

Abstract
Programmed Cell Death (PCD) plays a fundamental role in animal development and tissue homeostasis. Abnormal regulation of this process is associated with a wide variety of human diseases, including immunological and developmental disorders, neuro-degeneration, and cancer. Here, we provide a brief historical overview of the field and reflect on myriad functions carried out by PCD during development and explore how PCD is regulated. We also focus on the function and regulation of apoptotic proteins, including caspases, the key executioners of apoptosis, highlighting the non-lethal functions of these proteins in diverse developmental processes including cell differentiation and tissue remodeling. Finally, we explore a growing body of work about the connections between apoptosis, stem cells and cancer, focusing on how apoptotic cells release a variety of signals to communicate with their cellular environment, including factors that promote cell division, tissue regeneration, and wound healing.
Keywords: apoptosis, caspase, Bcl-2, IAPs, Reaper, ubiquitin-proteasome system, development, proliferation, regeneration, stem cells, cancer
Historical overview
Naturally occurring cell death was long considered a passive phenomenon that accounted for the inevitable end point of biological systems (reviewed in Glücksmann, 1951). This view began to change with studies of developmentally-timed cell death in the silkworm and tadpole. These early studies showed that cell death can be delayed with inhibitors of protein or RNA synthesis and that neuronal cell survival requires extracellular survival factors termed neurotrophins (Lockshin and Williams, 1965; Tata, 1966,). Although neurotrophins were initially seen as a form of special “nourishment” required for cell survival, it later became clear that these factors suppress the execution of an intrinsic cell suicide program (reviewed in Raff et al., 1993). Moreover, this mechanism is not restricted to the nervous system, and competition for a limiting supply of extracellular survival signals is a widely used general mechanism that regulates cell number in animals (reviewed in Jacobsen et al., 1997). Another major contribution to the cell death field is the ultrastructural study by Kerr, Wylie and Currie that defined a series of distinct morphological changes in cells dying under physiological conditions (Kerr et al., 1972). When cells die in response to overwhelming stress or injury, they swell and rupture in a process termed “necrosis”. In contrast, the majority of cells that die during normal development and homeostasis shrink, have condensed nuclei, retain membrane integrity and are rapidly eliminated by phagocytosis in a process termed apoptosis (reviewed in Jacobsen, 1997). As we will discuss in more detail below, more recent studies have uncovered other forms of programmed cell death (PCD), revealing that apoptosis is not the only form of developmental cell death, and that backup mechanisms likely compensate when it is prevented (reviewed in Yuan and Kroemer et al., 2010).
A breakthrough in elucidating the mechanism by which cells undergo PCD came from genetic studies in the nematode C. elegans. The identification of mutations with specific effects on programmed cell death and their ordering into a genetic pathway demonstrated that cell death is a developmental fate, with specific genes acting to initiate a program of cell suicide (Metzstein, 1998; Ellis and Horvitz, 1986). The subsequent molecular characterization of the corresponding genes led to the identification of a core cell death machinery that has been conserved in evolution and centers around a family of cysteine proteases, termed caspases, as key executioners of apoptosis (Fig. 1) (reviewed in Hengartner, 2000, Thornberry and Lazebnik, 1998).
Figure 1. Evolutionary conservation of the core apoptotic machinery.

A comparison between the apoptotic pathways in C. elegans, Drosophila and mammals reveals conservation and expansion of the apoptotic pathway during evolution. A. In C. elegans apoptotic signals regulate the interplay between Egl-1 (BH3-only homology) and CED-9 (Bcl-2 family homologue), liberating CED-4 (Apaf-1 homologue) to activate CED-3 (caspase-9 homologue) for programmed death of 131 cells. B. In Drosophila, many different signaling pathways regulate both the IAP antagonists Reaper, Hid, and Grim (RHG) and the apoptosome proteins Ark (Apaf-1 homologue) and Dronc (caspase-9 homologue). On the one hand, this causes the ubiquitin-mediated degradation of DIAP1 and de-repression of caspases, and on the other hand it enables Dronc (caspase-9 homologue) to associate with Ark, creating active apoptosomes and activation of the effector caspases DrICE and Dcp1. Both pathways are required for efficient caspase activation and are coordinately regulated, in analogy with driving a car with “gas” and “brake”. However, removal of the “brakes” is necessary for the efficient induction of apoptosis in vivo and often initiates it. The P35 protein can specifically inhibit the activity of Dcp-1 and DrICE. C. In mammals, the balance between pro-apoptotic and anti-apoptotic Bcl-2 family members is a key factor in the commitment to apoptosis by regulating the release of cytochrome c and IAP antagonists from mitochondria. Binding of cytochrome c to Apaf-1 promotes apoptosome assembly, which recruits and activates caspase-9. IAP antagonists liberate caspases from the inhibition of IAPs, most notably XIAP (X-linked inhibitor of apoptosis), which targets both initiator and effector caspases. The XIAP-antagonist ARTS is localized to the mitochondrial outer membrane and acts prior to the release of cytochrome c, Smac and other proteins released from the mitochondrial inter-membrane space.
Homologues proteins (by either function or sequence) are similarly illustrated.
Roles of Programmed Cell Death in development
Most of our knowledge regarding the role and regulation of PCD has come primarily from three model systems, the nematode C. elegans, the fruitfly Drosophila melanogaster, and the mouse. In C. elegans, the programmed death of somatic cells is an invariant fate that is strictly controlled by cell lineage (Ellis and Horvitz, 1986). During development of the hermaphrodite, 131 out of the total 1090 somatic cells die, mostly during embryogenesis and soon after cell division (Ellis et al., 1991). In loss of function mutants for egl-1, ced-3 or ced-4 (cell death is blocked leading to the survival of all 131 cells (Fig. 1). Despite the long-term persistence of undead cells, development proceeds normally and the lifespan, behavior and appearance of cell-death defective mutants is similar to wild-type worms. In contrast, loss of ced-9 function results in developmental lethality due to widespread ectopic cell death (Hengartner et al., 1992). For each of these C. elegans genes homologs have been identified in other organisms: CED-3 is a caspase, CED-4 is a homolog of the adapter protein, Apoptosis Activating Factor-1 (Apaf-1), which that promotes assembly and activation of caspases, CED-9 is a multi-domain Bcl-2 family member, and EGL-1 is similar to pro-apoptotic BH3-only proteins (Hengartner, 2000; Fig 1). Additional genes have been identified in C. elegans that affect the decision of cells to die, including the transcriptional regulators CES-1, CES-2 and CEH-30 (Metzstein et al., 1996, Thellmann et al., 2003, Schwarz and Horvitz, 2007). Finally, a non-apoptotic form of cell-autonomous PCD that is not mediated by caspases is responsible for the death of a specialized the linker cell during the larval/adult transition (Abraham et al., 2007).
Another important model to study PCD during development is the fruitfly, Drosophila melanogaster. This organism is comprised of over a 1000-fold more cells than C. elegans, and its total number of cells depends on environmental factors, including nutrient availability, DNA damage and environmental stress. In Drosophila, PCD is not an invariant fate specified by cell lineage but, like in vertebrates, it is regulated by a wide variety of stimuli originating from both within a cell, as well as from the environment (reviewed in Kornbluth and White, 2005). Drosophila has a well-defined mechanism of development, relatively simple and accessible anatomy, and is amenable to powerful genetics and molecular biology techniques. Therefore, it provides an important system for studying the role of PCD during development, and its regulation by different signaling pathways. Unlike the situation in C. elegans, PCD is required for the successful completion of development, and inhibition of PCD results in severe developmental defects and organismal lethality (White et al., 1994, Grether et al., 1995, Xu et al., 2005, Srivastava et al., 2007). Many of the genes that pattern the Drosophila embryo, including Hox genes, activate cell death by direct transcriptional regulation of the pro-apoptotic Reaper, Hid and Grim (RHG) genes (see, for example, Lohmann et al., 2002). These genes are also transcriptionally induced by many other signaling pathways, including the steroid hormone ecdysone. During metamorphosis, ecdysone induces the rapid destruction of larval tissues by activating transcriptional cascades that culminate in expression of RHG genes and caspase activation (Jiang et al., 2000). In contrast, ecdysone acts as a pro-survival factor for a set of adult neurons that survive through this transition but die soon after eclosion. In this case ecdysone represses the expression of reaper and grim (Robinow et al., 1997; Draizen et al., 1999). As discussed in more detail below, PCD by apoptosis contributes to the patterning and normal development of virtually all adult structures in the fly, including legs, wings, eyes, genitalia, digestive system and the nervous system. Also, defects in cell division, specification or differentiation almost invariantly cause apoptotic death, revealing a stringent quality control that removes defective and useless cells during development. In addition to apoptosis, other forms of PCD have been described in Drosophila as well, and several studies suggest that autophagy also contributes to the removal of superfluous cells during normal development (reviewed in Ryoo and Baehrecke, 2010).
As one may expect, the regulation of PCD in vertebrates appears considerably more complex and vast numbers of cells undergo PCD throughout development, from as early as inner cell mass differentiation in blastocysts to maintenance of tissue homeostasis in adulthood (Hardy et al., 1989). Therefore, it is somewhat surprising that the inactivation of mouse cell death genes leads to only relatively minor developmental defects and can often survive embryonic development (see, for example, Lindsten and Thompson 2006; Okamoto et al., 2006). One reason appears to be considerable redundancy within the caspase family, and the existence of multiple mechanisms for caspase activation. For example, some effector caspase can be activated in the absence of Apaf-1 function (Nagasaka et al., 2009). In addition, there is evidence for alternative back-up mechanisms that eliminate cells when apoptosis is defective (reviewed in Yuan and Kroemer, 2011). Despite the apparent robustness of cell death mechanisms in mammals, inhibition of apoptosis has been linked to several specific developmental abnormalities and also a variety of human pathologies, including cancer and degenerative disease (Thompson, 1995).
Studies in worms, flies and mice have been complemented and extended by work in many other systems, including Hydra, Manduca, Xenopus, zebrafish, chicken and the analysis of human patients. Collectively, this work has illustrated why cells need to be eliminated in different physiological contexts: (I) sculpting and (II) deleting structures, (III) regulating cell number and (IV) eliminating defective cells.
Sculpting structures and driving morphogenesis
PCD plays a crucial role in organogenesis and tissue remodeling. Perhaps, the best-known example is the formation of digits in higher vertebrates where PCD eliminates the interdigital webs primarily via the apoptotic machinery (Fig. 2A; Lindsten et al., 2000). Although apoptosis is the major cell death mechanism in developing limbs, inactivation of pro-apoptotic genes in the mouse only partially prevents the removal of the interdigital tissue, suggesting that backup mechanisms exist when apoptosis fails (Yuan and Kroemer, 2011). In Drosophila apoptosis plays a critical role in the formation of leg joints and for the morphogenesis of segments, in particular of the head, which all require RHG-mediated apoptosis (Lohman et al., 2002). Furthermore, apoptosis is also required to permit tissue rotation drives looping morphogenesis of male genitalia in the fly (Kuranaga et al., 2011).
Figure 2. Functions of PCD during development.
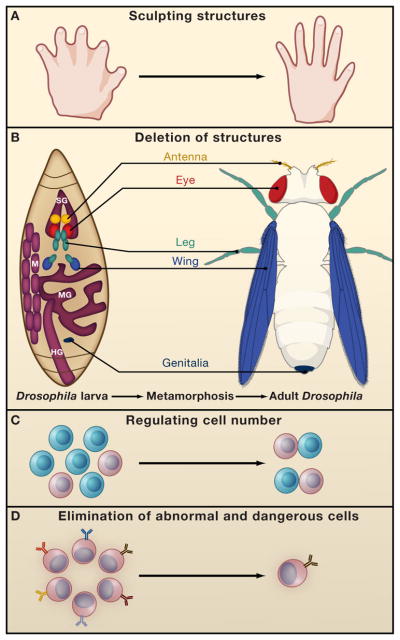
(A) PCD regulates proper structure sculpting by eliminating interdigital-webbings.
(B) During Drosophila metamorphosis nearly all larval structures are destroyed such as the Salivary glands (SG), Muscles (M), midgut (MG) and hindgut (HG). (depicted by purple), whereas novel structures are raised from undifferentiated cells termed imaginal discs (depicted by various colors). The locations and developmental fates of the imaginal discs are similarly illustrated.
PCD also controls cell number for example by deleting cells which fail to partner (C) and eliminates dangerous and abnormal cells such as autoreactive lymphocytes (D).
PCD is also involved in the conversion of solid structures to hollow tubes, thereby yielding a lumina such as in the creation of the proamniotic cavity (Coucouvanis and Martin 1995; Weil et al., 1997). It is observed when epithelial sheets invaginate, forming tubes or vesicles, for example in the establishment of the neural tube or lens and when epithelial sheets fuse to construct the mammalian palate (Glucksmann, 1951). In addition, PCD is involved in sculpting the future inner ear in chicks (Avallone et al., 2002) and essential for generating the four-chamber architecture of the heart (Abdelwahid et al., 2002).
Deleting Structures
During development, various structures that serve a transient function are removed by PCD when they are no longer required. Examples include evolutionary relics, structures that are required in only one sex, or structures that are transiently required. In fish and amphibians, pronerphric tubules form functioning kidneys, however they are not utilized in mammals and hence eliminated during embryogenesis. In female mammals the Müllerian duct forms the oviducts and uterus, but is deleted in males. Conversely, the Wolffian duct that forms the vas deferens, epididymis, and seminal vesicle in males is degraded in females (Jacobson et al., 1997). In metamorphosis, juvenile structures are removed by PCD. In amphibians, the tadpole tail and intestine are deleted and in insects most larval tissues by PCD (Fig. 2B, Baehrecke, 2002).
Regulating cell number
Developing tissues and organs rely heavily on an intricate balance between cell division and PCD to achieve appropriate cell numbers. In many organs, such as the nervous, immune and reproductive system, cells are overproduced and subsequently removed by PCD (Fig. 2C). In human females PCD is responsible for culling nearly 80% oocytes prior to birth and in almost all instances these eliminated cells are typified by apoptotic morphology (Reynaud and Driancourt 2000). It has been estimated that more than half of all neurons generated in the mammalian CNS are eliminated by PCD (Barres and Raff, 1999). Competition for limiting amounts of survival signals ensures proper matching of the numbers of different cell types in a tissue. This strategy is used in Drosophila where survival signaling via the Ras/EGFR pathway prevents expression of the pro-apoptotic hid gene (Bergmann et al., 1998). Competition also occurs between cells that proliferate at different rates. In this case, slower dividing cells are eliminated from the population by more rapidly dividing cells. This phenomenon was initially observed in Drosophila, but is also seen in mammals (Moreno, 2008; Bondar and Medzhitov, 2010). Cell competition is thought to contribute to growth homeostasis by adjusting for variations that might occur during normal growth, and selecting for the “fittest” cells is thought to optimize organ function (Moreno, 2008). “Looser cells” are eliminated by hid-mediated apoptosis, and genes that mediate cell engulfment are also required for this process (de la Cova et al., 2004; Li and Baker, 2007). Although cell competition is not essential under laboratory conditions, it appears to facilitate tissue repair and has been connected to oncogenic pathways (Moreno, 2008; Bondar and Medzhitov, 2010). All these observations reveal an intimate association among the processes of cell division, differentiation, and death, with mistakes often resolved through the induction of apoptosis.
Elimination of unwanted and potentially dangerous cells
PCD also serves as a protective process in both animal development and adult life by eliminating cells that are abnormal and potentially dangerous. One example is the human immune system, in which a highly stringent selection process dictates the survival of lymphocytes. In order to evade cell death, B and T lymphocytes have to pass both positive and negative selection demonstrating a functional antigen receptor that is not auto-reactive (Fig. 2D, Opferman and Korsmeyer 2003). In this manner PCD eliminates self-reactive cells that potentially could lead to autoimmunity. Other examples include the elimination of cells in response to viral infection, unrepaired DNA damage, cell cycle perturbations, and fate and differentiation defects (Abrams et al., 1993; Rossel and Capecchi, 1999; Derry et al., 2001, Vousden and Prives, 2009; Malumbres and Barbacid, 2009; Koto et al., 2011). In all these circumstances, apoptotic cell death serves as an important “quality control” mechanism for the elimination of faulty cells.
Regulation of Apoptosis
Apoptosis is the most studied and best understood form of PCD (Fig. 1). A central step in the execution of apoptosis is the activation of caspases, a family of cysteine proteases that are ubiquitously expressed as inactive precursors (zymogens) with little or no protease activity (Hengartner, 2000, Thornberry and Lazebnik, 1998). In response to death inducing stimuli, caspases are activated by cleavage at specific aspartic residues, resulting in removal of an inhibitory N-terminal domain and production of a large and a small subunit. Heterotetramers of these subunits form the active protease, leading to the demolition phase of apoptosis (Hengartner, 2000). Importantly, not all mammalian caspases participate in apoptosis; some are activated during innate immunity and function to regulate cytokine processing and maturation (Martinou and Tschopp, 2004).
The caspase family has been traditionally sub-divided into initiator and effector caspases (Hengartner, 2000, Thornberry and Lazebnik, 1998). Effector caspases have short pro-domains and are thought to execute apoptosis after they are proteolytic processed by initiator caspases. Initiator caspases have long pro-domains that bind large adapter molecules, which promote multimerization and caspase activation. In the case of mammalian caspase-9, the pro-domain associates with the adaptor protein Apaf-1 and cytochrome c to form the apoptosome complex and initiate apoptosis (Rodriguez and Lazebnik, 1999). However, it has also been suggested that the Apaf-1/caspase-9 system amplifies rather than initiates the mammalian caspase cascade (Adams and Cory, 2002). Amongst other things, this would explain why effector caspase activation can still occur in both Drosophila and mouse mutants lacking Apaf-1/Ark and caspase-9/Dronc function (Xu et al., 2005; Srivastava et al., 2007; Nagasaka et al., 2009). In addition, it would explain the requirement of two effector caspases, caspase-3 and 7, upstream of the mitochondrial outer membrane permeabilization (MOMP) that releases cytochrome c (Lakhani et al., 2006). Therefore, it is likely that in some conditions, low-level activation of effector caspases, perhaps via inactivation of their inhibitors, occurs upstream of “initiator” caspase activation.
A critical event for apoptosome assembly is the release of cytochrome c from the mitochondria, a step tightly regulated by members of the B-cell lymphoma-2 (Bcl-2) protein family (Fig. 1, Youle and Strasser, 2008). This family is comprised of three subfamilies depending on the number of Bcl-2 homologue (BH) domains they present. The anti-apoptotic subfamily consists of members with BH4 domain of which BCl-2 is most known. The two other subfamilies are pro-apoptotic in nature, lacking either the BH4 domain (BAX, BAK and BOK) or solely displaying BH3 (BH3-only). Once the BH3-only family is activated they overcome the inhibitory effect of the anti-apoptotic Bcl-2 family proteins, enabling the oligomerization of BAK–BAX within the mitochondrial outer membrane (MOM). This facilitates MOMP and allows the release of cytochrome c (and other proteins from the intermembrane space) into the cytosol, resulting in the formation of the apoptosome (Martinou and Youle, 2011). The apoptosome then promotes the cleavage and activation of executioner caspases, such as caspase-3 and caspase-7, which orchestrate the destruction of the cell by cleavage of many vital proteins (Hengartner, 2000). Caspase activation can also occur via association with death receptors, which leads to the oligomerization and activation of an initiator caspase, caspase-8 (Strasser et al., 2009).
An equally important layer of cell death regulation involves negative regulation of caspases. Pro-caspases are widely expressed in living cells and have low but significant protease activity. Despite this potential danger, healthy cells avoid activation of a caspase cascade and death and use effector caspases for non-apoptotic functions. Therefore, efficient mechanisms must exist that prevent unwanted caspase activation. One important family of caspase inhibitors are the Inhibitor of Apoptosis (IAP) Proteins, which can bind to and inhibit caspases via their baculovirus inhibitory repeat (BIR) domain (Vaux and Silke, 2005). IAPs were first discovered in insect viruses where they prevent apoptosis of infected cells (Crook et al., 1993). Subsequently, a family of related proteins has been described in both Drosophila and mammalian genomes (Vaux and Silke, 2005). IAPs are characterized by at least one BIR domain, which can directly bind and inhibit caspases (Vaux and Silke, 2005). The Drosophila IAP family member, Diap1, is essential for preventing inappropriate caspase activation and ubiquitous apoptosis (Goyal et al., 2000; Ryoo et al., 2004). Diap1 functions as an E3-ubiquitin ligase, targeting caspases in living cells, and promoting self-conjugation and degradation in apoptotic cells (Ryoo et al., 2002; Ryoo et al., 2004). The best studied mammalian IAP is the X-linked inhibitor of apoptosis (XIAP) protein which is considered the most potent caspase inhibitor in vitro (Eckelman and Salvesen, 2006). Like in Drosophila, XIAP functions as an E3 ubiquitin ligase to inhibit caspases in vivo (Schile et al., 2008). However, compared to Drosophila, inactivation of XIAP causes only relatively “mild” phenotypes as there is some redundancy among mammalian IAPs (Schile, 2008).
In cells committed to apoptosis, IAPs are inactivated by specific antagonists originally discovered in Drosophila (reviewed in Kornbluth and White, 2005). Deletion of three closely linked genes, reaper, hid and grim (RHG) blocks virtually all apoptotic cell death in Drosophila (White et al., 1994). On the other hand, ectopic expression of these genes can lead to potent induction of apoptosis (White et al., 1996). Although the proteins encoded by these genes share very little overall similarity, they all contain a short N-terminal peptide motif, termed IBM (IAP-Binding-Motif). The IBM is structurally conserved between Drosophila and mammalian IAP-antagonists and is required for IAP-binding and cell killing (Shi, 2002).
The reaper gene, and to some extent grim, and hid, are transcriptionally induced by multiple death-inducing signals, including developmental signals mediated by segmentation genes, steroid hormones, Dpp, Notch signaling, JNK, and various forms of cellular stress or injury (Fig. 3, Steller, 2008). These genes share a very large transcriptional control region that is the target for numerous transcription factors conveying these signals, such as various Hox transcription factors, nuclear hormone receptors, AP-1, Polycomb, p53 and histone modifying enzymes (Brodsky et al., 2000; Jiang et al., 2000; Christich et al., 2002; Lohmann et al., 2002; Zhang et al., 2008; Tan et al., 2011). Therefore, one major mechanism by which different signaling pathways converge in Drosophila is through transcriptional activation of reaper, hid and grim.
Figure 3. Integration of different signaling pathways by RHG proteins.
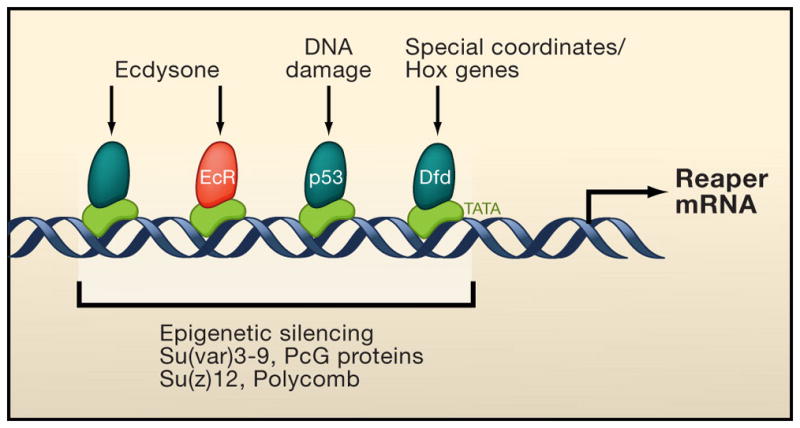
The genes encoding Reaper, Hid and Grim (RHG) are the targets for regulation of many signaling pathways that influence the decision between cell death and survival. The transcriptional control region of RHG genes, exemplified here by reaper, contains binding sites for many different transcription factors that are the downstream targets of different signaling pathways, including for the steroid hormone ecdysone, patterning signals and Hox genes, and DNA-damage/p53. Examples for both activators (green) and repressors (red) have been described. The steroid hormone ecdysone can either induce or repress reaper transcription depending at different stages of development. RHG genes are clustered and, to some extent, co-regulated at the transcriptional level, and the locus can be silenced by histone-modifying enzymes and Polycomb.
The pro-apoptotic activity of Hid is inhibited upon phosphorylation by MAP-kinase, and its transcription is repressed by the pro-survival the EGF-receptor/Ras-pathway in Drosophila (Bergmann et al., 1998; Bergmann et al., 2002). RHG genes are also the target for microRNA regulation (Brennecke et al., 2003). Finally, for effective cell killing, the Reaper protein has to localize to the mitochondrial outer membrane (MOM) (Sandu et al., 2010). This is achieved through multimerization with Hid, which localizes to the MOM via its C-terminal transmembrane domain (Haining et al., 1999). Taken together, the activity of RHG proteins is regulated by many different signaling pathways at both the transcriptional and posttranscriptional level, and these proteins serve as “integrators” to connect diverse signaling pathways with the core cell death program.
Mammalian IAP antagonists containing an IBM have also been discovered, including Smac/Diablo and HtrA2/Omi (Verhagen et al., 2000; Suzuki et al., 2001). In contrast to RHG proteins, these proteins are localized within the mitochondrial intermembrane space and require MOMP to access and bind cytosolic IAPs (Green and Kroemer, 2004). Another mammalian IAP-antagonist is ARTS (Larisch et al., 2000; Gottfried et al., 2004). Although ARTS does not contain a recognizable IBM it is localized, like RHG proteins, to the MOM and is a specific and physiological inhibitor of XIAP (Gottfried et al., 2004; Garcia-Fernandez et al., 2010; Edison et al., 2011). Therefore, ARTS does not require MOMP and acts upstream of cytoC and Smac/Diablo (Edison et al., 2011). Mice deficient for the Sept4 gene, which encodes ARTS, have elevated XIAP protein levels, are defective in the caspase-mediated elimination of bulk cytoplasm during spermiogenesis, and have elevated numbers of hematopoietic stem and progenitor cells (Kissel et al., 2005; Garcia-Fernandez et al., 2010). In addition, these mice demonstrate increased susceptibility towards developing malignancies (Garcia-Fernandez et al., 2010). Importantly, these phenotypes are suppressed by inactivation of XIAP, suggesting that it is a major physiological target for ARTS. These observations support a physiological role of ARTS and XIAP in the regulation of stem cell apoptosis.
In the end, the activation of caspases is under dual control by both activators and inhibitors, which in turn are subject to many layers of regulation. In analogy with driving a car, a combination of “gas” and “brakes” ensures that apoptosis only proceeds when multiple checkpoints are passed. The complexity of caspase regulation increases as the number of cells increase from worms to flies to mammals. As organismal complexity and lifespan increase, additional backup mechanisms are in place, presumably due to the elevated danger that unwanted cells pose to a long-lived animal.
Engulfment
The removal of apoptotic cells is the final step in the execution of apoptosis. Historically, this processes has presented a significant hurdle in evaluating the amount of apoptosis, as the dying cells are rapidly cleared by phagocytes, making it very difficult to detect apoptotic cells animal tissues. The engulfment process can be divided into the following stages: (I) sensing of the apoptotic cell, (II) recognition by the phagocyte, (III) internalization of target cell, (IV) ingestion and (V) post-engulfment response of the phagocyte (Ravichandran and Lorenz, 2007).
Dying apoptotic cells secrete “find me” and “eat me” signals that attract and recruit phagocytes. The first “find me” signal to be identified is Lysophospatidylcholine (LPC). LPC is released by apoptotic cells, in a caspase-3 dependant fashion. Caspase-3 activates the calcium independent pospholipase A2 (iPLA2), which converts phophatidylcholine to LPC (Lauber et al., 2003). Several other molecules have also been proposed to portray the “find me” role such as S1P (sphingosine-1-phosphate) and CXCL1 (Gude et al., 2008; Truman et al., 2008). In addition, ATP and UTP nucleotides, released by the PANX1 channel, have been suggested to act as “find me” signals (Elliott et al., 2009). PANX1 is a target of effector caspase-3 and -7, where a specific caspase-cleavage site is essential for PANX1 function in mediating release of “find me” signals during apoptosis (Chekeni et al., 2010).
The best studied “eat me” signal is phosphatidylserine (PS), a component retained exclusively on the inner leaflet of the plasma membrane which is only exposed to the extracellular surface when cells apoptose (Fadok et al., 1992). The exposure of PS on apoptotic cells is a conserved feature demonstrated by C. elegans, Drosophila and mammals (Nagata et al., 2010). This process is caspase-dependent (Martin et al., 1996), but the exact mechanism remains controversial. PS can be bound directly by a phagocyte receptor or indirectly by binding to bridging molecules that recognize PS, thus mediating the interaction with the phagocyte receptor (Nagata et al., 2010).
Apoptotic cell engulfment is regulated by the Rho family of GTPases such as Rac1, RhoA and Rab5 (Nakaya et al., 2006). Cells are then converted into their basic building blocks: amino acids, nucleotides, fatty acids and monosccharides in the lysosomes. Engulfment is not merely the concluding step of cell death, but also plays a decision-making role in cell death (Li and Baker, 2007). Engulfment is also required for cell competition in Drosophila. The engulfment genes -- Draper, wasp, PS receptor, mbc/dock180 and Rac1-- are required for the execution of a neighboring cell..
A promising avenue for future research will be to examine the engulfment/cell-competition process in cancer (Moreno, 2008). As tumors are comprised of cells with distinct genotypes, it is highly attractive to speculate that they can out-compete wild-type cells. Interestingly, in Drosophila, elimination of oncogenic neighbors is mediated by a JNK-dependent engulfment mechanism (Ohsawa et al., 2011).
Non-apoptotic Programmed Cell Death
Although apoptosis represents the most prominent and best studied mode of PCD in development, it is clear that alternative mechanisms exist (Yuan and Kroemer, 2010). In C. elegans, nearly all PCD is achieved by apoptosis, but death of the linker cell occurs in a caspase-independent fashion (Abraham et al., 2007). In addition, when apoptosis of mammalian cells is blocked cells often die by other mechanisms (Yuan and Kroemer, 2010). However, the importance of these alternative pathways during normal development remains to be critically examined. In addition, as many of these experiments utilize caspase inhibitors to prevent apoptosis, it is not clear whether complete inhibition of caspases has been achieved, and to what extent low caspase activity may compromise cell function.
Historically two types of PCD were defined based on morphological criteria: type I cell death known today as apoptosis (detailed above), and type II, now often referred to as autophagic cell death. Although necrosis has been traditionally seen as a passive form of cell death, proteins that regulate this process have been identified recently. This form of cell death is termed “necroptosis”, and it can be inhibited by a small molecule inhibitor necrostatin-1 (Nec-1) (Yuan and Kroemer, 2010).
Autophagy is a catabolic process that disposes of various cytoplasmic components, including protein aggregates and organelles. These components are marked for autophagy and then engulfed by autophagosomes, which fuse with lysosmes to be degraded. This process has been extensively studied in the yeast Saccharomyces cerevisiae in response to starvation. In this context, it protects the cell by recycling of its content. This is dependent upon a large group of ATG (autophagy-related) genes, which are conserved from yeast to human (Nakatogawa et al., 2009). Under most conditions, autophagy is a survival mechanism that can sustain cell viability for weeks in response to growth factor withdrawal or nutrient deprivation (Levine and Yuan, 2005). However, studies on the metamorphic death of Drosophila salivary glands suggest that autophagy contributes to the programmed death of this tissue (Berry and Baehrecke, 2007). At the same time, several apoptotic genes, including caspases participate in the death of salivary glands as well, and the exact contribution of each mechanism awaits further clarification (Lee et al., 2003). Another system involving both caspase activation and autophagy is the death of Drosophila larval midgut cells (Denton et al., 2009).
In mice, some experiments suggest that autophagy can serve as an alternate killing mechanism in situations where the apoptotic program cannot be executed. Mouse embryonic fibroblasts (MEFs) isolated from BAX−/−BAK−/− mice are resistant to a variety of apoptotic inducers but can undergo cell death in an autophagy-mediated manner (Maiuri et al., 2007). Knockdown of Beclin/ATG6 reduces cell death in BAX−/−BAK−/− MEFs (Shimizo et al., 2004). Together, these observations link caspases and autophagy, and one of the important questions in the field is understanding the mechanistic link between apoptosis and autophagy.
Non-apoptotic roles of caspases
The first caspase to be characterized, caspase-1 (then termed “interleukin converting enzyme”, ICE) was studied for its role in processing of the inflammatory cytokine proIL-1β into mature IL-1β (Thornberry et al., 1992). Therefore, it should not come as a surprise that some caspases have functions outside apoptosis. However, what was somewhat unexpected is that apoptotic effector caspases, together with their upstream regulators normally used in apoptotic cell death (e.g., Dronc/caspase-9, Apaf-1, and cytochrome C), are also used to eliminate portions of a cell, sculpting or dramatically altering cyto-architecture.
Caspases in immunity
The link between apoptosis and the immune response is well conserved among metazoans. For example, when C. elegans are fed Salmonella typhimurium, the bacteria inhabit the intestine, triggering cell death in the gonad and untimely death of the organism. Although ced-3 or ced-4 mutant worms develop normally, they are hypersenstive to S. typhimurium mediated killing, suggesting that that these apoptotic proteins play an important role in defending worms against pathogens (Aballay and Ausubel 2001).
In Drosophila, the innate immune system is comprised of two signaling networks. The Toll pathway recognizes gram-positive bacteria and fungi, and the IMD (immune deficiency) pathway is responsible for responding to gram-negative bacteria (Ferrandon et al., 2007). The IMD signaling cascade activates the NF-κB-related transcription factor Relish, an essential regulator of antimicrobial peptide gene induction. Activation of Relish requires phosphorylation by the Drosophila IκB kinase (IKK) and then cleavage by the death-related ced-3/Nedd2-like protein (DREDD), an orthologue of caspase-8 (Ertürk-Hasdemir et al., 2009). DREDD not only cleaves Relish but also activates the IKK complex, functioning as a key component of the defense response (Ertürk-Hasdemir et al., 2009). It is therefore, not surprising that mutant dredd flies are highly susceptible to E. Coli infection failing to produce antimicrobial peptides (Leulier et al., 2002).
In mammals, external death stimuli, such as those generated by bacterial infections, trigger the formation of to distinct caspase complexes the DISC, death inducing signaling complex, and the inflammasome, which is essential for the maturation of cytokines (Mace and Riedl 2010). When a death ligand, such as Fas, binds its receptor, the death receptors oligomerize and subsequently recruit FADD and initiator procaspase-8 or -10, forming the DISC (Yu and Shi, 2008). Originally described for its role in mediating cell death, recent data also uncover non-apoptotic functions for the DISC complex, demonstrating that caspase-8 functions are conserved between mammals and flies. Similar to DREDD, caspase-8 associates with the IKK complex and induces NF-κB transcriptional activity (Lemmers et al., 2007). In addition, two independent groups demonstrate that caspase-8 regulates the immune response, as deficient mice were unable to clear a viral infection or efficiently resist liver colonization of listeria monocytogenes (Salmena et al. 2003; Ben Moshe et al., 2007).
Some caspases regulate the immune system by promoting the maturation of inflammatory cytokines. Caspase-1, also known as Interleukin-1β converting enzyme or ICE, is the mammalian homologue of ced-3 and is the founding member of the pro-inflammatory caspase family (Miura, 2011). Originally known for its ability to regulate the conversion of pro-interleukin1β (IL-1β) into mature IL-1β, we now know that it regulates other cytokines, such as IL-18 and IL-33 (Miura, 2011; Yuan, 1993). In addition to caspase-1, caspases-11 and -12 act as pro-inflammatory caspases in mice; caspases-4 and -5 carryout this function in humans (Martinon and Tschopp, 2004). Mice deficient for either caspase-1 or caspase-11 display a remarkably similar phenotype. They are resistant to LPS induced shock and exhibit impaired IL-1 production in this context (Wang et al., 1998). Implying the need for a caspase complex to facilitate IL-1β, these two caspases can hetro-dimerize (Yi and Yuan, 2009). The complex that activates caspase-1 in response to pathogens is named the inflammasome. To date, four distinct inflammasomes have been described. The NLRP3 inflammasome, which has been most extensively characterized is composed of an NLRP3 scaffold, the ASC adaptor, and caspase-1 and responds to a variety of pathogens. In this context, autocleavage of pro-caspase-1 is stimulated, leading to the production of the active p10/p20 tetramer, which processes immature cytokines and generate active molecules (Schroder and Tscopp, 2010).
Cellular remodeling by caspases
In mammals, caspase-1 regulates the maturation and release of pro-inflammatory cytokines, as well as the secretion of FGF-2 and thioredoxin (Keller et al., 2008). Secretion of these caspase-1 mediated paracrinic and autocrinic factors can increase cell proliferation, indicating that caspase-1 might have a crucial role in homeostasis and reconstruction of tissues under stress or damage conditions. In this regard, it is noteworthy that caspases can induce mitogenic pathways, thereby facilitating proper wound repair and regeneration. During apoptosis, caspase activity causes rapid and complete destruction of the entire cell. This lethal activity can be refocused and restrained for the specific demolition of particular cellular structures during cellular remodeling and differentiation. Although distinct from apoptotic cell death, there are striking morphological and biochemical similarities and this process can be viewed as sub-cellular apoptosis.
Enucleation
The nucleus is a critical target in apoptosis, and elimination of the nucleus is also critical for the terminal differentiation of certain cell types, including megakaryocytes, erythrocytes keratinocytes and epithelial cells in the lens. The vertebrate lens functions to focus incoming light, which is possible by virtue of its transparency. During embryonic development lens fibers achieve transparency by degrading all membrane bound organelles in the center of the lens (Feinstein-Rotkopf and Arama, 2009). The morphological features of this terminal differentiation process highly resemble that of apoptosis, demonstrating chromatin condensation, nuclear pore clustering, and staining for TUNEL (Feinstein-Rotkopf and Arama, 2009). The enucleation process can be blocked with caspase inhibitors, and overexpression of Bcl-2 in both mice and chicken lenses resulted in retention of the nuclei (Feinstein-Rotkopf and Arama, 2009). However, mice deficient for caspase-3, 6, or 7 as well as mice lacking both caspase-3 and -6 do not demonstrate lens defects, indicating that no single executioner caspase (nor a combination of caspase-3 and 6) is required for organelle loss (Zandy et al, 2005). Interestingly, caspase-3 activity has been found to be required for the maintenance of lens transparency, as caspase-3 null mice get cataracts (Zandy et al, 2005). Given the functional redundancy between caspases 3 and 7, it will be interesting to investigate lens differentiation in these animals. Limiting this type of analysis, however, the inactivation of both of these caspases prevents mitochondrial and death receptor-mediated apoptosis, and these double mutant animals die immediately after birth due to defective cardiac development (Lakhani et al., 2006).
Another epithelial cell type that relies on a caspase-mediated process for enucleation is the keratinocyte, which forms the major component of the skin epidermis. The epidermis undergoes constant cell turn over with the keratinocytes in the basal layer being continuously pushed away from the basement membrane toward the outer differentiated layers. Upon detachment from the basement membrane, keratinocytes withdraw from the cell cycle and undergo a cornification, a specialized form of differentiation. This process is typified by expression and processing of caspase-14, which in contrast to ubiquitously expressed caspases, is only expressed in the differentiating and cornifying layers of the epidermis. Caspase-14 has been found to localize to the nuclear remnants of corneocytes and is associated with the nucleus in the precursor granular layer. Although these findings support the hypothesis that caspase-14 is required for nuclear degradation, caspase-14 deficient mice show no defects in cornification (Denecker et al., 2007).
Red blood cells and megakaryocytes also loose their nuclei as they mature. Erythroblasts mature into anucleate red blood cells in a process termed erythropoiesis, which also requires caspase activity. Caspases -2, -3 and -9 have been shown to be active during early differentiation, and inhibitors of these caspases interfere with the maturation of erythroid progenitors (Lamkanfi et al., 2007). These caspases cleave nuclear proteins Lamin B, PARP and Acinus during erythrocyte differentiation (Zermati et al., 2001). Similarly, the differentiation of megakaryocytes into mature enucleate platelets requires activation of caspases-3 and -9 (De Botton et al., 2002).
Spermatogenesis
In insects and mammals, spermatogenesis yields mature (yet short-lived) germ cells with limited cytoplasm and very few organelles. In Drosophila, this process is associated with the appearance of several apoptotic markers and caspase activation (Arama et al., 2003). Inhibition of caspase activity interferes with the removal of cytoplasm and causes sterility (Arama et al., 2003; Huh et al., 2004), and caspase activation in this system strictly depends on a testis-specific cytochrome c gene, cyt-c-d. Mutations in cyt-c-d cause male-sterility due to impaired Dronc activation, (Arama et al., 2006). Other components of the apoptosome complex, ark and dronc, are also involved in caspase activation during spermatogenesis (Huh et al., 2004; Arama et al., 2006). Finally, mutations in the giant IAP-like protein, dBruce, cause nuclear degeneration spermatid death, indicating that this protein plays a role in protecting the nucleus from unwanted caspase activity (Arama et al., 2003).
During spermatogenesis, regulation of apoptosis and control of non-apoptotic functions of caspases relies on the UPS. (Bader and Steller, 2010). A testis-specific ubiquitin ligase complex consisting of Cullin-3, Roc1b and Klhl10, regulates caspase activation in spermatids by targeting dBruce for local degradation (Arama et al., 2007; Kaplan et al., 2010). A recently identified inhibitor of this Cullin-3 complex, called Soti, competes with dBruce for biding to Klhl10, and Soti mutants exhibit elevated levels of active effector caspase. Interestingly, Soti protein is distributed as a subcellular gradient, in the inverse direction of caspase activation. This provides a mechanism to explain how spatial regulation of caspases can be achieved to drive differentiation instead of cell death execution (Kaplan et al., 2010).
Another ubiquitin E3-ligase complex critical for caspase activation in spermatids contains the Cullin-1 and the F-box protein, Nutcracker (Bader et al., 2010). Surprisingly, Nutcracker is not only required for caspase activation but also regulates proteasome activity (Bader et al., 2011). In particular, Nutracker promotes the stability of DmPI31, which in turn serves as an activator of the 26S proteasome. Proteaseome function is required for sperm differentiation and has been previously linked to the apoptotic pathway (Zhong and Belote, 2007). Taken together, these findings reveal the coordinate use of two major proteolytic systems – caspases and the 26S proteasome – to localized cell demolition and the removal of unwanted organelles.
In mammalian spermatogenesis, similar to Drosophila, caspase-3 is also activated during late stages of spermatogenesis (Kissel et al., 2005). Targeted deletion of the Sept4 locus, which encodes the IAP-antagonist ARTS protein, causes male infertility and structural abnormalities in sperm, including defects in the bulk removal of cytoplasm (Kissel et al., 2005). Furthermore, like in Drosophila, mammalian KLHL10 interacts with Cullin-3 and is exclusively expressed in spematids (Wang et al., 2006; Yan et al., 2004). Inactivation of KLHL10 in the mouse results in complete sterility, with spermiogenesis arrested during the elongating stage. Further supporting a conserved function for caspases in spermatid remodeling, a high frequency of mutations in KLHL10 is associated with human male infertility (Yan et al., 2004; Yatenesko et al., 2006). A direct role for KLHL10 in caspase regulation remains to be established in mammals. Therefore, it appears that caspases play a role in remodeling of spermatids that has been conserved in evolution from flies to humans.
Neurite pruning
Pruning of axon and dendrites is a crucial process that sculpts neuronal connections during development as it removes excessive or inaccurate projections without resulting in the death of the cell (Luo and O’leary, 2005). During insect metamorphosis, larval neurons undergo massive pruning via a multistep process of that includes destabilization of the cytoskeleton, fragmentation and then clearance of cellular remnants (Awasaki and Ito, 2004). Several groups have demonstrated that caspases promote the removal of dendrites during Drosophila metamorphosis. In Dronc deficient flies, pruning of dendrites is suppressed, with ectopic branches present after puparium formation (Williams et al., 2006). Furthermore over-expression of DIAP1, p35, or a dominant-negative form of Dronc resulted in suppression of pruning (Williams et al., 2006). Modulation of this process requires the ubiquitin-proteasome system (UPS) UPS promotes the degradation of Diap1 to unleash Dronc activity (Kuo et al., 2006). In addition, a ubiquitin-selective chaperone, VCP, binds Diap1 in a ubiquitin and BIR domain dependent manner, facilitating Diap1 degradation (Rumpf et al., 2011). Inhibition of VCP leads to elevated Diap1 levels, reduced caspase activation, and impairs the dendritic pruning process. Finally, inactivation of the Drosophila effector caspase Drice can also result in suppression of branch removal (Schoenmann et al., 2010).
In the mouse, effector caspases are also used for neurite pruning during development (Nikolaev et al., 2009). The death receptor DR6 protein is localized to both the cell bodies and axon of many neurons. Significantly, blocking DR6 function delays pruning of sensory axons in vitro and retinocollicular axons in vivo. Furthermore, chemical inhibition of Bax and knockdown of caspase-6 were also shown to inhibit sensory axon degeneration in Campenot chambers (Nikolaev et al., 2009). Remarkably, this study also showed that the amyloid precursor protein (APP), a protein widely studied for its role in Alzheimer’s Disease, serves as a regulated ligand of DR6 in this system. In this context, it collaborates with DR6 to initiate caspase activation, which may differentially trigger axonal degeneration and neuronal cell death. These findings reveal a mechanism that may contribute to shaping neuronal circuits during development and in adult plasticity, and they raise the possibility that abnormal regulation of this process may contribute to Alzheimer’s disease.
Caspases in learning and memory
There is also emerging evidence that caspases serve a role in learning and memory. In the zebra-finch auditory forebrain, the concentration of active caspase-3 is highly induced within minutes after exposure to recorded birdsong. Furthermore, caspase-3 is present in the dendritic spines in an inactive state through interaction with XIAP and is required for memory consolidation. Birds were repetitively trained with a song stimulus, testing the habitual response the following day, employing a habituation marker. In the control placebo group birds acknowledged the trained song. In striking contrast, the experimental group that was treated with a cell-permeable caspase-3 inhibitor did not recognize the song, reacting as if it were novel (Huesmann et al., 2006). These results, combined with the aforementioned studies on neuronal remodeling, lead to the obvious hypothesis that caspase activity may be important for the structural modifications required for memory.
Caspases may facilitate learning and memory by influencing the trafficking and internalization of neurotransmitter receptors. Many forms of learning and memory require experience-dependent synaptic adjustments in the hippocampus (Bateup and Sabatini, 2010). For example, the NMDA receptor-dependant synaptic modifications such as long–term potentiation (LTP) and long-term depression (LTD), correlate with an increase or decrease in the number of receptors, respectively. LTD occurs mainly by removal of AMPA receptors from the post-synaptic membrane. Neurons in the hippocapmus, an area of the brain critical for learning and memory, the activity of initiator and executioner caspases is essential for LTD and AMPA receptor internalization (Li et al., 2010b). Blocking caspase activity with caspase inhibitors, overexpression of XIAP or Bcl-2 inhibits LTD and AMPA receptor internalization. Furthermore, LTD is abolished in the hippocampus of caspase-3 deficient mice (Li et al., 2010b). In a subsequent study, caspase-3 was shown to be activated by BAD and BAX, but not Bid. BAD is activated in a transient and moderate fashion and BAX does not translocate to the mitochondria. This appears to fine-tune the activation intensity of caspase-3, thereby favoring LTD instead of cell death (Jiao and Li, 2011). Further studies are needed to establish the molecular targets linking caspases with AMPA receptor internalization, and it remains to be seen whether this caspase-dependent mechanism is responsible for the apparent effect on learning and memory in zebra-finches.
Role of caspases in cell differentiation
Besides promoting the destruction of specific cellular compartments, caspases also promote cell differentiation in more subtle ways. One example is for the specification of external sensory organs in Drosophila. Flies mutant for Ark, Dronc or cyto-c-d, contain a number of additional bristles, which are part of a mechano-sensory organ, (Chew et al., 2004; Kanuka et al., 2005; Mendes et al., 2006). In this system, caspases promote activation of Sgg46, the Drosophila orthologue of gsk3-β, an antagonist of the Wnt pathway (Kanuka et al., 2005). Activation of sgg46 is required for negative regulation of Wg signaling, indicating that caspases can regulate neural development by antagonizing Wg signaling. In a screen for caspase suppressors, dmIKKε, the Drosophila ortholugue of IKKε or NAK was identified (Kuranaga et al., 2006). dmIKKε was found to regulate Dronc activity by promoting phosphorilation and subsequent degradation of Diap1. Silencing of dmIKKε resulted in attenuation of caspase-3 activity in SOP cells and the appearance of additional macrochaetes (Kuranaga et al., 2006). Furthermore, mammalian IKKε plays a conserved role in phosphorilation-dependant degradation of XIAP, indicating a similar role in caspase regulation in mammals (Kuranaga et al., 2006).
Caspases have also been implicated in skewing monocyte differentiation towards to macrophages rather than dendritic cells (Sordet et al., 2002). As monocytes differentiate into macrophages, they activate both caspase-9 and -3 but do not exhibit apoptotic features. Inhibition of caspase activity with p35, zVAD, or over expression of Bcl-2 inhibited the macrophage differentiation (Sordet et al., 2002). Furthermore, a screen for caspase substrates during cell differentiation processes, uncovers 38 differentially expressed caspase targets (Cathelin et al., 2006). Caspase-8 may also play a role in this process as its deletion leads to arrest of differentiation into macrophages (Kang et al., 2004). Moreover, caspase-8 binds and cleaves RIP1, preventing sustained activation of NF-κB in monocytes undergoing macrophagic differentiation (Rebe et al., 2007).
Caspases also play crucial, non-apoptotic roles in the differentiation of various stem cells (SCs). For example, caspase-3 plays a crucial role in osteogenic differentiation of bone marrow stromal SCs, neural SC differentiation, differentiation of embryonic SCs, as well as in regulating the differentiation and proliferation in hematopoietic SCs (Feinstein-Rotkopf and Arama, 2009).
Apoptotic cells can stimulate proliferation, wound healing and tissue regeneration
Regeneration, a process of re-growth or repair, equips animals with the capacity to maintain homeostasis, even after severe injury. Remarkable examples for regeneration can be seen in various organisms. In Hydra, decapitation leads to the production of a new head, and planaria have the ability to regenerate complete individuals from small body fragments (Birnbaum and Sanchez Alvarado, 2008). In Drosophila, imaginal discs can give rise to normal wings, legs and eyes even after more than 50% of their cells have been killed (Haynie and Bryant 1976). In mammals, the regenerative capacity of the liver enables restoration of full organ mass within a very short time frame, even after 70% of the liver has been removed (Taub, 2004). Depending on the type of tissue damage, the regenerative process includes several steps, including wound repair, formation of highly proliferative blastema cells, differentiation and patterning (Gurtner et al., 2008). Work in many model systems reveals that apoptosis may be the driving force behind regeneration. Specifically, the proliferative aspect of regeneration, including blastema formation, is stimulated by signals from apoptotic cells. This phenomenon is termed “apoptosis-induced compensatory proliferation” (Bergmann and Steller, 2010).
Cells undergoing apoptosis in response to stress or injury can stimulate the proliferation of neighboring cells. A series of experiments in Drosophila first demonstrated this phenomenon (Pérez-Garijo et al., 2004; Ryoo et al., 2004). These studies had to overcome a fundamental problem in studying apoptotic-stimulated cells, namely their relatively short life span and rapid clearance. To circumvent this problem, the potent caspase inhibitor p35 was used to block the execution of apoptosis and sustain cells in an “undead” state. Under these conditions, the apoptotic program is induced but cannot be executed. Because p35 specifically inhibits effector caspases, Dronc is active in “undead cells” and is able to perform non-apoptotic functions. This system allowed the identification of genes and mechanisms that govern compensatory proliferation. p53 and Dronc collaborate to induce compensatory proliferation and stimulate blastema formation (Wells et al., 2006). Because the pro-apoptotic genes reaper and hid are direct transcriptional targets of p53, a positive feedback loop involving Dronc, Reaper and Hid proteins may operate in this system. Such a mechanism underscores the similarities between compensatory proliferation and hyperplastic overgrowth.
The JNK signaling plays a critical role in compensatory proliferation and wound healing in Drosophila (Ryoo et al., 2004; Bosch et al., 2005). Different reports have indicated that JNK is either a downstream target of Dronc, or that it acts independently from the apoptosis program (Ryoo et al., 2004; Pérez-Garijo et al., 2009). A recent study reconciles these different observations. It, demonstrates that p53 and JNK act upstream and downstream of the pro-apoptotic RHG genes and the initiator caspase Dronc, establishing a positive feedback loop that amplifies initial apoptotic stimuli and facilitates the apoptotic stress response (Shlevkov and Morata, 2011).
In addition to the p53 and JNK pathways, compensatory proliferation requires mitogenic signals. “Undead cells” secret mitogens/morphogens, such as Wingless (Wg, Wnt orthologue) and Decapentaplegic (Dpp, TGF-β ortholuge), stimulating proliferation of surrounding cells (Fig. 4A) (Ryoo et al., 2004). These factors play key signaling roles during early development, promote the self-renewal of stem cells, and stimulate tissue regeneration in vertebrates and insects (Bergmann and Steller, 2010). These mechanisms are not an artifact of p35-overexpression: active JNK signaling induces transcription of wingless in “normal” apoptotic cells (Ryoo et al., 2004), and Wg and JNK are required for regeneration in p35-independent paradigms (Fig. 4B) (Bergantiños et al., 2010; Smith-Bolton et al., 2009). For example, in the differentiating Drosophila retina, Hehdgehog (Hh) signaling is required for apoptosis-induced compensatory proliferation. In response to stress, post-mitotic photoreceptor neurons secrete Hh, stimulating the proliferation of nearby cells (Fig. 4C, Fan and Bergmann, 2008). Contrary to the Wg, Dpp, and JNK pathways, which require p53 and Dronc, hh activation occurs downstream of the effector caspases DrICE and Dcp-1.
Figure 4. Apoptosis induced compensatory proliferation (CP) in various organisms.

In different model organisms apoptosis and caspase activity have been observed to induce secretion of mitogenic factors, thereby promoting hyperplastic overgrowth or tissue regeneration. Mitogenic factors are indicated in pink and question marks indicate uncertainty. A. In Drosophila inhibition of caspases by P35 renders cells in an “undead” state unable to complete apoptosis. This results in the activation of p53 and JNK, triggering the release of the Wg and Dpp mitogens, thereby promoting hyperplasic overgrowth. B. In Drosophila, temporal and spatial apoptosis (in-dependently of p35) induces tissue regeneration via compensatory proliferation by secretion of Wg. A dP53/JNK positive feedback loop is essential for the apoptotic response. C. In Drosophila differentiating neurons induce a different compensatory proliferation pathway, via hedghog (Hh), in a manner requiring both DrICE and Dcp-1. Hh stimulates the proliferation of non-neuronal cells. D. In Hydra, head regeneration post midgut bisection is dependent upon caspase activity, where apoptotic cells secrete Wnt3 promoting CP. E. In newts and planaria, amputation is characterized by apoptosis and caspase activity in the wound site. However it still unknown whether this apoptotic response is responsible for the release of Wnt and Hh. F. In Xenopus, amputation of the tail results in caspase activity, whereas inhibition of caspase-9 and 3 prohibits cell proliferation and the regenerative process. It remains to established whether this form of CP is mediated by Wnt signaling. G. In mice, wound repair and liver regeneration is dependant upon caspase -3 and -7 which are necessary for proper induction of these processes. Caspase-3 mediates the proteolytic processing of iPLA2, which in turn produces archidonic acid the precursor of PGE2, a known stimulator of stem cell proliferation, tissue regeneration and wound repair.
Compensatory proliferation also occurs in other organisms including Hydra, Xenopus, Planaria, newts and mice. In the freshwater polyp Hydra, apoptosis is both necessary and sufficient for head regeneration post mid-gastric bisection (Chera et al., 2009). Similar to Drosophila, caspases stimulate release of Wnt3, promoting proliferation and facilitating regeneration (Fig. 4D). Although planarians and newts also demonstrate massive apoptosis at the site of amputation, the molecular connections with regeneration await discovery (Fig. 4E). In the Xenopus tadpole, a large number of apoptotic cells are seen in the nascent regeneration bud within 12 hours post amputation (Tseng et al., 2007), and apoptosis is important for the initiation of tail regeneration. Although Wnt signaling is crucial for regeneration in Xenopus, it remains to be seen whether it is secreted by the dying cells (Fig. 4F). Studies in mice also support a link between apoptosis and regeneration. For example, mice lacking either caspase-3 or 7 have impaired skin wound healing and liver regeneration. Interestingly, a downstream target of these caspases is Prostaglandin E2, known to work in tandem with the Wnt pathway to promote stem cell maintenance and tissue regeneration (Fig. 4G, Li et al., 2010a; Goessling et al., 2009).
These studies of apoptosis-induced compensatory proliferation illustrate that both initiator and executioner caspase influence the release of mitogens from stressed or injured cells, thereby promoting regeneration. This body of work also reveals striking similarities between tissue regeneration and cancer. Malignant tumors often develop at sites of chronic injury, and tissue injury has an important role in the pathogenesis of malignant disease (Schäfer and Werner, 2008). If compensatory proliferation operates in tumor cells, inducing apoptosis in cancer cells may actually stimulate proliferation of surviving cells. Even if cancer therapies kill the majority of cells and decrease tumor size, mitogenic signaling may contribute to relapse, secondary tumors, or metastases by stimulating proliferation of a small number cells, capable of seeding a new tumor. In support of this scenario, cancer cells demonstrate some level of apoptotic resistance, a feature they share with artificially generated “undead” cells.
Apoptosis and stem cells
Stem cells (SCs) are defined by their virtually unlimited proliferative potential and multi-lineage differentiation capacity and are required throughout animal development. In tissues, SCs are maintained for long periods of time, undergoing multiple rounds of self-renewal. As such, they are at risk of accumulating potentially deleterious mutations. The combined consequence of their longevity and proliferation potential enables the propagation of these mutations to downstream progeny in a manner that could lead to oncogenesis (Rossi et al., 2008).
The hematopoietic system provides clear examples of how changes in programmed cell death in a small number of hematopoietic stem cells (HSCs) can lead to differentiation defects and increased cancer risk. Over-expression of BCL-2 increases of HSC number and enhancement in HSC repopulation potential, while displaying resistance to various chemotherapeutic agents (Domen et al., 2000; Domen and Weissman, 2003). Another member of the BCL-2 family, Mcl-1, is important for HSC regulation. Silencing of Mcl-1 reduced the self-renewal of human HSC in vivo, whereas knock down of Mcl-1 in human pluripotent SC almost completely ablated SC self-renewal (Campbell et al., 2010a). Furthermore, mice overexpressing Mcl-1 in the hematopoietic compartment develop stem/progenitor cell tumors (Cambpell et al., 2010b). BCL-2 proteins protect other SCs from apoptosis. In human embryonic SCs (ESCs), BCL-2 enhances survival, and ESCs over-expressing BCL-2 displayed normal growth in the absence of serum (Ardehali et al., 2011).
HSC survival also requires additional layers of regulation. Mice lacking the IAP antagonist, ARTS, develop spontaneous hematopoietic malignancies and have increased number of functional hematopoietic stem and progenitor cells (HSPCs) in the bone marrow. These phenotypes can be suppressed by inactivation of XIAP, indicating that XIAP is a physiological target for the pro-apoptotic activity of ARTS (García-Fernández et al., 2010). p53 is also required for HSC survival and homeostasis. Upon irradiation, HSPCs lacking p53 have a selective advantage, which leads to long term clone expansion and lymphoma development (Marusyk et al., 2010). Interestingly, p53-mediated HSPC cell competition depends on the relative, not absolute, level of p53 in competing cells (Bondar and Medzhitov et al., 2010).
Given the potentially dire consequences of accumulating mutations in SCs, these cells are thought to employ mechanisms to limit DNA damage or respond rapidly to such damage (Seita et al., 2010). One protective feature may be the quiescent state in which many SCs are retained minimizing the chance for replication errors and endogenous ROS-mediated DNA damage (Yamazaki et al., 2006; Tothova et al., 2007). This quiescent state also raises a challenge for adult SCs as the majority of DNA repair pathways are cell-cycle dependant (Mandal et al., 2011). When mice are subjected to ionizing radiation (IR), their rapidly proliferating short-lived progenitors (SLPs) are quickly eliminated by apoptosis but their HSPCs are resistant. This differential resistance requires the ATM kinase, which induces cell cycle arrest and DNA repair in HSPCs (Mohrin et al., 2010). Interestingly, both quiescent and proliferating HSPCs demonstrate equal radio-resistance but employ different types of DNA repair mechanisms. Quiescent HSPCs preferentially utilize the error prone non-homologous end joining (NHEJ) DNA repair pathway, which renders them intrinsically vulnerable to mutagenesis. Whereas, proliferating HSPCs use the high fidelity homologous recombination mechanism and have significantly decreased risk of acquiring mutations (Mohrin et al., 2010). This demonstrates that HSPC quiescence serves as a double-edged sword, which protects against endogenous stress but also renders HSPCs susceptible to genomic instability, thus contributing to pre-malignant transformation.
In striking contrast, irradiated human HSPCs isolated from umbilical cord blood exhibit significantly increased apoptosis and delayed DNA repair compared to downstream progeny. IR-induced apoptosis is blocked by knockdown of p53 (p53KD) or overexpression of BCL-2 (BCL-2OE), suggesting that the p53-BCL-2 pathway regulates SCs response to radiation and DNA damage. Despite providing similar protection from irradiation-induced apoptosis, only BCL-2OE favors self-renewal (Milyavsky et al., 2010). It remains to be seen whether these contrasting conclusions about control of apoptosis in HSCs in mice and humans are due to differences between species or the anatomical location from which the cells were isolated. Future work in this area will provide important new insights into how apoptosis regulates SC homeostasis and differentiation, and how perturbations in survival and cell death signals transform SCs into tumor SCs. Experiments that address these questions will yield better understanding of basic biology and may have practical implications for in the areas of regenerative medicine and cancer treatment.
Concluding remarks
It has been almost 40 years since Kerr, Wyllie and Currie stated: “We should now like to speculate that hyperplasia might sometimes result from decreased apoptosis rather than increased mitosis, although we emphasize that we know of no definitive studies that support such an hypothesis”. Since then, tremendous progress has been made in identifying the core mechanism underlying apoptotic cell death. At the same time, it has become clear that other forms of cell death occur under physiological conditions, and much remains to be learned about their molecular mechanisms and connections to apoptosis. Also, apart from a few specific models, we still know very little about precisely how cells are selected for death in vivo, and how different signaling pathways with sometimes opposing functions are integrated in the decision between cell death and survival under physiological conditions. Another area of surprising complexity is the rich variety of signals released by apoptotic cells. In addition to signals mediating the attraction and recognition of phagocytes, apoptotic cells also secrete factors that stimulate cell proliferation, differentiation and response of competent neighboring cells. Furthermore, core cell death proteins, including apoptotic effector caspases, can also function in diverse non-apoptotic processes, such as the elimination of unwanted cellular structures and organelles. A major unresolved question is how the potentially lethal activity of effector caspases is directed to specific sub-cellular compartments without triggering a full-blown apoptotic response. A better understanding of cell death regulation is likely to provide the basis for treating a variety of human disorders associated with abnormal cell death, such as cancer, auto-immunity, viral infection, AIDS, sepsis, ischemia, neuro-degeneration, impaired healing and tissue regeneration.
Figure 5. Non-apoptotic function of caspases.
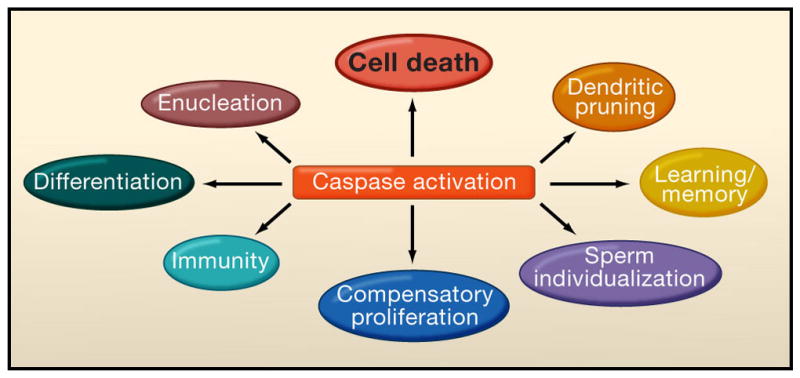
In addition to their central role in apoptosis, caspases are involved in many other vital process including: differentiation, enucleation, pruning of axons and dendrites, sperm differentiation, immunity, compensatory proliferation and even learning and memory.
It has been almost 40 years since Kerr, Wyllie and Currie stated: “We should now like speculate that hyperplasia might sometimes result from decreased apoptosis rather than increased mitosis, although we emphasize that we know of no definitive studies that support such an hypothesis”. Since then, tremendous progress has been made identifying the core mechanism underlying apoptotic cell death. At the same time, it has become clear that other forms of cell death occur under physiological conditions, and much remains to be learned about their molecular mechanisms and connections apoptosis. Also, apart from a few specific models, we still know very little about precisely how cells are selected for death in vivo, and how different signaling pathways with sometimes opposing functions are integrated in the decision between cell death and survival under physiological conditions. Another area of surprising complexity is rich variety of signals released by apoptotic cells. In addition to signals mediating attraction and recognition of phagocytes, apoptotic cells also secrete factors that stimulate cell proliferation, differentiation and response of competent neighboring cells Furthermore, core cell death proteins, including apoptotic effector caspases, can also function in diverse non-apoptotic processes, such as the elimination of unwanted cellular structures and organelles. A major unresolved question is how the potentially lethal activity of effector caspases is directed to specific sub-cellular compartments without triggering a full-blown apoptotic response. A better understanding of cell death regulation is likely to provide the basis for treating a variety of human disorders associated with abnormal cell death, such as cancer, auto-immunity, viral infection, AIDS, sepsis, ischemia, neuro-degeneration, impaired healing and tissue regeneration.
Acknowledgments
We would like to apologize to all our colleagues whose work we could not adequately present here due to space constraints. We would like to thank Yaniv Fuchs for the illustrations. HS is an Investigator of the Howard Hughes Medical Institute and was supported by NIH grant R01GM60124.
References
- Aballay A, Ausubel FM. Programmed cell death mediated by ced-3 and ced-4 protects Caenorhabditis elegans from Salmonella typhimurium-mediated killing. Proc Natl Acad Sci U S A. 2001;98:2735–2739. doi: 10.1073/pnas.041613098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelwahid E, Pelliniemi LJ, Jokinen E. Cell death and differentiation in the development of the endocardial cushion of the embryonic heart. Microsc Res Tech. 2002;58:395–403. doi: 10.1002/jemt.10159. [DOI] [PubMed] [Google Scholar]
- Abdelwahid E, Yokokura T, Krieser RJ, Balasundaram S, Fowle WH, White K. Mitochondrial disruption in Drosophila apoptosis. Dev Cell. 2007;12:793–806. doi: 10.1016/j.devcel.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Abraham MC, Lu Y, Shaham S. A morphologically conserved nonapoptotic program promotes linker cell death in Caenorhabditis elegans. Dev Cell. 2007;12:73–86. doi: 10.1016/j.devcel.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Abraham MC, Shaham S. Death without caspases, caspases without death. Trends Cell Biol. 2004;14:184–193. doi: 10.1016/j.tcb.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Abrams JM, White K, Fessler LI, Steller H. Programmed cell death during Drosophila embryogenesis. Development. 1993;117:29–43. doi: 10.1242/dev.117.1.29. [DOI] [PubMed] [Google Scholar]
- Adams JM, Cory S. Apoptosomes: engines for caspase activation. Curr Opin Cell Biol. 2002;14:715–720. doi: 10.1016/s0955-0674(02)00381-2. [DOI] [PubMed] [Google Scholar]
- Arama E, Agapite J, Steller H. Caspase activity and a specific cytochrome C are required for sperm differentiation in Drosophila. Dev Cell. 2003;4:687–697. doi: 10.1016/s1534-5807(03)00120-5. [DOI] [PubMed] [Google Scholar]
- Arama E, Bader M, Rieckhof GE, Steller H. A ubiquitin ligase complex regulates caspase activation during sperm differentiation in Drosophila. PLoS Biol. 2007;5:e251. doi: 10.1371/journal.pbio.0050251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arama E, Bader M, Srivastava M, Bergmann A, Steller H. The two Drosophila cytochrome C proteins can function in both respiration and caspase activation. EMBO J. 2006;25:232–243. doi: 10.1038/sj.emboj.7600920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardehali R, Inlay MA, Ali SR, Tang C, Drukker M, Weissman IL. Overexpression of BCL2 enhances survival of human embryonic stem cells during stress and obviates the requirement for serum factors. Proc Natl Acad Sci U S A. 2011;108:3282–3287. doi: 10.1073/pnas.1019047108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avallone B, Balsamo G, Trapani S, Marmo F. Apoptosis during chick inner ear development: some observations by TEM and TUNEL techniques. Eur J Histochem. 2002;46:53–59. doi: 10.4081/1654. [DOI] [PubMed] [Google Scholar]
- Awasaki T, Ito K. Engulfing action of glial cells is required for programmed axon pruning during Drosophila metamorphosis. Curr Biol. 2004;14:668–677. doi: 10.1016/j.cub.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Bader M, Arama E, Steller H. A novel F-box protein is required for caspase activation during cellular remodeling in Drosophila. Development. 2010;137:1679–1688. doi: 10.1242/dev.050088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader M, Benjamin S, Wapinski OL, Smith DM, Goldberg AL, Steller H. A conserved F box regulatory complex controls proteasome activity in Drosophila. Cell. 2011;145:371–382. doi: 10.1016/j.cell.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader M, Steller H. Regulation of cell death by the ubiquitin-proteasome system. Curr Opin Cell Biol. 2009;21:878–884. doi: 10.1016/j.ceb.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehrecke EH. How death shapes life during development. Nat Rev Mol Cell Biol. 2002;3:779–787. doi: 10.1038/nrm931. [DOI] [PubMed] [Google Scholar]
- Barres BA, Raff MC. Axonal control of oligodendrocyte development. J Cell Biol. 1999;147:1123–1128. doi: 10.1083/jcb.147.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateup HS, Sabatini BL. For synapses, it’s depression not death. Cell. 2010;141:750–752. doi: 10.1016/j.cell.2010.05.013. [DOI] [PubMed] [Google Scholar]
- Ben Moshe T, Barash H, Kang TB, Kim JC, Kovalenko A, Gross E, Schuchmann M, Abramovitch R, Galun E, Wallach D. Role of caspase-8 in hepatocyte response to infection and injury in mice. Hepatology. 2007;45:1014–1024. doi: 10.1002/hep.21495. [DOI] [PubMed] [Google Scholar]
- Bergantinos C, Corominas M, Serras F. Cell death-induced regeneration in wing imaginal discs requires JNK signalling. Development. 2010;137:1169–1179. doi: 10.1242/dev.045559. [DOI] [PubMed] [Google Scholar]
- Bergmann A, Agapite J, McCall K, Steller H. The Drosophila gene hid is a direct molecular target of Ras-dependent survival signaling. Cell. 1998;95:331–341. doi: 10.1016/s0092-8674(00)81765-1. [DOI] [PubMed] [Google Scholar]
- Bergmann A, Steller H. Apoptosis, stem cells, and tissue regeneration. Sci Signal. 2010;3:re8. doi: 10.1126/scisignal.3145re8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann A, Tugentman M, Shilo BZ, Steller H. Regulation of cell number by MAPK-dependent control of apoptosis: a mechanism for trophic survival signaling. Dev Cell. 2002;2:159–170. doi: 10.1016/s1534-5807(02)00116-8. [DOI] [PubMed] [Google Scholar]
- Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131:1137–1148. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum KD, Sanchez Alvarado A. Slicing across kingdoms: regeneration in plants and animals. Cell. 2008;132:697–710. doi: 10.1016/j.cell.2008.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondar T, Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010;6:309–322. doi: 10.1016/j.stem.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch M, Serras F, Martin-Blanco E, Baguna J. JNK signaling pathway required for wound healing in regenerating Drosophila wing imaginal discs. Dev Biol. 2005;280:73–86. doi: 10.1016/j.ydbio.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113:25–36. doi: 10.1016/s0092-8674(03)00231-9. [DOI] [PubMed] [Google Scholar]
- Brodsky MH, Nordstrom W, Tsang G, Kwan E, Rubin GM, Abrams JM. Drosophila p53 binds a damage response element at the reaper locus. Cell. 2000;101:103–113. doi: 10.1016/S0092-8674(00)80627-3. [DOI] [PubMed] [Google Scholar]
- Campbell CJ, Lee JB, Levadoux-Martin M, Wynder T, Xenocostas A, Leber B, Bhatia M. The human stem cell hierarchy is defined by a functional dependence on Mcl-1 for self-renewal capacity. Blood. 2010a;116:1433–1442. doi: 10.1182/blood-2009-12-258095. [DOI] [PubMed] [Google Scholar]
- Campbell KJ, Bath ML, Turner ML, Vandenberg CJ, Bouillet P, Metcalf D, Scott CL, Cory S. Elevated Mcl-1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells, and enhances drug resistance. Blood. 2010b;116:3197–3207. doi: 10.1182/blood-2010-04-281071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathelin S, Rebe C, Haddaoui L, Simioni N, Verdier F, Fontenay M, Launay S, Mayeux P, Solary E. Identification of proteins cleaved downstream of caspase activation in monocytes undergoing macrophage differentiation. J Biol Chem. 2006;281:17779–17788. doi: 10.1074/jbc.M600537200. [DOI] [PubMed] [Google Scholar]
- Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chera S, Ghila L, Dobretz K, Wenger Y, Bauer C, Buzgariu W, Martinou JC, Galliot B. Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev Cell. 2009;17:279–289. doi: 10.1016/j.devcel.2009.07.014. [DOI] [PubMed] [Google Scholar]
- Chew SK, Akdemir F, Chen P, Lu WJ, Mills K, Daish T, Kumar S, Rodriguez A, Abrams JM. The apical caspase dronc governs programmed and unprogrammed cell death in Drosophila. Dev Cell. 2004;7:897–907. doi: 10.1016/j.devcel.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Christich A, Kauppila S, Chen P, Sogame N, Ho SI, Abrams JM. The damage-responsive Drosophila gene sickle encodes a novel IAP binding protein similar to but distinct from reaper, grim, and hid. Curr Biol. 2002;12:137–140. doi: 10.1016/s0960-9822(01)00658-3. [DOI] [PubMed] [Google Scholar]
- Coucouvanis E, Martin GR. Signals for death and survival: a two-step mechanism for cavitation in the vertebrate embryo. Cell. 1995;83:279–287. doi: 10.1016/0092-8674(95)90169-8. [DOI] [PubMed] [Google Scholar]
- Crook NE, Clem RJ, Miller LK. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J Virol. 1993;67:2168–2174. doi: 10.1128/jvi.67.4.2168-2174.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Botton S, Sabri S, Daugas E, Zermati Y, Guidotti JE, Hermine O, Kroemer G, Vainchenker W, Debili N. Platelet formation is the consequence of caspase activation within megakaryocytes. Blood. 2002;100:1310–1317. doi: 10.1182/blood-2002-03-0686. [DOI] [PubMed] [Google Scholar]
- de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila myc regulates organ size by inducing cell competition. Cell. 2004;117:107–116. doi: 10.1016/s0092-8674(04)00214-4. [DOI] [PubMed] [Google Scholar]
- Denecker G, Hoste E, Gilbert B, Hochepied T, Ovaere P, Lippens S, Van den Broecke C, Van Damme P, D’Herde K, Hachem JP, et al. Caspase-14 protects against epidermal UVB photodamage and water loss. Nat Cell Biol. 2007;9:666–674. doi: 10.1038/ncb1597. [DOI] [PubMed] [Google Scholar]
- Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, Kumar S. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19:1741–1746. doi: 10.1016/j.cub.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derry WB, Putzke AP, Rothman JH. Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science. 2001;294:591–595. doi: 10.1126/science.1065486. [DOI] [PubMed] [Google Scholar]
- Domen J, Cheshier SH, Weissman IL. The role of apoptosis in the regulation of hematopoietic stem cells: Overexpression of Bcl-2 increases both their number and repopulation potential. J Exp Med. 2000;191:253–264. doi: 10.1084/jem.191.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domen J, Weissman IL. Hematopoietic stem cells and other hematopoietic cells show broad resistance to chemotherapeutic agents in vivo when overexpressing bcl-2. Exp Hematol. 2003;31:631–639. doi: 10.1016/s0301-472x(03)00084-5. [DOI] [PubMed] [Google Scholar]
- Domingos PM, Steller H. Pathways regulating apoptosis during patterning and development. Curr Opin Genet Dev. 2007;17:294–299. doi: 10.1016/j.gde.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draizen TA, Ewer J, Robinow S. Genetic and hormonal regulation of the death of peptidergic neurons in the Drosophila central nervous system. J Neurobiol. 1999;38:455–465. doi: 10.1002/(sici)1097-4695(199903)38:4<455::aid-neu2>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Eckelman BP, Salvesen GS. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J Biol Chem. 2006;281:3254–3260. doi: 10.1074/jbc.M510863200. [DOI] [PubMed] [Google Scholar]
- Edison N, Zuri D, Maniv I, Bornstein B, Lev T, Gottfried Y, Kemeny S, Garcia-Fernandez M, Kagan J, Larisch S. The IAP-antagonist ARTS initiates caspase activation upstream of cytochrome C and SMAC/Diablo. Cell Death Differ. 2011 doi: 10.1038/cdd.2011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–286. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- Ellis RE, Yuan JY, Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol. 1991;7:663–698. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- Erturk-Hasdemir D, Broemer M, Leulier F, Lane WS, Paquette N, Hwang D, Kim CH, Stoven S, Meier P, Silverman N. Two roles for the Drosophila IKK complex in the activation of Relish and the induction of antimicrobial peptide genes. Proc Natl Acad Sci U S A. 2009;106:9779–9784. doi: 10.1073/pnas.0812022106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- Fan Y, Bergmann A. Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev Cell. 2008;14:399–410. doi: 10.1016/j.devcel.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein-Rotkopf Y, Arama E. Can’t live without them, can live with them: roles of caspases during vital cellular processes. Apoptosis. 2009;14:980–995. doi: 10.1007/s10495-009-0346-6. [DOI] [PubMed] [Google Scholar]
- Ferrandon D, Imler JL, Hetru C, Hoffmann JA. The Drosophila systemic immune response: sensing and signalling during bacterial and fungal infections. Nat Rev Immunol. 2007;7:862–874. doi: 10.1038/nri2194. [DOI] [PubMed] [Google Scholar]
- Garcia-Fernandez M, Kissel H, Brown S, Gorenc T, Schile AJ, Rafii S, Larisch S, Steller H. Sept4/ARTS is required for stem cell apoptosis and tumor suppression. Genes Dev. 2010;24:2282–2293. doi: 10.1101/gad.1970110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glücksmann A. Cell deaths in normal vertebrate ontogeny. Biol Rev Camb Philos Soc. 1951;26:59–86. doi: 10.1111/j.1469-185x.1951.tb00774.x. [DOI] [PubMed] [Google Scholar]
- Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, Weidinger G, Puder M, Daley GQ, Moon RT, et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. 2009;136:1136–1147. doi: 10.1016/j.cell.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottfried Y, Rotem A, Lotan R, Steller H, Larisch S. The mitochondrial ARTS protein promotes apoptosis through targeting XIAP. EMBO J. 2004;23:1627–1635. doi: 10.1038/sj.emboj.7600155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal L, McCall K, Agapite J, Hartwieg E, Steller H. Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J. 2000;19:589–597. doi: 10.1093/emboj/19.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Grether ME, Abrams JM, Agapite J, White K, Steller H. The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev. 1995;9:1694–1708. doi: 10.1101/gad.9.14.1694. [DOI] [PubMed] [Google Scholar]
- Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, Barbour SE, Milstien S, Spiegel S. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 2008;22:2629–2638. doi: 10.1096/fj.08-107169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- Haining WN, Carboy-Newcomb C, Wei CL, Steller H. The proapoptotic function of Drosophila Hid is conserved in mammalian cells. Proc Natl Acad Sci U S A. 1999;96:4936–4941. doi: 10.1073/pnas.96.9.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hardy K, Handyside AH, Winston RM. The human blastocyst: cell number, death and allocation during late preimplantation development in vitro. Development. 1989;107:597–604. doi: 10.1242/dev.107.3.597. [DOI] [PubMed] [Google Scholar]
- Haynie JL, Bryant PJ. Intercalary regeneration in imaginal wing disk of Drosophila melanogaster. Nature. 1976;259:659–662. doi: 10.1038/259659b0. [DOI] [PubMed] [Google Scholar]
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Hengartner MO, Ellis RE, Horvitz HR. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature. 1992;356:494–499. doi: 10.1038/356494a0. [DOI] [PubMed] [Google Scholar]
- Huesmann GR, Clayton DF. Dynamic role of postsynaptic caspase-3 and BIRC4 in zebra finch song-response habituation. Neuron. 2006;52:1061–1072. doi: 10.1016/j.neuron.2006.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JR, Vernooy SY, Yu H, Yan N, Shi Y, Guo M, Hay BA. Multiple apoptotic caspase cascades are required in nonapoptotic roles for Drosophila spermatid individualization. PLoS Biol. 2004;2:E15. doi: 10.1371/journal.pbio.0020015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Jiang C, Lamblin AF, Steller H, Thummel CS. A steroid-triggered transcriptional hierarchy controls salivary gland cell death during Drosophila metamorphosis. Mol Cell. 2000;5:445–455. doi: 10.1016/s1097-2765(00)80439-6. [DOI] [PubMed] [Google Scholar]
- Jiao S, Li Z. Nonapoptotic function of BAD and BAX in long-term depression of synaptic transmission. Neuron. 2011;70:758–772. doi: 10.1016/j.neuron.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LA. Competitive interactions between cells: death, growth, and geography. Science. 2009;324:1679–1682. doi: 10.1126/science.1163862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TB, Ben-Moshe T, Varfolomeev EE, Pewzner-Jung Y, Yogev N, Jurewicz A, Waisman A, Brenner O, Haffner R, Gustafsson E, et al. Caspase-8 serves both apoptotic and nonapoptotic roles. J Immunol. 2004;173:2976–2984. doi: 10.4049/jimmunol.173.5.2976. [DOI] [PubMed] [Google Scholar]
- Kanuka H, Kuranaga E, Takemoto K, Hiratou T, Okano H, Miura M. Drosophila caspase transduces Shaggy/GSK-3beta kinase activity in neural precursor development. EMBO J. 2005;24:3793–3806. doi: 10.1038/sj.emboj.7600822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan Y, Gibbs-Bar L, Kalifa Y, Feinstein-Rotkopf Y, Arama E. Gradients of a ubiquitin E3 ligase inhibitor and a caspase inhibitor determine differentiation or death in spermatids. Dev Cell. 19:160–173. doi: 10.1016/j.devcel.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Keller M, Ruegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–831. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissel H, Georgescu MM, Larisch S, Manova K, Hunnicutt GR, Steller H. The Sept4 septin locus is required for sperm terminal differentiation in mice. Dev Cell. 2005;8:353–364. doi: 10.1016/j.devcel.2005.01.021. [DOI] [PubMed] [Google Scholar]
- Kornbluth S, White K. Apoptosis in Drosophila: neither fish nor fowl (nor man, nor worm) J Cell Sci. 2005;118:1779–1787. doi: 10.1242/jcs.02377. [DOI] [PubMed] [Google Scholar]
- Koto A, Kuranaga E, Miura M. Apoptosis ensures spacing pattern formation of Drosophila sensory organs. Curr Biol. 2011;21:278–287. doi: 10.1016/j.cub.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Martin SJ. Caspase-independent cell death. Nat Med. 2005;11:725–730. doi: 10.1038/nm1263. [DOI] [PubMed] [Google Scholar]
- Kuo CT, Zhu S, Younger S, Jan LY, Jan YN. Identification of E2/E3 ubiquitinating enzymes and caspase activity regulating Drosophila sensory neuron dendrite pruning. Neuron. 2006;51:283–290. doi: 10.1016/j.neuron.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Kuranaga E, Kanuka H, Tonoki A, Takemoto K, Tomioka T, Kobayashi M, Hayashi S, Miura M. Drosophila IKK-related kinase regulates nonapoptotic function of caspases via degradation of IAPs. Cell. 2006;126:583–596. doi: 10.1016/j.cell.2006.05.048. [DOI] [PubMed] [Google Scholar]
- Kuranaga E, Matsunuma T, Kanuka H, Takemoto K, Koto A, Kimura K, Miura M. Apoptosis controls the speed of looping morphogenesis in Drosophila male terminalia. Development. 2011;138:1493–1499. doi: 10.1242/dev.058958. [DOI] [PubMed] [Google Scholar]
- Lakhani SA, Masud A, Kuida K, Porter GA, Jr, Booth CJ, Mehal WZ, Inayat I, Flavell RA. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Festjens N, Declercq W, Vanden Berghe T, Vandenabeele P. Caspases in cell survival, proliferation and differentiation. Cell Death Differ. 2007;14:44–55. doi: 10.1038/sj.cdd.4402047. [DOI] [PubMed] [Google Scholar]
- Larisch S, Yi Y, Lotan R, Kerner H, Eimerl S, Tony Parks W, Gottfried Y, Birkey Reffey S, de Caestecker MP, Danielpour D, et al. A novel mitochondrial septin-like protein, ARTS, mediates apoptosis dependent on its P-loop motif. Nat Cell Biol. 2000;2:915–921. doi: 10.1038/35046566. [DOI] [PubMed] [Google Scholar]
- Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113:717–730. doi: 10.1016/s0092-8674(03)00422-7. [DOI] [PubMed] [Google Scholar]
- Lee CY, Clough EA, Yellon P, Teslovich TM, Stephan DA, Baehrecke EH. Genome-wide analyses of steroid- and radiation-triggered programmed cell death in Drosophila. Curr Biol. 2003;13:350–357. doi: 10.1016/s0960-9822(03)00085-x. [DOI] [PubMed] [Google Scholar]
- Lee TV, Fan Y, Wang S, Srivastava M, Broemer M, Meier P, Bergmann A. Drosophila IAP1-Mediated Ubiquitylation Controls Activation of the Initiator Caspase DRONC Independent of Protein Degradation. PLoS Genet. 2011;7:e1002261. doi: 10.1371/journal.pgen.1002261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmers B, Salmena L, Bidere N, Su H, Matysiak-Zablocki E, Murakami K, Ohashi PS, Jurisicova A, Lenardo M, Hakem R, et al. Essential role for caspase-8 in Toll-like receptors and NFkappaB signaling. J Biol Chem. 2007;282:7416–7423. doi: 10.1074/jbc.M606721200. [DOI] [PubMed] [Google Scholar]
- Leulier F, Vidal S, Saigo K, Ueda R, Lemaitre B. Inducible expression of double-stranded RNA reveals a role for dFADD in the regulation of the antibacterial response in Drosophila adults. Curr Biol. 2002;12:996–1000. doi: 10.1016/s0960-9822(02)00873-4. [DOI] [PubMed] [Google Scholar]
- Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Huang Q, Chen J, Peng Y, Roop DR, Bedford JS, Li CY. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci Signal. 2010a;3:ra13. doi: 10.1126/scisignal.2000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Baker NE. Engulfment is required for cell competition. Cell. 2007;129:1215–1225. doi: 10.1016/j.cell.2007.03.054. [DOI] [PubMed] [Google Scholar]
- Li Z, Jo J, Jia JM, Lo SC, Whitcomb DJ, Jiao S, Cho K, Sheng M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell. 2010b;141:859–871. doi: 10.1016/j.cell.2010.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, Thompson CB. Cell death in the absence of Bax and Bak. Cell Death Differ. 2006;13:1272–1276. doi: 10.1038/sj.cdd.4401953. [DOI] [PubMed] [Google Scholar]
- Lockshin RA, Williams CM. Programmed Cell Death--I. Cytology of Degeneration in the Intersegmental Muscles of the Pernyi Silkmoth. J Insect Physiol. 1965;11:123–133. doi: 10.1016/0022-1910(65)90099-5. [DOI] [PubMed] [Google Scholar]
- Lohmann I, McGinnis N, Bodmer M, McGinnis W. The Drosophila Hox gene deformed sculpts head morphology via direct regulation of the apoptosis activator reaper. Cell. 2002;110:457–466. doi: 10.1016/s0092-8674(02)00871-1. [DOI] [PubMed] [Google Scholar]
- Luo L, O’Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–156. doi: 10.1146/annurev.neuro.28.061604.135632. [DOI] [PubMed] [Google Scholar]
- Mace PD, Riedl SJ. Molecular cell death platforms and assemblies. Curr Opin Cell Biol. 2010;22:828–836. doi: 10.1016/j.ceb.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- Mandal PK, Blanpain C, Rossi DJ. DNA damage response in adult stem cells: pathways and consequences. Nat Rev Mol Cell Biol. 2011;12:198–202. doi: 10.1038/nrm3060. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Finucane DM, Amarante-Mendes GP, O’Brien GA, Green DR. Phosphatidylserine externalization during CD95-induced apoptosis of cells and cytoplasts requires ICE/CED-3 protease activity. J Biol Chem. 1996;271:28753–28756. doi: 10.1074/jbc.271.46.28753. [DOI] [PubMed] [Google Scholar]
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561–574. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Youle RJ. Mitochondria in apoptosis: bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21:92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusyk A, Porter CC, Zaberezhnyy V, DeGregori J. Irradiation selects for p53-deficient hematopoietic progenitors. PLoS Biol. 2010;8:e1000324. doi: 10.1371/journal.pbio.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes CS, Arama E, Brown S, Scherr H, Srivastava M, Bergmann A, Steller H, Mollereau B. Cytochrome c-d regulates developmental apoptosis in the Drosophila retina. EMBO Rep. 2006;7:933–939. doi: 10.1038/sj.embor.7400773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzstein MM, Hengartner MO, Tsung N, Ellis RE, Horvitz HR. Transcriptional regulator of programmed cell death encoded by Caenorhabditis elegans gene ces-2. Nature. 1996;382:545–547. doi: 10.1038/382545a0. [DOI] [PubMed] [Google Scholar]
- Metzstein MM, Stanfield GM, Horvitz HR. Genetics of programmed cell death in C. elegans: past, present and future. Trends Genet. 1998;14:410–416. doi: 10.1016/s0168-9525(98)01573-x. [DOI] [PubMed] [Google Scholar]
- Milyavsky M, Gan OI, Trottier M, Komosa M, Tabach O, Notta F, Lechman E, Hermans KG, Eppert K, Konovalova Z, et al. A distinctive DNA damage response in human hematopoietic stem cells reveals an apoptosis-independent role for p53 in self-renewal. Cell Stem Cell. 2010;7:186–197. doi: 10.1016/j.stem.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Miura M. Active participation of cell death in development and organismal homeostasis. Dev Growth Differ. 2011;53:125–136. doi: 10.1111/j.1440-169X.2010.01228.x. [DOI] [PubMed] [Google Scholar]
- Mohrin M, Bourke E, Alexander D, Warr MR, Barry-Holson K, Le Beau MM, Morrison CG, Passegue E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. 2010;7:174–185. doi: 10.1016/j.stem.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morata G, Martin FA. Cell competition: the embrace of death. Dev Cell. 2007;13:1–2. doi: 10.1016/j.devcel.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Morata G, Ripoll P. Minutes: mutants of drosophila autonomously affecting cell division rate. Dev Biol. 1975;42:211–221. doi: 10.1016/0012-1606(75)90330-9. [DOI] [PubMed] [Google Scholar]
- Moreno E. Is cell competition relevant to cancer? Nat Rev Cancer. 2008;8:141–147. doi: 10.1038/nrc2252. [DOI] [PubMed] [Google Scholar]
- Nagasaka A, Kawane K, Yoshida H, Nagata S. Apaf-1-independent programmed cell death in mouse development. Cell Death Differ. 2009;17:931–941. doi: 10.1038/cdd.2009.186. [DOI] [PubMed] [Google Scholar]
- Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619–630. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458–467. doi: 10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- Nakaya M, Tanaka M, Okabe Y, Hanayama R, Nagata S. Opposite effects of rho family GTPases on engulfment of apoptotic cells by macrophages. J Biol Chem. 2006;281:8836–8842. doi: 10.1074/jbc.M510972200. [DOI] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ohsawa S, Sugimura K, Takino K, Xu T, Miyawaki A, Igaki T. Elimination of oncogenic neighbors by JNK-mediated engulfment in Drosophila. Dev Cell. 2011;20:315–328. doi: 10.1016/j.devcel.2011.02.007. [DOI] [PubMed] [Google Scholar]
- Okamoto H, Shiraishi H, Yoshida H. Histological analyses of normally grown, fertile Apaf1-deficient mice. Cell Death Differ. 2006;13:668–671. doi: 10.1038/sj.cdd.4401806. [DOI] [PubMed] [Google Scholar]
- Opferman JT, Korsmeyer SJ. Apoptosis in the development and maintenance of the immune system. Nat Immunol. 2003;4:410–415. doi: 10.1038/ni0503-410. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Prevette D, Yin QW, Collins F, MacDonald J. Control of embryonic motoneuron survival in vivo by ciliary neurotrophic factor. Science. 1991;251:1616–1618. doi: 10.1126/science.2011743. [DOI] [PubMed] [Google Scholar]
- Pearson BJ, Sanchez Alvarado A. A planarian p53 homolog regulates proliferation and self-renewal in adult stem cell lineages. Development. 2010;137:213–221. doi: 10.1242/dev.044297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Garijo A, Martin FA, Morata G. Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development. 2004;131:5591–5598. doi: 10.1242/dev.01432. [DOI] [PubMed] [Google Scholar]
- Perez-Garijo A, Shlevkov E, Morata G. The role of Dpp and Wg in compensatory proliferation and in the formation of hyperplastic overgrowths caused by apoptotic cells in the Drosophila wing disc. Development. 2009;136:1169–1177. doi: 10.1242/dev.034017. [DOI] [PubMed] [Google Scholar]
- Raff MC. Social controls on cell survival and cell death. Nature. 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival: lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol. 2007;7:964–974. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- Rebe C, Cathelin S, Launay S, Filomenko R, Prevotat L, L’Ollivier C, Gyan E, Micheau O, Grant S, Dubart-Kupperschmitt A, et al. Caspase-8 prevents sustained activation of NF-kappaB in monocytes undergoing macrophagic differentiation. Blood. 2007;109:1442–1450. doi: 10.1182/blood-2006-03-011585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Reynaud K, Driancourt MA. Oocyte attrition. Mol Cell Endocrinol. 2000;163:101–108. doi: 10.1016/s0303-7207(99)00246-4. [DOI] [PubMed] [Google Scholar]
- Robinow S, Draizen TA, Truman JW. Genes that induce apoptosis: transcriptional regulation in identified, doomed neurons of the Drosophila CNS. Dev Biol. 1997;190:206–213. doi: 10.1006/dbio.1997.8696. [DOI] [PubMed] [Google Scholar]
- Rodriguez J, Lazebnik Y. Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev. 1999;13:3179–3184. doi: 10.1101/gad.13.24.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossel M, Capecchi MR. Mice mutant for both Hoxa1 and Hoxb1 show extensive remodeling of the hindbrain and defects in craniofacial development. Development. 1999;126:5027–5040. doi: 10.1242/dev.126.22.5027. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Jamieson CH, Weissman IL. Stems cells and the pathways to aging and cancer. Cell. 2008;132:681–696. doi: 10.1016/j.cell.2008.01.036. [DOI] [PubMed] [Google Scholar]
- Rumpf S, Lee SB, Jan LY, Jan YN. Neuronal remodeling and apoptosis require VCP-dependent degradation of the apoptosis inhibitor DIAP1. Development. 2011;138:1153–1160. doi: 10.1242/dev.062703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo HD, Baehrecke EH. Distinct death mechanisms in Drosophila development. Curr Opin Cell Biol. 2010;22:889–895. doi: 10.1016/j.ceb.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo HD, Bergmann A, Gonen H, Ciechanover A, Steller H. Regulation of Drosophila IAP1 degradation and apoptosis by reaper and ubcD1. Nat Cell Biol. 2002;4:432–438. doi: 10.1038/ncb795. [DOI] [PubMed] [Google Scholar]
- Ryoo HD, Gorenc T, Steller H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell. 2004;7:491–501. doi: 10.1016/j.devcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Salmena L, Lemmers B, Hakem A, Matysiak-Zablocki E, Murakami K, Au PY, Berry DM, Tamblyn L, Shehabeldin A, Migon E, et al. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 2003;17:883–895. doi: 10.1101/gad.1063703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvesen GS, Abrams JM. Caspase activation - stepping on the gas or releasing the brakes? Lessons from humans and flies. Oncogene. 2004;23:2774–2784. doi: 10.1038/sj.onc.1207522. [DOI] [PubMed] [Google Scholar]
- Sandu C, Ryoo HD, Steller H. Drosophila IAP antagonists form multimeric complexes to promote cell death. J Cell Biol. 2010;190:1039–1052. doi: 10.1083/jcb.201004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9:628–638. doi: 10.1038/nrm2455. [DOI] [PubMed] [Google Scholar]
- Schile AJ, Garcia-Fernandez M, Steller H. Regulation of apoptosis by XIAP ubiquitin-ligase activity. Genes Dev. 2008;22:2256–2266. doi: 10.1101/gad.1663108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenmann Z, Assa-Kunik E, Tiomny S, Minis A, Haklai-Topper L, Arama E, Yaron A. Axonal degeneration is regulated by the apoptotic machinery or a NAD+-sensitive pathway in insects and mammals. J Neurosci. 2010;30:6375–6386. doi: 10.1523/JNEUROSCI.0922-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Schwartz HT, Horvitz HR. The C. elegans protein CEH-30 protects male-specific neurons from apoptosis independently of the Bcl-2 homolog CED-9. Genes Dev. 2007;21:3181–3194. doi: 10.1101/gad.1607007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seita J, Rossi DJ, Weissman IL. Differential DNA damage response in stem and progenitor cells. Cell Stem Cell. 2010;7:145–147. doi: 10.1016/j.stem.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Shi Y. A conserved tetrapeptide motif: potentiating apoptosis through IAP-binding. Cell Death Differ. 2002;9:93–95. doi: 10.1038/sj.cdd.4400957. [DOI] [PubMed] [Google Scholar]
- Shlevkov E, Morata G. A dp53/JNK-dependant feedback amplification loop is essential for the apoptotic response to stress in Drosophila. Cell Death Differ. 2011 doi: 10.1038/cdd.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- Smith-Bolton RK, Worley MI, Kanda H, Hariharan IK. Regenerative growth in Drosophila imaginal discs is regulated by Wingless and Myc. Dev Cell. 2009;16:797–809. doi: 10.1016/j.devcel.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sordet O, Rebe C, Plenchette S, Zermati Y, Hermine O, Vainchenker W, Garrido C, Solary E, Dubrez-Daloz L. Specific involvement of caspases in the differentiation of monocytes into macrophages. Blood. 2002;100:4446–4453. doi: 10.1182/blood-2002-06-1778. [DOI] [PubMed] [Google Scholar]
- Sotiropoulou PA, Candi A, Mascre G, De Clercq S, Youssef KK, Lapouge G, Dahl E, Semeraro C, Denecker G, Marine JC, et al. Bcl-2 and accelerated DNA repair mediates resistance of hair follicle bulge stem cells to DNA-damage-induced cell death. Nat Cell Biol. 2010;12:572–582. doi: 10.1038/ncb2059. [DOI] [PubMed] [Google Scholar]
- Srivastava M, Scherr H, Lackey M, Xu D, Chen Z, Lu J, Bergmann A. ARK, the Apaf-1 related killer in Drosophila, requires diverse domains for its apoptotic activity. Cell Death Differ. 2007;14:92–102. doi: 10.1038/sj.cdd.4401931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steller H. Regulation of apoptosis in Drosophila. Cell Death Differ. 2008;15:1132–1138. doi: 10.1038/cdd.2008.50. [DOI] [PubMed] [Google Scholar]
- Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity. 2009;30:180–192. doi: 10.1016/j.immuni.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell. 2001;8:613–621. doi: 10.1016/s1097-2765(01)00341-0. [DOI] [PubMed] [Google Scholar]
- Tan Y, Yamada-Mabuchi M, Arya R, St Pierre S, Tang W, Tosa M, Brachmann C, White K. Coordinated expression of cell death genes regulates neuroblast apoptosis. Development. 2011;138:2197–2206. doi: 10.1242/dev.058826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tata JR. Requirement for RNA and protein synthesis for induced regression of the tadpole tail in organ culture. Dev Biol. 1966;13:77–94. doi: 10.1016/0012-1606(66)90050-9. [DOI] [PubMed] [Google Scholar]
- Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836–847. doi: 10.1038/nrm1489. [DOI] [PubMed] [Google Scholar]
- Thellmann M, Hatzold J, Conradt B. The Snail-like CES-1 protein of C. elegans can block the expression of the BH3-only cell-death activator gene egl-1 by antagonizing the function of bHLH proteins. Development. 2003;130:4057–4071. doi: 10.1242/dev.00597. [DOI] [PubMed] [Google Scholar]
- Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, Melville L, Melrose LA, Ogden CA, Nibbs R, et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. 2008;112:5026–5036. doi: 10.1182/blood-2008-06-162404. [DOI] [PubMed] [Google Scholar]
- Tseng AS, Adams DS, Qiu D, Koustubhan P, Levin M. Apoptosis is required during early stages of tail regeneration in Xenopus laevis. Dev Biol. 2007;301:62–69. doi: 10.1016/j.ydbio.2006.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux DL, Silke J. IAPs, RINGs and ubiquitylation. Nat Rev Mol Cell Biol. 2005;6:287–297. doi: 10.1038/nrm1621. [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell. 1998;92:501–509. doi: 10.1016/s0092-8674(00)80943-5. [DOI] [PubMed] [Google Scholar]
- Wang S, Zheng H, Esaki Y, Kelly F, Yan W. Cullin3 is a KLHL10-interacting protein preferentially expressed during late spermiogenesis. Biol Reprod. 2006;74:102–108. doi: 10.1095/biolreprod.105.045484. [DOI] [PubMed] [Google Scholar]
- Weil M, Jacobson MD, Raff MC. Is programmed cell death required for neural tube closure? Curr Biol. 1997;7:281–284. doi: 10.1016/s0960-9822(06)00125-4. [DOI] [PubMed] [Google Scholar]
- Wells BS, Yoshida E, Johnston LA. Compensatory proliferation in Drosophila imaginal discs requires Dronc-dependent p53 activity. Curr Biol. 2006;16:1606–1615. doi: 10.1016/j.cub2006.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K, Grether ME, Abrams JM, Young L, Farrell K, Steller H. Genetic control of programmed cell death in Drosophila. Science. 1994;264:677–683. doi: 10.1126/science.8171319. [DOI] [PubMed] [Google Scholar]
- White K, Tahaoglu E, Steller H. Cell killing by the Drosophila gene reaper. Science. 1996;271:805–807. doi: 10.1126/science.271.5250.805. [DOI] [PubMed] [Google Scholar]
- Williams DW, Kondo S, Krzyzanowska A, Hiromi Y, Truman JW. Local caspase activity directs engulfment of dendrites during pruning. Nat Neurosci. 2006;9:1234–1236. doi: 10.1038/nn1774. [DOI] [PubMed] [Google Scholar]
- Xu D, Li Y, Arcaro M, Lackey M, Bergmann A. The CARD-carrying caspase Dronc is essential for most, but not all, developmental cell death in Drosophila. Development. 2005;132:2125–2134. doi: 10.1242/dev.01790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki S, Iwama A, Takayanagi S, Morita Y, Eto K, Ema H, Nakauchi H. Cytokine signals modulated via lipid rafts mimic niche signals and induce hibernation in hematopoietic stem cells. EMBO J. 2006;25:3515–3523. doi: 10.1038/sj.emboj.7601236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Ma L, Burns KH, Matzuk MM. Haploinsufficiency of kelch-like protein homolog 10 causes infertility in male mice. Proc Natl Acad Sci U S A. 2004;101:7793–7798. doi: 10.1073/pnas.0308025101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsenko AN, Roy A, Chen R, Ma L, Murthy LJ, Yan W, Lamb DJ, Matzuk MM. Non-invasive genetic diagnosis of male infertility using spermatozoal RNA: KLHL10 mutations in oligozoospermic patients impair homodimerization. Hum Mol Genet. 2006;15:3411–3419. doi: 10.1093/hmg/ddl417. [DOI] [PubMed] [Google Scholar]
- Yi CH, Yuan J. The Jekyll and Hyde functions of caspases. Dev Cell. 2009;16:21–34. doi: 10.1016/j.devcel.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Yu JW, Shi Y. FLIP and the death effector domain family. Oncogene. 2008;27:6216–6227. doi: 10.1038/onc.2008.299. [DOI] [PubMed] [Google Scholar]
- Yuan J, Kroemer G. Alternative cell death mechanisms in development and beyond. Genes Dev. 2010;24:2592–2602. doi: 10.1101/gad.1984410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- Zandy AJ, Lakhani S, Zheng T, Flavell RA, Bassnett S. Role of the executioner caspases during lens development. J Biol Chem. 2005;280:30263–30272. doi: 10.1074/jbc.M504007200. [DOI] [PubMed] [Google Scholar]
- Zermati Y, Garrido C, Amsellem S, Fishelson S, Bouscary D, Valensi F, Varet B, Solary E, Hermine O. Caspase activation is required for terminal erythroid differentiation. J Exp Med. 2001;193:247–254. doi: 10.1084/jem.193.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lin N, Carroll PM, Chan G, Guan B, Xiao H, Yao B, Wu SS, Zhou L. Epigenetic blocking of an enhancer region controls irradiation-induced proapoptotic gene expression in Drosophila embryos. Dev Cell. 2008;14:481–493. doi: 10.1016/j.devcel.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Belote JM. The testis-specific proteasome subunit Prosalpha6T of D. melanogaster is required for individualization and nuclear maturation during spermatogenesis. Development. 2007;134:3517–3525. doi: 10.1242/dev.004770. [DOI] [PubMed] [Google Scholar]