

ABSTRACT
ToxR is a major virulence gene regulator in Vibrio cholerae. Although constitutively expressed under many laboratory conditions, our previous work demonstrated that the level of ToxR increases significantly when cells are grown in the presence of the 4 amino acids asparagine, arginine, glutamate, and serine (NRES). We show here that the increase in ToxR production in response to NRES requires the Var/Csr global regulatory circuit. The VarS/VarA two-component system controls the amount of active CsrA, a small RNA-binding protein involved in the regulation of a wide range of cellular processes. Our data show that a varA mutant, which is expected to overproduce active CsrA, had elevated levels of ToxR in the absence of the NRES stimulus. Conversely, specific amino acid substitutions in CsrA were associated with defects in ToxR production in response to NRES. These data indicate that CsrA is a positive regulator of ToxR levels. Unlike previously described effects of CsrA on virulence gene regulation, the effects of CsrA on ToxR were not mediated through quorum sensing and HapR. CsrA is likely essential in V. cholerae, since a complete deletion of csrA was not possible; however, point mutations in CsrA were tolerated well. The CsrA Arg6His mutant had wild-type growth in vitro but was severely attenuated in the infant mouse model of V. cholerae infection, showing that CsrA is critical for pathogenesis. This study has broad implications for our understanding of how V. cholerae integrates its response to environmental cues with the regulation of important virulence genes.
IMPORTANCE
In order to colonize the human host, Vibrio cholerae must sense and respond to environmental signals to ensure appropriate expression of genes required for pathogenesis. Uncovering how V. cholerae senses its environment and activates its virulence gene repertoire is critical for our understanding of how V. cholerae transitions from its natural aquatic habitat to the human host. Here we demonstrate a previously unknown link between the global regulator CsrA and the major V. cholerae virulence gene regulator ToxR. The role of CsrA in the cell is to receive input from the environment and coordinate an appropriate cellular response. By linking environmental sensing to the ToxR regulon, CsrA effectively acts as a switch that controls pathogenesis in response to specific signals. We demonstrate that CsrA is critical for virulence in the infant mouse model of V. cholerae infection, consistent with its role as an in vivo regulator of virulence gene expression.
INTRODUCTION
The severe diarrheal disease cholera is caused by pathogenic strains of the Gram-negative bacterial species Vibrio cholerae. All V. cholerae strains are native inhabitants of marine and estuarine environments, but a distinguishing feature of the pathogenic strains is their ability to adapt to growth in the human host, producing toxins that facilitate shedding in the stool and reentry into the environment. The transition of V. cholerae from its natural aquatic habitat to the human host requires a rapid response to radical environmental changes. How V. cholerae senses its environment and incorporates these signals into the regulation of its gene and protein expression profiles is a topic of active research.
One of the primary regulators of gene expression leading to survival, colonization, and production of disease in the mammalian host is the inner membrane-spanning DNA-binding protein ToxR (1–3). ToxR is at the top of an extensive hierarchy of V. cholerae virulence gene regulatory networks (reviewed in references 4 and 5). ToxR, together with the transcriptional regulator TcpP, induces expression of the central virulence gene regulator ToxT (6). ToxT, in turn, activates the genes encoding the toxin-coregulated pilus and the cholera toxin, both of which are required for pathogenesis (4). ToxR also directly induces expression of the ctx genes in the presence of specific compounds such as bile acids (7), which may act to stimulate the activity of the ToxR protein at its target promoters (8).
In addition to regulating virulence genes, ToxR is critical for controlling the protein composition of the outer membrane. In response to environmental stimuli, V. cholerae remodels its outer membrane to take advantage of different available nutrients and to increase the barrier function of the outer surface. Two of the major outer membrane proteins (OMPs) produced by V. cholerae are the porins OmpT and OmpU, both of which are controlled by ToxR (8–10). ToxR binds directly to the promoter regions of both of these genes, but with opposite effects: whereas ompU expression is upregulated by ToxR, ompT expression is repressed (10–12). OmpT has greater overall permeability than OmpU (13) and is thought to be the dominant porin during growth in the aquatic environment, where the concentration of nutrients and osmolytes may be relatively low. OmpU, although it has a slightly larger pore size than OmpT, is less permeable to negatively charged compounds (13). Consequently, OmpU is essential for resistance to bile acids and organic acids, as well as to many antimicrobial peptides, present in the digestive tract of the host (14, 15). OmpU is expressed at high levels, both during infection of the human host and during shedding in the stool of cholera patients (16, 17).
The mechanism of ToxR-dependent transcriptional regulation in response to environmental cues is not completely understood. Previous studies have shown that toxR expression is not generally responsive to environmental conditions that favor virulence gene expression in vitro, suggesting that toxR expression may be constitutive under most laboratory conditions (8, 18–20). We have demonstrated, however, that ToxR levels are modulated in vitro in response to specific environmental cues. ToxR protein levels increase rapidly in minimal medium upon addition of a mix of asparagine, arginine, glutamate and serine (NRES), and this increase in the level of ToxR is sufficient to promote production of OmpU while reducing synthesis of OmpT (8). In this study, we demonstrate that modulation of ToxR levels in response to the NRES mix is dependent upon the two-component system VarS/VarA and the global regulator CsrA.
Homologs of the VarS/VarA system are found in many bacterial species, including Escherichia coli (BarA/UvrY) and Pseudomonas species (GacS/GacA). These systems regulate processes as diverse as central carbon metabolism, motility, biofilm formation, quorum sensing, production of virulence determinants, and oxidative stress (reviewed in references 21 and 22). In V. cholerae, the VarS/VarA system is involved in carbon metabolism (23), quorum sensing and biofilm formation (24, 25), expression of virulence determinants (26, 27), fitness in infant mice and rabbits (23, 27), and survival in pond water after being shed from the mammalian host (23). VarS is predicted to be a sensor kinase that phosphorylates and activates its cognate response regulator VarA in response to an unknown signal. Activated VarA induces transcription of three noncoding small RNAs (sRNAs), CsrB/C/D (25), which bind the regulatory protein CsrA and sequester it in an inactive state (Fig. 1). Each of the three V. cholerae Csr sRNAs contains multiple CsrA recognition sequences and is predicted to bind 15 to 20 CsrA molecules (25), as has been shown for the CsrA-antagonizing sRNA CsrB in E. coli (28). CsrA is a global, posttranscriptional regulator that acts by binding to mRNA targets and affecting their translation or stability. CsrA is likely the primary target of signaling through the VarS/A system in V. cholerae (25), but it is not known whether the CsrA/B/C/D system is subject to VarS/A-independent regulation.
FIG 1 .
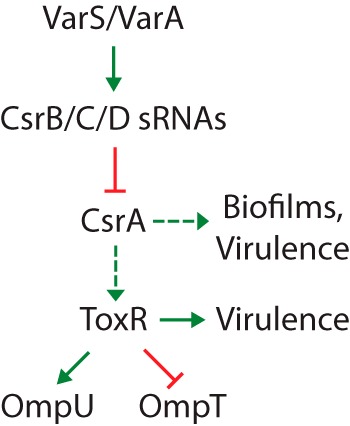
Overview of the VarA/S-CsrB/C/D-CsrA pathway. Dashed arrows indicate that the exact mechanism of regulation is currently unknown. We show here that CsrA positively regulates the level of ToxR protein in response to amino acids. Var/Csr-mediated regulation of biofilm and virulence gene expression via the quorum sensing pathway is described by Lenz et al. (25). The regulation of omp gene expression by ToxR is well established (8, 10–12, 14).
We show here that a varA mutant of V. cholerae, which is predicted to overproduce active CsrA due to poor expression of the CsrB/C/D sRNAs, had elevated levels of ToxR and OmpU in the absence of the NRES signal. This suggests that CsrA has a stimulatory effect on ToxR levels. Consistent with this, we identified a series of CsrA point mutants that failed to increase ToxR levels or produce OmpU in response to the NRES mix. One of these point mutants, CsrA Arg6His, was tested in vivo and failed to colonize infant mice, showing that CsrA is critical for the virulence of V. cholerae. The interconnection of the CsrA global regulatory circuit with the ToxR regulon has important implications for how V. cholerae incorporates environmental cues into the complex regulation of its virulence genes.
RESULTS
The transcriptional profile of V. cholerae in response to NRES.
Our previous studies demonstrated that growth of V. cholerae in a minimal medium containing the NRES amino acid mix caused ToxR levels to increase, resulting in a switch in the dominant porin from OmpT to OmpU (8). To better understand the nature of the response to amino acid stimulation in V. cholerae, changes in V. cholerae gene expression in response to the NRES mix were determined by microarray analysis. A list of V. cholerae genes for which the transcript level was altered more than 2-fold in response to NRES can be found in Table S1 in the supplemental material. These include genes involved in amino acid and carbon metabolism, peptide transporter genes, genes related to quorum sensing, biofilm formation, and chemotaxis, virulence genes, including toxR, toxS, and varA, iron transport genes, a putative σ54 modulation factor gene, the porin genes ompT and ompU, genes encoding hypothetical proteins, and many others. Several of the NRES-responsive genes were tested by mutant analysis to determine whether they played a role in the NRES-dependent ToxR increase and OMP switching (see Table S2 in the supplemental material). As expected, toxR and toxS mutants did not produce OmpU under any condition (8); however, only one additional NRES-regulated gene tested in this study, varA, displayed a mutant phenotype with respect to its OMP profile. Other genes known to play a role in the virulence of V. cholerae were tested as well, but none were found to be required for OMP switching in response to NRES (Table S2), making it unlikely that they participate in NRES-mediated regulation of ToxR levels.
VarA plays a role in the NRES response.
The VarS/A system is involved in quorum sensing and virulence gene regulation in V. cholerae (25–27). Interestingly, the gene encoding the response regulator VarA showed a 2- to 3-fold induction in the presence of NRES. Therefore, a varA mutant was constructed in V. cholerae El Tor strain N16961 and tested for its ability to respond to the NRES mix.
The N16961 varA mutant (NvarA) produced a mixture of large and small colonies. The small-colony variant, NvarAS, exhibited reduced growth in liquid culture compared with the wild-type parental strain, especially in a minimal medium (Fig. 2A). Growth was restored to wild-type levels when pVarA, a low-copy-number plasmid containing the cloned varA gene, was introduced into the NvarAS strain (Fig. 2A), showing that the poor growth of NvarAS was due solely to the lack of a functional VarA.
FIG 2 .
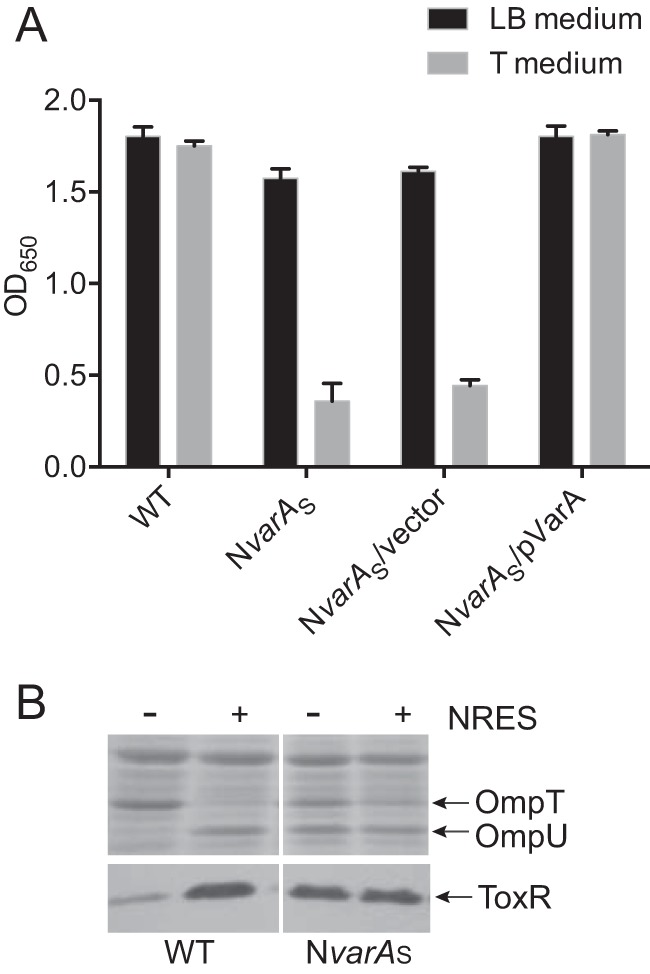
Mutation of varA causes growth defects and results in an abnormal pattern of OMP and ToxR expression. (A) N16961 (WT), the ΔvarA::cam mutant (NvarAS), the vector control strain NvarAS/pWKS30, and the complemented strain NvarAS/pVarA were grown overnight in LB broth and then subcultured 1:100 into fresh LB or T medium and grown for 6 h. (B) N16961 and NvarAS were grown overnight in LB broth and then subcultured 1:100 into T medium with or without 12.5 mM NRES mix. Cells were harvested in mid-log phase, and whole-cell preparations were resolved by SDS-PAGE (10%) and stained by Coomassie blue (top panel) or immunoblotted using polyclonal anti-ToxR antisera (bottom panel).
When grown in a minimal medium, the NvarAS strain had an abnormal OMP profile, producing both OmpT and OmpU, whether or not NRES was present (Fig. 2B). Alterations in the OMP profile often indicate misregulation of ToxR levels. Indeed, in the absence of amino acids, the level of ToxR was much higher in the NvarAS strain than in the parental strain (Fig. 2B). This increase in ToxR could reasonably account for the presence of OmpU in the NvarAS strain grown without any added NRES, as an increase in ToxR protein levels above basal levels is sufficient to induce ompU expression (8).
In contrast to NvarAS, the large-colony NvarA strain (NvarAL) had a normal growth phenotype compared with its wild-type parental strain (see Fig. S1 in the supplemental material) but did not produce any OmpU in response to NRES when grown in a minimal medium (Fig. 3A). This was not due to an inherent inability to synthesize OmpU, since NvarAL made OmpU exclusively when grown in medium containing the bile salt cholate (Fig. 3B), a condition known to promote the production of OmpU and repress the synthesis of OmpT (8). These data suggest that NvarAL specifically cannot respond to NRES. The switch from ompT to ompU expression in response to NRES requires an increase in the level of ToxR, unlike the response to bile acids, in which the ToxR level is unchanged (8). To determine whether the inability to produce OmpU in the NvarAL strain is due to failure to upregulate the production of ToxR in response to the NRES mix, ToxR levels were assessed. The results show that ToxR levels did not increase in NvarAL grown with NRES (Fig. 3A), suggesting that the level of ToxR is insufficient to promote a switch in omp gene expression from ompT to ompU under these conditions.
FIG 3 .
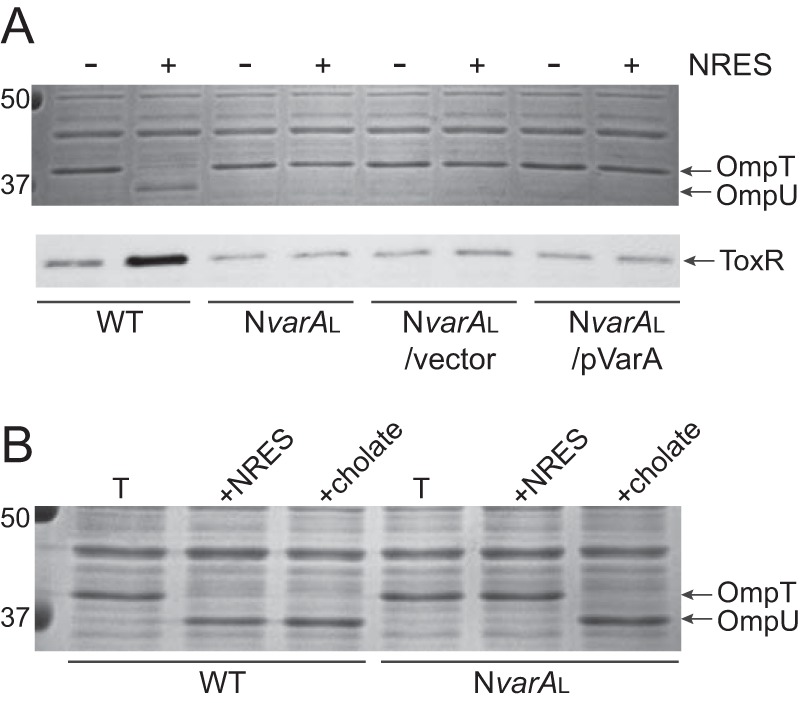
(A) The NvarAL mutant did not increase OmpU or ToxR levels in response to NRES. This phenotype could not be complemented with pVarA, suggesting a suppressor mutation. Strains N16961 (WT), NvarAL, NvarAL/pWKS30 (vector control), and NvarAL/pVarA (complemented strain) were grown overnight in LB broth and then subcultured 1:100 into T medium with or without 12.5 mM NRES mix. Cells were harvested in the mid-log phase, and whole-cell preparations were resolved by SDS-PAGE (10%) and stained by Coomassie blue (top panel) or immunoblotted using polyclonal anti-ToxR antisera (bottom panel). (B) The NvarAL strain did not have an inherent defect in OmpU production, since OmpU was made exclusively in response to the bile acid cholate. Strains N16961 (WT) and NvarAL were grown overnight in LB broth and then subcultured 1:100 into T medium with or without 0.1% cholate. Cells were harvested in the mid-log phase, and whole-cell preparations were resolved by SDS-PAGE (10%) and stained with Coomassie blue to visualize the OMPs.
When pVarA was introduced into NvarAL for complementation, it did not restore NRES-mediated OmpU production, nor did it cause an increase in ToxR levels in response to NRES (Fig. 3A). This suggested that the NvarAL strain contains a suppressor mutation that allows for relatively robust growth compared with the NvarAS strain but which disrupts the induction of ToxR and OmpU levels in response to NRES.
Loss of VarA results in suppressor mutations in csrA.
Since loss of VarA is predicted to result in overaccumulation of active CsrA due to low levels of the CsrA-sequestering sRNAs (Fig. 1), we reasoned that suppressor mutations could arise in the csrA gene itself. The csrA gene from the NvarAL strain was sequenced and was found to contain a point mutation that replaces the arginine residue at amino acid position 6 with a histidine (R6H). Several independently derived NvarAL isolates were analyzed to determine whether they also carried suppressor mutations in csrA and whether there was variation in the type of csrA point mutation that could arise in the absence of functional VarA. All of the NvarAL isolates tested carried point mutations affecting csrA. In contrast, none of the small colony phenotype NvarAS strains had mutations in csrA. Most of the NvarAL strain mutations were in the csrA coding region, but several isolates carried mutations in the predicted Shine-Dalgarno (SD) sequence upstream of the csrA translational start (Fig. 4). All coding region mutations resulted in a single amino acid substitution in CsrA (Fig. 4). The OMP profiles of several NvarAL suppressor mutants were analyzed. Many of these mutants did not produce OmpU in response to the NRES mix (Fig. 5A). Rather, they produced OmpT at high levels in minimal medium both with and without NRES supplementation. Thus, these mutants were not stimulated to switch OMPs in the presence of NRES. The amino acid substitutions in this group of mutants generally clustered in the N-terminal half of CsrA (R6H, R6L, T11P, I14T, and T19P) (Fig. 4). Another class of point mutants produced both OmpT and OmpU in T medium alone, and this pattern did not change with the addition of NRES, suggesting defects in OMP regulation in response to environmental cues (Fig. 5A). Several mutants of this class had amino acid substitutions localizing to a region within the C-terminal half of the CsrA protein (A36S and P37L) (Fig. 4). Some of the suppressor strains had mutations in the putative SD sequence (Fig. 4). These mutants were phenotypically similar to the class of mutants exhibiting no OmpU production in response to NRES (Fig. 5A; also data not shown).
FIG 4 .

Sequence conservation, structural features, and locations of suppressor mutations in V. cholerae CsrA. (A) An alignment of the V. cholerae CsrA protein with CsrA from additional Gram-negative species, E. coli, Salmonella enterica serovar Typhimurium, and Pseudomonas aeruginosa, showing a high degree of conservation among these species. The positions of the predicted β sheets (β1 to β5) and α helix (α1) in the CsrA protein are indicated above the alignment. The colored arrows point to the positions of the CsrA suppressor mutations relative to the secondary structural elements. The angled black arrow shows the translational start of the csrA open reading frame. The mutated residues in the suppressor strains are highlighted in the V. cholerae CsrA protein sequence. The thick arrows above the alignment show the positions of the transposon insertions within the published E. coli csrA::kan (open arrow) (29) and V. cholerae csrA::Tn5 (closed arrow) (25) mutant strains. (B) List of the CsrA point mutations identified in the NvarAL and the NtoxR, varAL suppressor strains (color coded to match the arrows in panel A), their phenotypes with respect to OMP and ToxR production, and their frequency of isolation. The ARM759L strains were not tested for their NRES response, since they are toxR mutants and do not make OmpU. The NvarAL suppressor mutants are in the V. cholerae N16961 ΔvarA::cam background, and the ARM759L suppressor mutants are in the N16961 ΔtoxR::kan ΔvarA::cam background.
FIG 5 .
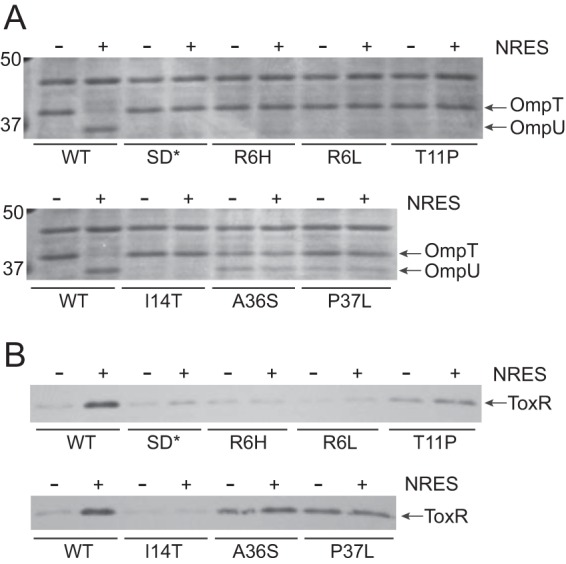
The NvarAL suppressor strains show abnormal responses to the NRES mix. Strains are labeled according to the csrA suppressor mutation present, but all mutant strains additionally carry the ΔvarA::cam mutation. Strains N16961 (wild type [WT]), NvarAL-00 (SD*), NvarAL-1 (R6H), NvarAL-2 (R6L), NvarAL-3 (T11P), NvarAL-4 (I14T), NvarAL-6 (A36S), and NvarAL-7 (P37L) were grown overnight in LB broth and then subcultured 1:100 into T medium with or without 12.5 mM NRES mix. Cells were harvested in mid-log phase, and whole-cell preparations were resolved by SDS-PAGE (10%) and stained by Coomassie blue (A) or immunoblotted using polyclonal anti-ToxR antisera (B).
The level of ToxR was determined in each of these suppressor mutants. In the mutants producing no detectable OmpU in response to NRES, the level of ToxR was found to be correspondingly low, indicating that the point mutations in CsrA abolished the NRES-mediated increase in ToxR protein levels (Fig. 5B). In the strains that produced a small, constitutive amount of OmpU with or without NRES, the level of ToxR protein was higher than normal in the absence of NRES but lower than normal in the presence of NRES (Fig. 5B); thus, the amount of ToxR produced was large enough to promote some OmpU synthesis in the absence of NRES but not sufficient to cause a complete switch in OMP production from OmpT to OmpU (Fig. 5A). The inappropriate production of ToxR and OmpU in the absence of NRES seen in this group of NvarAL suppressor strains might result from constitutively high levels of free (albeit less active) mutant CsrA, since the varA mutant strains do not produce the CsrA-sequestering sRNAs (Fig. 1).
High levels of active CsrA cause growth defects and overproduction of ToxR and OmpU in the absence of NRES.
Complementation experiments were carried out in order to rescue the csrA defect of the NvarAL mutant strains by supplying the csrA gene on a low-copy-number vector; however, the mutant strains did not tolerate the csrA plasmid well, and most of the strains failed to maintain the plasmid. This is not surprising, since the varA mutation impairs the ability of the strain to control the level of free CsrA, which is likely detrimental for growth, as shown above. Only one of the NvarAL strains, NvarAL-3 (ΔvarA::cam csrA.T11P), maintained the pWCsrA plasmid well enough for analysis. This strain exhibited very poor growth when carrying the pWCsrA plasmid but normal growth when carrying the vector alone (Fig. 6A). The presence of the pWCsrA plasmid did not restore the OMP profile to normal. Rather, OmpU was inappropriately expressed when the mutant strain carrying this plasmid was grown in T medium alone (Fig. 6B), suggesting overproduction of ToxR, even in the absence of an environmental cue. This is likely due to constitutively high levels of free CsrA in the absence of a functional VarA system and suggests that CsrA acts as a positive regulator of ToxR synthesis. The pWCsrA plasmid had no effect on the OMP profile of the wild-type strain grown with or without the NRES supplement (see Fig. S2 in the supplemental material). This is consistent with tight regulation of CsrA levels by the VarS/A-CsrB/C/D system in the wild-type strain to prevent toxic levels of free CsrA.
FIG 6 .
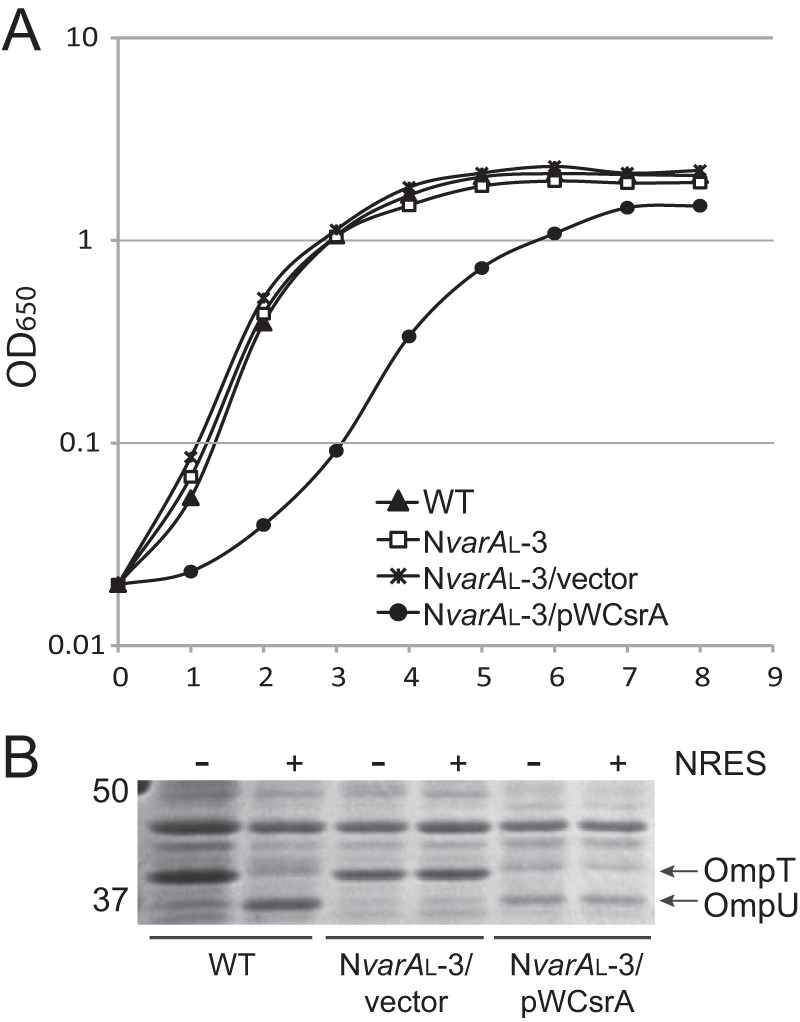
An NvarAL suppressor mutant cannot be complemented by supplying wild-type csrA on a plasmid. Expression of wild-type CsrA in the absence of functional VarA is toxic to the cells, and leads to production of OmpU in the absence of NRES. (A) Strains N16961 (WT), NvarAL-3 (ΔvarA::cam csrA.T11P), NvarAL-3/pWKS30, and NvarAL-3/pWCsrA were grown overnight in LB broth and then subcultured 1:100 into fresh LB broth and grown to the stationary phase. (B) Strains N16961 (wild type [WT]), NvarAL-3/pWKS30, and NvarAL-3/pWCsrA were grown overnight in LB broth and then subcultured 1:100 into T medium with or without 12.5 mM NRES mix. Cells were harvested in the mid-log phase, and whole-cell preparations were resolved by SDS-PAGE (10%) and stained by Coomassie blue.
An Arg6His substitution in CsrA is sufficient to abolish NRES-mediated ToxR production.
To test directly the role of CsrA in the regulation of ToxR protein levels, attempts were made to delete the entire csrA gene in strain N16961; however, none of these attempts was successful, suggesting that csrA is essential in V. cholerae. Instead, we tested whether a point mutation in CsrA alone is sufficient to abolish the ToxR/OMP response to the NRES mix. The csrA R6H point mutation, which was one of the most frequently isolated mutations, was constructed in the wild-type genetic background. The NcsrA.R6H strain exhibited wild-type growth (data not shown) but produced only OmpT, and no OmpU, in the presence of the NRES mix (Fig. 7A), indicating that the csrA point mutation, and not the varA deletion, caused the failure to respond to the NRES mix in the original NvarAL-1 strain. As in the NvarAL-1 strain, the ToxR levels in the NcsrA.R6H point mutant did not increase in the presence of NRES (Fig. 7B). This shows that changing just a single amino acid in CsrA profoundly affects the ability of V. cholerae to respond to amino acids. To verify that the NcsrA.R6H point mutant did not contain any additional suppressor mutations that might be responsible for the observed phenotype, the point mutant was complemented. A csrA plasmid clone was created in a single-copy-number vector, pCC1. When pFCsrA was introduced into NcsrA.R6H, both the OMP profile and the ToxR level were restored to wild type (Fig. 7A and B). This shows that the R6H point mutation in CsrA is solely responsible for the defect in ToxR/OmpU production. Expression of csrA from the pFCsrA plasmid did not interfere with regulation of the OMPs in the wild-type strain (see Fig. S2 in the supplemental material), most likely due to tight regulation of free CsrA levels by the VarS/A-CsrB/C/D system.
FIG 7 .
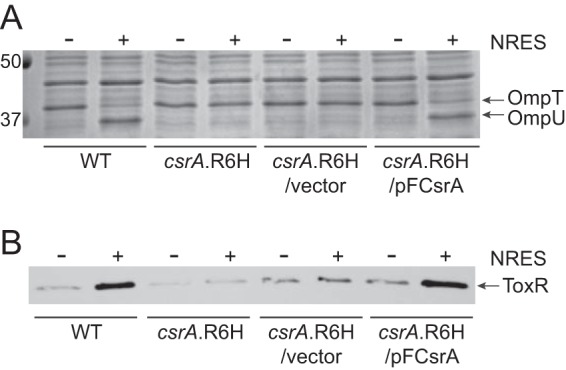
An Arg6His substitution in CsrA is sufficient to abolish the response to NRES, even when the Var system is functional. The wild-type strain N16961, the NcsrA.R6H mutant, the vector control strain NcsrA.R6H/pCC1, and the complemented strain NcsrA.R6H/pFCsrA were grown overnight in LB broth and then subcultured 1:100 into T medium with or without 12.5 mM NRES mix. Cells were harvested in mid-log phase, and whole-cell preparations were resolved by SDS-PAGE (10%) and stained by Coomassie blue (A) or immunoblotted using polyclonal anti-ToxR antisera (B).
A csrA::Tn5 carboxy-terminal insertion mutant is defective in NRES-mediated ToxR production.
Although we were unable to create a csrA deletion mutant in V. cholerae, a csrA::Tn5 mutant has been isolated and shown to affect quorum sensing pathways in V. cholerae strain C6706str2 (25). Interestingly, when the csrA::Tn5 allele was sequenced, we found that the site of the Tn5 insertion was at the end of the csrA gene, in a position nearly identical to the transposon insertion in a published, viable E. coli csrA::kan mutant (29, 30) (Fig. 4). This suggests that the V. cholerae csrA::Tn5 mutant, like the E. coli csrA::kan mutant, may produce a partially functional CsrA. This is further supported by the absence of any growth defect in the V. cholerae csrA::Tn5 mutant (25; also data not shown). An identical csrA::Tn5 insertion mutation was created in strain N16961. As shown in Fig. 4, this insertion results in the loss of the C-terminal 12 amino acid residues of V. cholerae CsrA. NcsrA::Tn5 exhibited wild-type growth in rich medium (data not shown), suggesting that the C-terminally truncated CsrA protein retains the activities needed for viability; however, similar to the NcsrA.R6H strain, NcsrA::Tn5 produced very low levels of ToxR and OmpU in response to NRES (see Fig. S3A and B in the supplemental material). Wild-type OMP and ToxR levels were fully restored when csrA was supplied on plasmid pFCsrA (see Fig. S3A and B).
The outgrowth of csrA suppressor mutants is not due to overproduction of a ToxR-regulated target.
As described in Fig. 1, mutation of varA leads to an increase in the level of free CsrA and therefore in the level of ToxR. Thus, the selective pressure to mutate csrA in a varA mutant strain background could be a result of overproduction of a ToxR-activated target, such as OmpU, in the varA mutant. To investigate this, a varA deletion was created in a toxR-defective strain. Both large and small colonies were observed for the resulting toxR varA mutants, suggesting the outgrowth of suppressor mutants (large colonies). The csrA gene was sequenced in a number of these double mutants. All large colonies had csrA point mutations (Fig. 4), whereas the small colonies were wild type for csrA. This demonstrates that suppressor mutations leading to improved growth of the varA mutant are selected for even in the absence of toxR, showing that misregulation of a ToxR-activated gene is unlikely to be the selective pressure responsible for the outgrowth of csrA suppressors in the varA mutant. Thus, the pressure to mutate csrA in the absence of varA may be due to misregulation of another, unknown, CsrA target.
HapR and LuxO are not needed for the NRES response.
V. cholerae CsrA has been shown to regulate the expression of genes involved in quorum sensing, biofilm formation, and virulence, primarily through the regulatory protein HapR, which integrates signals from multiple pathways, including the Lux quorum sensing pathway and the VarS/A-CsrA system (25, 31). The strain used in these studies, N16961, is naturally defective for hapR (32), suggesting that HapR is not needed for NRES-mediated regulation. Although N16961 does not produce functional HapR, it may produce another factor with a redundant function. To determine directly whether either quorum sensing or HapR plays a role in the CsrA-mediated regulation of ToxR levels in response to NRES, we tested the effects of various mutations in a HapR+ strain, C6706, which responds similarly to NRES as N16961 (see Fig. S4 in the supplemental material). Similar to the NvarAS strain, a C6706 varA mutant grew poorly and produced ToxR and OmpU in the absence of NRES, all of which could be remedied by supplying wild-type varA on a plasmid (data not shown). These data suggest that C6706 does not differ significantly from the HapR-defective N16961 strain in its response to NRES and the role of VarA in this; however, it did not rule out that HapR is needed for some aspect of this response in C6706. Therefore, a C6706 hapR mutant was tested for its role in the NRES regulation. No differences were detected between the wild-type parental strain and the hapR mutant with respect to OMP and ToxR levels in the presence of NRES (see Table S2 in the supplemental material), indicating that HapR is not required for the NRES response. The quorum sensing regulator, LuxO, which has been shown to play a role in relaying input from the Var/Csr pathway into the quorum sensing pathway (25), was not necessary for the NRES response either (see Table S2). This suggests that the mechanism of CsrA regulation of ToxR levels does not require either the Lux quorum sensing pathway or the HapR transcriptional regulator.
CsrA is required for virulence.
ToxR is a critical virulence factor within the mammalian host (4, 33, 34), suggesting that functional CsrA may also be necessary for the pathogenesis of V. cholerae. To test this, we assessed the ability of the NcsrA.R6H mutant to colonize infant mice. The infant mouse model has been shown to be effective in demonstrating the importance of many major virulence factors in this pathogen (35). A competition assay was performed to compare the in vivo fitness of the NcsrA.R6H point mutant to that of the wild-type strain. As described earlier, the NcsrA.R6H strain was not impaired in its in vitro growth but failed to increase ToxR and OmpU levels in response to the NRES mix. Five-day-old infant mice were inoculated intragastrically with equal numbers of the wild-type strain and the NcsrA.R6H mutant. The ability of the mutant strain to compete with the wild-type strain was assessed by determining the ratio of viable mutant cells to wild-type cells recovered after 18 h. The median competitive index (CI [i.e., the output ratio normalized to the input ratio]) for the NcsrA.R6H mutant was <0.01, showing a severe defect in the ability to colonize the infant mouse (Fig. 8). Four of the nine animals yielded results that were below the limit of detection (CI, <0.005), and therefore the median CI is likely even lower than what was computed. These data point to an overall decrease in fitness of more than 100-fold for the mutant strain, showing that CsrA is critical for colonization of the small intestine.
FIG 8 .
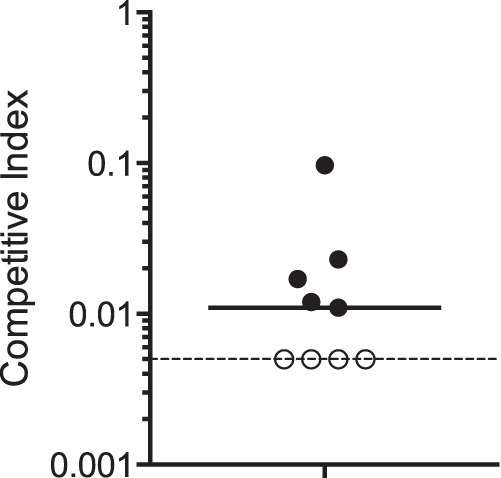
CsrA is required for V. cholerae virulence in infant mice. The CsrA Arg6His point mutant NcsrA.R6H was competed against the wild-type strain N16961G. Five-day-old suckling BALB/c mice were inoculated intragastrically with an equal number of each competing strain as described in Materials and Methods. The competitive index (CI) was calculated by normalizing the output ratio to the input ratio of the two competing strains. Each data point represents one mouse, and the median CI (0.01) is represented by the solid horizontal line. A CI value below 1 indicates that the mutant is at a competitive disadvantage. The dotted line shows the limit of detection for the CI in this experiment, which was 0.005. In 4 out of 9 mice, the mutant was not detected during analysis of more than 200 recovered colonies, and thus the CI falls below the limit of detection. These data points are included in the analysis as having a value of 0.005, which is the most conservative estimate for the CI. The actual CI values are likely to be lower. The results were statistically significant (P < 0.05) by the Mann-Whitney nonparametric test.
DISCUSSION
V. cholerae is a facultative human pathogen that spends most of its life cycle in aquatic habitats. The transition to the human host represents a major environmental shift that includes changes in temperature, pH, osmolarity, and the level of both nutrients and antimicrobial compounds. V. cholerae must sense and respond to these environmental cues in order to survive the initial transit through the upper portions of the digestive tract and become oriented toward the most favorable microenvironment for successful colonization and production of the cholera toxin within the lower part of the small intestine.
In V. cholerae, as well as in other human pathogens, the global regulator CsrA plays an important role in coordinating the cellular response to multiple environmental signals, including nutritional status and cell density (23, 25, 36). In this study, we demonstrate a link between CsrA and regulation of ToxR levels in response to an environmental signal. CsrA was essential for the increase in ToxR that occurred when V. cholerae grew in the presence of the NRES amino acids and for the subsequent alterations in the outer membrane protein profile. We show further that CsrA is critical for the virulence of V. cholerae; this is the first definitive study of the role of V. cholerae CsrA in vivo.
CsrA was shown previously to affect pathways leading to virulence gene expression in V. cholerae, but not via ToxR. Most of the effects of the CsrA signaling cascade on virulence gene expression in V. cholerae have been found to be relayed through HapR, either via quorum sensing and the Lux pathway (25) or through Lux-independent effects on HapR activity (37). HapR-independent regulation of a virulence gene by CsrA has not been demonstrated. However, in at least one HapR− strain of V. cholerae, O395 (38), VarA is required for production of virulence factors and for pathogenesis in mice (27). This suggests that there is HapR-independent regulation of the V. cholerae virulence regulon through the Var/Csr system. Here, we show that the interconnection between the CsrA pathway and the ToxR regulon occurs independently of both HapR and the central quorum sensing regulator LuxO.
Our data suggest a model in which CsrA acts as a positive regulator of ToxR synthesis. A V. cholerae VarA system mutant, which is predicted to produce too much active CsrA (25), exhibited abnormally high levels of ToxR and OmpU in minimal medium without amino acid supplementation. Overexpression of csrA from a plasmid in the absence of functional VarA caused a similar increase in ToxR and OmpU levels in the absence of the NRES signal. In addition, mutation of csrA, either through a point mutation or a transposon insertion, was associated with failure to upregulate ToxR levels in the presence of NRES, further establishing the requirement for CsrA in the ToxR-mediated response to amino acids.
CsrA can act as a positive or a negative regulator, depending on whether it increases or decreases either translation or stability of the target mRNA (reviewed in reference 22). Studies are under way to determine whether the observed positive effects of CsrA on ToxR regulation are direct or indirect. Putative binding sites for CsrA can be found in the region upstream of the predicted start of translation of the toxR mRNA (A. R. Mey, unpublished results), suggesting that CsrA may bind directly to the toxR transcript and increase its stability or rate of translation. The exact mechanism for this proposed regulation is not clear, since the putative CsrA binding sites are in close proximity to the proposed Shine-Dalgarno sequence in the predicted toxR transcript, which is more typical of translational repression. It should be noted, however, that experimental verification of the predicted start sites for toxR transcription and translation in V. cholerae has not been published. At least one study has suggested an alternative start of translation for ToxR (39), and this could have implications for the mechanism by which CsrA influences ToxR levels. In addition, new mechanisms of regulation by CsrA are still being uncovered, and there is much still to learn about how this important global regulator operates at its various mRNA targets.
Our studies suggest that csrA is essential in V. cholerae, as we were unable to construct a complete deletion of the csrA gene. In E. coli, CsrA is essential under laboratory conditions (30), and most published studies of csrA function in E. coli have been carried out using a strain carrying a transposon insertion at codon 51 out of 65 (Fig. 4), resulting in a partially active CsrA protein (29, 30). The published, viable V. cholerae csrA::Tn5 mutant carries an insertion in a similar position, suggesting that it may also retain partial CsrA activity. It has been proposed that the lethality of a csrA null mutant could be due in part to accumulation of excess glycogen (30), since CsrA is known to repress the glycogen biosynthetic pathway (29); however, we were unable generate a complete csrA deletion mutant in a glycogen synthesis-deficient strain of N16961 (40), suggesting that csrA is essential for the growth of V. cholerae for reasons other than or in addition to regulation of glycogen biosynthesis (H. A. Butz, unpublished results).
While loss of csrA is harmful to the cell, too much CsrA is detrimental as well. The level of active CsrA is tightly controlled through the CsrB/C/D sRNAs and by the VarS/VarA two-component system that regulates their expression. Our studies suggest that a constitutively high level of active CsrA through loss of VarA in V. cholerae causes growth defects, resulting in the accumulation of suppressor mutations in csrA. The varA csrA* suppressor strains exhibited normal growth compared with the varA mutant alone, but poor growth returned when wild-type csrA was supplied on a plasmid, further evidence that overproduction of active CsrA is damaging to the cell. A suppressor mutation in the csrA gene was identified also by Kamp et al. (23) during their studies of a V. cholerae El Tor strain E7946 varS mutant. The mutation was mapped to the start codon of csrA and was predicted to decrease the amount of CsrA made. Kamp et al. noted that the varS csrA* suppressor strain survived nutrient-limiting conditions far better than the varS mutant alone, suggesting that high levels of active CsrA impaired growth in this strain as well. A high rate of suppressor mutations in response to misregulation of CsrA levels has not been reported in other bacterial species. In E. coli, mutants with mutations in the BarA/UvrY two-component system do not grow poorly except when glycolytic carbon sources are used (41), and no csrA suppressor mutations have been reported for these mutant strains. Not surprisingly, active CsrA levels are tightly controlled in E. coli and subject to multiple checkpoints and feedback loops (42, 43).
An important question that remains is how the NRES amino acids control the level of active CsrA. The specific sensing mechanism for the NRES mix is not known, and it is unclear whether the VarS/A system is responsible for relaying the NRES signal. The transcriptional profile of V. cholerae in response to NRES showed no changes in the level of the varS and csrA transcripts, while the level of the varA transcript was increased. An increase in VarA should lower the level of active CsrA, but instead, active CsrA levels were higher in the presence of NRES. The increase in varA transcript levels could be due to feedback regulation from CsrA, since CsrA has been shown to positively affect transcription of the varA homolog uvrY in E. coli (43). In E. coli, CsrB sRNA levels were greatly reduced when cells were grown in the presence of amino acids (44), which could lead to an increase in active CsrA levels. We are currently investigating whether this may also be the case in V. cholerae.
V. cholerae CsrA is a small, 65-amino-acid protein with 84% amino acid identity to E. coli CsrA (Fig. 4). The three-dimensional structure of CsrA (or its homologs RsmA/RsmE) has been solved in E. coli (45), Pseudomonas putida (46), Pseudomonas fluorescens (47), and Yersinia enterocolitica (48). Based on the high level of amino acid conservation between these proteins and V. cholerae CsrA, it is likely that V. cholerae CsrA is very similar in structure. In these structures, CsrA is a homodimeric barrel-shaped protein made up of intertwined β sheets from each of the CsrA subunits, with two wing-like α-helical extensions. Alanine-scanning mutagenesis identified two regions of CsrA that are in close proximity to each other and that are critical for both RNA binding and regulatory activity (49). These regions encompass the first (amino acid residues 2 to 7) and the fifth (amino acid residues 40 to 47) β strands of CsrA (Fig. 4). β strand 1 is the most highly conserved region among CsrA proteins from a diverse range of bacterial species, and several residues in β strand 5 are invariable as well (49) (Fig. 4). β strand 1 from one CsrA subunit interdigitates with β strand 5 from the other subunit to form one of the two positively charged, solvent-accessible faces of the dimer. These are the regions of the CsrA dimer believed to be primarily involved in binding RNA. A large proportion of the point mutants isolated in this study clustered within the first β strand or in the region immediately following, confirming the importance of the N-terminal domain for CsrA function (Fig. 4). Arg6 was one of the most commonly mutated residues in V. cholerae CsrA in response to misregulation of CsrA levels in the varA mutant. It was shown in Pseudomonas fluorescens that an R6A substitution in the CsrA homolog RsmE, which has been cocrystallized with its target mRNA, hcnA (47), was associated with a complete failure to repress translation of the hcnA transcript (50), pointing to the critical nature of this residue. It was also demonstrated by structural analyses of Y. enterocolitica RsmE that Arg6 forms an important salt bridge with Glu46 which helps to stabilize the α-helical extension from each face of the dimer (48). Not surprisingly, a truncation of the C terminus was associated with defects in CsrA function, likely due to effects on the α helix and the adjacent RNA-binding domain. It is important to reiterate that the point mutants, as well as the csrA::Tn5 insertion mutant, obtained in this study retained some vital functions of CsrA, since the strains did not exhibit the growth defects associated with either overproduction or the complete absence of active CsrA.
Because csrA is essential, it has been difficult to study the role of CsrA in virulence. We show here that mutation of a single amino acid residue of CsrA is sufficient to abolish its ability to mediate an increase in the level of a major virulence gene regulator, ToxR, in response to amino acids. The NcsrA.R6H point mutant exhibited no growth defects in vitro compared with its wild-type parental strain; however, the mutation had a profound effect on the ability of V. cholerae to colonize infant mice. ToxR is one of the most important regulators of virulence gene expression in this pathogen. ToxR controls expression of the major V. cholerae virulence factors (4, 5) and is essential for causing disease in both humans and mice (33, 34). This might suggest that the ability to regulate ToxR levels in response to certain nutrients could be important in vivo; however, it is important to recognize that CsrA is a global regulator with pleiotropic effects on carbon metabolism, quorum sensing, biofilm production, and other processes that could influence the ability of V. cholerae to cause disease in the mammalian host. Our studies cannot distinguish between the CsrA-mediated increase in ToxR levels and another function of CsrA needed for colonization and virulence; nevertheless, these studies have demonstrated a clear requirement for CsrA in V. cholerae pathogenesis separate from its essential viability functions, and we have uncovered a previously unknown relationship between CsrA and ToxR. The regulation of ToxR levels by CsrA in response to amino acids adds another dimension to the complex network of transcriptional, translational, and posttranslational mechanisms for ensuring maximal ToxR activity at the appropriate times within the diverse environments occupied by this important pathogen.
MATERIALS AND METHODS
Bacterial strains and plasmids, and growth conditions.
Bacterial strains and plasmids used in this study are listed in Table S3 in the supplemental material. All strains were maintained at −80°C in tryptic soy broth (TSB) plus 20% glycerol. Strains were routinely grown at 37°C in Luria-Bertani (LB) broth (1% tryptone, 0.5% yeast extract, 1% NaCl [wt/vol]) (51) or in T medium (52) modified to contain 0.2% wt/vol sucrose, 20 µM FeSO4, and a mixture of vitamins (recipe for 100× VA vitamin solution at http://www.genome.wisc.edu/resources/protocols/ezmedium.htm). Amino acids (Sigma) for the NRES supplement were dissolved in water and used at a final concentration of 12.5 mM total amino acids (3.125 mM each amino acid), unless otherwise indicated. Cholate was used at 0.1% (wt/vol). Antibiotics were used at the following concentrations for E. coli strains: 50 µg/ml ampicillin, 50 µg/ml kanamycin, and 30 µg/ml chloramphenicol. For V. cholerae strains, the concentrations used were 25 µg/ml ampicillin, 25 µg/ml kanamycin, 6 µg/ml chloramphenicol, 30 µg/ml gentamicin, and 20 µg/ml polymyxin B.
Plasmid clones were transferred to V. cholerae strains by electroporation or bacterial conjugation, as described previously (53).
PCR.
The oligonucleotide primers for PCR were purchased from Sigma-Aldrich (St. Louis, MO) and Invitrogen (Carlsbad, CA). Primers used for cloning are listed in Table S4 in the supplemental material. PCR was performed using KOD Hot Start DNA polymerase (Novagen, EMD Chemicals, San Diego, CA) to create fragments for cloning and Taq DNA polymerase (Qiagen, Valencia, CA) for all general verification purposes. PCRs were carried out according to the manufacturer’s instructions, using the primers described below. Unless otherwise indicated, the template for PCR was bacterial whole-cell suspensions in water. All clones derived from PCR products were verified by DNA sequencing.
Sequence analysis.
DNA sequencing was performed by the University of Texas, Institute for Cellular and Molecular Biology DNA Core Facility using the capillary-based 3730 DNA analyzer from Applied Biosystems (Foster City, CA). Analysis of DNA sequences was carried out using MacVector 12.0.6. BLAST searches and other bioinformatic analyses were done using the National Center for Biotechnology Information (NCBI) and the Comprehensive Microbial Resource (CMR) databases. Pairwise alignments were carried out using ClustalW from within MacVector 12.0.6.
Construction of plasmids, strains, and chromosomal mutations.
To create pVarA, the varA gene (VC1213) was amplified from V. cholerae strain C6706 by PCR using primers varA.F and varA.R. The resulting PCR product was cloned into pWKS30 digested with SmaI to yield pVarA. The V. cholerae csrA gene (VC0548) was cloned by PCR amplification of the csrA gene from strain N16961 using primers csrA3 and csrA4. The resulting fragment was cloned into the PmlI site of the single copy vector pCC1 to create pFcsrA, or into the SmaI site of pWKS30 to create pWCsrA.
To create strain N16961G (the wild-type strain, N16961, carrying a gentamicin cassette in the Tn7att site), we used the protocol described by McKenzie and Craig (54). pGRGgent1 (E. E. Wyckoff) was transferred to N16961 by bacterial conjugation, and transconjugants were selected at 30°C on polymyxin B and ampicillin for maintenance of the temperature-sensitive plasmid. Strains were grown at 42°C in order to facilitate loss of the plasmid and then screened on gentamicin and ampicillin to identify candidates that had lost the plasmid, but retained the gentamicin cassette. Genr Amps candidate N16961G strains were screened by PCR with primers IntF1 and IntR1 to verify insertion of the gentamicin cassette in the Tn7att site.
To create a chromosomal deletion of the varA gene, two fragments with a short overlap were generated by PCR using primer sets varA.F/VarA.Sma.1 and VarA.Sma.2/varA.R. The overlapping fragments were then used as the template to generate a splice overlap extension (SOE) PCR product using primers varA.F and varA.R. The final PCR fragment, ΔvarA::SmaI, was cloned into the SmaI site of pWKS30, and the resulting plasmid was digested with SmaI to insert a chloramphenicol resistance cassette, yielding pWKS30 carrying ΔvarA::cam. The ΔvarA::cam fragment was subcloned as an XbaI/EcoRV fragment into pHM5 digested with XbaI/EcoRV to yield the allelic exchange construct pAMS30. To create the V. cholerae csrA.R6H point mutation, the csrA.R6H allele was amplified from strain NvarAL-1 using KOD Hot Start polymerase and primers csrA3.Sal and csrA4.Bam. The PCR product was digested with SalI and BamHI and ligated into pHM5 digested with SalI and BglII to create allelic exchange construct pAMS31. To create the Tn5 insertion in csrA, fragments with a short overlap were generated using primers csrA3 and csrA2 to amplify one segment from V. cholerae strain dl2395 (which carries the csrA::Tn5 allele [generously provided by B. Bassler]), and primers csrA7 and csrA4 to amplify the other segment from the wild-type strain N16961. The overlapping segments were used as the template for SOE PCR with primers csrA3 and csrA4 to generate the complete csrA::Tn5 fragment with flanking sequences. The csrA::Tn5 fragment was cloned as a blunt fragment into pCVD442N digested with SmaI to create allelic exchange construct pAMS32. Allelic exchange constructs were transferred to V. cholerae strains via bacterial conjugation, and allelic exchange was carried out as described previously (55, 56).
SDS-PAGE and immunoblotting.
Cultures were grown overnight in LB medium and diluted 1:100 into fresh medium, as described above and in the figure legends. Cultures were grown to the mid-log phase (optical density at 650 nm [OD650] of ≈0.5), and samples containing an equivalent number of cells were resuspended in Laemmli solubilization buffer (57). Whole-cell extracts were resolved by SDS-PAGE (10%) and visualized by Coomassie brilliant blue staining or electroblotted for 1.5 h at 45 V onto Hybond ECL (enhanced chemiluminescence) nitrocellulose (Amersham Pharmacia Biotech, Little Chalfont, Buckinghamshire, England). The positions of OmpT and OmpU were determined through comparisons of the wild-type strain with ompT and ompU mutants and by immunodetection of OmpT and OmpU (58) (data not shown). Immunodetection of ToxR was carried out using rabbit polyclonal anti-ToxR antiserum (diluted 1:1,000) (generous gift of R. K. Taylor), and horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Bio-Rad Laboratories, Hercules, CA) secondary antibody (diluted 1:10,000). Equal loading of the samples for immunoblotting was confirmed by evaluating the corresponding Coomassie-stained gel.
RNA isolation and microarray analysis.
Strains were grown to mid-log phase (OD650 of ≈0.5) in T medium supplemented with 0.2% sucrose, 20 µM FeSO4, and a mix of vitamins, as described above. The cultures were then divided, and amino acid solutions were added to a final concentration of 50 mM total amino acids. Cultures were grown for an additional 15 min and then treated with an RNase-free solution of 95% absolute ethanol–5% phenol (pH 4.5), used at 20% vol/vol, and kept on ice. RNA was isolated from 109 cells per sample using the RNeasy minikit (Qiagen) as per the manufacturer’s instructions for isolation of total RNA from bacterial cells. Following purification, each RNA sample was treated with DNase I (Invitrogen) as per the manufacturer’s protocol. The RNA samples were ethanol precipitated, dried, resuspended in RNase-free water, and quantified using an ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). For microarray analysis, approximately 20 µg RNA per sample was used to generate cDNA for hybridization to V. cholerae NimbleChip_X4 microarray slides (NimbleGen Systems, Roche Diagnostics Operations, Inc., Indianapolis, IN). The microarray slides were processed by the Genomic Sequencing and Analysis Facility at the University of Texas, Austin, according to the NimbleGen system’s protocols. The slides were scanned with GenePix 4000B Scanner (Axon Instruments, Molecular Devices, Sunnyvale, CA) and normalized using the GenePix software.
In vivo competition assays.
Animal experiments were performed according to protocols approved by the University of Texas Institutional Animal Care and Use Committee. In vivo competition assays were performed using 5-day-old BALB/c mice in a modified version of the protocol described by Taylor et al. (33). The two competing strains were the wild-type strain N16961G, containing an insertion of a gentamicin resistance cassette in the Tn7att site, and the NCsrA.R6H point mutant strain. We chose to mark the wild-type strain rather than the CsrA point mutant strain in order to avoid any adverse effects of the gentamicin cassette on the fitness of the mutant strain, which might skew the results. The infant mice were inoculated intragastrically with 50 µl saline containing 0.02% Evan’s blue dye and approximately 105 CFU of each competing strain grown to the mid-log phase in LB medium. The inoculation mixture was also diluted and plated, first on medium containing an antibiotic permissive for all V. cholerae El Tor stains (polymyxin B) and then on selective medium by patching in order to assess the input ratio of mutant to wild-type bacteria in the inoculum. The mice were sacrificed after 18 h, and the intestines were removed. The intestines of all the animals were visibly distended, consistent with significant fluid accumulation due to V. cholerae infection. The intestines were homogenized in 10 ml of sterile saline, and serial dilutions were plated on medium allowing recovery of both strains. Replica plating on differential medium by patching was then performed to determine the viable counts for each competing strain. The output ratios were normalized to the input ratios to determine the competitive index (CI): CI = (mutant output/wild-type output)/(mutant input/wild-type input). Differential medium patching was performed on up to 230 recovered colonies from each mouse, setting the limits of detection for the competitive index at approximately 0.005. Examples of both gentamicin-sensitive and -resistant colonies recovered from infection were analyzed by sequencing to verify the presence or absence, respectively, of the point mutation in the csrA gene.
SUPPLEMENTAL MATERIAL
The wild-type strain N16961 (WT) and strain NvarAL-1 (ΔvarA::cam; csrA.R6H) were grown overnight in LB broth and then subcultured 1:100 into fresh LB broth and grown to stationary phase. The experiment was performed at least three times, but only one representative experiment is shown. Download
Expression of wild-type csrA in the wild-type (VarA+) background does not disrupt the normal OMP expression pattern in response to the NRES mix. The wild-type parental strain N16961 was transformed with either pWCsrA or pFCsrA, and the resulting strains were assayed for their OMP profile. Strains N16961 (WT), N16961/pWKS30 (vector control strain), N16961/pWCsrA, N16961/pCC1 (vector control strain), and N16961/pFCsrA were grown overnight in LB broth and then subcultured 1:100 into T medium with or without 12.5 mM NRES mix. Cells were grown to the mid-log phase, and whole-cell preparations were resolved by SDS-PAGE (10%) and stained with Coomassie blue. Download
A Tn5 insertion at the 3′ end of the csrA gene abolishes the ToxR and OMP response to NRES. (A) The wild-type strain N16961 (WT) and the csrA::Tn5 derivative were grown overnight in LB broth and then subcultured 1:100 into T medium with or without 12.5 mM NRES mix. Cells were harvested in the mid-log phase, and whole-cell preparations were resolved by SDS-PAGE (10%) and stained by Coomassie blue. (B) The whole-cell preparations from panel A were resolved by SDS-PAGE and immunoblotted using polyclonal anti-ToxR antisera. Download
Response of strain C6706 to NRES. C6706 was grown overnight in LB broth and then subcultured 1:100 into T medium with or without 12.5 mM NRES mix. Cells were harvested in the mid-log phase, and whole-cell preparations were resolved by SDS-PAGE (10%) and stained by Coomassie blue (top panel) or immunoblotted using polyclonal anti-ToxR antisera (bottom panel). Download
Microarray analysis of gene expression by V. cholerae grown in T medium, T medium plus NRES, and T medium plus Asp. Shown are the V. cholerae genes for which the transcript level was altered more than 2-fold in response to NRES.
Effect of mutations on the OMP response to NRES.
Bacterial strains and plasmids used in this study.
PCR primers used in this study.
ACKNOWLEDGMENTS
We gratefully acknowledge Ron Taylor for the ToxR antiserum and Bonnie Bassler, Andy Camilli, Tony Romeo, and Bryan Davies for helpful discussions and for generously sharing strains and plasmids. We thank Scott Hunicke-Smith for expertise with microarray analysis and Martha Thomas for technical assistance. We also thank Mary Lozano and Eric Peng for invaluable assistance with the mouse experiments. Finally, we are greatly indebted to Liz Wyckoff for many insightful discussions and critical reading of the manuscript.
This work was supported by grants AI091957 and AI050669 from the National Institutes of Health.
Footnotes
Citation Mey AR, Butz HA, Payne SM. 2015. Vibrio cholerae CsrA regulates ToxR levels in response to amino acids and is essential for virulence. mBio 6(4):e01064-15. doi:10.1128/mBio.01064-15.
REFERENCES
- 1.Miller VL, Taylor RK, Mekalanos JJ. 1987. Cholera toxin transcriptional activator ToxR is a transmembrane DNA binding protein. Cell 48:271–279. doi: 10.1016/0092-8674(87)90430-2. [DOI] [PubMed] [Google Scholar]
- 2.Ottemann KM, DiRita VJ, Mekalanos JJ. 1992. ToxR proteins with substitutions in residues conserved with OmpR fail to activate transcription from the cholera toxin promoter. J Bacteriol 174:6807–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DiRita VJ, Mekalanos JJ. 1991. Periplasmic interaction between two membrane regulatory proteins, ToxR and ToxS, results in signal transduction and transcriptional activation. Cell 64:29–37. doi: 10.1016/0092-8674(91)90206-E. [DOI] [PubMed] [Google Scholar]
- 4.Cotter PA, DiRita VJ. 2000. Bacterial virulence gene regulation: an evolutionary perspective. Annu Rev Microbiol 54:519–565. doi: 10.1146/annurev.micro.54.1.519. [DOI] [PubMed] [Google Scholar]
- 5.Matson JS, Withey JH, DiRita VJ. 2007. Regulatory networks controlling Vibrio cholerae virulence gene expression. Infect Immun 75:5542–5549. doi: 10.1128/IAI.01094-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krukonis ES, Yu RR, Dirita VJ. 2000. The Vibrio cholerae ToxR/TcpP/ToxT virulence cascade: distinct roles for two membrane-localized transcriptional activators on a single promoter. Mol Microbiol 38:67–84. doi: 10.1046/j.1365-2958.2000.02111.x. [DOI] [PubMed] [Google Scholar]
- 7.Hung DT, Mekalanos JJ. 2005. Bile acids induce cholera toxin expression in Vibrio cholerae in a ToxT-independent manner. Proc Natl Acad Sci U S A 102:3028–3033. doi: 10.1073/pnas.0409559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mey AR, Craig SA, Payne SM. 2012. Effects of amino acid supplementation on porin expression and ToxR levels in Vibrio cholerae. Infect Immun 80:518–528. doi: 10.1128/IAI.05851-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Provenzano D, Schuhmacher DA, Barker JL, Klose KE. 2000. The virulence regulatory protein ToxR mediates enhanced bile resistance in Vibrio cholerae and other pathogenic Vibrio species. Infect Immun 68:1491–1497. doi: 10.1128/IAI.68.3.1491-1497.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller VL, Mekalanos JJ. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol 170:2575–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crawford JA, Kaper JB, DiRita VJ. 1998. Analysis of ToxR-dependent transcription activation of ompU, the gene encoding a major envelope protein in Vibrio cholerae. Mol Microbiol 29:235–246. doi: 10.1046/j.1365-2958.1998.00925.x. [DOI] [PubMed] [Google Scholar]
- 12.Li CC, Crawford JA, DiRita VJ, Kaper JB. 2000. Molecular cloning and transcriptional regulation of ompT, a ToxR-repressed gene in Vibrio cholerae. Mol Microbiol 35:189–203. doi: 10.1046/j.1365-2958.2000.01699.x. [DOI] [PubMed] [Google Scholar]
- 13.Duret G, Delcour AH. 2010. Size and dynamics of the Vibrio cholerae porins OmpU and OmpT probed by polymer exclusion. Biophys J 98:1820–1829. doi: 10.1016/j.bpj.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Provenzano D, Klose KE. 2000. Altered expression of the ToxR-regulated porins OmpU and OmpT diminishes Vibrio cholerae bile resistance, virulence factor expression, and intestinal colonization. Proc Natl Acad Sci U S A 97:10220–10224. doi: 10.1073/pnas.170219997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mathur J, Waldor MK. 2004. The Vibrio cholerae ToxR-regulated porin OmpU confers resistance to antimicrobial peptides. Infect Immun 72:3577–3583. doi: 10.1128/IAI.72.6.3577-3583.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sperandio V, Girón JA, Silveira WD, Kaper JB. 1995. The OmpU outer membrane protein, a potential adherence factor of Vibrio cholerae. Infect Immun 63:4433–4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LaRocque RC, Krastins B, Harris JB, Lebrun LM, Parker KC, Chase M, Ryan ET, Qadri F, Sarracino D, Calderwood SB. 2008. Proteomic analysis of Vibrio cholerae in human stool. Infect Immun 76:4145–4151. doi: 10.1128/IAI.00585-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DiRita VJ, Neely M, Taylor RK, Bruss PM. 1996. Differential expression of the ToxR regulon in classical and E1 Tor biotypes of Vibrio cholerae is due to biotype-specific control over toxT expression. Proc Natl Acad Sci U S A 93:7991–7995. doi: 10.1073/pnas.93.15.7991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanjilal S, Citorik R, LaRocque RC, Ramoni MF, Calderwood SB. 2010. A systems biology approach to modeling Vibrio cholerae gene expression under virulence-inducing conditions. J Bacteriol 192:4300–4310. doi: 10.1128/JB.00182-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Medrano AI, DiRita VJ, Castillo G, Sanchez J. 1999. Transient transcriptional activation of the Vibrio cholerae El Tor virulence regulator toxT in response to culture conditions. Infect Immun 67:2178–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Timmermans J, Van Melderen L. 2010. Post-transcriptional global regulation by CsrA in bacteria. Cell Mol Life Sci 67:2897–2908. doi: 10.1007/s00018-010-0381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romeo T, Vakulskas CA, Babitzke P. 2013. Post-transcriptional regulation on a global scale: form and function of Csr/Rsm systems. Environ Microbiol 15:313–324. doi: 10.1111/j.1462-2920.2012.02794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamp HD, Patimalla-Dipali B, Lazinski DW, Wallace-Gadsden F, Camilli A. 2013. Gene fitness landscapes of Vibrio cholerae at important stages of its life cycle. PLoS Pathog 9:e1003800. doi: 10.1371/journal.ppat.1003800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jang J, Jung KT, Yoo CK, Rhie GE. 2010. Regulation of hemagglutinin/protease expression by the VarS/VarA-CsrA/B/C/D system in Vibrio cholerae. Microb Pathog 48:245–250. doi: 10.1016/j.micpath.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 25.Lenz DH, Miller MB, Zhu J, Kulkarni RV, Bassler BL. 2005. CsrA and three redundant small RNAs regulate quorum sensing in Vibrio cholerae. Mol Microbiol 58:1186–1202. doi: 10.1111/j.1365-2958.2005.04902.x. [DOI] [PubMed] [Google Scholar]
- 26.Jang J, Jung KT, Park J, Yoo CK, Rhie GE. 2011. The Vibrio cholerae Vars/VarA two-component system controls the expression of virulence proteins through ToxT regulation. Microbiology 157:1466–1473. doi: 10.1099/mic.0.043737-0. [DOI] [PubMed] [Google Scholar]
- 27.Wong SM, Carroll PA, Rahme LG, Ausubel FM, Calderwood SB. 1998. Modulation of expression of the ToxR regulon in Vibrio cholerae by a member of the two-component family of response regulators. Infect Immun 66:5854–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu MY, Gui G, Wei B, Preston JF III, Oakford L, Yüksel U, Giedroc DP, Romeo T. 1997. The RNA molecule CsrB binds to the global regulatory protein CsrA and antagonizes its activity in Escherichia coli. J Biol Chem 272:17502–17510. doi: 10.1074/jbc.272.28.17502. [DOI] [PubMed] [Google Scholar]
- 29.Romeo T, Gong M, Liu MY, Brun-Zinkernagel AM. 1993. Identification and molecular characterization of csrA, a pleiotropic gene from Escherichia coli that affects glycogen biosynthesis, gluconeogenesis, cell size, and surface properties. J Bacteriol 175:4744–4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Timmermans J, Van Melderen L. 2009. Conditional essentiality of the csrA gene in Escherichia coli. J Bacteriol 191:1722–1724. doi: 10.1128/JB.01573-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bardill JP, Zhao X, Hammer BK. 2011. The Vibrio cholerae quorum sensing response is mediated by Hfq-dependent sRNA/mRNA base pairing interactions. Mol Microbiol 80:1381–1394. doi: 10.1111/j.1365-2958.2011.07655.x. [DOI] [PubMed] [Google Scholar]
- 32.Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, Gill SR, Nelson KE, Read TD, Tettelin H, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonald L, Utterback T, Fleishmann RD, Nierman WC, White O, Salzberg SL, Smith HO, Colwell RR, Mekalanos JJ, Venter JC, Fraser CM. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406:477–483. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. 1987. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci U S A 84:2833–2837. doi: 10.1073/pnas.84.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herrington DA, Hall RH, Losonsky G, Mekalanos JJ, Taylor RK, Levine MM. 1988. Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med 168:1487–1492. doi: 10.1084/jem.168.4.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ritchie JM, Waldor MK. 2009. Vibrio cholerae interactions with the gastrointestinal tract: lessons from animal studies. Curr Top Microbiol Immunol 337:37–59. doi: 10.1007/978-3-642-01846-6_2. [DOI] [PubMed] [Google Scholar]
- 36.Vakulskas CA, Potts AH, Babitzke P, Ahmer BM, Romeo T. 2015. Regulation of bacterial virulence by Csr (Rsm) systems. Microbiol Mol Biol Rev 79:193–224. doi: 10.1128/MMBR.00052-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsou AM, Liu Z, Cai T, Zhu J. 2011. The VarS/VarA two-component system modulates the activity of the Vibrio cholerae quorum-sensing transcriptional regulator HapR. Microbiology 157:1620–1628. doi: 10.1099/mic.0.046235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, Mekalanos JJ. 2002. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci U S A 99:3129–3134. doi: 10.1073/pnas.052694299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pfau JD, Taylor RK. 1998. Mutations in toxR and toxS that separate transcriptional activation from DNA binding at the cholera toxin gene promoter. J Bacteriol 180:4724–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bourassa L, Camilli A. 2009. Glycogen contributes to the environmental persistence and transmission of Vibrio cholerae. Mol Microbiol 72:124–138. doi: 10.1111/j.1365-2958.2009.06629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pernestig AK, Georgellis D, Romeo T, Suzuki K, Tomenius H, Normark S, Melefors O. 2003. The Escherichia coli BarA-UvrY two-component system is needed for efficient switching between glycolytic and gluconeogenic carbon sources. J Bacteriol 185:843–853. doi: 10.1128/JB.185.3.843-853.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suzuki K, Wang X, Weilbacher T, Pernestig AK, Melefors O, Georgellis D, Babitzke P, Romeo T. 2002. Regulatory circuitry of the CsrA/CsrB and BarA/UvrY systems of Escherichia coli. J Bacteriol 184:5130–5140. doi: 10.1128/JB.184.18.5130-5140.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Camacho MI, Alvarez AF, Chavez RG, Romeo T, Merino E, Georgellis D. 2015. Effects of the global regulator CsrA on the BarA/UvrY two-component signaling system. J Bacteriol 197:983–991. doi: 10.1128/JB.02325-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jonas K, Melefors O. 2009. The Escherichia coli CsrB and CsrC small RNAs are strongly induced during growth in nutrient-poor medium. FEMS Microbiol Lett 297:80–86. doi: 10.1111/j.1574-6968.2009.01661.x. [DOI] [PubMed] [Google Scholar]
- 45.Gutiérrez P, Li Y, Osborne MJ, Pomerantseva E, Liu Q, Gehring K. 2005. Solution structure of the carbon storage regulator protein CsrA from Escherichia coli. J Bacteriol 187:3496–3501. doi: 10.1128/JB.187.10.3496-3501.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rife C, Schwarzenbacher R, McMullan D, Abdubek P, Ambing E, Axelrod H, Biorac T, Canaves JM, Chiu HJ, Deacon AM, DiDonato M, Elsliger MA, Godzik A, Grittini C, Grzechnik SK, Hale J, Hampton E, Han GW, Haugen J, Hornsby M, Jaroszewski L, Klock HE, Koesema E, Kreusch A, Kuhn P, Lesley SA, Miller MD, Moy K, Nigoghossian E, Paulsen J, Quijano K, Reyes R, Sims E, Spraggon G, Stevens RC, van den Bedem H, Velasquez J, Vincent J, White A, Wolf G, Xu Q, Hodgson KO, Wooley J, Wilson IA. 2005. Crystal structure of the global regulatory protein CsrA from Pseudomonas putida at 2.05-A resolution reveals a new fold. Proteins 61:449–453. doi: 10.1002/prot.20502. [DOI] [PubMed] [Google Scholar]
- 47.Schubert M, Lapouge K, Duss O, Oberstrass FC, Jelesarov I, Haas D, Allain FH. 2007. Molecular basis of messenger RNA recognition by the specific bacterial repressing clamp RsmA/CsrA. Nat Struct Mol Biol 14:807–813. doi: 10.1038/nsmb1285. [DOI] [PubMed] [Google Scholar]
- 48.Heeb S, Kuehne SA, Bycroft M, Crivii S, Allen MD, Haas D, Cámara M, Williams P. 2006. Functional analysis of the post-transcriptional regulator RsmA reveals a novel RNA-binding site. J Mol Biol 355:1026–1036. doi: 10.1016/j.jmb.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 49.Mercante J, Suzuki K, Cheng X, Babitzke P, Romeo T. 2006. Comprehensive alanine-scanning mutagenesis of Escherichia coli CsrA defines two subdomains of critical functional importance. J Biol Chem 281:31832–31842. doi: 10.1074/jbc.M606057200. [DOI] [PubMed] [Google Scholar]
- 50.Lapouge K, Schubert M, Allain FH, Haas D. 2008. Gac/Rsm signal transduction pathway of gamma-proteobacteria: from RNA recognition to regulation of social behaviour. Mol Microbiol 67:241–253. doi: 10.1111/j.1365-2958.2007.06042.x. [DOI] [PubMed] [Google Scholar]
- 51.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 52.Simon EH, Tessman I. 1963. Thymidine-requiring mutants of phage T4. Proc Natl Acad Sci U S A 50:526–532. doi: 10.1073/pnas.50.3.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Occhino DA, Wyckoff EE, Henderson DP, Wrona TJ, Payne SM. 1998. Vibrio cholerae iron transport: haem transport genes are linked to one of two sets of tonB, exbB, exbD genes. Mol Microbiol 29:1493–1507. doi: 10.1046/j.1365-2958.1998.01034.x. [DOI] [PubMed] [Google Scholar]
- 54.McKenzie GJ, Craig NL. 2006. Fast, easy and efficient: site-specific insertion of transgenes into enterobacterial chromosomes using Tn7 without need for selection of the insertion event. BMC Microbiol 6:39. doi: 10.1186/1471-2180-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mey AR, Payne SM. 2001. Haem utilization in Vibrio cholerae involves multiple TonB-dependent haem receptors. Mol Microbiol 42:835–849. doi: 10.1046/j.1365-2958.2001.02683.x. [DOI] [PubMed] [Google Scholar]
- 56.Mey AR, Craig SA, Payne SM. 2005. Characterization of Vibrio cholerae RyhB: the RyhB regulon and role of ryhB in biofilm formation. Infect Immun 73:5706–5719. doi: 10.1128/IAI.73.9.5706-5719.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 58.Craig SA, Carpenter CD, Mey AR, Wyckoff EE, Payne SM. 2011. Positive regulation of the Vibrio cholerae porin OmpT by iron and Fur. J Bacteriol 193:6505–6511. doi: 10.1128/JB.05681-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The wild-type strain N16961 (WT) and strain NvarAL-1 (ΔvarA::cam; csrA.R6H) were grown overnight in LB broth and then subcultured 1:100 into fresh LB broth and grown to stationary phase. The experiment was performed at least three times, but only one representative experiment is shown. Download
Expression of wild-type csrA in the wild-type (VarA+) background does not disrupt the normal OMP expression pattern in response to the NRES mix. The wild-type parental strain N16961 was transformed with either pWCsrA or pFCsrA, and the resulting strains were assayed for their OMP profile. Strains N16961 (WT), N16961/pWKS30 (vector control strain), N16961/pWCsrA, N16961/pCC1 (vector control strain), and N16961/pFCsrA were grown overnight in LB broth and then subcultured 1:100 into T medium with or without 12.5 mM NRES mix. Cells were grown to the mid-log phase, and whole-cell preparations were resolved by SDS-PAGE (10%) and stained with Coomassie blue. Download
A Tn5 insertion at the 3′ end of the csrA gene abolishes the ToxR and OMP response to NRES. (A) The wild-type strain N16961 (WT) and the csrA::Tn5 derivative were grown overnight in LB broth and then subcultured 1:100 into T medium with or without 12.5 mM NRES mix. Cells were harvested in the mid-log phase, and whole-cell preparations were resolved by SDS-PAGE (10%) and stained by Coomassie blue. (B) The whole-cell preparations from panel A were resolved by SDS-PAGE and immunoblotted using polyclonal anti-ToxR antisera. Download
Response of strain C6706 to NRES. C6706 was grown overnight in LB broth and then subcultured 1:100 into T medium with or without 12.5 mM NRES mix. Cells were harvested in the mid-log phase, and whole-cell preparations were resolved by SDS-PAGE (10%) and stained by Coomassie blue (top panel) or immunoblotted using polyclonal anti-ToxR antisera (bottom panel). Download
Microarray analysis of gene expression by V. cholerae grown in T medium, T medium plus NRES, and T medium plus Asp. Shown are the V. cholerae genes for which the transcript level was altered more than 2-fold in response to NRES.
Effect of mutations on the OMP response to NRES.
Bacterial strains and plasmids used in this study.
PCR primers used in this study.