

Abstract
Mathematical models of tumor size (TS) dynamics and tumor growth inhibition (TGI) need to place more emphasis on resistance development, given its relevant implications for clinical outcomes. A deeper understanding of the underlying processes, and effective data integration at different complexity levels, can foster the incorporation of new mechanistic aspects into modeling approaches, improving anticancer drug effect prediction. As such, we propose a general framework for developing future semi-mechanistic TS/TGI models of drug resistance.
PROBLEM STATEMENT
The intrinsic and negative connotation of the term "resistance" prevails when referring to one of the leading and key obstacles to successful cancer treatment. Actually, resistance to anticancer drugs is deemed as therapy's shadow remaining an established hindrance in the management of the recurrent disease and prolongation of patient overall survival.
The advent of the genomics era and the subsequent introduction of targeted cancer therapies, accompanied by the technological development and adoptions of new clinical measurement tools and approaches, have tremendously influenced the research on therapy resistance. The investigation of underlying mechanisms responsible for resistance appearance reveals a historical evolution in the level of complexity at which specific molecular pathways have been studied.1 Moreover, in addition to the knowledge derived from preclinical systems, the recognition of clinical trials as pivotal in generating new information unveils the need for considering resistance as an essential part of the therapy and of the clinical efficacy evaluation.
Anticancer treatments are undergoing significant improvements, thanks to molecular targeted drugs; furthermore, the identification of some specific oncogene mutations, as mechanisms of innate resistance, allows the selection of patients who would optimally benefit from tailored therapies. Nevertheless, much more progress remains to be done.
On the one hand, the success of a therapeutic approach is not guaranteed by patient selection and might fail after an initial response due to other factors responsible for the so-called acquired resistance. On the other hand, uncovering the resistance mechanisms in nonresponders is urgently required. Defining a tumor resistance profile of cancer patients remains a great challenge for fostering an improved use of personalized medicine in anticancer therapies.
In this context, Modeling & Simulation has remarkable resources for providing quantitative insights into the dogma of resistance by looking at this phenomenon with magnifying glasses of different resolutions.2
CHALLENGES OF INCORPORATING DRUG RESISTANCE DEVELOPMENT INTO MODELING OF TUMOR GROWTH INHIBITION
Recently, the adoption of mathematical models of tumor dynamics based on longitudinal TS data has been increasingly promoted as a means of improving quantitative informed decision-making in the drug development process and regulatory evaluations.3 Indeed, using TS data as a continuous variable to model the tumor growth dynamics overcomes the large loss of information and limitations resulting from evaluating the categorical RECIST tumor response. (According to the Response Evaluation Criteria in Solid Tumors [RECIST], the TS measurements recorded along clinical trials are classified and then transformed into a discrete response variable of four categories: complete response [CR], partial response [PR], stable disease [SD], and progressive disease [PD].) Furthermore, it has the potential of providing improved predictive metrics of long-term clinical outcomes.4 However, most existing TS/TGI models disregard drug resistance appearance or consider it in a purely empirical formulation, thus lacking a mechanistic basis and pharmacological interpretation.
Conversely, resistance-related mechanisms have been mathematically studied and incorporated in several mechanistic fundamental models, based on different methodological approaches, in order to provide new quantitative insights in the field (see refs.5,6). Such models, by dealing with the complexity of drug resistance evolution, have focused on specific factors responsible for primary or intrinsic resistance (e.g., host-related factors) and for secondary or acquired resistance (e.g., point mutations, cancer-clone evolution, selective microenvironmental pressure).
Even though the adoption of these models might be discouraged by their complex formulation or highly theoretical nature, and limited availability of experimental data, their contribution to drug resistance understanding is remarkable. Addressing these biological and pharmacological complexities, extrapolating the mechanisms of interest, and including them in a TS/TGI model in a simplified manner would pave the way for developing new, convincing models of tumor dynamics and, in turn, new treatment paradigms.
GENERAL FRAMEWORK FOR BUILDING SEMI-MECHANISTIC TS/TGI MODELS OF DRUG RESISTANCE
The evidence for intratumoral heterogeneity and, notably, the existence of cell subpopulations, responding differently to treatment, is one of the crucial aspects to contemplate when developing semi-mechanistic TS/TGI models of drug resistance.
Tumors can be considered as composed of sensitive cells on which the drug exerts its action, and of cells holding innate or acquired resistance, that survive chemotherapy and proliferate. As tumor cells can be resistant to a single drug or multiple drugs simultaneously, different stages of resistance can be associated with cells. Therefore, a resistant cell population can be further divided into a group of fully resistant cells, in the case of multidrug resistance, and groups of partially resistant cells: resistant to one or more structurally/functionally related drugs, but still sensitive to others.
Besides intrinsic factors, tumor cells can have resistance properties coming from genetic and/or epigenetic alterations due to prior anticancer drug exposure, or independent of any treatment, e.g., cross-talk with the tumor microenvironment and its “favorable” conditions.
Actually, in accordance with the conventional model of clonal evolution, cancer can be considered “a complex Darwinian adaptive system.”7 Before and after treatment, cancer clones with improved fitness and a malignant phenotype are sequentially selected, thus they evolve and expand with resistant features (Figure 1). Although these processes are very complex, it is likely that by killing sensitive cells most anticancer drugs play a significant role in changing the selective pressure on tumors: selected resistant cell populations occupy space and exploit resources without any sensitive cell population as competitor.
Figure 1.
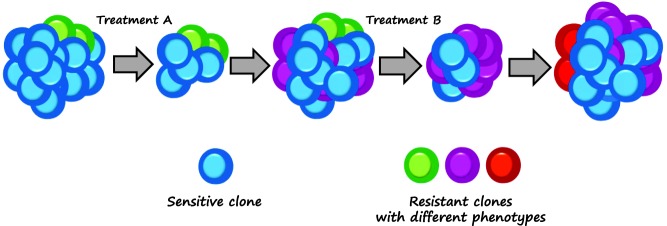
In the conventional clonal evolution model, tumor cells may hold intrinsic drug resistance as well as acquire resistance under selective pressure due to anticancer therapy and “favorable” microenvironmental conditions.
This framework, sufficiently general and flexible for representing various experimental evidence, can be further specified for accounting specific case studies.
For instance, several retrospective analyses of metastatic colorectal cancer (mCRC) trials have reported rat sarcoma viral oncogene (RAS) mutations as a leading mechanism of resistance to anti-EGFR (epidermal growth factor receptor) targeted agents.8 Clinically meaningful benefit is observed in patients bearing all-RAS wild-type tumors treated with these inhibitors, even though some patients often invariably acquire secondary resistance during treatment. Hence, acquisition of secondary Kirsten-RAS (KRAS) mutations not detectable before therapy initiation has been recently reported.9 It is still controversial whether the acquisition of KRAS mutations is due to continuous mutagenesis or such mutations were already present in low abundance, undetectable by current screening methods.
To illustrate the effect of resistance acquisition, we considered the coadministration of an anti-EGFR agent and a cytotoxic drug to mCRC patients.
As previously discussed, tumor can be assumed as consisting of a group of sensitive cells (S), fully resistant cells (R), and two groups of partially resistant cells: sensitive to the EGFR inhibitor but resistant to the cytotoxic drug and vice versa (PRCy and PRaE, respectively). Based on the following assumptions, the resulting compartmental model can be graphically represented as in Figure 2, where the correspondent system of ordinary differential equations (ODE) is also reported.
Figure 2.

Graphical representation and related ODE system of the resulting TS model of drug resistance in mCRC patients receiving an anti-EGFR agent in combination with a cytotoxic drug.
An exponential tumor growth, described by the parameters λS, λR, λPRCy, and λPRaE, is assumed for all the defined cell groups.
Drug actions on cells sensitive to them are included in the model by assuming: 1) a killing effect of the cytotoxic drug, proportional to the cell amount and its exposure through parameter KDcyto; 2) an inhibitory effect of the anti-EGFR agent on tumor proliferation described by an Imax model. A synergistic interaction is assumed to enhance the cytotoxic killing effect, proportionally to the anti-EGFR exposure through an interaction term γ. No pharmacokinetic interaction is assumed.
The existence of different cell subpopulations allows accounting for primary resistance, whereas secondary resistance can be translated into the model by assuming possible transitions of tumor cells to more resistant subgroups. Hence, this entails the definition of the transition rates kSaE, kSCy, kaER, and kCyR. No back transition to more sensitive subgroups is assumed so far, thus excluding mechanisms of restoring sensitivity.
Further assumptions on resistance mechanisms might be accommodated by the model.
For example, the emergence of acquired partial resistance can be defined as a mechanism driven by the drug that exerts a selective pressure of resistant clones, thus, assuming that the transition rates kSaE and kSCy are proportional to the exposure of the drug driving this selection, through a constant parameter (ka and kb, respectively).
Given this scenario and the parameter configuration reported in Supplementary Table S1, different TS profiles have been simulated with the mlxR package in R (Supplementary Figure S1). Tumor dynamics of cell subpopulations for different KDcyto values, and TS profiles simulated by one-at-a-time variation of any other model parameter, are reported in Supplementary Figures S2 and S3, respectively.
Initial amounts of tumor cells in the compartments, here set as portions (A, B, C, D) of the initial TS, are in reality expected to be related to some conditions at therapy initiation (e.g., mutational status, target expression) and to information on the patient's therapeutic history.
CONCLUSIONS AND PERSPECTIVES
The proposed framework aims to provide a reliable basis for building new TS/TGI mechanistic-based models in light of enriched data from future-designed trials. Currently available data would require a lumped model representation, which still preserves its mechanistic/biological interpretation and allows assessing the subsequent information loss.
Starting from this framework, additional information (e.g., intertumor heterogeneity, new lesion appearance) can be integrated, and other emerging mechanistic aspects (e.g., mechanisms of restoring sensitivity in refractory settings) can be included and further specified depending on the focus of the modeling approach.
For instance, the presence of tumor cells endowed with self-renewal and multilineage differentiation capacity (known as cancer stem cells) and, then, the existence of a hierarchical cell organization, need to be considered when modeling the dynamics of hematopoietic malignancies.
Moreover, mechanisms of cancer dormancy (cellular, angiogenic, or by immune-surveillance) and the existence of hypoxic regions, characterized by an inadequate supply of oxygen, play a critical role in promoting drug resistance by limiting drug penetration and distribution, and enhancing genomic instability.10
Current efforts in developing new compounds targeting the tumor microenvironment (e.g., hypoxia-activated drugs, antiangiogenic drugs, agents restoring/augmenting tumor immunity), associated with the adoption of advanced imaging techniques and newly designed biomarkers, highlight additional forthcoming challenges to be engaged by future modeling initiatives for efficiently informing the drug development process in oncology and immuno-oncology.
Acknowledgments
The research leading to these results has received support from the Innovative Medicines Initiative Joint Undertaking under grant agreement no 115156, resources of which are composed of financial contributions from the European Union's Seventh Framework Programme (FP7/2007–2013) and EFPIA companies in kind contribution. The DDMoRe project is also supported by financial contribution from Academic and SME partners. This work does not necessarily represent the view of all DDMoRe partners. We thank Michael Zuehlsdorf for helpful discussion and comments during the model-building process.
Author Contributions
N.T., P.G., U.K., and A.M. wrote manuscript; N.T., P.G., U.K., and A.M. designed research; N.T. performed research; N.T. analyzed data.
Conflict of Interest
N.T., P.G., and A.M. are employees of Merck Serono S.A., Switzerland, a Subsidiary of Merck KGaA, Darmstadt, Germany. U.K., now at CureVac GmbH, when this work was carried out was an employee of Merck KGaA Darmstadt, Germany.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
References
- 1.Keating P, Cambrosio A, Nelson NC, Mogoutov A. Cointet JP. Therapy's shadow: a short history of the study of resistance to cancer chemotherapy. Front. Pharmacol. 2013;4:58. doi: 10.3389/fphar.2013.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lavi O, Gottesman MM. Levy D. The dynamics of drug resistance: a mathematical perspective. Drug Resist. Updates. 2012;15:90–97. doi: 10.1016/j.drup.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mould DR, Walz AC, Lave T, Gibbs JP. Frame B. Developing exposure/response models for anticancer drug treatment: special considerations. CPT Pharmacometrics Syst. Pharmacol. 2015;4:e16. doi: 10.1002/psp4.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ribba B, et al. A review of mixed-effects models of tumor growth and effects of anticancer drug treatment used in population analysis. CPT Pharmacometrics Syst. Pharmacol. 2014;3:e113. doi: 10.1038/psp.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim M, Gillies RJ. Rejniak KA. Current advances in mathematical modeling of anti-cancer drug penetration into tumor tissues. Front. Oncol. 2013;3:278. doi: 10.3389/fonc.2013.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foo J. Michor F. Evolution of acquired resistance to anti-cancer therapy. J. Theor. Biol. 2014;355:10–20. doi: 10.1016/j.jtbi.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greaves M. Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaib W, Mahajan R. El-Rayes B. Markers of resistance to anti-EGFR therapy in colorectal cancer. J. Gastrointest. Oncol. 2013;4:308–318. doi: 10.3978/j.issn.2078-6891.2013.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Misale S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer. 2007;7:834–846. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information