J, Katz M, Gottschalk LA, Gulasekaram B, Marshall JF, Lottenberg S, Teng CY. Striatal metabolic rate and clinical response to neuroleptics in schizophrenia. Arch Gen Psychiatry. 1992;49:966–974. doi: 10.1001/archpsyc.1992.01820120054008. [DOI] [PubMed] [Google Scholar]
- Buchsbaum MS, Shihabuddin L, Brickman AM, Miozzo R, Prikryl R, Shaw R, Davis K. Caudate and putamen volumes in good and poor outcome patients with schizophrenia. Schizophr Res. 2003;64:53–62. doi: 10.1016/s0920-9964(02)00526-1. [DOI] [PubMed] [Google Scholar]
- Carlsson A. Antipsychotic drugs, neurotransmitters, and schizophrenia. Am J Psychiatry. 1978;135:165–173. doi: 10.1176/ajp.135.2.165. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Lindqvist M. Effect of chlorpromazine or haloperidol on formation of 3methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol. 1963;20:140–144. doi: 10.1111/j.1600-0773.1963.tb01730.x. [DOI] [PubMed] [Google Scholar]
- Cavelier L, Jazin EE, Eriksson I, Prince J, Bave U, Oreland L, Gyllensten U. Decreased cytochrome-c oxidase activity and lack of age-related accumulation of mitochondrial DNA deletions in the brains of schizophrenics. Genomics. 1995;29:217–224. doi: 10.1006/geno.1995.1234. [DOI] [PubMed] [Google Scholar]
- Chakos MH, Lieberman JA, Bilder RM, Borenstein M, Lerner G, Bogerts B, Wu H, Kinon B, Ashtari M. Increase in caudate nuclei volumes of first-episode schizophrenic patients taking antipsychotic drugs. Am J Psychiatry. 1994;151:1430–1436. doi: 10.1176/ajp.151.10.1430. [DOI] [PubMed] [Google Scholar]
- Chang DT, Reynolds IJ. Mitochondrial trafficking and morphology in healthy and injured neurons. Prog Neurobiol. 2006;80(5):241–268. doi: 10.1016/j.pneurobio.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Conley RR, Kelly DL. Pharmacologic treatment of schizophrenia, 1st edition. Professional Communications. 2000 [Google Scholar]
- Conley RR, Kelly DL. Management of treatment resistance in schizophrenia. Biol Psychiatry. 2001;50:898–911. doi: 10.1016/s0006-3223(01)01271-9. [DOI] [PubMed] [Google Scholar]
- Coppens HJ, Slooff CJ, Paans AM, Wiegman T, Vaalburg W, Korf J. High central D2-dopamine receptor occupancy as assessed with positron emission tomography in medicated but therapy-resistant schizophrenic patients. Biol Psychiatry. 1991;29:629–634. doi: 10.1016/0006-3223(91)90132-6. [DOI] [PubMed] [Google Scholar]
- Duchen MR, Verkhratsky A, Muallem S. Mitochondria and calcium in health and disease. Cell Calcium. 2008;44:1–5. doi: 10.1016/j.ceca.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Geinisman Y, Gundersen HJ, van der Zee E, West MJ. Unbiased stereological estimation of the total number of synapses in a brain region. J Neurocytol. 1996;25:805–819. doi: 10.1007/BF02284843. [DOI] [PubMed] [Google Scholar]
- Gemmell HG, Evans NT, Besson JA, Roeda D, Davidson J, Dodd MG, Sharp PF, Smith FW, Crawford JR, Newton RH, Kulkarni V, Mallard JR. Regional cerebral blood flow imaging: a quantitative comparison of technetium-99m-HMPAO SPECT with C15O2 PET. J Nucl Med. 1990;31:1595–1600. [PubMed] [Google Scholar]
- Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry. 2001;158:1367–1377. doi: 10.1176/appi.ajp.158.9.1367. [DOI] [PubMed] [Google Scholar]
- Gunduz H, Wu H, Ashtari M, Bogerts B, Crandall D, Robinson DG, Alvir J, Lieberman J, Kane J, Bilder R. Basal ganglia volumes in first-episode schizophrenia and healthy comparison subjects. Biol Psychiatry. 2002;51:801–808. doi: 10.1016/s0006-3223(01)01345-2. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Gunter KK, Sheu SS, Gavin CE. Mitochondrial calcium transport: physiological and pathological relevance. Am J Physiol. 1994;267:313–339. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- Gur RE, Maany V, Mozley PD, Swanson C, Bilker W, Gur RC. Subcortical MRI volumes in neuroleptic-naive and treated patients with schizophrenia. Am J Psychiatry. 1998;155:1711–1717. doi: 10.1176/ajp.155.12.1711. [DOI] [PubMed] [Google Scholar]
- Harrison PJ. The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain. 1999;122:593–624. doi: 10.1093/brain/122.4.593. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Heath PR, Eastwood SL, Burnet PW, McDonald B, Pearson RC. The relative importance of premortem acidosis and postmortem interval for human brain gene expression studies: selective mRNA vulnerability and comparison with their encoded proteins. Neurosci Lett. 1995;200:151–154. doi: 10.1016/0304-3940(95)12102-a. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- Isaacs KR, Anderson BJ, Alcantara AA, Black JE, Greenough WT. Exercise and the brain: angiogenesis in the adult rat cerebellum after vigorous physical activity and motor skill learning. J Cereb Blood Flow Metab. 1992;12:110–119. doi: 10.1038/jcbfm.1992.14. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20:201–25. doi: 10.1016/S0893-133X(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Johnson SA, Morgan DG, Finch CE. Extensive postmortem stability of RNA from rat and human brain. J Neurosci Res. 1986;16:267–80. doi: 10.1002/jnr.490160123. [DOI] [PubMed] [Google Scholar]
- Johnston NL, Cervenak J, Shore AD, Torrey EF, Yolken RH. Multivariate analysis of RNA levels from postmortem human brains as measured by three different methods of RT-PCR. Stanley Neuropathology Consortium. J Neurosci Methods. 1997;77:83–92. doi: 10.1016/s0165-0270(97)00115-5. [DOI] [PubMed] [Google Scholar]
- Kane JM, Assunção-Talbott S, Eudicone JM, Pikalov A, Whitehead R, Crandall DT. The efficacy of aripiprazole in the treatment of multiple symptom domains in patients with acute schizophrenia: a pooled analysis of data from the pivotal trials. Schizophr Res. 2008;105:208–215. doi: 10.1016/j.schres.2008.06.018. [DOI] [PubMed] [Google Scholar]
- Kane JM, Honigfeld G, Singer J, Meltzer H. Clozapine in treatment-resistant schizophrenics. Psychopharmacol Bull. 1988;24:62–67. [PubMed] [Google Scholar]
- Karry R, Klein E, Ben Shachar D. Mitochondrial complex I subunits expression is altered in schizophrenia: a postmortem study. Biol Psychiatry. 2004;55:676–684. doi: 10.1016/j.biopsych.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Kolomeets NS, Uranova N. Ultrastructural abnormalities of astrocytes in the hippocampus in schizophrenia and duration of illness: A postortem (sic) morphometric study. World J Biol Psych. 2009;9:1–11. doi: 10.1080/15622970902806124. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr., Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- Kung L, Roberts RC. Mitochondrial pathology in human schizophrenic striatum: a postmortem ultrastructural study. Synapse. 1999;31:67–75. doi: 10.1002/(SICI)1098-2396(199901)31:1<67::AID-SYN9>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Kvajo M, Dhilla A, Swor DE, Karayiorgou M, Gogos JA. Evidence implicating the candidate schizophrenia/bipolar disorder susceptibility gene G72 in mitochondrial function. Mol Psychiatry. 2008;13:685–696. doi: 10.1038/sj.mp.4002052. [DOI] [PubMed] [Google Scholar]
- Lahti AC, Holcomb HH, Medoff DR, Tamminga CA. Ketamine activates psychosis and alters limbic blood flow in schizophrenia. NeuroReport. 1995;6:869–872. doi: 10.1097/00001756-199504190-00011. [DOI] [PubMed] [Google Scholar]
- Lahti AC, Weiler MA, Tamara Michaelidis BA, Parwani A, Tamminga CA. Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology. 2001;25:455–467. doi: 10.1016/S0893-133X(01)00243-3. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol Psychiatry. 1999;46:56–72. doi: 10.1016/s0006-3223(99)00067-0. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A, van Dyck CH, Gil R, D'Souza CD, Erdos J, McCance E, Rosenblatt W, Fingado C, Zoghbi SS, Baldwin RM, Seibyl JP, Krystal JH, Charney DS, Innis RB. Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Natl Acad Sci. 1996;93:9235–9240. doi: 10.1073/pnas.93.17.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Lieberman JA. Catching up on schizophrenia: natural history and neurobiology. Neuron. 2000;28:325–334. doi: 10.1016/s0896-6273(00)00111-2. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz BD, Severe J, Hsiao JK. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- Ligon LA, Steward O. Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J Comp Neurol. 2000;427:351–361. doi: 10.1002/1096-9861(20001120)427:3<351::aid-cne3>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Marchbanks RM, Ryan M, Day IN, Owen M, McGuffin P, Whatley SA. A mitochondrial DNA sequence variant associated with schizophrenia and oxidative stress. Schizophr Res. 2003;65:33–38. doi: 10.1016/s0920-9964(03)00011-2. [DOI] [PubMed] [Google Scholar]
- Maurer I, Moller HJ. Inhibition of complex I by neuroleptics in normal human brain cortex parallels the extrapyramidal toxicity of neuroleptics. Mol Cell Biochem. 1997;174:255–259. [PubMed] [Google Scholar]
- Maurer I, Zierz S, Moller H. Evidence for a mitochondrial oxidative phosphorylation defect in brains from patients with schizophrenia. Schizophr Res. 2001;48:125–136. doi: 10.1016/s0920-9964(00)00075-x. [DOI] [PubMed] [Google Scholar]
- McEvoy JP. An overview of the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study. CNS Spectr. 2006;11:4–8. doi: 10.1017/s1092852900026626. [DOI] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Mitelman SA, Canfield EL, Chu KW, Brickman AM, Shihabuddin L, Hazlett EA, Buchsbaum MS. Poor outcome in chronic schizophrenia is associated with progressive loss of volume of the putamen. Schizophr Res. 2009;113:241–245. doi: 10.1016/j.schres.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitelman SA, Shihabuddin L, Brickman AM, Hazlett EA, Buchsbaum MS. Volume of the cingulate and outcome in schizophrenia. Schizophr Res. 2005;72:91–108. doi: 10.1016/j.schres.2004.02.011. [DOI] [PubMed] [Google Scholar]
- Mjaatvedt AE, Wong-Riley MT. Relationship between synaptogenesis and cytochrome oxidase activity in Purkinje cells of the developing rat cerebellum. J Comp Neurol. 1988;277:155–182. doi: 10.1002/cne.902770202. [DOI] [PubMed] [Google Scholar]
- Perez-Costas E, Melendez-Ferro M, Roberts R. Microscopy techniques and the study of synapses. In: Mendez-Vilas A, Diaz J, editors. Modern Research and Educational Topics in Microscopy. Vol. 1. Badajoz; Spain Formatex: 2007. pp. 164–170. [Google Scholar]
- Powers RE. The neuropathology of schizophrenia. J Neuropathol Exp Neurol. 1999;58:679–90. doi: 10.1097/00005072-199907000-00001. [DOI] [PubMed] [Google Scholar]
- Prabakaran S, Swatton JE, Ryan MM, Huffaker SJ, Huang JT, Griffin JL, Wayland M, Freeman T, Dudbridge F, Lilley KS, Karp NA, Hester S, Tkachev D, Mimmack ML, Yolken RH, Webster MJ, Torrey EF, Bahn S. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol Psychiatry. 2004;9:684–97. doi: 10.1038/sj.mp.4001511. [DOI] [PubMed] [Google Scholar]
- Prince JA, Blennow K, Gottfries CG, Karlsson I, Oreland L. Mitochondrial function is differentially altered in the basal ganglia of chronic schizophrenics. Neuropsychopharmacology. 1999;21:372–379. doi: 10.1016/S0893-133X(99)00016-0. [DOI] [PubMed] [Google Scholar]
- Prince JA, Harro J, Blennow K, Gottfries CG, Oreland L. Putamen mitochondrial energy metabolism is highly correlated to emotional and intellectual impairment in schizophrenics. Neuropsychopharmacology. 2000;22:284–292. doi: 10.1016/S0893-133X(99)00111-6. [DOI] [PubMed] [Google Scholar]
- Prince JA, Yassin MS, Oreland L. Normalization of cytochrome-c oxidase activity in the rat brain by neuroleptics after chronic treatment with PCP or methamphetamine. Neuropharmacology. 1997;36:1665–1678. doi: 10.1016/s0028-3908(97)00152-4. [DOI] [PubMed] [Google Scholar]
- Resnick SM, Gur RE, Alavi A, Gur RC, Reivich M. Positron emission tomography and subcortical glucose metabolism in schizophrenia. Psychiatry Res. 1988;24:1–11. doi: 10.1016/0165-1781(88)90134-5. [DOI] [PubMed] [Google Scholar]
- Roberts RC, Conley R, Kung L, Peretti FJ, Chute DJ. Reduced striatal spine size in schizophrenia: a postmortem ultrastructural study. NeuroReport. 1996;7:1214–1218. doi: 10.1097/00001756-199604260-00024. [DOI] [PubMed] [Google Scholar]
- Roberts RC, Roche JK, Conley RR. Synaptic differences in the postmortem striatum of subjects with schizophrenia: a stereological ultrastructural analysis. Synapse. 2005a;56:185–197. doi: 10.1002/syn.20144. [DOI] [PubMed] [Google Scholar]
- Roberts RC, Roche JK, Conley RR. Synaptic differences in the patch matrix compartments of subjects with schizophrenia: a postmortem ultrastructural study of the striatum. Neurobiol Dis. 2005b;20:324–335. doi: 10.1016/j.nbd.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Roberts RC, Roche JK, Conley RR. Differential synaptic changes in the striatum of subjects with undifferentiated versus paranoid schizophrenia. Synapse. 2008;62:616–627. doi: 10.1002/syn.20534. [DOI] [PubMed] [Google Scholar]
- Roberts RC, Roche JK, Conley RR, Lahti AC. Dopaminergic synapses in the caudate of subjects with schizophrenia: relationship to treatment response. Synapse. 2009;63:520–530. doi: 10.1002/syn.20623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez VM, Andree RM, Castejon MJ, Zamora ML, Alvaro PC, Delgado JL, Vila FJ. Fronto-striato-thalamic perfusion and clozapine response in treatment-refractory schizophrenic patients. A 99mTc-HMPAO study. Psychiatry Res. 1997;76:51–61. doi: 10.1016/s0925-4927(97)00057-7. [DOI] [PubMed] [Google Scholar]
- Salzman S, Endicott J, Clayton P, Winokur G. National Institute of Mental Health. Neuroscience Research Branch; Rockville, MD: 1983. Diagnostic evaluation after death (DEAD). [Google Scholar]
- Sheitman BB, Lieberman JA. The natural history and pathophysiology of treatment resistant schizophrenia. J Psychiatr Res. 1998;32:143–150. doi: 10.1016/s0022-3956(97)00052-6. [DOI] [PubMed] [Google Scholar]
- Shihabuddin L, Buchsbaum MS, Hazlett EA, Haznedar MM, Harvey PD, Newman A, Schnur DB, Spiegel-Cohen J, Wei T, Machac J, Knesaurek K, Vallabhajosula S, Biren MA, Ciaravolo TM, Luu-Hsia C. Dorsal striatal size, shape, and metabolic rate in never-medicated and previously medicated schizophrenics performing a verbal learning task. Arch Gen Psychiatry. 1998;55:235–243. doi: 10.1001/archpsyc.55.3.235. [DOI] [PubMed] [Google Scholar]
- Somerville SM, Roche JK, Conley RR, Roberts RC. Ultrastructural study of mitochondria in postmortem striatal patch and matrix compartments in schizophrenia. Soc Neurosci Abstr. 2003;29:316.3. [Google Scholar]
- Spitzer RL, Williams JB, Gibbon M, First MB. The Structured Clinical Interview for DSM-III-R (SCID). I: History, rationale, and description. Arch Gen Psychiatry. 1992;49:624–629. doi: 10.1001/archpsyc.1992.01820080032005. [DOI] [PubMed] [Google Scholar]
- Staal WG, Hulshoff Pol HE, Schnack HG, van Haren NE, Seifert N, Kahn RS. Structural brain abnormalities in chronic schizophrenia at the extremes of the outcome spectrum. Am J Psychiatry. 2001;158:1140–1142. doi: 10.1176/appi.ajp.158.7.1140. [DOI] [PubMed] [Google Scholar]
- Stan AD, Ghose S, Gao X-M, Roberts RC, Lewis-Amezcua K, Hatanpaa KJ, Tamminga CA. Human postmortem tissue: What quality markers matter? Brain Research. 2006;1123:1–11. doi: 10.1016/j.brainres.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterio DC. The unbiased estimation of number and sizes of arbitrary particles using the disector. J Microsc. 1984;134:127–136. doi: 10.1111/j.1365-2818.1984.tb02501.x. [DOI] [PubMed] [Google Scholar]
- Stern RG, Kahn RS, Harvey PD, Amin F, Apter SH, Hirschowitz J. Early response to haloperidol treatment in chronic schizophrenia. Schizophr Res. 1993;10:165–171. doi: 10.1016/0920-9964(93)90052-k. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Tomiyama G. Chronic schizophrenia-like states in methamphetamine psychosis. Jpn J Psychiatry Neurol. 1990;44:531–539. doi: 10.1111/j.1440-1819.1990.tb01626.x. [DOI] [PubMed] [Google Scholar]
- Turkington TG, Jaszczak RJ, Pelizzari CA, Harris CC, MacFall JR, Hoffman JM, Coleman RE. Accuracy of registration of PET, SPECT and MR images of a brain phantom. J Nucl Med. 1993;34:1587–1594. [PubMed] [Google Scholar]
- Uranova NA, Casanova MF, DeVaughn NM, Orlovskaya DD, Denisov DV. Ultrastructural alterations of synaptic contacts and astrocytes in postmortem caudate nucleus of schizophrenic patients. Schizophr Res. 1996;22:81–83. doi: 10.1016/0920-9964(96)00059-x. [DOI] [PubMed] [Google Scholar]
- Uranova N, Orlovskaya D, Vikhreva O, Zimina I, Kolomeets N, Vostrikov V, Rachmanova V. Electron microscopy of oligodendroglia in severe mental illness. Brain Res Bull. 2001;55:597–610. doi: 10.1016/s0361-9230(01)00528-7. [DOI] [PubMed] [Google Scholar]
- Uranova NA, Vostrikiv VM, Vikhreva OV, Zimina IS, Kolomeets NS, Orlovskaya DD. The role of oligodendrocyte pathology in schizophrenia. Int J Neuropsychopharm. 2007;10:537–545. doi: 10.1017/S1461145707007626. [DOI] [PubMed] [Google Scholar]
- Wiesel FA, Wik G, Sjogren I, Blomqvist G, Greitz T, Stone-Elander S. Regional brain glucose metabolism in drug free schizophrenic patients and clinical correlates. Acta Psychiatr Scand. 1987;76:628–641. doi: 10.1111/j.1600-0447.1987.tb02933.x. [DOI] [PubMed] [Google Scholar]
- Wong-Riley MT. Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989;12:94–101. doi: 10.1016/0166-2236(89)90165-3. [DOI] [PubMed] [Google Scholar]