

Abstract
CBFβ-SMMHC, the fusion protein generated by the chromosome 16 inversion fusion gene, CBFB-MYH11, is known to initiate leukemogenesis. However, the mechanism through which CBFβ-SMMHC contributes to leukemia development is not well understood. Previously it was proposed that CBFβ-SMMHC acts by dominantly repressing the transcription factor RUNX1, but we recently showed that CBFβ-SMMHC has activities that are independent of RUNX1 repression. In addition, we showed that a modified CBFβ-SMMHC with decreased RUNX1 binding activity accelerates leukemogenesis. These results raise questions about the importance of RUNX1 in leukemogenesis by CBFβ-SMMHC. To test this, we generated mice expressing Cbfb-MYH11 in a Runx1 deficient background, resulting from either homozygous Runx1 null alleles (Runx1−/−) or a single dominant negative Runx1 allele (Runx1+/lz). We found that loss of Runx1 activity rescued the differentiation defects induced by Cbfb-MYH11 during primitive hematopoiesis. During definitive hematopoiesis, RUNX1 loss also significantly reduced the proliferation and differentiation defects induced by Cbfb-MYH11. Importantly, Cbfb-MYH11 induced leukemia had much longer latency in Runx1+/lz mice than in Runx1 sufficient mice. These data indicate that Runx1 activity is critical for Cbfb-MYH11 induced hematopoietic defects and leukemogenesis.
Keywords: AML, Runx1, Cbfb-MYH11, CBFβ-SMMHC, Inversion 16
Introduction
Acute myeloid leukemia (AML) is often characterized by the presence of specific, recurrent chromosomal abnormalities, many of which involve transcription factors important for normal hematopoiesis1. Inversion of chromosome 16, inv(16)(p13;q22) or the related t(16;16)(p13;q22) translocation, is found in nearly all patients with AML subtype M4 with eosinophilia2 and generates a fusion gene between the core-binding factor beta gene, CBFB, and MYH11, the gene for smooth muscle myosin heavy chain (SMMHC)3, 4. The fusion gene, CBFB-MYH11, which encodes the CBFβ-SMMHC protein, has been shown to be necessary, but not sufficient for leukemogenesis5, 6.
CBFβ-SMMHC is thought to initiate leukemogenesis by blocking normal hematopoietic differentiation through inhibition of the key hematopoietic transcription factor Runt-related protein 1 (RUNX1; AML1). RUNX1 is one of the three α subunits in the core binding factor (CBF) family, all of which have RUNT domains that mediate DNA binding and heterodimerization with the single β subunit in the CBF family, CBFβ7. Dimerization with CBFβ stabilizes binding of the α subunits to DNA and allows the α–β heterodimer to regulate gene expression through the transactivation domains of the α subunits8. CBFβ-SMMHC retains the RUNX binding domain from CBFβ, and also contains a second RUNX high affinity binding domain (HABD) in the SMMHC tail, resulting in a higher binding affinity for RUNX as compared to wildtype CBFβ9. In vitro work has shown that CBFβ-SMMHC may serve as a transcriptional repressor and that CBFβ-SMMHC may sequester RUNX1 in the cytoplasm10–12. Based on these findings, it has been proposed that CBFβ-SMMHC acts by dominantly repressing RUNX1.
Mouse models have provided evidence that CBFβ-SMMHC has dominant RUNX repressor activities in vivo. Previously, we generated Cbfb-MYH11 knockin mice (Cbfb+/MYH11) which express the fusion gene under the control of the endogenous Cbfb promoter. At embryonic day 12.5 (E12.5), heterozygous Cbfb+/MYH11 embryos have a complete block in definitive hematopoiesis and severe central nervous system hemorrhaging, which contributes to lethality by E13.513. This phenotype is very similar to that of Runx1 null (Runx1−/−) and Cbfb null (Cbfb−/−)14–18 mice, consistent with CBFβ-SMMHC acting as a dominant repressor of RUNX1 and CBFβ functions during embryogenesis.
More recent work has shown that CBFβ-SMMHC also has RUNX1-repression independent activities. We found that Cbfb+/MYH11 embryos have defects in differentiation and gene expression during primitive hematopoiesis that are not seen in Runx1−/− or Cbfb−/− embryos19. In addition, we found that many genes are uniquely deregulated in Cbfb+/MYH11 embryos (but not in Runx1−/− or Cbfb−/− embryos), and are expressed in leukemic cells from mice and humans. Together, these results imply that Cbfb-MYH11’s Runx1-repression independent activities play a role in leukemogenesis.
Our previous research using deletion mutants of the Cbfb-MYH11 fusion gene has raised further questions about Runx1’s role in Cbfb-MYH11 induced leukemogenesis. We generated knock-in mice expressing a CBFβ-SMMHC mutant lacking the HABD in the SMMHC tail. Consequently, the mutant CBFβ-SMMHC has lower RUNX1 binding affinity20. Unexpectedly, these mice showed accelerated leukemogenesis. We also generated knock-in mice with a deletion mutant of CBFβ-SMMHC lacking the C-terminal 95 amino acids (Cbfb+/ΔC95), a region not predicted to interact with RUNX1, but that contains multimerization and repression domains. These mice showed a rescue of the Runx1-repression independent defects in primitive hematopoiesis, and never developed leukemia implying that CBFβ-SMMHC has activities independent of RUNX1 binding that are critical for mediating differentiation defects and leukemogenesis21.
Together, these findings raise the possibility that Runx1 may be dispensable for Cbfb-MYH11 activity. To test this hypothesis, we generated mice expressing Cbfb-MYH11 but lacking Runx1 activity. We found that Runx1 activity is required for Cbfb-MYH11 induced differentiation defects during both primitive and definitive hematopoiesis. In addition, we found that insufficient Runx1 activity results in delayed leukemogenesis. These results indicate that Cbfb-MYH11 requires Runx1 activity for leukemogenesis and validates current efforts to develop inhibitors of the CBFβ-SMMHC:RUNX1 interaction for the treatment of inv(16) leukemia22.
Material and Methods
Animals
All animals used and the procedures performed in this study were approved by the National Human Genome Research Institute (NHGRI) Animal Care and Use Committee. Cbfb-MYH11 conditional knockin (Cbfb+/56M)23, Runx1 knockout (Runx1−/−)15, Runx1-lacZ knockin (Runx1+/lz)24, β-actin-Cre recombinase transgenic (Actb-Cre+)25 and Mx1-Cre recombinase transgenic (Mx1-Cre+) 26 mice have been described previously. All mice were maintained on a mixed C57BL/6;129/SvEv background and were genotyped by polymerase chain reaction (PCR) with gene-specific primers (sequences available upon request) using tail-snip DNA prepared using Gentra Puregene Mouse Tail Kit (Qiagen, Venlo, the Netherlands). Polyinosine-polycytidylic acid (pI:pC) (InvivoGen, San Diego, CA, USA) was used to induce Cbfb-MYH11 expression as described previously19. To accelerate leukemia development, mice were treated with N-ethyl-N-nitrosourea (ENU) as described previously5. All mice were observed for leukemia development for 12 months.
Peripheral Blood Counts
Peripheral blood was collected from adult mice and the complete blood counts were analyzed using an Abbott Cell-Dyn 3700 Hematology Analyzer (Abbott Park, IL, USA).
Flow Cytometry
Peripheral blood cells from embryos and lineage negative bone marrow cells were isolated and stained as described previously19. Data were acquired using a LSRII Flow Cytometer (BD Biosciences, SanJose, CA, USA) and analyzed using FlowJo software (Flowjo, LLC, Ashland, OR, USA).
Proliferation and Cell Number Assays
Cell proliferation was determined by BrdU incorporation using the BrdU Flow kit (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) according to the manufacturer’s instructions. Cell number was determined using a hemocytometer to count the number of lineage depleted cells extracted from both femurs of a mouse of the indicated genotype and resuspended in phosphate buffered saline with 5% fetal bovine serum.
Construction of Runx1-lz Expression Plasmid
Partial Runx1-lz cDNA was generated by RT-PCR with RNA from thymus tissue of Runx1+/lz mice and forward and reverse primers (sequences available upon request) in the Runx1 and lacZ domains, respectively, of the fusion gene. The RT-PCR fragment was cloned in frame into pSV-βgal (Promega, Madison, WI, USA) and the RT-PCR fragment with the remainder of the lacZ coding sequence was cloned into pCDNA-Runx1 to generate pCDNA-Runx1-lz. The coding frame of the final plasmid was verified by sequencing.
Immunoprecipitation and Western blot analysis
Cell lysates were prepared from primary mouse tissues or from 293HEK cells transfected with the indicated plasmids using Lipofectamine 2000 (LifeTechnologies, Grand Island, NY, USA). Immunoprecipitation and western blot analysis was performed as described previously19, 22 using antibodies against CBFβ and CBFβ-SMMHC (Aviva Biosystems, San Diego, CA, USA and anti-CBFβ14116), β-galactosidase (ab616, Abcam, Cambridge, MA, USA), RUNX1 (Active Motif, Carlsbad, CA, USA), and α-Tubulin (Abcam, Cambridge, MA, USA). Quantification of western blots was performed using ImageJ software27.
MCSFR Promoter Assay
The MCSFR promoter assay was performed as described previously21.
Statistical Analysis
Student t tests were performed using Excel (Microsoft, Redmond, WA, USA) to assess the significance of the differences in the indicated cell populations between samples. Log rank test was used (http://bioinf.wehi.edu.au/software/russell/logrank/) to assess the significance of the differences between the survival curves in Figure 4.
Figure 4. Impaired Runx1 activity delays leukemogenesis by Cbfb-MYH11.

Kaplan-Meier curve of mice with different genotypes during 12-month observation of leukemia development. Green line: Mx1-Cre+, Cbfb+l56M, Runx1+/lz, N=14; red line: Mx1-Cre+, Cbfb+l56M, Runx1+/l+, N=34; blue line: control mice (Runx1+/+, Runx1+/lz, and Cbfb+l56M), N=33. The p values were calculated with log rank test. The difference between any two survival curves in the panel was highly significant (p<0.0001).
Results
Runx1 is required for primitive hematopoietic defects induced by Cbfb-MYH11
Previously we showed that Cbfb-MYH11 induces defects in primitive hematopoiesis that are absent in Runx1 null embryos, suggesting such defects are Runx1-repression independent19. However, it is not clear if Runx1 is completely dispensable for these defects. To test this possibility, we generated mouse embryos homozygous for a null allele of Runx1 (Runx1−/−; Figure 1A)15 and heterozygous for a floxed allele of Cbfb-MYH11 (Cbfb56M)23. The mice also harbor the β-Actin Cre Recombinase transgene (Actb-Cre+)25 which allows for expression of Cbfb-MYH11 in all tissues of the embryo. Using cell surface staining for the differentiation markers Kit and Ter119, we found that peripheral blood from E10.5 Actb-Cre+, Cbfb+/56M, Runx1−/− embryos had an increase in mature Kit−, Ter119high cells coupled with a decrease in the less mature Kit−, Ter119low; Kit+, Ter119low and Kit+, Ter119− cells as compared to the blood from Actb-Cre+, Cbfb+/56M, Runx1+/− and Actb-Cre+, Cbfb+/56M, Runx1+/+ littermates (Figure 1B and D). We also performed staining for the cell surface marker Csf2rb, which we previously showed is expressed on immature primitive blood cells19. Consistent with the Kit and Ter119 staining, we found that Actb-Cre+, Cbfb+/56M, Runx1−/− embryos had fewer immature Csf2rb+ cells in the peripheral blood as compared to their Actb-Cre+, Cbfb+/56M, Runx1+/− and Actb-Cre+, Cbfb+/56M, Runx1+/+ littermates (Figure 1C and E). We did not observe any differences in cell surface staining between Actb-Cre+, Cbfb+/56M, Runx1+/− and Actb-Cre+, Cbfb+/56M, Runx1+/+ littermates. In addition, we did not observe any staining difference between Runx1+/+ and Runx1+/− embryos, but did observe a more subtle differentiation defect in the primitive blood of Runx1−/− embryos (Supplemental Figure 1a), consistent with previous reports19, 28. Together, these results show that genetic deletion of Runx1 partially rescued the primitive hematopoietic defects induced by Cbfb-MYH11, indicating that Cbfb-MYH11 has a genetic requirement for Runx1 for its blockage of primitive hematopoiesis.
Figure 1. Runx1 is required for Cbfb-MYH11 induced differentiation defects during primitive hematopoiesis.

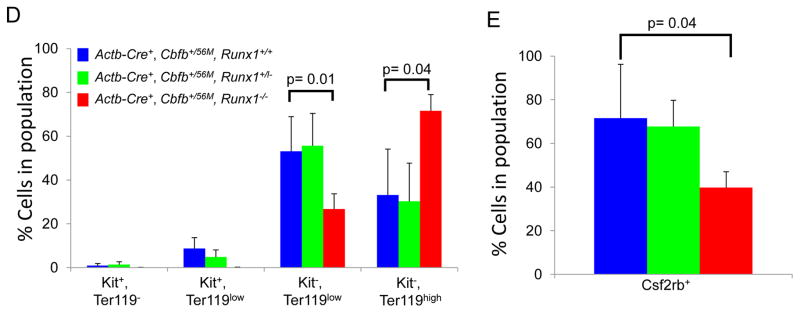
(A) Schematic representations of different Runx1 alleles used15, 24. (B and C) Representative FACS plots and (D and E) bar graphs of Terr119, Kit, and Csf2rb staining of peripheral blood cells from E10.5 embryos of the indicated genotype. Percentage of cells in each gate is given. Brackets in D and E indicate the samples being compared with the associated p-values. N≥3 for each genotype.
Rescue of Cbfb-MYH11 induced primitive hematopoietic defects by the Runx1lz allele
In order to confirm the requirement for Runx1 activity by Cbfb-MYH11, we used a mouse model with a different Runx1 allele in which exons 7 and 8 are replaced by the gene for β-galactosidase, lacZ (Runx1lz; Figure 1A). This allele has been shown to be non-functional, presumably due to the loss of the transactivation domain24. Interestingly, Runx1+/lz had defects not seen in Runx1+/− mice, but are able to survive to adulthood, unlike Runx1−/− mice. This implies that expression of one Runx1-lz allele results in a decrease in RUNX1 activity more severe than in Runx1+/− mice, but not a complete loss of RUNX1 activity as in Runx1−/− mice. Consequently, it is thought that the Runx1-lz allele acts as semi-dominant negative. To confirm this, we examined the platelet counts in the peripheral blood of adult Runx1+/+ and Runx1+/lz mice. Previous work has shown that homozygous loss of Runx1 causes a significant decrease in peripheral blood platelets, but that loss of a single Runx1 allele does not14, 29. We found that the number of platelets in the peripheral blood in Runx1+/lz mice was significantly decreased as compared to that in Runx1+/+ mice (Supplemental Figure 2), indicating that the Runx1-lz allele indeed has dominant negative activity.
To test whether the Runx1-lz allele also acts as a dominant negative in the context of Cbfb-MYH11, we generated embryos expressing the fusion gene and a single Runx1-lz allele. We found that the peripheral blood from Actb-Cre+, Cbfb+/56M, Runx1+/lz embryos had more mature Kit−, Ter119high cells and fewer immature Kit−, Ter119low; Kit+, Ter119low; and Kit+, Ter119− cells as compared to Actb-Cre+, Cbfb+/56M, Runx1+/+ littermates (Figure 2A and C). Similarly, Actb-Cre+, Cbfb+/56M, Runx1+/lz embryos had fewer Csf2rb+ cells as compared to Actb-Cre+, Cbfb+/56M, Runx1+/+ littermates (Figure 2B and D). These results indicate that a single Runx1-lz allele causes sufficient reduction in Runx1 activity to rescue Cbfb-MYH11 induced defects during primitive hematopoiesis. In addition, because Runx1+/lz mice are not embryonic lethal, these results indicate that the Runx1-lz allele can be used to study Cbfb-MYH11’s requirement for Runx1 during adult hematopoiesis.
Figure 2. Expression of one Runx1-lz allele rescues Cbfb-MYH11 induced defects in primitive blood differentiation.

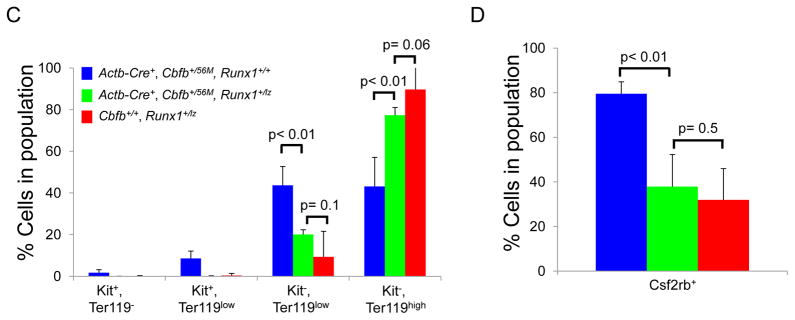
(A and B) Representative FACS plots and (C and D) bar graphs of Terr119, Kit, and Csf2rb staining of peripheral blood cells from E10.5 embryos of the indicated genotype. Percentage of cells in each gate is given. Brackets in C and D indicate the samples being compared with the associated p-values. N≥3 for each genotype.
Because RUNX1 is a transcription factor, it is possible that it is required for the expression of Cbfb-MYH11. To test this, we examined the level of CBFβ-SMMHC, the protein product of the Cbfb-MYH11 fusion gene, in E12.5 embryos by western blot (Supplemental Figure 3A). As shown in Supplemental Figure 3B, the relative CBFβ-SMMHC protein levels in Actb-Cre+, Cbfb+/56M, Runx1+/lz embryos was comparable to that in Actb-Cre+, Cbfb+/56M, Runx1+/+ embryos. This finding indicates that Runx1 activity is not required for the expression of CBFβ-SMMHC, implying that the Runx1 requirement is related to the activity of the fusion protein.
Runx1 is required for Cbfb-MYH11 induced proliferation defects in definitive hematopoiesis
We showed previously that expression of Cbfb-MYH11 in adult mice causes defects in definitive hematopoiesis prior to leukemic transformation19. These “pre-leukemic” defects include an increase in lineage negative (lin−) cells in the bone marrow shortly after induction of Cbfb-MYH11 in Cbfb+/56M mice with Cre expressed from the Mx-1 promoter, which allows for inducible expression of Cbfb-MYH11 in nearly all adult blood cells26 (Figure 3A and Supplemental Figure 4A). To test whether this increase in lin− cells is due to increased proliferation, we performed bromodeoxyuridine (BrdU) staining on lin− cells after pI:pC induction of Cbfb-MYH11 in Mx1-Cre+, Cbfb+/56M mice. One week after Cbfb-MYH11 induction, Mx1-Cre+, Cbfb+/56M, Runx1+/+ mice showed an increase of BrdU+, lin− cells. (Figure 3B and Supplemental Figure 4B). In contrast, Mx1-Cre+, Cbfb+/56M, Runx1+/lz mice showed significantly fewer BrdU+, lin− cells and no increase in total lin− cell number after Cbfb-MYH11 induction as compared to Mx1-Cre+, Cbfb+/56M, Runx1+/+ mice (Figure 3A and B, Supplemental Figure 4A and B). These results indicate that Cbfb-MYH11 induced proliferation and lin− cell population increase require Runx1 activity during adult hematopoiesis.
Figure 3. Runx1 activity is required for Cbfb-MYH11 induced defects during definitive hematopoiesis.
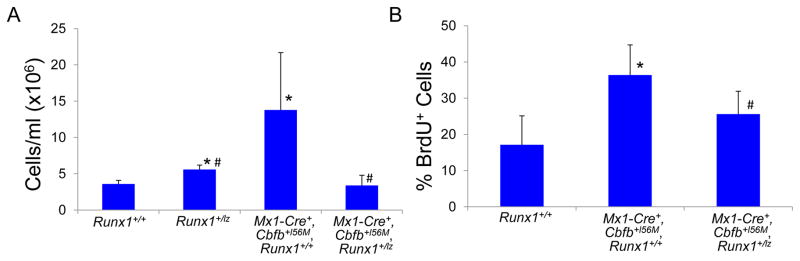


(A) Graph of the number of the lineage negative (lin-) cells per ml from the bone marrow of adult mice of the indicated genotypes 2 weeks after the induction of Cbfb-MYH11 expression. (B) Graph of percentage of lin-cells that were BrdU+ in the bone marrow of adult mice of the indicated genotypes 1 week after the induction of Cbfb-MYH11 expression. * indicates p≤0.05 as compared to Runx1+/+ mice. # indicates p≤0.05 as compared to Mx1-Cre+, Cbfb+/56M, Runx1+/+ mice. (C) Representative FACs plots of Il1rl1 and Csf2rb and (D) Kit, ScaI, CD34 and FcRγII/III staining of lin-bone marrow from adult mice of the indicated genotypes 2 weeks after the induction of Cbfb-MYH11 expression. N≥3 for each genotype.
Runx1 is required for the induction of the pre-leukemia cell population by Cbfb-MYH11
Cbfb-MYH11 expression in adult hematopoietic cells also induces an abnormal population of pre-leukemic cells expressing the cell surface markers Il1rl1 and Csf2rb19. As shown in Figure 3C, Mx1-Cre+, Cbfb+/56M, Runx1+/+ mice show an increase of Il1rl1+,Csf2rb− and Il1rl1+,Csf2rb+ cells two weeks after induction of Cbfb-MYH11 expression. Both of these populations were significantly reduced in Mx1-Cre+, Cbfb+/56M, Runx1+/lz mice, especially the Il1rl1+,Csf2rb+ cells (Figure 3C and Supplemental Figure 4C&D). These data suggest that the induction of abnormal pre-leukemic cells is impaired in Mx1-Cre+, Cbfb+/56M, Runx1+/lz mice.
Cbfb-MYH11 expression also induces defects in the lin− Sca1−, Kit+ myeloid progenitor compartment19. Consistent with our previous findings, Mx1-Cre+, Cbfb+/56M, Runx1+/+ showed a decrease in the proportion of Sca1−, Kit+ cells 2 weeks after induction of Cbfb-MYH11 (Fig. 3D, Supplemental Figure 4E). However, this decrease was not seen in Mx1-Cre+, Cbfb+/56M, Runx1+/lz mice indicating Runx1 activity is required for Cbfb-MYH11 induced decreases in myeloid progenitors.
Within the myeloid progenitor compartment, previous work showed that expression of Cbfb-MYH11 results in an accumulation of CD34−, FCγRII/III− cells that have leukemia initiating activity23. Similarly, we saw a statistically significant increase in the proportion of Sca1−, Kit+, CD34−, FCγRII/III− cells in the bone marrow of Mx1-Cre+, Cbfb+/56M, Runx1+/+ mice two weeks after induction of Cbfb-MYH11 (Fig. 3D, Supplemental Fig. 4F). In contrast, 2 weeks after induction of Cbfb-MYH11, the proportion of the Sca1−, Kit+, CD34−, FCγRII/III− cells was significantly lower in Mx1-Cre+, Cbfb+/56M, Runx1+/lz mice (Fig 3D, Supplemental Figure 4F). These results indicate that loss of Runx1 activity impairs Cbfb-MYH11 induced accumulation of Sca1−, Kit+, CD34−, FCγRII/III− cells, implying that Runx1 may be important for Cbfb-MYH11’s ability to induce leukemia initiating cells and consequently, leukemia.
Runx1 is required for efficient leukemogenesis by Cbfb-MYH11
To test whether Runx1 is required for Cbfb-MYH11 induced leukemia, we treated adult Mx1-Cre+, Cbfb+/56M, Runx1+/+ mice (N=34), Mx1-Cre+, Cbfb+/56M, Runx1+/− (N= 5), Mx1-Cre+, Cbfb+/56M, Runx1+/lz mice (N=14), and their litter mate control mice (WT; Mx1-Cre+, Runx1+/lz; Cbfb+/56M; Mx1-Cre+; Runx1+/lz; Cbfb+/56M, Runx1+/lz; N=33) with pI:pC to induce Cbfb-MYH11 expression. The mice were also treated with the mutagen N-ethyl-N-nitrosourea to induce cooperating mutations. We monitored these mice for leukemia development and progression for 12 months. Mice from both experimental cohorts developed myeloid leukemias similar to what has been described previously5, 23. The leukemia development was significantly delayed (p<0.0001) in Mx1-Cre+, Cbfb+/56M, Runx1+/lz mice as compared to Mx1-Cre+, Cbfb+/56M, Runx1+/+ mice (Figure 4). In addition, 21% (3 out of 14) of the Mx1-Cre+, Cbfb+/56M, Runx1+/lz mice never developed leukemia during the 12 months observation, while 100% of Mx1-Cre+, Cbfb+/56M, Runx1+/+ mice died from leukemia within 5 months after pI:pC and ENU treatment. We did not observe any difference in leukemia development between Mx1-Cre+, Cbfb+/56M, Runx1+/− and Mx1-Cre+, Cbfb+/56M, Runx1+/+ mice (Supplemental Figure 5), which is consistent with Runx1-lz allele having dominant negative activity. No leukemias were observed in the control mice, although solid tumors, including thymomas, were detected in some control mice upon dissection likely due to the ENU treatment. These data suggest that Runx1 strongly enhances Cbfb-MYH11 induced leukemogenisis.
RUNX1-βgal interacts with CBFβ-SMMHC and is more abundant than RUNX1 in the bone marrow
To explore the mechanism of Runx1-lz’s ability to block CBFβ-SMMHC activity, we tested whether the Runx1-lz protein product, RUNX1-βgal, is able to interact with CBFβ and CBFβ-SMMHC. HEK293 cells were transfected with constructs expressing RUNX1-βgal and either CBFβ or CBFβ-SMMHC. RUNX1-βgal containing complexes were immunoprecipitated with an antibody against β-galactosidase, and probed by western blot with an antibody against CBFβ. We found that RUNX1-βgal co-immunoprecipitated with both wildtype CBFβ and CBFβ-SMMHC (Figure 5A). β-galactosidase alone was not able to co-immunoprecipitate either wildtype CBFβ or CBFβ-SMMHC (data not shown) indicating that the RUNX1 portion of the RUNX1-βgal fusion protein is required for this interaction. These results indicate that RUNX1-βgal is capable of binding wildtype CBFβ and CBFβ-SMMHC, just as has been shown for wildtype RUNX1.
Figure 5. RUNX1-βgal binds CBFβ and CBFβ-SMMHC and is present at higher levels than RUNX1 in the bone marrow.
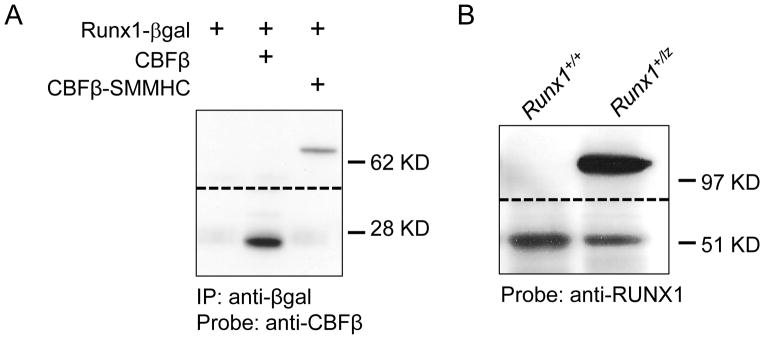
(A) Co-immunoprecipitation with anti-βgal and western blot with anti-CBFβ1411616 of lysate from 293HEK cells transfected with plasmids expressing the indicated proteins. Data shown is from two different regions of the same blot. The dashed line indicates the transition between the two sections. (B) Representative western blot of bone marrow of adult mice of the indicated genotypes probed with anti-RUNX1. Data shown is from two different regions of the same blot. The dashed line indicates the transition between the two sections.
To understand how RUNX1-βgal is capable of acting as a dominant negative, we performed a reporter assay with the MCSFR reporter construct, which has been demonstrated before as a RUNX1 target30. In this assay, RUNX1-βgal was nonfunctional but did not display significant dominant negative activity (Supplemental Figure 6). We then examined the relative expression levels of RUNX1 and RUNX1-βgal in the bone marrow of adult Runx1+/+ and Runx1+/lz mice. In Runx1+/lz mice, we found that the intensity of the RUNX1-βgal band was 2.22 fold (S.D.= 0.61, N=4) greater than the wildtype RUNX1 band (Figure 5B). The overabundance of the RUNX1-βgal protein provides a potential explanation for its semi-dominant negative behavior: the non-functional RUNX1-βgal protein may be able to outcompete wildtype RUNX1 for CBFβ and DNA target binding, leading to reduction of functional CBFβ-RUNX1 heterodimers and RUNX1-DNA complexes.
Discussion
It has been proposed that CBFβ-SMMHC, the protein product of the Cbfb-MYH11 fusion gene, acts by binding and dominantly inhibiting the activity of RUNX1, which leads to changes in gene expression that contribute to leukemogenesis. However, recent studies using knock-in mouse models have demonstrated that CBFβ-SMMHC also has RUNX1-repression independent activities that contribute to abnormal differentiation and leukemia development in Cbfb-MYH11 mice19–21. Notably we found that Cbfb-MYH11 is able to induce defects in primitive hematopoiesis, a stage not significantly affected by Runx1 knockout. Moreover, we found that mice expressing a modified CBFβ-SMMHC with reduced ability to bind RUNX1 developed leukemia faster than those expressing the full length CBFβ-SMMHC. These findings raised the possibility that CBFβ-SMMHC can induce leukemia through a RUNX1-independent mechanism. In this study we performed experiments to determine the importance of RUNX1 for leukemogenesis by CBFβ-SMMHC.
Using mouse models, we showed that Runx1 is genetically required for the differentiation block induced by Cbfb-MYH11 during primitive hematopoiesis, since this block is rescued in embryos either homozygous for a null allele of Runx1 (Runx1−/−) or with a single semi-dominant negative Runx1-lz allele (Runx1+/lz). These results, together with our previous work19–21, emphasize that CBFβ-SMMHC has important activities that don’t involve repression of RUNX1. In fact, it is likely that CBFβ-SMMHC binds RUNX1 to form a transcriptional activator complex. We previously showed that the majority of the gene expression changes induced by the fusion protein are increases in expression19. Similarly, Mandoli et al showed that the majority of genes bound by CBFβ-SMMHC are positively regulated by the fusion protein31. CBFβ-SMMHC cannot bind DNA without a RUNX protein, so homozygous deletion of Runx1 would severely impair target gene regulation by CBFβ-SMMHC. Because the Runx1-βgal protein encoded by the Runx1-lz allele is missing Runx1’s transactivation domain, we would predict that expression of this mutant inhibits CBFβ-SMMHC activity because of its inability to activate transcription. However, it is also possible that the β-gal domain interferes with DNA binding. By electrophoretic mobility shift assay, we were unable to detect a Runx1-βgal:CBFβ complex bound to DNA (data not shown).
We also found that loss of Runx1 activity significantly rescues Cbfb-MYH11 induced defects during definitive hematopoiesis. Expression of the dominant negative Runx1-lz allele markedly reduced the rate of proliferation and the number of pre-leukemic cells induced by Cbfb-MYH11. These data suggest that Runx1 is also important for Cbfb-MYH11 activities during definitive hematopoiesis. Importantly, we found that loss of Runx1 activity causes a significant delay in Cbfb-MYH11 induced leukemogenesis. Half of the Cbfb-MYH11 mice with wildtype Runx1 developed leukemia 2 months after pIpC injection, and all of them died from leukemia within 5 months. On the other hand, it took 5 months for half of the Cbfb-MYH11 mice with Runx1-lz to develop leukemia and 21% of them were still alive after 12 months. It is likely that Runx1 is required for efficient generation of the leukemia initiating population, since we observed a smaller population of lin− cells in the Cbfb-MYH11 mice with Runx1-lz. Our recent finding that the RUNX1 inhibitor Ro5-3335 decreases leukemic burden and increases survival of mice transplanted with CBFβ-SMMHC+ leukemic cells22, as well as a recent report of RUNX1 addiction of CBF leukemia cell lines in culture32, suggest that RUNX1 also has a role during leukemic maintenance, in addition to its role during initiation.
Currently, we can only speculate how RUNX1 is required for the leukemogenesis induced by CBFβ-SMMHC. Chromatin immunoprecipitation experiments with the inv(16) cell line ME-1 imply that RUNX1 binds DNA with both the fusion protein and wildtype CBFβ and that each complex regulates transcription of different target genes31, suggesting that direct interaction between RUNX1 and CBFβ-SMMHC is required. On the other hand, work with other leukemia fusion proteins shows that the CBFβ:RUNX1 dimer is required for the transcription of pro-survival factors in leukemia cells32, 33. It is possible that the CBFβ:RUNX1 dimer plays a similar role in inv(16) leukemia. In fact, Goyama et al propose that the accelerated leukemogenesis observed in mice expressing a Cbfb-MYH11 mutant with reduced RUNX1 binding may be due to a concomitant increase in the CBFβ:RUNX1 dimer, and consequently increased expression of pro-survival target genes20, 33. Ben-Ami et al also demonstrated addiction to normal RUNX1 in leukemia cells with the other common CBF fusion protein, RUNX1-RUNX1T1 (also known as AML1-ETO)32. In fact, RUNX1 mutations are rare in CBF leukemia patients, while such mutations are common in non-CBF leukemia cases, implying a requirement for the CBFβ;RUNX1 dimer in CBF leukemias34. We showed that the non-functional RUNX1-βgal protein bound both CBFβ-SMMHC and wildtype CBFβ, so RUNX1-βgal would be expected to reduce the activity of both complexes.
In this study, we showed that there is a genetic requirement for Runx1 activity for Cbfb-MYH11 induced defects during hematopoiesis and leukemogenesis. This work provides important insight into the mechanism of leukemogenesis associated with CBFβ-SMMHC and validates current efforts to develop inhibitors of the CBFβ-SMMHC: RUNX1 interaction for the treatment of inv(16) AML.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health, and NIH grant R00CA148963 (RKH)
Footnotes
Conflict of Interest
The authors have no conflicts of interest to disclose.
Supplemental information is available at Leukemia’s website.
References
- 1.Look AT. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278(5340):1059–1064. doi: 10.1126/science.278.5340.1059. [DOI] [PubMed] [Google Scholar]
- 2.Le Beau MM, Larson RA, Bitter MA, Vardiman JW, Golomb HM, Rowley JD. Association of an inversion of chromosome 16 with abnormal marrow eosinophils in acute myelomonocytic leukemia. A unique cytogenetic-clinicopathological association. N Engl J Med. 1983;309(11):630–636. doi: 10.1056/NEJM198309153091103. [DOI] [PubMed] [Google Scholar]
- 3.Liu P, Tarle SA, Hajra A, Claxton DF, Marlton P, Freedman M, et al. Fusion between transcription factor CBF beta/PEBP2 beta and a myosin heavy chain in acute myeloid leukemia. Science. 1993;261(5124):1041–1044. doi: 10.1126/science.8351518. [DOI] [PubMed] [Google Scholar]
- 4.Liu PP, Wijmenga C, Hajra A, Blake TB, Kelley CA, Adelstein RS, et al. Identification of the chimeric protein product of the CBFB-MYH11 fusion gene in inv(16) leukemia cells. Genes Chromosomes Cancer. 1996;16(2):77–87. doi: 10.1002/(SICI)1098-2264(199606)16:2<77::AID-GCC1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 5.Castilla LH, Garrett L, Adya N, Orlic D, Dutra A, Anderson S, et al. The fusion gene Cbfb-MYH11 blocks myeloid differentiation and predisposes mice to acute myelomonocytic leukaemia [letter] Nat Genet. 1999;23(2):144–146. doi: 10.1038/13776. [DOI] [PubMed] [Google Scholar]
- 6.Castilla LH, Perrat P, Martinez NJ, Landrette SF, Keys R, Oikemus S, et al. Identification of genes that synergize with Cbfb-MYH11 in the pathogenesis of acute myeloid leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(14):4924–4929. doi: 10.1073/pnas.0400930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chuang LS, Ito K, Ito Y. RUNX family: Regulation and diversification of roles through interacting proteins. Int J Cancer. 2013 Mar 15;132(6):1260–1271. doi: 10.1002/ijc.27964. [DOI] [PubMed] [Google Scholar]
- 8.Tang YY, Shi J, Zhang L, Davis A, Bravo J, Warren AJ, et al. Energetic and functional contribution of residues in the core binding factor beta (CBFbeta ) subunit to heterodimerization with CBFalpha. J Biol Chem. 2000;275(50):39579–39588. doi: 10.1074/jbc.M007350200. [DOI] [PubMed] [Google Scholar]
- 9.Lukasik SM, Zhang L, Corpora T, Tomanicek S, Li Y, Kundu M, et al. Altered affinity of CBF beta-SMMHC for Runx1 explains its role in leukemogenesis. Nat Struct Biol. 2002;9(9):674–679. doi: 10.1038/nsb831. [DOI] [PubMed] [Google Scholar]
- 10.Durst KL, Lutterbach B, Kummalue T, Friedman AD, Hiebert SW. The inv(16) fusion protein associates with corepressors via a smooth muscle myosin heavy-chain domain. Mol Cell Biol. 2003;23(2):607–619. doi: 10.1128/MCB.23.2.607-619.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lutterbach B, Hou Y, Durst KL, Hiebert SW. The inv(16) encodes an acute myeloid leukemia 1 transcriptional corepressor. Proc Natl Acad Sci U S A. 1999;96(22):12822–12827. doi: 10.1073/pnas.96.22.12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adya N, Stacy T, Speck NA, Liu PP. The leukemic protein core binding factor beta (CBFbeta)-smooth-muscle myosin heavy chain sequesters CBFalpha2 into cytoskeletal filaments and aggregates. Mol Cell Biol. 1998;18(12):7432–7443. doi: 10.1128/mcb.18.12.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castilla LH, Wijmenga C, Wang Q, Stacy T, Speck NA, Eckhaus M, et al. Failure of embryonic hematopoiesis and lethal hemorrhages in mouse embryos heterozygous for a knocked-in leukemia gene CBFB-MYH11. Cell. 1996;87( 4):687–696. doi: 10.1016/s0092-8674(00)81388-4. [DOI] [PubMed] [Google Scholar]
- 14.Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84(2):321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe AH, Speck NA. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci U S A. 1996;93( 8):3444–3449. doi: 10.1073/pnas.93.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Q, Stacy T, Miller JD, Lewis AF, Gu TL, Huang X, et al. The CBFbeta subunit is essential for CBFalpha2 (AML1) function in vivo. Cell. 1996;87(4):697–708. doi: 10.1016/s0092-8674(00)81389-6. [DOI] [PubMed] [Google Scholar]
- 17.Niki M, Okada H, Takano H, Kuno J, Tani K, Hibino H, et al. Hematopoiesis in the fetal liver is impaired by targeted mutagenesis of a gene encoding a non-DNA binding subunit of the transcription factor, polyomavirus enhancer binding protein 2/core binding factor. Proc Natl Acad Sci U S A. 1997;94(11):5697–5702. doi: 10.1073/pnas.94.11.5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sasaki K, Yagi H, Bronson RT, Tominaga K, Matsunashi T, Deguchi K, et al. Absence of fetal liver hematopoiesis in mice deficient in transcriptional coactivator core binding factor beta. Proc Natl Acad Sci U S A. 1996;93(22):12359–12363. doi: 10.1073/pnas.93.22.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hyde RK, Kamikubo Y, Anderson S, Kirby M, Alemu L, Zhao L, et al. Cbfb/Runx1 repression-independent blockage of differentiation and accumulation of Csf2rb-expressing cells by Cbfb-MYH11. Blood. 2010 Feb 18;115(7):1433–1443. doi: 10.1182/blood-2009-06-227413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamikubo Y, Zhao L, Wunderlich M, Corpora T, Hyde RK, Paul TA, et al. Accelerated leukemogenesis by truncated CBF beta-SMMHC defective in high-affinity binding with RUNX1. Cancer Cell. 2010 May 18;17(5):455–468. doi: 10.1016/j.ccr.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamikubo Y, Hyde RK, Zhao L, Alemu L, Rivas C, Garrett LJ, et al. The C-terminus of CBFbeta-SMMHC is required to induce embryonic hematopoietic defects and leukemogenesis. Blood. 2013 Jan 24;121(4):638–642. doi: 10.1182/blood-2012-06-434688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cunningham L, Finckbeiner S, Hyde RK, Southall N, Marugan J, Yedavalli VR, et al. Identification of benzodiazepine Ro5-3335 as an inhibitor of CBF leukemia through quantitative high throughput screen against RUNX1-CBFbeta interaction. Proceedings of the National Academy of Sciences of the United States of America. 2012 Sep 4;109(36):14592–14597. doi: 10.1073/pnas.1200037109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuo YH, Landrette SF, Heilman SA, Perrat PN, Garrett L, Liu PP, et al. Cbf beta-SMMHC induces distinct abnormal myeloid progenitors able to develop acute myeloid leukemia. Cancer Cell. 2006 Jan;9(1):57–68. doi: 10.1016/j.ccr.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 24.North T, Gu TL, Stacy T, Wang Q, Howard L, Binder M, et al. Cbfa2 is required for the formation of intra-aortic hematopoietic clusters. Development. 1999;126(11):2563–2575. doi: 10.1242/dev.126.11.2563. [DOI] [PubMed] [Google Scholar]
- 25.Lewandoski M, Meyers EN, Martin GR. Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harb Symp Quant Biol. 1997;62:159–168. [PubMed] [Google Scholar]
- 26.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269(5229):1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 27.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012 Jul;9(7):671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokomizo T, Hasegawa K, Ishitobi H, Osato M, Ema M, Ito Y, et al. Runx1 is involved in primitive erythropoiesis in the mouse. Blood. 2008 Apr 15;111(8):4075–4080. doi: 10.1182/blood-2007-05-091637. [DOI] [PubMed] [Google Scholar]
- 29.Growney JD, Shigematsu H, Li Z, Lee BH, Adelsperger J, Rowan R, et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood. 2005 Jul 15;106(2):494–504. doi: 10.1182/blood-2004-08-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rhoades KL, Hetherington CJ, Rowley JD, Hiebert SW, Nucifora G, Tenen DG, et al. Synergistic up-regulation of the myeloid-specific promoter for the macrophage colony-stimulating factor receptor by AML1 and the t(8;21) fusion protein may contribute to leukemogenesis. Proc Natl Acad Sci U S A. 1996;93(21):11895–11900. doi: 10.1073/pnas.93.21.11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mandoli A, Singh AA, Jansen PW, Wierenga AT, Riahi H, Franci G, et al. CBFB-MYH11/RUNX1 together with a compendium of hematopoietic regulators, chromatin modifiers and basal transcription factors occupies self-renewal genes in inv(16) acute myeloid leukemia. Leukemia. 2013 Sep 4; doi: 10.1038/leu.2013.257. [DOI] [PubMed] [Google Scholar]
- 32.Ben-Ami O, Friedman D, Leshkowitz D, Goldenberg D, Orlovsky K, Pencovich N, et al. Addiction of t(8;21) and inv(16) acute myeloid leukemia to native RUNX1. Cell reports. 2013 Sep 26;4(6):1131–1143. doi: 10.1016/j.celrep.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 33.Goyama S, Schibler J, Cunningham L, Zhang Y, Rao Y, Nishimoto N, et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J Clin Invest. 2013 Sep 3;123(9):3876–3888. doi: 10.1172/JCI68557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang JL, Hou HA, Chen CY, Liu CY, Chou WC, Tseng MH, et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009 Dec 17;114(26):5352–5361. doi: 10.1182/blood-2009-05-223784. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.