

Abstract
Chronic inflammation is associated with the development of human hepatocellular carcinoma (HCC), an essentially incurable cancer. Anti-inflammatory nutraceuticals have emerged as promising candidates against HCC, yet the mechanisms through which they influence the cell signaling machinery to impose phenotypic changes remain unresolved. Herein we implemented a systems biology approach in HCC cells, based on the integration of cytokine release and phospoproteomic data from high-throughput xMAP Luminex assays to elucidate the action mode of prominent nutraceuticals in terms of topology alterations of HCC-specific signaling networks. An optimization algorithm based on SigNetTrainer, an Integer Linear Programming formulation, was applied to construct networks linking signal transduction to cytokine secretion by combining prior knowledge of protein connectivity with proteomic data. Our analysis identified the most probable target phosphoproteins of interrogated compounds and predicted translational control as a new mechanism underlying their anticytokine action. Induced alterations corroborated with inhibition of HCC-driven angiogenesis and metastasis.
Study Highlights
• WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC? ☑ HCC is an inflammation-related cancer with no effective curative options. Nutraceuticals with established anti-inflammatory properties and low toxicity are considered an attractive alternative to develop new strategies against HCC. • WHAT QUESTION DID THIS STUDY ADDRESS? ☑ The question addressed is whether the applied network-based analysis connecting the inflammatory to phosphoproteomic response data from nutraceutical-treated HCC cells is a valuable systems strategy to assess the mode of action of most promising preclinical candidates. • WHAT THIS STUDY ADDS TO OUR KNOWLEDGE ☑ This study advances the mechanistic knowledge for HCC chemoprevention by the most efficient nutraceuticals (EGCG, FIS, and ERI), as it provides innovative data on mediated functional alterations (cytokine reduction), targeted phosphoproteins (signaling pathway nodes and effectors), and mode of anti-inflammatory action (negative translational control of cytokine expression). • HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS ☑ Providing a systems analysis of mechanisms underlying the actions of selected efficient anti-inflammatory nutraceuticals may open new perspectives for development of novel clinical chemopreventive/therapeutic modalities for human HCC.
Hepatocellular carcinoma (HCC) is the fifth most common malignancy worldwide, with a high rate of metastasis. Despite major improvements in HCC management (resection, transplantation, radiofrequency ablation, chemoembolization, sorafenib therapy), long-term survival remains poor.1,2 Therefore, there is an urgent need to identify novel HCC chemopreventive and/or therapeutic agents able to protect populations at high risk and/or improve prognosis of patients following curative treatment.3
Compelling evidence has established that inflammation plays a critical role in tumor progression.4,5 HCC, a typical example of inflammation-related cancer, slowly progresses on a background of chronic inflammation mainly triggered by exposure to infectious agents (hepatotropic viruses), toxic compounds (ethanol), or dietary carcinogens (aflatoxins, nitrosamines). Inflammatory cytokines have been shown to play a prominent role in mediating changes within the tumor or tumor microenvironment through abnormal regulation of signaling pathways that influence crucial cancer-promoting processes such as cell proliferation, survival, angiogenesis, and metastasis.6 On the other hand, the neoplastic cell itself can advance the development of local inflammation, as many of the most frequently activated oncogenes (RAS, MYC) could elicit a transcriptional program leading to the expression of various inflammatory mediators that lead to tumor immune escape and expansion.4
At the genome level, HCC is characterized by several aberrations which underlie dysregulation of multiple steps in cell signaling pathways.7 Therefore, modulation of a single gene product or signaling cascade is unlikely to mediate an efficient therapeutic outcome. Hence, current research efforts have been focused on developing multitargeted therapies using novel high-throughput technologies.8 Nutraceuticals (a term coined from "nutrition" and "pharmaceutical”) i.e., compounds in dietary sources with disease chemopreventive/chemotherapeutic activities,9 have been proved to possess such multitargeting properties and reduced side effects, thus providing a suitable alternative in achieving alleviation of various cancers.10,11 Despite the emerging social and economic interest in nutraceuticals,9 there is not yet a global mechanistic understanding of their action mode to provide a scientifically solid support for their clinical use.
Identifying the detailed mode of action of active compounds is a major endeavor today, as is evident by the large number of publications on off-target effects of drugs that made it to the market even decades ago and whose off-target effects remained unknown.12,13 Typically, the identification of a drug mode of action is carried out either via bioactivity assays, which screen the effect of the compound on key cellular processes (proliferation/viability, etc.) or via kinase assays, which screen the binding of the interrogated compound on a kinase panel.14–16 However, even if the drug interactions are easy to obtain, the chain effects happening in the cells because of the inhibition of a certain kinase is not trivial to predict, as it is orchestrated to a great extent by the properties and robustness of the signaling mechanisms of the specific cell type. Herein, we developed a novel methodology that leverages multicombinatorial proteomic data from multiplex antibody assays and screens the effects of selected nutraceuticals on three human HCC model cell lines both on the basal level of cytokines release and phosphoproteins activity, but also on perturbation with prototypical cell growth and inflammation signaling stimuli, in an attempt to capture alterations induced by each interrogated compound on key signaling pathways. To this end, we employ an optimization algorithm based on the SigNetTrainer software that combines the proteomic data with prior knowledge of protein interactions (e.g., from online pathway databases), identifies the signaling reactions that are functional based on the data at hand, thus deconvoluting compound effects, and facilitates their mechanistic interpretation as topology alterations of cell-type-specific signaling pathways.17 Furthermore, we use experimental models of cancer metastasis and angiogenesis to ensure the biological importance of compound-imposed alterations. Our integrated analysis presents new mechanistic insights into the targeted anti-inflammatory actions of three promising nutraceuticals, (–)-epigallocatechin gallate (EGCG), fisetin (FIS), and eriodictyol (ERI), thus setting the basis for innovative HCC chemopreventive and/or therapeutic interventions.
METHODS
Plant-derived nutraceuticals
Nutraceuticals (Supplementary Table S1) were purchased from Carl Roth (Karlsruhe, Germany), or Sigma (St. Louis, MO). Stocks were prepared in dimethyl sulfoxide (DMSO; Sigma) and stored at −20oC. Working dilutions contained up to 0.1% v/v DMSO.
Cell cultures
HEP3B, HEPG2 (ATCC, Manassas, VA), and HUH7 (provided by J. Wands, Brown University) cells were maintained as described before.18 To obtain HCC conditioned medium (HCC-CM), confluent cultures were treated with test compound or DMSO for 24 hours, thoroughly washed twice with phosphate-buffered saline (PBS) and incubated for 24 hours in starvation medium, i.e., serum-free medium containing 0.25% bovine serum albumin (BSA). Supernatant was then collected and stored at −80 °C. Human umbilical vein endothelial cells (HUVEC) were isolated and maintained up to passage four as previously described.19
To assess the potential cytotoxicity of test compounds, cells were treated as specified for each of the following biological assays and the number of viable cells was measured by the MTT method as described before.20
xMAP assays
HCC cells, seeded the day before at 3 × 104 cells/well into a 96-well plate, were serum-starved for 4 hours and then treated with selected nutraceuticals or DMSO at the indicated concentration (Table 1) for 2 hours. Cells were then exposed to stimuli (PeproTech, Rocky Hill, NJ) at saturated levels18: interleukin (IL)-6 (0.1 μg/ml), tumor necrosis factor A (TNFA) (0.1 μg/ml), IL1A (0.02 μg/ml), IL1B (0.01 μg/ml), tumor growth factor A (TGFA) (0.2 μg/ml), or insulin (INS) (1.72 μg/ml), for either 22 hours (cytokine measurements) or 15 minutes (phosphoprotein measurements) as indicated by previous studies18 and pilot experiments (Supplementary Figure S1). Cell supernatants and lysates were then collected and stored at −80˚C until use. Total protein concentrations were quantified using the BioRad Dc Protein Assay kit (Hercules, CA).
Table 1.
Selected nutraceuticals tested against HCC cells*
| Nutraceutical | Major functions | Reference | HCC-effective concentrations | xMAP assay selected concentrations |
|---|---|---|---|---|
| QUE (flavonol) | • antioxidant | 10, 21 | 10–50 μM | 25 μM |
| • antiproliferative | ||||
| • anti-inflammatory | ||||
| • antimetastatic | ||||
| POH (monoterpene) | • antiproliferative | 22 | 0.25–1 mM | 500 μM |
| • anti-inflammatory | ||||
| • proapoptotic | ||||
| • antiangiogenic | ||||
| FIS (flavonol) | • antiproliferative | 10 | 10–50 μM | 10 μM |
| • anti-inflammatory | ||||
| • antioxidant | ||||
| • antiangiogenic | ||||
| CUR (polyphenol) | • anti-inflammatory | 10 | 10–25 μM | 15 μM |
| • antioxidant | ||||
| EGCG (polyphenol) | • anti-inflammatory | 23–24 | 50–200 μM | 50–200 μM |
| • antioxidant | ||||
| • antiproliferative | ||||
| • antiangiogenic | ||||
| ERI (flavonone) | • anti-inflammatory | 25 | 10–50 μM | 25 μM |
| • antioxidant | ||||
| • antiproliferative | ||||
| NAR (flavonone) | • antiproliferative | 21 | 10–200 μM | 100 μM |
| • anti-inflammatory | ||||
| • proapoptotic |
A complete list of tested nutraceuticals is shown in Supplementary Table S1.
xMAP assays were performed on a Luminex-200 platform (Luminex, Austin, TX) 18 using custom cytokine/ phosphoprotein antibody-coupled beads (ProtATonce, Athens, Greece). A custom 28-plex was used to detect the levels of selected cytokines in cell supernatant (see Supplementary Table S2 for full name, classification, and biological role). A custom 15-plex was used to determine in cell lysates the levels of test phosphoproteins: ribosomal protein S6 kinase alpha-1 (RSK1), heat shock protein beta-1 (HSPB1, alternate name HSP27), cAMP-responsive element-binding protein-1 (CREB1), protein kinase B (AKT1), p38 mitogen-activated protein kinase (P38MAPK), mammalian target of rapamycin (TOR), glycogen synthase kinase-3 beta (GSK3B), dual specificity mitogen-activated protein kinase kinase-1 (MEK1), extracellular signal-regulated kinase-1 (ERK1), Src homology 2 domain-containing protein-tyrosine phosphatase-2 (SHP2), c-Jun N-terminal kinase-2 (JNK2), dual specificity mitogen-activated protein kinase kinase 6 (MP2K6), ribosomal protein S6 (RPS6), p70 ribosomal S6 kinase (P70S6K), and nuclear factor-kappa B (NFKB). Custom antibody-coupled beads were technically validated as described before26 (Supplementary Figures S2-S5, Supplementary Table S3).
xMAP data processing
Signaling datasets were analyzed and plotted using Datarail, an opensource MATLAB (MathWorks, Natick, MA) toolbox.27 For modeling, the fold change of the signals relatively to the unstimulated state, i.e., DMSO-MEM, was computed and then data were discretized to [0, −1,1], where 1 denotes signal increase ≥1.5 or 2-fold, −1 denotes signal decrease >1.5 or 2-fold, and 0 denotes any signal activation in between. The thresholds were selected based on the platforms sensitivity (see Supplementary Material) and previous studies.28,29 Data discretization is vital before proceeding with the construction of compound-specific signaling networks, since the applied SigNetTrainer methodology implements a qualitative approach in the modeling of signal transduction and can only handle the above-mentioned values for protein activation [−1, 0, 1].
Construction of compound-specific signaling networks
A canonical pathway was constructed downstream of the five stimuli and in the neighborhood of 15 measured phosphoproteins. First, the stimuli receptors were identified, and then canonical pathways downstream of these receptors were extracted from several online databases including Pathway Commons, KEGG, and Ingenuity, with most of the interactions obtained from Ingenuity. Subsequently, the receptor-specific pathways were merged together into a signaling network, and the Floyd-Warshall algorithm was used to identify the observable-controllable part of this network.27 Observables are defined as all network nodes that are upstream of the measured phosphoproteins (thus, their activation value can be inferred based on the value of the signals downstream). Controllables are defined as all network nodes that are downstream of stimuli used (thus, their activation value can be controlled by the stimuli upstream). All nodes and corresponding reactions that are either nonobservable or noncontrollable were removed from the network, because the Integer Linear Programming (ILP) algorithm cannot handle them properly.
Subsequently, the canonical pathway was augmented with compound→target interactions obtained from PubChem (Supplementary Table S5) and with hypothetical phosphoprotein→cytokine release interactions to result in an integrated network that describes both levels (intracellular and extracellular) of signal transduction. Finally, the signaling network was trained to compound-specific data via an adapted variant of SigNetTrainer,17 an ILP formulation that detects and removes inconsistencies between network predictions and data at hand. Details of the formulation, model code, and datasets are provided as Supplementary Material.
Migration, invasion, and tube-like formation assays
Cell invasion and migration were evaluated as described before20 using an 8 μm-pore size membrane BioCoat Matrigel Invasion (BD Biocoat, BD Biosciences, Franklin Lakes, NJ) and migration (ThinCerts, Greiner Bio-One International, Kremsmuenster, Austria) chambers, respectively. Serum-starved cells (3 − 5 × 104cells/well) migrated towards HCC-CM or starvation medium for 24 hours (HCC) or 6 hours (HUVEC). Migrating/invading cells were photographed and manually counted. Formation of HUVEC tube-like structures in the presence of HCC-CM or starvation medium was assessed on growth factor-reduced Matrigel and quantified as previously.30
Statistical analysis
Data were analyzed by Mann–Whitney test with significance levels of P < 0.05, using GraphPad Prism 5.00 for Windows (GraphPad Software, San Diego, CA). Data are presented as means ± SEM of the indicated number of observations.
RESULTS
Collection of released cytokine data
Since HCC cells have been shown to express a number of proinflammatory cytokines and growth factors,18 we examined whether nutraceuticals with known anti-inflammatory properties10 might be able to modulate their secretion pattern. HCC-effective compounds were chosen from a library of 23 nutraceuticals based initially on preliminary HCC cell proliferation/viability experiments determining the range of effective noncytotoxic concentrations (Supplementary Table S1) and next on pilot xMAP assays evaluating the effect of 14 chosen compounds (selected as potent HCC cell proliferation inhibitors that represented major structural classes of nutraceuticals), on the levels of 13 cytokines upon induction with a stimuli mixture (Supplementary Figure S6). Finally, nutraceuticals found to be active in at least one cell line were included in a more extensive xMAP analysis at selected concentrations (Table 1), which although high in some cases (POH, NAR, EGCG) did not affect normal endothelial cell proliferation (unpublished observations). Multiplex cytokine assays concomitantly determined the release of 28 cytokines (Supplementary Table S2), constitutively or upon stimulation with IL6, TNFA, IL1B, TGFA, or INS. As shown in Figure 1, some cytokines (CXCL16, IL8, GROA, CCL20, NGAL, HAVR1) were highly expressed by HCC cells, whereas administration of stimuli, especially IL1B and TNFA, further enhanced their basal levels or triggered the secretion of numerous other cytokines. EGCG treatment proved to be the most effective in reducing the expression (basal and/or induced) of 13 out of 28 test cytokines, without affecting others (e.g., CCL20, PLGF, HAVR1, MIA2, NGAL, PDGF), thus excluding nonspecific cytotoxicity. FIS, ERI, QUE, CUR, POH, and NAR displayed (in order of efficacy) a less broad inhibitory action. Overall, we noticed that HEP3B were the most sensitive cells to compound treatment (see Supplementary Table S4 for a summary of data).
Figure 1.

Cytokine xMAP datasets from (a) HEP3B, (b) HEPG2, and (c) HUH7 cells. Rows represent measurements (mean values) of secreted cytokines 24 hours posttreatment. Columns correspond to treatment with indicated stimulus or minimum essential medium (MEM). Column blocks designate treatment with indicated compound or DMSO vehicle. Black color highlights signals that exceeded a 2-fold threshold alteration compared to DMSO-MEM. Mean values ± SEM (2–3 experiments) and data transformation of fluorescence units to pg/ml are provided in Appendices I and II of the Supplementary Material, respectively.
Collection of signaling phosphoprotein data
Next, we analyzed the impact of EGCG, FIS, and ERI (selected based on their cytokine-reducing activities; Supplementary Table S4) on the levels of 15 phosphoproteins. As shown in Figure 2, (a) HEP3B, (b) HEPG2, and (c) HUH7 cells generally displayed a remarkable divergence regarding their response to both compounds and stimuli. In HEP3B cells, which were again found to be the most responsive, EGCG, FIS, and to a lesser extent ERI downregulated the basal levels of phosphorylated AKT1, TOR, GSK3B, P70S6K, RPS6, P38MAPK, JNK2, MP2K6, and NFKB. Furthermore, EGCG decreased both the constitutive and stimulated phosphorylation of SHP2, whereas it increased the basal and induced expression of phosphorylated CREB1 and HSPB1. In HEPG2, FIS was found to attenuate most of the phosphoproteins (SHP2, AKT1, GSK3B, TOR, P70S6K, RPS6, MP2K6, P38MAPK, JNK2, NFKB). Finally, in HUH7 only a weak inhibition in certain phosphoproteins was observed in response to FIS and ERI, whereas, unexpectedly, EGCG upregulated the levels of phospho-AKT1, GSK3B, RPS6, P38MAPK, and NFKB. Immunoblotting performed in cell lysates from EGCG-treated HCC cells for phospho-AKT1 confirmed these multiplex data (Supplementary Figure S7a). Furthermore, as the active nonphosphorylated GSK3B is a known negative regulator of beta-catenin stability,31 we predicted and experimentally confirmed that the levels of the stable active form of beta-catenin were altered by EGCG according to the phospho-AKT1/GSK3B modification pattern (Supplementary Figure S7b) in each HCC cell line, thus indicating a differential targeting of the WNT/beta-catenin signaling.32
Figure 2.
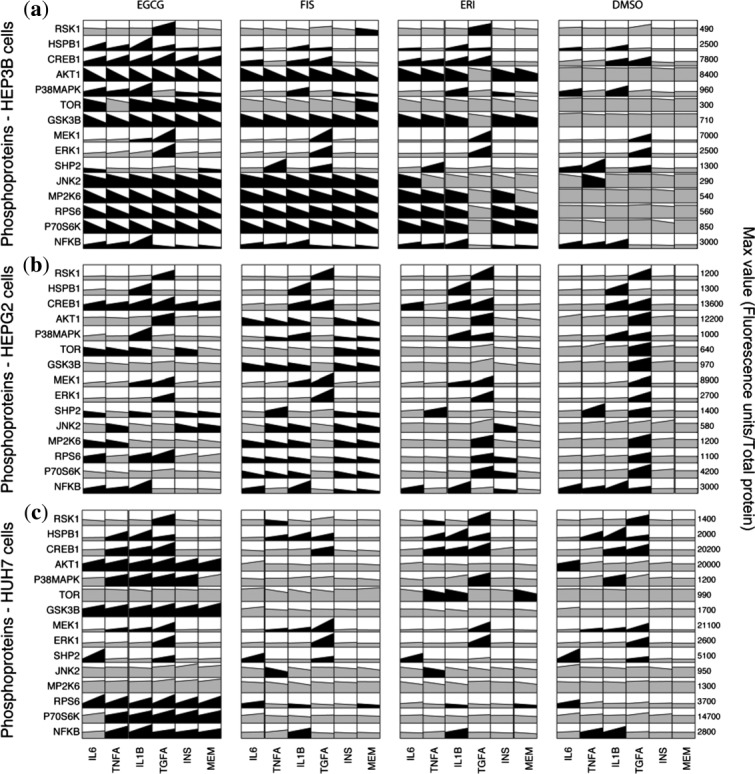
Phosphosignaling xMAP datasets from (a) HEP3B, (b) HEPG2, and (c) HUH7 cells. Rows represent measurements (mean values) of phosphoproteins at 15 minutes following stimulation. Columns correspond to treatment with indicated stimulus or minimum essential medium (MEM). Column blocks designate treatment with indicated compound or DMSO vehicle. Black color highlights signals that exceeded a 1.5-fold threshold alteration compared to DMSO-MEM. Mean values ± SEM (2–3 experiments) are provided in Appendix III of Supplementary Material.
Integrated data analysis to decipher compound action mechanisms
Since HEP3B cells provided the broadest datasets due to their high sensitivity to imposed perturbations, we subsequently constructed compound-specific signaling networks that combined existing knowledge with HEP3B phosphoproteomic and cytokine-release data employing an adapted version of our SigNetTrainer methodology and a sensitivity analysis for a range of thresholds (see Methods and Supplementary Material).
As a result, we visualized how EGCG (Figure 3), FIS (Figure 4), and ERI (Figure 5), modulated signal transduction and cytokine secretion in interrogated cells under basal conditions (Figures 3a, 4a, and 5a) or following subsequent exposure to stimuli (Figures 3b, 4b, and 5b). Our network-based analysis confirmed or rejected existing information (black and gray-colored edges, respectively) and furthermore enriched the canonical pathway with new interactions (blue-colored edges), thus predicting potential nutraceutical-targeted pathways. A major mechanism designated for EGCG and FIS is through inhibition of the PI3K/PIP3/PDK1/P70S6K/RPS6 pathway with potential involvement of AKT1/TOR signaling blockade (Figures 3, 4, solid and dashed black edges, respectively), in accordance with existing literature (Supplementary Table S5) and current data showing attenuation of AKT1, TOR, P70S6K, and RPS6 activation by these compounds. Regarding ERI, the inhibition of the same pathway downstream of PDK1 (blue edge, Figure 5a) emerged for the first time in HEP3B cells. Most important, the fact that the blockade of this particular signaling cascade by all test compounds is well correlated with their inhibitory effects on the release of a number of cytokines (EGCG: CXCL7, CXCL16, CXCL10, IL17A, FIS: CXCL7, CXCL10, CXCL11, ERI: CXCL10) pointed to the prediction of a common cytokine downregulating mechanism through eventual inhibition of the translation factor RSP6 (blue edges, Figures 3, 4, 5). This major network prediction was further validated by independent real-time quantitative reverse transcription-polymerase chain reaction (RT-PCR) and western blot analyses (Supplementary Figure S7c,d) focusing on CXCL10, as this cytokine was connected with RPS6 in the networks of all three compounds. We found that nutraceutical treatment: (a) did not reduce CXCL10 mRNAs levels, (b) did not further decrease compared to vehicle control the total CXCL10 protein levels under conditions of protein synthesis blockade, hence indicating no effect on protein degradation machinery, while (c) reduced the total CXCL10 levels under conditions of proteasome-mediated protein degradation blockade, thus indicating an effect on new protein synthesis. Overall, these results supported the conclusion that the observed reduction in CXCL10 protein levels is not attributed to either a decrease in gene transcription or protein degradation but is rather causally linked to a translational downregulation of cytokine expression via RPS6 inhibition, as predicted by our network analysis.
Figure 3.
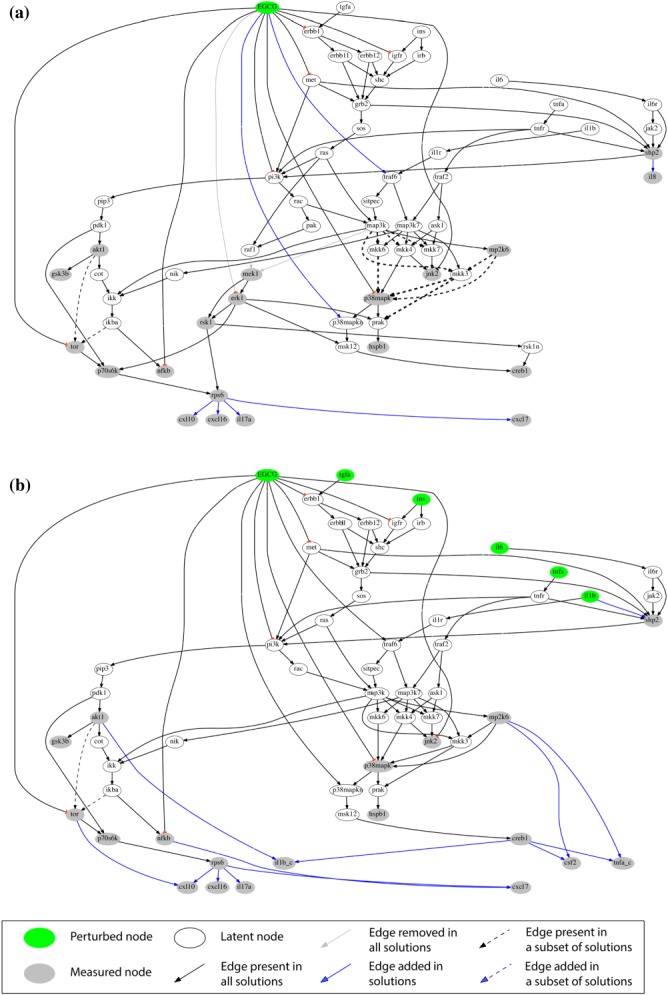
Compound-specific signaling networks in HEP3B cells treated with EGCG under (a) basal and (b) stimulated conditions. Compound→phosphoprotein expression→cytokine release pathways were constructed from xMAP data and a reference network with canonical pathways using an adapted SigNetTrainer method. Black, opaque edges correspond to interactions that were found to be functional based on the data at hand and were conserved in the solution. Gray edges correspond to interactions that were found to contradict with the data and were thus removed from the solution. Black dashed edges correspond to interactions that may be functional or not, depending on the data discretization threshold used. The thickness of the dashed lines corresponds to the number of solutions (for different thresholds) that support the respective interaction. Blue edges correspond to interactions that were added by the algorithm for explaining patterns in the data that could not be fitted with solely removing interactions from the canonical pathway. Abbreviations are explained in Appendix IV of the Supplementary Material.
Figure 4.
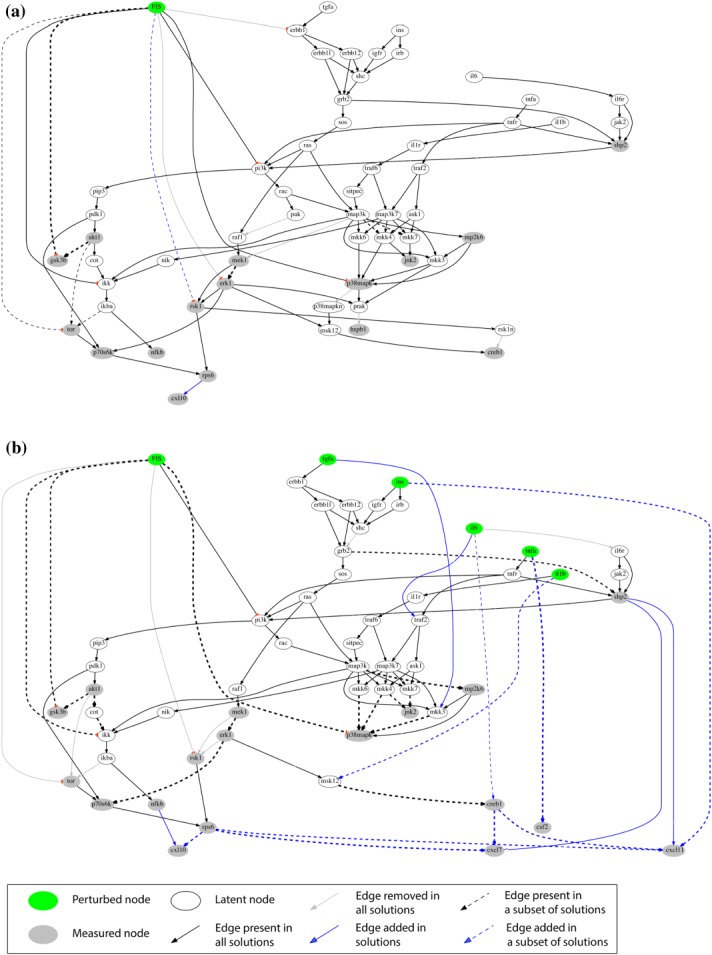
Compound-specific signaling networks in HEP3B treated with FIS under (a) basal and (b) stimulated conditions (see legend to Figure 3 for details).
Figure 5.
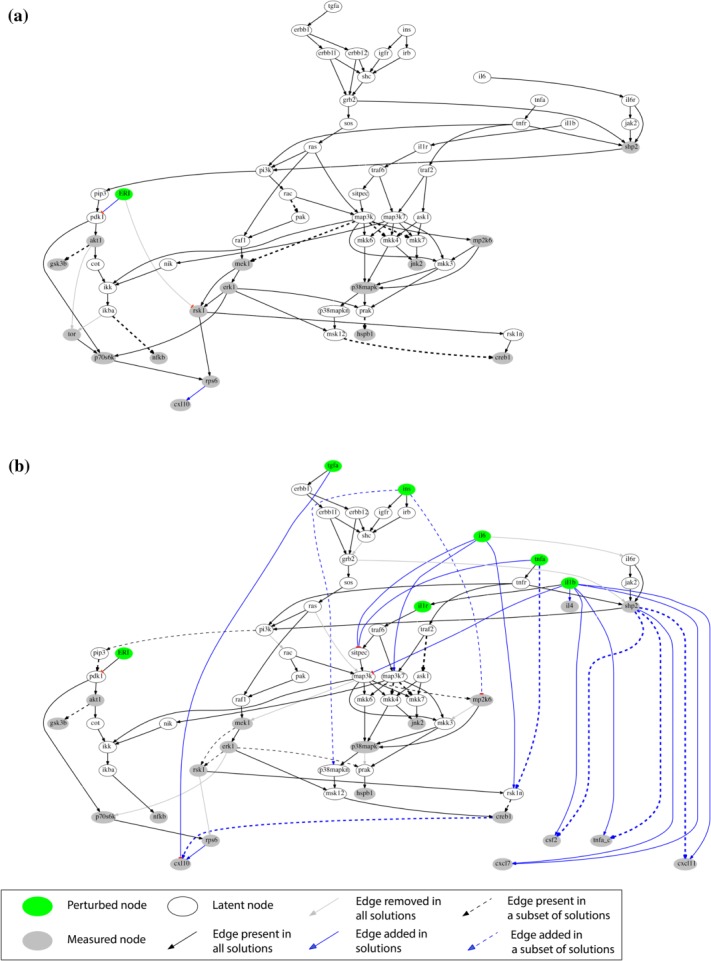
Compound-specific signaling networks in HEP3B treated with ERI under (a) basal and (b) stimulated conditions (see legend to Figure 3 for details).
On the other hand, only signaling alterations specific for the examined cancer cell type were maintained in the network. For instance, the PAK/RAF1/MEK1/ERK1 pathway was removed from the relevant network (Figure 3b) as not being involved in the action mode of EGCG in HEP3B cells because prior knowledge (Supplementary Table S5) was not confirmed by our data for MEK1 and ERK1 in the case of EGCG (gray edges, Figure 3a).
Prevention of HCC-induced metastasis and angiogenesis
To assess the biological significance of changes imposed by EGCG, FIS, and ERI on HCC-secreted factors (including cytokines shown in Figure 1, Supplementary Table S4), we next applied a chemotaxis model of metastasis, where naïve HCC cells were allowed to migrate and/or invade Matrigel-coated membranes towards HCC-CM collected from compound pretreated cultures. As shown in Figure 6a,b, HCC-CM from vehicle-treated cells (CTL) significantly induced cancer cell migration and Matrigel invasion, respectively, compared to basal starvation medium, thus confirming that factors released from HCC cells act as strong stimulators of the metastatic process. However, this effect was reversed in HCC-CM collected from cells preexposed to compounds. Likewise, we examined the responsiveness of HUVEC to HCC-secreted signaling mediators. We found that HCC-CM from vehicle-treated cells (CTL) strongly induced HUVEC motility (Figure 6c), invasiveness (Figure 6d) and differentiation into tube-like structures (Figure 6e), compared to basal starvation medium. However, these proangiogenic attributes were significantly abrogated when HUVEC were challenged with HCC-CM from compound pretreated cultures. Overall, the cytokine-reducing activity of EGCG, FIS, and ERI (Figure 1, Supplementary Table S4) correlated with their capability to inhibit tumor-driven prometastatic and proangiogenic phenotypes (Figure 6).
Figure 6.
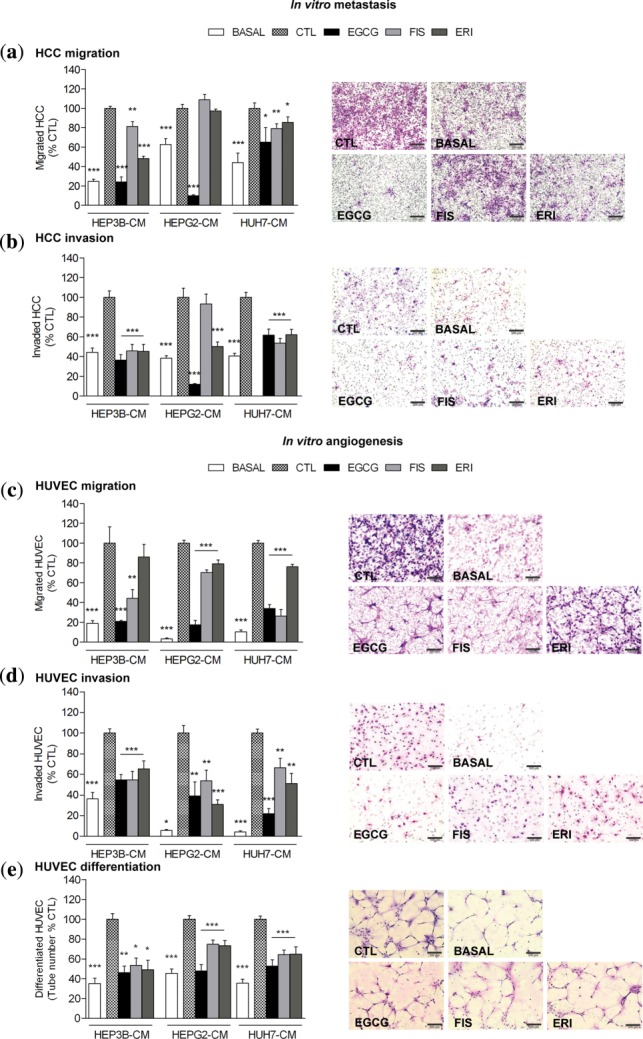
Preventive potential of test compounds on HCC-induced metastasis and angiogenesis. HCC cells (a,b) or HUVEC (c,d) loaded onto migration or Matrigel invasion chambers chemotactically moved towards starvation medium (BASAL) or HCC-CM from DMSO-vehicle (CTL), EGCG, FIS, or ERI pretreated cancer cells. (e) HUVEC differentiation on Matrigel-coated plates in presence of starvation medium (BASAL) or HCC-CM from DMSO-vehicle (CTL), EGCG, FIS, or ERI pretreated cells. Results show mean percentage of CTL ± SEM of migrating/invading cells (n = 10) or tube-like structures (n = 15; *P < 0.05; **P < 0.01; ***P < 0.001). Representative microphotographs from HEP3B metastasis (a,b) and HEP3B-induced angiogenesis (c–e) assays are shown on the right panels; magnification ×100 (a,b,e) or ×200 (c,d); scale bars = 200 μm (a,b,e) or 100 μm (c,d).
DISCUSSION
Plant-derived compounds have always been an important source for the development of new drugs in pharmaceutical research.33 However, their clinical use is limited by the fact that, in the majority, these compounds do not have a known set of target proteins but they act on many different levels, thus making the identification of their detailed action mechanism very difficult if at all possible. In this work by using the Luminex xMAP system as a drug-discovery platform, we screened several anti-inflammatory nutraceuticals for their ability to modulate the cytokine release and phosphoproteomic response of HCC cells, constitutively or upon perturbation with signaling stimulators that are actively involved in human hepatocarcinogenesis.34 Then, by applying an integrated multicombinatorial analysis of the high-throughput proteomic data we provided novel mechanistic insights into the mode of action of most promising compounds.
Like the majority of neoplastic cells, HCC secrete an array of inflammatory cytokines that exert their multiple, partially overlapping functions (mitogenic, motogenic, angiogenic) in a para-/autocrine manner via binding to their appropriate receptors.6 Our results demonstrated EGCG as the most effective modulator of inflammatory cytokine secretion (followed by FIS and ERI) and HEP3B cells as the best responders. Despite previous extensive literature,10,23 this is the first study to our knowledge showing the outstanding capability of this compound to concurrently reduce a wide range of HCC-secreted cytokines, including a group of C-X-C motif ELR-positive (CXCL7, CXL16, GROA, IL8) as well as ELR-negative (CXL10, CXCL11) chemokines and interleukins (IL4 and IL17A), all known to be critical modulators of tumor microenvironment. In fact, CXCL7, GROA, and IL8, identified as CXCR2 ligands, have been shown to strongly promote tumor growth and angiogenesis,35 whereas soluble CXCL16 acting through the CXCL16/CXCR6 axis has been proved to induce cancer invasion and metastasis.36 Overexpression of CXL10 by malignant cells has been reported to desensitize CXCR3 in lymphocytes from HCC patients resulting in tumor escaping from host defense mechanisms,37 while activation of the CXCL11/CXCR7 pathway has been related to HCC progression.38 Concerning IL4 and IL17A, they have been connected with suppression of cancer immunosurveillance and promotion of metastasis.39–41
At the phosphoproteomic level, xMAP measurements in treated cells, besides confirming previous knowledge, thus supporting the validity of the applied method, uncovered some new interesting molecular targets such as SHP2, CREB, and HSPB1. Particularly, SHP2 tyrosine phosphatase has emerged as a key nodal point in cytokine and growth factor-induced signaling; therefore, its inhibition by EGCG and FIS in HEP3B and HEPG2 cells is expected to drastically influence the activation state of several downstream effectors, including JAK, STAT, and PI3K.42 In consistency, EGCG and FIS were found capable of inhibiting the phosphorylation of important components of PI3K axis (AKT1, GSK3B, TOR, P70S6K, RPS6), which is known to be abnormally activated in various cancers, including HCC, promoting cell survival, invasion and angiogenesis.43 Regarding phospho-CREB and HSPB1 upregulation, although data interpretation merits further experimental investigation, the available literature supports that phosphorylated CREB can competitively inhibit NFKB activation, thereby restricting proinflammatory responses,44 whereas phosphorylated HSPB1 can mediate growth suppression in human HCC.45,46
At the computational front, we adapted our previously described SigNetTrainer formulation17 to construct compound-specific signaling networks in HEP3B cells that linked intracellular activity (phosphosignaling) to cellular function (cytokine release), thus revealing mechanisms of compound actions on the basis of topology alterations of key signaling pathways. In contrast to previous mechanistic studies using kinase assays,14–16 the proposed methodology is able to capture not only the compound effects on specific target kinases but also how the signaling machinery in its entirety is affected by this compound. A novel outcome of our computational analysis, which we further validated experimentally, is the prediction of a common negative control mechanism underlying the downregulation of several of the aforementioned cytokines, through ultimate inhibition of the translational factor RPS6 via PI3K/PIP3/PDK1/P70S6K and/or AKT1/TOR signaling obstruction. Since aberrant mRNA translation plays a pivotal role in cancer progression, modulating the activation status of essential components of the protein synthetic machinery, such as RPS6, is expected to have a more general impact on cancer homeostasis.47 Because the construction of networks was based on data restricted in the neighborhood of five receptors and 15 measured phosphoproteins, some potential nutraceutical targets with importance for cancer growth, such as, for example, the AMP-dependent protein kinase,48 were omitted. Despite this limitation, our proposed systems methodology is the first effort to obtain, on a systems scale, mechanistic cues into the mode of action of HCC inhibitory nutraceuticals.
At the phenotypic level, changes in HCC-secreted factors mediated by EGCG, FIS, and HES (including the above-mentioned reduction of crucial prometastatic and proangiogenic cytokines), were found eventually to be essential, as they restricted in vitro the capability of treated cancer cells to transmit prometastatic and proangiogenic signals to other cancer cells, as well as to their context tumor vasculature, respectively. These results further support the potential effectiveness of these compounds to prevent HCC expansion through restriction of tumor neovascularization and metastasis.49
In conclusion, in this study by combining high-throughput protein profiling and network-based analysis of inflammatory and phosphoproteomic HCC responses, we were able to provide important preclinical evidence and molecular insight for use of most promising nutraceuticals in novel chemopreventive and/or therapeutic interventions for HCC.
Acknowledgments
This work was supported by SYNERGASIA 2009 PROGRAMME (09SYN-21-1078) cofunded by the European Regional Development Fund and National Resources and by the John S. Latsis Public Benefit Foundation. We also acknowledge funding by the German Federal Ministry of Education and Research (‘‘Virtual Liver’’ project, grant 0315744) and the European Union (European Social Fund–ESF) - Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF)-Research Funding Program Thalis “Investing in knowledge society through the European Social Fund”. Finally, we thank E. Ladoukakis for his assistance in xMAP data processing.
Conflict of Interest
The authors declare no conflicts of interest.
Author Contributions
M.M., I.N.M. and H.L. wrote the article; L.G.A., F.N.K. and H.L. designed the research; M.M., I.N.M. and D.E.M. performed the research; M.M., I.N.M., D.E.M., L.G.A., F.N.K. and H.L. analyzed the data; I.N.M., S.K. and L.G.A. contributed new reagents/analytical tools.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
References
- 1.El-Serag HB. Hepatocellular carcinoma. N. Engl. J. Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 2.Villanueva A, Hernandez-Gea V. Llovet JM. Medical therapies for hepatocellular carcinoma: a critical view of the evidence. Nat. Rev. Gastroenterol. Hepatol. 2013;10:34–42. doi: 10.1038/nrgastro.2012.199. [DOI] [PubMed] [Google Scholar]
- 3.Singh S, Singh PP, Roberts LR. Sanchez W. Chemopreventive strategies in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2014;11:45–54. doi: 10.1038/nrgastro.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Candido J. Hagemann T. J. Clin. Immunol. 2013;33:79–84. doi: 10.1007/s10875-012-9847-0. Cancer-related inflammation. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D. Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Berasain C, Castillo J, Perugorria MJ, Latasa MU, Prieto J. Avila MA. cancer. Ann. N. Y. Acad. Sci. 2009;1155:206–221. doi: 10.1111/j.1749-6632.2009.03704.x. Inflammation and liver. [DOI] [PubMed] [Google Scholar]
- 7.Aravalli RN, Steer CJ. Cressman EN. Molecular mechanisms of hepatocellular carcinoma. Hepatology. 2008;48:2047–2063. doi: 10.1002/hep.22580. [DOI] [PubMed] [Google Scholar]
- 8.Llovet JM. Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–1327. doi: 10.1002/hep.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalra EK. Nutraceutical—definition and introduction. AAPS PharmSci. 2003;5:27–28. doi: 10.1208/ps050325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gupta S, Kim J, Prasad S. Aggarwal B. Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metast. Rev. 2010;29:405–434. doi: 10.1007/s10555-010-9235-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee KW, Bode AM. Dong Z. Molecular targets of phytochemicals for cancer prevention. Nat. Rev. Cancer. 2011;11:211–218. doi: 10.1038/nrc3017. [DOI] [PubMed] [Google Scholar]
- 12.Wang S, Wilkes MC, Leof EB. Hirschberg R. Imatinib mesylate blocks a non-Smad TGF-β pathway and reduces renal fibrogenesis in vivo. FASEB J. 2005;19:1–11. doi: 10.1096/fj.04-2370com. [DOI] [PubMed] [Google Scholar]
- 13.Denard B, Lee C. Ye J. Doxorubicin blocks proliferation of cancer cells through proteolytic activation of CREB3L1. ELife. 2012;1:e00090. doi: 10.7554/eLife.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma H, Deacon S. Horiuchi K. The challenge of selecting protein kinase assays for lead discovery optimization. Expert Opin. Drug Discov. 2008;3:607–621. doi: 10.1517/17460441.3.6.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaulton A, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, et al. PubChem BioAssay: 2014 update. Nucleic Acids Res. 2014;42:D1075–D1082. doi: 10.1093/nar/gkt978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melas IN, Samaga R, Alexopoulos LG. Klamt S. Detecting and removing inconsistencies between experimental data and signaling network topologies using integer linear programming on interaction graphs. PLoS Comput. Biol. 2013;9:e1003204. doi: 10.1371/journal.pcbi.1003204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alexopoulos L, Saez-Rodriguez J, Cosgrove BD, Lauffenburger DA. Sorger PK. Networks inferred from biochemical data reveal profound differences in toll-like receptor and inflammatory signaling between normal and transformed hepatocytes. Mol. Cell. Proteomics. 2010;9:1849–1865. doi: 10.1074/mcp.M110.000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hatziapostolou M, Katsoris P. Papadimitriou E. Different inhibitors of plasmin differentially affect angiostatin production and angiogenesis. Eur. J. Pharmacol. 2003;460:1–8. doi: 10.1016/s0014-2999(02)02868-6. [DOI] [PubMed] [Google Scholar]
- 20.Loutrari H, Magkouta S, Papapetropoulos A. Roussos C. Mastic oil inhibits the metastatic phenotype of mouse lung adenocarcinoma cells. Cancers. 2011;3:789–801. doi: 10.3390/cancers3010789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erlund I. Review of the flavonoids quercetin, hesperetin, and naringenin. Dietary sources, bioactivities, bioavailability, and epidemiology. Nutr. Res. 2004;24:851–874. [Google Scholar]
- 22.Ong TP, Testoni Cardozo M, de Conti A. Moreno FS. Chemoprevention of hepatocarcinogenesis with dietary isoprenic derivatives: cellular and molecular aspects. Curr. Cancer Drug Targets. 2012;12:1173–1190. doi: 10.2174/156800912803987986. [DOI] [PubMed] [Google Scholar]
- 23.Darvesh A. Bishayee A. Chemopreventive and therapeutic potential of tea polyphenols in hepatocellular cancer. Nutr. Cancer. 2013;65:329–344. doi: 10.1080/01635581.2013.767367. [DOI] [PubMed] [Google Scholar]
- 24.Khan N, Afaq F, Saleem M, Ahmad N. Mukhtar H. Targeting multiple signaling pathways by green tea polyphenol (–)-epigallocatechin-3-gallate. Cancer Res. 2006;66 doi: 10.1158/0008-5472.CAN-05-3636. [DOI] [PubMed] [Google Scholar]
- 25.Manach C, Scalbert A, Morand C, Rémésy C. Jiménez L. Polyphenols: food sources and bioavailability. Am. J. Clin. Nutr. 2004;79:727–747. doi: 10.1093/ajcn/79.5.727. [DOI] [PubMed] [Google Scholar]
- 26.Poussin C, et al. The species translation challenge—a systems biology perspective on human and rat bronchial epithelial cells. 2014;1 doi: 10.1038/sdata.2014.9. Sci. Data. doi: 10.1038/sdata.2014.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saez-Rodriguez J, et al. Discrete logic modelling as a means to link protein signalling networks with functional analysis of mammalian signal transduction. Mol. Syst. Biol. 2009;5:331. doi: 10.1038/msb.2009.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melas IN, Mitsos A, Messinis DE, Weiss TS. Alexopoulos LG. Combined logical and data-driven models for linking signalling pathways to cellular response. BMC Syst. Biol. 2011;5:107. doi: 10.1186/1752-0509-5-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Melas IN, Mitsos A, Messinis DE, Weiss TS, Saez-Rodriguez J. Alexopoulos LG. Construction of large signaling pathways using an adaptive perturbation approach with phosphoproteomic data. Mol. BioSyst. 2012;8:1571–1584. doi: 10.1039/c2mb05482e. [DOI] [PubMed] [Google Scholar]
- 30.Michailidou M, et al. Microvascular endothelial cell responses in vitro and in vivo: modulation by zoledronic acid and paclitaxel? J. Vasc. Res. 2010;47:481–493. doi: 10.1159/000313876. [DOI] [PubMed] [Google Scholar]
- 31.Ban KC, Singh H, Krishnan R. Seow HF. GSK-3b phosphorylation and alteration of b-catenin in hepatocellular carcinoma. Cancer Lett. 2003;199:201–208. doi: 10.1016/s0304-3835(03)00421-x. [DOI] [PubMed] [Google Scholar]
- 32.Polakis P. Drugging Wnt signalling in cancer. EMBO J. 2012;31:2737–2746. doi: 10.1038/emboj.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newman DJ. Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Capece D, et al. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor-associated macrophages. Biomed. Res. Int. 2013;2013:187204. doi: 10.1155/2013/187204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keeley EC, Mehrad B. Strieter RM. Chemokines as mediators of tumor angiogenesis and neovascularization. Exp. Cell Res. 2011;317:685–690. doi: 10.1016/j.yexcr.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Hayre M, Salanga CL, Handel TM. Allen SJ. Chemokines and cancer: migration, intracellular signalling and intercellular communication in the microenvironment. Biochem. J. 2008;409:635–649. doi: 10.1042/BJ20071493. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y-Q, et al. Desensitization of T lymphocyte function by CXCR3 ligands in human hepatocellular carcinoma. World J. Gastroenterol. 2005;11:164–170. doi: 10.3748/wjg.v11.i2.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monnier J, et al. CXCR7 is up-regulated in human and murine hepatocellular carcinoma and is specifically expressed by endothelial cells. Eur. J. Cancer. 2012;48:138–148. doi: 10.1016/j.ejca.2011.06.044. [DOI] [PubMed] [Google Scholar]
- 39.Hallett MA, Venmar KT. Fingleton B. Cytokine stimulation of epithelial cancer cells: the similar and divergent functions of IL4 and IL13. Cancer Res. 2012;72:6338–6343. doi: 10.1158/0008-5472.CAN-12-3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, et al. Interleukin 17A promotes hepatocellular carcinoma metastasis via NF-κB induced matrix metalloproteinases 2 and 9 expression. PLoS One. 2011;6:e21816. doi: 10.1371/journal.pone.0021816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma S, et al. IL17A produced by γδ T cells promotes tumor growth in hepatocellular carcinoma. Cancer Res. 2014;74:1969–1982. doi: 10.1158/0008-5472.CAN-13-2534. [DOI] [PubMed] [Google Scholar]
- 42.Qu CK. Role of the SHP-2 tyrosine phosphatase in cytokine-induced signaling and cellular response. Biochim. Biophys. Acta. 2002;1592:297–301. doi: 10.1016/s0167-4889(02)00322-1. [DOI] [PubMed] [Google Scholar]
- 43.Chen J-S, et al. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: association with MMP-9. Hepatol. Res. 2009;39:177–186. doi: 10.1111/j.1872-034X.2008.00449.x. [DOI] [PubMed] [Google Scholar]
- 44.Wen AY, Sakamoto KM. Miller LS. The role of the transcription factor CREB in immune function. J. Immunol. 2010;185:6413–6419. doi: 10.4049/jimmunol.1001829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsushima-Nishiwaki R, et al. Phosphorylated heat shock protein 27 represses growth of hepatocellular carcinoma via inhibition of extracellular signal-regulated kinase. J. Biol. Chem. 2008;283:18852–18860. doi: 10.1074/jbc.M801301200. [DOI] [PubMed] [Google Scholar]
- 46.Yasuda E, et al. Attenuated phosphorylation of heat shock protein 27 correlates with tumor progression in patients with hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2005;337:337–342. doi: 10.1016/j.bbrc.2005.08.273. [DOI] [PubMed] [Google Scholar]
- 47.Topisirovic I. Sonenberg N. mRNA translation and energy metabolism in cancer: the role of the MAPK and mTORC1 pathways. Cold Spring Harb. Symp. Quant. Biol. 2011;76:355–367. doi: 10.1101/sqb.2011.76.010785. [DOI] [PubMed] [Google Scholar]
- 48.Lea M, Pourat T, Patel R. desBordes C. Growth inhibition of colon cancer cells by compounds affecting AMPK activity. World J. Gastrointest. Oncol. 2014;6:244–252. doi: 10.4251/wjgo.v6.i7.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hernandez-Gea V, Toffanin S, Friedman SL. Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology. 2013;144:512–527. doi: 10.1053/j.gastro.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information