

Abstract
Post-traumatic stress disorder (PTSD) is characterized in part by impaired extinction of conditioned fear. Traumatic brain injury (TBI) is thought to be a risk factor for development of PTSD. We tested the hypothesis that controlled cortical impact (CCI) would impair extinction of fear learned by Pavlovian conditioning, in mice. To mimic the scenarios in which TBI occurs prior to or after exposure to an aversive event, severe CCI was delivered to the left parietal cortex at one of two time points: (1) Prior to fear conditioning, or (2) after conditioning. Delay auditory conditioning was achieved by pairing a tone with a foot shock in “context A”. Extinction training involved the presentation of tones in a different context (context B) in the absence of foot shock. Test for extinction memory was achieved by presentation of additional tones alone in context B over the following two days. In pre- or post-injury paradigms, CCI did not influence fear learning and extinction. Furthermore, CCI did not affect locomotor activity or elevated plus maze testing. Our results demonstrate that, within the time frame studied, CCI does not impair the acquisition and expression of conditioned fear or extinction memory.
Keywords: Traumatic brain injury, Fear learning, Extinction, Anxiety disorder, Dorsal hippocampus, Learning and memory, Rodent
1. Introduction
Traumatic brain injury (TBI) affects 1.7 million civilians (Selassie et al., 2008) and 125,000 soldiers each year (Hoge et al., 2008; Taylor et al., 2012) with approximately 50,000 cases classified as severe. TBI is often associated with mental health and anxiety disorders, such as post-traumatic stress disorder (PTSD) (Hoge et al., 2008; Taylor et al., 2012). As a mental health disorder, PTSD is unique inasmuch as it is the only one for which a proximal cause (e.g., an aversive event) is known (Pitman et al., 2012). Patients with PTSD have persistence of fear responses to cues that were present during an aversive experience, as well as emotional manifestations of fear responses in unrelated contexts (Milad et al., 2009; Rougemont-Bücking et al., 2011). Although epidemiological studies are strongly suggestive, experimental animal studies are needed to provide a mechanistic linkage between TBI and development of PTSD.
One experimental approach used to understand the biology of PTSD is Pavlovian fear conditioning, in which a neutral conditioned stimulus (CS, e.g., a tone) is learned to predict an aversive unconditioned stimulus (US, e.g., a foot shock) and elicits fear responses (LeDoux, 2000). Delay conditioning is a form of Pavlovian conditioning where the CS terminates with an overlapping US, and is largely considered to be independent of the dorsal hippocampus (dHPC) (Kim and Fanselow, 1992; Phillips and LeDoux, 1992). However, other reports using different methodological and conditioning parameters implicate the dHPC in delay conditioning (Maren, 2008). Thus, the role of the dHPC in delay fear conditioning remains to be clarified, particularly with models of brain injury.
Fear responses can be inhibited by extinction, which occurs when the CS is presented repeatedly without the unconditioned stimulus (Pavlov, 1927). Fear extinction requires an interaction between the amygdala, medial pre-frontal cortex, and hippocampus (HPC) in humans (Milad et al., 2006; Phelps et al., 2004) and in rodents (Quirk and Mueller, 2008; Sierra-Mercado et al., 2011), suggesting homology in the neural circuitry of extinction across species (Milad and Quirk, 2012). Interestingly, PTSD is characterized by deficiencies in the ability to extinguish fear responses (Rothbaum and Davis, 2003) suggesting dysfunction in the neural circuits of extinction (Milad et al., 2009).
Given recent reports documenting a strong association between TBI and development of PTSD in war fighters (Taylor et al., 2012), we aimed to test the hypothesis that TBI in mice would impair extinction of fear memories (Genovese et al., 2013; Meyer et al., 2012). To test this hypothesis we combined controlled cortical impact (CCI), a widely used model of cerebral contusion graded to produce significant dorsal hippocampal damage, with a model of Pavlovian fear conditioning and extinction in mice. CCI was delivered to the left parietal cortex at one of two time points: prior to fear conditioning and extinction, or after fear conditioning and prior to extinction (post-fear conditioning). The experimental design aimed to mimic the scenarios in which brain injury occurs prior to, or after, exposure to an aversive event associated with PTSD. Our results demonstrate that CCI has no affect on either the acquisition or expression of fear and extinction memories.
2. Results
All mice survived CCI and the post-injury period. Fig. 1 shows the histopathology of sham injury (Fig. 1A) and CCI (Fig. 1B). We performed histological analysis on a total of 12 mice, 6 shams and 6 CCI injured. CCI produced a cavitary lesion with significant loss of ipsilateral cortex and dorsal hippocampus; volume of remaining hippocampal tissue was 47.9±3.1% of the contralateral (uninjured) hemisphere, compared to 104.7±3% in shams (p<0.001, n=6 shams). Volume of the contralateral hippocampus in CCI mice (13.0±0.4 mm3) did not differ from that of sham injured mice (13.4±0.4 mm3), conforming no detectable brain tissue loss in contralateral hemispheres of injured mice. Anterior and ventral brain regions involved in fear learning and extinction were spared by CCI.
Fig. 1.

Histopathology of sham and unilateral controlled cortical impact (CCI). (A) Histopathology of sham injury showing representative brain sections corresponding to the extent of the lesion in CCI mice. (B) Histopathology of CCI in a representative mouse showing overt damage to the ipsilateral dorsal hippocampus. Arrows indicate regions involved in fear and extinction circuitry (prelimbic (PL) region of prefrontal cortex, dorsal hippocampus, amygdala, and ventral hippocampus).
2.1. No effect of CCI applied before fear conditioning on freezing during fear acquisition and extinction trials
We began by examining the effect of CCI on fear conditioning and extinction (Fig. 2A). During conditioning trials (Cond), mice subjected to CCI had freezing levels comparable to those of sham injured (sham CCI) mice (F1,22=0.74; p=0.40, Fig. 2B). Freezing levels on the first trial of extinction (Ext), which represents consolidation of fear learning, were also similar between CCI and sham-injured groups (CCI: 59.8±4.8%, CCI sham: 65.5±3.9%; t22=0.91; p=0.37). Both CCI and sham-injured groups showed comparable levels of freezing during the extinction trials (F1,22=0.44; p=0.52) as well as trials for extinction memory test (Test; F1,22=0.59; p=0.45). Moreover, the level of spontaneous recovery, (ratio of time spent freezing on the first trial of the test day and the last trial of conditioning), was similar between CCI and sham (t22=0.85; p=0.41). Thus, CCI did not affect acquisition or extinction of conditioned fear. Open field testing demonstrated no difference in locomotion in CCI mice compared to sham (t22=0.84; p=0.41, Fig. 2C). Furthermore, we did not observe any differences in center vs. periphery exploration between sham injured and CCI groups (t22=1.15; p=0.26). Fear to the context was similar between groups, as indicated by comparable levels of average freezing 30 s immediately prior to the first trial of extinction (CCI: 17.3±3.6%, sham: 14.8±5.1%; t22=0.40; p=0.69) and prior to the first trial of test (CCI: 16.2±3.9%, sham: 12.0±2.8%; t22=0.86; p=0.40).
Fig. 2.
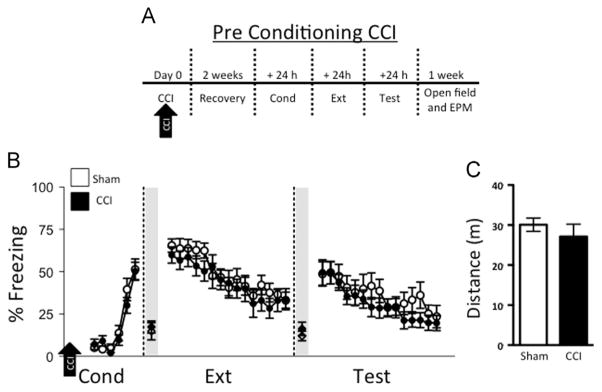
Controlled cortical impact (CCI) prior to conditioning does not affect fear conditioning or extinction. (A) Experimental timeline: mice were subjected to CCI or sham injury. After a two-week recovery period, all mice underwent fear conditioning (Cond), extinction training (Ext), and test for extinction memory (Test). (B) Sham controls (n=12) and CCI (n=12) display comparable levels of freezing 30 s immediately prior to the first tone of extinction (Ext; shaded region) and prior to the first tone of test (Test; shaded region), which is an index of context fear. Sham controls and CCI display comparable levels of freezing during Cond, Ext, and Test. (C) Total distance traveled in the open field between sham and CCI was similar, suggesting CCI does not affect locomotor activity.
2.2. No effect of CCI applied after fear conditioning on subsequent fear expression and extinction
We next aimed to determine the effects of CCI delivered after conditioning, akin to the scenario of TBI suffered after an aversive stimulus (Fig. 3A). Mice were subjected to CCI or sham injury 24 h after fear conditioning (Fig. 3B). As expected, no differences in fear conditioning were observed between groups prior to CCI. After a 2-week recovery period, freezing behavior on the first trial of extinction training was similar between CCI and sham groups (post-conditioning; p.c. CCI: 38.7±5.8%, p.c. CCI sham: 50.9±5.9%; t26=1.46; p=0.16), suggesting that consolidation of fear learning was not affected by CCI. This was likely due to the fact that injury occurred outside of the consolidation window, and therefore did not affect those processes. Freezing was comparable between groups during extinction training (Ext) (F1,26=2.52; p=0.12 for group). During extinction memory (Test) trials, freezing (F1,26=1.62; p=0.21) and levels of spontaneous recovery (t26=0.09; p=0.93) did not indicate impaired extinction. Thus, similar to the scenario in which CCI occurs before conditioning, CCI delivered after fear conditioning does not affect extinction learning or memory. Open field testing demonstrated no difference in locomotion in p.c. CCI mice compared to sham (t18=1.07; p=0.30, Fig. 3C). Moreover, we did not observe any differences in center vs. periphery exploration between sham injured and CCI groups (t18=1.71; p=0.10). Fear to the context was similar between groups, as indicated by comparable levels of average freezing 30 s immediately prior to the first trial of extinction (p.c. CCI: 18.0±5.4%, sham: 11.0±3.0%; t26=1.14; p=0.26) and prior to the first trial of test (p.c. CCI: 6.2±2.0%, CCI sham: 11.5±2.8%; t26=1.55; p=0.13).
Fig. 3.
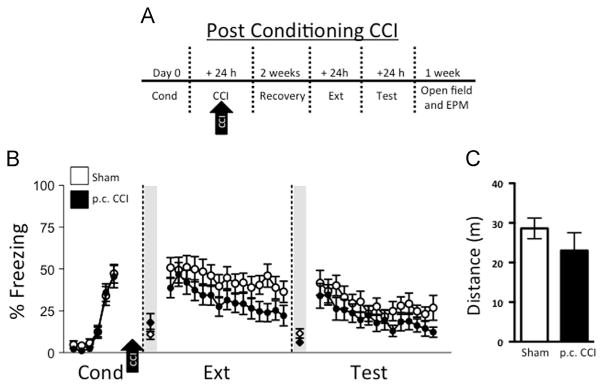
Controlled cortical impact (CCI) administered post-fear conditioning (p.c.) does not affect fear expression or extinction. (A) Experimental timeline: sham injury and CCI occurred 24 h after conditioning (Cond). Following a two-week recovery period, all mice underwent extinction training (Ext) and test for extinction memory (Test). (B) Sham controls (n=14) and p.c. CCI (n=14) displayed comparable levels of freezing 30 s immediately prior to the first tone of extinction (Ext; shaded region) and prior to the first tone of test (Test; shaded region). Sham controls and CCI had similar levels of freezing during Cond, Ext, and Test. (C) Total distance traveled in the open field between sham (n=10) and CCI (n=10) was also similar, suggesting that CCI does not affect locomotor activity.
2.3. No effect of CCI in elevated plus maze testing
Mice from each experiment were tested in the elevated plus maze. Time spent in the open arms was measured as an index of anxiety-like behavior. No pairs of sham and injured groups differed in the amount of time spent in the open arms of the elevated plus maze, suggesting that CCI does not induce anxiety under the experimental conditions and time frame herein (Supplementary Fig. A and B).
3. Discussion
We combined traumatic brain injury using a severe model of CCI that induces significant hippocampal damage and Morris Water Maze deficits (Yang et al, 2010), with auditory fear conditioning and extinction in mice. We observed that CCI did not affect the acquisition and expression of conditioned fear. In fact, freezing levels in sham and injured mice were comparable to those in other studies using C57BL/6 mice (Hefner et al., 2008). Notably, contrary to our a priori hypothesis, CCI delivered before or after fear conditioning did not impair extinction learning and memory. CCI also did not affect locomotor activity, time in the open arms of the elevated plus maze (a measure of anxiety), or gross brain anatomy in ventral brain regions involved in fear circuitry. These findings suggest that our published model of CCI (Yang et al., 2010) does not affect the neural mechanisms of fear and extinction within the first two weeks of injury, which represents the early phase of injury (Lowing et al., 2014).
We found that freezing levels were comparable during conditioning and the early part of extinction (trials 1 and 2 of extinction training), whether CCI was administered before or after conditioning. We interpret these data to mean that fear learning and memory are not affected by CCI at the level of injury used herein. This is likely because the amygdala and other anterior/ventral brain regions involved in fear learning (LeDoux, 2000) were not directly damaged by CCI. In contrast to these findings, Reger et al. (2012) reported increased strength of fear learning after unilateral fluid percussion TBI compared to sham injured controls (Reger et al., 2012). It is possible that fluid percussion injury, which models some aspects of concussion (Thompson et al., 2005), affects fear-learning circuitry differently than CCI. Some studies using lateral fluid percussion show impaired fear conditioning whereas others show no effect or increased fear conditioning in rodents (Abrous et al., 1999; Hogg et al., 1998; Reger et al., 2012; Witgen et al., 2005). Another difference between our study and that of Reger et al. (2012) is that the latter used a hippocampus/prefrontal cortex-dependent trace fear conditioning paradigm (Runyan et al., 2004), as well as a delay conditioning paradigm, whereas we used tone-shock parameters necessary for hippocampal-independent delay fear conditioning in the current study (Phillips and LeDoux, 1992; Quinn et al., 2008). We used this conditioning paradigm because we wanted to avoid effects of CCI on conditioning so that extinction data would be more readily interpretable given equal levels of fear conditioning between groups.
In the study of Reger et al., fear memory to the tone was tested but individual extinction trials were not reported, thus prohibiting analysis of time dependent effects of extinction trials on fear memory. It is possible that averaging the freezing data obtained over multiple extinction trials might have produced an apparent increase in freezing levels because of data compression, thus masking a potential effect on extinction learning. In our study, we elected to compare freezing levels across trials to dissociate any potential differences between fear and extinction memories.
The role of the hippocampus in auditory fear conditioning has been examined using different types of lesions, which has led to alternate interpretations. For example, electrolytic lesions of the dHPC, which destroy neurons and axons, have resulted in no effect on conditioned fear (Phillips and LeDoux, 1992). On the other hand, neurotoxic lesions of the dHPC that destroy neurons, but leave axons intact, impair fear acquisition (Maren et al., 1997). Other reports indicate that effects on fear conditioning are largely dependent on the volume of hippocampal tissue that is damaged, such that lesions including the ventral HPC (vHPC) impair fear expression (Maren and Holt, 2004; Richmond et al., 1999). The damage induced by CCI in the current study, as noted in Fig. 1B, spares portions of vHPC, suggesting that normal fear expression with CCI may be driven by a functional vHPC.
The mechanism by which the hippocampus contributes to fear expression likely involves an interaction with other brain regions that signal fear. Current models of fear circuitry suggest that various components of fear-related information are integrated in the prelimbic (PL) subregion of the medial prefrontal cortex (Sierra-Mercado et al., 2011; Sotres-Bayon and Quirk, 2010), which allows PL to regulate conditioned fear (Sierra-Mercado et al., 2011; Vidal-Gonzalez et al., 2006). In the current study, other brain regions not damaged by CCI likely contribute fear-related information to PL, thus resulting in normal levels of fear expression.
The degree to which CCI mimics hippocampal lesions may not be directly comparable when evaluating their effects on conditioned fear. Unlike unilateral neurotoxic and electrolytic lesions that do not impair performance in the Morris Water Maze, unilateral HPC injury induced by CCI results in a Morris Water Maze deficit (Yang et al., 2010). It is likely that the type of damage that is caused by lesions is functionally different than that of CCI. In electrolytic and chemical lesion studies, bilateral lesions of the hippocampus, but not unilateral lesions, impair performance in the Morris Water Maze (Devan et al., 1996; Devan and White, 1999; Morris et al., 1982; 1990). Thus, unilateral CCI resulting in a performance deficit in the Morris Water Maze suggests bilateral dysfunction of the hippocampus (Bermpohl et al., 2007; Yang et al., 2010). This result is somewhat surprising given the robust effects of unilateral CCI on Morris Water Maze performance. Nonetheless, the severity of CCI used in the present study did not yield a fear conditioning or extinction deficit, suggesting that the requisite HPC circuitry remains intact despite the apparent bilateral HPC dysfunction with respect to place learning (Devan et al., 1996; Devan and White, 1999; Morris et al., 1982; 1990). More work needs to be performed to determine whether unilateral CCI affects fear learning and extinction pathways in the contralateral hemisphere. Future studies using electrophysiology more precise brain mapping paradigms, such as optogenetics, are needed to fully understand the complex effects of lesioning vs. CCI on fear circuitry.
A striking and unexpected finding of the current study was lack of impairment of extinction by CCI. CCI to the parietal cortex resulted in structural damage to the dorsal hippocampus (dHPC), which is part of the circuitry required for extinction (Quirk and Mueller, 2008). In addition, mice were conditioned on a partial reinforcement rate, which reduces the strength of extinction learning (Bouton, 2004; Gallistel and Gibbon, 2000). Despite the increased difficulty in extinction that this paradigm imposes, CCI mice extinguished to floor levels (e.g., freezing eventually tapers off). Thus, contrary to our prediction, CCI does not impair extinction of conditioned fear.
The lack of impaired extinction in CCI may be due to lack of structural damage to other brain regions necessary for extinction, such as the amygdala (Sotres-Bayon et al., 2007) and infralimbic subregion of the ventral medial prefrontal cortex (Quirk et al., 2000; Sierra-Mercado et al., 2006). Recent evidence suggests that at least some regions of ventral hippocampus are also necessary for normal extinction (Farinelli et al., 2006; Sierra-Mercado et al., 2011; Sotres-Bayon et al., 2012). Furthermore, hippocampal lesions can result in compensatory mechanisms, such as the recruitment of additional brain structures that can participate in extinction (Anglada-Figueroa and Quirk, 2005; Maren et al., 1997), thus masking lesion deficits. Further studies are needed to assess function of brain regions left structurally intact after CCI.
There is a growing debate from epidemiological studies of soldiers returning from the battlefield as to whether TBI contributes to PTSD (Bahraini et al., 2014). Our results using CCI models do not demonstrate a direct relationship between contusion TBI and fear extinction, which is impaired in patients with PTSD (Milad et al., 2009). The lack of a relationship between TBI and extinction in the current study must be interpreted with caution with respect to relevance to combat-related TBI, particularly since we used a model of focal contusion, rather than a blast or concussive TBI model. While our results may appear to be consistent with the idea that focal brain damage does not contribute to PTSD, our findings do not address other behaviors related to anxiety that may be affected. Interestingly, we observed no effect of CCI in one measure of anxiety using the elevated plus maze. Additional measures of anxiety (McHugh et al., 2004) are needed to more fully characterize the effects of CCI on anxiety. Moreover, our study is limited by use of a single injury level and testing in the immediate period after TBI. Further studies are needed to probe the relationship between brain injury and sensitivity to development of PTSD in the short and long term after injury. These studies are currently underway in our laboratory.
4. Experimental procedures
4.1. Subjects
All procedures were approved by the Institutional Animal Care and Use Committee of the Massachusetts General Hospital and were in compliance with National Institutes of Health guidelines for the care and use of laboratory animals. A total of 52 male C57BL/6 mice (age 2–4 months; Jackson Laboratories, Bar Harbor, ME) were used in the studies. Mice were given access to food and water ad libitum and housed in a temperature-controlled room with a 12 h light/dark cycle. All experiments were performed by investigators blinded to the study groups.
4.2. Controlled cortical impact (CCI)
CCI was used to induce cerebral contusion as previously described (Yang et al., 2010). In separate groups, CCI was performed either 2 weeks prior to fear conditioning, or 24 h after fear conditioning (post-conditioning; p.c.). Mice were anesthetized with 4% isoflurane (Anaquest, Memphis, TN) in 70% N2O and 30% O2 using a Fluotec 3 vaporizer (Colonial Medical, Amherst, NH). Blow-by anesthesia was maintained with 3.5% isoflurane for the duration of surgery. This regimen produces unresponsiveness to tail and toe pinch and to surgical procedures, yet maintains blood pressure and blood gases within normal limits (Khuman et al., 2011; Yang et al., 2010). The head was positioned in a stereotaxic apparatus. Following a midline scalp incision, a 5 mm craniotomy was performed with a hand drill and trephine above the left parieto-temporal cortex. The bone flaps were removed and the dura mater was left intact. Impact was delivered using a 3 mm flat-tip pneumatic piston at a velocity of 6 m/s, a duration of 100 ms, and depth of 0.6 mm. The piston was centered on the dura and the brain compressed until the flat tip impactor was flat on the brain surface. Because the brain is deformed approximately 0.6 mm during centering of the impounder tip, the total depth of impact is 1.2 mm. This injury level results in significant loss of ipsilateral dorsal hippocampus, and robust cognitive deficits assessed by Morris Water Maze (Yang et al., 2010). Sham injured mice had craniotomy without CCI. The bone flaps were discarded and the scalp incision sutured closed. Mice were returned to their cages to recover from anesthesia.
4.3. Fear conditioning and extinction
Delay fear conditioning, which is largely hippocampal independent (Phillips and LeDoux, 1992), was performed in operant chambers (17 ×17 ×25 cm3; Ugo Basile, Model 46003, Varese, Italy) within sound-attenuating cubicles (Ugo Basile, Model 46000-595). The chamber floors were comprised of stainless steel bars that delivered a scrambled foot shock. The floors and walls were cleaned with soap and water and 1% vanilla extract between experiments. On the day of conditioning (Cond), mice were placed in Context “A” for 300 s to acclimate to the chamber prior to delay auditory fear conditioning. Next, mice received two habituation tones (30 s; 1.5 kHz; 75 dB; 1 min variable intertrial interval (ITI); Ugo Basile, Model 46000-165) to assess baseline levels of freezing. Habituation tones were immediately followed by three identical tones that co-terminated with a 2 s mild foot shock (0.6 mA) delivered by a digital shocker (Ugo Basile, Model 47554). One additional tone was then given without a foot shock resulting in a partial reinforcement rate of 75% of trials. Partially reinforced conditioning, which reduces the rate of subsequent extinction, was used for two reasons: (1) To more closely resemble fear-conditioning and extinction paradigms used in patients with PTSD (Milad et al., 2009); and (2) to maximize our chances of detecting potential impairments in extinction induced by CCI. Mice remained in the chamber for 90 s following the last tone before being returned to their home cages.
For extinction and extinction memory test the following day, mice were placed in a novel chamber (Context B) consisting of striped walls and a solid plastic floor with no odor. For experiments where CCI occurred after conditioning, extinction began 2 weeks after injury. Extinction (Ext), which is context- and hippocampus-dependent (Bouton, 2004; Corcoran et al., 2005; Milad et al., 2005), consisted of 15 tone presentations without foot shock. The test for extinction memory consisted of 15 additional tone-alone presentations the following day (Test). The strength of extinction memory was also determined by levels of spontaneous recovery, which was defined as the ratio of time spent freezing on the first trial of the test day and the last trial of conditioning (Quirk et al., 2000). Freezing during 30 s prior to the onset of the first tone of extinction training and test was measured as an index of context fear (Phillips and LeDoux, 1992).
4.4. Elevated plus maze testing and open field
Elevated plus maze and open-field testing were performed 1 week following tests for extinction memory. Assessment of anxiety-like behavior was performed in the elevated plus maze, which is comprised of two sets of arms, one opened and one closed. Behavior was quantified by observing the time spent in the open arms (Pellow, 1985; Walf and Frye, 2007), which was tracked using a video camera positioned over the maze and analyzed using ANY-Maze software. Locomotor activity was assessed upon completion of the experiments using the open-field test as described previously (Washington et al., 2012). Mice were tracked with ANYMaze video tracking software (Stoelting, Wood Dale, IL) and total distance traveled was measured over a five-minute period.
4.5. Histopathology of CCI
Upon completion of the experiments, mice were deeply anesthetized with isoflurane and decapitated. Brains were removed and frozen in nitrogen vapor and stored at −80 °C. Brain sections were cut on a cryostat, stained with hematoxylin, and photographed. The mouse with the median hippocampal tissue loss was selected as a representative for the CCI group. Brain tissue was intact in all sham-injured mice, therefore one was chosen at random to represent the sham group. Hippocampal volume was assessed using image analysis. Brains were cut on a cryostat every 500 μm, stained with hematoxylin, and photographed. The ipsilateral and contralateral hippocampi were outlined and area was determined using Image J software. Area measurements were summed and multiplied by 0.5 to determine volume measurements.
4.6. Data collection and analysis
Behavior was recorded with digital video cameras, and open field test data and freezing behavior were quantified from digitized video images using ANY-Maze. The amount of time freezing to the tone was expressed as a percentage of the 30 s tone presentation. Fear learning and extinction data were analyzed by repeated measures ANOVA (RM ANOVA; group x time) using custom-written software in MATLAB (Math-works), and Graphpad Prism VI (La Jolla, CA). The percent of time spent in the open arms of the elevated plus maze was used to assess anxiety, and total distance traveled in the open field was measured to determine locomotor activity, both of which were analyzed using Student’s t-test. Hippocampal lesion data were analyzed by t-test. For all tests, p<0.05 was regarded as significant.
Supplementary Material
Acknowledgments
We thank Jimmy Zhang for technical assistance. This study was supported in part by RCMI 8G12MD007600, a supplement from DA026297 and a Young Investigator Grant from the Brain & Behavior Research Foundation (NARSAD) to DS-M, MH097964 and MH097880 to MRM, MH086400 and NS086422 to ENE, DOD/CIMIT W81XWH-07-2-0011 to MJW, and Behavioral Core Facility (P01NS5510406).
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.brainres.2015.02.031
Footnotes
Financial disclosures
The authors do not have any financial interests or potential confiicts of interest.
Author contributions
DS-M, MJW and MRM designed the study, interpreted the data, and wrote the paper. DS-M, MJW, LMM and CCL collected and analyzed the data.
Contributor Information
Demetrio Sierra-Mercado, Email: demetrio.sierra@upr.edu.
Lauren M. McAllister, Email: LMCALLISTER1@mgh.harvard.edu.
Christopher C.H. Lee, Email: lee.chris.ch@gmail.com.
Mohammed R. Milad, Email: milad@nmr.mgh.harvard.edu.
Emad N. Eskandar, Email: eeskandar@mgh.harvard.edu.
Michael J. Whalen, Email: mwhalen@partners.org.
References
- Abrous DN, Rodriguez J, le Moal M, Moser PC, Barnéoud P. Effects of mild traumatic brain injury on immunoreactivity for the inducible transcription factors c-Fos, c-Jun, JunB, and Krox-24 in cerebral regions associated with conditioned fear responding. Brain Res. 1999;826:181–192. doi: 10.1016/s0006-8993(99)01259-7. [DOI] [PubMed] [Google Scholar]
- Anglada-Figueroa D, Quirk GJ. Lesions of the basal amygdala block expression of conditioned fear but not extinction. J Neurosci. 2005;25:9680–9685. doi: 10.1523/JNEUROSCI.2600-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahraini NH, Breshears RE, Hernández TD, Schneider AL, Forster JE, Brenner LA. Traumatic brain injury and posttraumatic stress disorder. Psychiatr Clin N Am. 2014;37:55–75. doi: 10.1016/j.psc.2013.11.002. http://dx.doi.org/10.1016/j.psc.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Bermpohl D, You Z, Lo EH, Kim HH, Whalen MJ. TNF alpha and Fas mediate tissue damage and functional outcome after traumatic brain injury in mice. J Cereb Blood Flow Metab. 2007;27:1806–1818. doi: 10.1038/sj.jcbfm.9600487. http://dx.doi.org/10.1038/sj.jcbfm.9600487. [DOI] [PubMed] [Google Scholar]
- Bouton ME. Context and behavioral processes in extinction. Learn Mem. 2004;11:485–494. doi: 10.1101/lm.78804. [DOI] [PubMed] [Google Scholar]
- Corcoran KA, Desmond TJ, Frey KA, Maren S. Hippocampal inactivation disrupts the acquisition and contextual encoding of fear extinction. J Neurosci. 2005;25:8978–8987. doi: 10.1523/JNEUROSCI.2246-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devan BD, Goad EH, Petri HL. Dissociation of hippocampal and striatal contributions to spatial navigation in the water maze. Neurobiol Learn Mem. 1996;66:305–323. doi: 10.1006/nlme.1996.0072. http://dx.doi.org/10.1006/nlme.1996.0072. [DOI] [PubMed] [Google Scholar]
- Devan BD, White NM. Parallel information processing in the dorsal striatum: relation to hippocampal function. J Neurosci. 1999;19:2789–2798. doi: 10.1523/JNEUROSCI.19-07-02789.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinelli M, Deschaux O, Hugues S, Thevenet A, Garcia R. Hippocampal train stimulation modulates recall of fear extinction independently of prefrontal cortex synaptic plasticity and lesions. Learn Mem. 2006;13:329–334. doi: 10.1101/lm.204806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallistel CR, Gibbon J. Time, rate, and conditioning. Psychol Rev. 2000;107:289–344. doi: 10.1037/0033-295x.107.2.289. [DOI] [PubMed] [Google Scholar]
- Genovese RF, Simmons LP, Ahlers ST, Maudlin-Jeronimo E, Dave JR, Boutte AM. Effects of mild TBI from repeated blast overpressure on the expression and extinction of conditioned fear in rats. Neuroscience. 2013;254:120–129. doi: 10.1016/j.neuroscience.2013.09.021. http://dx.doi.org/10.1016/j.neuroscience.2013.09.021. [DOI] [PubMed] [Google Scholar]
- Hefner K, Whittle N, Juhasz J, Norcross M, Karlsson RM, Saksida LM, Bussey TJ, Singewald N, Holmes A. Impaired fear extinction learning and cortico-amygdala circuit abnormalities in a common genetic mouse strain. J Neurosci. 2008;28:8074–8085. doi: 10.1523/JNEUROSCI.4904-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA. Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N Engl J Med. 2008;358:453–463. doi: 10.1056/NEJMoa072972. http://dx.doi.org/10.1056/NEJMoa072972. [DOI] [PubMed] [Google Scholar]
- Hogg S, Sanger DJ, Moser PC. Mild traumatic lesion of the right parietal cortex in the rat: characterisation of a conditioned freezing deficit and its reversal by dizocilpine. Behav Brain Res. 1998;93:157–165. doi: 10.1016/s0166-4328(97)00145-9. [DOI] [PubMed] [Google Scholar]
- Khuman J, Meehan WP, Zhu X, Qiu J, Hoffmann U, Zhang J, Giovannone E, Lo EH, Whalen MJ. Tumor necrosis factor alpha and Fas receptor contribute to cognitive deficits independent of cell death after concussive traumatic brain injury in mice. J Cereb Blood Flow Metab. 2011;31:778–789. doi: 10.1038/jcbfm.2010.172. http://dx.doi.org/10.1038/jcbfm.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. http://dx.doi.org/10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- Lowing JL, Susick LL, Caruso JP, Provenzano AM, Raghupathi R, Conti AC. Experimental traumatic brain injury alters ethanol consumption and sensitivity. J Neurotrauma. 2014;31:1700–1710. doi: 10.1089/neu.2013.3286. http://dx.doi.org/10.1089/neu.2013.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S. Pavlovian fear conditioning as a behavioral assay for hippocampus and amygdala function: cautions and caveats. Eur J Neurosci. 2008;28:1661–1666. doi: 10.1111/j.1460-9568.2008.06485.x. http://dx.doi.org/10.1111/j.1460-9568.2008.06485.x. [DOI] [PubMed] [Google Scholar]
- Maren S, Aharonov G, Fanselow MS. Neurotoxic lesions of the dorsal hippocampus and Pavlovian fear conditioning in rats. Behav Brain Res. 1997;88:261–274. doi: 10.1016/s0166-4328(97)00088-0. [DOI] [PubMed] [Google Scholar]
- Maren S, Holt WG. Hippocampus and Pavlovian fear conditioning in rats: muscimol infusions into the ventral, but not dorsal, hippocampus impair the acquisition of conditional freezing to an auditory conditional stimulus. Behav Neurosci. 2004;118:97–110. doi: 10.1037/0735-7044.118.1.97. http://dx.doi.org/10.1037/0735-7044.118.1.97. [DOI] [PubMed] [Google Scholar]
- McHugh SB, Deacon RMJ, Rawlins JNP, Bannerman DM. Amygdala and ventral hippocampus contribute differentially to mechanisms of fear and anxiety. Behav Neurosci. 2004;118:63–78. doi: 10.1037/0735-7044.118.1.63. http://dx.doi.org/10.1037/0735-7044.118.1.63. [DOI] [PubMed] [Google Scholar]
- Meyer DL, Davies DR, Barr JL, Manzerra P, Forster GL. Mild traumatic brain injury in the rat alters neuronal number in the limbic system and increases conditioned fear and anxiety-like behaviors. Exp Neurol. 2012;235:574–587. doi: 10.1016/j.expneurol.2012.03.012. http://dx.doi.org/10.1016/j.expneurol.2012.03.012. [DOI] [PubMed] [Google Scholar]
- Milad MR, Orr SP, Pitman RK, Rauch SL. Context modulation of memory for fear extinction in humans. Psychophysiology. 2005;42:456–464. doi: 10.1111/j.1469-8986.2005.00302.x. [DOI] [PubMed] [Google Scholar]
- Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, Zeidan MA, Handwerger K, Orr SP, Rauch SL. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry. 2009;66:1075–1082. doi: 10.1016/j.biopsych.2009.06.026. http://dx.doi.org/10.1016/j.biopsych.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Fear extinction as a model for translational neuroscience: ten years of progress. Annu Rev Psychol. 2012;63:129–151. doi: 10.1146/annurev.psych.121208.131631. http://dx.doi.org/10.1146/annurev.psych.121208.131631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Rauch SL, Pitman RK, Quirk GJ. Fear extinction in rats: implications for human brain imaging and anxiety disorders. Biol Psychol. 2006;73:61–71. doi: 10.1016/j.biopsycho.2006.01.008. http://dx.doi.org/10.1016/j.biopsycho.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, O’Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- Morris RGM, Schenk F, Tweedie F, Jarrard LE. Ibotenate lesions of hippocampus and/or subiculum: dissociating components of allocentric spatial learning. Eur J Neurosci. 1990;2:1016–1028. doi: 10.1111/j.1460-9568.1990.tb00014.x. [DOI] [PubMed] [Google Scholar]
- Pavlov I. Conditioned Reflexes. Oxford University Press; London: 1927. [Google Scholar]
- Pellow S. Can drug effects on anxiety and convulsions be separated? Neurosci Biobehav Rev. 1985;9:55–73. doi: 10.1016/0149-7634(85)90032-6. [DOI] [PubMed] [Google Scholar]
- Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: role of the amygdala and vmPFC. Neuron. 2004;43:897–905. doi: 10.1016/j.neuron.2004.08.042. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW, Milad MR, Liberzon I. Biological studies of post-traumatic stress disorder. Nat Rev Neurosci. 2012;13:769–787. doi: 10.1038/nrn3339. http://dx.doi.org/10.1038/nrn3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn JJ, Wied HM, Ma QD, Tinsley MR, Fanselow MS. Dorsal hippocampus involvement in delay fear conditioning depends upon the strength of the tone-footshock association. Hippocampus. 2008;18:640–654. doi: 10.1002/hipo.20424. http://dx.doi.org/10.1002/hipo.20424. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. http://dx.doi.org/10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Russo GK, Barron JL, Lebron K. The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci. 2000;20:6225–6231. doi: 10.1523/JNEUROSCI.20-16-06225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reger ML, Poulos AM, Buen F, Giza CC, Hovda DA, Fanselow MS. Concussive brain injury enhances fear learning and excitatory processes in the amygdala. Biol Psychiatry. 2012;71:335–343. doi: 10.1016/j.biopsych.2011.11.007. http://dx.doi.org/10.1016/j.biopsych.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond MA, Yee BK, Pouzet B, Veenman L, Rawlins JN, Feldon J, Bannerman DM. Dissociating context and space within the hippocampus: effects of complete, dorsal, and ventral excitotoxic hippocampal lesions on conditioned freezing and spatial learning. Behav Neurosci. 1999;113:1189–1203. doi: 10.1037/0735-7044.113.6.1189. [DOI] [PubMed] [Google Scholar]
- Rothbaum BO, Davis M. Applying learning principles to the treatment of post-trauma reactions. Ann N Y Acad Sci. 2003;1008:112–121. doi: 10.1196/annals.1301.012. [DOI] [PubMed] [Google Scholar]
- Rougemont-Bücking A, Linnman C, Zeffiro TA, Zeidan MA, Lebrón-Milad K, Rodriguez-Romaguera J, Rauch SL, Pitman RK, Milad MR. Altered processing of contextual information during fear extinction in PTSD: an fMRI study. CNS Neurosci Ther. 2011;17:227–236. doi: 10.1111/j.1755-5949.2010.00152.x. http://dx.doi.org/10.1111/j.1755-5949.2010.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runyan JD, Moore AN, Dash PK. A role for prefrontal cortex in memory storage for trace fear conditioning. J Neurosci. 2004;24:1288–1295. doi: 10.1523/JNEUROSCI.4880-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selassie AW, Zaloshnja E, Langlois JA, Miller T, Jones P, Steiner C. Incidence of long-term disability following traumatic brain injury hospitalization, United States, 2003. J Head Trauma Rehabil. 2008;23:123–131. doi: 10.1097/01.HTR.0000314531.30401.39. http://dx.doi.org/10.1097/01.HTR.0000314531.30401.39. [DOI] [PubMed] [Google Scholar]
- Sierra-Mercado D, Corcoran KA, Lebrón-Milad K, Quirk GJ. Inactivation of the ventromedial prefrontal cortex reduces expression of conditioned fear and impairs subsequent recall of extinction. Eur J Neurosci. 2006;24:1751–1758. doi: 10.1111/j.1460-9568.2006.05014.x. http://dx.doi.org/10.1111/j.1460-9568.2006.05014.x. [DOI] [PubMed] [Google Scholar]
- Sierra-Mercado D, Padilla-Coreano N, Quirk GJ. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology. 2011;36:529–538. doi: 10.1038/npp.2010.184. http://dx.doi.org/10.1038/npp.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotres-Bayon F, Bush DE, LeDoux JE. Acquisition of fear extinction requires activation of NR2B-containing NMDA receptors in the lateral amygdala. Neuropsychopharmacology. 2007;32:1929–1940. doi: 10.1038/sj.npp.1301316. [DOI] [PubMed] [Google Scholar]
- Sotres-Bayon F, Quirk GJ. Prefrontal control of fear: more than just extinction. Curr Opin Neurobiol. 2010 doi: 10.1016/j.conb.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotres-Bayon F, Sierra-Mercado D, Pardilla-Delgado E, Quirk GJ. Gating of fear in prelimbic cortex by hippocampal and amygdala inputs. Neuron. 2012;76:804–812. doi: 10.1016/j.neuron.2012.09.028. http://dx.doi.org/10.1016/j.neuron.2012.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BC, Hagel EM, Carlson KF, Cifu DX, Cutting A, Bidelspach DE, Sayer NA. Prevalence and costs of co-occurring traumatic brain injury with and without psychiatric disturbance and pain among Afghanistan and Iraq war Veteran V.A. users. Med Care. 2012;50:342–346. doi: 10.1097/MLR.0b013e318245a558. http://dx.doi.org/10.1097/MLR.0b013e318245a558. [DOI] [PubMed] [Google Scholar]
- Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, McIntosh TK. Lateral fluid percussion brain injury: a 15-year review and evaluation. J Neurotrauma. 2005;22:42–75. doi: 10.1089/neu.2005.22.42. http://dx.doi.org/10.1089/neu.2005.22.42. [DOI] [PubMed] [Google Scholar]
- Vidal-Gonzalez I, Vidal-Gonzalez B, Rauch SL, Quirk GJ. Microstimulation reveals opposing influences of prelimbic and infralimbic cortex on the expression of conditioned fear. Learn Mem. 2006;13:728–733. doi: 10.1101/lm.306106. http://dx.doi.org/10.1101/lm.306106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walf AA, Frye CA. The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat Protoc. 2007;2:322–328. doi: 10.1038/nprot.2007.44. http://dx.doi.org/10.1038/nprot.2007.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington PM, Forcelli PA, Wilkins T, Zapple DN, Parsadanian M, Burns MP. The effect of injury severity on behavior: a phenotypic study of cognitive and emotional deficits after mild, moderate, and severe controlled cortical impact injury in mice. J Neurotrauma. 2012;29:2283–2296. doi: 10.1089/neu.2012.2456. http://dx.doi.org/10.1089/neu.2012.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witgen BM, Lifshitz J, Smith ML, Schwarzbach E, Liang SL, Grady MS, Cohen AS. Regional hippocampal alteration associated with cognitive deficit following experimental brain injury: a systems, network and cellular evaluation. Neuroscience. 2005;133:1–15. doi: 10.1016/j.neuroscience.2005.01.052. http://dx.doi.org/10.1016/j.neuroscience.2005.01.052. [DOI] [PubMed] [Google Scholar]
- Yang J, You Z, Kim HH, Hwang SK, Khuman J, Guo S, Lo EH, Whalen MJ. Genetic analysis of the role of tumor necrosis factor receptors in functional outcome after traumatic brain injury in mice. J Neurotrauma. 2010;27:1037–1046. doi: 10.1089/neu.2009.1229. http://dx.doi.org/10.1089/neu.2009.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.