

Background: ATP regulates soluble guanylate cyclase (sGC), which mediates NO signaling.
Results: ATP inhibition of sGC is dependent on NO levels; the pseudosymmetric site is involved in ATP regulation.
Conclusion: A new kinetic model for sGC activity in the presence of excess NO is proposed.
Significance: Cooperativity between the pseudosymmetric and catalytic site results in a more specific cyclase in vivo.
Keywords: nitric oxide, guanylate cyclase (guanylyl cyclase), cyclic GMP (cGMP), ATP, signal transduction, heme
Abstract
Activation of soluble guanylate cyclase (sGC) by the signaling molecule nitric oxide (NO) leads to formation of the second messenger cGMP, which mediates numerous physiological processes. NO activates sGC by binding to the ferrous heme cofactor; the relative amount of NO with respect to sGC heme affects the enzyme activity. ATP can also influence the activity by binding to an allosteric site, most likely the pseudosymmetric site located in the catalytic domain. Here, the role of the pseudosymmetric site on nucleotide regulation was investigated by point mutations at this site. ATP inhibition kinetics of wild type and a pseudosymmetric site (α1-C594A/β1-D477A) variant of sGC was determined at various levels of NO. Results obtained show that in the presence of less than 1 eq of NO, there appears to be less than complete activation and little change in the nucleotide binding parameters. The most dramatic effects are observed for the addition of excess NO, which results in an increase in the affinity of GTP at the catalytic site and full activation of sGC. The pseudosymmetric site mutation only affected nucleotide affinities in the presence of excess NO; there was a decrease in the affinity for ATP in both the allosteric and catalytic sites. These observations led to a new kinetic model for sGC activity in the presence of excess NO. This model revealed that the active and allosteric sites show cooperativity. This new comprehensive model gives a more accurate description of sGC regulation by NO and nucleotides in vivo.
Introduction
Nitric oxide (NO) signaling mediates numerous physiological processes, including blood vessel relaxation, myocardial function, synaptic plasticity, and platelet aggregation (1–4). Soluble guanylate cyclase (sGC)4 is the primary receptor for NO. NO activation of sGC leads to formation of cyclic GMP (cGMP) (5). As a second messenger, cGMP interacts with protein kinases, phosphodiesterases, and ion channels; these interactions lead to the cellular effects of NO (6). Dysregulation of the NO/sGC/cGMP signaling pathway is linked to various disease states, including heart disease, hypertension, and neurodegeneration (7–9). Accordingly, sGC is an attractive therapeutic target for the treatment of these disorders, and the mechanisms of activation and regulation of sGC are an active area of research (10–12).
The most common isoform of sGC is composed of two homologous subunits, α1 and β1. Both sGC subunits consist of four separate domains: the N-terminal heme-NO/O2 binding (H-NOX) domain, the Per/Arnt/Sim (PAS) domain, the helical domain, and the C-terminal catalytic domain (13, 14). The H-NOX domain of β1 subunit binds heme, whereas the corresponding region in the α1 subunit does not. Given the homology, the N terminus of the α1 subunit may have similar domain structure as the β1 H-NOX domain, but it lacks key residues involved in heme binding. The β1 H-NOX domain is also called the sensor domain because NO binding to heme in the β1 subunit is directly involved in the activation mechanism. The PAS and helical domains are involved in heterodimer formation and activation of the catalytic domain (15–17).
The catalytic domain is composed of the C termini of both the α1 and β1 subunits. The active site is located at the subunit interface and consists of residues from both subunits (14, 18). Moreover, this domain shares significant homology with the catalytic domain of mammalian adenylate cyclases (ACs) (19, 20). In both guanylate cyclases (GCs) and ACs, the catalytic site contains two aspartate residues that coordinate two Mg2+ ions, which are involved in substrate binding and catalysis (21). In addition to the catalytic site, ACs contain a cavity that is pseudosymmetric to the catalytic site. This pseudosymmetric site lacks key catalytic residues but is the binding site of the AC activator forskolin (22). To date, all reported crystal structures of the sGC catalytic domain are in a putative inactive (open) conformation and lack bound nucleotides (Protein Data Bank entries 3UVJ, 2WZ1, and 4NI2) (23, 24). Modeling the active (closed) conformation of sGC based on AC structures also reveals a pseudosymmetric cavity (Fig. 1). Similar to ACs, this site lacks key residues required for catalysis, most importantly one of the aspartate residues (α1-Asp-485 in the active site) that coordinates one of the two Mg2+ ions. Nevertheless, the pseudosymmetric site does include several residues involved in nucleotide binding. One of these residues is β1-Asp-477, the pseudosymmetric site counterpart of α1-Asp-529. The residue α1-Asp-529 is located at the active site and is coordinated with the second Mg2+ ion involved in nucleotide binding and catalysis. In addition, the pseudosymmetric site residue α1-Cys-594 is the counterpart of β1-Cys-541, which is involved in GTP binding to the catalytic site (Fig. 1B) (25). Consequently, it was proposed that the pseudosymmetric site could be involved in nucleotide regulation of sGC (26). Binding of nucleotides to the pseudosymmetric site has not been directly observed, but previous studies have shown that sGC binds two GTPs (27) and that nucleotide binding to an allosteric site influences activity (28–30).
FIGURE 1.
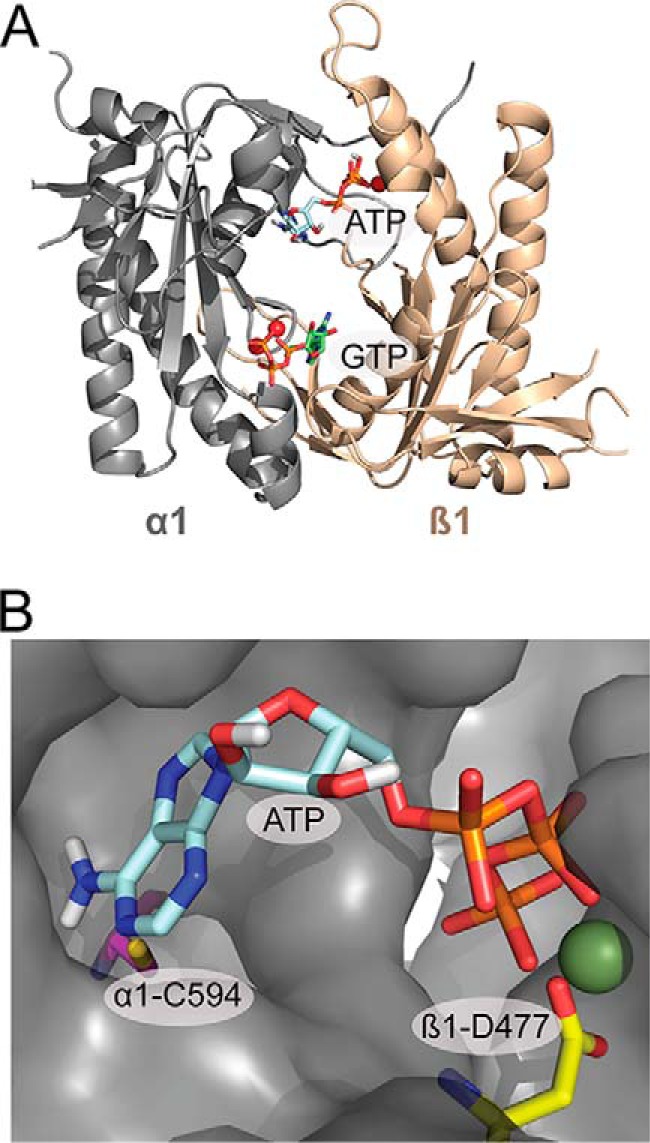
Model of the catalytic domain of sGC in the active confirmation based on the crystal structure of mammalian adenylate cyclase (21). A, Model of the catalytic domain with ATP docked at the pseudosymmetric site, and GTP docked at the catalytic site into the model using Autodock 4 (58). B, ATP docked in the pseudosymmetric site cavity. The residues mutated in the pseudosymmetric site, α1-Cys-549 (magenta) and β1-Asp-477 (yellow), and the Mg2+ ion (green) are highlighted.
The first model of NO activation of sGC posited that sGC is activated upon binding of NO to the ferrous heme cofactor, followed by the cleavage of the heme iron proximal histidine bond (31). The proposed steps involved NO binding to the open heme coordination site (distal pocket), axial to the histidine ligand, to form a 6-coordinate Fe2+-NO (6c NO) complex, followed by rupture of the Fe2+-His bond and conversion to the fully activated 5-coordinate Fe2+-NO (5c NO) complex. This complex can be formed using a stoichiometric amount of NO relative to the sGC heme. These steps would place NO on the distal pocket face of the heme. However, in the presence of an excess of NO relative to the heme, NO can also act as a catalyst and accelerate the rate of iron-histidine bond cleavage (31, 32). These observations were explained by a second molecule of NO binding to the open proximal heme site (in the distal 5c NO sGC) after the rupture of the Fe2+-His bond and formation of a transient dinitrosyl complex. Dissociation of the distal NO from the dinitrosyl complex would lead to a proximal 5c NO complex (33–35). A recent bacterial H-NOX structure has been reported utilizing excess NO during crystallization (36). This 5-coordinate structure showed NO in the proximal pocket. Therefore, NO can occupy the distal or proximal site in the 5c NO sGC.
Formation of the 5c NO complex is essential for activation of sGC, but it is not sufficient for full activation. Indeed, low activity and high activity NO-bound sGC species have been observed (35, 37). The low activity species is formed by either the addition of stoichiometric amounts of NO (1-NO per heme) or by removal of excess NO after formation of the ferrous nitrosyl complex. Although the nature of the 1-NO species is unclear (proximal or distal 5c NO), it is well established that the 1-NO state of sGC is not fully active. In addition, 1-NO sGC can be activated upon the addition of excess NO (excess over heme) irrespective of how it was formed (35). The sGC 5c NO complex is very stable (t½ ∼2 min) (38). The NO dissociation rate in vitro is inconsistent with the rapid deactivation profile observed in vivo, making it unlikely that dissociation of NO from the heme is the mechanism for sGC deactivation. Taken together, these results can be explained by additional NO binding to low affinity sites, such as cysteine residues in sGC, for full activation of the enzyme (39–41). It is possible that deactivation of sGC in vivo is characterized by conversion of the excess NO state to the 1-NO state of sGC rather than the basal (unliganded) state of the enzyme. Furthermore, NO signaling in cells does not follow a binary mechanism that only includes basal and active states (42, 43). In smooth muscle cells, tonic levels of NO lead to low levels of cGMP production in the resting state, whereas stimulation of NO synthase leads to acute levels of NO and relaxation of the cells (44, 45). Similar effects of NO have been observed in other tissues (2, 3, 46). Taken together, these observations are best explained by invoking three states of sGC in vivo: the basal (unliganded), the 5c NO (1-NO), and the excess NO (xsNO) state.
sGC activity is also directly influenced by nucleotides (28, 30, 47). The cellular ATP concentration is very high (∼3 mm) (48), and therefore even relatively weak effects are likely to be important in vivo. In addition, ATP was suggested to play a role in moderating the NO response with respect to the energy status of the cell (47). Although nucleotide regulation of sGC was previously reported, the relationship between NO activation states and ATP inhibition of sGC was unknown (28, 30, 47). In this paper, ATP inhibition of sGC at all three NO states, including the assessment of ATP binding kinetics of the 1-NO state, was investigated. Moreover, because the pseudosymmetric site is involved in the nucleotide regulation of sGC (26), we sought to probe the effects of ATP binding on the three activation states by designing an sGC variant with compromised pseudosymmetric nucleotide binding. Accordingly, ATP inhibition of the pseudosymmetric site variant α1-C594A/β1-D477A (double mutant (DM)) of sGC at various NO states was also investigated. The results obtained lead to a new model of ATP inhibition of sGC in the excess NO state.
Experimental Procedures
Materials
Wild type and α1-C594A/β1-D477A (DM) variant of full-length Rattus norvegicus sGC were expressed via the Bac-to-Bac baculovirus/Sf9 expression system and purified as described previously (49). Diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate (DEA NONOate) and 1-(hydroxy-NNO-azoxy)-l-proline (PROLI NONOate) were obtained from Cayman Chemical Co. All other reagents were purchased from Fisher, unless otherwise noted.
ATP Inhibition of sGC in the Basal and Excess NO State
sGC activity was determined in the presence of ATP by performing duplicate end point assays at 25 °C. sGC activity was determined in Buffer A (50 mm HEPES, pH 7.4, 6 mm MgCl2, 1 mm dithiothreitol (DTT)) with increasing concentrations (0.02–1 mm) of GTP. All assays were conducted in a final volume of 50 μl. sGC (0.1 μg) was incubated with varying concentrations (0–1 mm) of ATP for 2 min, before initiation of reactions by the addition of GTP. Reactions were quenched after 3 min by the addition of 200 μl of 125 mm Zn(CH3CO2)2 and 250 μl of 125 mm Na2CO3. To determine activity in the excess NO state, the assay was performed exactly the same except sGC was incubated with 0.1 mm DEA NONOate for 1 min before initiation of the reaction with GTP. Final cGMP was quantified by the cGMP EIA kit from Enzo Life Sciences, Inc. Each experiment was repeated at least twice to ensure reproducibility. Experiments were first analyzed on their own to obtain individual Vmax values, after which data were normalized (ν/Vmax). Normalized data from several experiments were combined and fit to models described below.
2′-Deoxy-ATP Inhibition of sGC
Wild type sGC (0.1 μg) activity was determined as described above in the absence and presence of 0.5 mm ATP and 0.5 mm 2′-deoxy-ATP.
ATP Inhibition of sGC in the 1-NO State
Ferrous nitrosyl sGC is obtained by titration of ferrous sGC (180–320 nm) in Buffer B (50 mm HEPES, pH 7.4, 6 mm MgCl2, 1 mm tris(2-carboxyethyl)phosphine) with 0.1 mm PROLI NONOate in 0.01 m NaOH. Electronic absorbance spectra were monitored during titration, and the addition of PROLI NONOate stopped shy of full conversion to 399 nm to ensure that no excess NO existed in solution. The concentration of ferrous nitrosyl sGC was determined by deconvolution of final spectra to known spectra of ferrous (ϵ431 nm = 148 mm−1 cm−1) and ferrous nitrosyl (ϵ399 nm = 110 mm−1 cm−1) sGC using the Solver function in Microsoft Excel (38). The stability of ferrous nitrosyl sGC was monitored by UV-visible spectroscopy. Spectra were taken every 20 min for 1 h. The activity of ferrous sGC was determined immediately after titration with PROLI NONOate and after 1 h incubation to test the stability of the enzyme. All 1-NO activity assays were conducted in a final volume of 10 μl. Ferrous nitrosyl sGC (180–320 nm) was incubated with varying concentrations (0–1 mm) of ATP for 1 min before initiation of reactions by the addition of GTP. Reactions were quenched after 3 min by the addition of 40 μl of 125 mm Zn(CH3CO2)2 and 50 μl of 125 mm Na2CO3. As described above, the final concentration of cGMP product was determined by the cGMP EIA kit from Enzo Life Sciences, Inc. Each experiment was repeated at least twice. The data from each experiment were first analyzed on their own to obtain individual Vmax values, after which data were normalized (ν/Vmax), combined, and fit to models described below.
Data Analysis
Data were first fit to a mixed type inhibition model with two independent binding sites (Scheme 1 and Equations 1–3) (50). When the cyclization reaction is at steady state, the binding of nucleotides is expected to be close to equilibrium (51); therefore, the rate equations were derived using rapid equilibrium treatment. Accordingly, it was assumed that the reaction between enzyme, substrate, and inhibitor remained in equilibrium and that the concentration of free substrate and inhibitor remained unchanged because the enzyme concentration was significantly lower than the concentration of nucleotides. The equilibrium constants (Ks, Kia, Kic, etc.) were determined following these assumptions (50). In the initial analysis, to reduce errors, Ks was obtained separately from the other variables. The difference in the Ks for WT and DM sGC was insignificant; therefore, for each NO state, a common Ks was obtained by combining WT and DM data at 0 mm ATP. Subsequently, these Ks values were used to solve the other variables. Other inhibition models used are described under “Discussion.”
SCHEME 1.
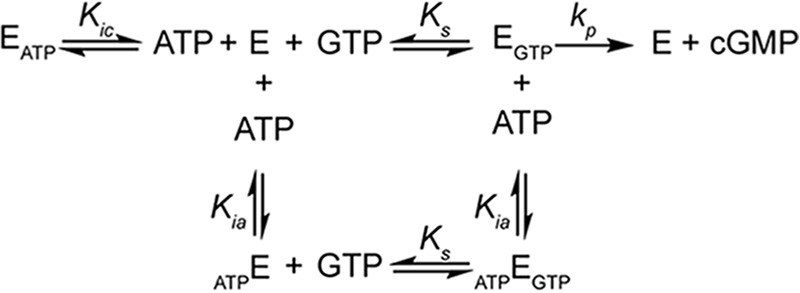
Kinetic scheme of ATP inhibition of sGC (E) by two independent sites. Equilibria scheme for an enzyme (E) with one active site and one regulating allosteric site, binding to substrate (GTP) and inhibitor (ATP). Binding of GTP to the active site composes an active complex (EGTP) producing product (cGMP). Ks, Kic, and Kia are the dissociation constants of the EGTP, EATP, and ATPE complexes.
Results
ATP Inhibition of sGC in the Basal State
ATP inhibition of wild type (WT) sGC (Fig. 2, A and C) and the pseudosymmetric site variant α1-C594A/β1-D477A (DM) (Fig. 2, B and D) of sGC was investigated. ATP is a mixed type inhibitor (Fig. 2) under these conditions for both WT and DM sGC. Previous studies have shown that ATP inhibits sGC by binding to both the catalytic site and an unidentified allosteric nucleotide binding site (27, 30). Therefore, ATP inhibition was modeled as mixed type inhibitor with two independent binding sites (Scheme 1 and Equations 1–3).
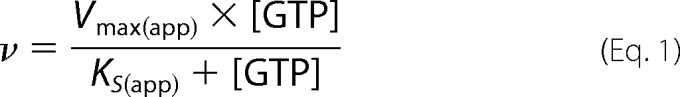 |
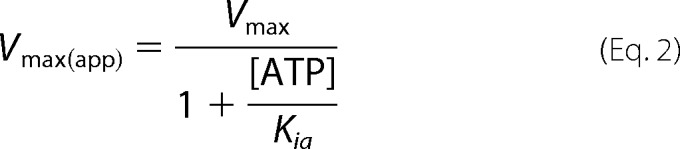 |
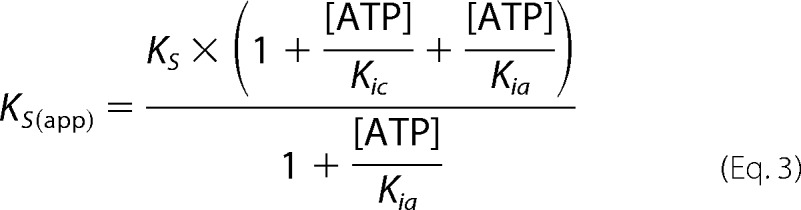 |
FIGURE 2.

ATP inhibition of sGC in the basal state for WT and DM. Changes in activity with increasing concentrations of ATP for WT (A) and DM (B) sGC. Data are replotted as double reciprocal plots for WT (C) and DM (D). sGC (0.1 μg) wild type or variant was assayed for cyclase activity in 50 mm HEPES, pH 7.4, 6 mm MgCl2, and 1 mm DTT. Each curve was fit to mixed type inhibition with two independent binding sites (Equations 1–3). Error bars, S.E.
Using this model, kinetic parameters were obtained for both WT and DM sGC inhibition. In the basal state, sGC activity and ATP inhibition were not significantly altered by the α1-C594A/β1-D477A mutation (Fig. 2) (Ks = 70 ± 14 μm for both WT and DM sGC). ATP inhibition by binding to the catalytic site (Kic) was also not significantly altered by mutation; Kic was 80 ± 20 μm and 60 ± 20 μm for WT and DM sGC, respectively. In addition, no significant change was observed for ATP inhibition at the allosteric site (Kia), 460 ± 110 and 510 ± 170 μm for WT and DM sGC, respectively. Basal state activity of DM sGC (kcat = 0.073 ± 0.003 s−1) was higher than the activity of WT (kcat = 0.029 ± 0.001 s−1) (Table 1).
TABLE 1.
Kinetic parameters of sGC activity and inhibition by ATP depending on NO state of the enzyme
| NO state |
kcat |
Ks, WT |
Kic |
Kia |
|||
|---|---|---|---|---|---|---|---|
| WT | DM | WT | DM | WT | DM | ||
| s−1 | μm | mm | mm | ||||
| Basal | 0.029 ± 0.001 | 0.073 ± 0.003 | 70 ± 14 | 0.08 ± 0.02 | 0.06 ± 0.02 | 0.46 ± 0.11 | 0.51 ± 0.17 |
| 1-NO | 1.9a ± 0.1 | 2.8a ± 0.2 | 80 ± 20 | 0.18 ± 0.07 | 0.19 ± 0.07 | 0.43 ± 0.1 | 0.44 ± 0.1 |
| xsNO | 15.0 ± 0.6 | 11.0 ± 0.5 | 30 ± 10 | 0.18 ± 0.07 | 0.30 ± 0.09 | 0.49 ± 0.08 | 1.40 ± 0.27 |
a In the 1-NO state, kcat values were determined based on the concentration of 5c NO complex estimated by deconvolution of final spectra as shown in Fig. 3A.
ATP Inhibition of sGC in the 1-NO State
The 1-NO state of sGC was obtained through titration of ferrous sGC with PROLI NONOate until only a small shoulder at 431 nm (ferrous sGC) remained, at which point the protein was assayed. The addition of PROLI NONOate was stopped short of full conversion to ferrous nitrosyl sGC (399 nm) to ensure that no excess NO was present (Fig. 3A). The stability of this complex was monitored by UV-visible spectroscopy, and the activity decrease was followed over time. No significant change was observed in ferrous nitrosyl sGC spectra, and the activity remained unchanged for at least 1 h, thus confirming the stability of the 1-NO state (Fig. 3B). The concentration of ferrous nitrosyl sGC was estimated by deconvolution of observed spectra to known spectra of ferrous sGC and ferrous nitrosyl sGC (Fig. 3A). Because the addition of PROLI NONOate was stopped before full conversion to 399 nm, the activity samples contain both ferrous and ferrous nitrosyl sGC. The contribution of each species can be determined using the estimated concentration of ferrous sGC and known kinetic parameters in the basal state (Table 1 and Fig. 3C). Based on this analysis, ATP inhibition of WT (Fig. 3, D and F) and DM (Fig. 3, E and G) 1-NO sGC was determined to be similar to that of the basal state of enzyme. ATP inhibition was again modeled as mixed type inhibition with two binding sites (Scheme 1 and Equations 1–3), and the kinetic parameters for sGC activity and inhibition for both WT and DM sGC were obtained for the 1-NO state. sGC activity and ATP inhibition were not significantly altered by the pseudosymmetric site mutation (Fig. 3) (Ks = 80 ± 20 μm for both WT and DM sGC). ATP inhibition by binding to the catalytic site (Kic) was also not significantly altered by mutation; Kic was 180 ± 7 μm and 190 ± 7 μm for WT and DM sGC, respectively. In addition, no significant change was observed for ATP inhibition at the allosteric site (Kia), 430 ± 100 and 440 ± 100 μm for WT and DM sGC, respectively. NO binding to the heme led to an ∼50-fold activation; turnover rate (kcat) for WT and DM was 1.9 ± 0.1 s−1 and 2.8 ± 0.2 s−1, respectively (Table 1).
FIGURE 3.

Characterization of 1-NO state of sGC. The 1-NO state of sGC is achieved by titration of ferrous sGC with PROLI NONOate (A), and the concentration of ferrous nitrosyl (Fe2+-NO) sGC is estimated by deconvolution of the final spectrum (red; 55% Fe2+-NO) to ferrous (blue) and Fe2+-NO (green) sGC spectra. The stability of 1-NO sGC was monitored by UV-visible spectroscopy and activity (B). sGC activity was tested immediately after the formation of Fe2+-NO (O) and after a 1-h incubation (B, inset). C, contribution of Fe2+ (basal) and Fe2+-NO (1-NO) sGC to the final activity observed in 1-NO samples. ATP inhibition of sGC in the 1-NO state for WT (D) and DM (E). Activity data are replotted as double reciprocal plots for WT (F) and DM (G). Ferrous nitrosyl sGC (100–200 nm) wild type or variant was assayed in 50 mm HEPES, pH 7.4, 6 mm MgCl2, and 1 mm tris(2-carboxyethyl)phosphine. Each curve was fit to mixed type inhibition with two binding sites (Equations 1–3). Error bars, S.E.
ATP Inhibition of sGC in the Excess NO State
ATP inhibition of WT (Fig. 4, A and C) and DM (Fig. 3, B and D) sGC was investigated in the presence of 0.1 mm DEA NONOate. ATP was again found to be a mixed type inhibitor (Fig. 4) (30, 47). ATP inhibition was modeled as mixed type inhibition with two binding sites (Scheme 1 and Equations 1–3). Mutation at the pseudosymmetric site did not significantly alter the Ks (30 ± 10 μm). Compared with the Ks in the basal and 1-NO states (∼80 μm), in the excess NO state, sGC shows a higher affinity for GTP in the active site. On the other hand, ATP inhibition of sGC in the excess NO state was significantly altered by the pseudosymmetric site mutation (Fig. 4). The affinity of ATP for the catalytic site was weaker for DM sGC; Kic was 300 ± 9 μm for DM compared with 180 ± 7 μm for WT. The ATP inhibition constant for the allosteric site (Kia) also increased from 490 ± 8 μm in WT to 1400 ± 270 μm in DM sGC. In accordance with previous results, sGC was activated around 200-fold in the presence of excess NO; turnover rates (kcat) for WT and DM sGC were 15 ± 0.6 s−1 and 11 ± 0.5 s−1, respectively (Table 1).
FIGURE 4.
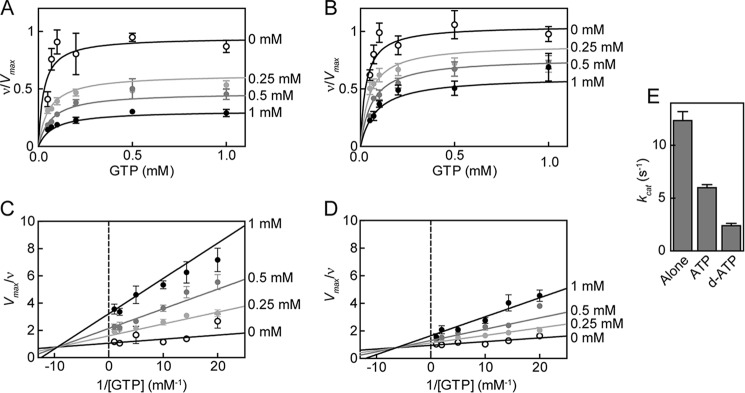
ATP inhibition of sGC in the xsNO state for WT and DM. Changes in activity with increasing concentrations of ATP for WT (A) and DM (B) sGC. Data are replotted as double reciprocal plots for WT (C) and DM (D). sGC (0.1 μg) wild type or variant was assayed in 50 mm HEPES, pH 7.4, 6 mm MgCl2, and 1 mm DTT in the presence of 0.1 mm DEA NONOate. Each curve was fit to mixed type inhibition with two binding sites (Equations 1–3). sGC inhibition of 2′-deoxy-ATP compared with ATP (E). sGC (0.1 μg) was assayed in 50 mm HEPES, pH 7.4, 6 mm MgCl2, and 1 mm DTT with 0.1 mm DEA NONOate in the absence and presence of 0.5 mm ATP and 0.5 mm 2′-deoxy-ATP (d-ATP). Error bars, S.E.
The hydroxyl group at the 2′-position of the ribose ring on ATP plays a role in regulation of particulate GCs by ATP (GC-A and GC-B) (53). Therefore, the role of the hydroxyl group at the 2′-position of the ribose ring on ATP in sGC inhibition was investigated. ATP lacking the 2′-OH group, 2′-deoxy-ATP, was a more potent inhibitor of sGC compared with ATP (Fig. 4E).
Discussion
Here, the ATP inhibition of sGC was investigated at the basal, 1-NO, and excess NO states. The equilibrium constants for nucleotide binding were obtained for each state. The equilibrium constants obtained distinguished the 1-NO state from the basal and excess NO states as an intermediate state in enzyme activation. The 1-NO state shows similar affinity for GTP as in the basal state but a decreased affinity for ATP. In addition, the role of pseudosymmetric site in nucleotide regulation was determined by making use of the pseudosymmetric site variant, α1-C594A/β1-D477A (DM). The results showed that mutation of the pseudosymmetric site resulted in a distinctive change in ATP inhibition mechanism in the excess NO state but not in the basal and 1-NO states of sGC. These observations can best be explained by invoking cooperativity between the catalytic and pseudosymmetric site of sGC. The analysis of ATP inhibition in the excess NO state with a more comprehensive model showed strong positive cooperativity between the pseudosymmetric site and the catalytic site, which is perturbed by mutations in the pseudosymmetric site.
The kinetic parameters of sGC inhibition were obtained by fitting enzyme activity at various concentrations of ATP and GTP to a mixed type inhibition model where the inhibitor binds at two independent sites (Equations 1–3). In previous studies, two models were used to analyze ATP (or ATP analogue) inhibition of sGC. Ruiz-Stewart et al. (47) fit their data to a model for mixed non-competitive allosteric inhibition. According to this model, ATP only binds to an allosteric site. ATP binding to the allosteric site induces a conformational change in the active site that completely inhibits cGMP formation and alters the affinity of the active site for GTP. On the other hand, Yazawa et al. (27) fit their data to a mixed inhibition model in which the inhibitor binds to two independent sites, competing with substrate at the active site and non-competitively inhibiting the enzyme at the allosteric site (Scheme 1). Later, Derbyshire et al. (30) showed that sGC can also cyclize ATP to cAMP, and this activity is inhibited by ATP via substrate inhibition. This observed substrate inhibition can only be explained by ATP binding to two sites on sGC. Due to the increased evidence for two nucleotide binding sites in sGC, the model from Yazawa et al. was used for kinetic fitting in this study (27, 28, 30, 52).
The effect of NO on the kinetic parameters of WT sGC are shown in Fig. 5, A and B. Consistent with previous observations, the addition of 1 eq of NO resulted in activation of the enzyme from the basal state, but the 1-NO state was still 10-fold less active than fully activated enzyme (xsNO state). The changes in kinetic parameters show that the 1-NO state of sGC is an intermediate step in full activation of the enzyme. Because the 1-NO and basal states exhibit the same Ks, GTP affinity of the active site is not altered by binding of NO at the ferrous heme. However, in the presence of excess NO, there is a significant decrease in Ks along with the large increase in kcat, resulting in a significant increase in the catalytic efficiency of the enzyme in the fully activated state (Table 1). Although the nature of the low activity ferrous nitrosyl sGC remains unclear, because NO can be bound at the proximal or distal site of heme, its kinetic parameters can be determined using limiting concentrations of NO. Examination of ATP inhibition at different NO levels demonstrates that the Kic of 1-NO and xsNO states are identical and larger than the Kic in the basal state. These results reveal a change in the binding of ATP at the catalytic site due to binding of NO to the ferrous heme. In contrast, ATP binding to the allosteric site seems to be independent of NO interaction, because Kia does not change significantly at any NO state. Taken together, these results show that NO binding to the ferrous heme and additional NO interacting with sGC have unique effects on the catalytic site of the enzyme; NO binding to the heme increases kcat but does not alter the affinity for GTP, whereas additional NO is necessary to tighten substrate binding to the enzyme.
FIGURE 5.
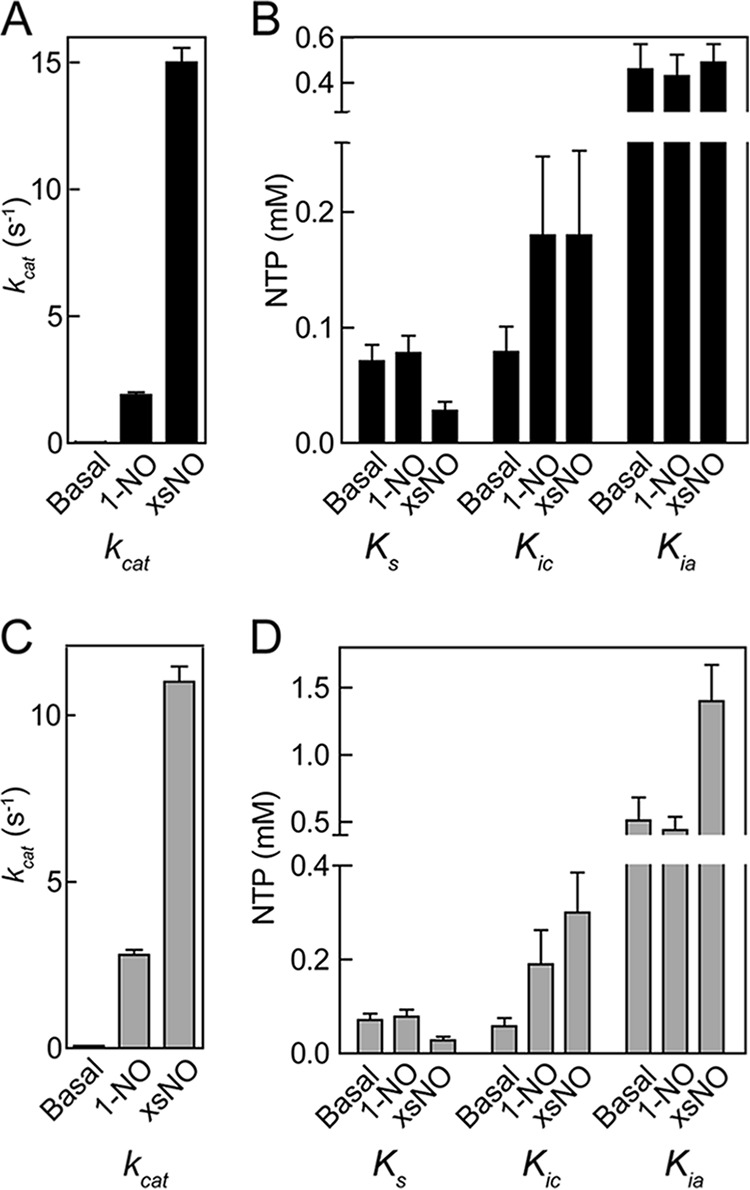
Changes in the kinetic parameters of WT (A and B) and DM (C and D) sGC with respect to NO levels. Values were obtained from data in Figs. 2–4 based on mixed type inhibition with two binding sites (Equations 1–3). Error bars, S.E.
Crystal structures of the sGC catalytic domain (Protein Data Bank entries 3UVJ, 2WZ1, and 4NI2) suggest two binding sites for nucleotides located in the interface between the two domains (23, 24). One of these sites is the active site, and the other is the pseudosymmetric site. Previous studies have implied that the pseudosymmetric site is involved in allosteric ATP regulation of sGC activity (26). Two residues in the pseudosymmetric site (α1-C594A and β1-D477A) were mutated (DM) to investigate the role of this site in ATP regulation. The change in kinetic parameters of ATP inhibition at various NO states of DM sGC is shown in Fig. 5, C and D. We expected these mutations to decrease the affinity of ATP in the allosteric site in all NO states. However, we observed that ATP affinity significantly changed only in the excess NO state, which showed a large increase in both Kia and Kic. The fact that mutations in the pseudosymmetric site affected binding of ATP to the catalytic site (Kic) shows that the catalytic site and pseudosymmetric site interact with each other in the excess NO state. These results are in contrast to the hypothesis by Yazawa et al. (27), who reported that nucleotide binding to the allosteric and catalytic site are independent. Indeed, in the excess NO state, ATP binding to the allosteric site changes the affinity of nucleotides in the catalytic site.
A complete kinetic scheme of sGC activity in the excess NO state must now incorporate cooperativity between the allosteric and catalytic site as well as possible product (cGMP) formation in the presence of inhibitor. In addition, due to the pseudosymmetry between the catalytic and allosteric site, GTP might also bind to the allosteric site on sGC. Incorporating these requirements into the kinetic scheme of sGC leads to a new model depicted in Scheme 2. It is not possible to determine both cooperativity between the two sites and the nucleotide affinity of each site by analyzing the activity of sGC in the presence of ATP alone. Therefore, GTP and ATP affinity of each site were independently determined, as described below.
SCHEME 2.
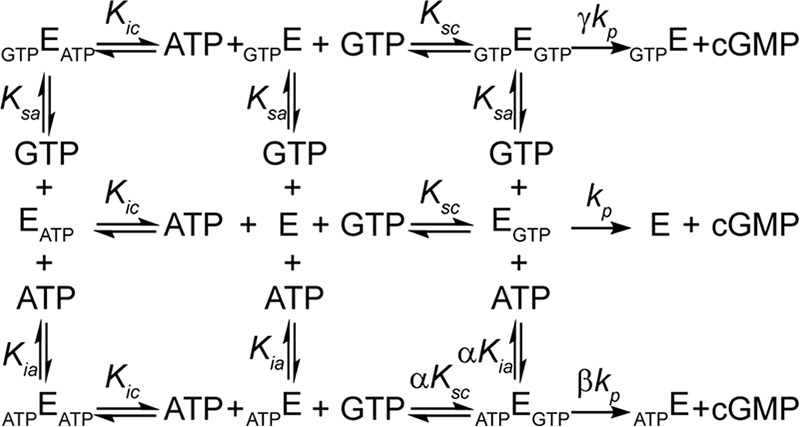
Kinetic scheme of nucleotide regulation of sGC. Equilibria scheme for sGC (E) with one active site and one regulating allosteric site, binding to substrate (GTP) and inhibitor (ATP). Binding of GTP to the active site composes an active complex (EGTP) producing product (cGMP). Binding of ATP to the allosteric site (ATPE) affects the affinity of the active site for GTP by a factor of α. ATPEGTP and GTPEGTP have a different rate of product formation (βkp and γkp, respectively) than EGTP (kp). Ksc, Ksa, Kic, and Kia are the dissociation constants of the EGTP, GTPE, EATP, and ATPE complexes.
Saturation plots of GTP analog GMP-CPP with ferrous sGC revealed two nucleotide binding sites, one with high and one with low affinity (27). Based on this result, the possibility of a second binding site for GTP was investigated. Fitting sGC activity at various concentrations of GTP to the Hill equation, in the presence of excess NO, yielded a Hill coefficient of 1.0 ± 0.2 (Fig. 6A); therefore, if there is an allosteric binding site for GTP, the two sites are not cooperative. Indeed, a double reciprocal plot of the same experimental data is linear, consistent with no cooperativity (Fig. 6B). Furthermore, sGC activity decreased at higher concentrations of GTP (Fig. 6C). Because there is no cooperativity between the two GTP binding sites, this can be described as non-competitive substrate inhibition,
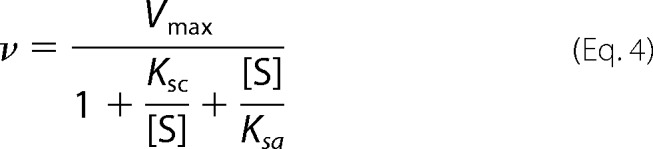 |
where [S] represents substrate concentration. The data from sGC activity were fit to Equation 4 (Fig. 6C) (30). This yielded a GTP affinity of 34 ± 7 μm for the active site (Ksc) and of 4.6 ± 1.3 mm for the allosteric site (Ksa). Non-competitive substrate inhibition assumes that GTP bound at the allosteric site (GTPEGTP) is inactive; hence, γ in Scheme 2 is zero. Fitting the data to a model that incorporated possible activity for GTP-bound sGC gave much larger errors; Ksc was 36 ± 9 μm, and Ksa was 3.5 ± 2.9 mm. In addition, the activity of sGC bound to GTP at the allosteric site was only around 9% of the activity of the free enzyme. Because the activity of GTP-bound sGC was so low and changing Ksa (between 1 and 1000 mm) had very little effect on the calculations and fits below, we continued with the initial assumption that sGC with GTP bound to the allosteric site has no activity, and affinity of GTP for the allosteric site (Ksa) is 4.6 ± 1.3 mm.
FIGURE 6.
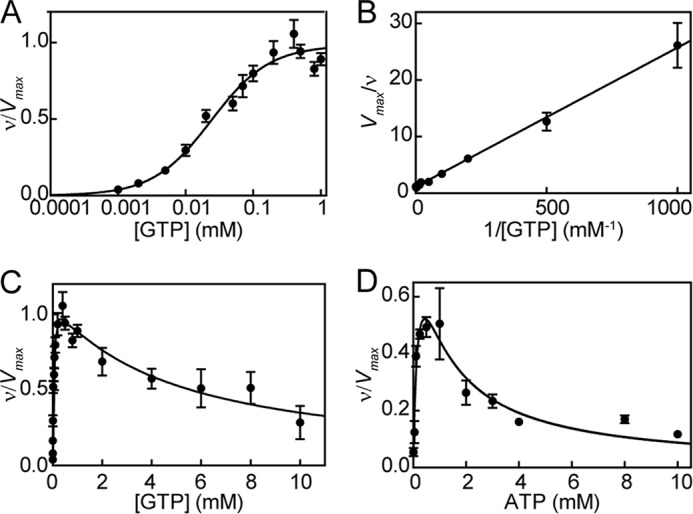
sGC activity with GTP (A–C) and ATP (D) as substrate in the presence of excess NO. A, guanylate cyclase activity at various concentrations of GTP (0.001–1 mm) were fit to the Hill equation. B, data were replotted as a double reciprocal plot. C, GTP (0.001–10 mm) cyclization activity fit to non-competitive substrate inhibition (Equation 4). D, adenylate cyclase activity of sGC at various concentrations of ATP (0.01–10 mm) in the presence of excess NO (from Derbyshire et al. (30)); data fit to Equation 4. Error bars, S.E.
Previous studies have shown that sGC exhibits low level adenylate cyclase activity with substrate inhibition at higher ATP concentrations (30, 52). It is assumed that cAMP is formed at the active site, and this process is inhibited by binding of ATP to the allosteric site (30). This property was used to study the behavior of sGC in the presence of ATP and the absence of GTP. The adenylate cyclase activity of sGC can also be described as non-competitive substrate inhibition. Therefore, the data from sGC adenylate cyclase activity from Derbyshire et al. (30) were fit to Equation 4 (Fig. 6D). This analysis yielded an ATP affinity of 0.2 ± 0.08 mm for the active site (Kic) and 1.0 ± 0.3 mm for the allosteric site (Kia).
Having derived affinities of the nucleotides to the active site (Ksc and Kic) and allosteric site (Ksa and Kia), the cooperativity coefficient (α) and the activity of sGC bound to ATP (βkp) can be determined by fitting sGC activity in the presence of GTP and ATP to the complete model (Equations 5–7).
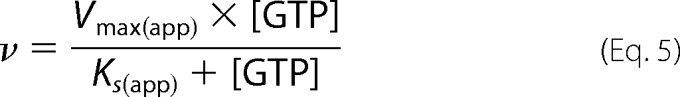 |
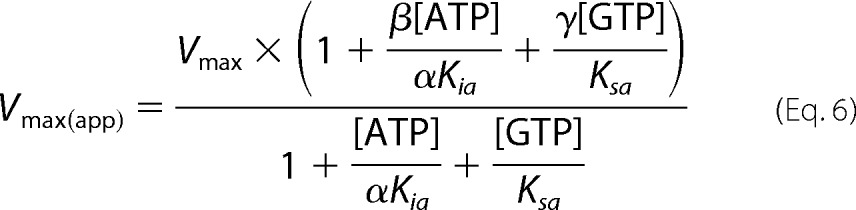 |
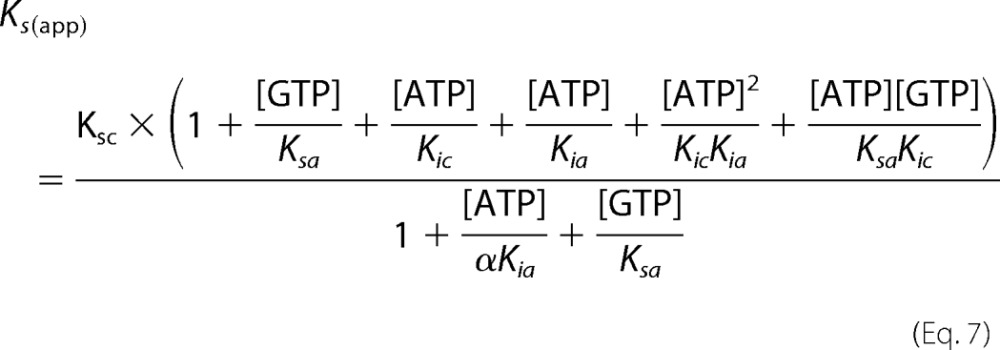 |
Using this model, all kinetic parameters of sGC inhibition by nucleotides were determined (Table 2). Integration of these values into Equations 5–7 was used to construct Lineweaver-Burk, Dixon, and Cornish-Bowden plots (Fig. 7). The three plots are based on the same data and equations, yet each plot has a different emphasis. For example, the Lineweaver-Burk plot highlights the data at low levels of GTP and high levels of ATP, whereas the Cornish-Bowden plot accentuates activity at high levels of both GTP and ATP. The high degree of consistency of the data with each of the curves validates the model and results obtained. In addition, if the previously proposed ATP inhibition model, shown in Scheme 1, was correct, then the plot of Vmax/ν (or Vmax[GTP]/ν) versus [ATP] (Dixon and Cornish-Bowden plots) would be linear according to Equation 8 (derived from Equations 1–3). Indeed, Dixon plots are linear for mixed inhibition systems as long as the enzyme bound to inhibitor is inactive (50).
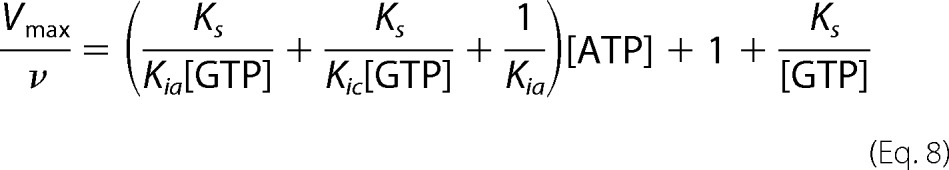 |
TABLE 2.
Kinetic parameters of ATP inhibition of sGC determined by fitting Equations 5–7 to ATP inhibition data
The parameter α is the cooperativity coefficient. It describes the changes in affinity at the active site due to binding of ATP at the allosteric site. The parameters β and γ denote the relative activity of sGC bound to ATP and GTP at the allosteric site, respectively (see Scheme 2).
| Parameter | WT | DM |
|---|---|---|
| Ksc (μm) | 34.0 ± 7 | 34.0 ± 7 |
| Ksa (mm) | 4.6 ± 1.3 | 4.6 ± 1.3 |
| Kic (mm) | 0.20 ± 0.08 | 0.20 ± 0.08 |
| Kia (mm) | 1.0 ± 0.3 | 1.0 ± 0.3 |
| α | 0.21 ± 0.04 | 0.35 ± 0.09 |
| β | 0.15 ± 0.03 | 0.45 ± 0.04 |
| γ | 0 | 0 |
FIGURE 7.
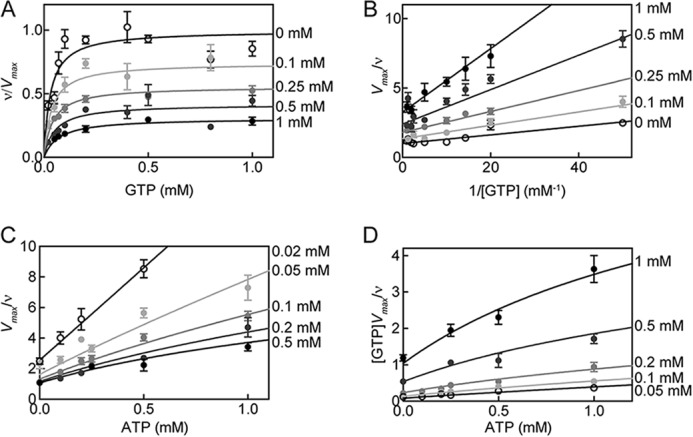
ATP inhibition of WT sGC activity in the excess NO state. The reaction rate of sGC as a fraction of the maximal reaction rate was plotted versus the concentration of substrate (GTP) in the presence of various ATP concentrations. Curves belonging to Equations 5–7 are depicted with data (A). B, Lineweaver-Burk plot of the data and curves; C, Dixon plot of the data and curves; D, Cornish-Bowden plot of the data and curves. Error bars, S.E.
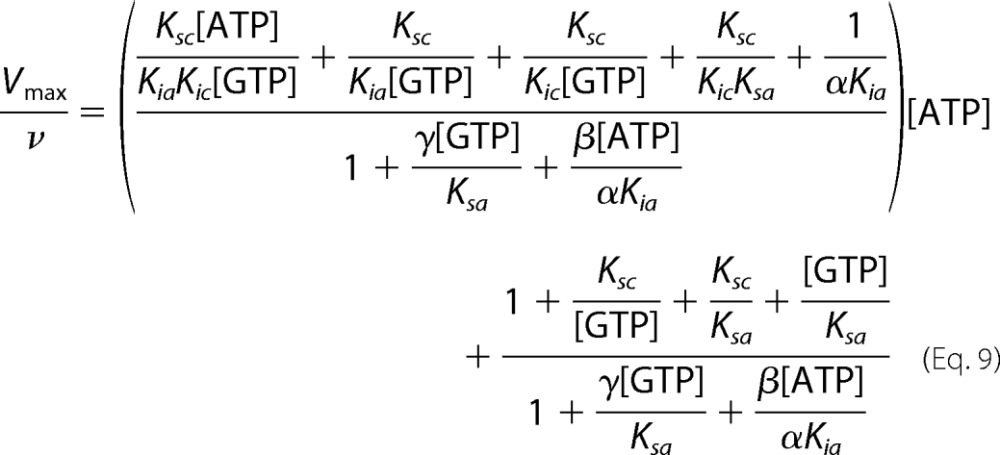
However, as seen in Fig. 7, C and D, the Dixon and Cornish-Bowden plots of ATP inhibition of sGC are nonlinear. This is expected for an enzyme described by the complete model (Scheme 2). Equation 9 (derived from Equations 5–7) describes the relationship between Vmax/ν and nucleotide concentrations for the complete model.
 |
In the excess NO state, sGC shows strong positive cooperativity between the catalytic and allosteric site, and α is much lower than 1 (Table 2). Upon binding of GTP to the catalytic site, there is a 5-fold increase in affinity of ATP for the allosteric site. This positive cooperative inhibitory effect of ATP is thought to be insignificant in the basal state (26). The lack of cooperativity in basal and 1-NO states can be explained by the fact that sGC must exhibit two states: an inactive, “open” state that, upon activation of the enzyme, changes into an active, “closed” state (Fig. 8) (49). A similar conclusion regarding “open” and “closed” states has been advanced with the ACs. The two crystal structures reported most likely represent the open state present in the basal and 1-NO states of sGC. Upon the addition of excess NO, the two subunits move toward one another to contract the active site and form the “closed” state (17). The changes in the relative positions of the two subunits in the “closed” state affect the active and pseudosymmetric site because they are located at the subunit interface. This may lead to the increase in cooperativity between the catalytic and pseudosymmetric site observed in the excess NO state.
FIGURE 8.
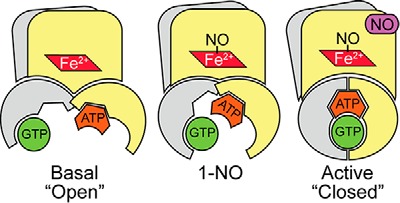
Schematic representation of different states of sGC. Previous biochemical and structural studies with adenylate cyclases and guanylate cyclases have shown that there is a low activity (open) conformation and a high activity (closed) conformation. Basal activity is shown in the open conformation, and the high activity closed conformation is shown the presence of excess NO. The 1-NO form is in an intermediate state. NO is shown bound to the distal site of the heme; however, it probably resides in the proximal site in the active (excess NO) state.
For DM sGC, there was no noticeable change in ATP inhibition kinetics compared with WT in the basal and 1-NO states (Figs. 2 and 3), meaning that the affinity of ATP binding to either of these sites was not affected by this mutation. Therefore, the significant difference observed in ATP inhibition of the pseudosymmetric site variant in the excess NO state must be due to changes in the cooperativity constant (α) and activity of the inhibitor-bound complex (βkp). Indeed, when the same constants (Ksc, Ksa, Kic, and Kia) were used and the activity of the pseudosymmetric site variant was fit to Equations 5–7, α and β were determined to be 0.35 ± 0.09 and 0.45 ± 0.04 (Fig. 9). Taken together, these results indicate that the mutations in the pseudosymmetric site did not alter the affinity for ATP sufficiently to inhibit binding; instead, they altered the cooperativity between the two sites and the activity of the ATP-bound enzyme. Therefore, in the closed state (Fig. 8), the allosteric communication between the catalytic site and pseudosymmetric site seems to be disrupted by the pseudosymmetric site mutations, revealing a new role for these residues in ATP regulation. A crystal structure of sGC catalytic domain in the active conformation is necessary to clearly identify the source of this effect.
FIGURE 9.
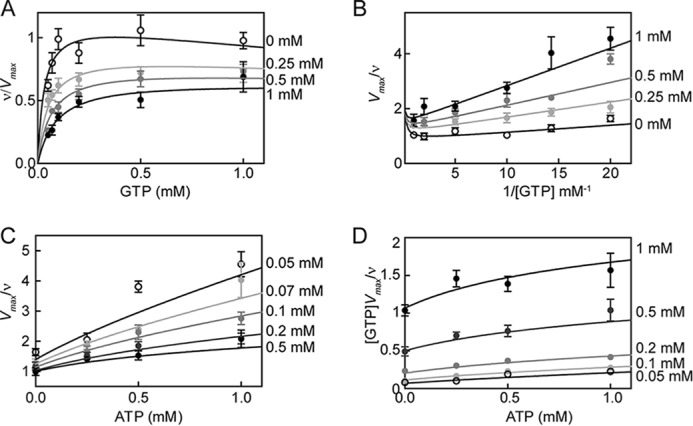
ATP inhibition of DM sGC activity in the excess NO state. The reaction rate of sGC variant as a fraction of the maximal reaction rate is plotted versus the concentration of substrate (GTP) in the presence of various ATP concentrations. Curves belonging to Equations 5–7 are depicted with data (A). B, Lineweaver-Burk plot of the data and curves; C, Dixon plot of the data and curves; D, Cornish-Bowden plot of the data and curves. Error bars, S.E.
In the excess NO state, sGC that is bound to the inhibitor ATP at the allosteric site is still active (Scheme 2, β > 0), albeit with 6-fold lower activity than in the absence of ATP. Taking into account the ATP (∼3 mm) and GTP (∼0.45 mm) concentrations in vivo and the strong positive cooperativity between the catalytic and allosteric site, this leads to the conclusion that most of the enzyme will exist in this low activity state (48). Because ATP levels in cells are much higher than GTP levels, and sGC can also accept ATP as a substrate, the decrease in Ksc for GTP in the presence of ATP results in much higher substrate selectivity for the enzyme, which is essential for its role in cellular signaling. The decrease in Ksc, combined with the increase in kcat, results in a much higher efficiency in catalysis at cellular levels of ATP and GTP in the presence of excess NO compared with basal and 1-NO states.
GCs are structurally similar to ACs, and together they are classified as class III nucleotide cyclases (21, 53). Indeed, mutation of substrate-specifying residues in sGC to corresponding residues in AC converts the enzyme to an AC and results in elimination of GTP as a substrate (54). However, similar mutations in AC resulted in an enzyme that could synthesize both cAMP and cGMP (54). Therefore, GCs are more specialized enzymes that evolved from refinement of the AC core (55, 56). The pseudosymmetric site is most likely a vestige from the early evolution of these enzymes from a symmetric to an asymmetric core. ATP regulation of sGC shows striking similarity to ATP regulation of particulate GCs.
Similar to sGC, ATP binding to a pseudosymmetric site in the catalytic domain of GC-A and GC-B leads to an increase in the affinity for GTP in the active site in the presence of natriuretic peptides, which activate GC-A and GC-B. (57) In addition, this positive cooperative effect of ATP is also not observed in the basal state for GC-A and GC-B. On the other hand, whereas ATP activates GC-A and GC-B, it inhibits sGC. The reason for the inhibition of sGC by ATP may lie in lowering the activity of the 1-NO enzyme to fine tune sGC activation by NO. In conclusion, for all GCs, the cooperativity between ATP binding at the pseudosymmetric site and GTP binding to the catalytic site conferred selectivity to sGC for cyclization of GTP (in an environment with high ATP concentrations) in vivo, which may have driven an increased affinity for ATP in the evolution of a pseudosymmetric site.
Acknowledgment
We thank Dr. Eric S. Underbakke for critical reading of the manuscript.
Footnotes
- sGC
- soluble guanylate cyclase
- GC
- guanylate cyclase
- H-NOX
- N-terminal heme-NO/O2 binding
- PAS
- Per/Arnt/Sim
- AC
- adenylate cyclase
- 6c NO and 5c NO
- 6- and 5-coordinate Fe2+-NO, respectively
- xsNO
- excess NO
- DM
- double mutant
- DEA NONOate
- diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate
- PROLI NONOate
- 1-(hydroxy-NNO-azoxy)-l-proline
- GMP-CPP
- α,β-methylene guanosine 5′-triphosphate.
References
- 1. Hill B. G., Dranka B. P., Bailey S. M., Lancaster J. R., Jr., Darley-Usmar V. M. (2010) What part of NO don't you understand? Some answers to the cardinal questions in nitric oxide biology. J. Biol. Chem. 285, 19699–19704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shah A. M., MacCarthy P. A. (2000) Paracrine and autocrine effects of nitric oxide on myocardial function. Pharmacol. Ther. 86, 49–86 [DOI] [PubMed] [Google Scholar]
- 3. Toda N., Okamura T. (2003) The pharmacology of nitric oxide in the peripheral nervous system of blood vessels. Pharmacol. Rev. 55, 271–324 [DOI] [PubMed] [Google Scholar]
- 4. Garthwaite J. (2008) Concepts of neural nitric oxide-mediated transmission. Eur. J. Neurosci. 27, 2783–2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Denninger J. W., Marletta M. A. (1999) Guanylate cyclase and the NO/cGMP signaling pathway. Biochim. Biophys. Acta 1411, 334–350 [DOI] [PubMed] [Google Scholar]
- 6. Waldman S. A., Murad F. (1987) Cyclic GMP synthesis and function. Pharmacol. Rev. 39, 163–196 [PubMed] [Google Scholar]
- 7. Bredt D. S. (2003) Nitric oxide signaling specificity: the heart of the problem. J. Cell Sci. 116, 9–15 [DOI] [PubMed] [Google Scholar]
- 8. Friebe A., Koesling D. (2009) The function of NO-sensitive guanylyl cyclase: what we can learn from genetic mouse models. Nitric Oxide 21, 149–156 [DOI] [PubMed] [Google Scholar]
- 9. Buys E. S., Potter L. R., Pasquale L. R., Ksander B. R. (2014) Regulation of intraocular pressure by soluble and membrane guanylate cyclases and their role in glaucoma. Front. Mol. Neurosci. 7, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ghofrani H.-A., Galiè N., Grimminger F., Grünig E., Humbert M., Jing Z.-C., Keogh A. M., Langleben D., Kilama M. O., Fritsch A., Neuser D., Rubin L. J., and PATENT-1 Study Group (2013) Riociguat for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 369, 330–340 [DOI] [PubMed] [Google Scholar]
- 11. Greene S. J., Gheorghiade M., Borlaug B. A, Pieske B., Vaduganathan M., Burnett J. C., Jr., Roessig L., Stasch J.-P., Solomon S. D., Paulus W. J., Butler J. (2013) The cGMP signaling pathway as a therapeutic target in heart failure with preserved ejection fraction. J. Am. Heart Assoc. 2, e000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Follmann M., Griebenow N., Hahn M. G., Hartung I., Mais F.-J., Mittendorf J., Schäfer M., Schirok H., Stasch J.-P., Stoll F., Straub A. (2013) The chemistry and biology of soluble guanylate cyclase stimulators and activators. Angew. Chem. Int. Ed. Engl. 52, 9442–9462 [DOI] [PubMed] [Google Scholar]
- 13. Karow D. S., Pan D., Davis J. H., Behrends S., Mathies R. A., Marletta M. A. (2005) Characterization of functional heme domains from soluble guanylate cyclase. Biochemistry 44, 16266–16274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Winger J. A., Marletta M. A. (2005) Expression and characterization of the catalytic domains of soluble guanylate cyclase: interaction with the heme domain. Biochemistry 44, 4083–4090 [DOI] [PubMed] [Google Scholar]
- 15. Rothkegel C., Schmidt P. M., Atkins D. J., Hoffmann L. S., Schmidt H. H. H. W., Schröder H., Stasch J. P. (2007) Dimerization region of soluble guanylate cyclase characterized by bimolecular fluorescence complementation in vivo. Mol. Pharmacol. 72, 1181–1190 [DOI] [PubMed] [Google Scholar]
- 16. Ma X., Sayed N., Baskaran P., Beuve A., van den Akker F. (2008) PAS-mediated dimerization of soluble guanylyl cyclase revealed by signal transduction histidine kinase domain crystal structure. J. Biol. Chem. 283, 1167–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Underbakke E. S., Iavarone A. T., Chalmers M. J., Pascal B. D., Novick S., Griffin P. R., Marletta M. A. (2014) Nitric oxide-induced conformational changes in soluble guanylate cyclase. Structure 22, 602–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tesmer J. J., Sprang S. R. (1998) The structure, catalytic mechanism and regulation of adenylyl cyclase. Curr. Opin. Struct. Biol. 8, 713–719 [DOI] [PubMed] [Google Scholar]
- 19. Tang W. J., Gilman A. G. (1992) Adenylyl cyclases. Cell 70, 869–872 [DOI] [PubMed] [Google Scholar]
- 20. Garbers D. L., Lowe D. G. (1994) Guanylyl cyclase receptors. J. Biol. Chem. 269, 30741–30744 [PubMed] [Google Scholar]
- 21. Tesmer J. J., Sunahara R. K., Johnson R. A., Gosslin G., Gilman A. G., Sprang S. R. (1999) Two-metal-ion catalysis in adenylyl cyclase. Science 285, 756–760 [DOI] [PubMed] [Google Scholar]
- 22. Tesmer J. J., Sunahara R. K., Gilman A. G., Sprang S. R. (1997) Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsα·GTPγS. Science 278, 1907–1916 [DOI] [PubMed] [Google Scholar]
- 23. Allerston C. K., von Delft F., Gileadi O. (2013) Crystal structures of the catalytic domain of human soluble guanylate cyclase. PLoS One 8, e57644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Seeger F., Quintyn R., Tanimoto A., Williams G. J., Tainer J. A., Wysocki V. H., Garcin E. D. (2014) Interfacial residues promote an optimal alignment of the catalytic center in human soluble guanylate cyclase: heterodimerization is required but not sufficient for activity. Biochemistry 53, 2153–2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu Y., Ruoho A. E., Rao V. D., Hurley J. H. (1997) Catalytic mechanism of the adenylyl and guanylyl cyclases: modeling and mutational analysis. Proc. Natl. Acad. Sci. U.S.A. 94, 13414–13419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chang F.-J., Lemme S., Sun Q., Sunahara R. K., Beuve A. (2005) Nitric oxide-dependent allosteric inhibitory role of a second nucleotide binding site in soluble guanylyl cyclase. J. Biol. Chem. 280, 11513–11519 [DOI] [PubMed] [Google Scholar]
- 27. Yazawa S., Tsuchiya H., Hori H., Makino R. (2006) Functional characterization of two nucleotide-binding sites in soluble guanylate cyclase. J. Biol. Chem. 281, 21763–21770 [DOI] [PubMed] [Google Scholar]
- 28. Ruiz-Stewart I., Kazerounian S., Pitari G. M., Schulz S., Waldman S. A. (2002) Soluble guanylate cyclase is allosterically inhibited by direct interaction with 2-substituted adenine nucleotides. Eur. J. Biochem. 269, 2186–2193 [DOI] [PubMed] [Google Scholar]
- 29. Suzuki T., Suematsu M., Makino R. (2001) Organic phosphates as a new class of soluble guanylate cyclase inhibitors. FEBS Lett. 507, 49–53 [DOI] [PubMed] [Google Scholar]
- 30. Derbyshire E. R., Fernhoff N. B., Deng S., Marletta M. A. (2009) Nucleotide regulation of soluble guanylate cyclase substrate specificity. Biochemistry 48, 7519–7524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stone J. R., Marletta M. A. (1996) Spectral and kinetic studies on the activation of soluble guanylate cyclase by nitric oxide. Biochemistry 35, 1093–1099 [DOI] [PubMed] [Google Scholar]
- 32. Zhao Y., Brandish P. E., Ballou D. P., Marletta M. A. (1999) A molecular basis for nitric oxide sensing by soluble guanylate cyclase. Proc. Natl. Acad. Sci. U.S.A. 96, 14753–14758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tsai A. L., Berka V., Sharina I., Martin E. (2011) Dynamic ligand exchange in soluble guanylyl cyclase (sGC): implications for sGC regulation and desensitization. J. Biol. Chem. 286, 43182–43192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martin E., Berka V., Sharina I., Tsai A. L. (2012) Mechanism of binding of NO to soluble guanylyl cyclase: implication for the second NO binding to the heme proximal site. Biochemistry 51, 2737–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Russwurm M., Koesling D. (2004) NO activation of guanylyl cyclase. EMBO J. 23, 4443–4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Herzik M. A., Jr., Jonnalagadda R., Kuriyan J., Marletta M. A. (2014) Structural insights into the role of iron-histidine bond cleavage in nitric oxide-induced activation of H-NOX gas sensor proteins. Proc. Natl. Acad. Sci. U.S.A. 111, E4156–E4164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cary S. P. L., Winger J. A., Marletta M. A. (2005) Tonic and acute nitric oxide signaling through soluble guanylate cyclase is mediated by nonheme nitric oxide, ATP, and GTP. Proc. Natl. Acad. Sci. U.S.A. 102, 13064–13069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brandish P. E., Buechler W., Marletta M. A. (1998) Regeneration of the ferrous heme of soluble guanylate cyclase from the nitric oxide complex: acceleration by thiols and oxyhemoglobin. Biochemistry 37, 16898–16907 [DOI] [PubMed] [Google Scholar]
- 39. Fernhoff N. B., Derbyshire E. R., Marletta M. A. (2009) A nitric oxide/cysteine interaction mediates the activation of soluble guanylate cyclase. Proc. Natl. Acad. Sci. U.S.A. 106, 21602–21607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bellamy T. C., Garthwaite J. (2001) Sub-second kinetics of the nitric oxide receptor, soluble guanylyl cyclase, in intact cerebellar cells. J. Biol. Chem. 276, 4287–4292 [DOI] [PubMed] [Google Scholar]
- 41. Bellamy T. C., Wood J., Goodwin D. A., Garthwaite J. (2000) Rapid desensitization of the nitric oxide receptor, soluble guanylyl cyclase, underlies diversity of cellular cGMP responses. Proc. Natl. Acad. Sci. U.S.A. 97, 2928–2933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kurtz A., Wagner C. (1998) Role of nitric oxide in the control of renin secretion. Am. J. Physiol. 275, F849–F862 [DOI] [PubMed] [Google Scholar]
- 43. Cals-Grierson M.-M., Ormerod A. D. (2004) Nitric oxide function in the skin. Nitric Oxide 10, 179–193 [DOI] [PubMed] [Google Scholar]
- 44. Martin W., Furchgott R. F., Villani G. M., Jothianandan D. (1986) Depression of contractile responses in rat aorta by spontaneously released endothelium-derived relaxing factor. J. Pharmacol. Exp. Ther. 237, 529–538 [PubMed] [Google Scholar]
- 45. Buvinic S., Huidobro-Toro J. P. (2001) Basal tonic release of nitric oxide coupled to cGMP production regulates the vascular reactivity of the mesenteric bed. Eur. J. Pharmacol. 424, 221–227 [DOI] [PubMed] [Google Scholar]
- 46. Cary S. P. L., Winger J. A., Derbyshire E. R., Marletta M. A. (2006) Nitric oxide signaling: no longer simply on or off. Trends Biochem. Sci. 31, 231–239 [DOI] [PubMed] [Google Scholar]
- 47. Ruiz-Stewart I., Tiyyagura S. R., Lin J. E., Kazerounian S., Pitari G. M., Schulz S., Martin E., Murad F., Waldman S. A. (2004) Guanylyl cyclase is an ATP sensor coupling nitric oxide signaling to cell metabolism. Proc. Natl. Acad. Sci. U.S.A. 101, 37–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Traut T. W. (1994) Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 140, 1–22 [DOI] [PubMed] [Google Scholar]
- 49. Underbakke E. S., Iavarone A. T., Marletta M. A. (2013) Higher-order interactions bridge the nitric oxide receptor and catalytic domains of soluble guanylate cyclase. Proc. Natl. Acad. Sci. U.S.A. 110, 6777–6782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Segel I. H. (1993) Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-state Enzyme Systems, Wiley-Interscience, New York [Google Scholar]
- 51. Roy B., Halvey E. J., Garthwaite J. (2008) An enzyme-linked receptor mechanism for nitric oxide-activated guanylyl cyclase. J. Biol. Chem. 283, 18841–18851 [DOI] [PubMed] [Google Scholar]
- 52. Beste K. Y., Burhenne H., Kaever V., Stasch J.-P., Seifert R. (2012) Nucleotidyl cyclase activity of soluble guanylyl cyclase α1β1. Biochemistry 51, 194–204 [DOI] [PubMed] [Google Scholar]
- 53. Tucker C. L., Hurley J. H., Miller T. R., Hurley J. B. (1998) Two amino acid substitutions convert a guanylyl cyclase, RetGC-1, into an adenylyl cyclase. Proc. Natl. Acad. Sci. U.S.A. 95, 5993–5997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sunahara R. K., Beuve A., Tesmer J. J. G., Sprang S. R., Garbers D. L., Gilman A. G. (1998) Exchange of substrate and inhibitor specificities between adenylyl and guanylyl cyclases. J. Biol. Chem. 273, 16332–16338 [DOI] [PubMed] [Google Scholar]
- 55. Shenroy A. R., Visweswariah S. S. (2004) Class III nucleotide cyclases in bacteria and archaebacteria: lineage-specific expansion of adenylyl cyclases and a dearth of guanylyl cyclases. FEBS Lett. 561, 11–21 [DOI] [PubMed] [Google Scholar]
- 56. Roelofs J., Van Haastert P. J. M. (2002) Deducing the origin of soluble adenylyl cyclase, a gene lost in multiple lineages. Mol. Biol. Evol. 19, 2239–2246 [DOI] [PubMed] [Google Scholar]
- 57. Robinson J. W., Potter L. R. (2012) Guanylyl cyclases A and B are asymmetric dimers that are allosterically activated by ATP binding to the catalytic domain. Sci. Signal. 5, ra65–ra65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]