

Abstract
Background
Avocado (Persea americana) is an economically important tropical fruit considered to be a good source of fatty acids. Despite its importance, the molecular and cellular characterization of biochemical and developmental processes in avocado is limited due to the lack of transcriptome and genomic information.
Results
The transcriptomes of seeds, roots, stems, leaves, aerial buds and flowers were determined using different sequencing platforms. Additionally, the transcriptomes of three different stages of fruit ripening (pre-climacteric, climacteric and post-climacteric) were also analyzed. The analysis of the RNAseqatlas presented here reveals strong differences in gene expression patterns between different organs, especially between root and flower, but also reveals similarities among the gene expression patterns in other organs, such as stem, leaves and aerial buds (vegetative organs) or seed and fruit (storage organs). Important regulators, functional categories, and differentially expressed genes involved in avocado fruit ripening were identified. Additionally, to demonstrate the utility of the avocado gene expression atlas, we investigated the expression patterns of genes implicated in fatty acid metabolism and fruit ripening.
Conclusions
A description of transcriptomic changes occurring during fruit ripening was obtained in Mexican avocado, contributing to a dynamic view of the expression patterns of genes involved in fatty acid biosynthesis and the fruit ripening process.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1775-y) contains supplementary material, which is available to authorized users.
Background
Avocado (Persea americana Mill.) is a crop plant with oleaginous fruits belonging to the Magnoliidae clade, a basal lineage of flowering plants. It is a member of Lauraceae, a large family of about 50 genera and approximately 2500–3000 species, mostly trees [1, 2]. Despite its recent introduction to international commerce, the avocado is no longer just an exotic fruit; it has been rapidly incorporated as a key component in the diet of many countries [3]. Although Mexico is the world’s largest producer and consumer of avocados (about 28 % of total world production), there are at least ten other countries with annual production of over 100,000 t of avocado fruit (FAOSTAT, 2011;http://faostat.fao.org/DesktopDefault.aspx?PageID=339&lang=es).
P. americana comprises no fewer than three well-recognized varieties, geographical ecotypes, or botanical races, also known as horticultural races. The Mexican race, P. americana var. drymifolia (Mexican avocado), is adapted to the tropical highlands and constitutes the most commonly used rootstock in Mexican orchards; P. americana var. guatemalensis (L.O. Williams), the Guatemalan race, which grows preferentially at medium elevations in the tropics, and the West-Indian race, P. americana var. americana, which is typically cultivated in the lowland humid tropics [2]. Commercial avocado production is based on grafting cultivars onto rootstocks of Mexican and Guatemalan races; the cultivars grown in subtropical climates are selections from these races or hybrids of them, with the Guatemalan genotypes being the dominant horticultural race among subtropical avocado cultivars [4]. However, the Mexican genetic background contributes to avocado diversity with a plethora of desirable, selected characteristics in commercial varieties, such as cold tolerance, smaller tree size, high oil content, early maturity, and smooth fruit skin. It is imperative to identify key genes and the signaling pathways associated with these traits, as well as to study the allelic diversity present among botanical races for these desirable traits.
The avocado fruit accumulates oil instead of sugar unlike most fruits, probably as a consequence of co-evolutionary processes developed with ancient neotropical megafauna that became extinct about 30,000–11,000 years ago [5]. Avocado has been described as the most nutritious of all fruits [3], as the mature fruit flesh of avocado contains about 20 % beneficial fatty acids, 6 % carbohydrates, 2 % protein, and vitamin precursors and antioxidants such as carotenoids and vitamins E, C, B2, B12, B1, K and D [6]. The mesocarp of the Mexican race avocado possesses up to 25–30 % oil content, of which nearly 90 % is mono-unsaturated oleic, palmitic and linoleic fatty acids. Although avocado is a strongly climacteric fruit, its ripening or softening process does not take place during maturation on the tree, but instead it starts several days after the fruit has been picked. In the avocado fruit oil content increases in the mesocarp a few weeks after the fruit sets, and healthy fruits on trees continue to grow and accumulate oil for several months after maturation [7]. Once an avocado fruit has been detached from the tree, an ethylene-dependent ripening process is triggered, orchestrating flesh softening, skin color change and lipid biosynthesis. Upon ripening completion, concentrations of unsaturated fatty acids increase and those of saturated fatty acids decrease [8].
The production of essential oils in avocado vegetative organs is influenced by environmental and developmental conditions. However, the distinctive characteristics of the avocado fruit, including secondary metabolite production, are genotype-dependent. For example, the chemical composition of the leaves of the Mexican race is distinctive in its anise scent, which is absent from the two other horticultural races. Estraole, which represents 60 % of the total essential oils in leaves of the Mexican race, is responsible for this trait [9, 10]. Essential oils contain a variety of volatile molecules such as terpenes and terpenoids, phenol-derived aromatic components, and aliphatic compounds; these are widely used for various pharmaceutical, sanitary, cosmetic, agricultural and food purposes, and in nature, they function as herbivore repellants and pollinator attractants [10, 11]. Understanding the biosynthetic pathways of such bioactive compounds by associating functional genomics information with the enzymes involved in the metabolic pathways is fundamental for their commercial production.
Because to date there are only limited avocado genomic resources, carrying out comprehensive gene expression profiling is a challenge requiring large-scale analysis of transcriptomic data. Breakthroughs in next generation sequencing technology and data analysis during the last 10 years have made it possible to generate reference transcriptomes in the absence of a reference genome at a relatively low cost [12]. Reference transcriptomes can be used to perform comparative expression profiling by methods such as digital gene expression profiling [13]. Here, we present the de novo assembly of the Mexican avocado (Persea americana var. drymifolia) transcriptome, based on hybrid sequencing datasets derived from GS-FLX+ Roche and MiSeq Illumina platforms. Additionally, using a high-throughput sequencing platform, we develop a gene expression atlas of the avocado transcriptome in which a total of six different avocado organs and three fruit ripening stages (pre-climacteric, climacteric and post-climacteric) were included. To confirm the utility of the avocado transcriptome atlas, we specifically analyzed the expression of genes involved in acyl-lipid metabolism, ripening processes, and organ-specificity. Our approach generated over 67,000 unigenes with high quality annotations, providing an unprecedented coverage of the avocado transcriptome. The availability of the avocado gene expression atlas should facilitate additional studies on the basic biology of avocado, while also supporting applied research to improve this increasingly important crop.
Results and discussion
Sequencing and assembly of the Mexican race avocado transcriptome
In order to obtain sequences for as many avocado (Persea americana var. drymifolia) genes as possible, a cDNA library from an RNA pool isolated from seeds, roots, stems, leaves, aerial buds, flowers and pre-climacteric, climacteric and post-climacteric fruits was generated and sequenced using the GS-FLX+ (Roche) and MiSeq (Illumina) sequencers. MiSeq produced paired-end reads of length 250 bases while GS-FLX+ sequencer generated less reads (unpaired) but the average length was ~3× longer. It is well known that a mixed platform approach (hybrid assembly) can improve the number of full length genes through the inclusion of longer reads, while a higher paired-read coverage increases the detection of low abundance transcripts [14]. Pre-climacteric, climacteric and post-climacteric fruit stages were determined according to their ethylene production (see Methods). It should be noted that unlike other commercial varieties such as ‘Fuerte’, which show the maximum ethylene production seven days after harvest [15], the climacteric physiological stage (which marks the end of fruit maturation and the beginning of fruit senescence) is detected 3 day after harvest in Mexican avocado (Fig. 1).
Fig. 1.
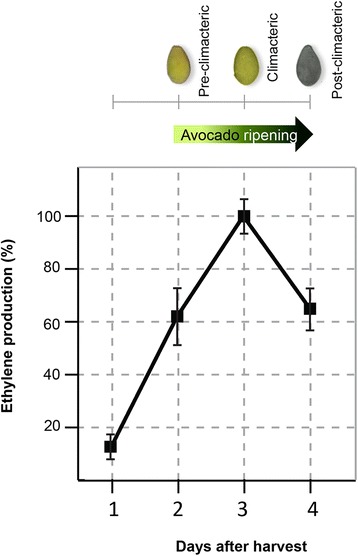
Ethylene production rises during avocado ripening. Ethylene was measured by gas chromatography during the subsequent 4 days after harvest. Maximum ethylene production was adjusted to be 100 %. Three independent biological replicates were analyzed
Reads generated by GS-FLX+ were masked using the SeqClean software pipeline to eliminate sequence regions that would cause incorrect assembly while the MiSeq read pairs (2 × 150 bp) were trimmed and/or merged together using the SeqPrep pipeline (see Methods for more details). To carry out the assembly process, 4,530,278 high quality reads (931,834 generated by GS-FLX+ and 3,598,444 paired-reads generated by MiSeq) were considered (Additional file 1: Table S1).
Many assemblers have been developed to assemble reads generated by Next Generation Sequencing platforms (NGS). The overlap layout algorithm is able to handle the longer reads of GS-FLX+, and programs such as MIRA [16] incorporate it. Trinity [12] in contrast is a de-Bruijn graph-based assembler developed for short reads. We compared the performance of the MIRA v3.4.1 and Trinity assemblers, both previously used in several analyses of plant transcriptomes [17, 18]. For each assembler, we used the default parameters recommended for transcriptome assembly. Standard metrics describing the assembly process, such as number of contigs ≥1 Kb, average contig lengths and maximum contig size were used to compare the assembly programs (Additional file 1: Table S2). Considering that many contigs representative of unique genes are often produced in de novo assemblies due to the presence of variant alleles, sequencing errors, and alternative splicing of transcripts, the resulting contigs (derived from both assemblies) were filtered to eliminate redundant sequences and then passed through a second assembly step using the CAP3 assembler [19] (see Methods for more details). A unigene set (83,650 sequences) from P. americana was generated including resulting contigs (25,665) and “singlets” (57,985; 64.2 % from MIRA and 33.8 % from Trinity) derived from the CAP3 run with a minimum size of 200 bp (Additional file 2). It should be noted that “singlets” are the contigs generated from the first steps of MIRA or Trinity assemblies that were not reassembled by CAP3. The average length of unigenes was 816.21 bp (ranging from 0.2 to 8.6 kb) (Additional file 1: Table S2). Considering the mean size of coding sequences (≈942.16 bp) in Amborella trichopoda, a basal angiosperm species [20, 21], it was expected that a large percentage of these avocado unigene transcripts may represent full-length cDNAs. A comparison of P. americana unigenes against the unpublished ca. 800 Mbp draft genome of P. americana var. drymifolia (unpublished data) using BLASTN (e-value 10−3) shows that 94.65 % of the transcripts had a significant hit against the genome (98 % of alignment length and minimal sequence identity of 90 % over the complete alignment).
To annotate the avocado transcriptome, we performed BLASTX alignments (e-value of ≤10−03 and a bit score ≥25) between the unigene set and several protein databases, including Arabidopsis thaliana, Amborella trichopoda and plant proteins available in the Reference Sequences (RefSeq) collection of NCBI. We found that 67,709 (80.94 %) unigenes of P. americana show high identity to at least one plant protein; the remaining (15,941 unigenes) had no function assigned (Additional file 1: Table S3). In a total of 14,845 avocado unigenes, an individual high-scoring segment pair (HSP) produced by BLASTX covered at least 80 % of the target protein. Results indicated that 34,218 distinct plant proteins could be identified among the 63,459 unigenes that showed significant similarities against RefSeq database (Additional file 1: Table S3). We further compared P. americana unigenes against the Pfam (Protein families) domain database (Additional file 1: Table S3; see Methods for more details) [22].
Functional annotation
The results of BLASTX searches against the Arabidopsis thaliana protein database were used for gene ontology (GO) mapping and annotation. Based on the Arabidopsis top hits, we obtained the GO annotations for the avocado unigenes, and WEGO software [23] was used to perform GO functional classification into the three major classes (Fig. 2; Additional file 1: Table S3). Among the unigenes with Arabidopsis hits, 63,430 (75.82 %) were assigned to gene ontology classes with 547,032 functional terms. Biological processes comprised the majority of the functional terms (259,327; 47.40 %), followed by cellular component (151,379; 27.67 %) and molecular functions (136,326; 24.92 %). Within the biological processes category, cellular (39,365 unigenes) and metabolic (37,208 unigenes) processes were prominently represented. To further predict the metabolic pathway in P. americana, the assembled unigenes were annotated with corresponding enzyme commission (EC) numbers in the KEGG automatic annotation server (KAAS; [24]) using Arabidopsis thaliana and Oryza sativa as references (Additional file 1: Table S3). A total of 2559 unigenes were mapped to 202 pathways corresponding to five KEGG modules: energy metabolism, carbohydrate and lipid metabolism, nucleotide and amino acid metabolism, genetic information processing, and environmental information processing. Additionally, the modules energy metabolism (structural complex) and metabolism (functional set) were also identified (Additional file 3: Figure S1 and Additional file 4: Table S4). Ribosome had the largest number of unigenes (78 members, M00177), followed by glycolysis (Embden-Meyerhof pathway; 62 members, M00001), reductive pentose phosphate cycle (Calvin cycle; 47 members, M00165), gluconeogenesis (40 members, M00003), and spliceosome (30 members, M00354) (Fig. 3; Additional file 4: Table S4).
Fig. 2.
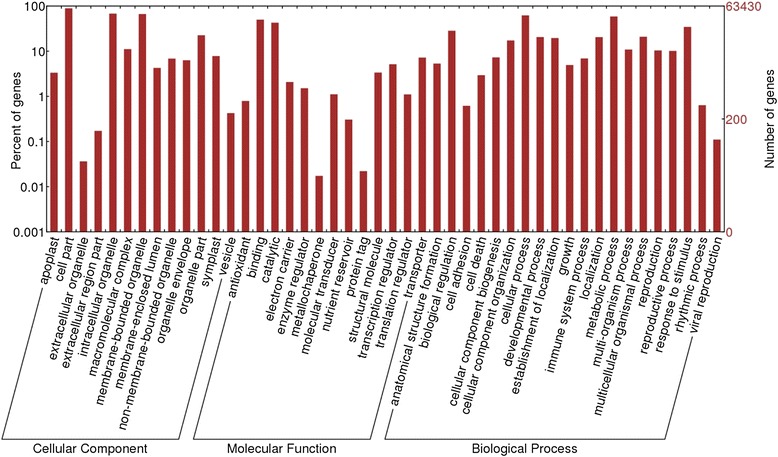
Gene ontology classification of P. americana transcriptome. Unigenes with BLASTX matches against the Arabidopsis proteins were classified into three main GO categories (cellular components, molecular functions and biological processes). The left-hand scale on the y-axis shows the percentage of unigenes belonged to each category. The right-hand scale on the y-axis indicates the number of unigenes in the same category
Fig. 3.
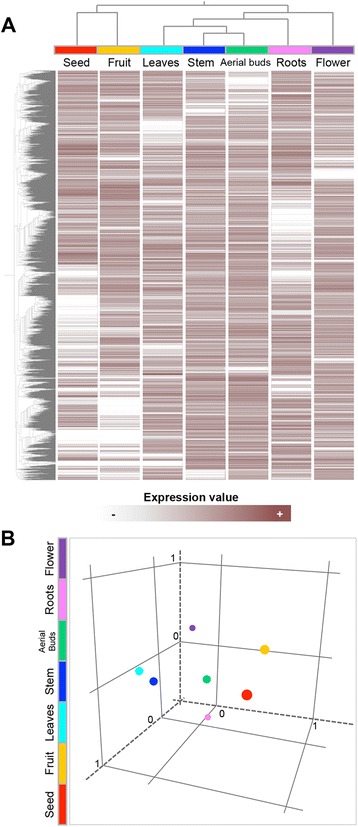
Profiling expression of P. americana transcriptome. a Hierarchical clustering shows expression levels of unigenes across different avocado organs. b Principal component analysis [seed (red), fruit (orange), leaves (cyan), stem (blue), aerial buds (green), roots (pink) and flower (purple)]
Expression map of the P. americana unigenes (the transcriptome atlas)
In the past, gene expression atlases from different plant species have been established by using massive parallel-signature sequencing and array-hybridization technologies [25–30]. Plant transcriptomes strongly vary from one tissue to another [25, 29], and it has been suggested that these variations are responsible, at least in part, for the identities of different plant organs [26]. We used the SOLiD v4.0-sequencing platform to quantify the expression of P. americana unigenes in seven different organs: seeds, roots, stem, leaves, aerial buds, flowers and fruits. Fruit was considered as the total of reads generated from pre-climacteric, climacteric and post-climacteric libraries. The resulting reads from these three libraries were also independently mapped. Between 4.18 and 6.84 million SOLiD reads were generated for each of the seven organ libraries; among them, 51.13 % were mapped to the avocado transcriptome (Additional file 4: Table S5). An expression profile matrix containing the unigenes (rows) and the number of mapped reads in each normalized organ-specific transcriptome (columns), was created. To allow for data comparison among samples, a normalization of the reads per Kb per million (RPKM) was performed. A threshold of RPKM ≥5 was used to distinguish expressed genes in at least one of the organs sampled from background [31] (Additional file 4: Table S6). We further investigated how the organs can be classified according to transcript levels using a principal component analysis (PCA) and hierarchical clustering (HC), by employing the Pearson metric on average expression levels for each organ (Fig. 3a and b).
For many of the organ types, the hierarchical plot and the PCA reflect the known similarity of biological functions among organs. For example, transcripts from leaves and stems clustered as a neighboring group, reflecting their physiological similarity (vegetative organs). Likewise, the storage organ transcriptomes (fruit and seed) were grouped together, whereas gene products from roots clustered separately from the rest (Fig. 3b). To assess the relative abundance of gene transcripts among the organ-specific transcriptomes, we used the log-likelihood ratio statistic, R [32], which scores reads by departures from the null hypothesis of equal counts in each library given the total number of reads sampled from each library. Higher R-values indicate a greater probability of differential expression, whereas R-values near zero represent constitutive expression (ubiquitous unigenes; see Methods). By considering as preferentially expressed genes the unigenes with R-values ≥15 (true positive rate of ~98 %), a total of 9357 P. americana unigenes were selected as preferentially expressed in at least one of the organ analyzed (Additional file 4: Table S7). Of these, 1968 unigenes (224 from seed, 192 from leaves, 22 from stem, 503 from roots, 56 from aerial buds, 654 from flowers and 313 from fruit) could be considered as organ-specific genes because the reads were derived from a single library (Additional file 4: Table S8). Although 33.63 % of organ-specific unigenes corresponded to sequences that could not be annotated, in the remaining unigenes, these organ-specific expression data are consistent with previously documented expression patterns of known genes. For example, the avocado unigene UN06501, a homolog of the Arabidopsis high-affinity phosphate transporter PHT1-3 gene (AT5G43360), is expressed in roots [33]. Four well-characterized floral pattern determination genes, AGL2 (AT5G15800; homologous to UN50397), AGL6 (AT2G45650; homologous to UN22054), AP3 (AT3G54340; homologous to UN19620) and PI (AT5G20240; homologous to UN19579), exhibited specific expression patterns among floral organs [34–37]. Likewise, in leaves, several avocado unigenes homologous to Arabidopsis light-harvesting chlorophyll a/b (LHCB) proteins were expressed in an organ-specific manner. This was expected, since in higher plants and algae, LHCB proteins are major components of the light-harvesting complex of photosystem II (PSII) in chloroplasts, which is responsible for light harvesting and energy transfer to reaction centers [38, 39]. Moreover, the fruit-specific avocado unigene UN44288 is homologous to AGL4 (AT3G02310), a transcription factor that has been recently characterized for its main role in apple fruit development and ripening [40]. Interestingly, an oleosin-encoding gene (UN35609) homologous to AT3G18570 showed a higher expression level in fruits than in seeds (Additional file 4: Table S7). Oleosins have been described as key structural and signaling components of seed oil bodies that form during the desiccation process and prevent coalescence of the oil [41]. This suggests that oleosins could be involved in avocado fruit development, beyond their known functions in seeds. In addition to its high lipid content, the avocado fruit contain several bioactive phytochemicals, including carotenoids [42]. The homolog of carotenoid biosynthesis-associated epoxycarotenoid dioxygenase NCED3 (AT3G14440), unigene UN43474, was highly expressed in the fruit transcriptome (Additional file 4: Table S7). Seven different avocado unigenes (UN26753, UN45556, UN14598, UN31059, UN39923, UN51437 and UN56049), among the highest expression levels from seed-specific genes, are homologs of PAP85 (AT3G22640), a member of the large superfamily of cupins, which are expressed during seed development and act as a nutrient reservoir [43]. The homolog to fasciclin-like arabinogalactan protein FLA12 (AT5G60490; unigene UN17820) exhibited one of the highest expression levels of stem-specific genes, which is consistent with previous reports showing that expression of some members of the FLA gene family is correlated with the onset of secondary-wall cellulose synthesis in Arabidopsis stems and with wood formation in the stems and branches of trees, suggesting a biological role in avocado stem development [44]. Together, these results indicate that the transcriptome atlas of P. americana presented here provides an accurate estimation of organ-specific gene expression patterns that may assist functional interpretations.
Transcriptome changes during avocado fruit development and ripening
We identified unigenes that are differentially expressed during fruit development and ripening using a similar approach to that described above (RPKM values derived from mapped reads and a threshold of R ≥15 to select differentially expressed genes; see Methods). First, the RPKM values between flower and fruit were compared in order to identify some genes that may play important roles during fruit development. A total of 382 unigenes were selected due to the significant increase of their transcripts in the fruit/flower comparison. We surveyed some of these genes in order to determine their potential roles during fruit development (Additional file 4: Table S9). A total of 10 avocado unigenes (UN54812, UN37501, UN61103, UN59855, UN53293, UN48116, UN58333, UN64333, UN64163 and UN68295), homologs of Arabidopsis metallothionein MT2A (AT3G09390), were identified. Metallothioneins are small cysteine-rich proteins required for heavy metal tolerance in animals and fungi. In plants, metallothionein genes are up-regulated in response to heavy metal stress [45] but also participate in natural and induced leaf senescence [46, 47], ethylene-induced abscission [48] and biotic and abiotic stress responses [49–51]. However, metallothioneins have been also identified as up-regulated genes during climacteric fruit development in banana [52], apple [53], and kiwifruit [54] and in non-climacteric fruit such as grape [55], Citrus unshiu [56], strawberry [57] and pineapple [58]. Despite their abundance, the function of metallothioneins during fruit development still remains largely unknown. Chitinases were also found to be a well-represented group of fruit specific unigenes (18 in total), most of them homologs of the basic chitinase CHIB/PR-3 (AT3G12500). Consistently, besides the role of chitinases in plant defense [59], these proteins have also been associated with fruit development in both climacteric [60] and non-climacteric fruits [61]. Pectate lyases constitute an additional protein family that has been suggested to play an important role in fruit ripening and softening [62]. This is consistent with the fact that eight avocado unigenes, homologous to four different members of the Arabidopsis pectin lyase-like superfamily (AT1G80170, AT3G07850, AT3G07970 and AT5G18990), showed significant increase in the frequency of transcriptional units in the flower/fruit comparison.
One-thousand-two-hundred-thirty unigenes out of 16,025 (RPKM values ≥5) were identified as differentially expressed during the ripening process of the avocado fruit (Additional file 4: Table S10). In order to find over-representation of a given function, GO categories were assigned to the unigenes differentially expressed in each ripening stage (pre-climacteric: 756 unigenes, climacteric: 841 unigenes, and post-climacteric: 812 unigenes). Only GO sub-categories that showed significant differences in the ‘molecular function’ and ‘biological process’ categories were analyzed. Figure 4a shows the differentially expressed unigenes within the ‘molecular function’ category. Sub-categories ‘Hydrolase activity, acting on glycosyl bonds’, ‘polysaccharide binding’ and ‘peptidase inhibitor activity’ showed a significant increase in the number of genes while ‘oxidoreductase activity, acting on the aldehyde or oxo group of donors’, ‘passive transmembrane transporter activity’, ‘water transporter activity’ and ‘chlorophyll binding’ showed a decrease. Regarding ‘biological process’ GO terms, the majority of differentially expressed unigenes appeared to be related to some major biological changes, including ‘monosaccharide metabolic process’, ‘carbohydrate catabolic process’, ‘cell wall macromolecule catabolic process’, ‘gene expression’, ‘cellular aldehyde metabolic process’, ‘organic acid metabolic process’, ‘vitamin metabolic process’, ‘photosynthesis’, ‘cellular lipid metabolic process’, ‘reproductive process in a multicellular organism’, ‘aging’, ‘defense response’, ‘cellular response to hormone stimulus’, ‘fluid transport’, and ‘secondary metabolic process’ (Fig. 4b). The complete list of GO categories is provided in Additional file 4: Table S11. These results are consistent with the notion that the principal changes associated with ripening include color (loss of green color and increase in non-photosynthetic pigments that vary depending on species), firmness (softening by cell wall degrading activities and alterations in cuticle properties), taste (increase in sugar and decline in organic acids), and flavor (production of volatile compounds providing characteristic aromas) [63].
Fig. 4.
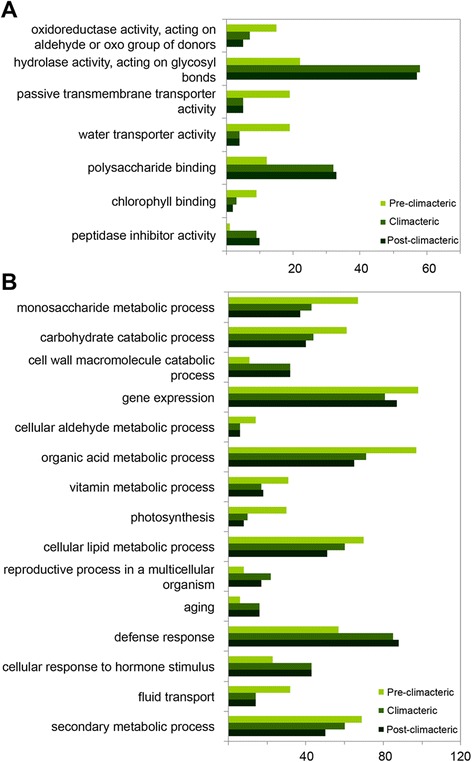
Functional categorization of differentially expressed genes during avocado fruit ripening. Genes were categorized based on GO annotation, and the number for each category is displayed based on molecular function (a), or biological process (b). Data presented represent GO terms at level 4
Differentially expressed unigenes during fruit ripening were also examined by cluster analysis of gene expression patterns, which arranged the gene products into 5 major groups named as classes I-V (Fig. 5a and b; Additional file 4: Table S10). The most represented classes comprised the unigenes whose expression increased (class III) or decreased (class IV) during fruit ripening. Both classes were subdivided into two sub-classes (A and B, respectively). Classes III-A and IV-A comprised the unigenes whose expression increased or decreased continuously during fruit ripening, while classes III-B and IV-B showed increase or decrease at the climacteric stage, with similar expression levels maintained at post-climacteric stages. Classes I and II represent the unigenes in which the highest or lowest expression levels were detected at the climacteric stage. Finally, class V represents the remaining unigenes that were not grouped into none of the classes mentioned above. Differential gene expression patterns observed in the RNA-seq experiments were validated by real-time PCR for eight avocado unigenes differentially expressed during fruit ripening (Additional file 3: Figure S2). These genes showed the same expression pattern in both RNA-seq (measured as RPKM values) and in the real-time PCR analyses.
Fig. 5.
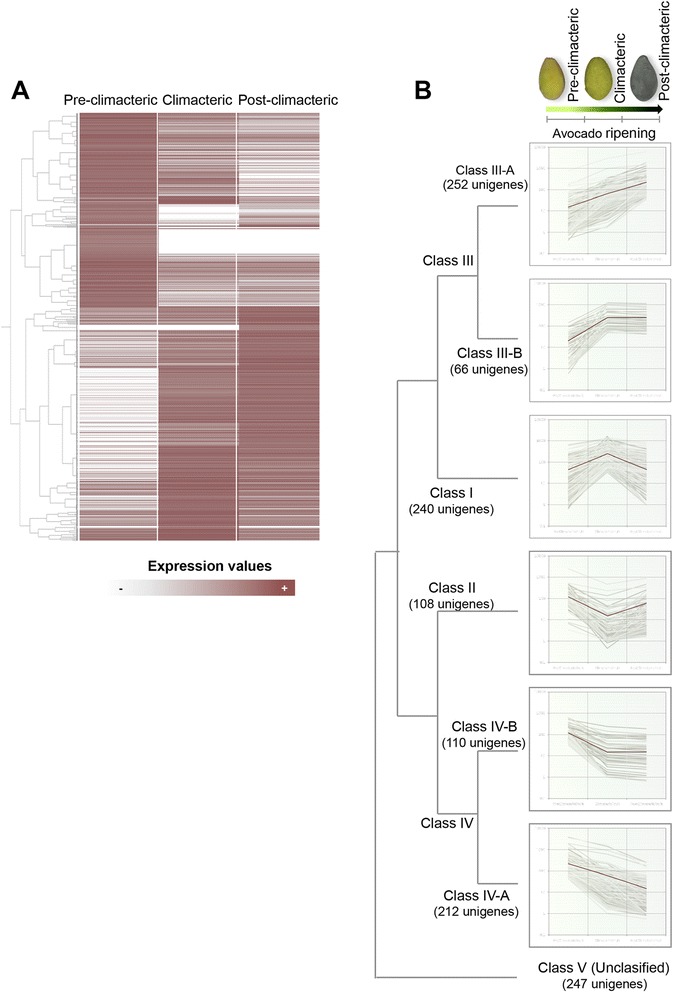
RNA-seq based transcriptome dynamics of avocado during fruit ripening. a The log2 of RPKM values for each gene was used for the hierarchical clustering analysis at each of the three selected ripening stages (pre-climacteric, climacteric and post-climacteric). b The 1235 differentially expressed unigenes were classified into 5 regulation patterns (classes I-V, respectively). Classes III and IV were subdivided into two sub-classes each. The graph shows the expression profile of unigenes for each class. Gray lines, expression profiles for individual unigenes. Brown lines represent the average intensities of unigene members of the clusters. For additional information, see Additional file 4: Table S10
Genes relevant to avocado fruit ripening
In ripening of climacteric fruits, the expression of some members of the gene families encoding ACC synthase and ACC oxidase is induced, regulating the biosynthesis of ethylene [64]. Two differentially expressed unigenes (UN03798 and UN39755) were found to be homologs of Arabidopsis ACC synthase (AT3G61510), and one, UN38306, a homolog of ACC oxidase (AT1G62380). In relation to their expression profiles, the unigenes homologous to the ACC synthase were classified into class III-A, while the homolog of ACC oxidase was classified into class II (Fig. 5b). Therefore, during avocado fruit ripening, the ethylene burst seems to be regulated mainly by ACC oxidase. This is consistent with the notion that in climacteric fruits the rate of ethylene production is well correlated with the pattern of accumulation of ACC synthase and ACC oxidase gene transcripts [65]. Avocado unigenes homologous to ethylene response factor ERF110 (AT5G50080; UN29560 and UN42714) and ethylene receptor EIN4 (AT3G04580; UN01530 and UN24061), which play key roles in the signaling of ethylene responses [66], were also identified as members of class III-A (Fig. 5b).
The genetic and physiological characterization of tomato ripening mutants, ripening-inhibitor (rin; [67]), non-ripening (nor; [68]) and colorless non-ripening (cnr; [69]), together with the molecular characterization of the mutated genes, have demonstrated that several important regulatory factors must be properly coordinated with the ethylene signal to properly activate and orchestrate the ripening program. RIN [70], NOR [71] and CNR [72] genes encode transcription factors belonging to the MADS-box, NAC-domain, and SBP-box families, respectively, that act upstream of ethylene biosynthesis and perform key functions in the control of fruit ripening [73]. RIN [74] and two regulatory proteins more recently identify as involved in fruit ripening, the TAGL1 MADS-box factor [75] and the HB-1 homeobox protein [76], are able to bind to the promoter region of ACS2 [74] and ACO1 [76] genes, respectively, demonstrating that transcription factors directly regulate the activity of ethylene biosynthesis genes in tomato. A bi-directional best BLAST hit approach was used to identify the avocado orthologs of tomato genes involved in ripening (see above). Using the SeaView program [77] the protein-coding nucleotide sequences were then aligned based on their corresponding amino acid translations to calculate the percent identity at nucleotide and amino acid levels (Additional file 5). The identities of avocado/tomato genes ranged from 48.82 to 63.53 % (Additional file 4: Table S12) and with only one exception (HB-1; UN22151), their expression levels were considerably higher in flowers than in the remaining organs sampled. Meanwhile, NOR and HB-1 avocado transcripts increased during fruit ripening while CNR decreased. RIN and TAGL1 showed very low transcript levels (RPKM <5) only at the preclimacteric stage (Additional file 3: Figure S3). Avocado is considered a basal angiosperms with origin near the split between monocot and eudicot plant species. The finding that some but not all transcription factors involved in tomato fruit ripening are up-regulated or even expressed during avocado fruit ripening suggests that part but not all of the transcription factor wiring was ancestrally present and that substantial rewiring occurred during the evolution of modern eudicots.
It has been reported that a thaumatin-like protein and a class I chitinase are some of the most abundant ripening-associated proteins in banana fruit [60]. Genes encoding these proteins were also found to have differential expression during ripening in avocado. Avocado unigene UN10274, a homolog of a pathogenesis-related thaumatin (AT1G20030), clustered into class III-A (transcripts continuously rising; Fig. 5b), the same as several unigenes (16 in total; see Additional file 4: Table S10) homologous to PR-3 (AT3G12500), a well-known class I chitinase strongly induced when plants respond to wounding or infection by fungal, bacterial, or viral pathogens [78]. Additional unigenes (UN43717 and UN45051) with different expression profiles (class I) were identified as homologs of AT4G01700 and AT2G43610, class II and IV chitinases, respectively. Therefore, comparisons with data reported for other climacteric fruits (including ethylene responsive genes) facilitated identification of putative conserved orthologous ripening-related genes, which serve as an initial set of candidates for assessing the conservation of gene activity during the evolution of fleshy-fruited plant species.
Acyl lipid metabolism of Persea americana fruits
Previously, an acyl lipid metabolism gene database was developed for Arabidopsis [79], and this was recently expanded by deep transcriptional profiling of developing seeds from four different oilseed species [80]. This significant effort resulted in detailed sequence information for over 740 genes encoding proteins involved in lipid metabolism, providing a useful resource for construction of additional databases of genes related to lipid biosynthesis from other oilseed or plant species with lipid-rich fruits (http://aralip.plantbiology.msu.edu/). After comparing our assembled transcriptome against the aforementioned database, we found that 1177 avocado unigenes had been annotated as homologous to lipid metabolism genes with transcriptional evidence for their expression (RPKM values ≥5) in at least one of the organ sampled (flower, leaves, roots, stem, aerial buds, seed and fruit) (Fig. 6; Additional file 6). A similar number of unigenes related to the ‘fatty acid synthesis’ category were detected in fruit, seed, flower and aerial bud organs, whereas stem, leaves, and root organs had a lower number of expressed unigenes annotated in the same category. The category ‘fatty acid elongation, desaturation & export from plastid’ shares a similar pattern (Fig. 6a). Interestingly, the number of genes detected in both categories decreases during fruit ripening (Fig. 6b). This was expected since after flowering and during avocado fruit development, the moisture content decreases while the lipid content steadily increases [81], and after the picking, and during the post-harvest ripening period, the avocado fruit does not show significant changes in the fatty acid composition [82]. Our data suggests that the initiation of fruit ripening marks the end of oil accumulation and fatty acid composition.
Fig. 6.
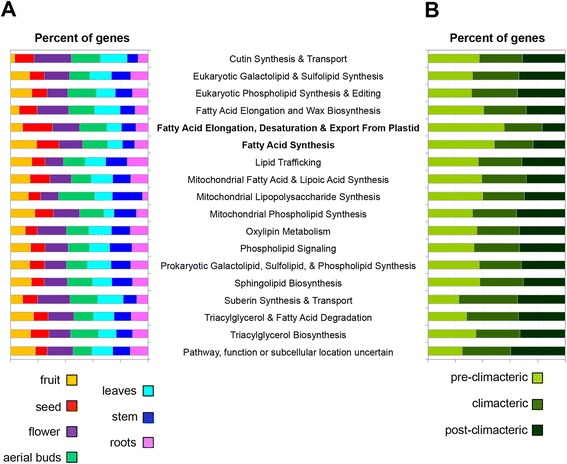
Totals of P. americana unigenes suspected to be involved in acyl-lipid metabolism. Unigenes were annotated with a translated BLAST (e-value ≤10−03 and a bit score ≥25) against Arabidopsis proteins and further annotated based on information at the Arabidopsis Lipid Gene Database (http://aralip.plantbiology.msu.edu/). For additional information, see Additional file 4: Table S11. a Percent of the genes detected in each of the organs sampled and (b) during fruit ripening
The average RPKM values of unigenes annotated as homologous to fatty acid biosynthesis genes were calculated in order to estimate expression profiles of their corresponding avocado genes (Additional file 4: Table S13). The transcription levels of the majority of genes involved in fatty acid biosynthesis are significantly higher in fruit than in all the other organs analyzed (Fig. 7); nonetheless, they decrease during fruit ripening (Fig. 8). In addition, according to their expression profiles, three homologs (UN06747, UN21149 and UN33083) of FAB2/SSI2 (AT2G43710) were identified as class IV-A differentially expressed unigenes. FAB2/SSI2, a soluble stearoyl-acyl carrier protein desaturase, is a major enzyme responsible for converting saturated stearic acid (C18:0) to monounsaturated oleic acid (C18:1) in chloroplasts [83]. Our data are also in agreement with the transcriptome of developing mesocarp of ‘Hass’ avocado that was generated in parallel to these studies where genes involved in acyl lipid metabolism were exclusively investigated [84]. Together, these results suggest that lipid accumulation and changes in fatty acid composition (for example, some fatty acid desaturations) occur during avocado fruit development, and probably both processes stop a few days after the fruit has been harvested (at the pre-climacteric stage). The noticeable decrease of the FAB2/SSI2 transcripts, as well as genes involved in fatty acid biosynthesis during fruit ripening, could explain the fact that no significant changes have been detected in avocado fatty acid composition during the post-harvest ripening period [7]. Considering that ethylene production starts at the pre-climacteric stage and quickly increases towards the climacteric state, it is tempting to hypothesize that in avocado fruit, ethylene is perhaps a signaling molecule that once perceived, halts lipid biosynthesis and programs future changes in fatty acid composition.
Fig. 7.
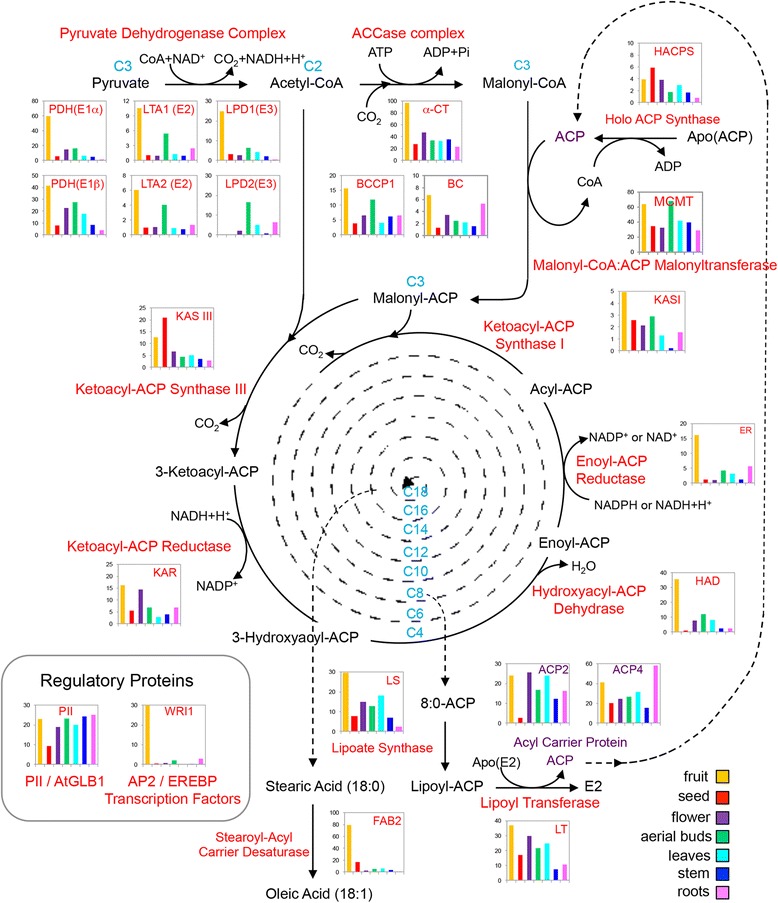
Expression profile of P. americana unigenes involved in the fatty acid biosynthetic pathway. The bar graphs show the frequency of transcriptional units as the average of RPKM values of all unigenes annotated as homologous to each Arabidopsis gene (represented by red letters in the figure). Each avocado organ analyzed is represented by different color: fruit (yellow), seed (red), flower (purple), aerial buds (green), leaves (cyan), stem (blue) and roots (pink). This figure was modified from the Arabidopsis Lipid Gene Database (http://aralip.plantbiology.msu.edu/)
Fig. 8.
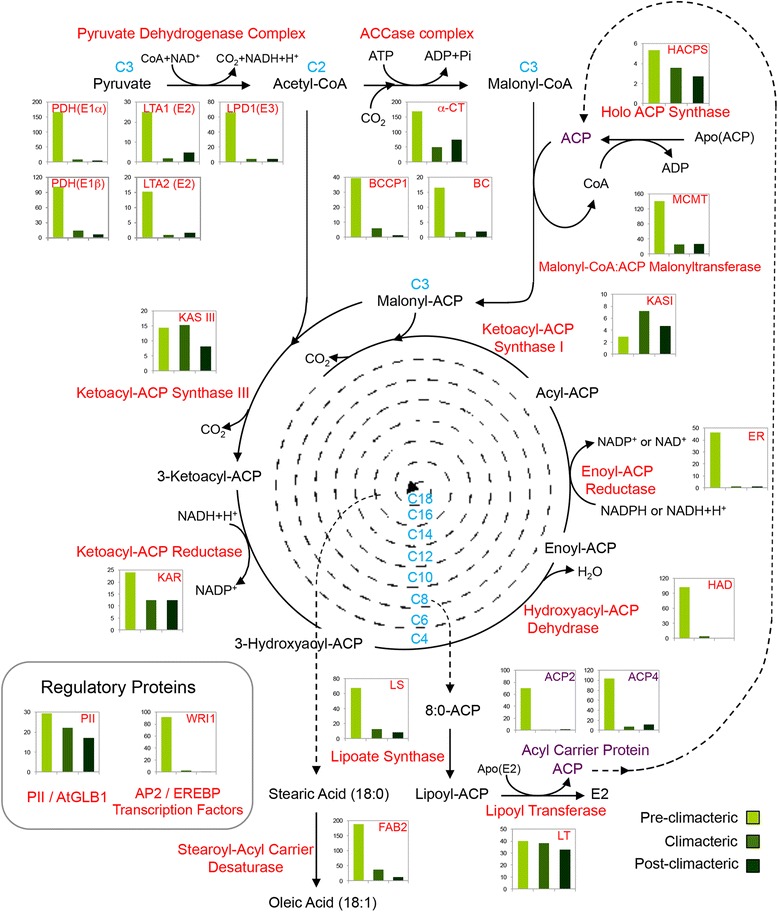
Expression profile of genes coding for fatty acid biosynthesis during avocado fruit-ripening. The bar graphs show the frequency of transcriptional units as the average of RPKM values of all unigenes annotated as homologous to each Arabidopsis gene (represented by red letters in the figure). Each avocado fruit ripening stage is represented by different color: pre-climacteric (light green), climacteric (green) and post-climacteric (dark green). This figure was modified from the Arabidopsis Lipid Gene Database (http://aralip.plantbiology.msu.edu/)
Although in most avocado varieties over half of the total fat present is in the form of oleic acid (C18:1), recent studies have shown that at least 22 different fatty acids can be detected in pulp and seed avocado oils [85]. Palmitic (C16:0) and linoleic (C18:2) fatty acids are the second major constituents of fruit oils while some others like stearic acid (C18:0) are present in trace amounts [85]. The benefits to nutrition and health of avocado fatty acids have been recently reviewed [86]). In addition, avocado acetogenins, such as persin [(+)-(Z,Z)-1-(acetyloxy)-2-hydroxy-12,15-heneicosadien-4-one], a deoxy-derivative of glyceride with close structural homology to the monoglyceride of linoleic acid, has been proposed as an alternative therapy against breast cancer due its necrotic effect in the mammary gland [87]. Considering the importance of avocado to nutrition and health, transcripts encoding enzymes of the biosynthetic pathway from palmitic to linoleic acid, passing through stearic and oleic acids, were reconstructed similarly to described above (Additional file 4: Table S13; Additional file 3: Figure S4). It is worth noting that genes involved in the biosynthesis of linoleic acid (downstream of oleic acid) showed lower expression levels in fruit than in seed. This is consistent with previous reports in which linoleic acid seems to accumulate in greater amounts (around 4-fold, [85]) in seeds than in fruits.
Conclusion
The avocado transcriptome reported in this study provides a foundation for the molecular genetics and functional genomics required to study the basis of the genetic diversity that determines the different levels and quality of oil accumulation in the fruit of different avocado cultivars, as well as other important agronomic traits for this increasingly important crop. Transcriptomic data will also facilitate the identification of the pathways involved in production of a wide variety of essential nutrients and phytochemicals beneficial for human health that are produced by the avocado. This data will also facilitate the study of early plant evolution since avocado is phylogenetically placed near the separation between monocot and eudicot plants.
Methods
Plant material
Avocado (Persea americana var. drymifolia) samples were obtained from the germplasm bank of the “Instituto Nacional de Investigaciones Forestales y Agropecuarias (INIFAP)” in Uruapan, Michoacán, México. Organs sampled [Leaves: mixed development stages of expanded leaves; Stem: segments from young branches, Aerial buds: developing buds emerging from shoot apical meristem; Flower: whole inflorescences with flowers at multiple stages of development (young, immature, and mature); Seed: isolated from harvested fruits at the mature-green state (approximately 8 months-old); roots: whole root system recovered from in vitro-propagated seedlings [88], or from cuttings grown in pots in a greenhouse]. Fruits in the mature-green state were also harvested for the ripening experiments. Seeds were removed once the fruit was frozen at each rippening stage. All organs/tissues sampled were obtained from a single reference tree (accession 001-01, which has been sequenced as part of the avocado whole-genome sequencing project).
Characterization of the climacteric behavior of avocado fruit
In avocado and other climacteric fruits such as apple, melon and banana, an ethylene burst is required for normal fruit ripening (reviewed in [89]). According to the method described by Hoffman and Yang [15], ethylene production of each fruit was monitored individually by gas chromatography during the course of ripening until they were sacrificed for RNA extraction.
RNA sequencing
Total RNA was isolated using the Trizol reagment (Invitrogen) and re-purified with the RNeasy kit (Qiagen) following the manufacturer’s instructions. Five micrograms of total RNA from each sample were pooled to produce sequencing libraries. Additionally, according to ethylene quantification (Additional file 3: Figure S1), three independent extractions were prepared from 8 months-old fruits (pericarp), at the second, third and fourth day after harvest (pre-climacteric, climacteric and post-climacteric stages respectively), allowing us to contrast changes in the transcriptome profile of the avocado fruit at different ripening stages. cDNA preparation, library construction, and sequencing were performed according to Illumina and Roche manufacturer instructions. From the RNA pool, two sequencing libraries were prepared and a single sequencing run was carried out on the GS-FLX+ (Roche) sequencer, and another on the Illumina MiSeq platform. A total of 4,674,756 reads were generated (1,055,903 reads from GS-FLX+ with an estimated average size of 462.27 bp and 3,618,853 paired reads, 2 × 150 bp, from MiSeq). Additionally, using the SOLiD system and the SOLiD RNA Barcoding Kit (v4 chemistry), 1/4 run, in which all RNA samples were independently represented using barcodes, was performed. A total of 70,650,328 short reads (up to 50 pb) were generated (Additional file 1: Table S1). Files containing sequence reads and quality scores were deposited in the Short Read Archive of the National Center for Biotechnology Information (NCBI) [Accession number SRS923862].
Assembly and sequence analysis
To carry out the assembly process only the reads generated with GS-FLX+ and Illumina-MiSeq sequencer were considered. Using the CDHIT program [90], natural and artificial duplicate reads were removed from the data set generated by the GS-FLX+ sequencer. Additionally, reads with an average quality less than 20 (phred score), were also removed. On the other hand, forward and reverse read pairs (generated by Illumina-MiSeq) were merged to form single “longer-reads” using the SeqPrep pipeline (https://github.com/jstjohn/SeqPrep), with default parameters (a quality score cutoff of phred 33, a minimum merged read length of 15 bp and no mismatches in the overlapping region). Paired-end reads that did not overlap were trimmed using a sliding window approach (window size 10 bases, shift 1 base). Reads were discarded if they were smaller than 30 bases after trimming, and orphan reads were also removed in order to keep pairs only. MIRA v3.4.1 [16] and Trinity [12] assemblers were used independently. Standard assembly metrics, such as number of contigs ≥1 Kb, average contig lengths, and maximum contig size were estimated (Additional file 1: Table S2). Resulting contigs (with a minimum length of 100 bp) derived from both assembly processes were merged into a single file, and those that were redundant were eliminated using the BlastClust program. Redundant contigs were defined as those having greater than 95 % identity over an area covering 95 % of the length of the sequence. Unique contigs were trimmed of low quality, low complexity and poly(A/T) tails using the SeqClean software (http://compbio.dfci.harvard.edu/tgi/), then, they were passed through a second assembly step using the CAP3 assembler [19]. CAP3 was run with default parameters (minimum overlap length of 40 bp and 95 % minimum sequence identity). A set of unigenes from P. americana were generated considering only resulting contigs with a minimum size of 200 bp (Additional file 1: Table S2). SOLiD sequence reads were only used for differential expression analysis and they were excluded from the assembly because direct conversion of sequencing reads is possible but not recommended because all bases that follow a single error in colorspace will create errors in all subsequent bases of a read. Additionally, it has been recently reported that a hybrid strategy generated high quality assemblies following three simple recommendations: (1) using a single individual with large representation of biological tissues, (2) merging both long reads and paired-end reads (derived from GS-FLX+ and Illumina platforms, respectively) and (3), using several assemblers in order to combine specific advantage of each [91].
Annotation of Persea americana var. drymifolia unigenes
To annotate sequences obtained by de novo assembly, we performed sequence similarity searches using the BLASTX algorithm (e-value 10−3, bit score ≥25) on Arabidopsis thaliana (TAIR v11; http://www.arabidopsis.org/) and Amborella trichopoda (http://www.amborella.org/) proteins, and plant proteins from other species available in the Reference Sequence (RefSeq) collection (NCBI; ftp://ftp.ncbi.nlm.nih.gov/refseq/release/plant/). Top protein matches from Arabidopsis, Amborella or other plant species were assigned to each of the avocado unigenes (Additional file 1: Table S3). The putative protein domains contained within the translated unigenes were identified using Hidden Markov Model (HMM)-based searches against Pfam database (e-value 10−3) [22]. The gene ontology (GO) functional classes and pathways for each avocado unigene were assigned based on Arabidopsis GO SLIM and pathway annotation (Additional file 1: Table S3). Association of unigenes with KEGG pathways was determined using a directional best hit (BBH) method (that is, top reciprocal BLAST hits) against the Kyoto Encyclopedia of Genes and Genomes database [92]. The KEGG pathways annotation (Additional file 3) was performed in the KEGG Automatic Annotation Server (KAAS) (http:/www.genome.jp/tools/kaas/) [24].
Expression profile analysis of the P. americana transcriptome
After assembling the P. americana transcriptome, reads generated by the SOLiD system were separated according to the barcode used, and then were separately aligned to the avocado unigene set using BioScope (Life Technologies). To run whole transcriptome analysis with BioScope, a reference genome is required. Using a custom Perl script, a reference genome proxy was created linking all unigenes derived from the assembly process (in which unigenes were separated by 50 N’s); at the same time, a GTF/GFF file was also created. The libraries were relatively uniform with respect to mapping efficiency (Additional file 4: Table S5). Gene expression levels were calculated by the RPKM (reads per kb per million reads) method, and preferentially expressed genes were selected according to the method described by Stekel [32]. Briefly, all unigenes were submitted to a log-likelihood ratio statistic that trends asymptotically to a χ2 distribution, as described by Stekel et al. [32]. We considered as preferentially expressed those unigenes with a value of R ≥15 among organs sampled (Additional file 3: Figure S5). This provides a single statistical test to describe the extent to which a gene is differentially expressed between libraries. This method permits identification of differentially expressed genes among any number of libraries. Hierarchical clustering was performed using the Pearson correlation coefficient and average linkage clustering [93]. Results were visualized using GeneSpring GX 7.3.1 software (Agilent Technologies).
Real-time quantitative PCR verification
Eight avocado unigenes identified as differentially expressed genes were aligned against their Arabidopsis homologues in order to identify the coding sequences in their correct open reading frame. Protein-coding nucleotide sequences were aligned based on their corresponding amino acid translations using the SeaView program (Additional file 4: Table S14). Gene-specific primer pairs (Additional file 4: Table S15), which were designed using the Primer3 v.0.4.0 web tool (http://bioinfo.ut.ee/primer3-0.4.0/primer3/), were used for real-time PCR.
A total of 10 μg of RNA was reverse transcribed for first-strand cDNA synthesis using SuperScript® III Reverse Transcriptase (Life Technologies) according to the manufacturer’s instructions. Reactions were performed with the SYBR Green PCR Master Mix in an ABI 7500 Fast Real-time system. Actin was used as the standard to normalize the content of cDNA, as described previously [94]. The thermal cycling program was set to 95 °C for 5 min, 40 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min. Results were analyzed using the ABI 7500 on-board software, version 2.0.5 (Applied Biosystems). The real-time PCR was conducted with at least three experimental replicates for each biological sample.
Availability of supporting data
All the supporting data are included as additional files, including the sequences of all assembled transcripts. The raw sequencing data was deposited in the NCBI and can be accesed under the following identifier: BioProject PRJNA282441.
Acknowledgements
This work was funded by SAGARPA, Grant 2009-C02-126261 (LH-E). We thank Flor Zamudio-Hernández and María de Jesus Ortega-Estrada for their technical support in the real-time PCR services, as well as INIFAP authorities for their permission to access the germoplasm bank. We also thank Alexandro Alonso-Sánchez and Emanuel Villafán de la Torre for discussion and critical comments on the manuscript, Araceli Fernández-Cortes who has responsibility for the high-performance computing system at LANGEBIO. Special thank goes to the staff of “Laboratorio de Servicios Genómicos” at LANBEBIO for their assistance with RNAseq library preparation and sequencing services. We thank Le-Shin Wu and the National Center for Genome Analysis Support at Indiana University for computing assistance. We also thank the anonymous reviewers for their positive and relevant comments, which have improved the quality of this manuscript.
Additional files
Summary of sequencing data generated from P. americana transcriptome. Table S2. Basic P. americana assembly metrics. Table S3. Annotation of P. americana unigenes. (ZIP 16319 kb)
P. americana unigenes (ZIP 20363 kb)
Complete metabolic network represented in the P. americana unigenes. Nodes in this figure are metabolic compounds. Edges are enzymatic transformations. The edges have been highlighted to indicate the modules: energy, carbohydrate and lipid metabolism (green), nucleotide and amino acid metabolism (orange) and genetic information processing (reed). The metabolic network was re-constructed using “Search & Color Pathway” tool from KEGG database (http://www.genome.jp/kegg/). Figure S2. Real-time PCR validation of differentially expressed genes. (A) RNA-Seq expression levels measured as reads per kb per million of reads (RPKM). (B) Real-time PCR expression levels given as 40-ΔCT, where ΔCT is the difference in threshold cycle number of the respective gene and the reference ACTIN; the number 40 was chosen because the PCR run stops after 40 cycles and a constant value was required (as calibrator) in order to see the differences existing in three ripening stages (see as example [95]). The results are shown as the averages ± SE of three biological replicates. Figure S3. Expression profiles of avocado orthologs to well characterized tomato ripening-associated genes. RNA-Seq expression for 5 different unigenes, measured as RPKM values (y axis) at specific-organs (A) and during fruit ripening (B) are shown. Figure S4. Metabolic pathway from palmitic to linoleic acids. The bar graphs show the frequency of transcriptional units as the average of RPKM values of all unigenes annotated as homologs to each of the Arabidopsis genes (represented by red letters in the figure). Avocado organs (to the left) are represented by different colors: fruit (yellow), seed (red), flower (purple), aerial buds (green), leaves (cyan), stem (blue) and roots (pink). Ripening stages (to the right) are shown in green scale (from light to dark; pre-climacteric, climacteric and post-climacteric respectively). This biosynthetic pathway was reconstructed based on information available for A. thaliana in BioCyc Database Collection (http://biocyc.org/). Figure S5. The number of unigenes for a given value of the test statistic R is plotted as a function of R. The data falling within 5 ≤ R ≤ 15 decrease exponentially with R. When R ≥15, the number of genes is above this exponential curve. (PPTX 896 kb)
Detailed information for unigenes mapping to KEGG pathways in P. americana. Table S5. Total number of reads mapped by BioScopeTM software (LifeTechnologies) against the P. americana unigene set. Table S6 . Expression profile matrix of P. americana unigenes. Table S7 . Preferentially expressed unigenes in P. americana by organ. Table S8 . P. americana unigenes selected as organ-specifc. Table S9 . P. americana unigenes selected as preferentially expresed in the fruit/flower comparison. Table S10 . Differentially expressed genes during avocado fruit ripening. Table S11 . Association of specific GO terms to preferentially expressed genes involved in the avocado ripening process. Table S12 . Identity percent between avocado (Persea americana var. drymifolia) and tomato (Solanum lycopersicum) genes involved in the ripening process. Table S13 . List of avocado unigenes annotated as homologous to Arabidopsis genes known or suspected to be involved in acyl-lipid metabolism. Table S14 . P. americana unigenes annotated as homologous to Arabidopsis genes involved in the fatty acid biosynthetic pathway. Table S15 . Primers employed in real-time PCR assay. (XLSX 15767 kb)
Pairwise sequence alignment (Rippening-like genes) (ZIP 7 kb)
Pairwise sequence alignment (qRT-PCR validation) (ZIP 8 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EI-L carried out the bioinformatics analysis, performed the statistical analysis and drafted the manuscript. EI-L, AM-B and CAP-T participated in experiment materials preparation and collected data. EI-L, AM-B, CAP-T, VA, KM, AK and LH-E analyzed data. RL-G carried out the gas chromatography assays. JIC-L participated in the sequence mapping. EI-L, AM-B, CAP-T, VA, KM, AK and LH-E participated in manuscript formatting and editing. LH-E conceived the study, and participated in its design and coordination. All authors read and approved the final manuscript.
References
- 1.Rohwer JC. Lauraceae. In: Kubitzki KRJ, Bittrich V, editors. The families and genera of flowering plants vol. 2. Berlin: Springer-Verlag; 1993. pp. 366–91. [Google Scholar]
- 2.Chanderbali AS, Albert VA, Ashworth VE, Clegg MT, Litz RE, Soltis DE, et al. Persea americana (avocado): bringing ancient flowers to fruit in the genomics era. Bioessays. 2008;30(4):386–96. doi: 10.1002/bies.20721. [DOI] [PubMed] [Google Scholar]
- 3.Schaffer B, Wolstenholme BN, Whiley AW. The avocado: botany, production, and uses. 2. Wallingford, UK: CAB International; 2013. [Google Scholar]
- 4.Scora RW, Wolstenholme BN, Lavi U. Taxonomy and botany. In: Whiley AW, Schaffer B, Wolstenholme BN, editors. The avocado: botany, production and uses. Wallingford, UK: CAB International; 2002. pp. 15–37. [Google Scholar]
- 5.Barlow CC. The ghosts of evolution: nonsensical fruit, missing partners, and other ecological anachronisms. New York, NY: Basic Books; 2000. [Google Scholar]
- 6.Knight RJ. History, distribution and uses. In: Whiley AW, Schaffer B, Wolstenholme BN, editors. The avocado: botany, production and uses. Wallingford, UK: CAB International; 2002. pp. 1–14. [Google Scholar]
- 7.Ozdemir F, Topuz A. Changes in dry matter, oil content and fatty acids composition of avocado during harvesting time and post-harvesting ripening period. Food Chem. 2004;86(1):79–83. doi: 10.1016/j.foodchem.2003.08.012. [DOI] [Google Scholar]
- 8.Martinez Nieto L, Moreno Romero MV. Paramètres de détermination de la maturité des avocats. Ind Aliment Agric. 1995;112(4):200–3. [Google Scholar]
- 9.Scora RW, Bergh B. The origins and taxonomy of avocado (Persea americana) Mill. Lauraceae. Acta Horticulturae (ISHS) 1990;275:387–94. [Google Scholar]
- 10.Scora RW, Scora PE. Essential oils of Persea subgenus Persea (Lauraceae) J Essent Oil Res. 2000;12(6):709–13. doi: 10.1080/10412905.2000.9712197. [DOI] [Google Scholar]
- 11.Bakkali F, Averbeck S, Averbeck D, Idaomar M. Biological effects of essential oils–a review. Food Chem Toxicol. 2008;46(2):446–75. doi: 10.1016/j.fct.2007.09.106. [DOI] [PubMed] [Google Scholar]
- 12.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotech. 2011;29(7):644–52. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reeksting BJ, Coetzer N, Mahomed W, Engelbrecht J, van den Berg N. De novo sequencing, assembly, and analysis of the root transcriptome of Persea americana (Mill.) in response to Phytophthora cinnamomi and flooding. PLoS One. 2014;9(2):e86399. doi: 10.1371/journal.pone.0086399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wall PK, Leebens-Mack J, Chanderbali A, Barakat A, Wolcott E, Liang H, et al. Comparison of next generation sequencing technologies for transcriptome characterization. BMC Genomics. 2009;10(1):347. doi: 10.1186/1471-2164-10-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffman NE, Yang SF. Changes in 1-aminocyclopropane-1-carboxylic acid content in ripening fruits in relation to their ethylene production rates. J Am Soc Hortic Sci. 1980;105(4):492–5. [Google Scholar]
- 16.Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WEG, Wetter T, et al. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004;14(6):1147–59. doi: 10.1101/gr.1917404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franssen S, Shrestha R, Brautigam A, Bornberg-Bauer E, Weber A. Comprehensive transcriptome analysis of the highly complex Pisum sativum genome using next generation sequencing. BMC Genomics. 2011;12(1):227. doi: 10.1186/1471-2164-12-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Azim MK, Khan IA, Zhang Y. Characterization of mango (Mangifera indica L.) transcriptome and chloroplast genome. Plant Mol Biol. 2014;85(1–2):193–208. doi: 10.1007/s11103-014-0179-8. [DOI] [PubMed] [Google Scholar]
- 19.Huang X, Madan A. CAP3: a DNA sequence assembly program. Genome Res. 1999;9(9):868–77. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amborella Genome Project The Amborella genome and the evolution of flowering plants. Science. 2013;342(6165):1241089. doi: 10.1126/science.1241089. [DOI] [PubMed] [Google Scholar]
- 21.Chanderbali AS, Albert VA, Leebens-Mack J, Altman NS, Soltis DE, Soltis PS. Transcriptional signatures of ancient floral developmental genetics in avocado (Persea americana; Lauraceae) Proc Natl Acad Sci. 2009;106(22):8929–34. doi: 10.1073/pnas.0811476106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42(D1):D222–30. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34(Web Server issue):W293–7. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35(Web Server issue):W182–5. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, et al. A gene expression map of Arabidopsis thaliana development. Nat Genet. 2005;37(5):501–6. doi: 10.1038/ng1543. [DOI] [PubMed] [Google Scholar]
- 26.Aceituno FF, Moseyko N, Rhee SY, Gutierrez RA. The rules of gene expression in plants: organ identity and gene body methylation are key factors for regulation of gene expression in Arabidopsis thaliana. BMC Genomics. 2008;9:438. doi: 10.1186/1471-2164-9-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nobuta K, Venu RC, Lu C, Belo A, Vemaraju K, Kulkarni K, et al. An expression atlas of rice mRNAs and small RNAs. Nat Biotechnol. 2007;25(4):473–7. doi: 10.1038/nbt1291. [DOI] [PubMed] [Google Scholar]
- 28.Jiao Y, Tausta SL, Gandotra N, Sun N, Liu T, Clay NK, et al. A transcriptome atlas of rice cell types uncovers cellular, functional and developmental hierarchies. Nat Genet. 2009;41(2):258–63. doi: 10.1038/ng.282. [DOI] [PubMed] [Google Scholar]
- 29.Benedito VA, Torres-Jerez I, Murray JD, Andriankaja A, Allen S, Kakar K, et al. A gene expression atlas of the model legume Medicago truncatula. Plant J. 2008;55(3):504–13. doi: 10.1111/j.1365-313X.2008.03519.x. [DOI] [PubMed] [Google Scholar]
- 30.Hogslund N, Radutoiu S, Krusell L, Voroshilova V, Hannah MA, Goffard N, et al. Dissection of symbiosis and organ development by integrated transcriptome analysis of Lotus japonicus mutant and wild-type plants. PLoS One. 2009;4(8) doi: 10.1371/journal.pone.0006556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 32.Stekel DJ, Git Y, Falciani F. The comparison of gene expression from multiple cDNA libraries. Genome Res. 2000;10(12):2055–61. doi: 10.1101/gr.GR-1325RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fizames C, Muños S, Cazettes C, Nacry P, Boucherez J, Gaymard F, et al. The Arabidopsis root transcriptome by serial analysis of gene expression. Gene identification using the genome sequence. Plant Physiol. 2004;134(1):67–80. doi: 10.1104/pp.103.030536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flanagan CA, Ma H. Spatially and temporally regulated expression of the MADS-box gene AGL2 in wild-type and mutant Arabidopsis flowers. Plant Mol Biol. 1994;26(2):581–95. doi: 10.1007/BF00013745. [DOI] [PubMed] [Google Scholar]
- 35.Yoo SK, Wu X, Lee JS, Ahn JH. AGAMOUS-LIKE 6 is a floral promoter that negatively regulates the FLC/MAF clade genes and positively regulates FT in Arabidopsis. Plant J. 2011;65(1):62–76. doi: 10.1111/j.1365-313X.2010.04402.x. [DOI] [PubMed] [Google Scholar]
- 36.Jack T, Brockman LL, Meyerowitz EM. The homeotic gene APETALA3 of Arabidopsis thaliana encodes a MADS box and is expressed in petals and stamens. Cell. 1992;68(4):683–97. doi: 10.1016/0092-8674(92)90144-2. [DOI] [PubMed] [Google Scholar]
- 37.Goto K, Meyerowitz EM. Function and regulation of the Arabidopsis floral homeotic gene PISTILLATA. Genes Dev. 1994;8(13):1548–60. doi: 10.1101/gad.8.13.1548. [DOI] [PubMed] [Google Scholar]
- 38.Jansson S. The light-harvesting chlorophyll a/b-binding proteins. Biochim Biophys Acta. 1994;1184(1):1–19. doi: 10.1016/0005-2728(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 39.Jansson S. A guide to the Lhc genes and their relatives in Arabidopsis. Trends Plant Sci. 1999;4(6):236–40. doi: 10.1016/S1360-1385(99)01419-3. [DOI] [PubMed] [Google Scholar]
- 40.Celton JM, Kelner JJ, Martinez S, Bechti A, Khelifi Touhami A, James MJ, et al. Fruit self-thinning: a trait to consider for genetic improvement of apple tree. PLoS One. 2014;9(3) doi: 10.1371/journal.pone.0091016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maurer S, Waschatko G, Schach D, Zielbauer BI, Dahl J, Weidner T, et al. The role of intact oleosin for stabilization and function of oleosomes. J Phys Chem B. 2013;117(44):13872–83. doi: 10.1021/jp403893n. [DOI] [PubMed] [Google Scholar]
- 42.Lu QY, Arteaga JR, Zhang Q, Huerta S, Go VL, Heber D. Inhibition of prostate cancer cell growth by an avocado extract: role of lipid-soluble bioactive substances. J Nutr Biochem. 2005;16(1):23–30. doi: 10.1016/j.jnutbio.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 43.Dunwell JM. Cupins: a new superfamily of functionally diverse proteins that include germins and plant storage proteins. Biotechnol Genet Eng Rev. 1998;15:1–32. doi: 10.1080/02648725.1998.10647950. [DOI] [PubMed] [Google Scholar]
- 44.MacMillan CP, Mansfield SD, Stachurski ZH, Evans R, Southerton SG. Fasciclin-like arabinogalactan proteins: specialization for stem biomechanics and cell wall architecture in Arabidopsis and Eucalyptus. Plant J. 2010;62(4):689–703. [DOI] [PubMed]
- 45.van Hoof NALM, Hassinen VH, Hakvoort HWJ, Ballintijn KF, Schat H, Verkleij JAC, et al. Enhanced copper tolerance in Silene vulgaris (Moench) Garcke populations from copper mines is associated with increased transcript levels of a 2b-type metallothionein gene. Plant Physiol. 2001;126(4):1519–26. doi: 10.1104/pp.126.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buchanan-Wollaston V. Isolation of cDNA clones for genes that are expressed during leaf senescence in Brassica napus (Identification of a gene encoding a senescence-specific metallothionein-like protein) Plant Physiol. 1994;105(3):839–46. doi: 10.1104/pp.105.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen HJ, Hou WC, Yang CY, Huang DJ, Liu JS, Lin YH. Molecular cloning of two metallothionein-like protein genes with differential expression patterns from sweet potato (Ipomoea batatas) leaves. J Plant Physiol. 2003;160(5):547–55. doi: 10.1078/0176-1617-01040. [DOI] [PubMed] [Google Scholar]
- 48.Coupe SA, Taylor JE, Roberts JA. Characterisation of an mRNA encoding a metallothionein-like protein that accumulates during ethylene-promoted abscission of Sambucus nigra L. leaflets. Planta. 1995;197(3):442–7. doi: 10.1007/BF00196665. [DOI] [PubMed] [Google Scholar]
- 49.Dunaeva M, Adamska I. Identification of genes expressed in response to light stress in leaves of Arabidopsis thaliana using RNA differential display. Eur J Biochem. 2001;268(21):5521–9. doi: 10.1046/j.1432-1033.2001.02471.x. [DOI] [PubMed] [Google Scholar]
- 50.Potenza C, Thomas SH, Sengupta-Gopalan C. Genes induced during early response to Meloidogyne incognita in roots of resistant and susceptible alfalfa cultivars. Plant Sci. 2001;161(2):289–99. doi: 10.1016/S0168-9452(01)00415-0. [DOI] [PubMed] [Google Scholar]
- 51.Oztur ZN, Talame V, Deyholos M, Michalowski CB, Galbraith DW, Gozukirmizi N, et al. Monitoring large-scale changes in transcript abundance in drought- and salt-stressed barley. Plant Mol Biol. 2002;48(5–6):551–73. doi: 10.1023/A:1014875215580. [DOI] [PubMed] [Google Scholar]
- 52.Clendennen SK, May GD. Differential gene expression in ripening banana fruit. Plant Physiol. 1997;115(2):463–9. doi: 10.1104/pp.115.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reid SJ, Ross GS. Up-regulation of two cDNA clones encoding metallothionein-like proteins in apple fruit during cool storage. Physiol Plant. 1997;100(1):183–9. doi: 10.1111/j.1399-3054.1997.tb03471.x. [DOI] [Google Scholar]
- 54.Ledger SE, Gardner RC. Cloning and characterization of five cDNAs for genes differentially expressed during fruit development of kiwifruit (Actinidia deliciosa var. deliciosa) Plant Mol Biol. 1994;25(5):877–86. doi: 10.1007/BF00028882. [DOI] [PubMed] [Google Scholar]
- 55.Davies C, Robinson SP. Differential screening indicates a dramatic change in mRNA profiles during grape berry ripening. Cloning and characterization of cDNAs encoding putative cell wall and stress response proteins. Plant Physiol. 2000;122(3):803–12. doi: 10.1104/pp.122.3.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moriguchi T, Kita M, Hisada S, Endo-Inagaki T, Omura M. Characterization of gene repertoires at mature stage of citrus fruits through random sequencing and analysis of redundant metallothionein-like genes expressed during fruit development. Gene. 1998;211(2):221–7. doi: 10.1016/S0378-1119(98)00138-3. [DOI] [PubMed] [Google Scholar]
- 57.Nam YW, Tichit L, Leperlier M, Cuerq B, Marty I, Lelievre JM. Isolation and characterization of mRNAs differentially expressed during ripening of wild strawberry (Fragaria vesca L.) fruits. Plant Mol Biol. 1999;39(3):629–36. doi: 10.1023/A:1006179928312. [DOI] [PubMed] [Google Scholar]
- 58.Moyle R, Fairbairn DJ, Ripi J, Crowe M, Botella JR. Developing pineapple fruit has a small transcriptome dominated by metallothionein. J Exp Bot. 2005;56(409):101–12. doi: 10.1093/jxb/eri015. [DOI] [PubMed] [Google Scholar]
- 59.Grover A. Plant chitinases: genetic diversity and physiological roles. Crit Rev Plant Sci. 2012;31(1):57–73. doi: 10.1080/07352689.2011.616043. [DOI] [Google Scholar]
- 60.Peumans WJ, Proost P, Swennen RL, Van Damme EJ. The abundant class III chitinase homolog in young developing banana fruits behaves as a transient vegetative storage protein and most probably serves as an important supply of amino acids for the synthesis of ripening-associated proteins. Plant Physiol. 2002;130(2):1063–72. doi: 10.1104/pp.006551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCollum TG, Doostdar H, Mayer RT, McDonald RE. Characterization of chitinases and β-1,3-glucanases in grapefruit flavedo during fruit development. Physiol Plant. 1997;99(3):486–94. doi: 10.1111/j.1399-3054.1997.tb00564.x. [DOI] [Google Scholar]
- 62.Marin-Rodriguez MC, Smith DL, Manning K, Orchard J, Seymour GB. Pectate lyase gene expression and enzyme activity in ripening banana fruit. Plant Mol Biol. 2003;51(6):851–7. doi: 10.1023/A:1023057202847. [DOI] [PubMed] [Google Scholar]
- 63.Seymour GB, Ostergaard L, Chapman NH, Knapp S, Martin C. Fruit development and ripening. Annu Rev Plant Biol. 2013;64:219–41. doi: 10.1146/annurev-arplant-050312-120057. [DOI] [PubMed] [Google Scholar]
- 64.Yang SF, Hoffman NE. Ethylene biosynthesis and its regulation in higher plants. Annu Rev Plant Physiol. 1984;35(1):155–89. doi: 10.1146/annurev.pp.35.060184.001103. [DOI] [Google Scholar]
- 65.Liu X, Shiomi S, Nakatsuka A, Kubo Y, Nakamura R, Inaba A. Characterization of ethylene biosynthesis associated with ripening in banana fruit. Plant Physiol. 1999;121(4):1257–65. doi: 10.1104/pp.121.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chaves ALS, Mello-Farias PC. Ethylene and fruit ripening: from illumination gas to the control of gene expression, more than a century of discoveries. Genet Mol Biol. 2006;29:508–15. doi: 10.1590/S1415-47572006000300020. [DOI] [Google Scholar]
- 67.Lincoln JE, Fischer RL. Regulation of gene expression by ethylene in wild-type and rin tomato (Lycopersicon esculentum) fruit. Plant Physiol. 1988;88(2):370–4. doi: 10.1104/pp.88.2.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu M, Chen G, Zhou S, Tu Y, Wang Y, Dong T, et al. A new tomato NAC (NAM/ATAF1/2/CUC2) transcription factor, SlNAC4, functions as a positive regulator of fruit ripening and carotenoid accumulation. Plant Cell Physiol. 2014;55(1):119–35. doi: 10.1093/pcp/pct162. [DOI] [PubMed] [Google Scholar]
- 69.Thompson AJ, Tor M, Barry CS, Vrebalov J, Orfila C, Jarvis MC, et al. Molecular and genetic characterization of a novel pleiotropic tomato-ripening mutant. Plant Physiol. 1999;120(2):383–90. doi: 10.1104/pp.120.2.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vrebalov J, Ruezinsky D, Padmanabhan V, White R, Medrano D, Drake R, et al. A MADS-box gene necessary for fruit ripening at the tomato ripening-inhibitor (rin) locus. Science. 2002;296(5566):343–6. doi: 10.1126/science.1068181. [DOI] [PubMed] [Google Scholar]
- 71.Giovannoni JJ. Genetic regulation of fruit development and ripening. Plant Cell. 2004;16(Suppl):S170–80. doi: 10.1105/tpc.019158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Manning K, Tor M, Poole M, Hong Y, Thompson AJ, King GJ, et al. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat Genet. 2006;38(8):948–52. doi: 10.1038/ng1841. [DOI] [PubMed] [Google Scholar]
- 73.Giovannoni JJ. Fruit ripening mutants yield insights into ripening control. Curr Opin Plant Biol. 2007;10(3):283–9. doi: 10.1016/j.pbi.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 74.Ito Y, Kitagawa M, Ihashi N, Yabe K, Kimbara J, Yasuda J, et al. DNA-binding specificity, transcriptional activation potential, and the rin mutation effect for the tomato fruit-ripening regulator RIN. Plant J. 2008;55(2):212–23. doi: 10.1111/j.1365-313X.2008.03491.x. [DOI] [PubMed] [Google Scholar]
- 75.Vrebalov J, Pan IL, Arroyo AJ, McQuinn R, Chung M, Poole M, et al. Fleshy fruit expansion and ripening are regulated by the tomato SHATTERPROOF gene TAGL1. Plant Cell. 2009;21(10):3041–62. doi: 10.1105/tpc.109.066936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lin Z, Hong Y, Yin M, Li C, Zhang K, Grierson D. A tomato HD-Zip homeobox protein, LeHB-1, plays an important role in floral organogenesis and ripening. Plant J. 2008;55(2):301–10. doi: 10.1111/j.1365-313X.2008.03505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27(2):221–4. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 78.Kombrink E, Somssich IE. Pathogenesis-related proteins and plant defense. In: Carroll G, Tudzynski P, editors. Plant relationships vol 5. Berlin Heidelberg: Springer; 1997. pp. 107–28. [Google Scholar]
- 79.Beisson F, Koo AJK, Ruuska S, Schwender J, Pollard M, Thelen JJ, et al. Arabidopsis genes involved in acyl lipid metabolism. A 2003 census of the candidates, a study of the distribution of expressed sequence tags in organs, and a web-based database. Plant Physiol. 2003;132(2):681–97. doi: 10.1104/pp.103.022988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Troncoso-Ponce MA, Kilaru A, Cao X, Durrett TP, Fan J, Jensen JK, et al. Comparative deep transcriptional profiling of four developing oilseeds. Plant J. 2011;68(6):1014–27. doi: 10.1111/j.1365-313X.2011.04751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gaydou EM, Lozano Y, Ratovohery J. Triglyceride and fatty acid compositions in the mesocarp of Persea americana during fruit development. Phytochemistry. 1987;26(6):1595–7. doi: 10.1016/S0031-9422(00)82251-7. [DOI] [Google Scholar]
- 82.Werman MJ, Neeman I. Avocado oil production and chemical characteristics. J Am Oil Chem Soc. 1987;64(2):229–32. doi: 10.1007/BF02542007. [DOI] [Google Scholar]
- 83.Shanklin J, Cahoon EB. Desaturation and related modifications of fatty acids1. Annu Rev Plant Physiol Plant Mol Biol. 1998;49:611–41. [DOI] [PubMed]
- 84.Kilaru A, Cao X, Dabbs PB, Sung H, Rahman MM, Thrower N, Zynda G, Podicheti R, Ibarra-Laclette E, Herrera-Estrella L, Mockaitis K, Ohlrogge JB. Oil Biosynthesis in a Basal Angiosperm: Transcriptome Analysis of Persea Americana Mesocarp. BMC Plant Biology 2015; in press. [DOI] [PMC free article] [PubMed]
- 85.Bora PS, Narain N, Rocha RVM, Paulo MQ. Characterization of the oils from the pulp and seeds of avocado (cultivar: Fuerte) fruits. Grasas Aceites. 2001;52(3/4):171–4.
- 86.Dreher ML, Davenport AJ. Hass avocado composition and potential health effects. Crit Rev Food Sci Nutr. 2012;53(7):738–50. doi: 10.1080/10408398.2011.556759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brooke DG, Shelley EJ, Roberts CG, Denny WA, Sutherland RL, Butt AJ. Synthesis and in vitro evaluation of analogues of avocado-produced toxin (+)-(R)-persin in human breast cancer cells. Bioorg Med Chem. 2011;19(23):7033–43. doi: 10.1016/j.bmc.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 88.Cortes-Rodriguez MA, Lopez-Gomez R, Martinez-Pacheco MM, Suarez-Rodriguez LM, Hernandez-Garcia A, Salgado-Garciglia R, et al. In vitro propagation of mexican race avocado (Persea americana Mill. var. drymifolia) Acta Horticulturae (ISHS) 2011;923:47–52. [Google Scholar]
- 89.Jiang Y, Fu J. Ethylene regulation of fruit ripening: molecular aspects. Plant Growth Regul. 2000;30(3):193–200. doi: 10.1023/A:1006348627110. [DOI] [Google Scholar]
- 90.Niu B, Fu L, Sun S, Li W. Artificial and natural duplicates in pyrosequencing reads of metagenomic data. BMC Bioinformatics. 2010;11:187. doi: 10.1186/1471-2105-11-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marchant A, Mougel F, Almeida C, Jacquin-Joly E, Costa J, Harry M. De novo transcriptome assembly for a non-model species, the blood-sucking bug Triatoma brasiliensis, a vector of Chagas disease. Genetica. 2015;143(2):225–39. doi: 10.1007/s10709-014-9790-5. [DOI] [PubMed] [Google Scholar]
- 92.Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32(Database issue):D277–80. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95(25):14863–8. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 95.Nischal L, Mohsin M, Khan I, Kardam H, Wadhwa A, Abrol YP, et al. Identification and comparative analysis of microRNAs associated with low-N tolerance in rice genotypes. PLoS One. 2012;7(12) doi: 10.1371/journal.pone.0050261. [DOI] [PMC free article] [PubMed] [Google Scholar]