

Abstract
Background
Lynch syndrome (LS) is associated with germline mutations in DNA mismatch repair (MMR) genes. The first “hit” to inactivate one allele of the predisposing MMR gene is present in every cell, contributing to accelerated tumorigenesis. Less information is available of the nature, timing, and order of other molecular “hits” required for tumor development. To this end, MMR protein expression and coordinated promoter methylation were examined in colorectal specimens prospectively collected from LS mutation carriers (n = 55) during colonoscopy surveillance (10/2011–5/2013), supplemented with retrospective specimens.
Results
Loss of MMR protein corresponding to the gene mutated in the germline increased with dysplasia, with frequency of 0 % in normal mucosa, 50–68 % in low-grade dysplasia adenomas, and 100 % in high-grade dysplasia adenomas and carcinomas. Promoter methylation as a putative “second hit” occurred in 1/56 (2 %) of tumors with silenced MMR protein. A general hypermethylation tendency was evaluated by two gene sets, eight CpG island methylator phenotype (CIMP) genes, and seven candidate tumor suppressor genes linked to colorectal carcinoma (CRC). Hypermethylation followed the same trend as MMR protein loss and was present in some low-grade dysplasia adenomas that still expressed MMR protein suggesting the absence of a “second hit.” To assess prospectively collected normal mucosa for carcinogenic “fields,” the specimen donors were stratified according to age at biopsy (50 years or below vs. above 50 years) and further according to the absence vs. presence of a (previous or concurrent) diagnosis of CRC. In mutation carriers over 50 years old, two markers from the candidate gene panel (SFRP1 and SLC5A8) revealed a significantly elevated average degree of methylation in individuals with CRC diagnosis vs. those without.
Conclusions
Our findings emphasize the importance and early appearance of epigenetic alterations in LS-associated tumorigenesis. The results serve early detection and assessment of progression of CRC.
Electronic supplementary material
The online version of this article (doi:10.1186/s13148-015-0102-4) contains supplementary material, which is available to authorized users.
Keywords: Lynch syndrome, Colorectal cancer, Adenoma-carcinoma sequence, DNA methylation, CpG island methylator phenotype, MS-MLPA
Background
Colorectal cancer (CRC) develops via multiple steps involving genetic and epigenetic changes. The majority of CRCs are sporadic. In Lynch syndrome (LS), inherited defects of the DNA mismatch repair (MMR) genes MLH1, MSH2, MSH6, and PMS2 confer high lifetime risks of CRC and extracolonic cancers [1]. Cancers arising in LS mutation carriers as well as some 12 % of sporadic CRCs exhibit microsatellite instability (MSI) [2]. Promoter methylation of MLH1 was recognized as a primary cause for sporadic MSI CRC [3, 4]. In LS, germline mutation combined with somatic mutations or loss of heterozygosity underlies biallelic inactivation of MMR genes. MMR defects together with other genetic and epigenetic changes accelerate neoplastic transformation of the normal colonic epithelium [5].
Aberrant CpG island methylation affecting multiple tumor suppressor genes is frequent in sporadic CRC and colonic adenomas [6, 7], giving rise to a CpG island methylator phenotype (CIMP). CIMP tumors form a subtype with distinct histology compared to tumors derived from the traditional adenoma carcinoma sequence [8]. The molecular basis of CIMP and its clinical implications are only beginning to be explored [5, 9]. In hereditary CRC, the importance of CIMP is largely unknown.
This investigation was undertaken to clarify the role of CIMP and its time of appearance in colorectal adenoma-carcinoma progression sequence in LS. LS mutation carriers are enrolled in lifelong colonoscopy surveillance with 2–3-year intervals [10], and we took advantage of the regular surveillance to obtain consecutive specimens. Furthermore, a previous mouse study implicated a number of candidate genes in association with MLH1 mutation and diet [11], prompting us to evaluate the respective genes as methylation targets in human LS.
Results
Study design
Biopsy specimens fresh frozen from different parts of the colon and rectum, together with blood samples, were obtained from 55 consecutive LS mutation carriers who underwent regular colonoscopy screening or colectomy at two Finnish hospitals during 10/2011–5/2013 (Table 1). This prospective series was used to study DNA methylation changes in normal colonic mucosa with respect to aging and previously diagnosed cancer. As only a few individuals developed colorectal lesions (mostly hyperplastic polyps) during the 1.5-year interval, all archival tubular and villous adenomas and carcinomas previously diagnosed in the same individuals were gathered (retrospective series in Table 1) and used to investigate DNA methylation changes occurring in the adenoma carcinoma progression sequence. Colonic and rectal biopsies from 22 familial adenomatous polyposis (FAP) mutation carriers who participated in colonoscopy screening (Table 1) were studied for comparison.
Table 1.
Characteristics of sample series
| Prospective series | Retrospective series | |
|---|---|---|
| No. of individuals or specimens | No. of individuals or specimens | |
| Lynch syndrome | ||
| • Individuals with mutation in | ||
| MLH1 | 39 (71 %) | 33 (77 %) |
| MSH2 | 13 (24 %) | 7 (16 %) |
| MSH6 | 3 (5 %) | 3 (7 %) |
| Total | 55 | 43 |
| • Colorectal specimens | ||
| Normal colonic mucosa | 55 | 24 |
| Hyperplastic polyp | 10 | – |
| Low dysplasia adenoma | 5 | 27 |
| High dysplasia adenoma | 3 | 13 |
| Carcinoma | 3 | 20 |
| Total | 76 | 84 |
| Familial adenomatous polyposis | ||
| • Individuals with mutation in | ||
| APC | 22 (100 %) | – |
| • Colorectal specimens | ||
| Normal colonic mucosa | 22 | – |
| Adenoma (all dysplasia grades) | 23 | – |
| Carcinoma | – | – |
| Total | 45 | – |
Note: three carcinomas, three low-grade dysplasia adenomas, and two high-grade dysplasia adenomas were available as fresh frozen and FFPE samples and were simultaneously included in both prospective and retrospective series
MMR status and analysis of MMR gene promoter methylation as the “second hit”
To test if the predisposing MMR gene had undergone somatic inactivation of the remaining wild-type allele, colorectal specimens were evaluated for MMR protein expression by immunohistochemical (IHC) analysis. All adenomas with high-grade dysplasia and all carcinomas from LS patients showed loss of MMR protein corresponding to the gene mutated in the germline whereas only 68 %, 67 % and 50 % of adenomas with low-grade dysplasia showed loss of expression in MLH1, MSH2, and MSH6 mutation carriers, respectively (Table 2). The difference (adenomas with low-grade dysplasia vs. adenomas with high-grade dysplasia and adenomas with low-grade dysplasia vs. carcinomas) was statistically significant for MLH1-associated tumors. Overall, the results suggest that silencing of the relevant MMR protein expression is a relatively late event in LS tumorigenesis. MSI analysis with the mononucleotide repeat markers BAT25 and BAT26 showed that all adenomas and carcinomas with absent MMR protein were microsatellite unstable with one exception, an adenoma with low-grade dysplasia from a MSH2 mutation carrier. The low tumor cell percentage (20 %) in that particular sample was likely to explain stable microsatellites. All eight low-grade dysplasia adenomas retaining MMR protein expression were microsatellite stable.
Table 2.
Proportion of decreased MMR protein expression in Lynch syndrome adenomas and carcinomas
| Proportion (%) with decreased MMR protein corresponding to the gene mutated in germline | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MLH1 | p value vs. normal colon | p value vs. low dysplasia | MSH2 | p value vs. normal colon | p value vs. low dysplasia | MSH6 | p value vs. normal colon | p value vs. low dysplasia | Total | p value vs. normal colon | p value vs. low dysplasia | |
| Normal colon | 0/33 (0 %) | 0/7 (0 %) | 0/3 (0 %) | 0/43 (0 %) | ||||||||
| Adenoma low dysplasia | 17/25 (68 %) | <0.0001 | 4/6 (67 %) | 0.021 | 1/2 (50 %) | NS | 22/33 (67 %) | <0.0001 | ||||
| Adenoma high dysplasia | 13/13 (100 %) | <0.0001 | 0.034 | 1/1 (100 %) | NS | NS | – | NS | 14/14 (100 %) | <0.0001 | 0.020 | |
| Carcinoma | 12/12 (100 %) | <0.0001 | 0.036 | 3/3 (100 %) | 0.008 | NS | 5/5 (100 %) | 0.018 | NS | 20/20 (100 %) | <0.0001 | 0.003 |
NS, no statistical significance
Promoter methylation as a possible “second hit” was assessed in tumors lacking MMR protein. Methylation of MLH1 was mostly detected in the distal promoter (region A [12]). This region was methylated in 30 %, 31 % and 60 % of adenomas with low-grade dysplasia, adenomas with high-grade dysplasia and carcinomas, respectively (Fig. 1). On the contrast, methylation of the proximal promoter (region C), most commonly associated with MLH1 protein loss, was only observed in one adenoma having high-grade dysplasia. No methylation was detected in promoter regions of MSH2 or MSH6. Taken together, promoter methylation constituted a putative “second hit” in 1/56 (2 %) tumors with silenced MMR protein.
Fig. 1.
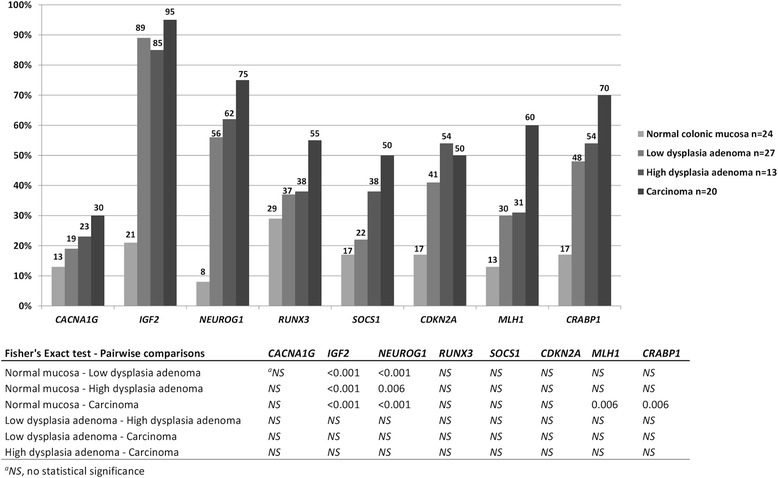
Frequency of hypermethylated CIMP markers in the LS retrospective series. Numerical values of percentages are given above each bar. Hypermethylation thresholds were calculated according to stringency level I. Pairwise comparisons were calculated by Fisher’s exact test (two-sided p values), and the p values were adjusted for multiple comparisons by Bonferroni correction
CIMP
To assess whether coordinated methylation of multiple CpG islands that are normally unmethylated plays a role in colorectal tumorigenesis, methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) was used to study eight genes firmly associated with CIMP (CACNA1G, IGF2, NEUROG1, RUNX3, SOCS1, CDKN2A, MLH1, and CRABP1). The MS-MLPA CIMP probe mix contains 3–6 probes for each CIMP marker gene, and the average methylation dosage ratios (Dm) obtained for each probe and type of specimen are given in Additional file 1: Table S1. Hypermethylation in tumor tissues was evaluated relative to probe-specific thresholds derived from normal mucosa. The hypermethylation thresholds for the retrospective (FFPE) series are given in Additional file 2: Table S2 and those for the prospective (fresh frozen) series in the legend of Additional file 3: Figure S1. A gene was considered hypermethylated when at least one fourth (25 %) or more of probe target sites were methylated [13].
The prospective LS series revealed increased methylation in adenomas and carcinomas vs. normal colonic mucosa for the CIMP markers IGF2, NEUROG1, and CRABP1 (p values mostly non-significant due to small numbers of specimens; Additional file 3: Figure S1A). Hyperplastic polyps, too, showed frequent hypermethylation with IGF2 and NEUROG1. LS vs. FAP-associated adenomas and matching normal mucosa showed comparable frequencies of hypermethylation.
The analyses were extended to the retrospective LS series with higher number of tumors available (Fig. 1). Adenomas with high-grade dysplasia and carcinomas showed the highest frequencies of hypermethylation (defined with stringency level I of Additional file 2: Table S2). The frequencies of hypermethylation for IGF2 and NEUROG1 were significantly increased in all tumor types when compared to normal colon. Notably, the difference was significant already in adenomas with low-grade dysplasia (89 % vs. 21 %, p < 0.001, for IGF2 and 56 % vs. 8 %, p < 0.001, for NEUROG1). Only carcinomas showed significantly elevated hypermethylation frequencies for MLH1 and CRABP1.
As no consensus exists on how to score CIMP and no single panel is superior to others [13], three different marker panels were considered. Colorectal specimens from the retrospective series were divided into CIMP(+) or CIMP(−) categories by using the Ogino 5/8, Ogino 6/8, and Weisenberger 3/5 panels (“Methods” section). Here, stringency level II (Additional file 2: Table S2) was used to calculate probe-specific thresholds to avoid hypermethylation in normal mucosa. The frequency of CIMP(+) specimens increased from normal mucosa to adenomas with low-grade dysplasia to adenomas with high-grade dysplasia to carcinomas regardless of the marker system used (Fig. 2). The Weisenberger panel yielded somewhat higher CIMP(+) frequencies compared to the Ogino panels. Accordingly, 15 %, 23 % and 50 % of the adenomas with low-grade dysplasia, adenomas with high-grade dysplasia, and carcinomas, respectively, were CIMP(+) when using the Weisenberger 3/5 panel, compared to 7 %, 23 % and 40 % with the Ogino 5/8 criteria, and 4 %, 23 % and 25 % with the Ogino 6/8 criteria (Fig. 2). Formal statistical significance was reached in the normal mucosa vs. carcinomas comparison according to Ogino 5/8 (p = 0.006) and Weisenberger 3/5 (p < 0.001) criteria and borderline significance in the adenomas with low-grade dysplasia vs. carcinomas comparison according to Ogino 5/8 (p = 0.054) criteria. CIMP(+) vs. CIMP(−) tumors were diagnosed at similar average ages (48 vs. 49 years for adenomas and 49 vs. 48 years for carcinomas) excluding age as a possible confounding factor in the analyses (see below).
Fig. 2.
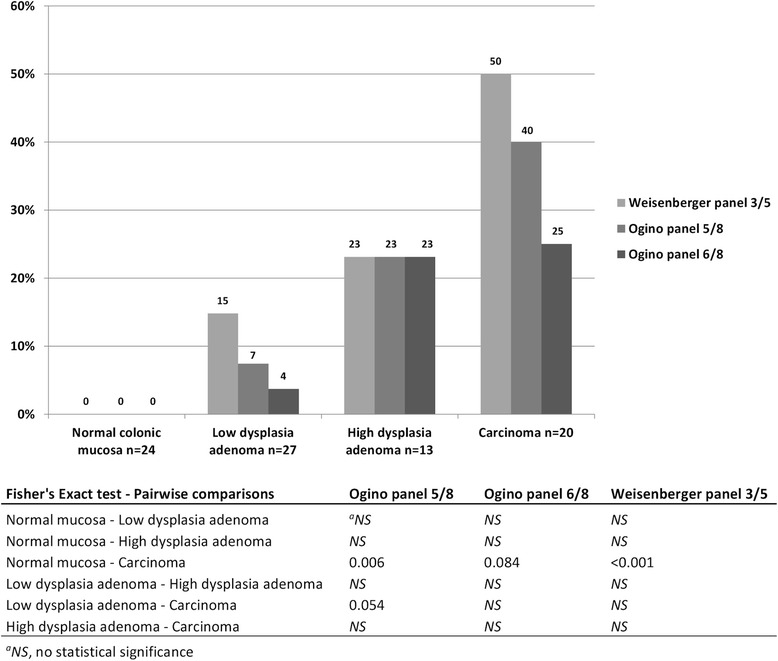
Frequency of CIMP(+) specimen calculated by three different criteria. Numerical values of percentages are given above each bar. Hypermethylation thresholds were calculated according to stringency level II. Pairwise comparisons were calculated by Fisher’s exact test (two-sided p values), and the p values were adjusted for multiple comparisons by Bonferroni correction
Methylation analysis of candidate genes
A custom MS-MLPA kit was designed to study methylation of seven candidate genes previously associated with early colon oncogenesis in an experimental mouse model (DKK1, SFRP1, SFRP2, SFRP5, CDH1, HOXD1, and SLC5A8 (Additional file 4: Table S3) [11]. The average Dm obtained for each probe and type of specimen are shown in Additional file 1: Table S1. Probe-specific hypermethylation thresholds were determined as described for CIMP markers (stringency level I), separately for the retrospective series (Additional file 2: Table S2) and the prospective series (legend of Additional file 3: Figure S1).
The prospective LS series indicated significantly higher frequencies of hypermethylation for SFRP1 (95 %, p = 0.006) and SFRP2 (67 %, p = 0.012) in carcinomas vs. normal colonic mucosa (Additional file 3: Figure S1B). Additionally, 50 % of hyperplastic polyps revealed hypermethylation for SFRP1 relative to normal mucosa (p = 0.030). Hypermethylation frequencies in colonic tissues were comparable in LS vs. FAP.
In the retrospective LS series, hypermethylation frequencies for SFRP2 were significantly higher in all tumor types when compared to normal mucosa (Fig. 3). Importantly, this included adenomas with low-grade dysplasia already (50 % vs. 13 %, p = 0.042). For SFRP1, significantly increased frequencies of hypermethylation were only observed in adenomas with high-grade dysplasia and carcinomas (Fig. 3).
Fig. 3.
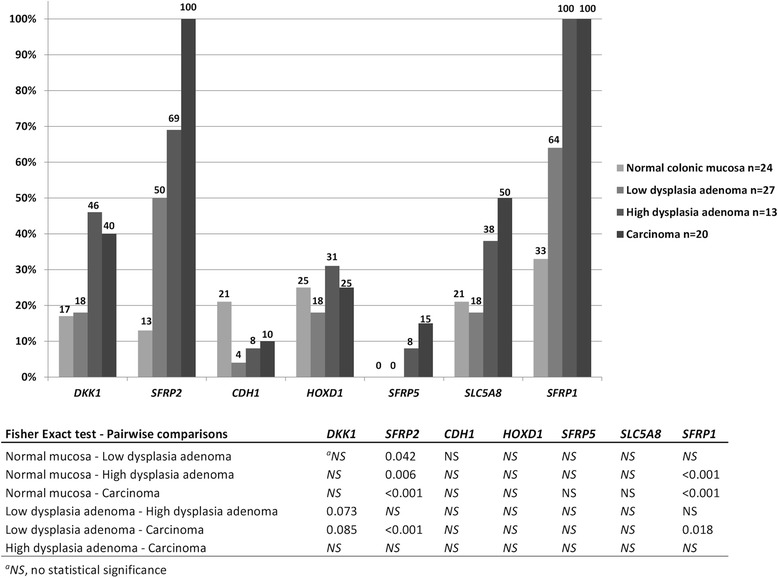
Frequency of hypermethylated candidate genes in the LS retrospective series. Numerical values of percentages are given above each bar. Hypermethylation thresholds were calculated according to stringency level I. Pairwise comparisons were calculated by Fisher’s exact test (two-sided p values), and the p values were adjusted for multiple comparisons by Bonferroni correction
Correlation of candidate gene methylation with mRNA expression in cancer cell lines
To evaluate functional significance of promoter methylation, MMR-deficient colorectal, endometrial, and ovarian cancer cell lines (Additional file 5: Table S4) were treated with the demethylating agent 5-aza-CdR and the histone deacetylase inhibitor TSA, followed by RNA profiling on microarrays. Consistent (1.8–7.2-fold) treatment-induced upregulation of SFRP1 was seen in HCT15, HCT116, and HEC59 analogous to LS-associated CRC and endometrial cancer. SFRP2 was significantly upregulated (1.9-fold) in HCT116. Upregulation was accompanied by reduced promoter methylation by MS-MLPA.
When methylation (Dm) values were plotted against mRNA expression in all studied cancer cell lines (treated and untreated) and the corresponding normal tissues, a significant inverse correlation was observed between SFRP1 (r = −0.688, p = 0.001) and SFRP2 (r = −0.657, p = 0.002) mRNA expression and methylation (Additional file 6: Figure S2). Moreover, SFRP1 and SFRP2 expressions were significantly lower in the untreated cancer cell lines compared to the corresponding normal controls (data not shown). Our data suggest that DNA methylation plays a significant role in the expressional regulation of these genes.
Hypermethylation of CIMP and candidate genes vs. expressional status of MMR proteins
Eleven adenomas with low-grade dysplasia retained MMR protein expression (Table 2) suggesting that the second hit to inactivate the responsible MMR gene had not yet occurred. DNA was available for eight such adenomas, and 3/8, 4/8, 6/8, and 2/8 revealed hypermethylation of IGF2, NEUROG1, SFRP1, and SFRP2, respectively. The hypermethylation frequencies were essentially comparable to those in low-grade dysplasia adenomas with silenced MMR protein (15/18, 4/18, 11/18, and 10/18 for the respective loci). The results suggest that hypermethylation of the investigated genes can occur prior to somatic inactivation of the predisposing MMR gene.
Effect of age at biopsy on normal colonic tissue methylation
Since DNA methylation tends to increase with age [14], the level of methylation in each prospectively collected colorectal mucosa specimen was evaluated against the chronological age of the individual at the time of biopsy. A moderate-to-strong positive correlation was detected between age at biopsy and normal colonic mucosa Dm values corresponding to IGF2 probes I (r = 0.694, p < 0.0001), II (r = 0.726, p < 0.0001), and III (r = 0.742, p < 0.0001) and NEUROG1 probes I (r = 0.566, p < 0.0001), III (r = 0.703, p < 0.0001), and IV (r = 0.655, p < 0.0001) (Additional file 7: Figure 3SA–B). Additionally, moderate correlation between age at biopsy and SFRP1 (r = 0.554, p < 0.0001), SFRP2 (r = 0.550, p < 0.0001), and SLC5A8 (r = 0.554, p < 0.0001) methylation was observed in normal colonic mucosa (Additional file 7: Figure S3C). This indicates that aging itself increases methylation of the CIMP markers in the histologically normal mucosa. No age-related correlation was observed for MLH1 region C methylation.
Analysis of field defects in histologically normal colonic mucosa
To investigate if aberrant DNA methylation might form carcinogenic “fields” in the histologically normal mucosa, colonic mucosa biopsies of the prospective LS series were evaluated for hypermethylation of CIMP markers and candidate genes. The individuals were divided into four groups depending on age at biopsy and absence vs. presence of (previous or concurrent) CRC (groups 1–4). The first two groups included mutation carriers 50 years old and below and consisted of 22 individuals without CRC (group 1) and 6 individuals with CRC (group 2). The remaining two groups included mutation carriers above 50 years and consisted of 17 individuals without CRC (group 3) and 10 individuals with CRC (group 4). The interval between CRC diagnosis and time of biopsy was 5.1 years (range 0–11.5) in group 2 and 5.5 years (range 0–12.4) in group 4. Average age at biopsy was comparable in group 1 (35, range 26–50) vs. group 2 (43, range 39–48) and in group 3 (61, range 51–75) vs. group 4 (63, range 51–74), excluding the age effect as a possible confounder in the respective comparisons.
When the effect of CRC on CIMP marker methylation was examined within the age groups (≤50 and above 50), no significant differences were observed (Additional file 8: Figure S4). However, age effect was evident from comparisons of the “under 50, no CRC” with the “over 50, no CRC” groups and “under 50, CRC” with the “over 50, CRC” groups, which revealed significant increases in methylation for several IGF2 and NEUROG1 probes (Additional file 8: Figure S4).
The candidate genes, too, showed an age effect, but despite it, significant increases in methylation were observed for SFRP1 (p < 0.0001) and SLC5A8 (p = 0.007) in the “over 50 group,” when stratified by the presence vs. absence of previously diagnosed CRC (Fig. 4). Moreover, our results from duplicate MS-MLPA assays (“Methods” section) as well as from dilution experiments (Additional file 9: Figure S5) showed that the observed changes in methylation were unlikely to be explained by technical variation or other similar reasons. Thus, our finding may indicate a potential field defect in normal mucosa.
Fig. 4.
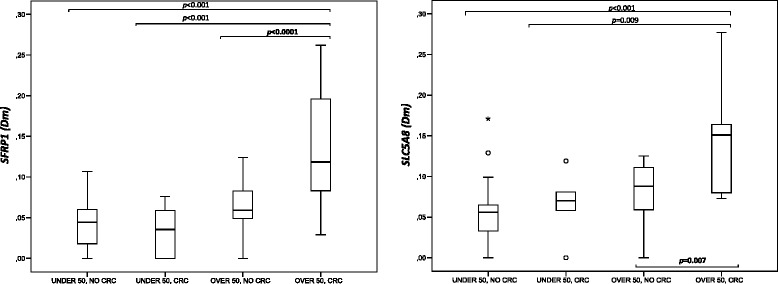
Effect of aging and previously diagnosed CRC on SFRP1 and SLC5A8 normal colonic mucosa Dm values. Study groups: patients ≤50 years without (n = 22) and with (n = 6) a CRC diagnosis, and patients >50 years without (n = 17) and with (n = 10) a CRC diagnosis. Statistical testing was performed with one-way ANOVA and Tukey’s post hoc test or Kruskal-Wallis one-way ANOVA
Discussion
Studies on sporadic CRC have demonstrated that promoter hypermethylation can act as an alternative mechanism to mutations, having a causal role in colorectal tumorigenesis [5, 3]. Promoter methylation can be detected in CRC-associated normal colonic mucosa and aberrant crypt foci (ACF), the earliest identifiable neoplastic lesions of the colon [15, 16]. Aging increases genomic DNA methylation [14] and is therefore particularly relevant to sporadic CRC which develops two to three decades later than hereditary CRC. We utilized unique features of LS, including increased tumor incidence and the availability of multiple specimens per individual (normal mucosa, adenoma, and carcinoma, taken at different time points), to gain insights into the role of DNA methylation alterations in hereditary CRC.
Loss of MMR protein corresponding to the predisposing MMR gene increased with the degree of dysplasia (Table 2), in agreement with results from a smaller series of screen-detected LS-adenomas [17]. In addition, we demonstrate that inactivation of MLH1 by somatic methylation can constitute a “second hit” in LS-associated tumorigenesis. The method of methylation analysis (notably, the ability for quantification of methylation) and the particular region(s) of MMR gene promoters evaluated are important factors to take into account in “second hit” analyses. While one previous study [18] detected MLH1 methylation in 10/18 (56 %) of LS-adenomas by methylation-specific PCR, which is not a quantitative method, our frequency (1/30, 3 %, for region C methylation in adenomas with silenced MLH1 protein) complies with other quantitative studies for the same region suggesting that in a small but definite proportion (2–15 %) of tumors from LS patients the wild-type allele is inactivated by MLH1 promoter hypermethylation [19–22]. Hypermethylation of region A of MLH1 was relatively common (up to 60 %) in the LS samples (Fig. 1). Methylation of region A correlates with age and does not silence gene expression, whereas methylation of region C (located closely upstream to the transcription start site) correlates with gene silencing [12, 21, 23]. Moreover, the single adenoma with MLH1 promoter methylation in region C in our investigation was CIMP(+) by all three criteria tested, supporting previous evidence of MMR gene methylation as the “second hit” being associated with a more general CIMP in the tumors [19, 24].
The fact that almost half of the low-grade dysplasia adenomas retained MMR protein expression suggests that somatic inactivation of the wild-type allele may not always precede polyp formation, consistent with Yurgelun et al. [25]. In such cases, the existence of other somatic driver events has been hypothesized [26]. Moreover, non-tumorous mucosa from resections for intestinal cancer revealed a frequent occurrence of MMR-deficient crypt foci in LS mutation carriers, which contrasts with the low number of adenomas and carcinomas becoming clinically manifested and underlines the necessity of other tumorigenic events before or after MMR gene inactivation [27]. DNA methylation changes are excellent candidates for such events. The exact step at which the most abundant or important DNA methylation changes may occur remains unsettled. In the sporadic setting, a number of studies conclude that most alterations disrupting the normal patterns of DNA methylation occur in precursor lesions (ACF and polyps) rather than at more advanced stages (carcinoma and metastasis) [28]. On the other hand, Beggs et al. [7] detected the bulk of promoter hypermethylation in the transition from adenoma to carcinoma, rather than from normal tissue to adenoma. Ibrahim et al. [29] found that DNA methylation changes take place sequentially at specific transition points involving RUNX3 in normal colonic mucosa, NEUROG1 and CACNA1G in hyperplastic polyps, SFRP2 and IGF2-DMR0 in adenomatous polyps, and CDKN2A and MLH1 in the adenocarcinoma stage. We chose to use previously established CIMP markers and panels and found that CpG island methylation in LS adenomas and carcinomas increases with dysplasia (Figs. 1 and 2). Among the individual markers, the frequency of specimens with hypermethylation of IGF2 and NEUROG1 was significantly increased in adenomas with low-grade dysplasia already (Fig. 1), including some with the predisposing MMR gene not silenced, yet, indicating that methylation of IGF2 and NEUROG1 may provide a marker of early colon oncogenesis.
In sporadic colorectal tumors, MMR deficiency is mainly proposed to serve cancer progression rather than initiation [30]. Moreover, analysis of microdissected glands revealed significant heterogeneity for MSI and promoter methylation of MMR genes within individual polyps [31]. Our IHC and MSI results (Table 2), like those by Yurgelun et al. [25], suggest the possibility that some LS adenomas might be initiated by mechanisms other than MMR deficiency and that the MMR and non-MMR pathways may converge in later stages (adenomas with high-grade dysplasia and carcinomas). CIMP and candidate genes were often hypermethylated in low-grade dysplasia adenomas from our series regardless of MMR status of the tumors; thus, aberrant methylation might be among the earliest events in colorectal tumors developing in MMR gene mutation carriers.
Apart from adenomatous polyps that are viewed as precursors of CRC, LS mutation carriers may also exhibit polyps that develop along the serrated pathway, including hyperplastic polyps [32]. Hyperplastic polyps are traditionally considered to be non-neoplastic, lacking potential for malignant progression [33]. The latter view is supported by observations of a virtual absence of MMR defects in hyperplastic polyps from LS mutation carriers [25, 34]. A gland-level analysis of hyperplastic polyps from sporadic cases has shown frequent MSI and methylation aberrations, and the possibility that such polyps might be precursors to MSI CRC cannot be ruled out [31]. In light of the findings described above, the frequent hypermethylation we observed for the markers from the CIMP panel (Additional file 3: Figure S1A) and candidate gene panel (Additional file 3: Figure S1B) in hyperplastic polyps from LS mutation carriers is interesting and warrants further studies to evaluate the significance of such methylation events in tumorigenesis.
Specific dietary compounds are known to act as important modifiers of the methylation patterns of the colon, and DNA methylation of the intestinal mucosa can thus link nutrition to cancer [35]. Here, we report significantly increased hypermethylation frequencies for two secreted frizzled-related proteins, SFRP1 and SFRP2, in LS-associated adenomas and carcinomas when compared to normal colon (Fig. 3). These genes emerged from our previous dietary intervention study on the Mlh1 mouse model [11]. In the normal colon epithelial cells, SFRPs function as Wnt signaling antagonists and compete with Wnt proteins for binding to their receptor, frizzled. Transcriptional silencing of these genes through promoter hypermethylation (epigenetic gatekeeper) activates the APC protein complex which further promotes cell proliferation and ACF formation [36]. Interestingly, hypermethylation of these genes with concomitant reduction in gene expression was reported to apply to both CIMP-high and non-CIMP tumors, which indicates that aberrant methylation of these genes may occur in colorectal tumors irrespective of their subtype [37].
Field defects are clonal abnormalities in the epithelial gene expression that precede cancer development and predispose to it, occasionally causing the simultaneous occurrence of multiple tumors within a field [38]. Age-related DNA methylation changes have been proposed as potential sources of field defects in the colon [16, 14]. Observations that DNA methylation in normal mucosa may associate with pathway-specific susceptibility to CRC [39, 40] and that synchronous cancer pairs share epigenetic features such as CIMP and LINE-1 methylation status [41] support the existence of epigenetic field defects in CRC development. In our investigation, mutation carriers over 50 years old with a previous CRC diagnosis showed a significantly elevated average degree of methylation of SFRP1 and SLC5A8 vs. cancer-free individuals of a comparable age. Differences between the two groups of mucosa were small though statistically significant. SFRP1 was discussed above; SLC5A8 encodes a sodium transporter that directly influences the absorption of short-chain fatty acids (e.g., butyrate, a histone deacetylase inhibitor) from the apical membrane of the intestinal tract into the colon [42] and is frequently silenced in ACF and CRC by promoter methylation [43]. Our findings are consistent with a recent epigenome-wide study by Luo et al. who identified 65 loci with higher methylation in (sporadic) CRC-associated mucosa vs. colon mucosa from cancer-free individuals [44].
Conclusions
We show that increased DNA methylation of CIMP markers and candidate gene loci accompanies tumor progression in LS. Methylation alterations may form carcinogenic fields in histologically normal mucosa and occur in adenomas at a stage where MMR protein expression is still intact. When affecting the predisposing MMR gene, promoter methylation can constitute the somatic “second hit”. Our results provide new insights into the multistep colorectal tumorigenesis in LS and CRC in general [29, 36, 43]. The findings also pinpoint potential markers for early detection and the assessment of progression of CRC. Altered methylation at such marker loci may help identify individuals prone to develop CRC through CIMP who might benefit from demethylating agents for chemoprevention or treatment [45].
Methods
Patient samples
CRC families represented the nationwide Hereditary Colorectal Cancer Registry of Finland. Colorectal fresh frozen biopsies (prospective series, Table 1) were gathered from LS and FAP patients during colonoscopy screenings and colectomies performed at the Helsinki University Central Hospital and Jyväskylä Central Hospital during 10/2011–5/2013. Normal mucosa biopsies were collected from one to four distinct colonic regions. In addition, blood was drawn to provide another source of normal cells for comparison. Forty-three individuals from the prospective series also contributed archival specimens (retrospective series, Table 1; predisposing MMR gene mutations specified in Additional file 10: Table S5). The histology of adenomas and carcinomas was verified by one of the authors (A.R). Carcinoma sections for DNA extractions contained 30–80 % tumor epithelium (average 50 %). The Institutional Ethics Board of Central Finland Health Care District approved the collection of biopsies during surveillance (K-S shp Dnro 10U/2011). The National Authority for Medicolegal Affairs (Dnro 1272/04/044/07) approved the collection of archival specimens.
Immunohistochemistry (IHC) for MMR protein expression
IHC was performed by standard procedures [46] with the following primary antibodies: MLH1 (clone ES05; 75 mg/l; Dako North America, Inc. CA), MSH2 (clone G219-1129; 0.5 mg/ml; BD Pharmingen), MSH6 (clone EP49, AC00-47, Epitomics, Burlingame, CA), and PMS2 (clone EPR3947, 0.324 mg/ml, Abcam, Cambridge, UK). Negative cancer cell immunostaining was interpreted to indicate inactivation of the respective MMR gene.
Microsatellite instability (MSI) analysis
MSI analysis was based on the mononucleotide repeat markers BAT25 and BAT26, which are sensitive and specific indicators of the MSI-high phenotype [47, 48]. Tumors with unstable BAT25 or BAT26 were considered to have MSI, whereas those with normal BAT25 and BAT26 were microsatellite-stable.
Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) for methylation analysis
MS-MLPA probes contain a recognition sequence (GCGC) for the methylation-sensitive endonuclease HhaI, and methylated template DNA generates a signal peak [49]. MS-MLPA was performed according to the manufacturer’s instructions [50]. The methylation dosage ratios (Dm) were calculated separately for each normal mucosa and tumor sample (Additional file 1: Table S1) as described [51]. The Dm value of 0.15 or above (corresponding to 15 % of methylated DNA) was treated as the conservative technical threshold for methylation detection [49]. Since the baseline level for methylation in normal tissue may vary between probes (Additional file 1: Table S1), normal mucosa specimens were used to determine thresholds for hypermethylation in tumor tissues (Additional file 2: Table S2). Dm values in each tumor were compared to these thresholds to determine whether or not the tumor was hypermethylated at the respective locus. The results from individual tumors were then combined to calculate the hypermethylation frequency for each tumor type. The hypermethylation threshold (separate for fresh frozen and archival tissue-derived DNA) was defined as the mean Dm in normal mucosa plus 1 standard deviation (stringency level I) or 2 standard deviations (stringency level II) with the purpose to achieve an optimal discrimination between normal and tumor tissues. Stringency level I was used in all contexts except for classification of tissue specimens into CIMP(+) and CIMP(−) categories where stringency level II provided a better distinction (Fig. 2). In the prospective series, an average Dm value incorporating 1–4 colorectal regions was calculated to describe the normal mucosa Dm of each individual.
MS-MLPA was found to be highly reproducible (an average difference of Dm = 0.029 was observed between 288 replicate measurements) and sensitive (even 5 % methylation could be reliably detected in most cases; Additional file 9: Figure S5).
MMR gene promoter methylation
For studies of the “second hit,” the methylation status of MMR genes was analyzed by SALSA MLPA probemix ME011 (MRC Holland, Amsterdam, The Netherlands). Analyzed MLH1 promoter regions corresponded to regions A, B, and C as described by Deng et al. [12].
CpG island methylator phenotype (CIMP)
Promoter methylation of CACNA1G, IGF2, NEUROG1, RUNX3, SOCS1, CDKN2A, MLH1, and CRABP1 was studied by SALSA MLPA probemix ME042 (MRC Holland, Amsterdam, The Netherlands). The probemix contains 3–6 probes for each CIMP marker gene. A gene was considered methylated when one fourth (25 %) or more probes were methylated [13]. Two alternative marker panels were utilized to classify colorectal tumors as CIMP(+) or CIMP(−). The Weisenberger panel [52, 53] includes five genes (CACNA1G, SOCS1, RUNX3, IGF2, and NEUROG1), at least three of which should be methylated for CIMP(+) [13]. The Ogino panel includes three additional genes (CDKN2A, MLH1, and CRABP1) [52, 53], and five or more methylated genes out of eight (Ogino 5/8, CIMP-low) or six or more methylated genes out of eigth (Ogino 6/8, CIMP-high) were regarded to indicate CIMP(+).
Custom MS-MLPA for methylation analysis of candidate genes
To study promoter methylation of DKK1, SFRP1, SFRP2, SFRP5, CDH1, HOXD1, and SLC5A8 [11], a custom MS-MLPA assay was designed. CpG islands (CGI) were identified by EMBOSS CpGplot [54] and CpG Island Searcher [55]. DNAs from cancer cell lines and normal tissues were bisulfite sequenced (with primers specified in Additional file 4: Table S3A) to determine the methylation statuses of the CpG sites within the CGIs. Custom MS-MLPA probes were designed to target GCGC sites (Additional file 4: Table S3B). Salsa MLPA P300-A2 Human DNA Reference-2 (MRC Holland, Amsterdam, The Netherlands) was added to the custom designed MS-MLPA probe mix. The custom assay was optimized against bisulfite sequencing as described [56], resulting in the conservative technical threshold of Dm ≥ 0.15 for methylation detection.
Epigenetic drug treatments and analysis of genome-wide mRNA expression
Cancer cell lines (Additional file 5: Table S4) were treated with epigenetic drugs and RNA expression was profiled on Affymetrix Human Genome U133 Plus 2.0 GeneChip® microarrays (Affymetrix, Santa Clara, CA) as described [56]. Normal tissue DNA and RNA were purchased from Amsbio (Abingdon, UK). Microarray data were analyzed by GeneSpring GX software, version 12 (Agilent Technologies, Santa Clara, CA) using RMA normalization. Statistically significant gene expression changes were identified by moderated t test combined with the Benjamini and Hochberg correction for multiple testing and by using filters based on p value cutoff of 0.05 and fold change cutoff of +/−1.5. The mRNA expression profiling data have been submitted to GEO (accession number: GSE58058).
Statistical analysis
Statistical analysis of methylation data was performed using the SPSS software, version 20.0 (IBM SPSS Inc. Chicago, IL, USA). Frequency of methylated target sites in each type of tissue specimen was calculated separately for each gene using the probe-specific threshold values. Two-sided p values were calculated for each pairwise comparison by Fisher’s exact test and adjusted for multiple comparisons by Bonferroni correction. Pearson product-moment correlation coefficient (r) was used to study the correlation of methylation and expression and the association between age at biopsy and methylation in normal colonic mucosa (individual average normal colon Dm values). Statistical significance of methylation differences between groups studied for field defects was tested by one-way ANOVA, and Tukey’s post hoc test was used for pairwise comparisons. Alternatively, the non-parametric test Kruskal-Wallis one-way ANOVA (k samples, pairwise comparisons) was utilized for series not normally distributed. Homogeneity of variances was tested by Levene’s test and normality by Shapiro-Wilk test. P values <0.05 were considered significant.
Acknowledgements
The authors wish to thank all patients contributing to this study. We thank Tuula Lehtinen, Kirsi Pylvänäinen and Sanna Vainionpää for acquiring the samples and patients information. Saila Saarinen, Erkki Hänninen, Carita Liikanen, and Juha Manninen are thanked for laboratory assistance and Godfrey Lowndes for statistical advice.
This study was supported by the Integrative Life Science (ILS) doctoral program of the University of Helsinki (SV) and grants from the Academy of Finland (No. 257795 to PP), the Cancer Society of Finland (SV, AR, J-PM, and PP), the Sigrid Juselius Foundation (AR, J-PM, MN, and PP), the Nordic Cancer Union (PP), the Päivikki and Sakari Sohlberg Foundation (PP), Biocentrum Helsinki (PP), and the European Research Council (FP7-ERC-232635 to MN and PP).
Abbreviations
- 5-AZA-CdR
5-aza-2′ deoxycytidine
- ACF
aberrant crypt foci
- CGI
CpG island
- CIMP
CpG island methylator phenotype
- CRC
colorectal cancer
- FAP
familial adenomatous polyposis
- FFPE
formalin-fixed paraffin-embedded
- LINE
long interspersed element
- LS
Lynch syndrome
- MMR
mismatch repair
- MSI
microsatellite instability
- MS-MLPA
methylation-specific multiplex ligation-dependent probe amplification
- MSS
microsatellite stable
- SFRP
secreted frizzled-related protein
- TSA
trichostatin A
Additional files
Average methylation dosage ratios (Dm) and standard deviations for normal colonic mucosa and each tumor type.
Threshold values for hypermethylation at each gene locus in the Lynch syndrome retrospective series calculated based on normal colonic mucosa specimens.
Frequency of hypermethylated samples in the LS and FAP prospective series.
(A) Bisulfite primer sequences and (B) MS-MLPA probe design for candidate gene panel.
Cell line characteristics.
Correlation between SFRP1 and SFRP2 mRNA expression and methylation.
Correlation of age and normal colonic mucosa methylation. (A) IGF2 probes I, II, and III. (B) NEUROG1 probes I, III, and IV. (C) SFRP1, SFRP2, and SLC5A8.
Effect of aging and previously diagnosed CRC on normal colonic mucosa methylation. (A) IGF2 (probes I–III). (B) NEUROG1 (probes I, III, IV, and VI).
MS-MLPA sensitivity for (A) CIMP and (B) candidate gene panel. Results obtained with a gradient of decreasing amount of methylated control DNA diluted into a solution of unmethylated control DNA indicate that even 5 % of methylated DNA can be reliably detected.
Germline mutation specifications for patients (retrospective series).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SV, J-PM, MN, and PP designed the study. LR-S, HJJ, and J-PM provided the biopsy samples. AR performed the histopathological analyses. SV and SK performed the molecular analyses. SV and SK performed the data analyses. SV and PP wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Satu Valo, Phone: +358 2941 25795, Email: satu.valo@helsinki.fi.
Sippy Kaur, Email: sippy.kaur@helsinki.fi, Email: paivi.peltomaki@helsinki.fi.
Ari Ristimäki, Email: ari.ristimaki@hus.fi.
Laura Renkonen-Sinisalo, Email: laura.renkonen-sinisalo@hus.fi, Email: heikki.jj.jarvinen@gmail.com.
Jukka-Pekka Mecklin, Email: jukka-pekka.mecklin@ksshp.fi.
References
- 1.Vasen HF, Blanco I, Aktan-Collan K, Gopie JP, Alonso A, Aretz S, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut. 2013;62(6):812–823. doi: 10.1136/gutjnl-2012-304356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geiersbach KB, Samowitz WS. Microsatellite instability and colorectal cancer. Arch Pathol Lab Med. 2011;135(10):1269–1277. doi: 10.5858/arpa.2011-0035-RA. [DOI] [PubMed] [Google Scholar]
- 3.Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57(5):808–811. [PubMed] [Google Scholar]
- 4.Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci U S A. 1998;95(15):8698–8702. doi: 10.1073/pnas.95.15.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lao VV, Grady WM. Epigenetics and colorectal cancer. Nat Rev Gastroenterol Hepatol. 2011;8(12):686–700. doi: 10.1038/nrgastro.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim YH, Petko Z, Dzieciatkowski S, Lin L, Ghiassi M, Stain S, et al. CpG island methylation of genes accumulates during the adenoma progression step of the multistep pathogenesis of colorectal cancer. Genes, Chromosomes Cancer. 2006;45(8):781–789. doi: 10.1002/gcc.20341. [DOI] [PubMed] [Google Scholar]
- 7.Beggs AD, Jones A, El-Bahrawy M, Abulafi M, Hodgson SV, Tomlinson IP. Whole-genome methylation analysis of benign and malignant colorectal tumours. J Pathol. 2013;229(5):697–704. doi: 10.1002/path.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nosho K, Irahara N, Shima K, Kure S, Kirkner GJ, Schernhammer ES, et al. Comprehensive biostatistical analysis of CpG island methylator phenotype in colorectal cancer using a large population-based sample. PLoS One. 2008;3(11):e3698. doi: 10.1371/journal.pone.0003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCabe MT, Brandes JC, Vertino PM. Cancer DNA methylation: molecular mechanisms and clinical implications. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15(12):3927–3937. doi: 10.1158/1078-0432.CCR-08-2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mecklin JP, Aarnio M, Laara E, Kairaluoma MV, Pylvanainen K, Peltomaki P, et al. Development of colorectal tumors in colonoscopic surveillance in Lynch syndrome. Gastroenterology. 2007;133(4):1093–1098. doi: 10.1053/j.gastro.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 11.Pussila M, Sarantaus L, Dermadi Bebek D, Valo S, Reyhani N, Ollila S, et al. Cancer-predicting gene expression changes in colonic mucosa of Western diet fed Mlh1+/− mice. PLoS One. 2013;8(10):e76865. doi: 10.1371/journal.pone.0076865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng G, Chen A, Hong J, Chae HS, Kim YS. Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer Res. 1999;59(9):2029–2033. [PubMed] [Google Scholar]
- 13.Berg M, Hagland HR, Soreide K. Comparison of CpG island methylator phenotype (CIMP) frequency in colon cancer using different probe- and gene-specific scoring alternatives on recommended multi-gene panels. PLoS One. 2014;9(1):e86657. doi: 10.1371/journal.pone.0086657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Issa JP. CpG-island methylation in aging and cancer. Curr Top Microbiol Immunol. 2000;249:101–118. doi: 10.1007/978-3-642-59696-4_7. [DOI] [PubMed] [Google Scholar]
- 15.Chan AO, Broaddus RR, Houlihan PS, Issa JP, Hamilton SR, Rashid A. CpG island methylation in aberrant crypt foci of the colorectum. Am J Pathol. 2002;160(5):1823–1830. doi: 10.1016/S0002-9440(10)61128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst. 2005;97(18):1330–1338. doi: 10.1093/jnci/dji275. [DOI] [PubMed] [Google Scholar]
- 17.De Jong AE, Morreau H, Van Puijenbroek M, Eilers PH, Wijnen J, Nagengast FM, et al. The role of mismatch repair gene defects in the development of adenomas in patients with HNPCC. Gastroenterology. 2004;126(1):42–48. doi: 10.1053/j.gastro.2003.10.043. [DOI] [PubMed] [Google Scholar]
- 18.Kaz A, Kim YH, Dzieciatkowski S, Lynch H, Watson P, Kay Washington M, et al. Evidence for the role of aberrant DNA methylation in the pathogenesis of Lynch syndrome adenomas. International journal of cancer Journal international du cancer. 2007;120(9):1922–1929. doi: 10.1002/ijc.22544. [DOI] [PubMed] [Google Scholar]
- 19.Ollikainen M, Hannelius U, Lindgren CM, Abdel-Rahman WM, Kere J, Peltomaki P. Mechanisms of inactivation of MLH1 in hereditary nonpolyposis colorectal carcinoma: a novel approach. Oncogene. 2007;26(31):4541–4549. doi: 10.1038/sj.onc.1210236. [DOI] [PubMed] [Google Scholar]
- 20.Rahner N, Friedrichs N, Steinke V, Aretz S, Friedl W, Buettner R, et al. Coexisting somatic promoter hypermethylation and pathogenic MLH1 germline mutation in Lynch syndrome. J Pathol. 2008;214(1):10–16. doi: 10.1002/path.2263. [DOI] [PubMed] [Google Scholar]
- 21.Parsons MT, Buchanan DD, Thompson B, Young JP, Spurdle AB. Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: a literature review assessing utility of tumour features for MMR variant classification. J Med Genet. 2012;49(3):151–157. doi: 10.1136/jmedgenet-2011-100714. [DOI] [PubMed] [Google Scholar]
- 22.Moreira L, Munoz J, Cuatrecasas M, Quintanilla I, Leoz ML, Carballal S, et al. Prevalence of somatic mutl homolog 1 promoter hypermethylation in Lynch syndrome colorectal cancer. Cancer. 2015;121(9):1395–1404. doi: 10.1002/cncr.29190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakagawa H, Chadwick RB, Peltomaki P, Plass C, Nakamura Y, de La Chapelle A. Loss of imprinting of the insulin-like growth factor II gene occurs by biallelic methylation in a core region of H19-associated CTCF-binding sites in colorectal cancer. Proc Natl Acad Sci U S A. 2001;98(2):591–596. doi: 10.1073/pnas.98.2.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagasaka T, Rhees J, Kloor M, Gebert J, Naomoto Y, Boland CR, et al. Somatic hypermethylation of MSH2 is a frequent event in Lynch syndrome colorectal cancers. Cancer Res. 2010;70(8):3098–3108. doi: 10.1158/0008-5472.CAN-09-3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yurgelun MB, Goel A, Hornick JL, Sen A, Turgeon DK, Ruffin MT, et al. Microsatellite instability and DNA mismatch repair protein deficiency in Lynch syndrome colorectal polyps. Cancer prevention research. 2012;5(4):574–582. doi: 10.1158/1940-6207.CAPR-11-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. Milestones of Lynch syndrome: 1895–2015. Nat Rev Cancer. 2015;15(3):181–194. doi: 10.1038/nrc3878. [DOI] [PubMed] [Google Scholar]
- 27.Kloor M, Huth C, Voigt AY, Benner A, Schirmacher P, von Knebel Doeberitz M, et al. Prevalence of mismatch repair-deficient crypt foci in Lynch syndrome: a pathological study. Lancet Oncol. 2012;13(6):598–606. doi: 10.1016/S1470-2045(12)70109-2. [DOI] [PubMed] [Google Scholar]
- 28.Carmona FJ, Esteller M. Epigenomics of human colon cancer. Mutat Res. 2010;693(1–2):53–60. doi: 10.1016/j.mrfmmm.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 29.Ibrahim AE, Arends MJ, Silva AL, Wyllie AH, Greger L, Ito Y, et al. Sequential DNA methylation changes are associated with DNMT3B overexpression in colorectal neoplastic progression. Gut. 2011;60(4):499–508. doi: 10.1136/gut.2010.223602. [DOI] [PubMed] [Google Scholar]
- 30.Homfray TF, Cottrell SE, Ilyas M, Rowan A, Talbot IC, Bodmer WF, et al. Defects in mismatch repair occur after APC mutations in the pathogenesis of sporadic colorectal tumours. Hum Mutat. 1998;11(2):114–120. doi: 10.1002/(SICI)1098-1004(1998)11:2<114::AID-HUMU3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 31.Beggs AD, Domingo E, Abulafi M, Hodgson SV, Tomlinson IP. A study of genomic instability in early preneoplastic colonic lesions. Oncogene. 2013;32(46):5333–5337. doi: 10.1038/onc.2012.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burt RW. Colonic polyps in Lynch syndrome. Dis Colon Rectum. 2015;58(4):371–372. doi: 10.1097/DCR.0000000000000332. [DOI] [PubMed] [Google Scholar]
- 33.Noffsinger AE, Hart J. Serrated adenoma: a distinct form of non-polypoid colorectal neoplasia? Gastrointest Endosc Clin N Am. 2010;20(3):543–563. doi: 10.1016/j.giec.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 34.Rijcken FE, van der Sluis T, Hollema H, Kleibeuker JH. Hyperplastic polyps in hereditary nonpolyposis colorectal cancer. Am J Gastroenterol. 2003;98(10):2306–2311. doi: 10.1111/j.1572-0241.2003.07629.x. [DOI] [PubMed] [Google Scholar]
- 35.Arasaradnam RP, Commane DM, Bradburn D, Mathers JC. A review of dietary factors and its influence on DNA methylation in colorectal carcinogenesis. Epigenetics: official journal of the DNA Methylation Society. 2008;3(4):193–198. doi: 10.4161/epi.3.4.6508. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36(4):417–422. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]
- 37.Hinoue T, Weisenberger DJ, Lange CP, Shen H, Byun HM, Van Den Berg D, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012;22(2):271–282. doi: 10.1101/gr.117523.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Braakhuis BJ, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63(8):1727–1730. [PubMed] [Google Scholar]
- 39.Worthley DL, Whitehall VL, Buttenshaw RL, Irahara N, Greco SA, Ramsnes I, et al. DNA methylation within the normal colorectal mucosa is associated with pathway-specific predisposition to cancer. Oncogene. 2010;29(11):1653–1662. doi: 10.1038/onc.2009.449. [DOI] [PubMed] [Google Scholar]
- 40.Svrcek M, Buhard O, Colas C, Coulet F, Dumont S, Massaoudi I, et al. Methylation tolerance due to an O6-methylguanine DNA methyltransferase (MGMT) field defect in the colonic mucosa: an initiating step in the development of mismatch repair-deficient colorectal cancers. Gut. 2010;59(11):1516–1526. doi: 10.1136/gut.2009.194787. [DOI] [PubMed] [Google Scholar]
- 41.Nosho K, Kure S, Irahara N, Shima K, Baba Y, Spiegelman D, et al. A prospective cohort study shows unique epigenetic, genetic, and prognostic features of synchronous colorectal cancers. Gastroenterology. 2009;137(5):1609–1620. doi: 10.1053/j.gastro.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thangaraju M, Cresci G, Itagaki S, Mellinger J, Browning DD, Berger FG, et al. Sodium-coupled transport of the short chain fatty acid butyrate by SLC5A8 and its relevance to colon cancer. Journal of gastrointestinal surgery: official journal of the Society for Surgery of the Alimentary Tract. 2008;12(10):1773–1781. doi: 10.1007/s11605-008-0573-0. [DOI] [PubMed] [Google Scholar]
- 43.Li H, Myeroff L, Smiraglia D, Romero MF, Pretlow TP, Kasturi L, et al. SLC5A8, a sodium transporter, is a tumor suppressor gene silenced by methylation in human colon aberrant crypt foci and cancers. Proc Natl Acad Sci U S A. 2003;100(14):8412–8417. doi: 10.1073/pnas.1430846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo Y, Wong CJ, Kaz AM, Dzieciatkowski S, Carter KT, Morris SM, et al. Differences in DNA methylation signatures reveal multiple pathways of progression from adenoma to colorectal cancer. Gastroenterology. 2014;147(2):418–429. doi: 10.1053/j.gastro.2014.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kodach LL, Jacobs RJ, Voorneveld PW, Wildenberg ME, Verspaget HW, van Wezel T, et al. Statins augment the chemosensitivity of colorectal cancer cells inducing epigenetic reprogramming and reducing colorectal cancer cell ‘stemness’ via the bone morphogenetic protein pathway. Gut. 2011;60(11):1544–1553. doi: 10.1136/gut.2011.237495. [DOI] [PubMed] [Google Scholar]
- 46.Thiel A, Heinonen M, Kantonen J, Gylling A, Lahtinen L, Korhonen M, et al. BRAF mutation in sporadic colorectal cancer and Lynch syndrome. Virchows Archiv: an international journal of pathology. 2013;463(5):613–621. doi: 10.1007/s00428-013-1470-9. [DOI] [PubMed] [Google Scholar]
- 47.Loukola A, Eklin K, Laiho P, Salovaara R, Kristo P, Jarvinen H, et al. Microsatellite marker analysis in screening for hereditary nonpolyposis colorectal cancer (HNPCC) Cancer Res. 2001;61(11):4545–4549. [PubMed] [Google Scholar]
- 48.Esemuede I, Forslund A, Khan SA, Qin LX, Gimbel MI, Nash GM, et al. Improved testing for microsatellite instability in colorectal cancer using a simplified 3-marker assay. Ann Surg Oncol. 2010;17(12):3370–3378. doi: 10.1245/s10434-010-1147-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nygren AO, Ameziane N, Duarte HM, Vijzelaar RN, Waisfisz Q, Hess CJ, et al. Methylation-specific MLPA (MS-MLPA): simultaneous detection of CpG methylation and copy number changes of up to 40 sequences. Nucleic Acids Res. 2005;33(14):e128. doi: 10.1093/nar/gni127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MRC Holland. http://mrc-holland.com. Accessed 5 March 2015.
- 51.Pavicic W, Perkio E, Kaur S, Peltomaki P. Altered methylation at microRNA-associated CpG islands in hereditary and sporadic carcinomas: a methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA)-based approach. Mol Med. 2011;17(7–8):726–735. doi: 10.2119/molmed.2010.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38(7):787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 53.Ogino S, Kawasaki T, Kirkner GJ, Kraft P, Loda M, Fuchs CS. Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample. The Journal of molecular diagnostics: JMD. 2007;9(3):305–314. doi: 10.2353/jmoldx.2007.060170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.EMBOSS CpGplot. http://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot. Accessed 5 March 2015.
- 55.CpG Island Searcher. http://cpgislands.usc.edu. Accessed 5 March 2015.
- 56.Niskakoski A, Kaur S, Staff S, Renkonen-Sinisalo L, Lassus H, Jarvinen HJ, et al. Epigenetic analysis of sporadic and Lynch-associated ovarian cancers reveals histology-specific patterns of DNA methylation. Epigenetics: official journal of the DNA Methylation Society. 2014;9(12):1577–1587. doi: 10.4161/15592294.2014.983374. [DOI] [PMC free article] [PubMed] [Google Scholar]