

Abstract
Inorganic phosphate (Pi) is abundant in cells and tissues as an important component of nucleic acids and phospholipids, a source of high-energy bonds in nucleoside triphosphates, a substrate for kinases and phosphatases, and a regulator of intracellular signaling. The majority of the body’s Pi exists in the mineralized matrix of bones and teeth. Systemic Pi metabolism is regulated by a cast of hormones, phosphatonins, and other factors via the bone-kidney-intestine axis. Mineralization in bones and teeth is in turn affected by homeostasis of Pi and inorganic pyrophosphate (PPi), with further regulation of the Pi/PPi ratio by cellular enzymes and transporters. Much has been learned by analyzing the molecular basis for changes in mineralized tissue development in mutant and knock-out mice with altered Pi metabolism. This review focuses on factors regulating systemic and local Pi homeostasis and their known and putative effects on the hard tissues of the oral cavity. By understanding the role of Pi metabolism in the development and maintenance of the oral mineralized tissues, it will be possible to develop improved regenerative approaches.
Keywords: tooth development, tooth root, cementum, dentin, periodontal ligament, phosphate metabolism, fibroblast growth factor 23, Phex, tissue nonspecific alkaline phosphatase, progressive ankylosis protein
I. Introduction
a. Overview of phosphate metabolism
Phosphorus is abundant in all cells and tissues as an important component of DNA, RNA and phospholipids, a source of high-energy bonds in adenosine triphosphates (ATP), a substrate for various kinases and phosphatases, and a regulator of intracellular signaling. Phosphate homeostasis on a cellular level is therefore a significant aspect of normal function for most tissues and organs. Approximately 85% of phosphorus, the second most abundant mineral in the human body, is in bone, primarily compounded with calcium (Ca2+), the most abundant mineral, in hydroxyapatite (HAP) crystals deposited on the collagen matrix (Broadus, 2003). Other mineralized tissues such as teeth also contain calcium phosphate as HAP. The remainder is in soft tissue with only about 1% in extracellular fluids (Drezner, 2002). Therefore, maintenance of “normal” phosphate (inorganic or orthophosphate, Pi) homeostasis is essential for normal development, maintenance, and repair of teeth and skeletal tissues.
Natural foods contain substantial quantities of phosphorus. Deficiency can occur as a result of severe starvation, intake of Pi binders that prevent absorption in the gut, or in diseases associated with renal Pi wasting. Dietary Pi is absorbed in the small intestine where the impact of hormonal regulation, mediated by the active form of vitamin D, 1,25 (OH)2 vitamin D3 (referred to herein as Vit D), is minor relative to dietary load. From blood, phosphorus is taken into cells, incorporated into mineralized tissue matrices, or excreted from the body in urine. Hormonal regulation is critical to the homeostasis of absorbed Pi, with the primary locus being the kidney, as much of the absorbed Pi is excreted in the urine. Consequently, hormonal regulation of Pi excretion and reabsorption, more so than absorption, maintains circulating plasma concentrations (Drezner, 2002). This “parathyroid-kidney-intestine-bone/tooth” axis of Ca2+ and Pi balance is exhibited in Figure 1, with further description of the factors of interest featured in section II.
Figure 1. Serum calcium (Ca2+) and phosphate (Pi) levels regulate gene expression in the parathyroid-kidney-intestine- tooth axis.
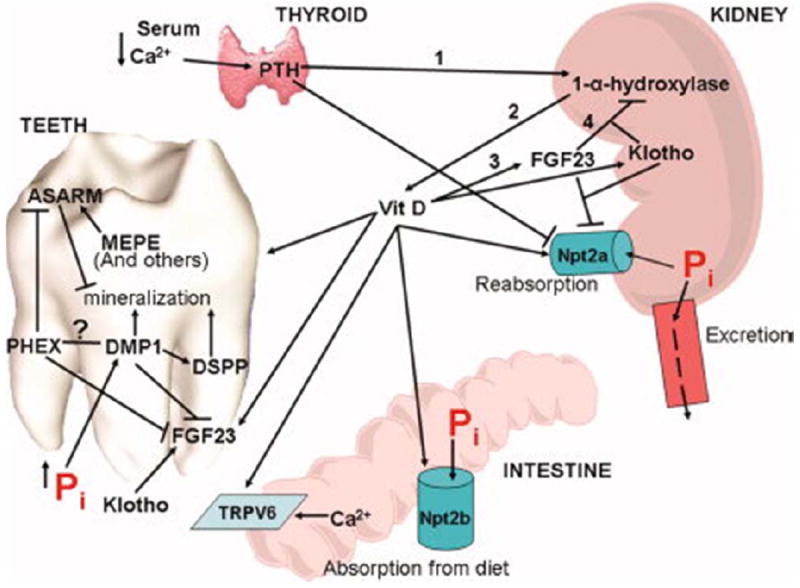
Decreases in serum Ca2+ induce the calcium sensing receptor in the parathyroid glands, embedded in the thyroid gland, to secrete PTH into the bloodstream. PTH stimulates the activity of 1-α-hydroxylase in the kidney which catalyzes the formation of the active 1,25 dihydroxy form of Vit D (line 1). PTH potently stimulates osteoclast activity to release Ca2+ from bone. Active Vit D increases intestinal absorption of Ca2+ via the TRPV6 Ca2+ channel and of Pi through the Npt2b ion channel. Renal reabsorption of Pi is increased by Vit D through increased Npt2c activity (line 2). PTH acts to reduce Pi reabsorbtion by down-regulation of Npt2a, while the induced increase in serum Ca2+ reduces secretion of PTH. Vit D has effects on tooth mineralization as evidenced by dentin defects under Vit D deficient states. Vit D acts to increase the expression of FGF23 in bone and the FGF23 receptor binding partner Klotho in the kidney (line 3). FGF23 reduces the activity of 1-α-hydroxylase, decreasing the formation of active Vit D (line 4), closing the loop began by PTH demonstrated by the lines numbered 1-4. SIBLING protein expression in bones and teeth are affected by Pi levels. Mutations in Phex induce expression of FGF23 in osteocytes and ameloblasts and odontoblasts, with concurrent increases in MEPE expression and ASARM formation. Loss of function mutations in Dmp1 induce an increase in FGF23 and decrease in DSPP expression levels. The gene expression changes in these mutations result in decreased mineralization in bones and teeth. The hyperphosphatemia in the FGF23 loss of function mutant may induce the observed increased expression of DMP1 in bone and tooth in a compensatory attempt to increase mineralization.
Renal Pi reabsorption, typically 80-90% of the glomerular filtrate, is mediated principally by the sodium-phosphate co-transporter Type 2a (NPT2a) in the proximal tubules, the expression of which regulates reabsorption (Murer et al., 1999; Murer et al., 1998). The expression and subcellular distribution of co-transporter protein is substantially affected by dietary phosphorus load; hyperphosphatemia decreases expression of NPT2a and hypophosphatemia increases expression. Tubular Pi reabsorption is enhanced by insulin-like growth factor (IGF-1), insulin, thyroid hormone, epidermal growth factor (EGF), Pi depletion, and Vit D, and inhibited by transforming growth factor alpha (TGFα), calcitonin, parathyroid hormone (PTH), parathyroid hormone related peptide (PTHrP), and glucocorticoids. PTH increases endocytosis of NPT2a from the cell membrane (Nashiki et al., 2005). In the proximal tubule, PTH induces mRNA expression of 25-hydroxy-vitamin D 1-α hydroxylase, a key enzyme in the production of Vit D, which will in turn mobilize Pi stored in the bone matrix. However, effects on Pi reabsorption are typically seen as secondary to the primary functions of these hormones. Whereas PTH is considered an important physiological regulator of renal Pi excretion, the balance between dietary intake and renal excretion is maintained in hyper-and hypoparathyroidism, suggesting that the proximal tubule intrinsically regulates excretion in response to dietary load (Levi et al., 1994). The two major classical Pi regulating hormones, PTH and Vit D, are discussed in detail in Section IIa and summarized in Table 1.
Table 1.
“Classical” regulators of calcium (Ca2+) and phosphate (Pi) homeostasis.
| Factor | Expression Pattern Cells/Tissues | Known/Putative Function | Murine KO/Mutation Models | Phenotype: | |
|---|---|---|---|---|---|
| General | Teeth | ||||
|
| |||||
| PTH: Parathyroid hormone | Secretion from parathyroid gland based on serum Ca2+ levels. | Secreted into circulation in response to low serum Ca2+ in order to increase osteoclast activity, releasing Ca2+ from bone stores. Acts indirectly by increasing RANKL expression in osteoblasts to stimulate osteoclasts in turn. | PTH receptor null | Mineralized tissues: Enhanced mineralization of chondrocytes during long bone formation | Alveolar bone: Osteoclast activity reduced, delayed eruption of teeth |
| Cells with PTH receptors include: Osteoblasts, odontoblasts, cementoblasts | Acts in kidney to stimulate formation of active Vit D (see below) | ||||
| Clinically, PTH has anabolic activity when delivered intermittently at low dose | |||||
|
| |||||
| Vit D: 1,25-dihyrdoxy vitamin D3 | Kidney | Stimulates, indirectly, increased Ca2+ and Pi absorption through intestine, and increased Ca2+ and Pi reabsorption in kidneys. Stimulates bone marrow stromal cell RANKL expression to free Ca2+ from bone matrix via osteoclast activity. | Nutritional deficiency | Mineralized tissues: Rickets/osteomalacia, hypocalcemia, hyperparathyroidism | Reduced expression of amelogenin and enamelin, disrupted enamel formation. Dentin mineralization defects including decreases in OCN and DPP |
|
|
|||||
| Stimulates osteoblast development by regulating expression of non-collagenous marker proteins | VDR null | Mineralized tissues: Rickets/osteomalacia, hypocalcemia, hyperparathyroidism | Thin dentin, widened and irregular predentin, increased pulp chamber | ||
|
|
|||||
| 25-hydroxyvitamin D-1-α-hydroxylase null | Mineralized tissues: Same as above, also secondary hyperparathyroidism, hypophosphatemia | Not reported | |||
Maintenance of normal circulating Pi levels is critical to the normal development, maintenance, healing, and repair (and presumably regeneration) of mineralized tissues, including teeth. For example, hypophosphatemia during development leads to dental and skeletal deformities (rickets) (Millan, 2006; Whyte, 2002). As stated above, the major determinants of serum Pi levels are dietary load and reabsorption of the renal glomerular filtrate in the proximal tubule. Additionally, other conditions, such as sepsis and insulin therapy, affect circulating Pi levels. Certain diseases, such as tumor-induced osteomalacia (TIO), X-linked hypophosphatemic rickets (XLH), and autosomal dominant hypophosphatemia (ADHR), which involve renal Pi wasting and affect mineralized tissues, have stimulated the search for hormones that specifically regulate Pi homeostasis (the so-called “phosphatonins”). Current candidate phosphatonins include fibroblast growth factor 23 (FGF23), matrix extracellular phosphoglycoprotein (MEPE), and secreted frizzled related peptide-4 (FRP4). The potential roles of phosphatonins during tooth development are discussed under Section IIb and summarized in Table 2.
Table 2.
Systemic/humoral regulators of phosphate (Pi) homeostasis: Phosphatonins, phosphatonin-like factors, and co-factors
| Factor | Expression Pattern Cells/Tissues | Known/Putative Function | Murine KO/Mutation Models | Phenotype: | |
|---|---|---|---|---|---|
| General | Teeth | ||||
|
| |||||
| FGF23: Fibroblast growth factor 23 | Highest expression in bone and highly expressed during active bone remodeling. Noted in osteocytes, osteoblasts, cementoblasts, odontoblasts, and low levels in chondrocytes, cementocytes, and osteoclasts | Regulates Pi homeostasis by controlling renal reabsorption via modulation of expression of Pi co-transporters (i.e., NPT2a, NPT2c). Inhibits expression of α-hydroxylase required for formation of Vit D. DMP1 may modulate Fgf23 expression. Vit D regulates Fgf23 expression. | Fgf23 KO | General: Premature aging-like features, e.g., shortened lifespan, hypogonadism, emphysema, organ atrophy | Alveolar bone: Accumulation of unmineralized osteoid associated with ankylosis, apoptotic cells |
| Lower levels in fetal vs. postnatal and adult tissues | Reports indicate that FGF23 signals through FGF type I receptors and requires Klotho binding for activation. Low FGF23 levels in fetal tissues, with high levels in young adult and adult tissues, suggests a role in mineral homeostasis vs. skeletal development. | Mineralized Tissues: Disorganized growth plate, accumulation of unmineralized osteoid, suppressed bone turnover, ectopic calcification, decreased mass & volume in long bones | Enamel: Defective in continually erupting rodent incisors, cyst-like appearance and lack of polarity to ameloblasts | ||
| Altered circulating levels used to diagnose Pi-wasting diseases. | Pulp: Narrow chamber, ectopic matrix formation | ||||
| Cementum: Normal appearance | |||||
| PDL: Disorganized, and decreased width | |||||
| Dentin: Normal appearance | |||||
|
| |||||
| Klotho | Expressed predominantly in kidney, also in choroid plexus, parathyroid gland, and other tissues. | Single-pass transmembrane domain and short cytoplasmic domain having b-glucuronidase activity | Klotho KO | General: Similar to Fgf23 KO mouse, e.g., rapid aging syndrome including shortened lifespan, skin and muscle atrophy, osteoporosis, and emphysema. | Incisors: Ectopic calcification, altered odontoblast morphology (with defects in predentin and dentin) |
| Soluble form in circulation | Extracellular domain: Shed and secreted in blood, considered a cofactor of FGF-23 signaling and suppressor of insulin/IGF-1 signaling. | Mineralized tissues: Accelerated osteoblast aging, ectopic calcification | Molars: Ankylosis with alveolar bone | ||
|
| |||||
| PHEX: Phosphate regulating gene with homology to endopeptidases on the X chromosome | Highest expression in osteoblasts, osteocytes, and odontoblasts | Interacts with (cleaves) small circulating factors outside of the kidney to control renal Pi homeostasis and mineralization (e.g., MEPE, DMP-1, and other SIBLINGs), releasing acidic serine and aspartic acid-rich MEPE-associated motif (ASARM) peptides (“minhibins”), potent inhibitors of mineralization | Hyp mutant mouse (3’ deletion in Phex gene) | Growth retardation, osteomalacia, reduced mineralization in growth plate, reduced bone volume and osteoclasts in long bone | Alveolar bone: Hypomineralization, increased osteoid |
| Regulates expression of Fgf-23 (may be indirect) | Dentin: Hypomineralization, interglobular dentin, widened predentin, irregular dentinal tubules | ||||
| Pulp chamber: Enlarged | |||||
| Enamel: Appears normal | |||||
| Cementum: Disrupted globular appearance at SEM level (appears normal by H&E) | |||||
|
| |||||
| DMP1: Dentin matrix protein 1 | High levels in osteocytes, also found in odontoblasts, cementoblasts/cytes | Considered to be required for mineralization; may regulate odontoblast and osteoblast/cyte specific genes; in osteocytes, considered to have a role in protection during mechanical loading; processed to NH2 and COOH fragments that are distributed differently in dentin and may have unique roles; suggested role as a transcription factor (regulates Dspp); may regulate Fgf-23 expression | Dmp1 KO | Increased osteoid, hypomineralized bone, defective osteocyte maturation, osteomalacia rickets, decreased bone mineral, increased crystal size | Dentin: Similar to Hyp mouse, e.g., hypomineralized with widened predentin |
| PDL: Disorganized, with detachment from cementum | |||||
No tooth phenotype reported for Mepe KO mouse; No animal models for loss of FRP4, FGF7.
Another important molecule that is pivotal for regulation of Pi metabolism is pyrophosphate (PPi). PPi is composed of two molecules of Pi, which are released upon hydrolysis, in addition to being produced by numerous other physiological reactions. PPi is ubiquitous in the body, being present in numerous bodily fluids (e.g., blood, urine, saliva, synovial fluid), at the cell and tissue level, and in the mineralized matrices of bones and teeth. The numerous biosynthetic reactions producing PPi, notably including synthesis reactions coupled to hydrolysis of nucleoside triphosphates (NTPs) such as ATP, have been extensively reviewed (Heinonen, 2001). Measurements of PPi synthesis and concentrations in vivo are difficult due to instability in aqueous solution and rapid turnover by numerous enzymes with PPi-ase activity. In vitro and in vivo data strongly support a role for PPi as a potent inhibitor of HAP crystal growth. The suggested mechanism for inhibition is the binding of PPi to HAP crystals, inhibiting growth and dissolution of the crystals (Fleisch and Bisaz, 1962; Fleisch et al., 1966; Meyer, 1984; Termine et al., 1970). Interestingly, PPi is included in tartar control toothpastes to inhibit dental calculus, which results from calcification of dental plaque, a mixture of microorganisms, their metabolic products, and components of saliva, including Ca2+ and Pi (Netuveli and Sheiham, 2004; Zacherl et al., 1985).
The metabolic reactions governing Pi and PPi in the body are inextricably linked—at times concerted, other times antagonistic, but always intimately connected by the close chemical nature of Pi and PPi, and by enzymes, transporters, and feedback mechanisms that dictate the locations and concentrations of Pi and PPi in and around the cell. Several proteins have been associated with local/cellular and systemic/humoral regulation of Pi/PPi homeostasis and these are introduced below and discussed in greater detail in section III.
b. The tooth: A phosphate sensitive organ
The teeth and their supporting tissues are complex epithelial-mesenchymal organs produced by several unique cell types and composed of four mineralized tissues, namely, the highly mineralized enamel covering the crown, the resilient dentin surrounding the pulp chamber and forming the bulk of the crown and root, the thin layer of cementum that covers the root and is essential for attachment of the tooth via the periodontal ligament (PDL), and the metabolically active alveolar bone comprising the bony socket and anchoring the tooth via the PDL (Nanci, 2003). The close approximation of four Pi-rich mineralized tissues (the only such occurrence in the body), as well as the important soft tissue interfaces, makes the tooth an intriguing organ for study of the developmental and homeostatic ramifications of Pi metabolism disorders. This reality is emerging as more studies indicate that the various mineralized tissues of the teeth are differentially regulated by prevailing Pi conditions (Boukpessi et al., 2006; Fong et al., 2008a; Foster et al., 2007; Nociti et al., 2002; Onishi et al., 2007; Toyosawa et al., 2004; van den Bos et al., 2005; Ye et al., 2004; Ye et al., 2008). In other words, systemic and local factors modulating Pi homeostasis seem to differentially control formation of oral mineralized tissues (e.g., pulp/dentin/bone versus follicle-PDL/cementum regions) based on a number of developmental disorders in humans, and as a result of gene mutation and knock out (KO) in mouse models, which will be described in detail in the text below and in the included Tables. These models will be described to provide insight into the specific roles of key regulators of Pi homeostasis during development, maintenance, disease, and repair/regeneration of tooth tissues.
c. Potential role for phosphate in regeneration of oral mineralized tissues
Oral health is essential for general health and nutrition as well as being a major determinant for quality of life. While there has been much progress in recent decades in prevention and treatment of oral-dental-craniofacial diseases, pathologies, and disorders, unfortunately they continue to be prevalent global health problems (WHO, 2003). Among approaches to treat congenital or acquired loss of mineralized tissues, repair or therapeutic regeneration may be second only to prevention in terms of importance. The major goal of regenerative therapy is to predictably restore normal tissue structure and function. Regeneration of mineralized tissues such as bones and teeth recapitulates development, at least in part, so that full understanding of the regulation of normal development will provide information key to designing effective therapies for the regeneration of teeth and associated tissues. Recognizing that regeneration of oral tissues is possible has resulted in increased attempts to identify factors and to understand their roles in controlling formation of oral tissues during development and regeneration (Bartold et al., 2000; Bartold et al., 2006a; Bartold et al., 2006b; Fong et al., 2005; Foster et al., 2007; Hu et al., 2006; Papaccio et al., 2006; Popowics et al., 2005; Saygin et al., 2000).
Clues to identifying candidate molecules for use in regeneration of oral tissues have come from studies focused on defining the factors involved in development of these tissues. Expression of specific genes/proteins have been mapped over time during tooth development using rodent models under normal conditions, as well as animal models where genes have been mutated, deliberately knocked out, knocked in, or conditionally expressed. In this regard, much has been learned from detailed analyses of the molecular basis of certain mutant animals where alterations in tooth development have resulted in abnormal adult teeth that significantly affect normal dental/oral function and health. This review is limited to factors involved in controlling Pi metabolism and their known/putative effects on the hard tissues of the oral cavity.
Regulators of Pi metabolism have received considerable attention over the last decade due to the role of Pi as a component of HAP mineralization and because of evidence that Pi may regulate cell behavior and mineralization as a signaling molecule. This review aims to consolidate and summarize the most exciting research on Pi metabolism and mineralized tissues from recent years, focusing especially on the association with the hard tissues of the oral cavity. The next section, Section II, and Tables 1 and 2 will cover hormones governing serum Pi levels, followed by Section III and Table 3, which introduce local, microenvironmental Pi/PPi regulatory elements, meaning those elements located in, on, or around the cell that contribute to Pi metabolism. Section IV will discuss accumulating evidence that Pi may act as a cell signaling molecule, modulating gene expression and cell function. Current therapeutics and materials with a basis in Pi metabolism will then be discussed in Section V. Lastly, under Section VI, we will attempt to synthesize this information and provide a vision for applying knowledge of Pi metabolism to regenerative approaches for the oral mineralized tissues.
Table 3.
Local/cellular regulators of phosphate (Pi) and pyrophosphate (PPi) metabolism.
| Factor | Expression Pattern Cells/Tissues | Known/Putative Function | Murine KO/Mutations Models | Phenotype: | |
|---|---|---|---|---|---|
| General | Teeth | ||||
|
| |||||
| TNAP: Tissue non-specific alkaline phosphatase (ALP, TNSALP, ALKP) | Expressed by diverse cells including osteoblasts, PDL cells, odontoblasts, follicle cells, cementoblasts, and chondrocytes, among others. Also present in matrix vesicles (MVs). | Marker of osteoblast differentiation, catalytic function in mineralization, cleaves the mineralization inhibitor, PPi | Tnap mutation/KO | Hypophosphatasia: Increased local levels of PPi, osteopenia | Cementum: Hypoplasia or aplasia with compromised PDL attachment, exfoliation of teeth |
| Gene: Akp2 | |||||
|
| |||||
| ANK: Progressive ankylosis gene/protein. (Human homologue, ANKH) | Expressed by diverse cells including osteoblasts, PDL cells, odontoblasts, follicle cells, cementoblasts, and chondrocytes, among others. | Transporter/co-transporter of PPi from intracellular to extracellular space; inhibits HAP deposition via increased extracellular PPi | Ank mutation/KO | Decreased extracellular PPi, ectopic calcification | Cementum: Hypercementosis (increase ~10 fold), unusually cellular cementum may result from rapid matrix accumulation |
|
| |||||
| PC-1: Plasma cell membrane glycoprotein 1 (NPP1-nucleotide pyrophosphatase phosphodiesterase–1) | Expressed by diverse cells including osteoblasts, PDL cells, odontoblasts, follicle cells, cementoblasts, and chondrocytes, among others. Also present in matrix vesicles (MVs). | Increases intra/extracellular and MV PPi, inhibits HAP deposition via increased extracellular PPi | Enpp1 mutation (tiptoe walking, ttw/ttw) | Decreased extracellular PPi, ectopic calcification | Cementum: Similar to Ank mutant and KO (e.g., hypercementosis) |
| Gene: Enpp1 | |||||
II. Systemic hormone regulators of phosphate metabolism (Figure 1)
Some Pi metabolism regulating factors, discussed briefly in the introduction, exert their influence by modulating circulating Pi by actions in the kidneys and gastrointestinal tract. These systemic or humoral regulators of Pi include the “classical” regulators of systemic Ca2+ and Pi, Vit D, and parathyroid hormone (PTH). Systemic acting factors discovered and characterized more recently include endopeptidases on the X-chromosome (PHEX), and a collection of phosphaturic peptides identified in sera of individuals with disorders/diseases associated with renal Pi wasting. High levels of these peptides have been associated with hypophosphatemia and osteomalacia. These include the phosphatonin-like factors FGF23 (Quarles, 2003; Rowe, 2004; White et al., 2006), FRP4 (Berndt et al., 2003; Berndt et al., 2006; Berndt et al., 2005; Vaes et al., 2005; White et al., 2006), matrix extracellular phosphoglycoprotein (MEPE) (Argiro et al., 2001; Fisher and Fedarko, 2003; MacDougall et al., 2002; Quarles, 2003; Rowe, 2004; Rowe et al., 2000; Rowe et al., 2004; White et al., 2006), and FGF7 (Berndt et al., 2005; Carpenter et al., 2005). Both FGF23 and SFRP4 have been shown to inhibit 25(OH)D3 1 α-hydroxylase activity (normally increased in conditions causing hypophosphatemia) and are thus considered “phosphatonins,” i.e., a label attributed to factors that increase renal loss of Pi and inhibit Vit D synthesis (Berndt et al., 2005; Econs and Drezner, 1994). Importantly, factors that alter Ca2+/Pi in the sera affect both PTH and Vit D levels, adding an additional complexity to studies targeted at defining the mechanisms by which factors regulating circulating Pi levels modulate mineral homeostasis. Several publications provide excellent reviews of these factors (Berndt et al., 2005; Razzaque et al., 2005; Renkema et al., 2008; Rowe, 2004; Schiavi, 2006; Schiavi and Kumar, 2004; Yu and White, 2005b) and Figure 1 and Tables 1 and 2 provide an overview of the actions of the above-mentioned systemic factors.
a. “Classical” hormonal regulators of phosphate metabolism: 1, 25 (OH)2D3 and parathyroid hormone (Table 1)
The regulation of serum levels of electrolytes, especially Ca2+ and Pi, is of great importance for physiological functions such as nerve impulses, blood clotting, contraction of muscles (Ca2+), and as constituents of cellular membranes, DNA, and signal transduction (Pi). Hypocalcemia may result in seizures and muscle cramping while symptoms of hypercalcemia may include kidney stones, nausea, and fatigue (Gunn and Gaffney, 2004). Hypophosphatemia results in lowered bone mineralization and rickets, and hyperphosphatemia is characterized by soft tissue mineralization and excess Vit D levels. The appropriate levels of Ca2+ and Pi are maintained by the manipulation of a parathyroid-intestinal-kidney-bone axis under the influence of multiple endocrine system effector molecules. These players are featured in Figure 1, with an inclusion of tooth rather than bone as per the focus of this review. The molecules which have been classically believed to regulate Ca2+ and Pi levels are PTH, Vit D, and calcitonin, acting via a self-limiting feedback loop mechanism. It had long been perceived that Ca2+ levels were paramount in influencing the actions of these molecules, and that Pi levels simply followed the flow of Ca2+ in and out of the body, to maintain electrical balance. However, it has now become apparent that the regulation of Pi levels is under as strict control as Ca2+ by newly discovered agents such as FGF23 and others.
i. Parathyroid hormone (PTH)
Overview
The primary structure of PTH was first identified in 1970 as an 84 amino acid protein (Niall et al., 1970), of which the biological activity was found to reside in the amino terminal 34 residues (Rosenblatt et al., 1978; Tregear et al., 1973). The PTH receptor was identified in 1991, and was found to be a G-protein coupled receptor using a cAMP second messenger system (Henderson et al., 1992). PTH is secreted from the parathyroid gland based on serum Ca2+ levels which are detected by the calcium sensing receptor (CR). Low levels of Ca2+ inhibit CR activity, allowing PTH to be released into the circulation to achieve its downstream effects (Riccardi et al., 1995). A major action of PTH is to increase osteoclast activity in order to release Ca2+ from bone. This action is indirect in that osteoblasts respond to PTH signals by increasing their production of receptor activator of nuclear factor-kβ ligand (RANKL) (Suda et al., 1999). RANKL binds to RANK on osteoclast precursor cells and directs their differentiation (Kondo et al., 2002). PTH also induces osteoblasts to secrete osteoprotegerin (OPG), a RANK decoy ligand and regulator of osteoclast activation (Fu et al., 2002). In addition, PTH acts in the kidney to stimulate the formation of active Vit D which results in both increased Ca2+ absorption from the intestine and reabsorption in the kidneys, while reducing the reabsorption of Pi in the kidney by endocytic removal of NaPi2a transporters (Nashiki et al., 2005). High levels of Ca2+ stimulate the CR which inhibits PTH release, and increases calcitonin release from the thyroid, inhibiting osteoclast activity (Fudge and Kovacs, 2004). The importance of PTH functioning in development was highlighted in the PTH receptor null mouse, which had a phenotype exhibiting enhanced mineralization of the chondrocytes during long-bone formation (Lanske et al., 1996). This in part contributes to the ability of PTH-associated peptides to act in both anabolic and catabolic fashions depending on dose and method of delivery, both in vitro and in vivo (Jilka, 2007; Pettway et al., 2008; Poole and Reeve, 2005).
Tooth-specific findings
There is some evidence that PTH has direct effects on dental cells. Both odontoblasts and cementoblasts are reported to have cell surface PTH receptors, and odontoblast cells responded to PTH treatment with increased expression of tissue nonspecific alkaline phosphatase (TNAP), in vitro (Lundgren et al., 1998; Tenorio and Hughes, 1996). Rats fed a diet which resulted in increased in vivo PTH levels had an increase in the width of the predentin, the unmineralized dentin matrix of the tooth, compared to controls (Engstrom et al., 1978; Engstrom et al., 1977). Several studies have been performed either over-expressing or eliminating the function of the PTH receptor which resulted in dental phenotypes. The PTHR null mouse exhibited a phenotype in which osteoclast activity was greatly reduced with a resultant arrest in the eruption of teeth of normal morphology (Kitahara et al., 2002). Expression of constitutively active PTH receptors invoked abnormal tooth development (Calvi et al., 2004). However, this may not be due to PTH activity per se; PTHrP is a peptide with amino terminal homology to PTH and shares the same receptor (Juppner et al., 1991). PTHrP was initially found as the cause of hypocalcemia of malignancy, and has been since reported to be involved in epithelial-mesenchymal signaling in organ development, including teeth (Suva et al., 1987; Wysolmerski et al., 2001). PTHrP has been shown to have direct effects on cementoblast cells in vitro, reducing the mRNA expression of both bone sialoprotein (Bsp) and osteocalcin (Ocn), and inhibiting mineralization (Ouyang et al., 2000).
ii. 1, 25 (OH)2D3 (Vit D)
Overview
Vitamin D was first discovered in 1920, with its chemical structure elucidated in 1932, and was soon understood to be pivotal for proper bone formation and Ca2+ regulation (Brockman, 1936; Mellanby, 1921). The vitamin D precursor cholecalciferol can be produced in response to sunlight (UV light) in the skin, or as part of dietary intake (Goldblatt and Soames, 1923). However, this pre-vitamin D3 molecule must undergo two sequential hydroxylations in order to produce the active metabolite Vit D (1,25 (OH)2D3 or calcitriol). Cholecalciferol first moves through the bloodstream to the liver with the vitamin D binding protein (DBP) due to its high degree of hydrophobicity (Cooke and Haddad, 1989). In the liver, cholecalciferol receives a hydroxyl group at carbon 25 from the action of cytochrome P450 25-hydroxylase enzymes such as CYP27A1 (Oftebro et al., 1981). 25(OH)D3 then, again with DBP, moves to the proximal kidney tubule to receive another hydroxylation at the 1-α position via cytochrome P450 25-hydroxyvitamin D-1-α-hydroxylase (CYP27B1) (Henry and Norman, 1974). This renal enzyme is stimulated by PTH in response to low Ca2+ levels in the blood, or low Vit D levels (Brown et al., 1995). The active metabolite, Vit D, moves with DBP to its target tissues, one of which is the parathyroid gland to inhibit the release of PTH in a negative feedback loop (Russell et al., 1993).
Due to its hydrophobic nature, Vit D readily crosses the cell membrane, and the Vit D receptor (VDR) is located in the cytosol of target cells, similar to receptors for other steroids (Haussler, 1986). In addition to the main Vit D target organs, kidney, intestine, and bone, many other tissues/cell types express the VDR, suggesting Vit D has direct effects in addition to its electrolyte balancing function (Bouillon et al., 1995). Once inside the cell, Vit D binds to VDR which then forms a heterodimer with the retinoid X receptor, which is required for its actions (Sutton and MacDonald, 2003). This complex then moves to the nucleus and binds to the DNA of Vit D response elements present in the promoters of Vit D target genes, acting as either enhancers or repressors (Prufer et al., 2000). Vit D can also act indirectly by reducing the effects of other transcription factors such as NF-kβ (Harant et al., 1997).
Vit D exerts its effects on mineral homeostasis in the intestine to increase the absorption of Ca2+ and Pi, and in the kidney to increase their reabsorption in the kidneys from the glomerular filtrate. In the intestine, Vit D stimulates the expression of the Ca2+ channel TRPV6, which moves Ca2+ into the cell, and also of calbindin-D9K which moves it across the cell and PMCA1b which allows Ca2+ into the bloodstream (Hoenderop et al., 2005; Peng et al., 1999). In the kidney, Vit D increases Ca2+ reabsorption via up-regulating the expression of TRPV5, calbindin-D28k, and PMCA1b Ca2+ transport proteins (Hoenderop et al., 2005). Vit D increases the expression levels of the sodium dependant Pi transporter IIb (NaPi2b) in the intestine and NaPi2c in the kidney, which increases reabsorption (Barthel et al., 2007; Capuano et al., 2005). Vit D also stimulates the bone marrow stromal cells to produce RANKL, which induces osteoclastogenesis, thus indirectly increasing the body Ca2+ levels (Udagawa et al., 1990).
Apart from its role in mineral homeostasis, Vit D has direct effects on developing bone cells, stimulating the expression of OCN, TNAP, and osteopontin (OPN) in osteoblasts (Chang and Prince, 1993; Mulkins et al., 1983; Yoon et al., 1988). Vit D may also work synergistically with the Wnt and Notch signaling proteins in directing osteoblast development (Fretz et al., 2007; Shen and Christakos, 2005).
With the importance that Vit D plays in both mineral metabolism and osteoblast development, it is not surprising that these systems are affected in Vit D deficiency states. Nutritional deficiency in Vit D is characterized by rickets/osteomalacia, hypocalcemia, and secondary hyperparathyroidism (Nagpal et al., 2005). In mice lacking the VDR, the same findings were present and the mice did not survive past 4-6 months (Nagpal et al., 2005). Mice lacking the 25-hydroxyvitamin D-1-α-hydroxylase gene had a phenotype characterized by rickets at birth, and then osteomalacia upon aging, with secondary hyperparathyroidism, hypocalcemia, and hypophosphatemia (Panda et al., 2001).
Tooth findings
As mineralized structures, the teeth have long been considered as potential target organs for the actions of Vit D, as a consequence of Ca2+/Pi homeostasis abnormalities, as well as via direct effects. The VDR was identified by immunohistochemistry to be present in differentiating ameloblasts and odontoblasts in rat incisors, and was increased following a single dose administration of Vit D (Berdal et al., 1995). Vit D target genes, the calbindins, are expressed in rat incisor ameloblasts and odontoblasts and the expression of these genes also rose following a dose of Vit D. Studies using Vit D deficient animals have shown a reduced expression of amelogenin and enamelin, resulting in a disruption of enamel formation with an increase in interprismatic enamel, which was restored with Vit D treatment (Papagerakis et al., 2002). In Vit D deficient rats, the dentin had mineralization defects which may have resulted from noted decreases in the OCN and dentin phosphoprotein (DPP) in the rachitic animals (Berdal et al., 1991). In vitro studies have shown that dentinogenic cells exposed to Vit D exhibited increased expression of OPN and OCN, and TNAP, while the expression of dentin sialophosphoprotein (DSPP) was not affected (Bronckers et al., 1998; Ritchie et al., 2004; Tsukamoto et al., 1992). Mice lacking VDR expression had a tooth phenotype of thin dentin walls in the incisor, with an increase in pulp chamber space, and a widened predentin layer with an irregular border to the mineralized dentin (Zhang et al., 2007b). Humans with familial hypophosphatemic rickets, characterized by low circulating Vit D concentrations, have teeth with defects in mineralized dentin resulting from unmerged calcospherites (Chaussain-Miller et al., 2007). Treatment of these patients with Vit D and increased dietary Pi resulted in a normalization of dentin mineralization. Interestingly, as described below (and in Table 2), mice and humans with loss of PHEX or dentin matrix protein 1 (DMP-1) are hypophosphatemic and exhibit a similar tooth phenotype.
b. Phosphatonins: Emerging systemic regulators of phosphate metabolism (Table 2; Figures 1 and 2)
Figure 2. Phosphate disregulation and tooth root tissues.
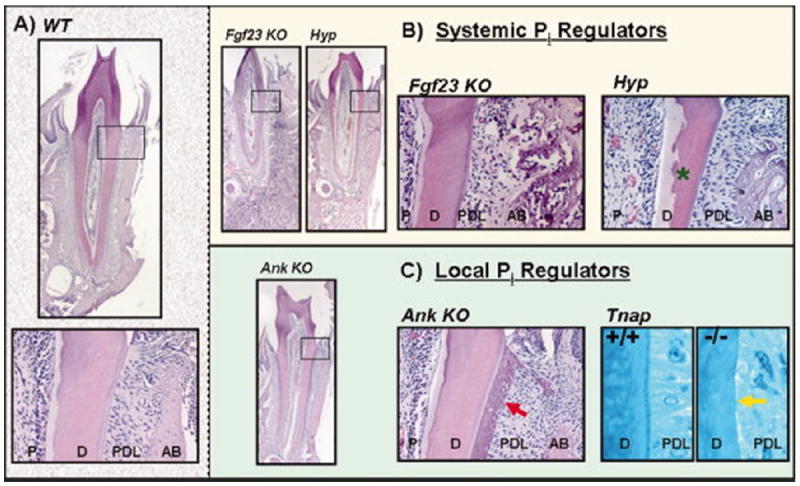
Mouse first mandibular molars here serve as models for effects of Pi disregulation on tooth roots and supporting tissues. All buccal-lingual sections shown are from 45 dpc mice, except Tnap images, which are from mice age 61 dpc. (A) Wild-type (WT) mouse shown for comparison. Top: Low magnification, Bottom: High magnification of the cementum-enamel junction (CEJ). (B) Systemic Pi regulation: Ablation of Fgf23 resulted in narrowed PDL space and a marked increase in alveolar bone. Alveolar bone exhibited high cell density and appeared to be primarily composed of osteoid. Phex mutations (Hyp mouse) resulted in enlarged pulp chambers, widened predentin, reduction in mineralized dentin, interglobular dentin (green star), and an increase in alveolar bone with pockets of osteoid. (C) Local Pi regulation: Loss of Ank resulted in dramatically thicker cementum (red arrow) while dentin, PDL, and alveolar bone appeared normal. Alternately, Tnap mutations (-/-) resulted in a failure to form cementum (yellow arrow). Tnap images courtesy of Dr. Wouter Beertsen. P=pulp, D=dentin, PDL=periodontal ligament, AB=alveolar bone.
The existence and function of endocrine regulators of Ca2+ such as PTH and Vit D have been known and investigated for many decades, with anionic Pi levels being believed to have a somewhat “passive” regulation to maintain electrical balance with cationic Ca2+. There exists, however, a variety of human phosphate wasting conditions characterized by reduced reabsorption of Pi from the urine and mineralization defects of varying degree. These conditions include X-linked hypophosphatemic rickets (XLH), autosomal dominant hypophosphatemic rickets (ADHR), autosomal recessive hypophosphatemic rickets (ARHR), and tumor-induced osteomalacia (TIO) (Bielesz, 2006; Quarles, 2003). The existence of these conditions led to the hypothesis that there is a class of molecules termed “phosphatonins”, whose function was to regulate Pi levels in the body (Berndt et al., 2005; Schiavi and Kumar, 2004). Molecular biology techniques have allowed for the elucidation of the genetic aberrations and proteins responsible for the pathological handling of Pi levels in these conditions, and have opened new avenues in research into the endocrine regulation of Ca2+/Pi levels and closed the functional loop began by the PTH/Vit D bone-kidney axis. The primary phosphatonins, to date, include, fibroblast growth factor 23 (FGF23), matrix extracellular phosphoglycoprotein (MEPE), and FRP4, as well as molecules that regulate, albeit indirectly, FGF23 or MEPE function including Pi-regulating gene with homologies to PHEX and (DMP1). As investigations continue, other phosphatonin-like molecules are likely to be identified. This review is limited to phosphatonins and associated factors that have a known tooth phenotype when function is lost, i.e., PHEX, DMP-1, FGF-23, KLOTHO, and MEPE. Actions of these phosphatonins and related factors in conjunction with the classical Ca2+ and Pi regulators are summarized in Figure 1.
i. Phosphate regulating gene with homology to endopeptidases on the X-chromosome (PHEX)
General
PHEX is an endopeptidase whose physiological substrates are currently unknown. PHEX expression appears to be limited to osteoblasts, osteocytes, and odontoblasts, suggesting a role in the mineralization process (Ruchon et al., 2000). Positional cloning was used to determine that Phex mutations were responsible for XLH, the most common form of rickets in humans (HYP_Consortium, 1995), which is characterized by rickets and osteomalacia, hypophosphatemia, reduced growth, and altered Vit D levels (Rasmussen and Tenenhouse, 1995). There is a naturally occurring mouse mutation homologous to human XLH, the so-called Hyp mouse (Eicher et al., 1976). The Hyp mouse has a reduced body weight compared to wild type (WT), with lower serum Pi levels, greatly reduced Vit D levels, and increased PTH levels, yet is normo-calcemic (Liu et al., 2006). Morphologically, the Hyp mouse has shortened bones with rachitic splaying at the epiphysis, and a widening of the growth plate due to an expansion of the hypertrophic zone, and an increase in unmineralized osteoid compared to WT. Paradoxically, transgenic expression of Phex in the Hyp mouse restored the bone phenotype, but did not affect the Pi metabolism deficiencies (Erben et al., 2005). It has been determined that in the Hyp mouse there is a dramatic increase in the expression levels of FGF23 in osteocytes (Liu et al., 2003). The over-expression of FGF23 has been shown to be responsible for much of the pathology in the Hyp mouse (Liu et al., 2006; Sitara et al., 2004). Further discussion of PHEX and its relationship to MEPE and FGF23 will be discussed in detail in sections II.b.ii and II.b.iii below.
Tooth findings
There is a record in the dental literature of dentin/pulp related disorders in XLH patients, including increased predentin, globular dentin, abnormal dentinal tubule distribution, and enlarged pulp chambers (Abe et al., 1988; Baroncelli et al., 2006; Cohen and Becker, 1976; Murayama et al., 2000; Shields et al., 1990). Importantly, Hyp mouse molar teeth exhibit a similar defect, as shown in Figure 2 (Abe et al., 1992; Abe et al., 1989; Ogawa et al., 2006; Sofaer and Southam, 1982). In the Hyp mouse tooth, an increase in Ocn mRNA was noted in the odontoblasts (Onishi et al., 2005). The expression of FGF23 was higher in both developing ameloblasts and odontoblasts in Hyp mouse, and the expression of NaPi2b transporter protein was reduced (Onishi et al., 2007; Onishi et al., 2008). Additionally, in human XLH subjects, there have been some reports of periodontal involvement, including cementum abnormalities, periodontal abscesses, and loss of lamina dura indicating alveolar bone disruption (Cohen and Becker, 1976; Murayama et al., 2000). These reports in humans parallel our recent findings that demonstrate, in addition to defects in dentin as described above, using SEM, a globular cementum morphology was apparent in Hyp versus WT, as well as a similar pattern in incisor cementum from a 5 yr old girl diagnosed with hypophosphatemic rickets (Chu et al., 2007; Fong et al., 2008a).
ii. Matrix extracellular phosphoglycoprotein (MEPE)
MEPE was identified in a tumor-induced osteomalacia (TIO) patient, and is a member of the SIBLING (Small Integrin-Binding Ligand N-linked Glycoprotein) family, a group also including DMP1, dentin sialophosphoprotein (DSPP), bone sialoprotein (BSP), and osteopontin (OPN), which are implicated in mineralization (Fisher and Fedarko, 2003; Rowe et al., 2000). Physiologic expression of MEPE is seen in osteocytes, odontoblasts, hypertrophic chondrocytes, and in the callus of bone fractures, which suggests it has a function in bone and dentin mineralization (Argiro et al., 2001; Fisher and Fedarko, 2003; Lu et al., 2004; MacDougall et al., 2002; Nampei et al., 2004; Rowe et al., 2000). MEPE has been shown to be suppressed by an injection of Vit D in mice, and it is up-regulated in the VDR null mouse (Rowe et al., 2004). Increased expression of MEPE protein has been noted in XLH patients and the homologous Hyp mice (Argiro et al., 2001; Guo et al., 2002; Liu et al., 2005; Rowe, 2004; Rowe et al., 2004). The MEPE null mouse had an increase in both cortical and trabecular bone, indicating that MEPE functions as an inhibitor of mineralization under conditions of normal Pi and Vit D (Gowen et al., 2003). A tooth-specific phenotype has not been identified in Mepe null mice. MEPE may play a role in the hypophosphatemia in TIO patients as it has been shown to reduce NaPi expression in opossum kidney cells in vitro, and lowers serum Pi in mice when injected (Gowen et al., 2003; Rowe et al., 2000). The functional mineralizing portion of MEPE is the acidic serine-aspartate rich MEPE associated Motif (ASARM) (Martin et al., 2008). It was formerly thought that MEPE was a PHEX substrate that could release the ASARM moiety upon cleavage, however, further studies showed that MEPE is not a substrate of PHEX, and in fact PHEX seems to protect MEPE (and possibly DMP1) from cleavage by cathepsin B, which frees ASARM (Campos et al., 2003; Rowe et al., 2004). In fact, newer investigations suggest that functional PHEX is capable of cleaving ASARM peptides, thus regulating the inhibitory effect of ASARM on HAP crystal growth (Addison et al., 2008). The decrease in mineralization noted in the Hyp mouse may therefore be a combination of increased MEPE expression, coupled with a lack of PHEX-mediated protection from degradation of MEPE and inability to control ASARM degradation, producing an excess of ASARM that may in turn play a major role in the decreased mineralization characterizing the Hyp mouse (Rowe, 2004). Mice null for Phex (Hyp) and for Mepe were crossed, and the resultant offspring did not have a correction of the mineralization inhibition, or hypophosphatemia and low Vit D levels seen in the Hyp mouse (Liu et al., 2005). This indicated that MEPE was not the prime phosphatonin defining the mineralization phenotype in Hyp mice. Dental findings from mice lacking MEPE have not been reported to date.
iii. Fibroblast growth factor 23 (FGF23) and Klotho
General
FGF23 is a member of the FGF family, a group of 22 proteins having various effects on cellular development, differentiation, and function. The Fgf23 gene localized to human chromosome 12p13 encodes the 251 amino acid FGF23 protein (Yamashita et al., 2000). Autosomal dominant hypophosphatemic rickets (ADHR) is a disorder characterized by low serum Pi, low Vit D levels, rickets and osteomalacia, and normal PTH levels. FGF23 was identified as the target of the genetic defect responsible for ADHR, and also found to be highly expressed in tumors responsible for TIO, another condition characterized by Pi wasting (ADHR_Consortium, 2000; Shimada et al., 2001; White et al., 2001). FGF23 was over-expressed in Chinese hamster ovary (CHO) cells that were implanted into SCID mice. The resulting tumors generated FGF23 and these mice developed the ADHR/TIO phenotype of osteomalacia, low serum Pi, low Vit D (with low levels of 25-hydroxyvitamin D-1-α-hydroxylase in the kidney) (Shimada et al., 2001). These data suggested that FGF23 functions as a phosphatonin, affecting Pi levels directly or by regulating the actions of Vit D. FGF23 has since been documented to negatively regulate renal expression of NaPi2a and NaPi2c, thereby decreasing Pi reabsorption in the kidney, and decreasing expression of 25-hydroxyvitamin D-1-α-hydroxylase in the kidney, thus reducing the levels of active Vit D as well as stimulating expression of 24-hydroxylase, which is catabolic for Vit D (Roy et al., 1994; Segawa et al., 2003; Shimada et al., 2004).
FGF23 expression has been detected by RT-PCR in multiple organs including heart, skeletal muscle, intestine, and liver, but it has been determined that the primary source is bone forming cells, especially the osteocytes, and in the bone fracture repair callus (ADHR_Consortium, 2000; Riminucci et al., 2003). Immunohistochemical results suggest that FGF23 expression is mainly in osteoblasts and osteocytes in bone, odontoblasts and cementoblasts in teeth, and in growth plate chondrocytes (Yoshiko et al., 2007). This study also indicated that the expression of FGF23 was greater in adult than fetal tissues. Evidence for responsiveness of FGF23 levels to serum Pi levels has been inconsistent, with reports showing little if any response by FGF23 to increases in dietary Pi in human subjects, to several reports in which FGF23 levels increased with dietary Pi load in mice (Berndt and Kumar, 2007; Ferrari et al., 2005; Larsson et al., 2003; Perwad et al., 2005). The expression of FGF23 has been shown to be increased by Vit D, although this action may be indirect, as the increase could be blocked by the administration of cycloheximide (Kolek et al., 2005). An FGF-inducing factor may also be produced by chondrocytes in response to Vit D signaling (Masuyama et al., 2006).
The pathologic over-expression of FGF23 is responsible for the development of multiple disease states characterized by Pi wasting, low levels of Vit D, and bone mineralization defects including rickets/osteomalacia, such as ADHR, TIO, XLH, and ARHR in humans, and the Hyp phenotype in mice with a Phex mutation. In ADHR, FGF23 is expressed in a mutated form resistant to proteolytic degradation (White et al., 2001). When full length FGF23 or either of its two breakdown fragments were injected into rats, only the full length form reduced serum Pi levels, suggesting that an accumulation of active FGF23 in ADHR patients results in the observed pathology (Shimada et al., 2002). The tumors present in TIO have been determined to frequently express high levels of FGF23, and conditioned media from the tumors injected into mice caused increased renal Pi clearance, and the Pi wasting in patients was resolved following resection of the tumors (Shimada et al., 2001). In XLH patients and Hyp mice, there is a mutation in Phex which appears to cause an increase in FGF23 levels. It was hypothesized that FGF23 was a PHEX substrate, and the lack of PHEX degradation of FGF23 led to its increase. However, FGF23 does not appear to be a PHEX substrate, and there is a frank over-expression of FGF23 in osteocytes examined from Hyp mice (Benet-Pages et al., 2004; Quarles, 2003). It is hypothesized that a natural substrate of PHEX regulates FGF23 expression and the buildup of this factor in the absence of PHEX results in the higher expression levels of FGF23 seen in the Hyp mouse. It is unclear if FGF23 has direct actions on bone forming cells, but FGF23 expression has been noted in the repairing callus of fractured bones, and Hyp osteoblasts transplanted into WT mice demonstrated mineralization defects (Ecarot et al., 1992; Liu et al., 2003; Riminucci et al., 2003). Autosomal recessive hypophosphatemic rickets (ARHR) is a newly classified Pi wasting disorder, in which mutations have been found in the Dmp1 gene, following the recognition that the Dmp1 null mouse has a phenotype similar to the Hyp mouse and to that of individuals afflicted with ADHR and XLH (Farrow et al., 2007). Osteocyte production of FGF23 is increased in the Dmp1 null mouse, which could account for some of the observed phenotype (Feng et al., 2006). DMP1 and its role in mineralization and effects of its deletion in mice will be discussed under section II.b.iv below.
Human tumoral calcinosis (TC) is the result of the loss of function of FGF23. Patients having TC are reported to have high serum Pi levels, increased Vit D levels, calcification in vascular and soft tissues, and mineralized tumor masses (Mitnick et al., 1980; Prince et al., 1982). There are two different mutations which result in the observed reduced FGF23 function. One is a mutation in the Galnt3 gene, which encodes an enzyme responsible for protein glycosylation, suggesting that inappropriate glycosylation of FGF23 inhibits its function (Topaz et al., 2004). A mutation in Fgf23 at serine residues, different from that in ADHR, also results in the development of TC (Araya et al., 2005). Serine residues are potential sites for glycosylation, which strengthens the notion that proper glycosylation is a requirement for FGF function.
Molecular biology techniques have been employed to further understand the function of FGF23 and the pathology associated with gain or loss of function. Several groups have generated transgenic mice that over-expressed Fgf23. These mice recapitulated the TIO phenotype of rickets and osteomalacia, abnormally low levels of Vit D, low serum Pi with renal Pi wasting, and growth reduction, confirming the involvement of FGF23 in the pathology seen in TIO, XLH, ADHR, and ARHR patients, and in Hyp mice (Larsson et al., 2004; Shimada et al., 2004). Other groups have produced mice in which the Fgf23 gene has been removed (Fgf23 KO). These null mice had some characteristics of TC with soft tissue calcifications, hyperphosphatemia, high levels of Vit D, as well as multiple atrophic organs such as thymus, testis, uterus, and skin, shortened lifespan, reduced growth, and a reduction in the hypertrophic zone of the growth plate (which remained at normal width) of endochondral bones (Liu et al., 2006; Shimada et al., 2004; Sitara et al., 2004). These null models were further investigated to determine the participation of FGF23 in the Hyp mouse by producing a Hyp/Fgf23 KO cross (Liu et al., 2006; Sitara et al., 2004). As described under section II.b.i., the Hyp mouse has a phenotype of rickets and osteomalacia, shortened long bones with rachitic splaying, increased FGF23 and PTH, low Vit D, low serum Pi, and normal Ca2+, and expanded growth plate due to an expansion of the hypertrophic zone (Liu et al., 2006). The compound mice had no detectable expression of FGF23, high levels of Vit D, low PTH, hyperphosphatemia, normal Ca2+, shortened bones, and a resolution of rickets, but not osteomalacia, with growth plates similar to the Fgf23 KO (Liu et al., 2006; Sitara et al., 2004). It is clear that much of the phenotype in the Hyp mouse is due to the Fgf23 over-expression, and its removal superimposes the Fgf23 KO phenotype onto the Hyp mouse. It was then determined that much of the phenotype of the Fgf23 KO is the result of the over-expression of Vit D when the Fgf23 KO was crossed with a mouse null for 25-hydroxyvitamin D-1-α-hydroxylase (Sitara et al., 2006). The resultant mouse showed a change from hyperphosphatemia in the Fgf23 KO mouse to hypophosphatemia in the double KO, with a reduction in renal expression of NaPi2a, and loss of the ectopic calcifications noted in the Fgf23 KO, and a slight restoration in size of the animal. The phenotype of the 25-hydroxyvitamin D-1-α-hydroxylase null mouse of hypophosphatemia and rickets seemed to be superimposed on the Fgf23 KO phenotype, suggesting that the excess Vit D in the Fgf23 KO is a major contributor to the noted pathology (Dardenne et al., 2001). An additional study crossed the Fgf23 KO with a VDR null mouse. In addition to the resultant phenotype just noted, this group also reported a recovery in the organ atrophy in the Fgf23 KO (Hesse et al., 2007). It is interesting to note that in the various endocrine conditions characterized by mishandling of Pi metabolism, existing evidence suggests that the observed phenotypes may not result from direct effects of the mutated genes in question. The similar phenotypes resulting from loss of PHEX in humans (XLH) and mice (Hyp mutants) and loss of DMP1 in humans (ARHR) may have a stronger association with noted high levels of FGF23 (as opposed to loss of PHEX or DMP1). Additionally, the phenotype expressed in the absence of FGF23 in the Fgf23 KO mouse may be in large part a consequence of the increase in active Vit D.
The signal transduction mechanisms by which FGF23 exerts its effects have begun to be dissected. Signaling by FGFs is mediated by FGF receptors (FGFR), which are type I transmembrane proteins that dimerize upon binding their cognate FGF ligand extracellularly and then autophosphorylate intracellularly and propagate a signal cascade which stimulates the FGF effector genes in the nucleus. There are four FGFRs with alternative splice types a-c, with the “c” type believed to be limited to mesenchymal cells (Ornitz et al., 1996; Werner et al., 1992). FGF23 has shown affinity to FGFR 1c, 3c, and 4c, and activates the MAP kinase (ERK1/2) system (Urakawa et al., 2006; Yamashita et al., 2002; Yu et al., 2005). The binding of FGF23 to FGFRs is enhanced by klotho, and may be required for its actions (Kurosu et al., 2006; Urakawa et al., 2006). Klotho is a transmembrane protein which may be related to glucuronidsases and is expressed in the kidney, parathyroid, pituitary gland, and choroid plexus, but may also exist in a circulating form (Liu and Quarles, 2007; Xiao et al., 2004). Mice null for klotho have a phenotype of advanced aging with defects in hearing, osteoporosis, ectopic calcifications, and skin atrophy (Kuro-o et al., 1997). The phenotype of the klotho null and Fgf23 KO, including hyperphosphatemia and increased Vit D, and ectopic tissue calcification, closely resemble each other; lending credence to the idea that klotho plays an integral role in FGF23 signal transduction (Memon et al., 2008).
Tooth findings
The lower incisor of the Klotho null shows a disturbance in the dentin development on the labial side, with an apparent defect in odontoblast function (Suzuki et al., 2008). The continually erupting mouse incisor tooth features a crown analogue on its labial aspect and a root analogue on its lingual aspect (Ohshima et al., 2005; Tummers and Thesleff, 2008). The labial incisor dentin had a reduction in the predentin width, with the development of an irregular bone-like mass of matrix having cells entrapped within. Many of these entrapped cells were shown immunohistochemically to contain high levels of DMP1 protein, and an increase in apoptosis was noted in these cells. A similar phenotype has been noted in incisors and molars obtained from Fgf23 KO mice (Figure 2), including narrow pulp chambers with ectopic matrix deposition, as well as accumulation of osteoid, increased incidence of apoptosis in cells, and disorganization and decreased width of the PDL (Blethen et al., 2008; Chu et al., 2007). Detailed descriptions of these findings will be featured in a forthcoming publication from our lab.
iv. Dentin matrix protein 1 (DMP1)
General
DMP1, another member of the SIBLING protein family (Fisher and Fedarko, 2003), is a highly phosphorylated acidic protein, initially identified as a product of odontoblasts. However, DMP1 has been identified as highly expressed in mature osteoblasts and in the bone-encased osteocytes (George et al., 1994; Toyosawa et al., 2001). DMP1 expression has also been noted in soft tissues, where it has been co-localized with MMP-9 for functions currently unknown (Ogbureke and Fisher, 2007). The acidic domains of DMP1 were seen in vitro to form β-like sheets capable of binding Ca2+ ions and initiating crystal nucleation, and in vivo could play a major role in the mineralization of predentin to dentin (He et al., 2003). In addition to its role in mineralization, DMP1 has also been implicated as a signaling molecule. Overexpression of Dmp1 in mesenchymal cells induced Dspp mRNA expression and an odontoblast phenotype, and DMP1 protein was observed to be localized to the nucleus (Narayanan et al., 2003; Narayanan et al., 2001). Intact DMP1 protein has not been recovered, only an amino and carboxyl terminal fragment, suggesting that DMP1 requires proteolytic cleavage to become active or is rapidly cleaved; however, the protease responsible for this is still unknown (Qin et al., 2003). The exact nature of DMP1 regulation of expression remains unknown, but many transcription factor response elements have been identified in the Dmp1 promoter region, and it is reported that in a cementoblast cell line treated with Pi, Dmp1 expression was up-regulated by 30-fold (Foster et al., 2006b; Narayanan et al., 2002). When mouse ulna were placed under strain, the osteocytes expressed high levels of DMP1, suggesting DMP1 plays a role in osteocyte function in the response to mechanical forces in bone (Yang et al., 2004).
The Dmp1 null mouse has both dental and skeletal phenotypes. Postnatal Dmp1 null mice develop skeletal defects including rickets and osteomalacia/mineralization defects, a widened hypertrophic zone resulting in an enlarged growth plate (Feng et al., 2006; Ling et al., 2005). Many of the defects observed in the Dmp1 null mouse may be attributed to the surprising finding that the Dmp1 null mouse is hypophosphatemic, and was a mouse model for ARHR (Feng et al., 2006; Lorenz-Depiereux et al., 2006; Ye et al., 2005). The hypophosphatemia is the result of an increase in the expression of FGF23 by osteocytes, by an unknown mechanism, similar to the Hyp mouse (Feng et al., 2006). This suggests that FGF 23 is under the control, directly or indirectly, of DMP1. The connection, if any, between DMP1 and PHEX is unknown. However, it is worth noting that the many factors discovered so far to play a role in Pi regulation, such as FGF23, MEPE, PHEX, and DMP1, are all expressed in osteocytes in normal or pathological conditions. Thus, it may be hypothesized that the osteocytes have an as yet undiscovered role in regulating Pi levels in the body.
Tooth findings
In the tooth of the Dmp1 KO mouse, there is a reduction in mineralization of dentin, with a widened predentin layer, thin dentin walls with an enlargement of the pulp space, dentinal tubule abnormalities, a reduced expression of Dspp, and periodontal defects, including changes in the thickness of alveolar bone, periodontal ligament, and cementum (Ye et al., 2004; Ye et al., 2008). Tooth abnormalities in ADHR patients have been reported for one individual and were described as observed defects of dentin and multiple caries in a 5-year-old subject (Lorenz-Depiereux et al., 2006).
v. Other factors
FRP4 was found to be a highly expressed gene in many tumors of TIO patients (Berndt et al., 2003; Kumar, 2002). FRP4 is structurally similar to the extracellular portion of the Wnt receptor, frizzled, and functions as an antagonist to Wnt binding to frizzled (Hsieh et al., 1999). Mounting data support a role for Wnt signaling in bone formation and remodeling (Gaur et al., 2005; Glass et al., 2005; Li et al., 2005; Vaes et al., 2005), as well as tooth development (Nadiri et al., 2004; Pispa and Thesleff, 2003). Gain of function in Wnt signaling mutations has been noted in patients with an increase in bone mass (Boyden et al., 2002). The injection of recombinant FRP4 into rats or mice resulted in an increase in Pi excretion and hypophosphatemia as early as 2 and 4 hrs, respectively, in rats, and in mice at 60 minutes (Berndt et al., 2003; Berndt et al., 2005). Opossum kidney cells treated with FRP4 in vitro exhibited reduced Pi uptake (Berndt et al., 2003). In addition to induction of phosphaturia and hypophosphatemia, FRP4 induced 25-hydroxy-vitamin D1-α hydroxylase activity (Berndt et al., 2003; Berndt et al., 2006).
McCune-Albright syndrome (MAS) is a skeletal disease caused by Gnas1 mutation and characterized by polyostotic fibrous bone dysplasia, endocrine hyperfunction, and café-au-lait skin pigmentation. Oral manifestations of MAS reported include enamel hypoplasia and hypomineralization, delayed tooth eruption, taurodontism, and craniofacial bone (mandible and maxilla) dysplasia (Akintoye et al., 2003; Akintoye et al., 2004; Gomes et al., 2002). MAS is sometimes complicated by hypophosphatemia and abnormally low Vit D levels, resembling the presentation of XLH and OOM. Increased plasma levels of FGF23 suggest a mechanism underlying the hypophosphatemia and abnormal Vit D metabolism (Yamamoto, 2006; Yamamoto et al., 2005).
c. Closing the Ca2+/Pi endocrine loop
The data derived from FGF23 studies have allowed for the endocrine regulatory loop of Ca2+/Pi homeostasis to be closed (as outlined in Figure 1). Low Ca2+ or low Pi stimulates PTH expression. PTH drives osteoclastogenesis, mobilizing Ca2+ (and Pi) from bone and the production of active Vit D, which allows for increased absorption of Ca2+ and Pi from the intestine and reabsorption in the kidney. PTH reduces the reabsorption of Pi in the kidney, but PTH is soon down-regulated as Ca2+ levels approach normal, so has a short term role in regulating Pi. The increase in Vit D directs an increase in the level of FGF23, which feeds back to reduce both Vit D levels and levels of NaPi Type 2a and 2c Pi transporters in the kidney to act in a more long term fashion, to balance the actions of Vit D, to ultimately prevent the development of a hyperphosphatemic state.
III. Local cellular regulators/ion transporters regulating Pi/PPi homeostasis (Table 3 and Figure 2)
While Pi metabolism functions on a systemic level via modulation of circulating concentrations, there are also several cell-localized regulators of both Pi and PPi that strongly influence local, microenvironmental concentrations. These factors transport Pi and PPi across cell membranes and govern the Pi/PPi ratio by generation and hydrolysis of PPi. The primary regulators of Pi metabolism at the microenvironmental level include mouse progressive ankylosis protein (ANK, as well as human homolog, ANKH), a putative transporter of PPi from the intracellular compartment to the extracellular space (Gurley et al., 2006a; Harmey et al., 2004; Ho et al., 2000; Johnson et al., 2003; Nociti et al., 2002; Nurnberg et al., 2001; Pendleton et al., 2002; Reichenberger et al., 2001; Sweet and Green, 1981; Terkeltaub, 2001), the PPi-generating nucleoside triphosphate pyrophosphohydrolase plasma cell membrane glycoprotein-1 (NPP1 or PC-1) (Fong et al., 2005; Goding et al., 1998; Harmey et al., 2004; Johnson et al., 2003; Murshed et al., 2005; Nociti et al., 2002; Okawa et al., 1998; Rutsch et al., 2000; Rutsch et al., 2001; Terkeltaub, 2001; van den Bos et al., 2005), and tissue nonspecific alkaline phosphatase (TNAP), an enzyme proposed to cleave PPi substrate to its Pi constituents (Beertsen et al., 1999; Chapple, 1993; Fedde et al., 1999; Groeneveld et al., 1996; Hessle et al., 2002; Murshed et al., 2005; Narisawa et al., 1997; van den Bos et al., 2005; Whyte, 2002; Whyte et al., 1995). Details regarding the functions of these factors are provided in Table 3, including implications for their roles based on mouse models where genes have been mutated or deleted. Existing data indicate that local control of Pi/PPi is critical for normal cementum/periodontal tissue development, an idea that will be explored in more detail in the sections below.
The importance of maintaining appropriate concentrations of Pi in the extracellular environment for regulation of mineralization was highlighted by Murshed and colleagues (Murshed et al., 2005). Examining a variety of KO mice, the modulation of extracellular Pi concentrations was found to be critical in both regulating physiological mineralization and preventing pathological calcification. However, these studies lacked details related to teeth and surrounding tissues, essential for defining the precise role of Pi and its regulators in controlling mineralization. Results from studies to date suggest that local control of PPi/Pi is critical for normal root/periodontal tissue development, and further, that cementum may be a uniquely sensitive tissue to PPi and Pi in the local area.
a. Tissue nonspecific alkaline phosphatase (TNAP)
General
TNAP is one of four alkaline phosphatase isozymes expressed in mammals, and is found in liver, kidney, and bone, as well as tooth (for an excellent and extensive review see Millan, 2006). TNAP activity produces Pi from hydrolysis of PPi, and other natural substrates (e.g., pyridoxal 5’-phosphate (PLP) and possibly phospoethanolamine (PEA)). TNAP function is important in skeletal mineralization by removing a mineralization inhibitor, PPi, and increasing a required HAP mineral building block, Pi (Murshed et al., 2005). In this sense, the tendency towards biological mineralization may be regulated by the ratio of Pi:PPi, with regulators like TNAP, ANK, and PC-1 (the latter two are discussed below) central in determining microenvironmental Pi and PPi levels. TNAP is present on cell membranes of osteoblasts and matrix vesicles (MVs), and circulating levels are also measurable.
The condition hypophosphatasia (HPP) results from dysfunctional or low levels of the TNAP causing poor mineralization and the conditions rickets and osteomalacia (Whyte, 1994; 2002). In Tnap (Akp2) KO mice, there was decreased mineral density in the bones and dentition (Beertsen et al., 1999; Millan, 2006).
Mice deficient in ANK or PC-1 exhibit decreased extracellular PPi, resulting in pathological calcification in the soft tissues (see Gurley et al., 2006a; Ho et al., 2000; Okawa et al., 1998, and below). Combinations of deficiencies in ANK or PC-1 with lack of TNAP resulted in partial improvements to the hypophosphatasia and mineralization defects (Anderson et al., 2005; Harmey et al., 2004; Hessle et al., 2002). Tooth phenotypes have not been described in these compound mutants. These data underscore the concerted nature of the interactions between PC-1, ANK, and TNAP in regulating Pi and PPi in the cellular milieu. The authors also described an apparent signaling effect of PPi in regulating gene expression of Ank, Enpp1, and the osteopontin (Opn) gene. This is discussed in more detail in section IV below.
A recombinant TNAP enzyme replacement therapy approach targeting bone was able to rescue skeletal (and tooth) developmental defects in Tnap null mice (Millan et al., 2007). This success, when contrasted to failures of previous IV infusions of TNAP into HPP patients (Weninger et al., 1989; Whyte et al., 1984; Whyte et al., 1982), suggests that local, pericellular function of TNAP around bones and teeth is more physiologically critical than circulating serum levels during development of mineralized tissues. This conclusion, however, is at odds with findings from another study in which increased circulating TNAP produced by the liver rescued the bone mineralization phenotype in Tnap KO mice (Murshed et al., 2005).
Tooth
TNAP is highly expressed in the periodontia, including cementoblasts, osteoblasts, and PDL fibroblasts (Groeneveld et al., 1995). HPP afforded the first linkage of a Pi metabolism disorder with a developmental cementum phenotype, and has been consistently linked to premature loss of deciduous teeth (Bruckner et al., 1962; Chapple, 1993; van den Bos et al., 2005). TNAP deficiency causes cementum aplasia or severe hypoplasia, especially in the more coronal acellular cementum region, compromising periodontal attachment due to defects in attachment of Sharpey’s fibers, ultimately resulting in premature exfoliation of teeth (Bruckner et al., 1962). Reports of effects of HPP on cellular cementum have varied, with some indication for hypoplasia (van den Bos et al., 2005).
In addition to decreased mineral density in the teeth, the Tnap (Akp2) KO mouse was reported to feature delayed incisor eruption (Beertsen et al., 1999; Millan, 2006). Molars developed relatively normally, though a mild enamel hypoplasia and slightly delayed root dentin mineralization was reported. The most striking defect noted was aplasia or severe hypoplasia of acellular cementum, including both matrix and mineral phase, accompanied by a suggestion of defective PDL attachment (Figure 2). No overt differences were described in cellular cementum structure. To understand the mild effect or lack of effect of TNAP deficiency on dentin, van den Bos et al. (2005) assessed PPi levels and PPi-associated enzyme regulators in dentin/pulp versus PDL of normal teeth. The authors reported decreased expression of Enpp1 and also decreased NPP1 (PC-1) activity in pulp versus PDL, and also lower levels of PPi in pulp versus PDL, reinforcing findings of others regarding differences in the role of regulators of Pi metabolism among a variety of tissues, and highlighting the need to understand genes dictating differences between hard tissues.
An increase in TNAP levels and activity has been linked to other human mineralized tissue diseases via case reports in the literature, some including dental phenotypes. Generalized hypercementosis has long been associated with Paget’s disease (osteitis deformans). Paget’s disease is a typically late onset disorder of focally increased bone turnover resulting in disorganized, sclerotic, and weak bone (Helfrich and Hocking, 2008). Paget’s disease has been linked to a number of gene mutations, all involved in osteoclast function. Serum calcium and Pi have been reported to lie within normal limits, but TNAP levels are highly elevated, usually attributed to rapid turnover of bone. Pendred Syndrome is an autosomal recessive inherited disorder of the thyroid gland. In addition to goiter, hearing impairment, and an apparent neutrophil defect, a case report has linked Pendred Syndrome with an oral manifestation of cementum hyperplasia (based on assessment of radiographs) and a thickened lamina dura (Sharma and Pradeep, 2007). These findings are intriguing in light of the observed elevated serum TNAP, Pi, and Ca2+, though a single case report must be considered cautiously. While the underlying mechanism for cementum phenotypes associated with these conditions is unclear, increased TNAP activity in the periodontia may be related to an altered Pi/PPi ratio and consequent tissue changes.
b. Progressive ankylosis protein (ANK, ANKH)
General
The mouse progressive ankylosis gene (Ank), analogous to Ankh in humans, transports intracellular PPi to the extracellular space (Ho et al., 2000). Loss of ANK function results in low levels of PPi in the local extracellular environment, with high intracellular PPi, in contrast to TNAP deficiency, which causes increased PPi outside cells (Ho et al., 2000; Terkeltaub, 2001). The ank/ank mutant mice have been described as a model for a progressive arthritis-like condition characterized by ectopic calcifications in cartilage and joint tissues, with mice exhibiting degeneration in joints, tendons, and ligaments, and impaired mobility manifested as an arthritis-like condition (Ho et al., 2000; Sweet and Green, 1981; Terkeltaub, 2001). While the phenotype had been characterized since 1981 (Sweet and Green, 1981), the gene and mutation were not identified until 2000, when a publication by the Kingsley laboratory described the mechanism of the observed pathology (Ho et al., 2000).
Mutation in the human ortholog of the mouse gene, Ankh, has been associated with a variety of skeletal defects and human disease. While PPi acts as a potent inhibitor for growth of HAP crystals, excess levels (as in conditions such as adult hypophosphatasia) result in pathological calcium pyrophosphate dihydrate (CPPD) elaboration (Netter et al., 2004; Ryan, 2001). The condition chondrocalcinosis (CC) is caused by deposition of CPPD within articular cartilage and causes joint pain and arthritis. Mutations associated with CCAL2 cluster to the N terminus of ANKH and result in alterations of 1-4 amino acids (Pendleton et al., 2002). Like ank/ank and Ank KO mice, CCAL2 patients experience crystal deposition in the articular cartilage and synovial fluid, however the crystals are CPPD rather than HAP. Evidence suggests gain of function of ANKH as a result of the CCAL2 mutations, causing excess extracellular PPi and pathological CPPD deposition (Gurley et al., 2006b; Pendleton et al., 2002; Ryan, 2001).
Additional dominant mutations in Ankh have been linked to the condition craniometaphyseal dysplasia (CMD), a congenital disorder resulting in cranial and long bone mineralization defects (Nurnberg et al., 2001; Reichenberger et al., 2001). CMD is characterized by overgrowth and sclerosis of the craniofacial bones and abnormal modeling of long bone metaphyses, and in contrast to CCAL2 patients and Ank mutation/KO in mice, there is no apparent joint phenotype. Ankh mutations associated with CMD affect single amino acids in a region distinct from the mutations that cause CCAL2, and have been linked to significantly less PPi transport (Gurley et al., 2006b).
Tooth findings
Our group became interested in the periodontal status of the ank/ank mice, hypothesizing that the PDL space perhaps exhibited some ectopic ossicles or even ankylosis. However, an unexpected and intriguing tooth phenotype was observed in the ank/ank mice, and more recently in Ank KO mice (Figure 2). Rather than observing ossicles or ankylosis as expected, a marked increase in cementum formation was instead exhibited, while PDL, dentin, and alveolar bone appeared unaffected (Nociti et al., 2002). An increased rate of cementogenesis and increased cellularity of the cementum (in the normally acellular cervical region) were observed over the entire course of root formation, from initiation of cementogenesis (26 dpc) through adulthood. The same hypercementosis phenotype was identified in adult mice with a Pc-1/Enpp1 mutation (see more on this factor below), suggesting that the increased cementum was a direct result of the decreased extracellular PPi common to both mutations. Studies to characterize the mechanical properties for the ank/ank mutant versus WT tissues have not identified any significant difference in the hardness, elastic modulus, or structure, as observed by SEM and TEM (Fong et al., 2008b).
The first detailed dental clinical report regarding CMD was recently published and described the dental features of a 3½ yr old patient (Zhang et al., 2007a). In this case, primary teeth were marked by dysmorphic and discolored surfaces, especially mandibular central incisors and maxillary 1st molars. The only indication for excess mineralization was an observation of enamel pearl-like material on the buccal surfaces of maxillary 2nd molars. The enamel pearls were evaluated at a gross level but their appearance was not inconsistent with the mineral accumulation observed in mice lacking ANK function; this is an area for further investigation to determine if there is a cementum phenotype and if there are any clinical dental ramifications. A previous CMD case report of a 10 yr old patient reported delayed tooth eruption, but did not report malformation or discoloration (Hayashibara et al., 2000). No tooth phenotype has been reported for CPPD, though cementum hypoplasia or aplasia might be expected in light of observations from HPP patients.
c. Plasma cell membrane glycoprotein 1 (PC-1)
General
Like ANK, the protein PC-1 (ectonucleotide pyrophosphatase/phosphodiesterase 1, ENPP1) regulates extracellular PPi. Mutations in the gene for PC-1, a membrane-bound enzyme that generates PPi from triphosphates, result in low extracellular PPi, and as a result, a joint phenotype in mice resembling those with ANK deficiency (Johnson et al., 2000; Terkeltaub, 2001). The hypermineralization observed in PC-1 deficient mice (tiptoe walking, or ttw mice), while similar to that described in ank/ank mice, has been reported to be more severe, and one explanation for this may be localization of PC-1 but not ANK in matrix vesicles (MVs), cell buds proposed to serve as focal points for mineralization (Hessle et al., 2002; Johnson et al., 2001; Vaingankar et al., 2004). Humans with mutations in these genes also present pathologies resulting from deficient PPi, including craniometaphyseal dysplasia (CMD) and idiopathic infantile arterial calcification (IIAC) (Nurnberg et al., 2001; Reichenberger et al., 2001; Rutsch and Terkeltaub, 2005; Rutsch et al., 2001).
While ANK and PC-1 functions increase PPi in cell environment, TNAP activity decreases PPi to release Pi. In this sense, PC-1 and ANK can be thought of as natural antagonists to TNAP in terms of regulating Pi/PPi levels and mineralization (Harmey et al., 2004). Crossing Ank mutant mice with Akp2 null mice resulted in partial correction of both PPi levels and mineralization defects.
Tooth findings
The tooth phenotype of ttw mice was described alongside ank/ank mice and presented a strikingly similar hypercementosis phenotype at 77 dpc (Nociti et al., 2002). This is presumably the result of relatively lower levels of extracellular PPi and/or the altered ratio of Pi/PPi. The tooth phenotype of ttw mice has not been more fully characterized.
d. Phosphate transporters: PiT1 and 2
Sodium dependent Pi transporters types I and II have been identified in several organs; type II NaPi2a and NaPi2b are the predominant transporters responsible for Pi reabsorption in the kidneys (as described above). Type III sodium-dependent Pi transporters, PiT-1 and PiT-2, are widely expressed and likely perform housekeeping functions in regulating cellular Pi entry (Kavanaugh and Kabat, 1996; Li et al., 2006; Ravera et al., 2007). However, Pit-1 expression in osteoblasts and other cells is regulated by Pi availability and other factors (Foster et al., 2006b; Suzuki et al., 2006; Suzuki et al., 2001; Zoidis et al., 2004), suggesting that PiT-1, but not PiT-2, may be of more physiological importance due to responsiveness to physiological cues (Zoidis et al., 2004). This hypothesis on PiT-2 was supported by a survey of the temporospatial expression during mouse tooth development, which mapped Pit-2 to secretory ameloblasts, dental papilla, and stratum intermedium, but not odontoblasts or cementoblasts (Zhao et al., 2006). Using tooth bud organ culture, it was found that Pit-2 expression does not respond to Pi starvation or treatment with PTH or Vit D. No tooth defect has been identified with loss of PiT function.
e. Other phosphatases
Other regulators of Pi/PPi levels include those more recently identified and of unknown importance in mineralized tissue formation. There is evidence that TNAP is not the sole enzyme with phosphatase activity in the osteoblast; in Tnap null mice, initial skeletal mineralization proceeds normally, and only later are hypomineralization defects manifested (Hessle et al., 2002; Narisawa et al., 1997). TNAP has been reported to be restricted to the basolateral domain of the osteoblast, and a phosphatase localized to the osteoidal aspect was characterized as a plasma membrane Ca2+ transport ATPase (PMCA, or PMCA1) (Francis et al., 2002; Kumar et al., 1993; Meszaros and Karin, 1993; Nakano et al., 2004; Nakano et al., 2003; Prasad et al., 2004; Stains et al., 2002), a protein identified previously in osteoblasts (Meszaros and Karin, 1993; Stains et al., 2002).
PHOSPHO1 is a cytoplasmic phosphatase linked to bone and cartilage mineralization (Houston et al., 2002; Houston et al., 2004; Stewart et al., 2006; Stewart et al., 2003) and hypothesized to contribute to Pi requirements for mineralization by cleaving phospholipid metabolites (Roberts et al., 2004; Roberts et al., 2005). PHOSPHO1 is associated with matrix vesicles of murine mineralizing cells, including osteoblasts and chondrocytes (Roberts et al., 2007; Stewart et al., 2006). No tooth phenotypes have been described to date in animal models, making it unclear if these phosphatases play a significant role in tooth mineralized tissue development.
IV. Phosphate as a signaling molecule
Disruptions in Pi metabolism leading to hyperphosphatemia, hypophosphatemia, or local disruptions in Pi/PPi ratio clearly have a considerable effect on bone and tooth development and maintenance, as outlined in the previous sections. Studies of mouse models are yielding a great deal of information about how local and systemic factors interact and have an impact on developing tissues. In parallel, in vitro experiments focused on effects of Pi may provide us with information of a more unambiguous and mechanistic nature. This section reviews the evidence for signaling properties of Pi in studies predominately done using in vitro culture systems. The combination of knowledge from such in vitro studies and in vivo observations in animal models should provide powerful and incisive insights into the role of Pi metabolism in health and disease, as well as potential for use in regeneration.
a. Phosphate regulates mineralized tissue cells
Phosphate present in the extracellular milieu of mineralizing cells may serve as a signal for a shift in cell function/differentiation, in addition to acting as one of the building blocks for hydroxyapatite mineral. The ability of Pi to regulate gene expression may represent an extracellular signaling mechanism, which subsequently informs the cell about its environment and provides cues for directing cell functions. Studies by several groups have begun to explore the mechanisms of Pi regulation of cell gene expression. The enzyme TNAP is up-regulated early in osteoblast differentiation, and a requirement of TNAP activity for Opn expression in MC3T3-E1 pre-osteoblast cells was observed by Beck et al. (1998). In a subsequent study exploring the TNAP-Opn link, Pi was shown to be a direct inducer of Opn expression in MC3T3-E1 cells (Beck et al., 2000), and was hypothesized to act as a signal to cue these precursor cells to undergo the coordinated program of cellular changes necessary for differentiation and matrix mineralization (Beck, 2003). The ability of 300 μM foscarnet to block Opn induction suggested that Pi internalization was necessary for signaling activity. Regulation of Pi-mediated Opn expression was further indicated to be ERK 1/2 and PKC mediated (Beck and Knecht, 2003). Subsequent in vitro studies employing microarray analyses identified several genes up- and down-regulated by 10 mM Pi, including those for extracellular matrix proteins and transcription factors, like Nrf2 (Beck et al., 2003). The increase in Nrf2 expression occurred by 2 hrs and did not require protein synthesis, suggesting it is an early response gene in MC3T3-E1 cells.
A standard procedure for promoting mineral nodule formation by osteoblast-like cells in vitro is by adding Pi or β-glycerophosphate. Pi is actively transported into osteoblasts via transporters, including PiT-1/PiT-2 (Guicheux et al., 2000; Nielsen et al., 2001; Palmer et al., 1999; Suzuki et al., 2000). In addition to the critical role of Pi for normal mineralization, it has become apparent that Pi also plays a major role in several other biological processes, including enzyme regulation, nucleotide metabolism and signal transduction (Beck et al., 2003; Berndt et al., 2005; Foster et al., 2006b; Fujita et al., 2001; Rutherford et al., 2006). New and established data provide evidence that calcitropic hormones, growth factors, and cytokines have the ability to modulate Pi/PPi levels, with significant impact on the extent of mineral formation (Bielesz, 2006; Rowe, 2004; Yu and White, 2005a). A recent example demonstrating the role of growth factors comes from the studies of Suzuki et al. (2006) where it was reported bone morphogenetic protein (BMP)-2 enhanced Pit-1 mRNA and functions in MC3T3-E1 cells, a murine osteogenic cell line.
Shapiro et al. suggested elevated Pi may signal stage-specific apoptosis of terminally differentiated chondrocytes (Mansfield et al., 2003; Mansfield et al., 1999; Mansfield et al., 2001; Teixeira et al., 2001), and additional studies showed 5 and 7 mM Pi significantly promoted apoptosis in human alveolar bone osteoblasts as well as MC3T3-E1 cells in culture (Adams et al., 2001; Meleti et al., 2000). These studies also indicated the requirement for Pi transport across the cell membrane by using foscarnet. The authors hypothesized that under normal physiologic conditions osteoblasts may encounter high Pi in areas of local bone resorption and be directed toward apoptosis as a result. Of course, the ramifications of such programming should be considered under pathological conditions as well. In the Hyp mice, which are hypophosphatemic as a result of Phex mutation (described in detail in section II.b.i), a widened and irregular hypertrophic zone was described in the growth plate cartilage (Miao et al., 2004). In contrast, Klotho KO mice (described in detail in section II.b.iii), which are hyperphosphatemic due to lack of Fgf-23 signaling, feature positive staining for apoptosis in odontoblasts and pulp cells in the incisor (Suzuki et al., 2008). Our studies of the similarly hyperphosphatemic Fgf-23 KO mice indicate not only apoptosis in odontoblasts embedded in matrix in the labial aspect of the incisor, but also widespread apoptosis in the alveolar bone osteoblasts (personal observations to be published in forthcoming manuscript).
Studies in the Shapiro laboratory showing that 10 mM Pi is sufficient to promote apoptosis in MC3T3-E1 cells must be reconciled with studies by Beck et al. treating the same cells with 10 and even 20 mM Pi for up to 96 hrs without reports of apoptosis or cell death. One possible explanation lies with the amount of fetal bovine serum (FBS) used in cell culture; FBS is known to contain proteins that can bind and prevent precipitation if Ca2+ and Pi even under supersaturated conditions. It is plausible that interactions between these binding proteins and Pi effectively limit the amount available to cells and reduce the tendency of Pi to induce apoptosis. Other factors to consider in this matter may include pH of Pi used, concentration of Ca 2+ in the media, and stage of cell differentiation (Beck, 2003; Mansfield et al., 2003). The role for Ca2+ ions in conjunction with Pi for signaling events has been discussed by George (Narayanan et al., 2003) and Shapiro (Adams et al., 2001; Mansfield et al., 2003), and is a facet that we did not examine here.
b. Effect of phosphate signaling on tooth cells
In light of the dramatic tooth root phenotypes resulting from disruption of local Pi metabolism regulators, we have undertaken studies to understand the role of Pi in tooth root development and its potential use in therapeutic regenerative applications. In vitro studies employed an immortalized mouse cementoblast cell line, OCCM-30 (D’Errico et al., 2000), to identify downstream events resulting from exposure of cementoblasts to elevated Pi. Exposure of cementoblasts to 5 mM Pi regulated several genes known to be involved in formation of cementum (as well as other mineralized tissues). Some of the most significantly regulated genes included members of the SIBLING family. Opn and Dmp1 were both strongly up-regulated by Pi, while Bsp was down-regulated (Foster et al., 2005; Foster et al., 2006b). The SIBLING family includes multiple genes having in common chromosomal location (4q21 in mice), gene structure, and post-translational modifications, and all were initially identified by their association with mineralized tissues (Fisher and Fedarko, 2003; Qin et al., 2004). Originally described as extracellular matrix proteins and regulators of mineralization, SIBLING family proteins have since been linked with several additional functions. In particular, BSP, OPN, and DMP1 partner with and activate in vitro matrix metalloproteinases (MMP)-2, -3, and -9, respectively, suggesting an additional tissue turnover function for these SIBLINGs (Fedarko et al., 2004). All three MMPs are regulated by Pi in these cementoblasts in vitro (Foster et al., 2006a). Interestingly, Opn and Dmp1, up-regulated by Pi in vitro, have been reported to be up-regulated in the hyperphosphatemic Fgf-23 KO mice. In OCCM-30 cementoblasts, Pi also regulated gene expression of Ank, Pc-1 (Enpp1), Pit-1, and Tnap/Akp2, suggesting that the cementoblasts were sensitive and responsive to Pi levels.
Further studies of the effect of Pi signaling on cementoblasts have focused on cellular response mechanisms. Microarray time course studies identified early response elements to elevated Pi in OCCM-30 cells, including transcription factors (TF) and Wnt pathway factors (Rutherford et al., 2006) that are the subjects of current research. Intriguingly, cementoblasts exposed to Pi up-regulated the zinc TF Egr1 (Egr1 was the TF found to be up-regulated by FGF23 in the presence of Klotho), known to be expressed in teeth, by 1 hr, and Yasutake et al. (2006) and Yu et al. (2006) have shown FGF23 up-regulates Egr1 mRNA in kidney cells and modulates expression of Klotho. Further, the dramatic response of Opn to elevated Pi has been studied by employing murine Opn promoter constructs, revealing a necessity for the glucocorticoid response element for the Opn response (Fatherazi et al., 2008; Fatherazi et al., 2006).
Studies of direct effects of Pi on other tooth mineralized tissue cells are relatively uncommon. Lundquist et al. characterized Ca2+ and Pi uptake in rat pulp MRPC-1 cells as they were stimulated to differentiate to an odontoblast-like phenotype (Lundquist, 2002; Lundquist et al., 2002). While Ca2+ uptake was at a maximum during proliferation, Pi uptake increased four-fold following 4 mM Pi addition and just prior to mineral nodule formation. Although it was hypothesized that Na-Pi transport was increased in order to translocate Pi ions for matrix mineralization, it is possible that Pi may have modulated Na-Pi uptake and gene expression in relation to differentiation and mineralization in these cells.
c. Effect of phosphate on osteoclasts
Alveolar bone has been described as featuring the highest degree of turnover of bone turnover in the human body. At sites of mineralized tissue resorption, active osteoclasts are likely exposed to high concentrations of Pi (and Ca2+) as HAP is dissolved in the acidic environment under the osteoclast ruffled border. The mechanism of Pi transport in osteoclasts has also been a topic of interest due to the presumably large concentrations of Pi generated under the ruffled border during active resorption. Gupta et al. (1997) reported the presence of a Na-dependent Pi transporter in rabbit osteoclasts. The transporter was localized in intracellular vesicles in undifferentiated osteoclasts, but upon polarization was found in the basolateral membrane (not in contact with bone) opposite the H-ATPase. This suggests that this Na-Pi transporter is not the primary element responsible for removing Pi after bone mineral dissolution. An acid-dependent high capacity Pi transport system was later identified in activated mouse RAW 264.7 osteoclasts (Ito et al., 2007; Ito et al., 2005). The unique Pi uptake was found to be regulated by intracellular Ca2+ and efflux was stimulated by Pi or its analogs.
Effects of alterations in extracellular Pi have also been studied for possible effects on osteoclasts and osteoclastogenesis. Inhibition of osteoclast formation and activity by elevated Pi has been observed in vitro using bone marrow derived precursors and cell lines from several species (Kanatani et al., 2003; Takeyama et al., 2001; Yates et al., 1991). One study suggested that differentiating osteoblasts may specifically down-regulate osteoclastogenesis via increased TNAP and subsequent increase in local Pi, however no mechanistic data were presented to support this and other factors could have been responsible for the in vitro observations (Takeyama et al., 2001). Doi et al. (1999), in studies of osteoclastic ability to resorb various types of calcium phosphate substrates, hypothesized that increases of Ca2+ and Pi in the culture media may have down-regulated osteoclast activity, though observed increases were relatively small (0.5 to a maximum of about 2.5 mM Pi) and no mechanism for Pi regulation of the rabbit osteoclasts was discussed. In further in vitro studies on human and mouse osteoclast precursors, it was demonstrated that increased Pi (1.5-4.5 mM) inhibits osteoclastic differentiation in the presence of RANKL and M-CSF, possibly by disrupting DNA binding of critical transcription factors AP-1 and NF-κB (Mozar et al., 2008). In addition to findings that high Pi inhibits osteoclastogenesis and function, studies on mice under hypophosphatemic conditions have suggested that low Pi may also inhibit osteoclasts. By employing both Hyp mice and mice maintained on a low Pi diet (low, 0.02%; control, 0.6%), Hayashibara et al. (2007) demonstrated fewer osteoclasts by histomorphometric measurements. By putting Hyp mice on a high Pi diet (3.0 %), the authors demonstrated partial correction in numbers of osteoclasts as well as bone mineralization. The authors tested whether osteoclast formation in vitro paralleled observations in hypophosphatemic mice. Osteoclastogenesis from WT marrow cells was decreased under both low (0.5, 0.75 mM) and high (1.5-2.5 mM) Pi conditions, while osteoclastogenesis was not impaired in Hyp mouse marrow cells under normal Pi conditions (1 mM). In contrast to studies already discussed (Mozar et al., 2008), Pi levels (low and high) did not significantly affect osteoclastogenesis when directly induced by RANKL and M-CSF addition. Therefore, to date there has been evidence of Pi effects on osteoclasts and their precursors, including in hypo- and hyperphosphatemic mouse models, however the extent to which this impacts mineralized tissues, especially those of the tooth, remains to be seen.
d. Pyrophosphate (PPi) and polyphosphates (polyP) as signaling molecules
More recently, a potential signaling role for PPi distinct from that of Pi has been proposed. While data compiled to date are intriguing, it is difficult to conclusively show PPi specifically is eliciting cellular effects when the possibility for hydrolysis to Pi is so likely (in the presence or absence of TNAP and other enzymes). Moreover, the hypothesis for PPi signaling also currently lacks an identified channel or cell surface sensor to convey PPi concentration information to the cell interior to effect expression changes. Nonetheless, further study is warranted to clarify initial data in this area of study.
PPi is a profound inhibitor of HAP mineral formation, but in vivo and in vitro studies have been cited for evidence that PPi additionally regulates cell function. Opn levels were altered in primary calvarial osteoblasts from both Ank mutant and Akp2 null mice (Harmey et al., 2004). Loss of ANK function decreased Opn, while loss of TNAP increased Opn. Crossing these two mutants results in partial normalization of Opn expression, in addition to mineralization. Addition of μM amounts of exogenous PPi was reported to increase Opn mRNA in calvarial osteoblasts. The authors hypothesized the effect must be from PPi rather than a Pi hydrolysis product because the effective dose was in the μM range. Regulation of Ank and Enpp1 by PPi was interpreted as evidence for a concerted feedback loop in which ANK and ENPP1/PC-1 regulate OPN expression by modulating PPi levels, and thus control mineralization. While this model is compelling, it is difficult to separate out Pi and PPi effects, and unclear why Opn should be lower in Ank mutant mice where intracellular PPi is elevated.
MC3T3-E1 cells were shown to respond to added μM-mM levels of PPi by increasing Opn expression in an ERK1/2 dependent response (Addison et al., 2007). This regulation of gene expression was not sensitive to foscarnet or levamisol (a TNAP inhibitor), again suggesting a role for PPi distinct from Pi. PPi also successfully blocked the ability of TNAP to release Pi from β-glycerophosphate (βGP) and OPN protein, in vitro. The authors thus suggest PPi may serve as a negative regulator of mineralization by multiple mechanisms, including direct binding to mineral, increase of OPN expression, and inhibition of TNAP activity.
Polyphosphates (polyP) are long polymers of Pi that have been found in cells and plasma that are suggested to have myriad functions including energy storage, nucleotide regulator, and Pi donor for kinases. Like PPi, polyP may also serve as a regulator of calcification. PolyP and exopolyphosphatase activity was detected in primary human osteoblasts in relatively high concentrations compared to non-mineralizing cell types (Leyhausen et al., 1998). In studies with MC3T3 E1 pre-osteoblasts, polyP has been cited as contributing to an osteoblast-like phenotype (Hacchou et al., 2007; Kawazoe et al., 2004), and polyP effects are being further explored for therapeutic potential (Kawazoe et al., 2008; Yamaoka et al., 2008). It remains unclear how much of the polyP effect may be attributed to liberation of Pi and PPi from the longer polymer chains.
V. Phosphate-based drugs and materials
The rationale for using Pi directly or regulating interstitial Pi for the therapeutic regeneration of mineralized tissues is partially informed by the extant phosphate-based therapeutics. These include the bisphosphonates, calcium phosphate bone cements, and bioactive glass. Of these, the bone cements and bioactive glasses have utility in the promotion of tissue repair, particularly bone. These compounds appear to function as scaffolds supporting tissue formation, i.e., osteoconductive rather than osteoinductive, in the case of bone. Broader utility would be obtained if a phosphate-based therapeutic device could provide a signal which initiated a regeneration cascade. Today this vision is an as yet unrealized goal.
a. Bisphosphonates
Current treatments for osteoporosis, metastatic disease in bone, Paget’s disease of bone, and hypercalcemia of malignancy frequently include bisphosphonates, originally conceived of as synthetic analogs of PPi. The amino bisphosphonates, e.g., zolendronate, pamidronate, and alendronate, are currently preferred due to faster onset, greater potency, and longer duration of action. The amino bisphosphonates are also reported to have anti-neoplastic activity (Santini et al., 2003).
Bisphosphonates (BPs) are synthetic analogues of PPi in which the pyrophosphate (P-O-P) bond has been replaced by a phosphoether (P-C-P) bond, essentially making a non-hydrolyzable surrogate for PPi. BPs were observed to strongly bind hydroxyapatite of bone, and had the effect of reducing bone resorption by osteoclasts. Early generation BPs were simple in structure, resembling PPi with simple side-chains at the R2 position which tuned their pharmacological properties. Later generation BPs incorporated bulkier side chains often containing nitrogen (known as amino-BPs), exhibiting much greater potency and persistence in inhibiting osteoclastic resorption (Rogers, 2003).
While BPs potently block bone resorption, the molecular mechanism of action was only elucidated recently. Convincing X-ray diffraction data have now demonstrated amino-BPs form an inhibitory complex with the enzyme farnesyl pyrophosphate synthase (FPPS) (Kavanagh et al., 2006; Rondeau et al., 2006). FPPS catalyzes a key step in the mevalonate synthesis pathway, and is also part of the larger HMG CoA reductase pathway for steroid and cholesterol synthesis. The mevalonate synthesis pathway provides isoprenoids for protein prenylation, essential for proper function of small GTPases such as Ras and Raf. These GTPases play important roles in cellular functions such as intracellular signaling, and intra/extracellular trafficking. By homing to mineralized surfaces, BPs become concentrated in osteoclasts during active resorption and are thought to severely impair osteoclast function.
One of the earliest clinical applications of BPs was to use etidronate as an inhibitor of calcification in fibrodysplasia ossificans progressiva and in patients who had total hip replacement to prevent heterotopic ossification (Russell, 2007). Due to their high affinity to bone mineral, especially at locations of increased bone turnover rate, bisphosphonates are also used as agents for “bone scanning” to detect bone metastasis and other bone lesions (Francis and Fogelman, 1987). However, the most extensive clinical applications of BPs relate to their antiresorption property.
Paget’s disease has been treated with BPs for many years (Devogelaer, 2000). A dose-dependent inhibition of bone resorption by using BPs was observed in human (Delmas and Meunier, 1997). Pamidronate infusion was used initially and later replaced by a new and more potent zoledronate with even more profound suppression of disease activity (Siris et al., 2006).
The use of BPs is popular in oncology since many human cancers are associated with hypercalcemia and/or bone destruction. The goals of such treatments are to prevent skeletal complications, palliate pain, and maintain quality of life (Russell, 2007). BP treatment (e.g., intravenous administration of ZA) has been demonstrated to be effective in multiple myeloma (Sirohi and Powles, 2004) and in patients with bone metastases from breast cancer (Coleman, 2005; Pavlakis et al., 2005) and prostate cancer (Parker, 2005).
The other successful clinical application for BPs is treatment of osteoporosis, a major public health problem (Rodan and Reszka, 2003). BPs are one of the major classes of drugs in treating postmenopausal and other forms of osteoporosis at present. Etidronate was the first BP used in osteoporosis treatment (Storm et al., 1990; Watts et al., 1990). Other BPs, including alendronate and risedronate, were shown to be effective in reducing fracture rates in osteoporosis patients by Meta-analysis (Boonen et al., 2005). To increase patient compliance to the therapy, recent research has been focused on improving formulations to reduce administration frequency, from a once-daily (alendronate, risedronate), to a once-weekly (alendronate, risedronate), to a once-monthly (ibandronate) tablet (Chesnut, 2006; Reginster et al., 2006). Now with new and more potent BPs and new routes of administration (IV), the treatment could be done even once-quarterly (ibandronate) (Croom and Scott, 2006) or once-yearly (ZA) (Reid et al., 2002).
Recently, bisphosphonate treatment of bone neoplasia has been associated with osteonecrosis of the jaws (ONJ) (Marx et al., 2005; Migliorati et al., 2005; Ruggiero et al., 2004; Woo et al., 2005). ONJ is relatively rare with an apparently increasing incidence rate which presents as a serious clinical complication to routine oral/dental procedures such as tooth extraction. Jaw bones are affected primarily, and various reasons for jaw selectivity have been proposed. Hypotheses include impaired healing of soft and hard tissues after trauma or periodontal (alveolar) procedures [as reviewed in (Bertoldo et al., 2007; Woo et al., 2006)], accumulated microcracks in jaw bones (Li et al., 2001; Mashiba et al., 2000; Mashiba et al., 2001), changes in vascularity in jaw bones (Wood et al., 2002), and association with infection (Marx et al., 2005). The exact reason for this selective and severe effect of BPs on jaw bones remains unknown and highlights the phosphate sensitivity of the dental-oral-craniofacial complex.
b. Bioceramics
Calcium phosphate bioceramics are salts of orthophosphoric acid and constitute a wide group of compounds, including dicalcium phosphate dihydrate (DCPD), octacalcium phosphate (OCP), tricalcium phosphate (TCP), and HAP (Barrere et al., 2006). Each calcium phosphate bioceramic has a different chemical composition and crystal structure, resulting in specific physicochemical properties. These calcium phosphate bioceramics could be either of natural origin (freeze-dried bone or derived coral HA), or synthetic origin (obtained after aqueous precipitation or after sintering) (Barrere et al., 2006). Due to their superior osteoconductivity, calcium phosphate bioceramics have been successfully used in cranio-maxillofacial, dental, and orthopedic surgery as bone fillers (cement or granules), coating materials on metallic orthopedic and dental implants (de Jonge et al., 2008; Le Guehennec et al., 2007), or scaffold materials for bone tissue regeneration (Barrere et al., 2006; El-Ghannam, 2005; Matsumoto et al., 2007; Warren et al., 2004).
To date, HAP has been the most widely used bioceramic material for bone substitution (Hak, 2007; Kokubo et al., 2003; Matsumoto et al., 2007; Warren et al., 2004). HAP can be fabricated to low porosity blocks and granules by sintering, or to high porosity structure by either a low heat alkaline extraction process producing xenograft (Caffesse et al., 2002), or by hydrothermal conversion of coral (Roy and Linnehan, 1974). The high porosity HAP structures have been cited for their advantages of favorable bone ingrowth and increased resorption in comparison to low porosity counterparts (Garg, 1999). However, since strength is inversely related to porosity, highly porous HAP structures are only suitable for non-load bearing conditions (Garg, 1999). An analogue of HAP bioceramic being used as a bone graft material is biphasic calcium phosphate (BCP), which is a mixture of HAP and TCP. Studies have established BCP as a viable graft material for use in treating periodontal osseous defects (Garg, 1999; Nery et al., 1992).
Porous HAP and BCP have been studied as a scaffold material for use in guided tissue regeneration (to exclude epithelial down-growth). These materials are desirable for scaffolds for their effectiveness in promoting cell/bone ingrowth and integration, and their bioresorbable properties. The use of collagen and other resorbable barrier membranes in conjunction with xenografts or porous BCP has demonstrated success in periodontal tissue repair and regeneration (Caffesse et al., 2002; Reynolds et al., 2003; Wang and Cooke, 2005; Zafiropoulos et al., 2007).
Calcium phosphate bioceramics are too brittle to be used as direct load bearing prostheses. Therefore, they are frequently used as coating materials on metal prosthesis surfaces to provide biological compatibility (de Jonge et al., 2008). Calcium phosphate coatings for metal implants were introduced about 2 decades ago due to their osteoconductive property (de Groot et al., 1987; Geesink et al., 1987). These coatings are demonstrated to increase bone-to-implant contact (Barrere et al., 2003a; Barrere et al., 2003b; Leeuwenburgh et al., 2006), to improve the implant fixation (Soballe et al., 2003), and to facilitate the bridging of small gaps between implant and surrounding bone (Soballe et al., 1991).
Another class of bioceramics is bioactive glass consisting of calcium oxide, sodium oxide, and phosphate mixed in a silicon oxide matrix. Bioactive glass is not porous and therefore is not susceptible to any tissue ingrowth. However, its highly bioactive surface is favorable for cell and tissue attachment. When introduced to the wound site, the glass surface is readily colonized by osteogenic cells, then by a collagen network attaching the particulate to the surrounding tissue, enabling further bone growth and attachment. Bioactive glass has been reported to be of value in treating intrabony periodontal defects when applied alone, or in combination with resorbable membranes or enamel matrix proteins (EMD) (Garg, 1999; Kuru et al., 2006; Mengel et al., 2006; Mengel et al., 2003; Miliauskaite et al., 2007; Park et al., 2001).
VI. Discussion and future directions (Figure 3)
Figure 3. Phosphate: Applying knowledge gained from developmental studies toward designing therapies to regenerate periodontal tissues.

TOP PANEL (Development): As discussed throughout this chapter, an appropriate balance of Pi/PPi locally, coupled with physiological serum levels of Pi, are required for development of teeth and supporting tissues.
MIDDLE BOX: Alterations in Pi metabolism, summarized in this box, result in defective tooth development
BOTTOM PANEL (Regeneration): Potential approaches for regenerating oral tissues include increasing levels of Pi at local sites.
* Note: Ank/PC-1 KO/mutant teeth with increased Pi/PPi suggest that increasing levels of Pi locally during early phases of wound healing may promote new cementum formation and subsequently promote new ligament attachment and new bone formation.
Currently, most regenerative therapeutic regimens for mineralized tissues employ some variation of the principles of tissue engineering as articulated by Langer and Vacanti (Langer and Vacanti, 1993; Lanza et al., 2007), e.g., local implantation of combinations of biological factors (cells, proteins) with engineered polymer-based scaffolds (e.g., poly-esters). Variations on this basic approach can range from employing scaffolds without added biological factors to cells, and/or proteins added to engineered scaffolds. Whereas stem cell therapy may restore a partial loss of organ function, the initial goal of tissue engineering is complete conversion of the engineered therapeutic combination to new tissue(s) or organs, e.g., bone (Herford and Boyne, 2008). The long-term goal is for regenerated tissues or organs to redress acquired or congenital deficiencies and become fully responsive to all the normal relevant physiological homeostatic mechanisms, i.e., become fully functional within the context of complete organism. Predictably, the development, structural and functional maturation, and maintenance of such tissue/organs would be adversely affected by malfunctions in key homeostatic regulatory systems. A case in point is the abnormalities in tooth development associated with alterations in local or serum Pi levels as described above. Hence, understanding of such systems is key to diagnosing, and if necessary, clinically managing any such malfunctioning systems to attempt to insure optimal outcomes to regeneration therapy.
Dental tissues and teeth form through a complex series of temporally and spatially regulated reciprocal interactions between oral epithelium and neural crest ectomesenchyme (Thesleff, 2003; Thesleff and Sharpe, 1997). Such interactions require like-cells to aggregate and join to interact with the other cell types for tissue formation to proceed normally. Gene expression studies of developing teeth, principally in mice, using normal and genetically altered animals has produced a large database (http://bite-it.helsinki.fi/) of genes implicated in the regulation of these processes. Members of major developmental signaling pathways, including FGF, sonic hedgehog (Shh), BMP, and Wnt signaling networks are involved at various and multiple stages (Cobourne and Sharpe, 2003; Dassule et al., 2000; Jarvinen et al., 2006; Jernvall and Thesleff, 2000; Laurikkala et al., 2003). The molecular basis of tooth agenesis has recently been reviewed (Matalova et al., 2008), identifying several genes, the loss of which can lead to agenesis of one or more teeth.
Strategies for whole tooth regeneration commonly start by separating tooth buds into epithelial and mesenchymal tissue components at the correct stage of development. Dental epithelium may be recombined with a competent dental mesenchyme (or mesenchymal substitute) prior to culture or implantation (Ohazama et al., 2004), the epithelial and mesenchymal tissues disaggregated into cell suspensions, mixed, added to engineered scaffolds and these constructs implanted in vivo (Young et al., 2002). Alternately, the separate epithelial and mesenchymal layers may be dissociated into high-density cell suspensions and placed in close approximation, permitting like-cells to reaggregate and the two cell types to interact and organize themselves (Nakao et al., 2007). Such recombined tooth buds have the potential to develop into structures resembling whole teeth. These data reveal that whole tooth regeneration will likely require cells, competent by nature or by manipulation, to interact in order to initiate the development cascade. It seems highly unlikely that manipulation of a single factor will provide sufficient signal to initiate whole tooth regeneration. The next challenge will be to produce a fully functioning tooth using readily available substitutes for competent dental epithelia and ectomesenchyme. Ultimately, to have real world applications, the bioengineered tooth will need to be “tunable” for characteristics of tooth-type (molar, incisor, etc.), size, shape, color, and must be integrated in its functional location.
To date, among the dental tissues, the periodontium comprising the cementum, PDL, and bone, has received the most effort toward developing regenerative therapy. Currently, partially characterized mixtures of enamel proteins, absent a clear developmentally based mechanism, are commercially available for regeneration of the periodontium (Esposito et al., 2005; Gestrelius et al., 2000; Giannobile and Somerman, 2003). Platelet-derived growth factor (PDGF), a mesenchymal cell mitogen and angiogenesis factor, combined with a calcium phosphate based carrier is also commercially available (Hollinger et al., 2008). Such first-generation products provide the “proof-of-principle” of dental tissue regeneration and inform refined subsequent efforts.
Cementum, based on the data reviewed above, appears to be particularly sensitive to local Pi concentration. TNAP deficiency results in increased local concentrations of PPi and cementum aplasia or severe hypoplasia. Mice with loss of function in ANK develop low local extracellular PPi, resulting in hypercementosis while other tissues apparently remain unaffected (Figure 2). In vitro data reveal that extracellular Pi can signal changes in gene expression in osteoblasts (Adams et al., 2001; Beck, 2003) and cementoblasts (Foster et al., 2006b; Rutherford et al., 2006), modulating cell phenotype. Taken together, these findings suggest that therapeutic regulation of extracellular Pi may have utility in the regeneration of the periodontium (Figure 3).
This idea immediately raises a number of practical questions. What is the optimum extracellular in vivo Pi concentration? What is the best method to achieve this concentration of extracellular Pi? How long must that concentration be sustained? Initially, in vitro washout experiments to determine the minimum time necessary for elevated Pi need to be undertaken. How could appropriate levels of Pi be maintained in the periodontal lesion for sufficient time to affect cells? Biodegradable thermoset or chemoset hydrogels containing the appropriate Pi concentration or active TNAP might be useful. Do sufficient populations of cementoblasts or their precursors exist in diseased periodontal tissues to enable cementogenesis? Would cementoblasts need to be included in the Pi/hydrogel implant? Would this signal be sufficient to induce regeneration of the PDL and bone to complete the periodontium? Would therapeutic stimulation of angiogenesis and neovascularization be necessary?
A concerted research program dedicated to these and other questions may bring a new facet to the fascinating and diverse biology of Pi. These types of studies may also further understanding of cementum and dentin formation and inform potential regenerative therapies for treatment of a broader group of diseases, including not only dental diseases, but also hypophosphatemic conditions, ectopic calcification, and osteoporosis.
Acknowledgments
The authors wish to thank Luke Wallace for the Figure 3 illustration. Special thanks to Dr. Wouter Beertsen for generously sharing histology images from TNAP null mice. This work and studies described herein were supported by NIH/NIDCR grant RO1 DE15109 (MJS), the Dean and Margaret Spencer Clinical Research Fund (HZ), and the University of Washington Research Advisory Committee (HZ).
Literature cited
- Abe K, Masatomi Y, Nakajima Y, Shintani S, Moriwaki Y, Sobue S, Ooshima T. The occurrence of interglobular dentin in incisors of hypophosphatemic mice fed a high-calcium and high-phosphate diet. J Dent Res. 1992;71(3):478–483. doi: 10.1177/00220345920710031101. [DOI] [PubMed] [Google Scholar]
- Abe K, Ooshima T, Lily TS, Yasufuku Y, Sobue S. Structural deformities of deciduous teeth in patients with hypophosphatemic vitamin D-resistant rickets. Oral Surg Oral Med Oral Pathol. 1988;65(2):191–198. doi: 10.1016/0030-4220(88)90165-x. [DOI] [PubMed] [Google Scholar]
- Abe K, Ooshima T, Masatomi Y, Sobue S, Moriwaki Y. Microscopic and crystallographic examinations of the teeth of the X-linked hypophosphatemic mouse. J Dent Res. 1989;68(11):1519–1524. doi: 10.1177/00220345890680111001. [DOI] [PubMed] [Google Scholar]
- Adams CS, Mansfield K, Perlot RL, Shapiro IM. Matrix regulation of skeletal cell apoptosis. Role of calcium and phosphate ions. J Biol Chem. 2001;276(23):20316–20322. doi: 10.1074/jbc.M006492200. [DOI] [PubMed] [Google Scholar]
- Addison W, Nakano Y, Loisel T, Crine P, McKee M. MEPE-ASARM Peptides Control Extracellular Matrix Mineralization by Binding to Hydroxyapatite - An Inhibition Regulated by PHEX Cleavage of ASARM. J Bone Miner Res. 2008 doi: 10.1359/jbmr.080601. [DOI] [PubMed] [Google Scholar]
- Addison WN, Azari F, Sorensen ES, Kaartinen MT, McKee MD. Pyrophosphate inhibits mineralization of osteoblast cultures by binding to mineral, up-regulating osteopontin, and inhibiting alkaline phosphatase activity. J Biol Chem. 2007;282(21):15872–15883. doi: 10.1074/jbc.M701116200. [DOI] [PubMed] [Google Scholar]
- ADHR_Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26(3):345–348. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- Akintoye SO, Lee JS, Feimster T, Booher S, Brahim J, Kingman A, Riminucci M, Robey PG, Collins MT. Dental characteristics of fibrous dysplasia and McCune-Albright syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;96(3):275–282. doi: 10.1016/s1079-2104(03)00225-7. [DOI] [PubMed] [Google Scholar]
- Akintoye SO, Otis LL, Atkinson JC, Brahim J, Kushner H, Robey PG, Collins MT. Analyses of variable panoramic radiographic characteristics of maxillo-mandibular fibrous dysplasia in McCune-Albright syndrome. Oral Dis. 2004;10(1):36–43. doi: 10.1046/j.1354-523x.2003.00971.x. [DOI] [PubMed] [Google Scholar]
- Anderson HC, Harmey D, Camacho NP, Garimella R, Sipe JB, Tague S, Bi X, Johnson K, Terkeltaub R, Millan JL. Sustained osteomalacia of long bones despite major improvement in other hypophosphatasia-related mineral deficits in tissue nonspecific alkaline phosphatase/nucleotide pyrophosphatase phosphodiesterase 1 double-deficient mice. Am J Pathol. 2005;166(6):1711–1720. doi: 10.1016/S0002-9440(10)62481-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya K, Fukumoto S, Backenroth R, Takeuchi Y, Nakayama K, Ito N, Yoshii N, Yamazaki Y, Yamashita T, Silver J, Igarashi T, Fujita T. A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J Clin Endocrinol Metab. 2005;90(10):5523–5527. doi: 10.1210/jc.2005-0301. [DOI] [PubMed] [Google Scholar]
- Argiro L, Desbarats M, Glorieux FH, Ecarot B. Mepe, the gene encoding a tumor-secreted protein in oncogenic hypophosphatemic osteomalacia, is expressed in bone. Genomics. 2001;74(3):342–351. doi: 10.1006/geno.2001.6553. [DOI] [PubMed] [Google Scholar]
- Baroncelli GI, Angiolini M, Ninni E, Galli V, Saggese R, Giuca MR. Prevalence and pathogenesis of dental and periodontal lesions in children with X-linked hypophosphatemic rickets. Eur J Paediatr Dent. 2006;7(2):61–66. [PubMed] [Google Scholar]
- Barrere F, van Blitterswijk CA, de Groot K. Bone regeneration: molecular and cellular interactions with calcium phosphate ceramics. Int J Nanomedicine. 2006;1(3):317–332. [PMC free article] [PubMed] [Google Scholar]
- Barrere F, van der Valk CM, Dalmeijer RA, Meijer G, van Blitterswijk CA, de Groot K, Layrolle P. Osteogenecity of octacalcium phosphate coatings applied on porous metal implants. J Biomed Mater Res A. 2003a;66(4):779–788. doi: 10.1002/jbm.a.10454. [DOI] [PubMed] [Google Scholar]
- Barrere F, van der Valk CM, Meijer G, Dalmeijer RA, de Groot K, Layrolle P. Osteointegration of biomimetic apatite coating applied onto dense and porous metal implants in femurs of goats. J Biomed Mater Res B Appl Biomater. 2003b;67(1):655–665. doi: 10.1002/jbm.b.10057. [DOI] [PubMed] [Google Scholar]
- Barthel TK, Mathern DR, Whitfield GK, Haussler CA, Hopper HAt, Hsieh JC, Slater SA, Hsieh G, Kaczmarska M, Jurutka PW, Kolek OI, Ghishan FK, Haussler MR. 1,25-Dihydroxyvitamin D3/VDR-mediated induction of FGF23 as well as transcriptional control of other bone anabolic and catabolic genes that orchestrate the regulation of phosphate and calcium mineral metabolism. J Steroid Biochem Mol Biol. 2007;103(3-5):381–388. doi: 10.1016/j.jsbmb.2006.12.054. [DOI] [PubMed] [Google Scholar]
- Bartold PM, McCulloch CA, Narayanan AS, Pitaru S. Tissue engineering: a new paradigm for periodontal regeneration based on molecular and cell biology. Perio 2000. 2000;24:253–269. doi: 10.1034/j.1600-0757.2000.2240113.x. [DOI] [PubMed] [Google Scholar]
- Bartold PM, Shi S, Gronthos S. Stem cells and periodontal regeneration. Periodontol 2000. 2006a;40:164–172. doi: 10.1111/j.1600-0757.2005.00139.x. [DOI] [PubMed] [Google Scholar]
- Bartold PM, Xiao Y, Lyngstaadas SP, Paine ML, Snead ML. Principles and applications of cell delivery systems for periodontal regeneration. Periodontol 2000. 2006b;41:123–135. doi: 10.1111/j.1600-0757.2006.00156.x. [DOI] [PubMed] [Google Scholar]
- Beck GR., Jr Inorganic phosphate as a signaling molecule in osteoblast differentiation. J Cell Biochem. 2003;90(2):234–243. doi: 10.1002/jcb.10622. [DOI] [PubMed] [Google Scholar]
- Beck GR, Jr, Knecht N. Osteopontin regulation by inorganic phosphate is ERK1/2-, protein kinase C-, and proteasome-dependent. J Biol Chem. 2003;278(43):41921–41929. doi: 10.1074/jbc.M304470200. [DOI] [PubMed] [Google Scholar]
- Beck GR, Jr, Moran E, Knecht N. Inorganic phosphate regulates multiple genes during osteoblast differentiation, including Nrf2. Exp Cell Res. 2003;288(2):288–300. doi: 10.1016/s0014-4827(03)00213-1. [DOI] [PubMed] [Google Scholar]
- Beck GR, Jr, Sullivan EC, Moran E, Zerler B. Relationship between alkaline phosphatase levels, osteopontin expression, and mineralization in differentiating MC3T3-E1 osteoblasts. J Cell Biochem. 1998;68(2):269–280. doi: 10.1002/(sici)1097-4644(19980201)68:2<269::aid-jcb13>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Beck GR, Jr, Zerler B, Moran E. Phosphate is a specific signal for induction of osteopontin gene expression. Proc Natl Acad Sci U S A. 2000;97(15):8352–8357. doi: 10.1073/pnas.140021997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beertsen W, VandenBos T, Everts V. Root development in mice lacking functional tissue non-specific alkaline phosphatase gene: inhibition of acellular cementum formation. J Dent Res. 1999;78(6):1221–1229. doi: 10.1177/00220345990780060501. In Process Citation. [DOI] [PubMed] [Google Scholar]
- Benet-Pages A, Lorenz-Depiereux B, Zischka H, White KE, Econs MJ, Strom TM. FGF23 is processed by proprotein convertases but not by PHEX. Bone. 2004;35(2):455–462. doi: 10.1016/j.bone.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Berdal A, Gorter de Vries I, Hotton D, Cuisinier-Gleizes P, Mathieu H. The cellular and extracellular distribution of osteocalcin and dentin phosphoprotein in teeth of vitamin D-deficient rats. J Biol Buccale. 1991;19(1):45–53. [PubMed] [Google Scholar]
- Berdal A, Papagerakis P, Hotton D, Bailleul-Forestier I, Davideau JL. Ameloblasts and odontoblasts, target-cells for 1,25-dihydroxyvitamin D3: a review. The International journal of developmental biology. 1995;39(1):257–262. [PubMed] [Google Scholar]
- Berndt T, Craig TA, Bowe AE, Vassiliadis J, Reczek D, Finnegan R, Jan De Beur SM, Schiavi SC, Kumar R. Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J Clin Invest. 2003;112(5):785–794. doi: 10.1172/JCI18563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt T, Kumar R. Phosphatonins and the regulation of phosphate homeostasis. Annu Rev Physiol. 2007;69:341–359. doi: 10.1146/annurev.physiol.69.040705.141729. [DOI] [PubMed] [Google Scholar]
- Berndt TJ, Bielesz B, Craig TA, Tebben PJ, Bacic D, Wagner CA, O’Brien S, Schiavi S, Biber J, Murer H, Kumar R. Secreted frizzled-related protein-4 reduces sodium-phosphate co-transporter abundance and activity in proximal tubule cells. Pflugers Arch. 2006;451(4):579–587. doi: 10.1007/s00424-005-1495-2. [DOI] [PubMed] [Google Scholar]
- Berndt TJ, Schiavi S, Kumar R. “Phosphatonins” and the regulation of phosphorus homeostasis. Am J Physiol Renal Physiol. 2005;289(6):F1170–1182. doi: 10.1152/ajprenal.00072.2005. [DOI] [PubMed] [Google Scholar]
- Bertoldo F, Santini D, Lo Cascio V. Bisphosphonates and osteomyelitis of the jaw: a pathogenic puzzle. Nat Clin Pract Oncol. 2007;4(12):711–721. doi: 10.1038/ncponc1000. [DOI] [PubMed] [Google Scholar]
- Bielesz B. Emerging role of a phosphatonin in mineral homeostasis and its derangements. Eur J Clin Invest. 2006;36(Suppl 2):34–42. doi: 10.1111/j.1365-2362.2006.01659.x. [DOI] [PubMed] [Google Scholar]
- Blethen FA, Chu EY, Foster BL, Tompkins KA, Sitara D, Lanske B, Somerman MJ. The effect of mutations in the systemic phosphate regulating gene Fgf-23 on the dentoalveolar complex. Vancouver, Canada: 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonen S, Laan RF, Barton IP, Watts NB. Effect of osteoporosis treatments on risk of non-vertebral fractures: review and meta-analysis of intention-to-treat studies. Osteoporos Int. 2005;16(10):1291–1298. doi: 10.1007/s00198-005-1945-x. [DOI] [PubMed] [Google Scholar]
- Bouillon R, Okamura WH, Norman AW. Structure-function relationships in the vitamin D endocrine system. Endocr Rev. 1995;16(2):200–257. doi: 10.1210/edrv-16-2-200. [DOI] [PubMed] [Google Scholar]
- Boukpessi T, Septier D, Bagga S, Garabedian M, Goldberg M, Chaussain-Miller C. Dentin alteration of deciduous teeth in human hypophosphatemic rickets. Calcif Tissue Int. 2006;79(5):294–300. doi: 10.1007/s00223-006-0182-4. [DOI] [PubMed] [Google Scholar]
- Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, Wu D, Insogna K, Lifton RP. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346(20):1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- Broadus AE. Mineral Balance and Homeostasis. In: Favus MJ, editor. Primer on the Metabolic Bone Diseases and Disorder of Mineral Metabolism. 5. Washington D.C.: ASBMR; 2003. pp. 105–111. [Google Scholar]
- Brockman H. Die Isolierung des antirachitischen Vitamins aus Thunfischleberol. Hoppe Seylers Z Physiol Chem. 1936;24:11. [Google Scholar]
- Bronckers AL, Price PA, Schrijvers A, Bervoets TJ, Karsenty G. Studies of osteocalcin function in dentin formation in rodent teeth. Eur J Oral Sci. 1998;106(3):795–807. doi: 10.1046/j.0909-8836.1998.eos106306.x. [DOI] [PubMed] [Google Scholar]
- Brown AJ, Zhong M, Finch J, Ritter C, Slatopolsky E. The roles of calcium and 1,25-dihydroxyvitamin D3 in the regulation of vitamin D receptor expression by rat parathyroid glands. Endocrinology. 1995;136(4):1419–1425. doi: 10.1210/endo.136.4.7895652. [DOI] [PubMed] [Google Scholar]
- Bruckner RJ, Rickles NH, Porter DR. Hypophosphatasia with premature shedding of teeth and aplasia of cementum. Oral Surg Oral Med Oral Pathol. 1962;15:1351–1369. doi: 10.1016/0030-4220(62)90356-0. [DOI] [PubMed] [Google Scholar]
- Caffesse RG, de la Rosa M, Mota LF. Regeneration of soft and hard tissue periodontal defects. Am J Dent. 2002;15(5):339–345. [PubMed] [Google Scholar]
- Calvi LM, Shin HI, Knight MC, Weber JM, Young MF, Giovannetti A, Schipani E. Constitutively active PTH/PTHrP receptor in odontoblasts alters odontoblast and ameloblast function and maturation. Mech Dev. 2004;121(4):397–408. doi: 10.1016/j.mod.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Campos M, Couture C, Hirata IY, Juliano MA, Loisel TP, Crine P, Juliano L, Boileau G, Carmona AK. Human recombinant endopeptidase PHEX has a strict S1’ specificity for acidic residues and cleaves peptides derived from fibroblast growth factor-23 and matrix extracellular phosphoglycoprotein. Biochem J. 2003;373(Pt 1):271–279. doi: 10.1042/BJ20030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuano P, Radanovic T, Wagner CA, Bacic D, Kato S, Uchiyama Y, St-Arnoud R, Murer H, Biber J. Intestinal and renal adaptation to a low-Pi diet of type II NaPi cotransporters in vitamin D receptor- and 1alphaOHase-deficient mice. Am J Physiol Cell Physiol. 2005;288(2):C429–434. doi: 10.1152/ajpcell.00331.2004. [DOI] [PubMed] [Google Scholar]
- Carpenter TO, Ellis BK, Insogna KL, Philbrick WM, Sterpka J, Shimkets R. Fibroblast growth factor 7: an inhibitor of phosphate transport derived from oncogenic osteomalacia-causing tumors. J Clin Endocrinol Metab. 2005;90(2):1012–1020. doi: 10.1210/jc.2004-0357. [DOI] [PubMed] [Google Scholar]
- Chang PL, Prince CW. 1 alpha,25-Dihydroxyvitamin D3 enhances 12-O-tetradecanoylphorbol-13-acetate- induced tumorigenic transformation and osteopontin expression in mouse JB6 epidermal cells. Cancer Res. 1993;53(10 Suppl):2217–2220. [PubMed] [Google Scholar]
- Chapple IL. Hypophosphatasia: dental aspects and mode of inheritance. J Clin Periodontol. 1993;20(9):615–622. doi: 10.1111/j.1600-051x.1993.tb00705.x. [DOI] [PubMed] [Google Scholar]
- Chaussain-Miller C, Sinding C, Septier D, Wolikow M, Goldberg M, Garabedian M. Dentin structure in familial hypophosphatemic rickets: benefits of vitamin D and phosphate treatment. Oral Dis. 2007;13(5):482–489. doi: 10.1111/j.1601-0825.2006.01326.x. [DOI] [PubMed] [Google Scholar]
- Chesnut CH. Treating osteoporosis with bisphosphonates and addressing adherence: a review of oral ibandronate. Drugs. 2006;66(10):1351–1359. doi: 10.2165/00003495-200666100-00004. [DOI] [PubMed] [Google Scholar]
- Chu EY, Foster BL, Swanson EC, Rutherford RB, Sitara D, Lanske BKM, Somerman MJ. Local and systemic regulation of tooth root tissues. New Orleans, LA: 2007. [Google Scholar]
- Cobourne MT, Sharpe PT. Tooth and jaw: molecular mechanisms of patterning in the first branchial arch. Arch Oral Biol. 2003;48(1):1–14. doi: 10.1016/s0003-9969(02)00208-x. [DOI] [PubMed] [Google Scholar]
- Cohen S, Becker GL. Origin, diagnosis, and treatment of the dental manifestations of vitamin D-resistant rickets: review of the literature and report of case. J Am Dent Assoc. 1976;92(1):120–129. doi: 10.14219/jada.archive.1976.0327. [DOI] [PubMed] [Google Scholar]
- Coleman RE. Bisphosphonates in breast cancer. Ann Oncol. 2005;16(5):687–695. doi: 10.1093/annonc/mdi162. [DOI] [PubMed] [Google Scholar]
- Cooke NE, Haddad JG. Vitamin D binding protein (Gc-globulin) Endocr Rev. 1989;10(3):294–307. doi: 10.1210/edrv-10-3-294. [DOI] [PubMed] [Google Scholar]
- Croom KF, Scott LJ. Intravenous ibandronate: in the treatment of osteoporosis. Drugs. 2006;66(12):1593–1601. doi: 10.2165/00003495-200666120-00005. discussion 1602-1593. [DOI] [PubMed] [Google Scholar]
- D’Errico JA, Berry JE, Ouyang H, Strayhorn CL, Windle JJ, Somerman MJ. Employing a transgenic animal model to obtain cementoblasts in vitro. J Periodontol. 2000;71(1):63–72. doi: 10.1902/jop.2000.71.1.63. [DOI] [PubMed] [Google Scholar]
- Dardenne O, Prud’homme J, Arabian A, Glorieux FH, St-Arnaud R. Targeted inactivation of the 25-hydroxyvitamin D(3)-1(alpha)-hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D-deficiency rickets. Endocrinology. 2001;142(7):3135–3141. doi: 10.1210/endo.142.7.8281. [DOI] [PubMed] [Google Scholar]
- Dassule HR, Lewis P, Bei M, Maas R, McMahon AP. Sonic hedgehog regulates growth and morphogenesis of the tooth. Development. 2000;127(22):4775–4785. doi: 10.1242/dev.127.22.4775. [DOI] [PubMed] [Google Scholar]
- de Groot K, Geesink R, Klein CP, Serekian P. Plasma sprayed coatings of hydroxylapatite. J Biomed Mater Res. 1987;21(12):1375–1381. doi: 10.1002/jbm.820211203. [DOI] [PubMed] [Google Scholar]
- de Jonge LT, Leeuwenburgh SC, Wolke JG, Jansen JA. Organic-Inorganic Surface Modifications for Titanium Implant Surfaces. Pharmaceutical research. 2008 doi: 10.1007/s11095-008-9617-0. [DOI] [PubMed] [Google Scholar]
- Delmas PD, Meunier PJ. The management of Paget’s disease of bone. N Engl J Med. 1997;336(8):558–566. doi: 10.1056/NEJM199702203360807. [DOI] [PubMed] [Google Scholar]
- Devogelaer JP. Treatment of bone diseases with bisphosphonates, excluding osteoporosis. Curr Opin Rheumatol. 2000;12(4):331–335. doi: 10.1097/00002281-200007000-00017. [DOI] [PubMed] [Google Scholar]
- Doi Y, Iwanaga H, Shibutani T, Moriwaki Y, Iwayama Y. Osteoclastic responses to various calcium phosphates in cell cultures. J Biomed Mater Res. 1999;47(3):424–433. doi: 10.1002/(sici)1097-4636(19991205)47:3<424::aid-jbm19>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Drezner MK. Phosphorus Homeostasis and Related Disorders. In: Bilezikian JP, Raisz LG, Rodan GA, editors. Principles of Bone Biology. 2. San Diego: Academic Press; 2002. pp. 321–338. [Google Scholar]
- Ecarot B, Glorieux FH, Desbarats M, Travers R, Labelle L. Defective bone formation by Hyp mouse bone cells transplanted into normal mice: evidence in favor of an intrinsic osteoblast defect. J Bone Miner Res. 1992;7(2):215–220. doi: 10.1002/jbmr.5650070213. [DOI] [PubMed] [Google Scholar]
- Econs MJ, Drezner MK. Tumor-induced osteomalacia--unveiling a new hormone. N Engl J Med. 1994;330(23):1679–1681. doi: 10.1056/NEJM199406093302310. [DOI] [PubMed] [Google Scholar]
- Eicher EM, Southard JL, Scriver CR, Glorieux FH. Hypophosphatemia: mouse model for human familial hypophosphatemic (vitamin D-resistant) rickets. Proc Natl Acad Sci U S A. 1976;73(12):4667–4671. doi: 10.1073/pnas.73.12.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Ghannam A. Bone reconstruction: from bioceramics to tissue engineering. Expert Rev Med Devices. 2005;2(1):87–101. doi: 10.1586/17434440.2.1.87. [DOI] [PubMed] [Google Scholar]
- Engstrom C, Jontell M, Linde A. Odontoblast metabolism in rats deficient in vitamin D and calcium. III. Protein synthesis in vitro. J Oral Pathol. 1978;7(4):227–235. doi: 10.1111/j.1600-0714.1978.tb01597.x. [DOI] [PubMed] [Google Scholar]
- Engstrom C, Linde A, Magnusson BC. Odontoblast metabolism in rats deficient in vitamin D and calcium I: A histochemical survey. J Oral Pathol. 1977;6(6):359–366. doi: 10.1111/j.1600-0714.1977.tb01801.x. [DOI] [PubMed] [Google Scholar]
- Erben RG, Mayer D, Weber K, Jonsson K, Juppner H, Lanske B. Overexpression of human PHEX under the human beta-actin promoter does not fully rescue the Hyp mouse phenotype. J Bone Miner Res. 2005;20(7):1149–1160. doi: 10.1359/JBMR.050212. [DOI] [PubMed] [Google Scholar]
- Esposito M, Grusovin MG, Coulthard P, Worthington HV. Enamel matrix derivative (Emdogain) for periodontal tissue regeneration in intrabony defects. Cochrane Database Syst Rev. 2005;4 doi: 10.1002/14651858.CD003875.pub2. CD003875. [DOI] [PubMed] [Google Scholar]
- Farrow EG, Davis SI, Ward LM, White KE. The role of DMP1 in autosomal recessive hypophosphatemic rickets. J Musculoskelet Neuronal Interact. 2007;7(4):310–312. [PubMed] [Google Scholar]
- Fatherazi S, Matsa-Dunn D, Foster BL, Rutherford RB, Somerman MJ, Presland RB. Phosphate regulation of osteopontin gene transcription involves a glucocorticoid response element. 2008 doi: 10.1177/0022034508328072. Under review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatherazi S, Presland R, Dunn D, BL F, Somerman MJ. Regulation of osteopontin gene transcription by inorganic phosphate; ASBMR 28th Annual Meeting; Philadelphia, PA, USA. 2006. [Google Scholar]
- Fedarko NS, Jain A, Karadag A, Fisher LW. Three small integrin binding ligand N-linked glycoproteins (SIBLINGs) bind and activate specific matrix metalloproteinases. Faseb J. 2004;18(6):734–736. doi: 10.1096/fj.03-0966fje. [DOI] [PubMed] [Google Scholar]
- Fedde KN, Blair L, Silverstein J, Coburn SP, Ryan LM, Weinstein RS, Waymire K, Narisawa S, Millan JL, MacGregor GR, Whyte MP. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J Bone Miner Res. 1999;14(12):2015–2026. doi: 10.1359/jbmr.1999.14.12.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006 doi: 10.1038/ng1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab. 2005;90(3):1519–1524. doi: 10.1210/jc.2004-1039. [DOI] [PubMed] [Google Scholar]
- Fisher LW, Fedarko NS. Six genes expressed in bones and teeth encode the current members of the SIBLING family of proteins. Connect Tissue Res. 2003;44(Suppl 1):33–40. [PubMed] [Google Scholar]
- Fleisch H, Bisaz S. Mechanism of calcification: inhibitory role of pyrophosphate. Nature. 1962;195:911. doi: 10.1038/195911a0. [DOI] [PubMed] [Google Scholar]
- Fleisch H, Straumann F, Schenk R, Bisaz S, Allgower M. Effect of condensed phosphates on calcification of chick embryo femurs in tissue culture. Am J Physiol. 1966;211(3):821–825. doi: 10.1152/ajplegacy.1966.211.3.821. [DOI] [PubMed] [Google Scholar]
- Fong H, Chu EY, Tompkins KA, BL F, Nociti FHJ, Sitara D, Lanske B, Somerman MJ. Aberrant cementum phenotype associated with hypophosphatemic Hyp mouse. 2008a doi: 10.1902/jop.2009.090129. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong HK, Foster BL, Popowics TE, Somerman MJ. The crowning achievement: getting to the root of the problem. J Dent Educ. 2005;69(5):555–570. [PubMed] [Google Scholar]
- Fong HK, Foster BL, Sarikaya M, Somerman MJ. Ank/ank mutant mice roots as a model for periodontal tissue regeneration- a study of structure and mechanical properties. 2008b doi: 10.1016/j.archoralbio.2009.02.011. Under review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster B, Swanson E, Matsa-Dunn D, Sato S, Rutherford R, Somerman M. Phosphate regulates expression of SIBLINGs and MMPs in cementoblasts; Proceedings of Hiroshima Conference on Education and Science in Dentistry; 2006a. pp. 43–49. [Google Scholar]
- Foster BL, Nociti FH, Jr, Swanson EC, Matsa-Dunn D, Berry JE, Cupp CJ, Zhang P, Somerman MJ. Regulation of SIBLING family genes by phosphate in cementoblasts. Banff Centre, Alberta; Canada: 2005. [Google Scholar]
- Foster BL, Nociti FH, Jr, Swanson EC, Matsa-Dunn D, Berry JE, Cupp CJ, Zhang P, Somerman MJ. Regulation of cementoblast gene expression by inorganic phosphate, in vitro. Calcified Tissue International. 2006b;78:103–112. doi: 10.1007/s00223-005-0184-7. [DOI] [PubMed] [Google Scholar]
- Foster BL, Popowics TE, Fong HK, Somerman MJ. Advances in defining regulators of cementum development and periodontal regeneration. Curr Top Dev Biol. 2007;78:47–126. doi: 10.1016/S0070-2153(06)78003-6. [DOI] [PubMed] [Google Scholar]
- Francis M, Fogelman I. 99mTc-diphosphonate uptake mechanisms on bone. In: Fogelman I, editor. Bone Scanning in Clinical Practice. London: Springer; 1987. pp. 1–6. [Google Scholar]
- Francis MJ, Lees RL, Trujillo E, Martin-Vasallo P, Heersche JN, Mobasheri A. ATPase pumps in osteoclasts and osteoblasts. Int J Biochem Cell Biol. 2002;34(5):459–476. doi: 10.1016/s1357-2725(01)00142-x. [DOI] [PubMed] [Google Scholar]
- Fretz JA, Zella LA, Kim S, Shevde NK, Pike JW. 1,25-Dihydroxyvitamin D3 induces expression of the Wnt signaling co-regulator LRP5 via regulatory elements located significantly downstream of the gene’s transcriptional start site. J Steroid Biochem Mol Biol. 2007;103(3-5):440–445. doi: 10.1016/j.jsbmb.2006.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Jilka RL, Manolagas SC, O’Brien CA. Parathyroid hormone stimulates receptor activator of NFkappa B ligand and inhibits osteoprotegerin expression via protein kinase A activation of cAMP-response element-binding protein. J Biol Chem. 2002;277(50):48868–48875. doi: 10.1074/jbc.M208494200. [DOI] [PubMed] [Google Scholar]
- Fudge NJ, Kovacs CS. Physiological studies in heterozygous calcium sensing receptor (CaSR) gene-ablated mice confirm that the CaSR regulates calcitonin release in vivo. BMC Physiol. 2004;4:5. doi: 10.1186/1472-6793-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T, Izumo N, Fukuyama R, Meguro T, Nakamuta H, Kohno T, Koida M. Phosphate provides an extracellular signal that drives nuclear export of Runx2/Cbfa1 in bone cells. Biochem Biophys Res Commun. 2001;280(1):348–352. doi: 10.1006/bbrc.2000.4108. [DOI] [PubMed] [Google Scholar]
- Garg AK. Grafting Materials in Repair and Restoration. In: Lynch SE, Genco RJ, Marx RE, editors. Tissue engineering : applications in maxillofacial surgery and periodontics. Chicago: Quintessence Publishing Co., Inc.; 1999. pp. 83–101. [Google Scholar]
- Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PV, Komm BS, Javed A, van Wijnen AJ, Stein JL, Stein GS, Lian JB. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem. 2005;280(39):33132–33140. doi: 10.1074/jbc.M500608200. [DOI] [PubMed] [Google Scholar]
- Geesink RG, de Groot K, Klein CP. Chemical implant fixation using hydroxyl-apatite coatings. The development of a human total hip prosthesis for chemical fixation to bone using hydroxyl-apatite coatings on titanium substrates. Clin Orthop Relat Res. 1987;(225):147–170. [PubMed] [Google Scholar]
- George A, Gui J, Jenkins NA, Gilbert DJ, Copeland NG, Veis A. In situ localization and chromosomal mapping of the AG1 (Dmp1) gene. J Histochem Cytochem. 1994;42(12):1527–1531. doi: 10.1177/42.12.7983353. [DOI] [PubMed] [Google Scholar]
- Gestrelius S, Lyngstadaas SP, Hammarstrom L. Emdogain--periodontal regeneration based on biomimicry. Clin Oral Investig. 2000;4(2):120–125. doi: 10.1007/s007840050127. [DOI] [PubMed] [Google Scholar]
- Giannobile WV, Somerman MJ. Growth and amelogenin-like factors in periodontal wound healing. A systematic review Ann Periodontol. 2003;8(1):193–204. doi: 10.1902/annals.2003.8.1.193. [DOI] [PubMed] [Google Scholar]
- Glass DA, 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8(5):751–764. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- Goding JW, Terkeltaub R, Maurice M, Deterre P, Sali A, Belli SI. Ecto-phosphodiesterase/pyrophosphatase of lymphocytes and non-lymphoid cells: structure and function of the PC-1 family. Immunol Rev. 1998;161:11–26. doi: 10.1111/j.1600-065x.1998.tb01568.x. [DOI] [PubMed] [Google Scholar]
- Goldblatt H, Soames KN. A study of rats on a normal diet irradiated daily by mercury vapor quartz lamp or kept in darkness. Biochem J. 1923;17:13. doi: 10.1042/bj0170294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes MF, Camargo AM, Sampaio TA, Graziozi MA, Armond MC. Oral manifestations of Albright hereditary osteodystrophy: a case report. Rev Hosp Clin Fac Med Sao Paulo. 2002;57(4):161–166. doi: 10.1590/s0041-87812002000400006. [DOI] [PubMed] [Google Scholar]
- Gowen LC, Petersen DN, Mansolf AL, Qi H, Stock JL, Tkalcevic GT, Simmons HA, Crawford DT, Chidsey-Frink KL, Ke HZ, McNeish JD, Brown TA. Targeted disruption of the osteoblast/osteocyte factor 45 gene (OF45) results in increased bone formation and bone mass. J Biol Chem. 2003;278(3):1998–2007. doi: 10.1074/jbc.M203250200. [DOI] [PubMed] [Google Scholar]
- Groeneveld MC, Everts V, Beertsen W. Alkaline phosphatase activity in the periodontal ligament and gingiva of the rat molar: its relation to cementum formation. J Dent Res. 1995;74(7):1374–1381. doi: 10.1177/00220345950740070901. [DOI] [PubMed] [Google Scholar]
- Groeneveld MC, Van den Bos T, Everts V, Beertsen W. Cell-bound and extracellular matrix-associated alkaline phosphatase activity in rat periodontal ligament. Experimental Oral Biology Group. J Periodontal Res. 1996;31(1):73–79. doi: 10.1111/j.1600-0765.1996.tb00466.x. [DOI] [PubMed] [Google Scholar]
- Guicheux J, Palmer G, Shukunami C, Hiraki Y, Bonjour JP, Caverzasio J. A novel in vitro culture system for analysis of functional role of phosphate transport in endochondral ossification. Bone. 2000;27(1):69–74. doi: 10.1016/s8756-3282(00)00302-1. [DOI] [PubMed] [Google Scholar]
- Gunn IR, Gaffney D. Clinical and laboratory features of calcium-sensing receptor disorders: a systematic review. Ann Clin Biochem. 2004;41(Pt 6):441–458. doi: 10.1258/0004563042466802. [DOI] [PubMed] [Google Scholar]
- Guo R, Rowe PS, Liu S, Simpson LG, Xiao ZS, Darryl Quarles LD. Inhibition of MEPE cleavage by Phex. Biochem Biophys Res Commun. 2002;297(1):38–45. doi: 10.1016/s0006-291x(02)02125-3. [DOI] [PubMed] [Google Scholar]
- Gupta A, Guo XL, Alvarez UM, Hruska KA. Regulation of sodium-dependent phosphate transport in osteoclasts. J Clin Invest. 1997;100(3):538–549. doi: 10.1172/JCI119563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurley KA, Chen H, Guenther C, Nguyen ET, Rountree RB, Schoor M, Kingsley DM. Mineral Formation in Joints Caused by Complete or Joint-Specific Loss of ANK Function. J Bone Miner Res. 2006a;21(8):1238–1247. doi: 10.1359/jbmr.060515. [DOI] [PubMed] [Google Scholar]
- Gurley KA, Reimer RJ, Kingsley DM. Biochemical and genetic analysis of ANK in arthritis and bone disease. Am J Hum Genet. 2006b;79(6):1017–1029. doi: 10.1086/509881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacchou Y, Uematsu T, Ueda O, Usui Y, Uematsu S, Takahashi M, Uchihashi T, Kawazoe Y, Shiba T, Kurihara S, Yamaoka M, Furusawa K. Inorganic polyphosphate: a possible stimulant of bone formation. J Dent Res. 2007;86(9):893–897. doi: 10.1177/154405910708600917. [DOI] [PubMed] [Google Scholar]
- Hak DJ. The use of osteoconductive bone graft substitutes in orthopaedic trauma. J Am Acad Orthop Surg. 2007;15(9):525–536. doi: 10.5435/00124635-200709000-00003. [DOI] [PubMed] [Google Scholar]
- Harant H, Andrew PJ, Reddy GS, Foglar E, Lindley IJ. 1alpha,25-dihydroxyvitamin D3 and a variety of its natural metabolites transcriptionally repress nuclear-factor-kappaB-mediated interleukin-8 gene expression. Eur J Biochem. 1997;250(1):63–71. doi: 10.1111/j.1432-1033.1997.00063.x. [DOI] [PubMed] [Google Scholar]
- Harmey D, Hessle L, Narisawa S, Johnson KA, Terkeltaub R, Millan JL. Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: an integrated model of the pathogenesis of mineralization disorders. Am J Pathol. 2004;164(4):1199–1209. doi: 10.1016/S0002-9440(10)63208-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussler MR. Vitamin D receptors: nature and function. Annu Rev Nutr. 1986;6:527–562. doi: 10.1146/annurev.nu.06.070186.002523. [DOI] [PubMed] [Google Scholar]
- Hayashibara T, Hiraga T, Sugita A, Wang L, Hata K, Ooshima T, Yoneda T. Regulation of osteoclast differentiation and function by phosphate: potential role of osteoclasts in the skeletal abnormalities in hypophosphatemic conditions. J Bone Miner Res. 2007;22(11):1743–1751. doi: 10.1359/jbmr.070709. [DOI] [PubMed] [Google Scholar]
- Hayashibara T, Komura T, Sobue S, Ooshima T. Tooth eruption in a patient with craniometaphyseal dysplasia: case report. J Oral Pathol Med. 2000;29(9):460–462. doi: 10.1034/j.1600-0714.2000.290907.x. [DOI] [PubMed] [Google Scholar]
- He G, Dahl T, Veis A, George A. Dentin matrix protein 1 initiates hydroxyapatite formation in vitro. Connect Tissue Res. 2003;44(Suppl 1):240–245. [PubMed] [Google Scholar]
- Heinonen JK. Biological Role of Inorganic Pyrophosphate. Norwell: Kluwer Academic Publishers; 2001. [Google Scholar]
- Helfrich MH, Hocking LJ. Genetics and aetiology of Pagetic disorders of bone. Arch Biochem Biophys. 2008;473(2):172–182. doi: 10.1016/j.abb.2008.02.045. [DOI] [PubMed] [Google Scholar]
- Henderson JE, Kremer R, Rhim JS, Goltzman D. Identification and functional characterization of adenylate cyclase-linked receptors for parathyroid hormone-like peptides on immortalized human keratinocytes. Endocrinology. 1992;130(1):449–457. doi: 10.1210/endo.130.1.1309343. [DOI] [PubMed] [Google Scholar]
- Henry HL, Norman AW. Studies on calciferol metabolism. IX. Renal 25-hydroxy-vitamin D3-1 hydroxylase Involvement of cytochrome P-450 and other properties. J Biol Chem. 1974;249(23):7529–7535. [PubMed] [Google Scholar]
- Herford AS, Boyne PJ. Reconstruction of mandibular continuity defects with bone morphogenetic protein-2 (rhBMP-2) J Oral Maxillofac Surg. 2008;66(4):616–624. doi: 10.1016/j.joms.2007.11.021. [DOI] [PubMed] [Google Scholar]
- Hesse M, Frohlich LF, Zeitz U, Lanske B, Erben RG. Ablation of vitamin D signaling rescues bone, mineral, and glucose homeostasis in Fgf-23 deficient mice. Matrix Biol. 2007;26(2):75–84. doi: 10.1016/j.matbio.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A, Goding JW, Terkeltaub R, Millan JL. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci U S A. 2002;99(14):9445–9449. doi: 10.1073/pnas.142063399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AM, Johnson MD, Kingsley DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000;289(5477):265–270. doi: 10.1126/science.289.5477.265. [DOI] [PubMed] [Google Scholar]
- Hoenderop JG, Nilius B, Bindels RJ. Calcium absorption across epithelia. Physiol Rev. 2005;85(1):373–422. doi: 10.1152/physrev.00003.2004. [DOI] [PubMed] [Google Scholar]
- Hollinger JO, Hart CE, Hirsch SN, Lynch S, Friedlaender GE. Recombinant human platelet-derived growth factor: biology and clinical applications. J Bone Joint Surg Am. 2008;90(Suppl 1):48–54. doi: 10.2106/JBJS.G.01231. [DOI] [PubMed] [Google Scholar]
- Houston B, Paton IR, Burt DW, Farquharson C. Chromosomal localization of the chicken and mammalian orthologues of the orphan phosphatase PHOSPHO1 gene. Anim Genet. 2002;33(6):451–454. doi: 10.1046/j.1365-2052.2002.00900.x. [DOI] [PubMed] [Google Scholar]
- Houston B, Stewart AJ, Farquharson C. PHOSPHO1-A novel phosphatase specifically expressed at sites of mineralisation in bone and cartilage. Bone. 2004;34(4):629–637. doi: 10.1016/j.bone.2003.12.023. [DOI] [PubMed] [Google Scholar]
- Hsieh JC, Kodjabachian L, Rebbert ML, Rattner A, Smallwood PM, Samos CH, Nusse R, Dawid IB, Nathans J. A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature. 1999;398(6726):431–436. doi: 10.1038/18899. [DOI] [PubMed] [Google Scholar]
- Hu B, Nadiri A, Kuchler-Bopp S, Perrin-Schmitt F, Peters H, Lesot H. Tissue engineering of tooth crown, root, and periodontium. Tissue Eng. 2006;12(8):2069–2075. doi: 10.1089/ten.2006.12.2069. [DOI] [PubMed] [Google Scholar]
- HYP_Consortium. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium Nat Genet. 1995;11(2):130–136. doi: 10.1038/ng1095-130. [DOI] [PubMed] [Google Scholar]
- Ito M, Haito S, Furumoto M, Uehata Y, Sakurai A, Segawa H, Tatsumi S, Kuwahata M, Miyamoto K. Unique uptake and efflux systems of inorganic phosphate in osteoclast-like cells. Am J Physiol Cell Physiol. 2007;292(1):C526–534. doi: 10.1152/ajpcell.00357.2006. [DOI] [PubMed] [Google Scholar]
- Ito M, Matsuka N, Izuka M, Haito S, Sakai Y, Nakamura R, Segawa H, Kuwahata M, Yamamoto H, Pike WJ, Miyamoto K. Characterization of inorganic phosphate transport in osteoclast-like cells. Am J Physiol Cell Physiol. 2005;288(4):C921–931. doi: 10.1152/ajpcell.00412.2004. [DOI] [PubMed] [Google Scholar]
- Jarvinen E, Salazar-Ciudad I, Birchmeier W, Taketo MM, Jernvall J, Thesleff I. Continuous tooth generation in mouse is induced by activated epithelial Wnt/beta-catenin signaling. Proc Natl Acad Sci U S A. 2006;103(49):18627–18632. doi: 10.1073/pnas.0607289103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernvall J, Thesleff I. Reiterative signaling and patterning during mammalian tooth morphogenesis. Mech Dev. 2000;92(1):19–29. doi: 10.1016/s0925-4773(99)00322-6. [DOI] [PubMed] [Google Scholar]
- Jilka RL. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone. 2007;40(6):1434–1446. doi: 10.1016/j.bone.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K, Goding J, Van Etten D, Sali A, Hu SI, Farley D, Krug H, Hessle L, Millan JL, Terkeltaub R. Linked deficiencies in extracellular PP(i) and osteopontin mediate pathologic calcification associated with defective PC-1 and ANK expression. J Bone Miner Res. 2003;18(6):994–1004. doi: 10.1359/jbmr.2003.18.6.994. [DOI] [PubMed] [Google Scholar]
- Johnson K, Pritzker K, Goding J, Terkeltaub R. The nucleoside triphosphate pyrophosphohydrolase isozyme PC-1 directly promotes cartilage calcification through chondrocyte apoptosis and increased calcium precipitation by mineralizing vesicles. J Rheumatol. 2001;28(12):2681–2691. [PubMed] [Google Scholar]
- Johnson KA, Hessle L, Vaingankar S, Wennberg C, Mauro S, Narisawa S, Goding JW, Sano K, Millan JL, Terkeltaub R. Osteoblast tissue-nonspecific alkaline phosphatase antagonizes and regulates PC-1. Am J Physiol Regul Integr Comp Physiol. 2000;279(4):R1365–1377. doi: 10.1152/ajpregu.2000.279.4.R1365. [DOI] [PubMed] [Google Scholar]
- Juppner H, Abou-Samra AB, Freeman M, Kong XF, Schipani E, Richards J, Kolakowski LF, Jr, Hock J, Potts JT, Jr, Kronenberg HM, et al. A G protein-linked receptor for parathyroid hormone and parathyroid hormone-related peptide. Science. 1991;254(5034):1024–1026. doi: 10.1126/science.1658941. [DOI] [PubMed] [Google Scholar]
- Kanatani M, Sugimoto T, Kano J, Kanzawa M, Chihara K. Effect of high phosphate concentration on osteoclast differentiation as well as bone-resorbing activity. J Cell Physiol. 2003;196(1):180–189. doi: 10.1002/jcp.10270. [DOI] [PubMed] [Google Scholar]
- Kavanagh KL, Guo K, Dunford JE, Wu X, Knapp S, Ebetino FH, Rogers MJ, Russell RG, Oppermann U. The molecular mechanism of nitrogen-containing bisphosphonates as antiosteoporosis drugs. Proc Natl Acad Sci U S A. 2006;103(20):7829–7834. doi: 10.1073/pnas.0601643103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh MP, Kabat D. Identification and characterization of a widely expressed phosphate transporter/retrovirus receptor family. Kidney Int. 1996;49(4):959–963. doi: 10.1038/ki.1996.135. [DOI] [PubMed] [Google Scholar]
- Kawazoe Y, Katoh S, Onodera Y, Kohgo T, Shindoh M, Shiba T. Activation of the FGF signaling pathway and subsequent induction of mesenchymal stem cell differentiation by inorganic polyphosphate. Int J Biol Sci. 2008;4(1):37–47. doi: 10.7150/ijbs.4.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawazoe Y, Shiba T, Nakamura R, Mizuno A, Tsutsumi K, Uematsu T, Yamaoka M, Shindoh M, Kohgo T. Induction of calcification in MC3T3-E1 cells by inorganic polyphosphate. J Dent Res. 2004;83(8):613–618. doi: 10.1177/154405910408300806. [DOI] [PubMed] [Google Scholar]
- Kitahara Y, Suda N, Kuroda T, Beck F, Hammond VE, Takano Y. Disturbed tooth development in parathyroid hormone-related protein (PTHrP)-gene knockout mice. Bone. 2002;30(1):48–56. doi: 10.1016/s8756-3282(01)00669-x. [DOI] [PubMed] [Google Scholar]
- Kokubo T, Kim HM, Kawashita M. Novel bioactive materials with different mechanical properties. Biomaterials. 2003;24(13):2161–2175. doi: 10.1016/s0142-9612(03)00044-9. [DOI] [PubMed] [Google Scholar]
- Kolek OI, Hines ER, Jones MD, LeSueur LK, Lipko MA, Kiela PR, Collins JF, Haussler MR, Ghishan FK. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol. 2005;289(6):G1036–1042. doi: 10.1152/ajpgi.00243.2005. [DOI] [PubMed] [Google Scholar]
- Kondo H, Guo J, Bringhurst FR. Cyclic adenosine monophosphate/protein kinase A mediates parathyroid hormone/parathyroid hormone-related protein receptor regulation of osteoclastogenesis and expression of RANKL and osteoprotegerin mRNAs by marrow stromal cells. J Bone Miner Res. 2002;17(9):1667–1679. doi: 10.1359/jbmr.2002.17.9.1667. [DOI] [PubMed] [Google Scholar]
- Kumar R. New insights into phosphate homeostasis: fibroblast growth factor 23 and frizzled-related protein-4 are phosphaturic factors derived from tumors associated with osteomalacia. Curr Opin Nephrol Hypertens. 2002;11(5):547–553. doi: 10.1097/00041552-200209000-00011. [DOI] [PubMed] [Google Scholar]
- Kumar R, Haugen JD, Penniston JT. Molecular cloning of a plasma membrane calcium pump from human osteoblasts. J Bone Miner Res. 1993;8(4):505–513. doi: 10.1002/jbmr.5650080415. [DOI] [PubMed] [Google Scholar]
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390(6655):45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro OM. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281(10):6120–6123. doi: 10.1074/jbc.C500457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuru B, Yilmaz S, Argin K, Noyan U. Enamel matrix derivative alone or in combination with a bioactive glass in wide intrabony defects. Clin Oral Investig. 2006;10(3):227–234. doi: 10.1007/s00784-006-0052-5. [DOI] [PubMed] [Google Scholar]
- Langer R, Vacanti JP. Tissue Engineering. Science. 1993;260:920–926. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- Lanske B, Karaplis AC, Lee K, Luz A, Vortkamp A, Pirro A, Karperien M, Defize LHK, Ho C, Mulligan RC, Abou-Samra AB, Juppner H, Segre GV, Kronenberg HM. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273(5275):663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- Lanza R, Langer R, Vacanti JP. Principles of Tissue Engineering. 3. San Diego: Academic Press; 2007. [Google Scholar]
- Larsson T, Marsell R, Schipani E, Ohlsson C, Ljunggren O, Tenenhouse HS, Juppner H, Jonsson KB. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology. 2004;145(7):3087–3094. doi: 10.1210/en.2003-1768. [DOI] [PubMed] [Google Scholar]
- Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003;64(6):2272–2279. doi: 10.1046/j.1523-1755.2003.00328.x. [DOI] [PubMed] [Google Scholar]
- Laurikkala J, Kassai Y, Pakkasjarvi L, Thesleff I, Itoh N. Identification of a secreted BMP antagonist, ectodin, integrating BMP, FGF, and SHH signals from the tooth enamel knot. Dev Biol. 2003;264(1):91–105. doi: 10.1016/j.ydbio.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Le Guehennec L, Soueidan A, Layrolle P, Amouriq Y. Surface treatments of titanium dental implants for rapid osseointegration. Dent Mater. 2007;23(7):844–854. doi: 10.1016/j.dental.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Leeuwenburgh SC, Wolke JG, Siebers MC, Schoonman J, Jansen JA. In vitro and in vivo reactivity of porous, electrosprayed calcium phosphate coatings. Biomaterials. 2006;27(18):3368–3378. doi: 10.1016/j.biomaterials.2006.01.052. [DOI] [PubMed] [Google Scholar]
- Levi M, Lotscher M, Sorribas V, Custer M, Arar M, Kaissling B, Murer H, Biber J. Cellular mechanisms of acute and chronic adaptation of rat renal P(i) transporter to alterations in dietary P(i) Am J Physiol. 1994;267(5 Pt 2):F900–908. doi: 10.1152/ajprenal.1994.267.5.F900. [DOI] [PubMed] [Google Scholar]
- Leyhausen G, Lorenz B, Zhu H, Geurtsen W, Bohnensack R, Muller WE, Schroder HC. Inorganic polyphosphate in human osteoblast-like cells. J Bone Miner Res. 1998;13(5):803–812. doi: 10.1359/jbmr.1998.13.5.803. [DOI] [PubMed] [Google Scholar]
- Li J, Mashiba T, Burr DB. Bisphosphonate treatment suppresses not only stochastic remodeling but also the targeted repair of microdamage. Calcif Tissue Int. 2001;69(5):281–286. doi: 10.1007/s002230010036. [DOI] [PubMed] [Google Scholar]
- Li X, Liu P, Liu W, Maye P, Zhang J, Zhang Y, Hurley M, Guo C, Boskey A, Sun L, Harris SE, Rowe DW, Ke HZ, Wu D. Dkk2 has a role in terminal osteoblast differentiation and mineralized matrix formation. Nat Genet. 2005;37(9):945–952. doi: 10.1038/ng1614. [DOI] [PubMed] [Google Scholar]
- Li X, Yang HY, Giachelli CM. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ Res. 2006;98(7):905–912. doi: 10.1161/01.RES.0000216409.20863.e7. [DOI] [PubMed] [Google Scholar]
- Ling Y, Rios HF, Myers ER, Lu Y, Feng JQ, Boskey AL. DMP1 depletion decreases bone mineralization in vivo: an FTIR imaging analysis. J Bone Miner Res. 2005;20(12):2169–2177. doi: 10.1359/JBMR.050815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Brown TA, Zhou J, Xiao ZS, Awad H, Guilak F, Quarles LD. Role of matrix extracellular phosphoglycoprotein in the pathogenesis of X-linked hypophosphatemia. J Am Soc Nephrol. 2005;16(6):1645–1653. doi: 10.1681/ASN.2004121060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Guo R, Simpson LG, Xiao ZS, Burnham CE, Quarles LD. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem. 2003;278(39):37419–37426. doi: 10.1074/jbc.M304544200. [DOI] [PubMed] [Google Scholar]
- Liu S, Quarles LD. How fibroblast growth factor 23 works. J Am Soc Nephrol. 2007;18(6):1637–1647. doi: 10.1681/ASN.2007010068. [DOI] [PubMed] [Google Scholar]
- Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic Role of FGF23 in Hyp Mice. Am J Physiol Endocrinol Metab. 2006 doi: 10.1152/ajpendo.00008.2006. [DOI] [PubMed] [Google Scholar]
- Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH, Olivares JL, Loris C, Ramos FJ, Glorieux F, Vikkula M, Juppner H, Strom TM. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet. 2006;38(11):1248–1250. doi: 10.1038/ng1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Huang S, Miclau T, Helms JA, Colnot C. Mepe is expressed during skeletal development and regeneration. Histochem Cell Biol. 2004;121(6):493–499. doi: 10.1007/s00418-004-0653-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren T, Stenport V, Wetter A, Linde A. Parathyroid hormone (1-34) receptor-binding and second-messenger response in rat incisor odontoblasts. Calcif Tissue Int. 1998;62(3):255–259. doi: 10.1007/s002239900426. [DOI] [PubMed] [Google Scholar]
- Lundquist P. Odontoblast phosphate and calcium transport in dentinogenesis. Swed Dent J. 2002;(Suppl(154)):1–52. [PubMed] [Google Scholar]
- Lundquist P, Ritchie HH, Moore K, Lundgren T, Linde A. Phosphate and calcium uptake by rat odontoblast-like MRPC-1 cells concomitant with mineralization. J Bone Miner Res. 2002;17(10):1801–1813. doi: 10.1359/jbmr.2002.17.10.1801. [DOI] [PubMed] [Google Scholar]
- MacDougall M, Simmons D, Gu TT, Dong J. MEPE/OF45, a new dentin/bone matrix protein and candidate gene for dentin diseases mapping to chromosome 4q21. Connect Tissue Res. 2002;43(2-3):320–330. doi: 10.1080/03008200290000556. [DOI] [PubMed] [Google Scholar]
- Mansfield K, Pucci B, Adams CS, Shapiro IM. Induction of apoptosis in skeletal tissues: phosphate-mediated chick chondrocyte apoptosis is calcium dependent. Calcif Tissue Int. 2003;73(2):161–172. doi: 10.1007/s00223-002-1056-z. [DOI] [PubMed] [Google Scholar]
- Mansfield K, Rajpurohit R, Shapiro IM. Extracellular phosphate ions cause apoptosis of terminally differentiated epiphyseal chondrocytes. J Cell Physiol. 1999;179(3):276–286. doi: 10.1002/(SICI)1097-4652(199906)179:3<276::AID-JCP5>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Mansfield K, Teixeira CC, Adams CS, Shapiro IM. Phosphate ions mediate chondrocyte apoptosis through a plasma membrane transporter mechanism. Bone. 2001;28(1):1–8. doi: 10.1016/s8756-3282(00)00409-9. [DOI] [PubMed] [Google Scholar]
- Martin A, David V, Laurence JS, Schwarz PM, Lafer EM, Hedge AM, Rowe PS. Degradation of MEPE, DMP1, and release of SIBLING ASARM-peptides (minhibins): ASARM-peptide(s) are directly responsible for defective mineralization in HYP. Endocrinology. 2008;149(4):1757–1772. doi: 10.1210/en.2007-1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx RE, Sawatari Y, Fortin M, Broumand V. Bisphosphonate-induced exposed bone (osteonecrosis/osteopetrosis) of the jaws: risk factors, recognition, prevention, and treatment. J Oral Maxillofac Surg. 2005;63(11):1567–1575. doi: 10.1016/j.joms.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Mashiba T, Hirano T, Turner CH, Forwood MR, Johnston CC, Burr DB. Suppressed bone turnover by bisphosphonates increases microdamage accumulation and reduces some biomechanical properties in dog rib. J Bone Miner Res. 2000;15(4):613–620. doi: 10.1359/jbmr.2000.15.4.613. [DOI] [PubMed] [Google Scholar]
- Mashiba T, Turner CH, Hirano T, Forwood MR, Johnston CC, Burr DB. Effects of suppressed bone turnover by bisphosphonates on microdamage accumulation and biomechanical properties in clinically relevant skeletal sites in beagles. Bone. 2001;28(5):524–531. doi: 10.1016/s8756-3282(01)00414-8. [DOI] [PubMed] [Google Scholar]
- Masuyama R, Stockmans I, Torrekens S, Van Looveren R, Maes C, Carmeliet P, Bouillon R, Carmeliet G. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J Clin Invest. 2006;116(12):3150–3159. doi: 10.1172/JCI29463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matalova E, Fleischmannova J, Sharpe PT, Tucker AS. Tooth agenesis: from molecular genetics to molecular dentistry. J Dent Res. 2008;87(7):617–623. doi: 10.1177/154405910808700715. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Okazaki M, Nakahira A, Sasaki J, Egusa H, Sohmura T. Modification of apatite materials for bone tissue engineering and drug delivery carriers. Curr Med Chem. 2007;14(25):2726–2733. doi: 10.2174/092986707782023208. [DOI] [PubMed] [Google Scholar]
- Meleti Z, Shapiro IM, Adams CS. Inorganic phosphate induces apoptosis of osteoblast-like cells in culture. Bone. 2000;27(3):359–366. doi: 10.1016/s8756-3282(00)00346-x. [DOI] [PubMed] [Google Scholar]
- Mellanby E. Experimental rickets. Spec Rep Ser Med Res Council(SRS61) 1921;4 [Google Scholar]
- Memon F, El-Abbadi M, Nakatani T, Taguchi T, Lanske B, Razzaque MS. Does Fgf23-klotho activity influence vascular and soft tissue calcification through regulating mineral ion metabolism? Kidney Int. 2008 doi: 10.1038/ki.2008.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengel R, Schreiber D, Flores-de-Jacoby L. Bioabsorbable membrane and bioactive glass in the treatment of intrabony defects in patients with generalized aggressive periodontitis: results of a 5-year clinical and radiological study. J Periodontol. 2006;77(10):1781–1787. doi: 10.1902/jop.2006.060029. [DOI] [PubMed] [Google Scholar]
- Mengel R, Soffner M, Flores-de-Jacoby L. Bioabsorbable membrane and bioactive glass in the treatment of intrabony defects in patients with generalized aggressive periodontitis: results of a 12-month clinical and radiological study. J Periodontol. 2003;74(6):899–908. doi: 10.1902/jop.2003.74.6.899. [DOI] [PubMed] [Google Scholar]
- Meszaros JG, Karin NJ. Osteoblasts express the PMCA1b isoform of the plasma membrane Ca(2+)-ATPase. J Bone Miner Res. 1993;8(10):1235–1240. doi: 10.1002/jbmr.5650081011. [DOI] [PubMed] [Google Scholar]
- Meyer JL. Can biological calcification occur in the presence of pyrophosphate? Archives of biochemistry and biophysics. 1984;231(1):1–8. doi: 10.1016/0003-9861(84)90356-4. [DOI] [PubMed] [Google Scholar]
- Miao D, Bai X, Panda DK, Karaplis AC, Goltzman D, McKee MD. Cartilage abnormalities are associated with abnormal Phex expression and with altered matrix protein and MMP-9 localization in Hyp mice. Bone. 2004;34(4):638–647. doi: 10.1016/j.bone.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Migliorati CA, Schubert MM, Peterson DE, Seneda LM. Bisphosphonate-associated osteonecrosis of mandibular and maxillary bone: an emerging oral complication of supportive cancer therapy. Cancer. 2005;104(1):83–93. doi: 10.1002/cncr.21130. [DOI] [PubMed] [Google Scholar]
- Miliauskaite A, Selimovic D, Hannig M. Successful management of aggressive periodontitis by regenerative therapy: a 3-year follow-up case report. J Periodontol. 2007;78(10):2043–2050. doi: 10.1902/jop.2007.060492. [DOI] [PubMed] [Google Scholar]
- Millan JL. Mammalian Alkaline Phosphatases. Weinheim: Wiley-VCH Verlag GmbH & Co KGaA; 2006. p. 322. [Google Scholar]
- Millan JL, Narisawa S, Lemire I, Loisel TP, Boileau G, Leonard P, Gramatikova S, Terkeltaub R, Pleshko Camacho N, McKee MD, Crine P, Whyte MP. Enzyme Replacement Therapy for Murine Hypophosphatasia. J Bone Miner Res. 2007 doi: 10.1359/JBMR.071213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitnick PD, Goldfarb S, Slatopolsky E, Lemann J, Jr, Gray RW, Agus ZS. Calcium and phosphate metabolism in tumoral calcinosis. Ann Intern Med. 1980;92(4):482–487. doi: 10.7326/0003-4819-92-4-482. [DOI] [PubMed] [Google Scholar]
- Mozar A, Haren N, Chasseraud M, Louvet L, Maziere C, Wattel A, Mentaverri R, Morliere P, Kamel S, Brazier M, Maziere JC, Massy ZA. High extracellular inorganic phosphate concentration inhibits RANK-RANKL signaling in osteoclast-like cells. J Cell Physiol. 2008;215(1):47–54. doi: 10.1002/jcp.21283. [DOI] [PubMed] [Google Scholar]
- Mulkins MA, Manolagas SC, Deftos LJ, Sussman HH. 1,25-Dihydroxyvitamin D3 increases bone alkaline phosphatase isoenzyme levels in human osteogenic sarcoma cells. J Biol Chem. 1983;258(10):6219–6225. [PubMed] [Google Scholar]
- Murayama T, Iwatsubo R, Akiyama S, Amano A, Morisaki I. Familial hypophosphatemic vitamin D-resistant rickets: dental findings and histologic study of teeth. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90(3):310–316. doi: 10.1067/moe.2000.107522. [DOI] [PubMed] [Google Scholar]
- Murer H, Forster I, Hernando N, Lambert G, Traebert M, Biber J. Posttranscriptional regulation of the proximal tubule NaPi-II transporter in response to PTH and dietary P(i) Am J Physiol. 1999;277(5 Pt 2):F676–684. doi: 10.1152/ajprenal.1999.277.5.F676. [DOI] [PubMed] [Google Scholar]
- Murer H, Forster I, Hilfiker H, Pfister M, Kaissling B, Lotscher M, Biber J. Cellular/molecular control of renal Na/Pi-cotransport. Kidney Int Suppl. 1998;65:S2–10. [PubMed] [Google Scholar]
- Murshed M, Harmey D, Millan JL, McKee MD, Karsenty G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 2005;19(9):1093–1104. doi: 10.1101/gad.1276205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadiri A, Kuchler-Bopp S, Haikel Y, Lesot H. Immunolocalization of BMP-2/-4, FGF-4, and WNT10b in the developing mouse first lower molar. J Histochem Cytochem. 2004;52(1):103–112. doi: 10.1177/002215540405200110. [DOI] [PubMed] [Google Scholar]
- Nagpal S, Na S, Rathnachalam R. Noncalcemic actions of vitamin D receptor ligands. Endocr Rev. 2005;26(5):662–687. doi: 10.1210/er.2004-0002. [DOI] [PubMed] [Google Scholar]
- Nakano Y, Beertsen W, van den Bos T, Kawamoto T, Oda K, Takano Y. Site-specific localization of two distinct phosphatases along the osteoblast plasma membrane: tissue non-specific alkaline phosphatase and plasma membrane calcium ATPase. Bone. 2004;35(5):1077–1085. doi: 10.1016/j.bone.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Nakano Y, Kawamoto T, Oda K, Takano Y. Alkaline and acid phosphatases in bone cells serve as phosphohydrolases at physiological pH in vivo: a histochemical implication. Connect Tissue Res. 2003;44(Suppl 1):219–222. [PubMed] [Google Scholar]
- Nakao K, Morita R, Saji Y, Ishida K, Tomita Y, Ogawa M, Saitoh M, Tomooka Y, Tsuji T. The development of a bioengineered organ germ method. Nat Methods. 2007;4(3):227–230. doi: 10.1038/nmeth1012. [DOI] [PubMed] [Google Scholar]
- Nampei A, Hashimoto J, Hayashida K, Tsuboi H, Shi K, Tsuji I, Miyashita H, Yamada T, Matsukawa N, Matsumoto M, Morimoto S, Ogihara T, Ochi T, Yoshikawa H. Matrix extracellular phosphoglycoprotein (MEPE) is highly expressed in osteocytes in human bone. J Bone Miner Metab. 2004;22(3):176–184. doi: 10.1007/s00774-003-0468-9. [DOI] [PubMed] [Google Scholar]
- Nanci A. Chapter 5. Development of the Tooth and its Supporting Tissues. In: Nanci A, editor. Ten Cate’s Oral Histology: Development, Structure, and Function. 6. St. Louis: Mosby; 2003. [Google Scholar]
- Narayanan K, Ramachandran A, Hao J, George A. Transcriptional regulation of dentin matrix protein 1 (DMP1) by AP-1 (c-fos/c-jun) factors. Connect Tissue Res. 2002;43(2-3):365–371. doi: 10.1080/03008200290000592. [DOI] [PubMed] [Google Scholar]
- Narayanan K, Ramachandran A, Hao J, He G, Park KW, Cho M, George A. Dual functional roles of dentin matrix protein 1. Implications in biomineralization and gene transcription by activation of intracellular Ca2+ store J Biol Chem. 2003;278(19):17500–17508. doi: 10.1074/jbc.M212700200. [DOI] [PubMed] [Google Scholar]
- Narayanan K, Srinivas R, Ramachandran A, Hao J, Quinn B, George A. Differentiation of embryonic mesenchymal cells to odontoblast-like cells by overexpression of dentin matrix protein 1. Proc Natl Acad Sci U S A. 2001;98(8):4516–4521. doi: 10.1073/pnas.081075198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narisawa S, Frohlander N, Millan JL. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev Dyn. 1997;208(3):432–446. doi: 10.1002/(SICI)1097-0177(199703)208:3<432::AID-AJA13>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Nashiki K, Taketani Y, Takeichi T, Sawada N, Yamamoto H, Ichikawa M, Arai H, Miyamoto K, Takeda E. Role of membrane microdomains in PTH-mediated down-regulation of NaPi-IIa in opossum kidney cells. Kidney Int. 2005;68(3):1137–1147. doi: 10.1111/j.1523-1755.2005.00505.x. [DOI] [PubMed] [Google Scholar]
- Nery EB, LeGeros RZ, Lynch KL, Lee K. Tissue response to biphasic calcium phosphate ceramic with different ratios of HA/beta TCP in periodontal osseous defects. J Periodontol. 1992;63(9):729–735. doi: 10.1902/jop.1992.63.9.729. [DOI] [PubMed] [Google Scholar]
- Netter P, Bardin T, Bianchi A, Richette P, Loeuille D. The ANKH gene and familial calcium pyrophosphate dihydrate deposition disease. Joint Bone Spine. 2004;71(5):365–368. doi: 10.1016/j.jbspin.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Netuveli GS, Sheiham A. A systematic review of the effectiveness of anticalculus dentifrices. Oral Health Prev Dent. 2004;2(1):49–58. [PubMed] [Google Scholar]
- Niall HD, Keutmann H, Sauer R, Hogan M, Dawson B, Aurbach G, Potts J., Jr The amino acid sequence of bovine parathyroid hormone I. Hoppe Seylers Z Physiol Chem. 1970;351(12):1586–1588. [PubMed] [Google Scholar]
- Nielsen LB, Pedersen FS, Pedersen L. Expression of type III sodium-dependent phosphate transporters/retroviral receptors mRNAs during osteoblast differentiation. Bone. 2001;28(2):160–166. doi: 10.1016/s8756-3282(00)00418-x. [DOI] [PubMed] [Google Scholar]
- Nociti FH, Jr, Berry JE, Foster BL, Gurley KA, Kingsley DM, Takata T, Miyauchi M, Somerman MJ. Cementum: a phosphate-sensitive tissue. J Dent Res. 2002;81(12):817–821. doi: 10.1177/154405910208101204. [DOI] [PubMed] [Google Scholar]
- Nurnberg P, Thiele H, Chandler D, Hohne W, Cunningham ML, Ritter H, Leschik G, Uhlmann K, Mischung C, Harrop K, Goldblatt J, Borochowitz ZU, Kotzot D, Westermann F, Mundlos S, Braun HS, Laing N, Tinschert S. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat Genet. 2001;28(1):37–41. doi: 10.1038/ng0501-37. [DOI] [PubMed] [Google Scholar]
- Oftebro H, Saarem K, Bjorkhem I, Pedersen JI. Side chain hydroxylation of C27-steroids and vitamin D3 by a cytochrome P-450 enzyme system isolated from human liver mitochondria. J Lipid Res. 1981;22(8):1254–1264. [PubMed] [Google Scholar]
- Ogawa T, Onishi T, Hayashibara T, Sakashita S, Okawa R, Ooshima T. Dentinal defects in Hyp mice not caused by hypophosphatemia alone. Arch Oral Biol. 2006;51(1):58–63. doi: 10.1016/j.archoralbio.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Ogbureke KU, Fisher LW. SIBLING expression patterns in duct epithelia reflect the degree of metabolic activity. J Histochem Cytochem. 2007;55(4):403–409. doi: 10.1369/jhc.6A7075.2007. [DOI] [PubMed] [Google Scholar]
- Ohazama A, Modino SA, Miletich I, Sharpe PT. Stem-cell-based tissue engineering of murine teeth. J Dent Res. 2004;83(7):518–522. doi: 10.1177/154405910408300702. [DOI] [PubMed] [Google Scholar]
- Ohshima H, Nakasone N, Hashimoto E, Sakai H, Nakakura-Ohshima K, Harada H. The eternal tooth germ is formed at the apical end of continuously growing teeth. Arch Oral Biol. 2005;50(2):153–157. doi: 10.1016/j.archoralbio.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Okawa A, Nakamura I, Goto S, Moriya H, Nakamura Y, Ikegawa S. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat Genet. 1998;19(3):271–273. doi: 10.1038/956. [DOI] [PubMed] [Google Scholar]
- Onishi T, Ogawa T, Hayashibara T, Hoshino T, Okawa R, Ooshima T. Hyper-expression of osteocalcin mRNA in odontoblasts of Hyp mice. J Dent Res. 2005;84(1):84–88. doi: 10.1177/154405910508400115. [DOI] [PubMed] [Google Scholar]
- Onishi T, Okawa R, Ogawa T, Shintani S, Ooshima T. Phex mutation causes the reduction of npt2b mRNA in teeth. J Dent Res. 2007;86(2):158–162. doi: 10.1177/154405910708600210. [DOI] [PubMed] [Google Scholar]
- Onishi T, Umemura S, Shintani S, Ooshima T. Phex mutation causes overexpression of FGF23 in teeth. Arch Oral Biol. 2008;53(2):99–104. doi: 10.1016/j.archoralbio.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271(25):15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- Ouyang H, McCauley LK, Berry JE, D’Errico JA, Strayhorn CL, Somerman MJ. Response of immortalized murine cementoblasts/periodontal ligament cells to parathyroid hormone and parathyroid hormone-related protein in vitro. Arch Oral Biol. 2000;45(4):293–303. doi: 10.1016/s0003-9969(99)00142-9. [DOI] [PubMed] [Google Scholar]
- Palmer G, Zhao J, Bonjour J, Hofstetter W, Caverzasio J. In vivo expression of transcripts encoding the Glvr-1 phosphate transporter/retrovirus receptor during bone development. Bone. 1999;24(1):1–7. doi: 10.1016/s8756-3282(98)00151-3. [DOI] [PubMed] [Google Scholar]
- Panda DK, Miao D, Tremblay ML, Sirois J, Farookhi R, Hendy GN, Goltzman D. Targeted ablation of the 25-hydroxyvitamin D 1alpha -hydroxylase enzyme: evidence for skeletal, reproductive, and immune dysfunction. Proc Natl Acad Sci U S A. 2001;98(13):7498–7503. doi: 10.1073/pnas.131029498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaccio G, Graziano A, d’Aquino R, Graziano MF, Pirozzi G, Menditti D, De Rosa A, Carinci F, Laino G. Long-term cryopreservation of dental pulp stem cells (SBP-DPSCs) and their differentiated osteoblasts: a cell source for tissue repair. J Cell Physiol. 2006;208(2):319–325. doi: 10.1002/jcp.20667. [DOI] [PubMed] [Google Scholar]
- Papagerakis P, MacDougall M, Berdal A. Differential epithelial and mesenchymal regulation of tooth-specific matrix proteins expression by 1,25-dihydroxyvitamin D3 in vivo. Connect Tissue Res. 2002;43(2-3):372–375. doi: 10.1080/03008200290000655. [DOI] [PubMed] [Google Scholar]
- Park JS, Suh JJ, Choi SH, Moon IS, Cho KS, Kim CK, Chai JK. Effects of pretreatment clinical parameters on bioactive glass implantation in intrabony periodontal defects. J Periodontol. 2001;72(6):730–740. doi: 10.1902/jop.2001.72.6.730. [DOI] [PubMed] [Google Scholar]
- Parker CC. The role of bisphosphonates in the treatment of prostate cancer. BJU Int. 2005;95(7):935–938. doi: 10.1111/j.1464-410X.2005.05441.x. [DOI] [PubMed] [Google Scholar]
- Pavlakis N, Schmidt R, Stockler M. Bisphosphonates for breast cancer. Cochrane Database Syst Rev. 2005;(3) doi: 10.1002/14651858.CD003474.pub2. CD003474. [DOI] [PubMed] [Google Scholar]
- Pendleton A, Johnson MD, Hughes A, Gurley KA, Ho AM, Doherty M, Dixey J, Gillet P, Loeuille D, McGrath R, Reginato A, Shiang R, Wright G, Netter P, Williams C, Kingsley DM. Mutations in ANKH cause chondrocalcinosis. Am J Hum Genet. 2002;71(4):933–940. doi: 10.1086/343054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng JB, Chen XZ, Berger UV, Vassilev PM, Tsukaguchi H, Brown EM, Hediger MA. Molecular cloning and characterization of a channel-like transporter mediating intestinal calcium absorption. J Biol Chem. 1999;274(32):22739–22746. doi: 10.1074/jbc.274.32.22739. [DOI] [PubMed] [Google Scholar]
- Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology. 2005;146(12):5358–5364. doi: 10.1210/en.2005-0777. [DOI] [PubMed] [Google Scholar]
- Pettway GJ, Meganck JA, Koh AJ, Keller ET, Goldstein SA, McCauley LK. Parathyroid hormone mediates bone growth through the regulation of osteoblast proliferation and differentiation. Bone. 2008;42(4):806–818. doi: 10.1016/j.bone.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pispa J, Thesleff I. Mechanisms of ectodermal organogenesis. Dev Biol. 2003;262(2):195–205. doi: 10.1016/s0012-1606(03)00325-7. [DOI] [PubMed] [Google Scholar]
- Poole KE, Reeve J. Parathyroid hormone - a bone anabolic and catabolic agent. Curr Opin Pharmacol. 2005;5(6):612–617. doi: 10.1016/j.coph.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Popowics T, Foster BL, Swanson EC, Fong H, Somerman MJ. Defining the roots of cementum formation. Cells Tissues Organs. 2005;181(3-4):248–257. doi: 10.1159/000091386. [DOI] [PubMed] [Google Scholar]
- Prasad V, Okunade GW, Miller ML, Shull GE. Phenotypes of SERCA and PMCA knockout mice. Biochem Biophys Res Commun. 2004;322(4):1192–1203. doi: 10.1016/j.bbrc.2004.07.156. [DOI] [PubMed] [Google Scholar]
- Prince MJ, Schaeffer PC, Goldsmith RS, Chausmer AB. Hyperphosphatemic tumoral calcinosis: association with elevation of serum 1,25-dihydroxycholecalciferol concentrations. Ann Intern Med. 1982;96(5):586–591. doi: 10.7326/0003-4819-96-5-586. [DOI] [PubMed] [Google Scholar]
- Prufer K, Racz A, Lin GC, Barsony J. Dimerization with retinoid X receptors promotes nuclear localization and subnuclear targeting of vitamin D receptors. J Biol Chem. 2000;275(52):41114–41123. doi: 10.1074/jbc.M003791200. [DOI] [PubMed] [Google Scholar]
- Qin C, Baba O, Butler WT. Post-translational modifications of sibling proteins and their roles in osteogenesis and dentinogenesis. Crit Rev Oral Biol Med. 2004;15(3):126–136. doi: 10.1177/154411130401500302. [DOI] [PubMed] [Google Scholar]
- Qin C, Brunn JC, Cook RG, Orkiszewski RS, Malone JP, Veis A, Butler WT. Evidence for the proteolytic processing of dentin matrix protein 1. Identification and characterization of processed fragments and cleavage sites. J Biol Chem. 2003;278(36):34700–34708. doi: 10.1074/jbc.M305315200. [DOI] [PubMed] [Google Scholar]
- Quarles LD. FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am J Physiol Endocrinol Metab. 2003;285(1):E1–9. doi: 10.1152/ajpendo.00016.2003. [DOI] [PubMed] [Google Scholar]
- Rasmussen H, Tenenhouse HS. Mendelian Hypophosphatemias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. New York: McGraw Hill; 1995. p. 3717. [Google Scholar]
- Ravera S, Virkki LV, Murer H, Forster IC. Deciphering PiT transport kinetics and substrate specificity using electrophysiology and flux measurements. Am J Physiol Cell Physiol. 2007;293(2):C606–620. doi: 10.1152/ajpcell.00064.2007. [DOI] [PubMed] [Google Scholar]
- Razzaque MS, St-Arnaud R, Taguchi T, Lanske B. FGF-23, vitamin D and calcification: the unholy triad. Nephrol Dial Transplant. 2005;20(10):2032–2035. doi: 10.1093/ndt/gfh991. [DOI] [PubMed] [Google Scholar]
- Reginster JY, Felsenberg D, Cooper C, Stakkestad JA, Miller PD, Kendler DL, Adami S, McClung MR, Bolognese MA, Civitelli R, Dumont E, Bonvoisin B, Recker RR, Delmas PD. A new concept for bisphosphonate therapy: a rationale for the development of monthly oral dosing of ibandronate. Osteoporos Int. 2006;17(2):159–166. doi: 10.1007/s00198-005-1957-6. [DOI] [PubMed] [Google Scholar]
- Reichenberger E, Tiziani V, Watanabe S, Park L, Ueki Y, Santanna C, Baur ST, Shiang R, Grange DK, Beighton P, Gardner J, Hamersma H, Sellars S, Ramesar R, Lidral AC, Sommer A, Raposo do Amaral CM, Gorlin RJ, Mulliken JB, Olsen BR. Autosomal dominant craniometaphyseal dysplasia is caused by mutations in the transmembrane protein ANK. Am J Hum Genet. 2001;68(6):1321–1326. doi: 10.1086/320612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid IR, Brown JP, Burckhardt P, Horowitz Z, Richardson P, Trechsel U, Widmer A, Devogelaer JP, Kaufman JM, Jaeger P, Body JJ, Brandi ML, Broell J, Di Micco R, Genazzani AR, Felsenberg D, Happ J, Hooper MJ, Ittner J, Leb G, Mallmin H, Murray T, Ortolani S, Rubinacci A, Saaf M, Samsioe G, Verbruggen L, Meunier PJ. Intravenous zoledronic acid in postmenopausal women with low bone mineral density. N Engl J Med. 2002;346(9):653–661. doi: 10.1056/NEJMoa011807. [DOI] [PubMed] [Google Scholar]
- Renkema KY, Alexander RT, Bindels RJ, Hoenderop JG. Calcium and phosphate homeostasis: concerted interplay of new regulators. Ann Med. 2008;40(2):82–91. doi: 10.1080/07853890701689645. [DOI] [PubMed] [Google Scholar]
- Reynolds MA, Aichelmann-Reidy ME, Branch-Mays GL, Gunsolley JC. The efficacy of bone replacement grafts in the treatment of periodontal osseous defects. A systematic review. Ann Periodontol. 2003;8(1):227–265. doi: 10.1902/annals.2003.8.1.227. [DOI] [PubMed] [Google Scholar]
- Riccardi D, Park J, Lee WS, Gamba G, Brown EM, Hebert SC. Cloning and functional expression of a rat kidney extracellular calcium/polyvalent cation-sensing receptor. Proc Natl Acad Sci U S A. 1995;92(1):131–135. doi: 10.1073/pnas.92.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, Bianco P, Gehron Robey P. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112(5):683–692. doi: 10.1172/JCI18399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie HH, Park H, Liu J, Bervoets TJ, Bronckers AL. Effects of dexamethasone, vitamin A and vitamin D3 on DSP-PP mRNA expression in rat tooth organ culture. Biochim Biophys Acta. 2004;1679(3):263–271. doi: 10.1016/j.bbaexp.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Roberts S, Narisawa S, Harmey D, Millan JL, Farquharson C. Functional involvement of PHOSPHO1 in matrix vesicle-mediated skeletal mineralization. J Bone Miner Res. 2007;22(4):617–627. doi: 10.1359/jbmr.070108. [DOI] [PubMed] [Google Scholar]
- Roberts SJ, Stewart AJ, Sadler PJ, Farquharson C. Human PHOSPHO1 exhibits high specific phosphoethanolamine and phosphocholine phosphatase activities. Biochem J. 2004;382(Pt 1):59–65. doi: 10.1042/BJ20040511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SJ, Stewart AJ, Schmid R, Blindauer CA, Bond SR, Sadler PJ, Farquharson C. Probing the substrate specificities of human PHOSPHO1 and PHOSPHO2. Biochim Biophys Acta. 2005;1752(1):73–82. doi: 10.1016/j.bbapap.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Rodan GA, Reszka AA. Osteoporosis and bisphosphonates. J Bone Joint Surg Am. 2003;85-A(Suppl 3):8–12. [PubMed] [Google Scholar]
- Rogers MJ. New insights into the molecular mechanisms of action of bisphosphonates. Curr Pharm Des. 2003;9(32):2643–2658. doi: 10.2174/1381612033453640. [DOI] [PubMed] [Google Scholar]
- Rondeau JM, Bitsch F, Bourgier E, Geiser M, Hemmig R, Kroemer M, Lehmann S, Ramage P, Rieffel S, Strauss A, Green JR, Jahnke W. Structural basis for the exceptional in vivo efficacy of bisphosphonate drugs. ChemMedChem. 2006;1(2):267–273. doi: 10.1002/cmdc.200500059. [DOI] [PubMed] [Google Scholar]
- Rosenblatt M, Segre GV, Tregear GW, Shepard GL, Tyler GA, Potts JT., Jr Human parathyroid hormone: synthesis and chemical, biological, and immunological evaluation of the carboxyl-terminal region. Endocrinology. 1978;103(3):978–984. doi: 10.1210/endo-103-3-978. [DOI] [PubMed] [Google Scholar]
- Rowe PS. The wrickkened pathways of FGF23, MEPE and PHEX. Crit Rev Oral Biol Med. 2004;15(5):264–281. doi: 10.1177/154411130401500503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe PS, de Zoysa PA, Dong R, Wang HR, White KE, Econs MJ, Oudet CL. MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia. Genomics. 2000;67(1):54–68. doi: 10.1006/geno.2000.6235. [DOI] [PubMed] [Google Scholar]
- Rowe PS, Kumagai Y, Gutierrez G, Garrett IR, Blacher R, Rosen D, Cundy J, Navvab S, Chen D, Drezner MK, Quarles LD, Mundy GR. MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone. 2004;34(2):303–319. doi: 10.1016/j.bone.2003.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy DM, Linnehan SK. Hydroxyapatite formed from coral skeletal carbonate by hydrothermal exchange. Nature. 1974;247(438):220–222. doi: 10.1038/247220a0. [DOI] [PubMed] [Google Scholar]
- Roy S, Martel J, Ma S, Tenenhouse HS. Increased renal 25-hydroxyvitamin D3-24-hydroxylase messenger ribonucleic acid and immunoreactive protein in phosphate-deprived Hyp mice: a mechanism for accelerated 1,25-dihydroxyvitamin D3 catabolism in X-linked hypophosphatemic rickets. Endocrinology. 1994;134(4):1761–1767. doi: 10.1210/endo.134.4.8137741. [DOI] [PubMed] [Google Scholar]
- Ruchon AF, Tenenhouse HS, Marcinkiewicz M, Siegfried G, Aubin JE, DesGroseillers L, Crine P, Boileau G. Developmental expression and tissue distribution of Phex protein: effect of the Hyp mutation and relationship to bone markers. J Bone Miner Res. 2000;15(8):1440–1450. doi: 10.1359/jbmr.2000.15.8.1440. [DOI] [PubMed] [Google Scholar]
- Ruggiero SL, Mehrotra B, Rosenberg TJ, Engroff SL. Osteonecrosis of the jaws associated with the use of bisphosphonates: a review of 63 cases. J Oral Maxillofac Surg. 2004;62(5):527–534. doi: 10.1016/j.joms.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Russell J, Bar A, Sherwood LM, Hurwitz S. Interaction between calcium and 1,25-dihydroxyvitamin D3 in the regulation of preproparathyroid hormone and vitamin D receptor messenger ribonucleic acid in avian parathyroids. Endocrinology. 1993;132(6):2639–2644. doi: 10.1210/endo.132.6.8389284. [DOI] [PubMed] [Google Scholar]
- Russell RG. Bisphosphonates: mode of action and pharmacology. Pediatrics. 2007;119(Suppl 2):S150–162. doi: 10.1542/peds.2006-2023H. [DOI] [PubMed] [Google Scholar]
- Rutherford RB, Foster BL, Bammler T, Beyer RP, Sato S, Somerman MJ. Extracellular phosphate alters cementoblast gene expression. J Dent Res. 2006;85(6):505–509. doi: 10.1177/154405910608500605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutsch F, Schauerte P, Kalhoff H, Petrarulo M, August C, Diekmann L. Low levels of urinary inorganic pyrophosphate indicating systemic pyrophosphate deficiency in a boy with idiopathic infantile arterial calcification. Acta Paediatr. 2000;89(10):1265–1269. doi: 10.1080/080352500750027682. [DOI] [PubMed] [Google Scholar]
- Rutsch F, Terkeltaub R. Deficiencies of physiologic calcification inhibitors and low-grade inflammation in arterial calcification: lessons for cartilage calcification. Joint Bone Spine. 2005;72(2):110–118. doi: 10.1016/j.jbspin.2004.05.014. [DOI] [PubMed] [Google Scholar]
- Rutsch F, Vaingankar S, Johnson K, Goldfine I, Maddux B, Schauerte P, Kalhoff H, Sano K, Boisvert WA, Superti-Furga A, Terkeltaub R. PC-1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am J Pathol. 2001;158(2):543–554. doi: 10.1016/S0002-9440(10)63996-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan LM. The ank gene story. Arthritis Res. 2001;3(2):77–79. doi: 10.1186/ar143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini D, Vespasiani Gentilucci U, Vincenzi B, Picardi A, Vasaturo F, La Cesa A, Onori N, Scarpa S, Tonini G. The antineoplastic role of bisphosphonates: from basic research to clinical evidence. Ann Oncol. 2003;14(10):1468–1476. doi: 10.1093/annonc/mdg401. [DOI] [PubMed] [Google Scholar]
- Saygin NE, Giannobile WV, Somerman MJ. Molecular and cell biology of cementum. Periodontology 2000. 2000;24:73–98. doi: 10.1034/j.1600-0757.2000.2240105.x. [DOI] [PubMed] [Google Scholar]
- Schiavi SC. Fibroblast growth factor 23: the making of a hormone. Kidney Int. 2006;69(3):425–427. doi: 10.1038/sj.ki.5000168. [DOI] [PubMed] [Google Scholar]
- Schiavi SC, Kumar R. The phosphatonin pathway: new insights in phosphate homeostasis. Kidney Int. 2004;65(1):1–14. doi: 10.1111/j.1523-1755.2004.00355.x. [DOI] [PubMed] [Google Scholar]
- Segawa H, Kawakami E, Kaneko I, Kuwahata M, Ito M, Kusano K, Saito H, Fukushima N, Miyamoto K. Effect of hydrolysis-resistant FGF23-R179Q on dietary phosphate regulation of the renal type-II Na/Pi transporter. Pflugers Arch. 2003;446(5):585–592. doi: 10.1007/s00424-003-1084-1. [DOI] [PubMed] [Google Scholar]
- Sharma CG, Pradeep AR. Localized attachment loss in Pendred syndrome: incidental? J Periodontol. 2007;78(5):948–954. doi: 10.1902/jop.2007.060270. [DOI] [PubMed] [Google Scholar]
- Shen Q, Christakos S. The vitamin D receptor, Runx2, and the Notch signaling pathway cooperate in the transcriptional regulation of osteopontin. J Biol Chem. 2005;280(49):40589–40598. doi: 10.1074/jbc.M504166200. [DOI] [PubMed] [Google Scholar]
- Shields ED, Scriver CR, Reade T, Fujiwara TM, Morgan K, Ciampi A, Schwartz S. X-linked hypophosphatemia: the mutant gene is expressed in teeth as well as in kidney. Am J Hum Genet. 1990;46(3):434–442. [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113(4):561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98(11):6500–6505. doi: 10.1073/pnas.101545198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Muto T, Urakawa I, Yoneya T, Yamazaki Y, Okawa K, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology. 2002;143(8):3179–3182. doi: 10.1210/endo.143.8.8795. [DOI] [PubMed] [Google Scholar]
- Siris ES, Lyles KW, Singer FR, Meunier PJ. Medical management of Paget’s disease of bone: indications for treatment and review of current therapies. J Bone Miner Res. 2006;21(Suppl 2):P94–98. doi: 10.1359/jbmr.06s218. [DOI] [PubMed] [Google Scholar]
- Sirohi B, Powles R. Multiple myeloma. Lancet. 2004;363(9412):875–887. doi: 10.1016/S0140-6736(04)15736-X. [DOI] [PubMed] [Google Scholar]
- Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, Juppner H, Lanske B. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004;23(7):421–432. doi: 10.1016/j.matbio.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitara D, Razzaque MS, St-Arnaud R, Huang W, Taguchi T, Erben RG, Lanske B. Genetic ablation of vitamin D activation pathway reverses biochemical and skeletal anomalies in Fgf-23-null animals. Am J Pathol. 2006;169(6):2161–2170. doi: 10.2353/ajpath.2006.060329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soballe K, Hansen ES, Brockstedt-Rasmussen H, Hjortdal VE, Juhl GI, Pedersen CM, Hvid I, Bunger C. Gap healing enhanced by hydroxyapatite coating in dogs. Clin Orthop Relat Res. 1991;272:300–307. [PubMed] [Google Scholar]
- Soballe K, Mouzin OR, Kidder LA, Overgaard S, Bechtold JE. The effects of hydroxyapatite coating and bone allograft on fixation of loaded experimental primary and revision implants. Acta Orthop Scand. 2003;74(3):239–247. doi: 10.1080/00016470310014139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofaer JA, Southam JC. Naturally-occurring exposure of the dental pulp in mice with inherited hypophosphataemia. Arch Oral Biol. 1982;27(8):701–703. doi: 10.1016/0003-9969(82)90196-0. [DOI] [PubMed] [Google Scholar]
- Stains JP, Weber JA, Gay CV. Expression of Na(+)/Ca(2+) exchanger isoforms (NCX1 and NCX3) and plasma membrane Ca(2+) ATPase during osteoblast differentiation. J Cell Biochem. 2002;84(3):625–635. [PubMed] [Google Scholar]
- Stewart AJ, Roberts SJ, Seawright E, Davey MG, Fleming RH, Farquharson C. The presence of PHOSPHO1 in matrix vesicles and its developmental expression prior to skeletal mineralization. Bone. 2006 doi: 10.1016/j.bone.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Stewart AJ, Schmid R, Blindauer CA, Paisey SJ, Farquharson C. Comparative modelling of human PHOSPHO1 reveals a new group of phosphatases within the haloacid dehalogenase superfamily. Protein Eng. 2003;16(12):889–895. doi: 10.1093/protein/gzg126. [DOI] [PubMed] [Google Scholar]
- Storm T, Thamsborg G, Steiniche T, Genant HK, Sorensen OH. Effect of intermittent cyclical etidronate therapy on bone mass and fracture rate in women with postmenopausal osteoporosis. N Engl J Med. 1990;322(18):1265–1271. doi: 10.1056/NEJM199005033221803. [DOI] [PubMed] [Google Scholar]
- Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20(3):345–357. doi: 10.1210/edrv.20.3.0367. [DOI] [PubMed] [Google Scholar]
- Sutton AL, MacDonald PN. Vitamin D: more than a “bone-a-fide” hormone. Mol Endocrinol. 2003;17(5):777–791. doi: 10.1210/me.2002-0363. [DOI] [PubMed] [Google Scholar]
- Suva LJ, Winslow GA, Wettenhall RE, Hammonds RG, Moseley JM, Diefenbach-Jagger H, Rodda CP, Kemp BE, Rodriguez H, Chen EY. A parathyroid hormone-related protein implicated in malignant hypercalcemia: cloning and expression. Science. 1987;237(4817):893–896. doi: 10.1126/science.3616618. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Ghayor C, Guicheux J, Magne D, Quillard S, Kakita A, Ono Y, Miura Y, Oiso Y, Itoh M, Caverzasio J. Enhanced expression of the inorganic phosphate transporter Pit-1 is involved in BMP-2-induced matrix mineralization in osteoblast-like cells. J Bone Miner Res. 2006;21(5):674–683. doi: 10.1359/jbmr.020603. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Palmer G, Bonjour JP, Caverzasio J. Stimulation of sodium-dependent phosphate transport and signaling mechanisms induced by basic fibroblast growth factor in MC3T3-E1 osteoblast-like cells. J Bone Miner Res. 2000;15(1):95–102. doi: 10.1359/jbmr.2000.15.1.95. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Palmer G, Bonjour JP, Caverzasio J. Stimulation of sodium-dependent inorganic phosphate transport by activation of Gi/o-protein-coupled receptors by epinephrine in MC3T3-E1 osteoblast-like cells. Bone. 2001;28(6):589–594. doi: 10.1016/s8756-3282(01)00459-8. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Amizuka N, Oda K, Noda M, Ohshima H, Maeda T. Involvement of the klotho protein in dentin formation and mineralization. Anat Rec (Hoboken) 2008;291(2):183–190. doi: 10.1002/ar.20630. [DOI] [PubMed] [Google Scholar]
- Sweet HO, Green MC. Progressive ankylosis, a new skeletal mutation in the mouse. J Hered. 1981;72(2):87–93. doi: 10.1093/oxfordjournals.jhered.a109459. [DOI] [PubMed] [Google Scholar]
- Takeyama S, Yoshimura Y, Deyama Y, Sugawara Y, Fukuda H, Matsumoto A. Phosphate decreases osteoclastogenesis in coculture of osteoblast and bone marrow. Biochem Biophys Res Commun. 2001;282(3):798–802. doi: 10.1006/bbrc.2001.4652. [DOI] [PubMed] [Google Scholar]
- Teixeira CC, Mansfield K, Hertkorn C, Ischiropoulos H, Shapiro IM. Phosphate-induced chondrocyte apoptosis is linked to nitric oxide generation. Am J Physiol Cell Physiol. 2001;281(3):C833–839. doi: 10.1152/ajpcell.2001.281.3.C833. [DOI] [PubMed] [Google Scholar]
- Tenorio D, Hughes FJ. An immunohistochemical investigation of the expression of parathyroid hormone receptors in rat cementoblasts. Arch Oral Biol. 1996;41(3):299–305. doi: 10.1016/0003-9969(95)00113-1. [DOI] [PubMed] [Google Scholar]
- Terkeltaub RA. Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol Cell Physiol. 2001;281(1):C1–C11. doi: 10.1152/ajpcell.2001.281.1.C1. [DOI] [PubMed] [Google Scholar]
- Termine JD, Peckauskas RA, Posner AS. Calcium phosphate formation in vitro. II. Effects of environment on amorphous-crystalline transformation. Arch Biochem Biophys. 1970;140(2):318–325. doi: 10.1016/0003-9861(70)90072-x. [DOI] [PubMed] [Google Scholar]
- Thesleff I. Epithelial-mesenchymal signalling regulating tooth morphogenesis. J Cell Sci. 2003;116(Pt 9):1647–1648. doi: 10.1242/jcs.00410. [DOI] [PubMed] [Google Scholar]
- Thesleff I, Sharpe P. Signalling networks regulating dental development. Mech Dev. 1997;67(2):111–123. doi: 10.1016/s0925-4773(97)00115-9. [DOI] [PubMed] [Google Scholar]
- Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, Khamaysi Z, Behar D, Petronius D, Friedman V, Zelikovic I, Raimer S, Metzker A, Richard G, Sprecher E. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet. 2004;36(6):579–581. doi: 10.1038/ng1358. [DOI] [PubMed] [Google Scholar]
- Toyosawa S, Okabayashi K, Komori T, Ijuhin N. mRNA expression and protein localization of dentin matrix protein 1 during dental root formation. Bone. 2004;34(1):124–133. doi: 10.1016/j.bone.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Toyosawa S, Shintani S, Fujiwara T, Ooshima T, Sato A, Ijuhin N, Komori T. Dentin matrix protein 1 is predominantly expressed in chicken and rat osteocytes but not in osteoblasts. J Bone Miner Res. 2001;16(11):2017–2026. doi: 10.1359/jbmr.2001.16.11.2017. [DOI] [PubMed] [Google Scholar]
- Tregear GW, Van Rietschoten J, Greene E, Keutmann HT, Niall HD, Reit B, Parsons JA, Potts JT., Jr Bovine parathyroid hormone: minimum chain length of synthetic peptide required for biological activity. Endocrinology. 1973;93(6):1349–1353. doi: 10.1210/endo-93-6-1349. [DOI] [PubMed] [Google Scholar]
- Tsukamoto Y, Fukutani S, Shin-Ike T, Kubota T, Sato S, Suzuki Y, Mori M. Mineralized nodule formation by cultures of human dental pulp-derived fibroblasts. Arch Oral Biol. 1992;37(12):1045–1055. doi: 10.1016/0003-9969(92)90037-9. [DOI] [PubMed] [Google Scholar]
- Tummers M, Thesleff I. Observations on continuously growing roots of the sloth and the K14-Eda transgenic mice indicate that epithelial stem cells can give rise to both the ameloblast and root epithelium cell lineage creating distinct tooth patterns. Evolution & development. 2008;10(2):187–195. doi: 10.1111/j.1525-142X.2008.00226.x. [DOI] [PubMed] [Google Scholar]
- Udagawa N, Takahashi N, Akatsu T, Tanaka H, Sasaki T, Nishihara T, Koga T, Martin TJ, Suda T. Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc Natl Acad Sci U S A. 1990;87(18):7260–7264. doi: 10.1073/pnas.87.18.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444(7120):770–774. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- Vaes BL, Dechering KJ, van Someren EP, Hendriks JM, van de Ven CJ, Feijen A, Mummery CL, Reinders MJ, Olijve W, van Zoelen EJ, Steegenga WT. Microarray analysis reveals expression regulation of Wnt antagonists in differentiating osteoblasts. Bone. 2005;36(5):803–811. doi: 10.1016/j.bone.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Vaingankar SM, Fitzpatrick TA, Johnson K, Goding JW, Maurice M, Terkeltaub R. Subcellular targeting and function of osteoblast nucleotide pyrophosphatase phosphodiesterase 1. Am J Physiol Cell Physiol. 2004;286(5):C1177–1187. doi: 10.1152/ajpcell.00320.2003. [DOI] [PubMed] [Google Scholar]
- van den Bos T, Handoko G, Niehof A, Ryan LM, Coburn SP, Whyte MP, Beertsen W. Cementum and dentin in hypophosphatasia. J Dent Res. 2005;84(11):1021–1025. doi: 10.1177/154405910508401110. [DOI] [PubMed] [Google Scholar]
- Wang HL, Cooke J. Periodontal regeneration techniques for treatment of periodontal diseases. Dent Clin North Am. 2005;49(3):637–659. vii. doi: 10.1016/j.cden.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Warren SM, Nacamuli RK, Song HM, Longaker MT. Tissue-engineered bone using mesenchymal stem cells and a biodegradable scaffold. J Craniofac Surg. 2004;15(1):34–37. doi: 10.1097/00001665-200401000-00012. [DOI] [PubMed] [Google Scholar]
- Watts NB, Harris ST, Genant HK, Wasnich RD, Miller PD, Jackson RD, Licata AA, Ross P, Woodson GC, 3rd, Yanover MJ, et al. Intermittent cyclical etidronate treatment of postmenopausal osteoporosis. N Engl J Med. 1990;323(2):73–79. doi: 10.1056/NEJM199007123230201. [DOI] [PubMed] [Google Scholar]
- Weninger M, Stinson RA, Plenk H, Jr, Bock P, Pollak A. Biochemical and morphological effects of human hepatic alkaline phosphatase in a neonate with hypophosphatasia. Acta Paediatr Scand Suppl. 1989;360:154–160. doi: 10.1111/j.1651-2227.1989.tb11297.x. [DOI] [PubMed] [Google Scholar]
- Werner S, Duan DS, de Vries C, Peters KG, Johnson DE, Williams LT. Differential splicing in the extracellular region of fibroblast growth factor receptor 1 generates receptor variants with different ligand-binding specificities. Mol Cell Biol. 1992;12(1):82–88. doi: 10.1128/mcb.12.1.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KE, Jonsson KB, Carn G, Hampson G, Spector TD, Mannstadt M, Lorenz-Depiereux B, Miyauchi A, Yang IM, Ljunggren O, Meitinger T, Strom TM, Juppner H, Econs MJ. The autosomal dominant hypophosphatemic rickets (ADHR) gene is a secreted polypeptide overexpressed by tumors that cause phosphate wasting. J Clin Endocrinol Metab. 2001;86(2):497–500. doi: 10.1210/jcem.86.2.7408. [DOI] [PubMed] [Google Scholar]
- White KE, Larsson TM, Econs MJ. The Roles of Specific Genes Implicated as Circulating Factors Involved in Normal and Disordered Phosphate Homeostasis: Frp-4, MEPE, and FGF23. Endocr Rev. 2006 doi: 10.1210/er.2005-0019. [DOI] [PubMed] [Google Scholar]
- WHO. World Oral Health Report 2003. Geneza, Switzerland: Noncommunicable Disease Prevention and Health Promotion, Oral Health Programme; 2003. p. 45. [Google Scholar]
- Whyte MP. Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr Rev. 1994;15(4):439–461. doi: 10.1210/edrv-15-4-439. [DOI] [PubMed] [Google Scholar]
- Whyte MP. Hypophosphatasia Nature’s window to alkaline phosphatase in man. In: Bilezikian, Raisz, Rodan, editors. Principles of Bone Biology. 2. San Diego, CA: Academic Press; 2002. pp. 1229–1248. [Google Scholar]
- Whyte MP, Landt M, Ryan LM, Mulivor RA, Henthorn PS, Fedde KN, Mahuren JD, Coburn SP. Alkaline phosphatase: placental and tissue-nonspecific isoenzymes hydrolyze phosphoethanolamine, inorganic pyrophosphate, and pyridoxal 5’-phosphate. Substrate accumulation in carriers of hypophosphatasia corrects during pregnancy. J Clin Invest. 1995;95(4):1440–1445. doi: 10.1172/JCI117814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte MP, McAlister WH, Patton LS, Magill HL, Fallon MD, Lorentz WB, Jr, Herrod HG. Enzyme replacement therapy for infantile hypophosphatasia attempted by intravenous infusions of alkaline phosphatase-rich Paget plasma: results in three additional patients. J Pediatr. 1984;105(6):926–933. doi: 10.1016/s0022-3476(84)80079-7. [DOI] [PubMed] [Google Scholar]
- Whyte MP, Valdes R, Jr, Ryan LM, McAlister WH. Infantile hypophosphatasia: enzyme replacement therapy by intravenous infusion of alkaline phosphatase-rich plasma from patients with Paget bone disease. J Pediatr. 1982;101(3):379–386. doi: 10.1016/s0022-3476(82)80061-9. [DOI] [PubMed] [Google Scholar]
- Woo SB, Hande K, Richardson PG. Osteonecrosis of the jaw and bisphosphonates. N Engl J Med. 2005;353(1):99–102. discussion 199-102. [PubMed] [Google Scholar]
- Woo SB, Hellstein JW, Kalmar JR. Narrative [corrected] review: bisphosphonates and osteonecrosis of the jaws. Ann Intern Med. 2006;144(10):753–761. doi: 10.7326/0003-4819-144-10-200605160-00009. [DOI] [PubMed] [Google Scholar]
- Wood J, Bonjean K, Ruetz S, Bellahcene A, Devy L, Foidart JM, Castronovo V, Green JR. Novel antiangiogenic effects of the bisphosphonate compound zoledronic acid. J Pharmacol Exp Ther. 2002;302(3):1055–1061. doi: 10.1124/jpet.102.035295. [DOI] [PubMed] [Google Scholar]
- Wysolmerski JJ, Cormier S, Philbrick WM, Dann P, Zhang JP, Roume J, Delezoide AL, Silve C. Absence of functional type 1 parathyroid hormone (PTH)/PTH-related protein receptors in humans is associated with abnormal breast development and tooth impaction. J Clin Endocrinol Metab. 2001;86(4):1788–1794. doi: 10.1210/jcem.86.4.7404. [DOI] [PubMed] [Google Scholar]
- Xiao NM, Zhang YM, Zheng Q, Gu J. Klotho is a serum factor related to human aging. Chin Med J (Engl) 2004;117(5):742–747. [PubMed] [Google Scholar]
- Yamamoto T. Clinical approach to clarifying the mechanism of abnormal bone metabolism in McCune-Albright syndrome. J Bone Miner Metab. 2006;24(1):7–10. doi: 10.1007/s00774-005-0638-z. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Imanishi Y, Kinoshita E, Nakagomi Y, Shimizu N, Miyauchi A, Satomura K, Koshiyama H, Inaba M, Nishizawa Y, Juppner H, Ozono K. The role of fibroblast growth factor 23 for hypophosphatemia and abnormal regulation of vitamin D metabolism in patients with McCune-Albright syndrome. J Bone Miner Metab. 2005;23(3):231–237. doi: 10.1007/s00774-004-0589-9. [DOI] [PubMed] [Google Scholar]
- Yamaoka M, Uematsu T, Shiba T, Matsuura T, Ono Y, Ishizuka M, Naramoto H, Takahashi M, Sugiura-Tomita M, Iguchi K, Yamashita S, Furusawa K. Effect of inorganic polyphosphate in periodontitis in the elderly. Gerodontology. 2008;25(1):10–17. doi: 10.1111/j.1741-2358.2007.00185.x. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Konishi M, Miyake A, Inui K, Itoh N. Fibroblast growth factor (FGF)-23 inhibits renal phosphate reabsorption by activation of the mitogen-activated protein kinase pathway. J Biol Chem. 2002;277(31):28265–28270. doi: 10.1074/jbc.M202527200. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277(2):494–498. doi: 10.1006/bbrc.2000.3696. [DOI] [PubMed] [Google Scholar]
- Yang W, Kalajzic I, Lu Y, Guo D, Harris MA, Gluhak-Heinrich J, Bonewald LF, Feng JQ, Rowe DW, Harris SE. In vitro and in vivo study on osteocyte-specific mechanical signaling pathways. J Musculoskelet Neuronal Interact. 2004;4(4):386–387. [PubMed] [Google Scholar]
- Yasutake J, Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Iijima K, Fujita T, Fukumoto S, Yamashita T. Effects of FGF23 and soluble Klotho on mineralization of MC3T3-E1 osteoblastic cells; 28th ASBMR Annual Meeting; Philadelphia, PA, USA. 2006. [Google Scholar]
- Yates AJ, Oreffo RO, Mayor K, Mundy GR. Inhibition of bone resorption by inorganic phosphate is mediated by both reduced osteoclast formation and decreased activity of mature osteoclasts. J Bone Miner Res. 1991;6(5):473–478. doi: 10.1002/jbmr.5650060508. [DOI] [PubMed] [Google Scholar]
- Ye L, MacDougall M, Zhang S, Xie Y, Zhang J, Li Z, Lu Y, Mishina Y, Feng JQ. Deletion of dentin matrix protein-1 leads to a partial failure of maturation of predentin into dentin, hypomineralization, and expanded cavities of pulp and root canal during postnatal tooth development. J Biol Chem. 2004;279(18):19141–19148. doi: 10.1074/jbc.M400490200. [DOI] [PubMed] [Google Scholar]
- Ye L, Mishina Y, Chen D, Huang H, Dallas SL, Dallas MR, Sivakumar P, Kunieda T, Tsutsui TW, Boskey A, Bonewald LF, Feng JQ. Dmp1-deficient mice display severe defects in cartilage formation responsible for a chondrodysplasia-like phenotype. J Biol Chem. 2005;280(7):6197–6203. doi: 10.1074/jbc.M412911200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Zhang S, Ke H, Bonewald LF, Feng J. Periodontal breakdown in the dmp1 null mouse model of hypophosphatemic rickets. J Dent Res. 2008;87(7):624–629. doi: 10.1177/154405910808700708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KG, Rutledge SJ, Buenaga RF, Rodan GA. Characterization of the rat osteocalcin gene: stimulation of promoter activity by 1,25-dihydroxyvitamin D3. Biochemistry. 1988;27(23):8521–8526. doi: 10.1021/bi00423a003. [DOI] [PubMed] [Google Scholar]
- Yoshiko Y, Wang H, Minamizaki T, Ijuin C, Yamamoto R, Suemune S, Kozai K, Tanne K, Aubin JE, Maeda N. Mineralized tissue cells are a principal source of FGF23. Bone. 2007;40(6):1565–1573. doi: 10.1016/j.bone.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Young CS, Terada S, Vacanti JP, Honda M, Bartlett JD, Yelick PC. Tissue engineering of complex tooth structures on biodegradable polymer scaffolds. J Dent Res. 2002;81(10):695–700. doi: 10.1177/154405910208101008. [DOI] [PubMed] [Google Scholar]
- Yu X, Davis SI, Larsson T, White KE. Regulation of Klotho expression in distal convoluted tubule cells; 28th ASBMR Annual Meeting; Philadelphia, PA, USA. 2006. [Google Scholar]
- Yu X, Ibrahimi OA, Goetz R, Zhang F, Davis SI, Garringer HJ, Linhardt RJ, Ornitz DM, Mohammadi M, White KE. Analysis of the biochemical mechanisms for the endocrine actions of fibroblast growth factor-23. Endocrinology. 2005;146(11):4647–4656. doi: 10.1210/en.2005-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, White KE. FGF23 and disorders of phosphate homeostasis. Cytokine Growth Factor Rev. 2005a;16(2):221–232. doi: 10.1016/j.cytogfr.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Yu X, White KE. Fibroblast growth factor 23 and its receptors. Ther Apher Dial. 2005b;9(4):308–312. doi: 10.1111/j.1744-9987.2005.00287.x. [DOI] [PubMed] [Google Scholar]
- Zacherl WA, Pfeiffer HJ, Swancar JR. The effect of soluble pyrophosphates on dental calculus in adults. J Am Dent Assoc. 1985;110(5):737–738. doi: 10.14219/jada.archive.1985.0441. [DOI] [PubMed] [Google Scholar]
- Zafiropoulos GG, Hoffmann O, Kasaj A, Willershausen B, Weiss O, Van Dyke TE. Treatment of intrabony defects using guided tissue regeneration and autogenous spongiosa alone or combined with hydroxyapatite/beta-tricalcium phosphate bone substitute or bovine-derived xenograft. J Periodontol. 2007;78(11):2216–2225. doi: 10.1902/jop.2007.070146. [DOI] [PubMed] [Google Scholar]
- Zhang H, Somerman MJ, Berg J, Cunningham ML, Williams B. Dental anomalies in a child with craniometaphysial dysplasia. Pediatr Dent. 2007a;29(5):415–419. [PubMed] [Google Scholar]
- Zhang X, Rahemtulla FG, MacDougall MJ, Thomas HF. Vitamin D receptor deficiency affects dentin maturation in mice. Arch Oral Biol. 2007b;52(12):1172–1179. doi: 10.1016/j.archoralbio.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Zhao D, Vaziri Sani F, Nilsson J, Rodenburg M, Stocking C, Linde A, Gritli-Linde A. Expression of Pit2 sodium-phosphate cotransporter during murine odontogenesis is developmentally regulated. Eur J Oral Sci. 2006;114(6):517–523. doi: 10.1111/j.1600-0722.2006.00414.x. [DOI] [PubMed] [Google Scholar]
- Zoidis E, Ghirlanda-Keller C, Gosteli-Peter M, Zapf J, Schmid C. Regulation of phosphate (Pi) transport and NaPi-III transporter (Pit-1) mRNA in rat osteoblasts. J Endocrinol. 2004;181(3):531–540. doi: 10.1677/joe.0.1810531. [DOI] [PubMed] [Google Scholar]