

Abstract
Pathogenic bacteria must contend with immune systems that actively restrict the availability of nutrients and cofactors, and create a hostile growth environment. To deal with these hostile environments, pathogenic bacteria have evolved or acquired virulence determinants that aid in the acquisition of nutrients. This connection between pathogenesis and nutrition may explain why regulators of metabolism in nonpathogenic bacteria are used by pathogenic bacteria to regulate both metabolism and virulence. Such coordinated regulation is presumably advantageous because it conserves carbon and energy by aligning synthesis of virulence determinants with the nutritional environment. In Gram-positive bacterial pathogens, at least three metabolite-responsive global regulators, CcpA, CodY, and Rex, have been shown to coordinate the expression of metabolism and virulence genes. In this chapter, we discuss how environmental challenges alter metabolism, the regulators that respond to this altered metabolism, and how these regulators influence the host-pathogen interaction.
For prototrophic bacteria, central metabolism (i.e., glycolysis, the pentose phosphate pathway, and the Krebs cycle) supplies the 13 biosynthetic intermediates necessary to synthesize all biomolecules (Fig. 1). Gram-positive bacteria (i.e., Actinobacteria and Firmicutes) exhibit a diverse collection of central metabolic capabilities that have been shaped by reductive evolution. Some Gram-positive bacteria (e.g., Bacillus anthracis and Staphylococcus aureus) have complete central metabolic pathways, but others (e.g., Streptococcus pyogenes and Enterococcus faecium) have Krebs cycle deficiencies, and some have multiple central metabolism deficiencies (e.g., Mycoplasma genitalium and Ureaplasma parvum). These differences in central metabolic capabilities are also reflected in the bacteria’s ability to persist away from a host organism; specifically, the more metabolically impaired the bacterium, the more dependent it is on its host. In essence, hosts serve as a reservoir for metabolites that overcome deficiencies in central and intermediary metabolism. Metabolic deficiencies are not created by only reductive evolution; they are also created when bacteria encounter stressful environments (e.g., iron limitation or a host immune response) that alter carbon flux (1, 2). These changes in flux alter the metabolome, which can modulate the activity of metabolite-responsive global regulators such as CodY, CcpA, Rex, and RpiR. In the first portion of this chapter, we discuss how genetic, environmental, and nutritional conditions alter the metabolome, primarily central metabolism, and in the second part, how these metabolic changes influence the activity of metabolite-responsive regulators. Finally, we discuss how metabolism and metabolite-responsive global regulators influence the outcomes of host-pathogen interactions. This review references only those manuscripts published through December 2013.
FIGURE 1.

A simplified view of bacterial physiology. The 13 biosynthetic intermediates discussed in this chapter are all derived from the three metabolic pathways of central metabolism. Alterations in the availability of these biosynthetic intermediates always affect virulence factor synthesis. doi:10.1128/microbiolspec.MBP-0004-2014.f1
GRAM-POSITIVE METABOLISM
Glycolysis (Embden–Meyerhof–Parnas Pathway)
Glycolysis is the catabolic pathway that generates two molecules of pyruvate per molecule of glucose and, in the process, reduces two molecules of NAD+ and produces a net gain of two ATP molecules. In addition to providing a modest amount of ATP by substrate-level phosphorylation, glycolysis also generates seven (i.e., glucose-6-phosphate, fructose-6-phosphate, dihydroxy-acetone phosphate, glyceraldehyde-3-phosphate, phosphoenolpyruvate [PEP], pyruvate, and acetylCoenzyme A) of the 13 biosynthetic intermediates that are used to synthesize all other organic molecules in a bacterium. Glycolysis is largely fueled by the PEP-dependent phosphotransferase system (PTS), a transport system that bacteria use to bring sugars into the cytoplasm using energy transferred from PEP (3, 4). In its simplest form, the PTS uses three enzymes to transfer the high-energy phosphate from PEP to the sugar being translocated, enzyme I (EI), enzyme II (EII, the permease), and a histidine-containing phosphocarrier protein (HPr) (3). The transfer of the high-energy phosphate from PEP to a carbohydrate initiates with transfer of phosphate from PEP to a histidine residue on EI, which is then transferred to a histidine residue on HPr. The phosphorylated HPr transfers the high-energy phosphate to EII, which transfers it to the carbohydrate. Transfer of a phosphate to a carbohydrate has two primary consequences: first, the phosphorylated carbohydrate is no longer recognized by the EII permease, preventing efflux of the carbohydrate from the cytoplasm; and second, the phosphorylated carbohydrate is activated for glycolysis. Glucose and fructose can also be activated for glycolysis intracellularly by transfer of a phosphate from ATP by hexokinase (5).
In glycolysis, activated glucose (i.e., glucose-6-phosphate) is processed in a series of key enzymatic reactions by phosphoglucose isomerase, phosphofructokinase, and aldolase to generate glyceraldehyde-3-phosphate. The importance of these enzymatic reactions is evident from the complex feedback and feedforward allosteric regulation that controls their activity. For example, PEP, ATP, and citrate are inhibitors of phosphofructokinase (6), while ADP and GDP enhance its enzymatic activity (7). Because the activity of enzymes like phosphofructokinase is modulated by allosteric effectors, changes in the intracellular concentrations of these effectors will alter carbon flow through glycolysis. Alterations in carbon flow through glycolysis will change the intracellular concentrations of glycolytic intermediates such as fructose-1,6-bisphosphate (FBP). In other words, anything that can alter the concentration of an allosteric effector will alter glycolysis.
Free iron in a eukaryotic cell is present at a concentration of 10−18 M, meaning that free iron is unavailable to invading pathogens. Whereas the activity of phosphofructokinase is independent of iron, phosphofructokinase activity and carbon flux through glycolysis are nonetheless affected by growth in iron-limited conditions. Two possible explanations can be considered: First, variations in the availability of iron alter transcription of the phosphofructokinase gene in Mycobacterium smegmatis, Enterococcus faecalis, and S. aureus (8–10). Second, some bacteria, when cultivated in an iron-limiting medium, accumulate citric acid in the cytosol and the culture medium due to a metabolic block in the Krebs cycle at aconitase (2, 11–13). Because citrate is an allosteric inhibitor of phosphofructokinase, the accumulation of citrate should lead to an increased concentration of fructose-6-phosphate or metabolites derived from fructose-6-phosphate. When the Krebs cycle in Staphylococcus epidermidis is genetically inactivated or the bacteria are cultivated in iron-limited medium, glucose-6-phosphate and amino sugars accumulate, which is indicative of reduced phosphofructokinase activity (1, 2, 12). Decreased phosphofructokinase activity limits the availability of downstream biosynthetic intermediates and precursors, which decreases the bacterium’s ability to assemble macromolecules (Fig. 1). The allosteric and genetic regulation of phosphofructokinase provides an excellent example of the interconnection between metabolism and the bacterial environment, but these connections also rely on metabolite-responsive regulators to control the adaptive response to environmental changes (discussed section 2).
Pentose Phosphate Pathway (Warburg-Lipmann-Dickens-Horecker Shunt)
The processing of activated glucose through the pentose phosphate pathway (PPP) produces three of the 13 biosynthetic intermediates; specifically, ribose-5-phosphate, sedoheptulose-7-phosphate, and erythrose-4-phosphate (14, 15). Two of these biosynthetic intermediates, ribose-5-phosphate and erythrose-4-phosphate, are essential for the synthesis of purines, histidine, and aromatic amino acids. The third intermediate, sedoheptulose-7-phosphate, in conjunction with glyceraldehyde-3-phosphate, can be used by tranketolase to generate ribose-5-phosphate or by transaldolase to generate fructose-6-phosphate and erythrose-4-phosphate (16). In addition to providing biosynthetic intermediates, the PPP also generates two molecules of NADPH per molecule of glucose-6-phosphate, which can be used as electron donors in biosynthetic reactions such as fatty acid and glutamate biosynthesis. The enzymatic reactions that reduce NADP+ to NADPH/ H+ occur in the oxidative portion of the PPP that produces ribulose-5-phosphate from activated glucose (15, 17). This process starts with the oxidation of glucose-6-phosphate to 6-phosphogluconolactone catalyzed by glucose-6-phosphate dehydrogenase. In Gram-positive bacteria, reductive evolution has caused the loss of glucose-6-phosphate dehydrogenase (zwf) in the oxidative portion of the PPP in Mycoplasma sp., Streptococcus pyogenes, S. mutans, S. agalactiae, and Clostridium difficile [(18, 19), http://biocyc.org]. While these bacteria lack part of the oxidative portion of the PPP, most possess the nonoxidative portion. One notable exception is Mycoplasma suis, which lacks the PPP (20), and other Mycoplasma sp. that lack transaldolase (Somerville, unpublished observations). The metabolic consequences of the loss of glucose-6-phosphate dehydrogenase are a decreased ability to generate pentose sugars and reducing potential, while the loss of transaldolase prevents regeneration of fructose-6-phosphate from sedoheptulose-7-phosphate. Despite the fact that reductive evolution has resulted in PPP variation, it is interesting to note that pentose phosphate metabolism is frequently increased in Gram-positive pathogens in response to environmental stresses and in infection models (21–24).
Increased carbon flow through the oxidative portion of the PPP generates NADPH, while the nonoxidative branch produces fructose-6-phosphate. In addition to biosynthetic reactions, NADPH is required for the enzymatic reduction of oxidized glutathione, thioredoxin, bacillithiol, mycothiol, and coenzyme A (25–28). As an example, thioredoxin reductase catalyzes the transfer of electrons from NADPH to the active site of thioredoxin via flavin adenine dinucleotide (29, 30). Reduced thioredoxin, in concert with other low-molecular-weight thiols, is critical for reducing protein disulfides and providing electrons to ribonucleotide reductase, methionine sulfoxide reductase, mycothiol disulfide reductase, and other enzymes (31). While NADPH is necessary for the function of reductases, transaldolase in the PPP is also important for redox homeostasis because it produces fructose-6-phosphate from sedoheptulose-7-phosphate and glyceraldehyde-3-phosphate. Fructose-6-phosphate is a precursor for N-acetylglucosamine, which is required for bacillithiol and mycothiol biosynthesis (26, 32). These connections provide a rationale for the observation that increased carbon flow through the PPP is often associated with stressful conditions, environments, or infection (21–23, 33).
While induction of the PPP during an infection supports redox homeostasis, it also is important for intra-cellular pathogens like L. monocytogenes to overcome one of the deleterious effects of interferon-γ: namely, the indoleamine 2,3-dioxygenase-induced depletion of tryp-tophan (34). Interferon-γ-activated macrophages increase synthesis of indoleamine 2,3-dioxygenase, which cleaves the 2,3-double bond in the indole ring of tryptophan, effectively depleting the cell of tryptophan and depriving bacteria of an important amino acid (35). To counter the host-mediated depletion of tryptophan, intracellular bacteria like L. monocytogenes are able to synthesize tryptophan from the PPP intermediate erythrose-4-phosphate (36). Synthesis proceeds via the chorismate pathway to anthranilate and, subsequently, to tryptophan by enzymes coded within the trp operon (trpEGDCFBA). While the ability to synthesize tryptophan can rescue some intracellular bacteria, bacteria that synthesize tryptophan but live predominantly extracellularly, such as S. aureus or Streptococcus pneumoniae, remain sensitive to host-mediated depletion of tryptophan (37, 38). It is unclear why bacteria that can synthesize tryptophan remain sensitive to its depletion; however, addition of exogenous tryptophan to the tissue culture media relieves this bacteriostatic condition. In summary, the PPP is important for maintaining bacterial redox homeostasis and biosynthesis during a host-pathogen interaction.
Tricarboxylic acid cycle (Krebs cycle)
As mentioned earlier, reductive evolution has strongly influenced central metabolism in Gram-positive bacteria, and the Krebs cycle is the most prominent example of this evolution (39–41). Many Gram-positive pathogens lack all or most of the Krebs cycle. Most Streptococcus spp. (S. mutans being an exception), Enterococcus spp., Erysipelothrix rhusiopathiae, Mycoplasma spp., and Ureaplasma spp. lack the Krebs cycle, which prevents the pyruvate-derived synthesis of three of the 13 biosynthetic intermediates [i.e., oxaloacetate, α-ketoglutarate (aka 2-oxoglutarate), and succinate/succinyl-CoA; (41) and Somerville, unpublished observations]. These three biosynthetic intermediates are critical for the de novo synthesis of many amino acids and porphyrins; for example, oxaloacetate is a precursor for biosynthesis of aspartate, asparagine, lysine, cysteine, threonine, isoleucine, and methionine; α-ketoglutarate is a precursor of glutamate, glutamine, arginine, and proline; and succinate is used in porphyrin biosynthesis. Thus, the evolutionary loss of Krebs cycle genes is reflected in the complex amino acid and vitamin requirements necessary for cultivation of these bacteria (42–44).
It is hypothesized that the Krebs cycle evolved from two amino acid biosynthetic pathways: one oxidative pathway and one reductive pathway (45). This metabolic arrangement allows the formation of a bifurcated pathway starting at pyruvate, with branches leading to succinate/succinyl-CoA and α-ketoglutarate. This bifurcated configuration is found in several Gram-positive pathogens; for example, L. monocytogenes, Clostridium difficile, and Peptostreptococcus anaerobius lack the genes for α-ketoglutarate dehydrogenase, succinyl-CoA synthetase, and succinate dehydrogenase (46) and http://biocyc.org, while Corynebacterium diphtheriae lacks succinyl-CoA synthetase (this may be compensated for by a putative succinyl-CoA:coenzyme A transferase) (47). In these examples, bacteria have maintained the Krebs cycle in an incomplete format but one that still allows the generation of oxaloacetate, α-ketoglutarate, and succinate. In addition, these bacteria use anaerobic respiration to oxidize NADH and FADH by running part of the Krebs cycle backwards (i.e., oxaloacetate to succinate). The use of anaerobic respiration also underscores the fact that most Gram-positive pathogens using this bifurcated Krebs cycle are anaerobes, L. monocytogenes being the exception. That said, some facultative anaerobes (e.g., B. subtilis) with a complete Krebs cycle also bifurcate the pathway when grown anaerobically (48).
Though an incomplete Krebs cycle is common in Gram-positive bacteria, two of the most prevalent Gram-positive pathogens worldwide have complete Krebs cycles: namely, M. tuberculosis and S. aureus. In both of these bacteria, the Krebs cycle is important for pathogenesis (49–51); however, having complete Krebs cycles and being important for pathogenesis is where the similarities end. One major difference between M. tuberculosis and S. aureus is that M. tuberculosis has a glyoxylate cycle, which allows the conservation of carbon by bypassing the Krebs cycle decarboxylation reactions (52). In Gram-positive bacteria, the glyoxylate cycle is primarily restricted to Actinobacteria (i.e., Mycobacterium sp., Nocardia sp., and Rhodococcus sp.); however, it is also found in the Firmicute B. anthracis. The presence of the glyoxylate cycle in Actinobacteria is likely a reflection of the poor nutrient environment they encounter when residing inside of a phagocytic cell or when walled-off in a granuloma (53–55). A second major difference between M. tuberculosis and S. aureus is that staphylococci exhibit carbon catabolite repression of the Krebs cycle when cultivated in media containing glucose (56, 57). This does not appear to be the case with M. tuberculosis (58), which grows best on non-glucose carbon sources like glycerol, acetate, and fatty acids that are degraded to acetyl CoA (59). The utilization of acetate explains why M. tuberculosis uses the glyoxylate shunt. Doing so prevents the formation of a futile cycle in which two carbons enter the Krebs cycle and two carbons are lost through decarboxylation reactions. For the remainder of this chapter, discussion of the Krebs cycle will be kept to the Firmicutes.
In general, Gram-positive bacteria repress transcription of Krebs cycle genes when cultivated in media containing a readily catabolizable carbohydrate and glutamate or glutamine (46, 60–63). This catabolite repression leads to the accumulation of incompletely oxidized metabolites/fermentation products in the culture media, most commonly acetic acid and lactic acid (61, 64). Once carbohydrates are depleted from the medium, these metabolites can be re-imported and used to fuel the Krebs cycle and generate the three biosynthetic intermediates. Catabolism of acetate begins with the ATP-dependent formation of a thioester bond between acetate and coenzyme A catalyzed by acetyl-CoA synthetase/acetyl-CoA ligase. At this point, acetyl-CoA can enter into the Krebs cycle via a condensation reaction with oxaloacetate that is catalyzed by citrate synthase, a process using the energy of thioester hydrolysis to drive carbon-carbon bond formation to form citric acid. As stated above, most Gram-positive pathogens lack the glyoxylate shunt; hence, two carbons are lost as CO2 for every two carbons (i.e., acetyl-CoA) that enter the Krebs cycle. For this reason, when biosynthetic intermediates are withdrawn from the Krebs cycle for biosynthesis, anaplerotic reactions are required to maintain carbon flow through the Krebs cycle. The most commonly used substrates for the anaplerotic reactions are amino acids (50). For instance, conversion of aspartate to oxaloacetate can start a new round of the Krebs cycle, allowing continued drawing off of intermediates. In total, catabolism of incompletely oxidized metabolites through the Krebs cycle provides biosynthetic intermediates (i.e., α-ketoglutarate, succinyl-CoA, and oxaloacetate), ATP, and reducing potential but consumes amino acids in the process.
Not only do genetic variation and catabolite repression of the Krebs cycle affect the availability of biosynthetic intermediates and ATP in Gram-positive bacteria, but Krebs cycle activity can also be altered by environmental changes (11, 60, 65–67). Like glycolysis or the PPP, altering carbon flow through the Krebs cycle will affect the intracellular concentrations of biosynthetic intermediates and precursors, ATP, and redox homeostasis. Of importance, the activity of metabolite-responsive regulators is controlled by intracellular concentrations of biosynthetic intermediates (68), amino acids (69), nucleic acids (70), and cofactors (e.g., iron) (71). In other words, altering metabolism provides a means to transduce external environmental changes into internal metabolic signals that alter the activity of metabolite-responsive regulators, which facilitate adaptation to the altered environment (72). The function of metabolite-responsive regulators will be discussed in the second part of this chapter.
Amino acid biosynthesis
As previously discussed for tryptophan, amino acids can be important factors in the host-pathogen interaction, and two of the more important amino acids for bacteria are glutamate and glutamine. These amino acids are important because they serve as the nitrogen donors in most biosynthetic processes (73). Synthesis of glutamate is dependent on the nutritional environment (74) and usually involves one of two enzymes: glutamate dehydrogenase or glutamine oxoglutarate aminotransferase (aka GOGAT or glutamate synthase). Glutamate dehydrogenase catalyzes the reductive amination of the Krebs cycle intermediate α-ketoglutarate by using the oxidation of NADH to drive the assimilation of ammonia. Glutamate synthase converts glutamine and α-ketoglutarate into two molecules of glutamate by a transamidation reaction using NADPH/H+ as a reductant (75). While glutamate is the nitrogen donor for most amino acids (exceptions being asparagine and tryptophan and histidine, which uses both glutamate and glutamine), glutamine also has an essential function as a nitrogen donor for the synthesis of tryptophan, amino sugars, and nucleic acids (73). Glutamine is synthesized by the condensation of glutamate and NH3 by glutamine synthetase, using the energy of ATP hydrolysis to catalyze the reaction. In all, these three enzymes and the reactions that they catalyze constitute the primary means of nitrogen assimilation in bacteria.
The assimilation of nitrogen into glutamate, and to a lesser extent, glutamine, allows transamination reactions that transfer amino groups from glutamate to an amino acid precursor. While all amino acid biosynthesis is important, we briefly describe branched-chain amino acid (BCAA) biosynthesis because the valine pathway also leads to synthesis of pantothenate and because of the importance of BCAA in modulating the activity of the Gram-positive metabolite-responsive regulator CodY (76). Like central metabolism, BCAA biosynthesis has been strongly influenced by reductive evolution. For example, E. rhusiopathiae, Mycoplasma spp., Ureaplasma spp., P. anaerobius, S. pyogenes, and S. agalactiae, lack all or most, of the genes necessary for the de novo synthesis of isoleucine, leucine, and valine, specifically, the ilvleu operon (http://biocyc.org). Obviously, the loss of BCAA biosynthetic genes creates auxotrophies for isoleucine, leucine, and valine, but this also means that pantothenate and coenzyme A biosynthesis are dependent on exogenous sources of valine. For Gram-positive bacteria that have BCAA biosynthetic pathways (e.g., Staphylococcus sp., Bacillus sp.), synthesis of valine and leucine begins with one of the 13 biosynthetic intermediates of central metabolism; namely, pyruvate. Acetolactate synthase (ilvB) catalyzes the thiamine pyrophosphate-dependent conversion of two pyruvate molecules into acetolactate, which is the substrate for the ketol-acid reductoisomerase encoded by ilvC. The products of ketol-acid reductoisomerase are oxidized NADP+ and 2,3-dihydroxy-isovalerate, the substrate for dihydroxy-acid dehydratase (ilvD) (77). Dihydroxy-acid dehydratase produces 2-oxoisovalerate, a key intermediate that sits at a branch point between valine and leucine biosynthesis (78). From this branch point, leucine is synthesized by the enzymes coded within the leuABCD cluster, and 2-oxoisovalerate is converted to valine by a branched-chain amino acid aminotransferase (e.g., YbgE or YwaA in B. subtilis) using the amino group of glutamate as the nitrogen donor.
In contrast to leucine and valine biosynthesis, isoleucine synthesis begins by condensation of pyruvate and 2-oxobutyrate (79). 2-oxobutyrate is not itself one of the canonical 13 precursors but is made from threonine by threonine dehydratase (ilvA)-catalyzed deamination; threonine is made from aspartate, a product of transamination of oxaloacetate, thereby connecting isoleucine biosynthesis to central metabolism. From 2-oxobutyrate, the enzymes that catalyze the synthesis of isoleucine are the same ones that catalyze the synthesis of valine; namely, acetolactate synthase (ilvB), ketol-acid reductoisomerase (ilvC), dihydroxy-acid dehydratase (ilvD), and branched-chain amino acid aminotransferases (ybgE and ywaA).
All biosynthetic reactions in a bacterium are dependent on the biosynthetic intermediates produced by central metabolism, and BCAA biosynthesis is no different (Fig. 1). Any perturbation of central metabolism has the potential to disrupt biosynthetic reactions like BCAA biosynthesis (21, 80, 81), and these disruptions affect the polymerizing and assembly reactions. As mentioned earlier, iron limitation creates metabolic blocks in the Krebs cycle that limit the availability of Krebs cycle intermediates (i.e., α-ketoglutarate, succinate and oxaloacetate). This decreased availability of intermediates alters the synthesis of amino acids, such as aspartate, which reduces the synthesis of threonine and BCAA synthesis. Not only is BCAA synthesis limited by the availability of intermediates during iron-limited growth, but the dihydroxy-acid dehydratase (IlvD) contains a [4Fe-4S] iron-sulfur cluster, which is susceptible to inactivation by iron-limitation or oxidative inactivation. When IlvD is inactive, the metabolic block in BCAA biosynthesis induces BCAA auxotrophy. In this example, the common cofactor requirements and the interconnections of metabolism create a “ripple effect” that cause metabolic changes seemingly unrelated to the nature of the perturbation. The severity of this ripple effect is determined by the extent of the perturbation and the availability of exogenous metabolites that can compensate for the loss of biosynthetic intermediates and precursors. To overcome these perturbations, bacteria have evolved/acquired metabolite-responsive regulators that facilitate adaptation and survival.
METABOLITE-RESPONSIVE GLOBAL REGULATORS THAT INFLUENCE VIRULENCE
Regulatory proteins that coordinately control metabolic and virulence genes provide compelling evidence that, from the bacterium’s point of view, virulence is one of many interrelated responses to specific environmental conditions. Such conditions may reflect nutrient availability, temperature, pH, oxidative stress, osmotic stress, or other stresses that bacteria may encounter within and outside of host environments. In Gram-positive bacteria, several such global regulatory proteins have been studied in detail. All of these regulators are found predominantly, if not exclusively, in Gram-positive species.
CcpA
Acting as a global regulator of carbon metabolism genes in response to the availability of certain preferred carbon sources, CcpA regulates dozens of metabolism and virulence genes in Bacillus (82, 83), Clostridium (84, 85), Staphylococcus (86, 87), Streptococcus (88–91), Lactococcus (92), and Enterococcus (93, 94). (It is important to note that not all pathways for metabolism of sugars or other carbon sources are under CcpA control, even though some of these pathways are affected by the availability of glucose and other rapidly metabolized sugars (95). Moreover, inducer exclusion is a major component of carbon catabolite repression in most bacteria (96).) CcpA proteins are members of the LacI family and bind to a DNA sequence (cre site) that is conserved among the various species. B. subtilis CcpA, the group member studied in the greatest detail, is activated as a DNA-binding protein by interaction with a phosphorylated form of HPr (97). HPr has two potential sites of phosphorylation. When phosphorylated on histidine-15 by EI of the PTS system, HPr is specifically involved in transferring phosphate to PTS EII. Serine-46, however, is phosphorylated by HPr kinase/ phosphorylase, whose kinase activity is activated by binding of FBP. It is the HPr-Ser46~P form that binds to CcpA and increases its activity as a DNA-binding protein (98). Moreover, HPr-His15~P does not activate CcpA, and HPr-Ser46~P is inactive in the PTS. Since accumulation of FBP reflects the availability of glucose and other rapidly metabolized sugars, such sugars activate CcpA indirectly via HPr. In most other Gram-positive bacteria, the same complex of CcpA and HPr-Ser46~P is the active form of the regulator. For instance, in S. mutans, increased phosphorylation of HPr at serine-46 inhibits sugar uptake, presumably by preventing phosphorylation of HPr at histidine-15, thereby interrupting the PTS signaling pathway (99). In contrast, C. difficile CcpA appears to interact directly with FBP, bypassing the need for HPr (100).
CcpA activity responds to other signals as well. Phosphorylation of CcpA at two threonine residues by the S. aureus kinase PknB leads to loss of CcpA binding to cre sites and disruption of the CcpA regulon, as well as overexpression of the ccpA gene (101). The signals activating PknB are not known.
Some clinical isolates of S. aureus appear to be pro-line auxotrophs; mutations in the major proline transporter cause loss of virulence in several models of pathogenesis (102, 103). Mutations in ccpA or ptsH relieve the auxotrophy, suggesting that the auxotrophy is due to severe repression of the proline biosynthetic pathway by CcpA (104). Because S. aureus does not encode the conventional glutamate-to-proline pathway, proline can be made in these bacteria only as a by-product of arginine degradation (104). Arginine degradation is strongly repressed by CcpA/ HPr when cells are grown with glucose (104), explaining the apparent proline auxotrophy of glucose-grown cells.
The role of CcpA in virulence gene expression
Streptococcus
Multiple effects of CcpA on virulence gene expression have been observed in Streptococcus spp. A S. pneumoniae ccpA mutant shows reduced expression of the capsular polysaccharide locus (105) and is highly attenuated for mouse nasopharyngeal and lung infection (106). Indirect evidence that S. suis CcpA controls capsule biosynthesis came from analysis of the effect of a mutation in a virulence-associated surface protein on HPr phosphorylation at serine-46 (107). In S. mutans, the major cause of dental caries, a ccpA mutant produces more acid than its parent, grows better at low pH, and excretes acid more rapidly, indicating that CcpA normally holds the cariogenic potential of S. mutans in check when rapidly metabolizable carbon sources are in excess (108). Two other oral streptococci, S. gordonii and S. sanguis, help prevent caries by producing H2O2, an antagonist of other oral bacteria, including S. mutans, in response to carbon limitation and changes in oxygen tension. This H2O2 is produced by pyruvate oxidase, and the gene coding for pyruvate oxidase (spx or pox) is repressed by CcpA (109, 110). Thus, when rapidly metabolizable carbon sources become limiting, repression by CcpA of both caries-promoting genes and S. mutans-antagonizing factors is relieved. Autolysis of S. mutans, a factor in survival of biofilms, is also regulated by CcpA in response to the availability of glucose (111). In Group A Streptococcus, CcpA appears to activate transcription of mga, the gene that encodes a positive regulator of genes whose products mediate adhesion and invasion of host cells, as well as resistance to host defenses (112).
Clostridium
The major C. difficile toxins, TcdA and TcdB, are synthesized during the stationary phase in rich media, as long as the media do not contain glucose or other rapidly metabolizable carbon sources (113, 114). CcpA mediates the glucose-dependent repression of the major toxin locus indirectly by binding to the promoter region of the tcdR gene, which encodes the alternative sigma factor necessary for high-level toxin gene transcription (Fig. 2) (84). CcpA also represses many metabolic genes whose products may be important for growth in the GI tract (discussed later). The CcpA protein of C. perfringens is responsible for the glucose-dependent repression of two major toxins, alpha-toxin (phospholipase C) and theta-toxin (perfringolysin), which initiate gas gangrene (115). The inhibitory effect of glucose on type IV pilus-dependent gliding motility is also mediated by CcpA (116). Gliding motility is required for efficient biofilm formation, a process that increases the bacterium’s resistance to antibiotics and other environmental stresses (117). In contrast, production of the enterotoxin responsible for C. perfringens food poisoning is positively regulated by CcpA (118). Whether that effect is direct or indirect is uncertain.
FIGURE 2.
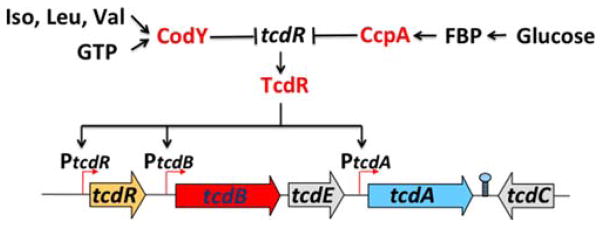
Synergistic repression of C. difficile toxin synthesis by CodY and CcpA. Responding independently to different nutritional signals, CcpA and CodY both bind to the regulatory region of the tcdR gene, repressing production of the alternative sigma factor necessary for high-level toxin gene (tcdA and tcdB) expression. doi:10.1128/microbiolspec.MBP-0004 -2014.f2
Staphylococcus
Interestingly, an S. aureus ccpA mutant shows decreased replication in the liver in a murine abscess model (104). This effect may be due in part to indirect positive regulation of immunodominant antigen B, α-hemolysin, and biofilm formation in the presence of glucose (119–121). On the other hand, synthesis of toxic shock syndrome toxin-1 and capsule is repressed by glucose via CcpA (121, 122).
Bacillus
Toxin gene expression in B. anthracis is subject to complex regulation involving an activator of toxin gene expression, AtxA, and multiple indirect regulators. CcpA, for instance, is needed for glucose-dependent induction of atxA transcription (123). Thus, a ccpA mutant is attenuated in virulence.
Other genera
In E. faecalis, CcpA activates expression of a collagen-binding colonization factor (124). In Listeria monocytogenes, effects of carbon availability on virulence gene expression are mediated by HPr independently of CcpA. Hyperphosphorylation of serine-46 leads to reduced activity of the global virulence gene regulator PrfA and, as a consequence, reduced expression of PrfA-dependent genes (125). This effect is not mediated by CcpA; instead, hyperphosphorylation of serine-46 interferes with phosphorylation of histidine-15, thereby inhibiting PTS-dependent transport, implying that sugar transporters or imported sugars are the direct modulators of PrfA activity (125, 126).
CodY
A global regulator of metabolism found in nearly all low G+C Gram-positive bacteria, CodY controls at least 200 genes in Bacillus (127), Clostridium (128), Staphylococcus (129, 130), Streptococcus (131, 132), Lactococcus (133), and Listeria (134). Lactobacillus spp. typically lack CodY, as do all high G+C Gram-positive bacteria. CodY activity is controlled by two types of ligands. The branched-chain amino acids (BCAAs) isoleucine, leucine, and valine are universal effectors of CodY activity (69, 133, 135), while GTP also activates CodY (136–138) in genera other than Streptococcus (139) and Lactococcus (140). The metabolic genes regulated by CodY, directly or indirectly, include those for amino acid biosynthetic pathways (typically isoleucine, leucine, valine, threonine, arginine, glutamate, and histidine), purine biosynthesis (particularly the steps from IMP to GMP), the Krebs cycle, energy metabolism, sugar and amino acid transport, carbon overflow metabolism, chemotaxis and motility, genetic competence, and sporulation. In all cases studied to date, most metabolism genes under CodY control are repressed by CodY; the major exceptions are carbon overflow pathways, which tend to be activated by CodY. In general, the genes repressed by CodY are expressed at low level during growth in rich medium until the cells reach the stationary phase but are expressed at high level during growth in a poor medium; conversely, genes whose transcription is activated by CodY are expressed during rapid growth.
The CodY ligands interact with the protein in different ways that are only partially understood. The BCAAs bind to an N-terminal GAF domain that is unstructured in the unliganded protein but folds around the BCAA side-chain, forming a hydrophobic pocket (141). Binding of the BCAA causes a conformational change in which the C-terminal winged helix-turn-helix motifs of the CodY dimer separate in a way that facilitates DNA binding. GTP also activates CodY as a DNA-binding protein but does so by an unknown mechanism. Whereas the BCAAs and GTP each activate CodY independently, the combination of ligands has a synergistic effect.
The bacterium’s rationale for using two unrelated types of effectors to modulate the activity of a single global regulator can only be surmised. One possibility is that the GTP pool is not only an indicator of ability to synthesize purine nucleotides, but is also a measure of protein synthetic capacity. That is, in addition to being a substrate for RNA synthesis, GTP is utilized as an energy source during protein synthesis. Perhaps most important, the GTP pool in Gram-positive bacteria is strongly affected by amino acid limitation. Limitation of any amino acid leads to accumulation of uncharged tRNA, a molecule sensed by the ribosome-bound stringency factor known as RelA or RSH. The RelA-catalyzed conversion of GTP to pppGpp reduces the GTP pool, but probably not enough to have a large impact on CodY activity. Instead, the major effect of pppGpp in B. subtilis and, presumably, other Gram-positive bacteria is to inhibit enzymes of GTP synthesis (142, 143), thereby linking CodY activity to availability of all amino acids. In fact, inducing the stringent response in B. subtilis or S. aureus leads to altered expression of most genes of the CodY regulon (143, 144). BCAAs are also abundant amino acids in proteins, but their special nature is that they are readily interconvertible with the branched-chain keto acids, which serve as precursors of pantothenate and coenzyme A, as described previously, and branched-chain fatty acids. In many Gram-positive bacteria, the branched-chain fatty acids are the predominant membrane fatty acids under normal growth conditions. Thus, by sensing the availability of BCAAs, CodY is monitoring the cell’s capacity to carry out essential reactions, including membrane biosynthesis.
In all species studied to date, the sites of CodY binding share a common 15-bp palindromic sequence, AATTTTCNGAAAATT (the “CodY box”) (139, 145); specific nucleotides within this sequence are essential for CodY binding (146), but the exact relationship between the 15-bp palindrome and affinity of CodY for DNA is not fully clear. It is very rare for any bacterium to have within its genome even one CodY box that matches the consensus sequence perfectly. When an imperfect B. subtilis CodY box was converted by mutation to a perfect box, the gene controlled by that site was barely expressed at all, even under conditions of very low CodY activity (147). Thus, it is not in the cell’s interest to have CodY boxes without mismatches. In fact, many sites of moderate-to-high affinity have 3–5 mismatches. In many of the latter cases, two overlapping copies of the palindromic sequence, each with multiple mismatches, serve as the binding site, suggesting that the protein can interact with DNA in multiple ways (148, 149).
CodY affects gene expression by multiple molecular mechanisms. As a positive regulator, CodY binds upstream of the promoter and, presumably, stabilizes the binding of RNA polymerase to promoters that are intrinsically weak (150). Like traditional repressors, CodY can inhibit transcription initiation by binding within or just downstream of a target promoter region (147), but CodY can also block transcription elongation by binding within a coding sequence, in some cases as far as 600 bp downstream of the start codon (149, 151). In at least one case, CodY acts as a negative regulator by inhibiting the binding of a positive regulator (152).
The Role of CodY in Virulence Gene Expression
CodY acts as a negative regulator of virulence in C. difficile and S. aureus but as a positive regulator of virulence in B. anthracis, B. cereus, and L. monocytogenes. In other species, CodY seems to act as an activator of some types of virulence genes and a repressor of others. The implication is that Gram-positive bacteria use CodY to regulate metabolism in a well-conserved manner but manipulate CodY to regulate virulence according to their niches within hosts and in accordance with whether they are virulent when growing rapidly or slowly. Moreover, depending on the environmental conditions in which a bacterium finds itself, it may be advantageous to activate some types of virulence genes while repressing others.
Clostridium
Proven virulence genes of C. difficile are restricted to the two major toxins that cause the inflammatory response and diarrheal disease characteristic of infection. It must be the case, however, that as-yet-unidentified colonization factors and defense mechanisms play critical roles in the infection process. CodY regulates many genes that may fit these latter categories, but proof of function for the target genes is lacking. Toxin synthesis is responsive to many nutrients, including glucose (noted previously) and BCAAs (114, 153). The latter effect raised the possibility that CodY functions in toxin gene regulation. In fact, CodY, like CcpA, represses the toxin genes indirectly by binding to sites upstream of tcdR, the gene that encodes the major sigma factor for toxin gene transcription (Figure 2). Consistent with CodY-dependent regulation, toxin gene transcription in bacteria growing in rich medium increases dramatically during the post-exponential phase, when CodY repression would be relieved (113). In a codY null mutant, expression of the toxin locus during the exponential growth phase is 50-fold higher than in wild-type cells (154), and toxin protein released into the culture fluid is 100-fold higher in the mutant strain after 24 hours of growth (Bouillaut, Sun, Tzipori, and Sonenshein, unpublished). Given these results, it is not surprising that a codY null mutant is hypervirulent compared with its parent strain (Bouillaut et al, unpublished).
The effect of CodY in C. perfringens is more subtle. A codY mutant of a type D strain shows reduced expression of enterotoxin (ETX), reduced sialidase (i.e., NanJ) in the culture fluid, and a reduced ability to attach to Caco-2 cells (155). By contrast, inactivation of CodY has no effect on production of phospholipase C or perfringolysin; however, it led to overexpression of NanH and increased efficiency of sporulation in a rich medium. CodY appears to bind to the etx promoter region, but other direct targets of CodY have not yet been defined (155).
Staphylococcus
A codY null mutant of S. aureus USA300 (the CA-MRSA prototype) is hypervirulent, correlating with increased transcription and protein accumulation for hemolysins, leukocidins, and proteases (156, 157). The situation in S. aureus is complex, however, because dozens of virulence-associated genes are known, and CodY directly or indirectly regulates most of them (129, 130, 157, 158). As reported by Novick (159), S. aureus virulence genes can be separated into two groups depending on whether their products are involved in colonization or serve as toxins, lipases, proteases, capsule-forming, biofilm-forming, or tissue-damaging proteins. Colonization genes are expressed in the laboratory during the exponential phase in rich medium, whereas the latter group of genes is expressed during the postexponential phase. CodY controls genes from both groups, activating the transcription of some growth and colonization genes (e.g., fnbA) and repressing the transcription of toxins (i.e., hla, hlb, hld, lukSF, tst-1, sak), capsule, biofilm (ica), lipase, nuclease, protease, and ROS detoxifying genes (129, 130, 156, 157). In some of these cases, CodY binds directly to the target gene region, but in other cases, it works indirectly through its repressive effect on expression of the quorum-sensing Agr system and the regulatory RNA, RNAIII (129). CodY also controls the agr locus indirectly by a mechanism that is not yet known (252). Moreover, the regulation may be reciprocal, as an S. epidermidis agr mutant showed reduced expression of codY and derepression of CodY-repressed genes (160).
The expression of many Staphylococcal virulence genes (e.g., fibronectin-binding protein, epidermin immunity proteins, enterotoxin B) is altered when the stringent response is induced by limitation of amino acids (leucine or valine) (144) or treatment with mupirocin, an inhibitor of isoleucyl-tRNA synthetase (161). Most of the affected genes are regulated by either CodY or the alternative sigma factor, σB. In addition, the expression of several virulence gene regulators is induced by stringency, including ArlRS, SarA, SarR, SarS, and σB (161). Like many other bacteria, S. aureus has several pppGpp-synthesizing enzymes but only one synthetase (RelA/ RSH), which also has pppGpp hydrolase activity. A relA/ rsh null mutant of S. aureus is not viable, presumably because it is unable to prevent blockage of GTP synthesis, but a mutant defective only in the synthetase function can be studied. Such a mutant is less virulent than its parent strain, but the defect can be suppressed by introducing a codY mutation, implying that CodY is hyperactive as a repressor of virulence in the relA/rsh mutant (144).
It is important to note that the first reported phenotype caused by an S. aureus codY null mutation was reduced biofilm formation (162); all subsequent reports based on mutations in other strains showed substantial increases in biofilm gene expression and biofilm formation (158). Thus, one must be open to the possibility that global regulators can be used by different species and different clones within species in different ways that allow adaptation to particular environmental conditions.
Bacillus
B. anthracis CodY promotes virulence by increasing the abundance of AtxA, the positive regulator of toxin genes; a codY null mutant shows greatly decreased virulence (163, 164). Again, this effect is indirect and appears to reflect repression by CodY of a gene that encodes a protease that degrades AtxA. A codY mutant of B. cereus, a close relative of B. anthracis, is also attenuated for virulence (165). In strain ATCC 14579, the reduced virulence phenotype could be explained by reduced expression of cytotoxin K, nonhemolytic enterotoxin and hemolysin BL (166). In the emetic strain F4810/72, CodY also appeared to activate expression of nonhemolytic enterotoxin as well as phospholipases and immune-inhibitor metalloprotease 1 (165). In the same strain, CodY represses indirectly the regulon controlled by the PlcR-PapR quorum-sensing system (165). CodY also appears to be an activator of biofilm formation in strain UW101C (167) but seems to repress biofilm formation in strain ATCC 14579 (166).
Listeria
In L. monocytogenes, CodY was initially postulated to be a negative regulator of virulence because a codY mutation partially restored virulence to a relA mutant (134). This conclusion was consistent with the idea that a relA mutant would be expected to have a higher-than-normal GTP pool and, as a result, accumulate more active CodY, leading to hyperrepression of CodY target genes. In subsequent work, CodY was shown to be a positive regulator of prfA, the gene that encodes a positive regulator of key virulence genes, such as those for listeriolysin and actin-polymerizing protein (168). When a global regulator can be a positive regulator of some genes and a negative regulator of others, it is sometimes inadvisable to draw too many conclusions about a complex phenomenon such as virulence based on the phenotype of a null mutant.
Streptococcus
In S. pyogenes, the first pathogen in which a codY mutant was isolated (169), the role of CodY is complex. By repressing some virulence genes and activating others, CodY appears to be used to express different classes of virulence genes under different environmental conditions. In part, this complexity may be accounted for by the effect of CodY on expression of the CovRS two-component regulatory system. That is, CovR can be a positive or negative regulator of some virulence-associated genes, depending on environmental conditions. By repressing the covRS operon, CodY seems to work at cross-purposes with CovRS and can therefore appear to be a reciprocal activator or inhibitor of virulence (132). How this complicated arrangement plays out in vivo is uncertain, but during growth of S. pyogenes in human blood in vitro, many genes are subject to regulation by CodY but not necessarily in the same way as in laboratory growth media (131).
The situation in S. pneumoniae is also complicated, in this case because of conflicting reports on the effects of a codY mutation. As originally reported, a codY mutant was defective in colonization (139). A later report challenged these findings, claiming that a codY null mutation in S. pneumoniae is lethal; the mutants previously isolated were likely to have had compensatory mutations that allowed the codY mutant to survive (170). Any apparent effects of a codY mutation on virulence, therefore, may be due to the compensatory mutations rather than the codY mutation itself.
PrdR
Proline is among the nutrients that severely inhibit C. difficile toxin synthesis (153); this inhibition is mediated via proline reductase, a “Stickland reaction” enzyme (171). C. difficile, like several other Clostridium spp., has the ability to cometabolize proline or glycine and certain other amino acids (172). That is, oxidative metabolism of isoleucine, leucine, valine, or alanine generates NADH and ATP; the NADH can then be used to reduce proline, producing 5-aminovalerate, or glycine, generating acetate and more ATP (Fig. 3). The proline reductase and glycine reductase selenoenzyme complexes are encoded in multigene operons (173). Transcription of the proline reductase operon, which has a σ54-type promoter, and inhibition of toxin synthesis by proline depend on PrdR, a σ54-activating regulatory protein encoded just upstream of the proline reductase genes (171). Addition of proline to rich medium has broad effects on metabolism in C. difficile; these effects are mostly related to reductive pathways and are dependent on PrdR, implying that PrdR is a global regulator (Bouillaut, Dubois, Monot, Dupuy, and Sonenshein, manuscript in preparation). This unexpected observation raises important questions, however: Is PrdR a global regulator or a specific regulator of the proline reductase operon? If the latter, why and how would C. difficile use proline availability and a proline-sensing regulator to control so many diverse pathways, including toxin production? The answer seems to be that proline has a favored role in NAD+ regeneration (see next section on Rex).
FIGURE 3.

Examples of oxidative and reductive metabolic pathways in C. difficile. NADH produced during glycolysis and other oxidative pathways is converted back to NAD+ by a series of reductive pathways. The proline pathway, catalyzed by proline reductase, appears to be the favored pathway. When proline is available, the other pathways shown are repressed by Rex. Repression by Rex is relieved when the ratio of NAD+ to NADH indicates the need for increased regeneration of NAD+. Additional repression by CcpA and CodY restricts maximal expression of the alternative pathways to conditions in which CcpA and CodY are relatively inactive. The bottom three pathways (effectively, acetyl-CoA to butyrate) are encoded in a single eight-gene operon. doi:10.1128 /microbiolspec.MBP-0004-2014.f3
Rex
First discovered in Streptomyces coelicolor (174), Rex is a global regulator of genes whose products interconvert NADH and NAD+. Rex is found in virtually all low G+C Gram-positive bacteria. Ironically, Streptomyces is the only high G+C Gram-positive genus that encodes Rex. By monitoring the ratio of reduced to oxidized forms of nicotinamide nucleotides, Rex helps to regulate pathways that regenerate NAD+ in cells that are actively oxidizing carbon-energy sources. For example, glycolysis, the Krebs cycle and the pentose phosphate pathway convert NAD+ and NADP+ to NADH and NADPH. Without adequate regeneration of oxidized co-factors, these metabolic pathways would come to a halt. Bacteria have three major ways of regenerating NAD+/NADP+: (i) NADH/NADPH-dependent biosynthesis; (ii) carbon overflow pathways (e.g., lactate dehydrogenase); and (iii) respiration. In some bacteria, respiration-independent NADH oxidase can also serve. Rex typically represses carbon overflow pathways and reductive pathways that are a form of respiration when the pool of NAD+ is adequate. Examples of Rex targets in S. coelicolor, S. aureus, C. acetobutylicum, B. subtilis, or E. faecalis include lactate dehydrogenase (LDH), lactate permease, alcohol dehydrogenase, butyryl-CoA synthesis, crotonase, acetyl-CoA acetyltransferase, pyruvate formate lyase, pyruvate oxidase, NADH dehydrogenase, and cytochrome bd (175–180).
Binding of Rex to its target sites (the well-conserved sequence (TTGTGAAa/t6TTCACAA) is inhibited by NADH; thus, Rex is active as a repressor only when the NAD+/NADH ratio indicates adequate NAD+ (174). Binding of S. aureus and C. difficile Rex proteins to their target sites is stimulated by NAD+; NADH competes with NAD+ for interaction with Rex, thereby inhibiting DNA binding [(178) and L. Bouillaut and A. L. Sonenshein, unpublished]. Thermus aquaticus Rex has served as the model for structural studies of Rex-NAD+/NADH interactions. When NADH binds, a conformational change ensues that causes Rex to lose its ability to interact with DNA (181).
In E. faecalis, deletion of the ldh gene causes the carbon overflow metabolism to be redirected toward acetoin, ethanol, and formate and, at the same time, causes changes in expression of many genes that have apparent Rex binding sites upstream (179). The implication is that LDH is the major regenerator of NAD+; in the absence of LDH, NADH accumulates, leading to derepression of Rex-repressed genes. A similar case of prioritization of NAD+-regenerating pathways is seen in C. difficile (discussed later).
The role of Rex in virulence gene regulation
Staphylococcus
The relationship between pathogenesis and the maintenance of an appropriate balance of reduced and oxidized NAD/NADP is not yet clear, but in some bacteria, metabolic pathways under Rex control are implicated in virulence. For instance, in S. aureus, Rex is intimately involved in survival of cells exposed to nitric oxide (NO). NO inhibits the activities of terminal respiratory oxidases, nitrate reductase, and pyruvate formate lyase, leaving lactate dehydrogenase, a target of Rex, as the major means of regenerating NAD+ (178). The known connection between LDH activity and resistance to host innate immunity means that modulating Rex function appropriately is a critical factor in pathogenesis (182).
Clostridium
In C. difficile, proline reductase appears to be the preferred pathway for NAD+ regeneration. If proline is limiting in the growth medium or if proline reductase or PrdR is inactive, alternative pathways for NAD+ regeneration [e.g., glycine reductase, alcohol dehydrogenase, succinate to crotonyl CoA, crotonyl CoA to butyrate (183, 184)] are induced (Bouillaut et al, manuscript in preparation). These alternative pathways (Fig. 3) are repressed by Rex in wild-type cells when proline is available but not in proline reductase or prdR mutants (Bouillaut and Sonenshein, manuscript in preparation). Since these pathways produce butyrate, a stimulator of toxin synthesis, one can imagine that C. difficile withholds toxin synthesis when proline is in excess, because the cells have an adequate supply of NAD+. If butyrate is being produced, the implication is that NAD+ is limiting. Alternatively, by sensing butyrate, the cells may be primed to detect the presence of other butyrate-producing bacteria in the intestinal tract. How the cell communicates its NAD+/NADH status to the toxin locus is unknown.
RpiR
The RpiR family of regulatory proteins controls carbohydrate metabolism in both Gram-negative and Gram-positive bacteria. First described as a regulator of ribose metabolism in Escherichia coli (185), members of the family have been shown to regulate N-acetylmuramic acid catabolism in E. coli (68), the pentose phosphate pathway in Pseudomonas aeruginosa (186), inositol catabolism in Sinorhizobium meliloti (187), maltose transport and metabolism in B. subtilis (188), and the pentose phosphate pathway in S. aureus (189). Family members typically have an apparent DNA binding domain near the N-terminus and a sugar isomerase binding domain near the C-terminus, suggesting that intermediates in pentose metabolism modulate the activity of at least some RpiR proteins. S. aureus encodes three RpiR homologs, of which two, RpiRb and RpiRc, are involved in pentose regulation (189). An rpiRc mutant shows increased levels of the regulatory RNA, RNAIII, during exponential growth, and an rpiRb mutant demonstrates a similar phenotype during the postexponential phase (189). This phenomenon may be attributable to increased synthesis of the regulatory protein SarA. Moreover, an rpiRc mutation causes increased capsule formation and increased resistance to peroxide (189). The coregulation of pentose metabolism and virulence factors by RpiR proteins suggests strongly that RpiR mediates one of several mechanisms by which virulence and metabolism are coupled.
Interactions Among the Global Regulators
Although each of these global regulators monitors a different intracellular metabolite pool, they interact with each other and with operon-specific regulators in multiple ways. For instance, in B. subtilis, ilvBHC leuABCD, the major operon for biosynthesis of BCAAs, is repressed by CodY and TnrA (the global regulator of nitrogen metabolism) and activated by CcpA (190–192). Since TnrA is active only under conditions of glutamine (nitrogen) limitation, and CodY is activated when BCAAs are in excess; these two regulators combine to reduce ilvB operon transcription when the cell has enough BCAAs or is so depleted of nitrogen that it is best to limit its consumption. When leucine is specifically in excess, most transcription that initiates is aborted before the beginning of the first coding sequence by a T-box–mediated termination/antitermination system that responds to uncharged tRNAleu (193). When cells are in carbon excess, CcpA stimulates transcription of the ilvB operon, thereby making CodY more active as a repressor of many pathways that draw intermediates away from glycolysis. On the other hand, since both CcpA and CodY activate carbon overflow pathways when glucose is in excess, the increased CodY activity generated by CcpA accentuates this cooperation. Finally, the decrease in the GTP pool caused by the stringent response also affects ilvB operon expression in two ways (i.e., by reducing CodY activity and the pool of GTP, the initiating nucleotide for ilvB transcription (143, 194)). Thus, expression of the ilvB operon responds positively to the pool of FBP and negatively to the pools of the three BCAAs, GTP, and glutamine.
Regulation of the Krebs cycle can also be mediated cooperatively by CcpA and CodY. In B. subtilis, CcpA represses the genes for citrate synthase and isocitrate dehydrogenase, whereas CodY represses the gene for aconitase. A third regulator, CcpC, is Krebs cycle-specific and represses the genes for all three enzymes (195). None of these repressors is fully effective by itself; therefore, complete repression requires high intracellular pools of FBP, BCAAs, and GTP and low citrate, the effector of CcpC. (When citrate accumulates to high level, CcpC is inactivated as a repressor of citrate synthase and isocitrate dehydrogenase but switches from a repressor to an activator of the aconitase gene (196)). A similar regulatory scheme is found in L. monocytogenes (196). S. aureus also uses CcpA and CcpE (a homolog of CcpC), as well as RpiRc, to regulate the tricarboxylic acid branch of the Krebs cycle. In this case, CcpE acts as a positive regulator of aconitase but not citrate synthase expression and binds to the aconitase promoter region, but its binding is not affected by citrate (197). CcpA represses the synthesis of citrate synthase, aconitase, and isocitrate dehydrogenase in glucose-containing medium (87). Unlike the situation in B. subtilis, an S. aureus codY mutation does not cause derepression of any TCA branch enzymes, at least not under the growth conditions tested (129, 130); of the Krebs cycle enzymes, only α-ketoglutarate dehydrogenase is derepressed in a codY mutant (129).
As mentioned previously, B. anthracis CodY and CcpA are both positive regulators of AtxA accumulation and toxin synthesis, but in C. difficile, both CcpA and CodY repress tcdR and thereby block toxin synthesis. In addition, C. difficile toxin synthesis is inhibited when proline or cysteine is in excess. The direct or indirect mediator of the proline effect on toxin gene expression is PrdR. Thus, C. difficile does not become fully virulent unless deprived of a rapidly metabolizable carbon source and sources of BCAAs, proline, and cysteine. Nonetheless, deprivation of any one of these nutrient sources leads to significant toxin production.
NAD+ regeneration is also subject to multiple overlapping controls. In S. aureus, a mutation in ccpA decreases expression of the LDH-1 gene, implying that CcpA is a positive regulator. But CcpA does not bind to the ldh1 regulatory region, and its effect on ldh1 expression depends on Rex. In contrast, Rex does bind to the ldh1 promoter region and represses transcription (198).
The CodY, CcpA, and Rex proteins of C. difficile all control reductive metabolism of acetyl-CoA, succinate, and glycine to butyryl-CoA. (In addition, PrdR appears to be an indirect regulator of these pathways through its direct effect on proline-dependent NAD+ regeneration, which was previously noted.) The three operons encoding these pathways appear to be direct targets of CodY and CcpA, as determined by in vivo and in vitro binding assays (84, 128). They also have apparent Rex binding sites (199). Moreover, the proline reductase operon is positively regulated by CcpA as well as by PrdR (84). One can speculate that these pathways play at least three roles: (i) They are designed to regenerate NAD+, hence their regulation by Rex; (ii) they are ATP-generating pathways, justifying their repression by CcpA when glucose is in excess; and, (iii) as producers of butyrate, an activator of toxin gene expression, they work at cross-purposes with repression of toxin synthesis by CcpA and CodY. Proline reductase is a special case; assuming it is the preferred pathway for NAD+ regeneration, it makes sense that it is positively regulated by CcpA as a means of balancing the redox state during growth on glucose. In summary, it is apparently in the interest of C. difficile to minimize toxin synthesis unless multiple checkpoints have been reached (i.e., lack of rapidly metabolizable carbon sources, limitation of BCAAs, and an unfavorable ratio of NAD+ to NADH). When all those conditions are met, butyrate can be made and can signal the cells to turn on toxin synthesis.
CodY and PrdR also appear to repress in partnership the utilization by C. difficile of ethanolamine as a carbon and nitrogen source (Bouillaut et al, manuscript in preparation). Ethanolamine is a relatively abundant nutrient in the GI tract by virtue of its abundance in food and its release from host and bacterial membrane phosphatidylethanolamine by phosphodiesterase activity produced by members of the normal microflora (200). The enzyme complex that deaminates ethanolamine and then converts the carbon skeleton to acetate while producing ATP by substrate-level phosphorylation is located in a microcompartment that appears to be necessary to avoid diffusion of the volatile intermediate acetaldehyde (201). The ability to metabolize ethanolamine may contribute to virulence by serving as a nutrient source or by acting as a signal to pathogens that they are in an appropriate environment for expressing virulence functions (202). It is well established that the eut genes of several bacterial species are induced during growth in the intestinal tract (202), but the evidence that ethanolamine metabolism is critical for pathogenesis is limited. A eut mutant strain of L. monocytogenes is defective in intracellular growth in vitro in Caco-2 cells (203), and an E. faecalis eut mutant strain is attenuated for virulence in a worm model of infection (204). No study has yet shown that the ability to metabolize ethanolamine is critical for successful infection of an animal.
METABOLISM AND HOST: PATHOGEN INTERACTIONS
It is becoming increasingly appreciated that host:pathogen interactions are not merely limited to physical contact between bacterial adhesins, toxins, and surface molecules with their cognate host cell receptors. Rather, the interconnected metabolic network encompassing both the host and the microbe can also have measurable effects on disease outcomes. Activated phagocytes responding to invading microbes must acquire nutrients and generate energy in order to produce an array of inflammatory mediators (reactive oxygen/nitrogen species, and antimicrobial peptides) as well as signaling molecules (e.g., cytokines, chemokines, and lipid mediators). The concerted effects of all of these immune functions are essential for clearing infections and restoring tissue homeostasis. In fact, activated phagocytes have energy demands similar to those of muscle cells and neurons, making them some of the most energetically active cells in the body (205). The metabolic pathways used by immune cells to meet the energy demands of activation depend on the immune cell type, the nature of the pathogen, and features of the infected tissue. On the other hand, for an invading microbial pathogen to succeed, it too must be able to acquire nutrients and generate energy for the purpose of persisting long enough to be transmitted to a new host. While the Gram-positive pathogens highlighted in this chapter have extensive interconnected webs of metabolic pathways available to them (reviewed in the first section), which ones are active and functional during various stages of infection are only recently becoming understood. The metabolic pathways used by a pathogen during infection are dictated not only by complex regulatory networks that sense both extra- and intra-cellular cues (reviewed in second section), but also by the compatibility of metabolic pathways with available nutrients and tissue environments, as well as the status of the host immune response. In this section, we will highlight some of the recent findings regarding the role of metabolism in the host:pathogen interaction, focusing both on the metabolic pathways used by the host as well as the diversity of pathways employed by various Gram-positive pathogens.
Host Immune Cell Metabolism During Infections
One of the first immune cells to infiltrate infected tissue is the polymorphonuclear monocyte (PMN or neutrophil). PMNs extravasate from the blood stream in response to a cytokine gradient generated from stromal and epithelial cells at sites of tissue damage/infection. PMNs are short-lived cells that become highly activated, engulf and kill infecting bacteria, and then undergo one of many forms of programmed cell death. They are by far the most abundant effector cell at sites of infection and are critical for rapid control of invading microbes. Another effector cell commonly found within infectious foci is the macrophage. Macrophages mature from circulating monocytes in the blood as they extravasate into infected tissue. Like PMNs, macrophages also engulf and kill invading bacteria, however these cells are generally much more long-lived than PMNs. This is due to the fact that macrophages serve additional roles beyond merely destroying microbes. First, macrophages (as well as dendritic cells) are efficient antigen-presenting cells (APCs), presenting processed antigen in the context of MHC to T-cells, thereby linking the innate and adaptive immune compartments. Furthermore, macrophages also play key roles in restoring tissue homeostasis as the infection resolves. Macrophages engulf and dispose of necrotic and apoptotic cell debris (such as dead neutrophils) and secrete proresolving cytokines and lipid mediators that signal to surrounding stromal and epithelial cells to restore tissue integrity (206). Depending on the state of the infection, the cytokine milieu, and the needs of the surrounding tissue, macrophages adopt distinct phenotypes that allow them to fulfill their wide array of roles within infected tissue. These phenotypes have been broadly categorized into classically or alternatively activated macrophages, the former being associated with inflammation and bacterial clearance (207).
Classically activated macrophages (aka M1 macrophages) resemble activated PMNs in many ways. Both cell types respond to proinflammatory cytokines such as TNF-α, IL-1β, and INF-γ, both are exquisitely sensitive to pathogen associated molecular patterns (PAMPs) sensed through toll-like receptors (TLRs), and both cell-types produce an array of antimicrobial effectors such as antimicrobial cationic peptides, reactive oxygen species (superoxide anion, ·O2−, hydrogen peroxide, H2O2, and hypochlorite HOCl), as well as nitric oxide (NO·). Together with PMNs, M1 macrophages produce most of the antimicrobial effectors that ensure the quick and efficient elimination of invading microbes. However, these effectors also damage host tissue, and prolonged inflammation can lead to the accumulation of damage associated molecular patterns (DAMPs). Within the site of infection, immune, stromal, and epithelial cells sense the accumulating DAMPs and, concomitant with diminishing levels of PAMPs, alter the cytokine and lipid mediator milieu towards a proresolving profile, including IL-4, IL-10, IL-13, and TGF-β (208). These signals alter the macrophage phenotypes to that of alternatively activated or M2-macrophages. The phenotype of an M2-macrophage differs from M1-macrophages or PMNs. M2-macrophages make no immune radicals and synthesize no antimicrobial peptides but are highly phagocytic to facilitate the elimination of cellular debris at the conclusion of an infection (209). They also produce numerous signals that promote cell proliferation and tissue regeneration. Thus, M2 macrophages are critical for the resolution or wound healing phase that follows an inflammatory response to invading microbes.
Despite the fact that PMNs, M1-, and M2-macrophages are all highly energetically active cells, the pathways they use to meet their energy demands differ greatly. For instance, PMNs and M1-macrophages rely heavily on rapid glucose consumption for energy (210). Activation of these phagocytes by inflammatory stimuli induces a form of metabolism reminiscent of that observed in cancer cells. Warburg metabolism was first described in cultured cancer cells and is a phenomenon whereby glucose is oxidized primarily to lactate with little flux into the Krebs cycle or mitochondrial oxidative phosphorylation (OxPhos) despite the abundance of oxygen (211). Indeed, activated PMNs and M1-macrophages show similar tendencies, consuming large quantities of glucose and acidifying the surrounding environment through the excretion of lactate (205, 212). This form of metabolism (also known as aerobic glycolysis) involves the import of glucose, primarily through the GLUT-1 transporter, after which it is phosphorylated to glucose-6-phosphate (G6P) via hexokinase-1 (HK–1) (205, 213). G6P is then oxidized to pyruvate, which is reduced to lactate and excreted (Fig. 4). This metabolism allows rapid ATP production through substrate-level phosphorylation, albeit less efficiently than through OxPhos, and maintains redox balance via lactate production. G6P is also a substrate for the first enzyme in the PPP. This pathway is important for the de novo synthesis of ribonucleotides, but, more importantly for activated phagocytes, this pathway provides most cellular reducing power in the form of NADPH (214). Electrons from NADPH are used to generate immune radicals such as ·O2− and NO·, thereby necessitating significant flux through PPP for an effective immune response (Fig. 4).
FIGURE 4.

Differences in M1- versus M2-macrophage fueling reactions. In response to inflammatory stimuli, M1-macrophages upregulate a pathway known as aerobic glycolysis. This involves the import of glucose through GLUT-1 and its phosphorylation by Hexokinase-1 (HK-1). The resulting glucose-6-phosphate (G6P) can be shuttled through the pentose phosphate pathway (PPP) for NADPH generation, which fuels immune radical production, including nitric oxide (NO). At the same time, G6P is also oxidized to pyruvate (PYR) for ATP synthesis, and this PYR is primarily reduced to lactate (LAC) to conserve redox balance. Very little PYR enters the Krebs cycle as acetyl-CoA (Ac-CoA) due to the phosphorylation and inactivation of pyruvate dehydrogenase (PDH). Genes activated/ repressed by HIF-1α are depicted as green/red. Upon stimulation with anti-inflammatory stimuli, M2-macrophages adopt an oxidative metabolism involving the import of free fatty acids and low-density lipoprotein (LDL)-associated lipids (fatty acids and LDL) by CD36. These fatty acids are linked to carnitine and shuttled to the mitochondria for β-oxidation, yielding ATP. In addition, some of the Ac-CoA is reused to synthesize new fatty acids. Rather than using tissue arginine for NO-production, these cells use the amino acid for proline and polyamine production, the former of which is critical for collagen synthesis. Features activated/repressed by PPAR-γ are depicted in green/red. doi:10.1128 /microbiolspec.MBP-0004-2014.f4
Thus, rapid consumption of glucose by activated PMNs and M1-macrophages allows rapid ATP production, allowing for chemotaxis and protein synthesis, as well as reducing power for the production of immune radicals. In contrast, M2-macrophages exhibit a markedly different metabolic profile upon stimulation. These cells exhibit drastic increases in fatty acid uptake and catabolism via β-oxidation, pathways not prevalent among activated PMNs or M1-macrophages (215). Curiously, significant increases in the expression of genes involved in fatty acid synthesis are also apparent in active M2-macrophages. The precise role for the balanced expression of fatty acid breakdown and synthesis programs in M2-macrophages is still unclear, but the fact remains that much more of the energy demand in M2-macrophages is met by β-oxidation rather than aerobic glycolysis (215) (Fig. 4).
Another metabolic feature that distinguishes M2-from M1-macrophages (and PMNs) is the fate of tissue arginine. L-arginine is considered a semi-essential amino acid in that it can be synthesized by mammalian cells, but the total body demand for arginine often outpaces the rate of de novo production, requiring dietary intake to maintain optimal levels (216). Arginine serves as a precursor to two important pathways during infection in addition to general protein synthesis. First, M1-macrophages and PMNs primarily use arginine for NO· synthesis via inducible NO·-synthase (iNOS) (208). This enzyme uses electrons from NADPH to convert arginine to NO· and citrulline. Consequently, iNOS expression is a hallmark of M1-macrophage activation and is required for the efficient clearance of a variety of microbial pathogens (217). Alternatively, M2-macrophages do not express iNOS; rather, arginine is metabolized to the amino acid ornithine by Arginase-1 (Arg-1) (208). Ornithine is a precursor to proline synthesis, the most abundant amino acid in collagen. Efficient collagen synthesis is critical to reestablishing basement membrane integrity for tissue regeneration. In addition, ornithine can be converted to a series of compounds known as polyamines, which promote cell proliferation. By rerouting arginine flux away from NO· and towards proline and polyamine production, M2-macrophages shift the tissue from an inflammatory, destructive environment to a resolving, profibrotic, and proliferative niche. Consequently, Arg-1 expression is a hallmark of M2 macrophages and is not abundantly produced by M1-macrophages or PMNs.
Just as distinct metabolic pathways fuel macrophages exhibiting M1 versus M2 phenotypes, the transcriptional regulators that drive these fueling reactions also differ between M1- and M2-macrophages. While the complex regulatory networks that differentiate M2-from M1-macrophages and/or PMNs are by no means completely understood, a few key regulators essential to each phenotype have been described. For instance, the aerobic glycolysis that fuels M1-macrophages requires the activity of the hypoxia-inducible factor 1α (HIF-1α) (213). Under normoxic or homeostatic conditions, HIF-1α is highly unstable stemming from its oxygen-dependent hydroxylation at proline residues 405 and 531 by prolyl-hydroxylases. This hydroxylation targets HIF-1α for proteosomal degradation (218). However, as oxygen tension drops, HIF-1α hydroxylation is inhibited, and the protein is therefore stabilized and efficiently translocates to the nucleus. There, it dimerizes with the constitutively expressed HIF-1β (aka aryl hydrocarbon nuclear translocator [ARNT]) to bind to HIF-responsive elements (HREs) and modulate gene expression. During inflammation, the massive consumption of oxygen for the production of reactive oxygen species results in hypoxic conditions sensed by infiltrating phagocytes (210). Thus, HIF-1α is highly stable within inflamed tissue. HIF-1α activates expression of genes involved in aerobic glycolysis, including the glucose importer GLUT-1, hexokinase-1 (HK-1), phosphoglycerate kinase (PGK), and lactate dehydrogenase (LDHA) (219) (Fig. 4). HIF-1α activity also limits the flux of pyruvate into the Krebs cycle by activating pyruvate dehydrogenase kinase (PDK) (219). PDK-mediated phosphorylation of pyruvate dehydrogenase (PDH) limits the conversion of pyruvate to acetyl-CoA, ensuring that glycolytic pyruvate is reduced to lactate via LDHA for maintaining redox balance. Thus, HIF-1α is essential for the metabolic priming of M1-macrophages, as well as PMNs. Mutant phagocytes lacking HIF-1α show diminished ATP levels, reduced chemotaxis, and limited immune effector production upon stimulation, demonstrating the importance of proper metabolic fueling to an effective immune response (213). Like M1-macrohpages and PMNs, M2-macrophages also express a regulator central to their metabolic strategies. Expression of Arg-1, as well as genes involved in β-oxidation and fatty acid synthesis, are all dependent on the activity of a nuclear receptor known as the peroxisome proliferator-activated receptor γ (PPAR-γ) (220). PPAR-γ is a member of a family of related nuclear receptors (PPAR-α and PPARβ/δ), all of which generally affect lipid homeostasis (221). These receptors enter the nucleus upon binding of their activator ligands and dimerize with the retinoid-x receptor (RXR) to bind PPAR-response elements (PPREs) and affect gene expression. In response to anti-inflammatory cytokines such as IL-4, macrophages stimulate PPAR-γ transcription in a STAT-6 dependent fashion (207). Elevated cytosolic PPAR-γ sensitizes M2-macrophages to the presence of activating ligands. The activating ligand for PPAR-γ is still unknown, but certain prostaglandins and oxidized fatty acids are known to bind and activate the receptor (222). These lipid-based signals accumulate in tissue following prolonged inflammation and oxidative/nitrosative radical production. Thus, PPAR-γ-activation serves as a negative feedback loop to limit the duration of inflammation and reprogram tissue to a resolution phenotype. Indeed, in addition to activating the metabolic fueling reactions of M2-macrophages, PPAR-γ is known to directly limit the activity of pro-inflammatory regulators, including NF-β and AP-1 (223). The central role of PPAR-γ in the M2-macrophage phenotype is exemplified by the fact that macrophages deficient in PPAR-γ cannot express Arg-1 and do not exhibit robust β-oxidation or fatty acid synthesis (220). Thus, PPAR-γ activity is essential for limiting inflammation and activating the metabolic fueling pathways that maintain the M2 macrophage phenotype. These cells are essential for wound healing and restoration of tissue homeostasis following an inflammatory response to invading microbes.
Pathogen metabolism during infections
Gram-positive pathogens represent a diverse group of bacteria that, in general, span two phyla, the Firmicutes and the Actinobacteria. Just as marked diversity exists among the different species of Gram-positive pathogens with respect to disease presentation and virulence factor production, the metabolic pathways used by these pathogens to establish infection and persist are also quite varied.
Some of the most in-depth knowledge of bacterial metabolism critical to disease progression has been gathered by studying the intracellular pathogen M. tuberculosis, the causative agent of tuberculosis. M. tuberculosis thrives within the phagosome of infected macrophages, which traffic the bacterium to multicellular caseous granulomas in the lung. Here, M. tuberculosis resides in a quiescent state in which it awaits activation for the elaboration of disease symptoms and transmission. In general, M. tuberculosis relies primarily on β-oxidation of host lipids, fatty acids, and cholesterol for energy and carbon sources (224). This generalization is supported by the presence of more than 36 acyl-CoA ligase and 35 acyl-CoA dehydrogenase genes in the M. tuberculosis genome (compared with two acyl-CoA ligase and one acyl-CoA dehydrogenase genes encoded by E. coli) (http://biocyc.org). Moreover, these genes are highly expressed within macrophages, and many are required for normal M. tuberculosis growth during infection (225–227). Fatty acids are oxidized to acetyl-CoA (and proprionyl-CoA in the case of cholesterol and odd-chained fatty acids) and therefore require both gluconeogenesis and the Krebs cycle to generate all precursor metabolites. This is supported by the requirement of a key gluconeogenic enzyme, pyruvate carboxykinase (PckA), for full virulence (227). M. tuberculosis does not flux lipid-derived carbon through the canonical Krebs pathway because it is incompatible with two-carbon nutrient sources (fatty acids are mainly oxidized to acetyl-CoA and can be thought of as polymers of two-carbon nutrients). As discussed in the first section, the two tandem decarboxylation steps in the Krebs cycle (isocitrate dehydrogenase followed by α-ketoglutarate dehydrogenase) would essentially oxidize both carbons of acetyl-CoA to CO2, precluding the synthesis of precursor metabolites. Instead, M. tuberculosis relies on the glyoxylate shunt, a pathway that involves isocitrate lyase and malate synthase; bypasses the two decarboxylation steps; and essentially combines two molecules of two-carbon acetyl-CoA into one molecule of four-carbon malate. The latter can be oxidized to oxaloacetate and serve as a gluconeogenic substrate via PckA. The importance of the glyoxylate shunt to M. tuberculosis pathogenesis is demonstrated by the requirement of both isoforms of isocitrate lyase (ICL1 and ICL2) for full virulence (228). However, ICL1 provides an additional role in M. tuberculosis metabolism, being critical in the methylcitrate cycle (MCC) as well as the glyoxylate shunt (229). Utilization of odd-chained fatty acids via β-oxidation results in the generation of a single proprionyl-CoA molecule, and oxidation of cholesterol results in the production of three proprionyl-CoA molecules. Propionyl-CoA can be toxic to cells in that it can inhibit pyruvate dehydrogenase (230). Accordingly, propionyl-CoA is shuttled through the MCC, a series of reactions analogous to the Krebs cycle, by ligation to oxaloacetate via methylcitrate synthase, generating methylcitrate. Methylcitrate accumulation, however, can also be toxic to cells relying on gluconeogenesis in that it can inhibit fructose bisphosphate phosphatase, an essential enzyme for complete gluconeogenesis (231). The fact that ICL1 uses methylcitrate as well as isocitrate as substrates allows M. tuberculosis to use ICL1 to reduce the toxic pool of methylcitrate. In addition, when proprionyl-CoA production outpaces ICL1 and the MCC, two other pathways assist in reducing propionyl CoA levels in M. tuberculosis: the vitamin B12-dependent methylmalonyl-CoA pathway and the direct incorporation of proprionyl-CoA into cell wall lipids (232, 233). The importance of the glyoxylate shunt, gluconeogenesis, the MCC, and cholesterol and fatty acid import to M. tuberculosis underscores the importance of host lipids and membrane cholesterol as fueling sources for this pathogen. However, M. tuberculosis does not rely on mere chance encounters with membrane lipids as nutrient sources. M. tuberculosis residing within granulomas comes in contact with macrophages that have accumulated high levels of lipids and cholesterol within large lipid droplets. These “foam cells” are thought to be major sources of nutrients for the infecting bacterium. Recent studies have suggested that direct interactions between macrophages and cell wall lipids of M. tuberculosis (particularly lipoarabanomannan, ManLAM) alter host cell gene expression, favoring the development of the foam cell phenotype (234). This interaction relies on PPAR-γ activation, which results in the robust fatty acid synthesis necessary for lipid droplet and foam cell formation (234, 235). PPAR-γ− acrophages (or macrophages treated with chemical PPAR-γ inhibitors) are far more bactericidal to intracellular M. tuberculosis. It is likely that some of this phenotype is due to the “starvation” of M. tuberculosis within macrophages defective in PPAR-γ activity and unable to perform robust lipid synthesis. However, ManLAM stimulation of PPAR-γ could have other important implications for M. tuberculosis infection outcomes. The PPAR-γ-dependent induction of Arg-1 competes for available arginine used by iNOS to generate NO·. This radical is a potent antituberculosis factor, and diminished NO·-production, as well as inhibition of iNOS expression mediated by PPAR-γ, likely contribute to enhanced M. tuberculosis survival within macrophages (236). In total, modulation of macrophage PPAR-γ activity by M. tuberculosis serves many functions in the pathogenesis of this organism, including the dampened production of inflammatory mediators as well as the activation of fueling pathways that “feed” the pathogen during infection.
Another Gram-positive pathogen that relies heavily on gluconeogenic carbon sources is the intracellular bacillus, L. monocytogenes. Unlike M. tuberculosis, however, L. monocytogenes does not reside within a phagosome, but rather escapes to replicate within the host cell cytosol. Here, the bacterium appears to primarily use host glycerol as a preferred source of carbon and energy. Because glycerol is a gluconeogenic carbon source, L. monocytogenes downregulates glycolytic enzyme expression and activates the synthesis of gluconeogenic genes during infection (237, 238). Moreover, the expression of genes involved in glycerol uptake and utilization are also induced intracellularly in L. monocytogenes, and mutations in these genes limit intracellular growth of the bacterium (237, 238). In addition, G6P can serve as a nutrient source for intracellular L. monocytogenes, albeit to a lesser extent than glycerol. Interestingly, the G6P uptake system is upregulated intracellularly, but the glucose transporter is not, suggesting that only carbohydrate phosphorylated by hexokinase is used by L. monocytogenes (239, 240). The reliance on glycerol and G6P rather than glucose intracellularly likely reflects the metabolic adaptation of infected host cells to L. monocytogenes. A marked increase in glycolysis and glutaminolysis accompanies infection of primary macrophages with the bacterium, which could lead to the intracellular accumulation of glycerol and G6P (241). While the regulatory pathways that control this host response are still unknown, HIF-1α induction of glycolysis may play an as-of-yet uncharacterized role.
In contrast to the obligate intracellular pathogens discussed previously that require gluconeogenesis for full virulence, S. aureus appears to require both glycolysis and gluconeogenesis to be fully pathogenic (242). This has been documented in an invertebrate infection model, and we have observed similar findings in murine infection models (253). Recent findings have shed light on this curious observation. While S. aureus can survive within phagocytes for prolonged periods, it often lyses host phagocytes and escapes. Within these phagocytes, however, it must endure an impressive onslaught of innate immune effectors, including immune radicals (·O2− and NO·). Interestingly, S. aureus is remarkably resistant to the effects of these immune radicals, particularly NO· (182, 243). Most bacteria are unable to replicate in the presence of NO· due to its propensity to attack redox centers of key metabolic enzymes (244). S. aureus elicits an incompletely defined metabolic state that allows the bacterium to circumvent the metabolic constraints imposed by host NO· (245). Of all the nutrients used by S. aureus, only certain glycolytic carbon sources support NO·-resistance (246). Therefore, glycolysis is essential for S. aureus to resist high-level NO· encountered within PMNs and M1-macrophages. Indeed, glycolytic mutants exhibit severe attenuation within mouse macrophages, but this defect is completely reversed upon inhibition of iNOS activity (253). Thus, because of host immune response, intracellular S. aureus relies on glycolysis to persist within the phagocyte until the bacterium can lyse the host cell and escape into the extracellular space. Once there, it then replicates to form bacterial aggregates at the center of tissue abscesses. Here, S. aureus resides within lysed host cell debris surrounded by newly infiltrating phagocytes. Inside these highly inflamed hypoxic abscesses, HIF-1α activity is high, thereby driving excessive glucose consumption by host cells (210). Therefore, glucose is likely scarce, and the bacterium must rely on abundant gluconeogenic substrates such as lactate resulting from host cell aerobic glycolysis. Accordingly, S. aureus encodes three independent enzymes that can use lactate as a carbon source (246). Furthermore, the low oxygen tension in this abscess environment likely limits the robust production of inflammatory radicals, including NO·, thereby relieving the strict dependence on glycolysis for S. aureus to thrive. Thus, lactate and host peptide-derived amino acids could serve as the major energy/ carbon source to S. aureus at the center of an abscess, necessitating gluconeogenesis. In essence, S. aureus relies on different nutrient sources throughout the course of animal infections, explaining the reliance on both glycolysis and gluconeogenesis for full virulence. However, more research is needed to solidify our understanding of the metabolic adaptions of S. aureus to the dynamic host immune environment.
The spore-forming Gram-positive pathogens, including Bacillus anthracis and Clostridium difficile, must germinate once inside the host in order to cause disease. Metabolite cues within host tissue interact with specific germinant receptors in the spore membrane and mechanically initiate the germination program. B. anthracis spores germinate once the nucleoside inosine and an accompanying cogerminant (usually an amino acid) are encountered (247). Ingested C. difficile spores germinate once they encounter bile salts such as taurocholate combined with glycine in the gut (248). Once germinated, little is known about the metabolism of either species. Curiously, as described above, glucose has opposing effects on the production of the major virulence toxins in the two species. Glucose stimulates the production of anthrax toxin, whereas it suppresses production of the C. difficile toxin. Both effects are mediated through CcpA (see section 2). It is tempting to hypothesize that C. difficile primarily relies on gluconeogenic substrates during infection; however, direct experimental confirmation of this is lacking. C. difficile replicates in the large intestine and, because most digestible carbohydrates are absorbed in the small intestine, the bacterium likely encounters low levels of simple sugars. Moreover, the organism encodes amino acid reductases (see second section) that allow C. difficile to thrive on peptide nutrient sources in anaerobic environments such as the gut. One of these reductases, the proline reductase, was induced upon inoculation into germ-free mice compared with laboratory-cultured bacteria (249). However, whether these systems are required for virulence has not been directly tested. In addition, it has long been appreciated that antibiotic treatment drastically predisposes patients to C. difficile infection. Thus, the normal gut microflora must provide an effective competitive barrier to C. difficile colonization. As mentioned earlier, the lumen of the large intestine is largely devoid of dietary carbohydrate, but it is rich in protein-associated sugars found in mucin and on enterocyte cell surfaces. These carbohydrates can be liberated by glycosidases expressed by various microflora and subsequently taken up by these symbiotic bacteria. Consequently, the steady-state levels of carbohydrate in the lumen of the large intestine are low. However, antibiotic treatment can interfere with the homeostasis of the gut flora and disrupt the balance between glycosidase-mediated carbohydrate liberation and consumption (250). This generally leads to a transient increase in luminal carbohydrate content in the large intestine, which may be exploited by C. difficile. Indeed, microflora-liberated sialic acid was shown to promote C. difficile expansion in a murine model of infection. Moreover, antibiotic treatment of mice resulted in elevated luminal sialic acid, and C. difficile mutants unable to metabolize sialic acid were defective in colonization of these antibiotic-treated animals (251). Thus, antibiotic treatment of the host may affect the balance of microflora-dependent carbohydrate metabolism in the large intestine, providing C. difficile with a glycolytic nutrient source during infection.
While we have gained significant insights into the roles of metabolism in the host:pathogen interaction, there is much still to be learned about what pathways fuel both host immune cells and invading pathogens. This information might yield possibilities for new therapeutics aimed at inhibiting key bacterial metabolic pathways as well as modulating host metabolism to boost immune production.
Acknowledgments
G.A.S. was supported by funds provided through the Hatch Act to the University of Nebraska Institute of Agriculture and Natural Resources and by funds provided through the National Institutes of Health (R01AI087668). Unpublished work from the A.L.S. laboratory was supported by a grant (R01GM042219) from the National Institutes of Health. A.R.R. was supported by funds provided through the National Institutes of Health (R01AI093613).
References
- 1.Sadykov MR, Olson ME, Halouska S, Zhu Y, Fey PD, Powers R, Somerville GA. Tricarboxylic acid cycle-dependent regulation of Staphylococcus epidermidis polysaccharide intercellular adhesin synthesis. J Bacteriol. 2008;190:7621–7632. doi: 10.1128/JB.00806-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sadykov MR, Zhang B, Halouska S, Nelson JL, Kreimer LW, Zhu Y, Powers R, Somerville GA. Using NMR metabolomics to investigate tricarboxylic acid cycle dependent signal transduction in Staphylococcus epidermidis. J Biol Chem. 2010;285:36616–36624. doi: 10.1074/jbc.M110.152843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kundig W, Ghosh S, Roseman S. Phosphate Bound to Histidine in a Protein as an Intermediate in a Novel Phospho-Transferase System. Proc Natl Acad Sci U S A. 1964;52:1067–1074. doi: 10.1073/pnas.52.4.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saier MH., Jr Protein phosphorylation and allosteric control of inducer exclusion and catabolite repression by the bacterial phosphoenolpyruvate: sugar phosphotransferase system. Microbiol Rev. 1989;53:109–120. doi: 10.1128/mr.53.1.109-120.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klein HP, Doudoroff M. The mutation of Pseudomonas putrefaciens to glucose utilization and its enzymatic basis. J Bacteriol. 1950;59:739–750. doi: 10.1128/jb.59.6.739-750.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanwal BD. Allosteric controls of amphilbolic pathways in bacteria. Bacteriological Reviews. 1970;34:20–39. doi: 10.1128/br.34.1.20-39.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blangy D, Buc H, Monod J. Kinetics of the allosteric interactions of phosphofructokinase from Escherichia coli. J Mol Biol. 1968;31:13–35. doi: 10.1016/0022-2836(68)90051-x. [DOI] [PubMed] [Google Scholar]
- 8.Lopez G, Latorre M, Reyes-Jara A, Cambiazo V, Gonzalez M. Transcriptomic response of Enterococcus faecalis to iron excess. Biometals. 2012;25:737–747. doi: 10.1007/s10534-012-9539-5. [DOI] [PubMed] [Google Scholar]
- 9.Ojha A, Hatfull GF. The role of iron in Mycobacterium smegmatis biofilm formation: the exochelin siderophore is essential in limiting iron conditions for biofilm formation but not for planktonic growth. Mol Micro. 2007;66:468–483. doi: 10.1111/j.1365-2958.2007.05935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedman DB, Stauff DL, Pishchany G, Whitwell CW, Torres VJ, Skaar EP. Staphylococcus aureus redirects central metabolism to increase iron availability. PLoS Pathog. 2006;2:e87. doi: 10.1371/journal.ppat.0020087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Somerville G, Mikoryak CA, Reitzer L. Physiological characterization of Pseudomonas aeruginosa during exotoxin A synthesis: glutamate, iron limitation, and aconitase activity. J Bacteriol. 1999;181:1072–1078. doi: 10.1128/jb.181.4.1072-1078.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang B, Halouska S, Schiaffo CE, Sadykov MR, Somerville GA, Powers R. NMR analysis of a stress response metabolic signaling network. J Proteome Res. 2011;10:3743–3754. doi: 10.1021/pr200360w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Craig JE, Ford MJ, Blaydon DC, Sonenshein AL. A null mutation in the Bacillus subtilis aconitase gene causes a block in Spo0A-phosphate-dependent gene expression. J Bacteriol. 1997;179:7351–7359. doi: 10.1128/jb.179.23.7351-7359.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dickens F. Oxidation of phosphohexonate and pentose phosphoric acids by yeast enzymes: Oxidation of phosphohexonate. II. Oxidation of pentose phosphoric acids. Biochem J. 1938;32:1626–1644. doi: 10.1042/bj0321626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warburg O, Christian W, Griese A. Hydrogen-transferring coenzyme, its composition and mode of action. Biochem Z. 1935;282:157–205. [Google Scholar]
- 16.Horecker BL, Smyrniotis PZ. Purification and properties of yeast transaldolase. J Biol Chem. 1955;212:811–825. [PubMed] [Google Scholar]
- 17.Scott DB, Cohen SS. The oxidative pathway of carbohydrate metabolism in Escherichia coli.1. The isolation and properties of glucose 6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase. Biochem J. 1953;55:23–33. doi: 10.1042/bj0550023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dutow P, Schmidl SR, Ridderbusch M, Stulke J. Interactions between glycolytic enzymes of Mycoplasma pneumoniae. J Mol Microbiol Biotechnol. 2010;19:134–139. doi: 10.1159/000321499. [DOI] [PubMed] [Google Scholar]
- 19.Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson RJ, Durkin AS, Gwinn M, Kolonay JF, Nelson WC, Peterson JD, Umayam LA, White O, Salzberg SL, Lewis MR, Radune D, Holtzapple E, Khouri H, Wolf AM, Utterback TR, Hansen CL, McDonald LA, Feldblyum TV, Angiuoli S, Dickinson T, Hickey EK, Holt IE, Loftus BJ, Yang F, Smith HO, Venter JC, Dougherty BA, Morrison DA, Hollingshead SK, Fraser CM. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science. 2001;293:498–506. doi: 10.1126/science.1061217. [DOI] [PubMed] [Google Scholar]
- 20.Guimaraes AM, Santos AP, SanMiguel P, Walter T, Timenetsky J, Messick JB. Complete genome sequence of Mycoplasma suis and insights into its biology and adaption to an erythrocyte niche. PloS One. 2011;6:e19574. doi: 10.1371/journal.pone.0019574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergman NH, Anderson EC, Swenson EE, Janes BK, Fisher N, Niemeyer MM, Miyoshi AD, Hanna PC. Transcriptional profiling of Bacillus anthracis during infection of host macrophages. Infect Immun. 2007;75:3434–3444. doi: 10.1128/IAI.01345-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chatterjee SS, Hossain H, Otten S, Kuenne C, Kuchmina K, Machata S, Domann E, Chakraborty T, Hain T. Intracellular gene expression profile of Listeria monocytogenes. Infect Immun. 2006;74:1323–1338. doi: 10.1128/IAI.74.2.1323-1338.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fleury B, Kelley WL, Lew D, Gotz F, Proctor RA, Vaudaux P. Transcriptomic and metabolic responses of Staphylococcus aureus exposed to supra-physiological temperatures. BMC Microbiol. 2009;9:76. doi: 10.1186/1471-2180-9-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weigoldt M, Meens J, Bange FC, Pich A, Gerlach GF, Goethe R. Metabolic adaptation of Mycobacterium avium subsp. paratuberculosis to the gut environment. Microbiol. 2013;159:380–391. doi: 10.1099/mic.0.062737-0. [DOI] [PubMed] [Google Scholar]
- 25.delCardayre SB, Stock KP, Newton GL, Fahey RC, Davies JE. Coenzyme A disulfide reductase, the primary low molecular weight disulfide reductase from Staphylococcus aureus. Purification and characterization of the native enzyme. J Biol Chem. 1998;273:5744–5751. doi: 10.1074/jbc.273.10.5744. [DOI] [PubMed] [Google Scholar]
- 26.Helmann JD. Bacillithiol, a new player in bacterial redox homeostasis. Antioxid Redox Signal. 2011;15:123–133. doi: 10.1089/ars.2010.3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Newton GL, Fahey RC, Rawat M. Detoxification of toxins by bacillithiol in Staphylococcus aureus. Microbiol. 2012;158:1117–1126. doi: 10.1099/mic.0.055715-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spies HS, Steenkamp DJ. Thiols of intracellular pathogens. Identification of ovothiol A in Leishmania donovani and structural analysis of a novel thiol from Mycobacterium bovis. Eur J Biochem. 1994;224:203–213. doi: 10.1111/j.1432-1033.1994.tb20013.x. [DOI] [PubMed] [Google Scholar]
- 29.Holmgren A. Thioredoxin. Annu Rev Biochem. 1985;54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 30.Moore EC, Reichard P, Thelander L. Enzymatic synthesis of deoxyribonucleotides. V. Purification and properties of thioredoxin reductase from Escherichia coli B. J Biol Chem. 1964;239:3445–3452. [PubMed] [Google Scholar]
- 31.Gustafsson TN, Sahlin M, Lu J, Sjoberg BM, Holmgren A. Bacillus anthracis thioredoxin systems, characterization and role as electron donors for ribonucleotide reductase. J Biol Chem. 2012;287:39686–39697. doi: 10.1074/jbc.M112.413427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Newton GL, Buchmeier N, Fahey RC. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol Mol Biol Rev. 2008;72:471–494. doi: 10.1128/MMBR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eisenreich W, Slaghuis J, Laupitz R, Bussemer J, Stritzker J, Schwarz C, Schwarz R, Dandekar T, Goebel W, Bacher A. 13C isotopologue perturbation studies of Listeria monocytogenes carbon metabolism and its modulation by the virulence regulator PrfA. Proc Natl Acad Sci U S A. 2006;103:2040–2045. doi: 10.1073/pnas.0507580103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfefferkorn ER, Rebhun S, Eckel M. Characterization of an indoleamine 2,3-dioxygenase induced by gamma-interferon in cultured human fibroblasts. J Interferon Res. 1986;6:267–279. doi: 10.1089/jir.1986.6.267. [DOI] [PubMed] [Google Scholar]
- 35.Daubener W, MacKenzie CR. IFN-gamma activated indoleamine 2,3-dioxygenase activity in human cells is an antiparasitic and an antibacterial effector mechanism. Adv Exp Med Biol. 1999;467:517–524. doi: 10.1007/978-1-4615-4709-9_64. [DOI] [PubMed] [Google Scholar]
- 36.Mraheil MA, Billion A, Mohamed W, Rawool D, Hain T, Chakraborty T. Adaptation of Listeria monocytogenes to oxidative and nitrosative stress in IFN-gamma-activated macrophages. Int J Med Microbiol. 2011;301:547–555. doi: 10.1016/j.ijmm.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 37.Hucke C, MacKenzie CR, Adjogble KD, Takikawa O, Daubener W. Nitric oxide-mediated regulation of gamma interferon-induced bacteriostasis: inhibition and degradation of human indoleamine 2,3-dioxygenase. Infect Immun. 2004;72:2723–2730. doi: 10.1128/IAI.72.5.2723-2730.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacKenzie CR, Hadding U, Daubener W. Interferon-gamma-induced activation of indoleamine 2,3-dioxygenase in cord blood monocyte-derived macrophages inhibits the growth of group B streptococci. J Infect Dis. 1998;178:875–878. doi: 10.1086/515347. [DOI] [PubMed] [Google Scholar]
- 39.Huynen MA, Dandekar T, Bork P. Variation and evolution of the citric-acid cycle: a genomic perspective. Trends Microbiol. 1999;7:281–291. doi: 10.1016/s0966-842x(99)01539-5. [DOI] [PubMed] [Google Scholar]
- 40.Srinivasan V, Morowitz HJ. Ancient genes in contemporary persistent microbial pathogens. Biol Bull. 2006;210:1–9. doi: 10.2307/4134531. [DOI] [PubMed] [Google Scholar]
- 41.Sonenshein AL. The Krebs citric acid cycle. In: Sonenshein AL, Hoch JA, Losick R, editors. Bacillus subtilis and its closest relatives: From genes to cells. ASM Press; Washington, D.C: 2002. pp. 151–162. [Google Scholar]
- 42.Bondi A, Kornblum J, De St Phalle M. The amino acid requirements of penicillin resistant and penicillin sensitive strains of Micrococcus pyogenes. J Bacteriol. 1954;68:617–621. doi: 10.1128/jb.68.5.617-621.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murray BE, Singh KV, Ross RP, Heath JD, Dunny GM, Weinstock GM. Generation of restriction map of Enterococcus faecalis OG1 and investigation of growth requirements and regions encoding biosynthetic function. J Bacteriol. 1993;175:5216–5223. doi: 10.1128/jb.175.16.5216-5223.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tourtellotte ME, Morowitz HJ, Kasimer P. Defined medium for Mycoplasma laidlawii. J Bacteriol. 1964;88:11–15. doi: 10.1128/jb.88.1.11-15.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Melendez-Hevia E, Waddell TG, Cascante M. The puzzle of the Krebs citric acid cycle: assembling the pieces of chemically feasible reactions, and opportunism in the design of metabolic pathways during evolution. J Mol Evol. 1996;43:293–303. doi: 10.1007/BF02338838. [DOI] [PubMed] [Google Scholar]
- 46.Kim HJ, Mittal M, Sonenshein AL. CcpC-dependent regulation of citB and lmo0847 in Listeria monocytogenes. J Bacteriol. 2006;188:179– 190. doi: 10.1128/JB.188.1.179-190.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cerdeno-Tarraga AM, Efstratiou A, Dover LG, Holden MT, Pallen M, Bentley SD, Besra GS, Churcher C, James KD, De Zoysa A, Chillingworth T, Cronin A, Dowd L, Feltwell T, Hamlin N, Holroyd S, Jagels K, Moule S, Quail MA, Rabbinowitsch E, Rutherford KM, Thomson NR, Unwin L, Whitehead S, Barrell BG, Parkhill J. The complete genome sequence and analysis of Corynebacterium diphtheriae NCTC13129. Nucleic Acids Res. 2003;31:6516–6523. doi: 10.1093/nar/gkg874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakano MM, Zuber P, Sonenshein AL. Anaerobic regulation of Bacillus subtilis Krebs cycle genes. J Bacteriol. 1998;180:3304–3311. doi: 10.1128/jb.180.13.3304-3311.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eoh H, Rhee KY. Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2013;110:6554–6559. doi: 10.1073/pnas.1219375110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Somerville GA, Chaussee MS, Morgan CI, Fitzgerald JR, Dorward DW, Reitzer LJ, Musser JM. Staphylococcus aureus aconitase inactivation unexpectedly inhibits post-exponential-phase growth and enhances stationary-phase survival. Infect Immun. 2002;70:6373–6382. doi: 10.1128/IAI.70.11.6373-6382.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mei JM, Nourbakhsh F, Ford CW, Holden DW. Identification of Staphylococcus aureus virulence genes in a murine model of bacteraemia using signature-tagged mutagenesis. Mol Microbiol. 1997;26:399–407. doi: 10.1046/j.1365-2958.1997.5911966.x. [DOI] [PubMed] [Google Scholar]
- 52.Kornberg HL, Krebs HA. Synthesis of cell constiuents from C2− units by a modified tricarboxylic acid cycle. Nature. 1957;179:988–991. doi: 10.1038/179988a0. [DOI] [PubMed] [Google Scholar]
- 53.Beaman BL, Beaman L. Nocardia species: hostparasite relationships. Clin Microbiol Rev. 1994;7:213–264. doi: 10.1128/cmr.7.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muscatello G. Rhodococcus equi pneumonia in the foal–part 1: pathogenesis and epidemiology. Vet J. 2012;192:20–26. doi: 10.1016/j.tvjl.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 55.Lorenz MC, Fink GR. Life and death in a macrophage: role of the glyoxylate cycle in virulence. Eukaryot. Cell. 2002;1:657–662. doi: 10.1128/EC.1.5.657-662.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Titgemeyer F, Hillen W. Global control of sugar metabolism: a gram-positive solution. Antonie Van Leeuwenhoek. 2002;82:59–71. [PubMed] [Google Scholar]
- 57.Warner JB, Lolkema JS. CcpA-dependent carbon catabolite repression in bacteria. Microbiol Mol Biol Rev. 2003;67:475–490. doi: 10.1128/MMBR.67.4.475-490.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pimentel-Schmitt EF, Thomae AW, Amon J, Klieber MA, Roth HM, Muller YA, Jahreis K, Burkovski A, Titgemeyer F. A glucose kinase from Mycobacterium smegmatis. J Mol Microbiol Biotechnol. 2007;12:75–81. doi: 10.1159/000096462. [DOI] [PubMed] [Google Scholar]
- 59.Bowles JA, Segal W. Kinetics of utilization of organic compounds in the growth of Mycobacterium tuberculosis. J Bacteriol. 1965;90:157–163. doi: 10.1128/jb.90.1.157-163.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Collins FM, Lascelles J. The effect of growth conditions on oxidative and dehydrogenase activity in Staphylococcus aureus. J Gen Microbiol. 1962;29:531–535. doi: 10.1099/00221287-29-3-531. [DOI] [PubMed] [Google Scholar]
- 61.Hanson RS, Srinivasan VR, Halvorson HO. Biochemistry of sporulation. I. Metabolism of acetate by vegetative and sporulating cells. J Bacteriol. 1963;85:451–460. doi: 10.1128/jb.85.2.451-460.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hanson RS, Blicharska J, Arnaud M, Szulmajster J. Observation on the regulation of the synthesis of the tricarboxylic acid cycle enzymes in Bacillus subtilis, Marburg. Biochem Biophys Res Commun. 1964;17:690–695. [Google Scholar]
- 63.Holmgren NB, Millman I, Youmans GP. Studies on the metabolism of Mycobacterium tuberculosis. VI. The effect of Krebs’ tricarboxylic acid cycle intermediates and precursors on the growth and respiration of Mycobacterium tuberculosis. J Bacteriol. 1954;68:405–410. doi: 10.1128/jb.68.4.405-410.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Somerville GA, Saïd-Salim B, Wickman JM, Raffel SJ, Kreiswirth BN, Musser JM. Correlation of acetate catabolism and growth yield in Staphylococcus aureus: Implications for host-pathogen interactions. Infect Immun. 2003;71:4724–4732. doi: 10.1128/IAI.71.8.4724-4732.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Somerville GA, Cockayne A, Dürr M, Peschel A, Otto M, Musser JM. Synthesis and deformylation of Staphylococcus aureus delta-toxin are linked to tricarboxylic acid cycle activity. J Bacteriol. 2003;185:6686–6694. doi: 10.1128/JB.185.22.6686-6694.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vuong C, Kidder JB, Jacobson ER, Otto M, Proctor RA, Somerville GA. Staphylococcus epidermidis polysaccharide intercellular adhesin production significantly increases during tricarboxylic acid cycle stress. J Bacteriol. 2005;187:2967–2973. doi: 10.1128/JB.187.9.2967-2973.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Varghese S, Tang Y, Imlay JA. Contrasting sensitivities of Escherichia coli aconitases A and B to oxidation and iron depletion. J Bacteriol. 2003;185:221–230. doi: 10.1128/JB.185.1.221-230.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jaeger T, Mayer C. The transcriptional factors MurR and catabolite activator protein regulate N-acetylmuramic acid catabolism in Escherichia coli. J Bacteriol. 2008;190:6598–6608. doi: 10.1128/JB.00642-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shivers RP, Sonenshein AL. Activation of the Bacillus subtilis global regulator CodY by direct interaction with branched-chain amino acids. Mol Microbiol. 2004;53:599–611. doi: 10.1111/j.1365-2958.2004.04135.x. [DOI] [PubMed] [Google Scholar]
- 70.Romling U, Galperin MY, Gomelsky M. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev. 2013;77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ernst JF, Bennett RL, Rothfield LI. Constitutive expression of the iron-enterochelin and ferrichrome uptake systems in a mutant strain of Salmonella typhimurium. J Bacteriol. 1978;135:928–934. doi: 10.1128/jb.135.3.928-934.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Somerville GA, Proctor RA. At the crossroads of bacterial metabolism and virulence factor synthesis in staphylococci. Microbiol Mol Biol Rev. 2009;73:233–248. doi: 10.1128/MMBR.00005-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reitzer L. Nitrogen assimilation and global regulation in Escherichia coli. Annu Rev Microbiol. 2003;57:155–176. doi: 10.1146/annurev.micro.57.030502.090820. [DOI] [PubMed] [Google Scholar]
- 74.Fisher SH. Regulation of nitrogen metabolism in Bacillus subtilis: vive la difference! Mol Microbiol. 1999;32:223–232. doi: 10.1046/j.1365-2958.1999.01333.x. [DOI] [PubMed] [Google Scholar]
- 75.Tempest DW, Meers JL, Brown CM. Synthesis of glutamate in Aerobacter aerogenes by a hitherto unknown route. Biochem J. 1970;117:405–407. doi: 10.1042/bj1170405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Slack FJ, Serror P, Joyce E, Sonenshein AL. A gene required for nutritional repression of the Bacillus subtilis dipeptide permease operon. Mol Microbiol. 1995;15:689–702. doi: 10.1111/j.1365-2958.1995.tb02378.x. [DOI] [PubMed] [Google Scholar]
- 77.Armstrong FB, Wagner RP. Biosynthesis of valine and isoleucine. IV. alpha-hydroxy-beta-keto acid reductoisomerase of Salmonella. J Biol Chem. 1961;236:2027–2032. [PubMed] [Google Scholar]
- 78.Myers JW. Dihydroxy acid dehydrase: an enzyme involved in the biosynthesis of isoleucine and valine. J Biol Chem. 1961;236:1414–1418. [PubMed] [Google Scholar]
- 79.Leitzmann C, Bernlohr RW. Threonine dehydratase of Bacillus licheniformis. I. Purification and properties. Biochim Biophys Acta. 1968;151:449–460. doi: 10.1016/0005-2744(68)90113-7. [DOI] [PubMed] [Google Scholar]
- 80.Nobre LS, Saraiva LM. Effect of combined oxidative and nitrosative stresses on Staphylococcus aureus transcriptome. Appl Microbiol Biotechnol. 2013;97:2563–2573. doi: 10.1007/s00253-013-4730-3. [DOI] [PubMed] [Google Scholar]
- 81.Thorsing M, Klitgaard JK, Atilano ML, Skov MN, Kolmos HJ, Filipe SR, Kallipolitis BH. Thioridazine induces major changes in global gene expression and cell wall composition in methicillin-resistant Staphylococcus aureus USA300. PloS One. 2013;8:e64518. doi: 10.1371/journal.pone.0064518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Moreno MS, Schneider BL, Maile RR, Weyler W, Saier MH., Jr Catabolite repression mediated by the CcpA protein in Bacillus subtilis: novel modes of regulation revealed by whole-genome analyses. Mol Microbiol. 2001;39:1366–1381. doi: 10.1111/j.1365-2958.2001.02328.x. [DOI] [PubMed] [Google Scholar]
- 83.Yoshida K, Kobayashi K, Miwa Y, Kang CM, Matsunaga M, Yamaguchi H, Tojo S, Yamamoto M, Nishi R, Ogasawara N, Nakayama T, Fujita Y. Combined transcriptome and proteome analysis as a powerful approach to study genes under glucose repression in Bacillus subtilis. Nucleic Acids Res. 2001;29:683–692. doi: 10.1093/nar/29.3.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Antunes A, Camiade E, Monot M, Courtois E, Barbut F, Sernova NV, Rodionov DA, Martin-Verstraete I, Dupuy B. Global transcriptional control by glucose and carbon regulator CcpA in Clostridium difficile. Nucleic Acids Res. 2012;40:10701–10718. doi: 10.1093/nar/gks864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ren C, Gu Y, Wu Y, Zhang W, Yang C, Yang S, Jiang W. Pleiotropic functions of catabolite control protein CcpA in Butanol-producing Clostridium acetobutylicum. BMC Genomics. 2012;13:349. doi: 10.1186/1471-2164-13-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jankovic I, Egeter O, Bruckner R. Analysis of catabolite control protein A-dependent repression in Staphylococcus xylosus by a genomic reporter gene system. J Bacteriol. 2001;183:580–586. doi: 10.1128/JB.183.2.580-586.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Seidl K, Muller S, Francois P, Kriebitzsch C, Schrenzel J, Engelmann S, Bischoff M, Berger-Bachi B. Effect of a glucose impulse on the CcpA regulon in Staphylococcus aureus. BMC Microbiol. 2009;9:95. doi: 10.1186/1471-2180-9-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zeng L, Choi SC, Danko CG, Siepel A, Stanhope MJ, Burne RA. Gene regulation by CcpA and catabolite repression explored by RNA-Seq in Streptococcus mutans. PloS One. 2013;8:e60465. doi: 10.1371/journal.pone.0060465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Willenborg J, Fulde M, de Greeff A, Rohde M, Smith HE, Valentin-Weigand P, Goethe R. Role of glucose and CcpA in capsule expression and virulence of Streptococcus suis. Microbiology. 2011;157:1823–1833. doi: 10.1099/mic.0.046417-0. [DOI] [PubMed] [Google Scholar]
- 90.Carvalho SM, Kloosterman TG, Kuipers OP, Neves AR. CcpA ensures optimal metabolic fitness of Streptococcus pneumoniae. PloS One. 2011;6:e26707. doi: 10.1371/journal.pone.0026707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kinkel TL, McIver KS. CcpA-mediated repression of streptolysin S expression and virulence in the group A streptococcus. Infect Immun. 2008;76:3451–3463. doi: 10.1128/IAI.00343-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zomer AL, Buist G, Larsen R, Kok J, Kuipers OP. Time-resolved determination of the CcpA regulon of Lactococcus lactis subsp. cremoris MG1363. J Bacteriol. 2007;189:1366–1381. doi: 10.1128/JB.01013-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Leboeuf C, Leblanc L, Auffray Y, Hartke A. Characterization of the ccpA gene of Enterococcus faecalis: identification of starvation-inducible proteins regulated by CcpA. J Bacteriol. 2000;182:5799–5806. doi: 10.1128/jb.182.20.5799-5806.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Asanuma N, Yoshii T, Hino T. Molecular characterization of CcpA and involvement of this protein in transcriptional regulation of lactate dehydrogenase and pyruvate formate-lyase in the ruminal bacterium Streptococcus bovis. Appl Environ Microbiol. 2004;70:5244–5251. doi: 10.1128/AEM.70.9.5244-5251.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fujita Y. Carbon catabolite control of the metabolic network in Bacillus subtilis. Biosci Biotechnol Biochem. 2009;73:245–259. doi: 10.1271/bbb.80479. [DOI] [PubMed] [Google Scholar]
- 96.Dahl MK. CcpA-independent carbon catabolite repression in Bacillus subtilis. J Mol Microbiol Biotechnol. 2002;4:315–321. [PubMed] [Google Scholar]
- 97.Deutscher J, Herro R, Bourand A, Mijakovic I, Poncet S. P-Ser-HPr–a link between carbon metabolism and the virulence of some pathogenic bacteria. Biochim Biophys Acta. 2005;1754:118–125. doi: 10.1016/j.bbapap.2005.07.029. [DOI] [PubMed] [Google Scholar]
- 98.Luesink EJ, Beumer CM, Kuipers OP, De Vos WM. Molecular characterization of the Lactococcus lactis ptsHI operon and analysis of the regulatory role of HPr. J Bacteriol. 1999;181:764–771. doi: 10.1128/jb.181.3.764-771.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zeng L, Burne RA. Seryl-phosphorylated HPr regulates CcpA-independent carbon catabolite repression in conjunction with PTS permeases in Streptococcus mutans. Mol Microbiol. 2010;75:1145–1158. doi: 10.1111/j.1365-2958.2009.07029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Antunes A, Martin-Verstraete I, Dupuy B. CcpA-mediated repression of Clostridium difficile toxin gene expression. Mol Microbiol. 2011;79:882–899. doi: 10.1111/j.1365-2958.2010.07495.x. [DOI] [PubMed] [Google Scholar]
- 101.Leiba J, Hartmann T, Cluzel ME, Cohen-Gonsaud M, Delolme F, Bischoff M, Molle V. A novel mode of regulation of the Staphylococcus aureus catabolite control protein A (CcpA) mediated by Stk1 protein phosphorylation. J Biol Chem. 2012;287:43607–43619. doi: 10.1074/jbc.M112.418913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bayer AS, Coulter SN, Stover CK, Schwan WR. Impact of the high-affinity proline permease gene (putP) on the virulence of Staphylococcus aureus in experimental endocarditis. Infect Immun. 1999;67:740–744. doi: 10.1128/iai.67.2.740-744.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schwan WR, Wetzel KJ, Gomez TS, Stiles MA, Beitlich BD, Grunwald S. Low-proline environments impair growth, proline transport and in vivo survival of Staphylococcus aureus strain-specific putP mutants. Microbiology. 2004;150:1055–1061. doi: 10.1099/mic.0.26710-0. [DOI] [PubMed] [Google Scholar]
- 104.Li C, Sun F, Cho H, Yelavarthi V, Sohn C, He C, Schneewind O, Bae T. CcpA mediates proline auxotrophy and is required for Staphylococcus aureus pathogenesis. J Bacteriol. 2010;192:3883–3892. doi: 10.1128/JB.00237-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Giammarinaro P, Paton JC. Role of RegM, a homologue of the catabolite repressor protein CcpA, in the virulence of Streptococcus pneumoniae. Infect Immun. 2002;70:5454–5461. doi: 10.1128/IAI.70.10.5454-5461.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Iyer R, Baliga NS, Camilli A. Catabolite control protein A (CcpA) contributes to virulence and regulation of sugar metabolism in Streptococcus pneumoniae. J Bacteriol. 2005;187:8340–8349. doi: 10.1128/JB.187.24.8340-8349.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang A, Chen B, Yuan Z, Li R, Liu C, Zhou H, Chen H, Jin M. HP0197 contributes to CPS synthesis and the virulence of Streptococcus suis via CcpA. PloS One. 2012;7:e50987. doi: 10.1371/journal.pone.0050987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Abranches J, Nascimento MM, Zeng L, Browngardt CM, Wen ZT, Rivera MF, Burne RA. CcpA regulates central metabolism and virulence gene expression in Streptococcus mutans. J Bacteriol. 2008;190:2340–2349. doi: 10.1128/JB.01237-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zheng L, Itzek A, Chen Z, Kreth J. Environmental influences on competitive hydrogen peroxide production in Streptococcus gordonii. Appl Environ Microbiol. 2011;77:4318–4328. doi: 10.1128/AEM.00309-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zheng L, Chen Z, Itzek A, Ashby M, Kreth J. Catabolite control protein A controls hydrogen peroxide production and cell death in Streptococcus sanguinis. J Bacteriol. 2011;193:516–526. doi: 10.1128/JB.01131-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ahn SJ, Rice KC, Oleas J, Bayles KW, Burne RA. The Streptococcus mutans Cid and Lrg systems modulate virulence traits in response to multiple environmental signals. Microbiology. 2010;156:3136–3147. doi: 10.1099/mic.0.039586-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Almengor AC, Kinkel TL, Day SJ, McIver KS. The catabolite control protein CcpA binds to Pmga and influences expression of the virulence regulator Mga in the Group A Streptococcus. J Bacteriol. 2007;189:8405–8416. doi: 10.1128/JB.01038-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dupuy B, Sonenshein AL. Regulated transcription of Clostridium difficile toxin genes. Mol Microbiol. 1998;27:107–120. doi: 10.1046/j.1365-2958.1998.00663.x. [DOI] [PubMed] [Google Scholar]
- 114.Karlsson S, Burman LG, Akerlund T. Suppression of toxin production in Clostridium difficile VPI 10463 by amino acids. Microbiology. 1999;145:1683–1693. doi: 10.1099/13500872-145-7-1683. [DOI] [PubMed] [Google Scholar]
- 115.Mendez MB, Goni A, Ramirez W, Grau RR. Sugar inhibits the production of the toxins that trigger clostridial gas gangrene. Microb Pathog. 2012;52:85–91. doi: 10.1016/j.micpath.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 116.Mendez M, Huang IH, Ohtani K, Grau R, Shimizu T, Sarker MR. Carbon catabolite repression of type IV pilus-dependent gliding motility in the anaerobic pathogen Clostridium perfringens. J Bacteriol. 2008;190:48–60. doi: 10.1128/JB.01407-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Varga JJ, Therit B, Melville SB. Type IV pili and the CcpA protein are needed for maximal biofilm formation by the gram-positive anaerobic pathogen Clostridium perfringens. Infect Immun. 2008;76:4944– 4951. doi: 10.1128/IAI.00692-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Varga J, Stirewalt VL, Melville SB. The CcpA protein is necessary for efficient sporulation and enterotoxin gene (cpe) regulation in Clostridium perfringens. J Bacteriol. 2004;186:5221–5229. doi: 10.1128/JB.186.16.5221-5229.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mackey-Lawrence NM, Jefferson KK. Regulation of Staphylococcus aureus immunodominant antigen B (IsaB) Microbiol Res. 2013;168:113–118. doi: 10.1016/j.micres.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Seidl K, Goerke C, Wolz C, Mack D, Berger-Bachi B, Bischoff M. Staphylococcus aureus CcpA affects biofilm formation. Infect Immun. 2008;76:2044–2050. doi: 10.1128/IAI.00035-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Seidl K, Stucki M, Ruegg M, Goerke C, Wolz C, Harris L, Berger-Bachi B, Bischoff M. Staphylococcus aureus CcpA affects virulence determinant production and antibiotic resistance. Antimicrob Agents Chemother. 2006;50:1183–1194. doi: 10.1128/AAC.50.4.1183-1194.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Seidl K, Bischoff M, Berger-Bachi B. CcpA mediates the catabolite repression of tst in Staphylococcus aureus. Infect Immun. 2008;76:5093–5099. doi: 10.1128/IAI.00724-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chiang C, Bongiorni C, Perego M. Glucose-dependent activation of Bacillus anthracis toxin gene expression and virulence requires the carbon catabolite protein CcpA. J Bacteriol. 2011;193:52–62. doi: 10.1128/JB.01656-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gao P, Pinkston KL, Bourgogne A, Cruz MR, Garsin DA, Murray BE, Harvey BR. Library screen identifies Enterococcus faecalis CcpA, the catabolite control protein A, as an effector of Ace, a collagen adhesion protein linked to virulence. J Bacteriol. 2013;195:4761–4768. doi: 10.1128/JB.00706-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Herro R, Poncet S, Cossart P, Buchrieser C, Gouin E, Glaser P, Deutscher J. How seryl-phosphorylated HPr inhibits PrfA, a transcription activator of Listeria monocytogenes virulence genes. J Mol Microbiol Biotechnol. 2005;9:224–234. doi: 10.1159/000089650. [DOI] [PubMed] [Google Scholar]
- 126.Mertins S, Joseph B, Goetz M, Ecke R, Seidel G, Sprehe M, Hillen W, Goebel W, Muller-Altrock S. Interference of components of the phosphoenolpyruvate phosphotransferase system with the central virulence gene regulator PrfA of Listeria monocytogenes. J Bacteriol. 2007;189:473–490. doi: 10.1128/JB.00972-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Molle V, Nakaura Y, Shivers RP, Yamaguchi H, Losick R, Fujita Y, Sonenshein AL. Additional targets of the Bacillus subtilis global regulator CodY identified by chromatin immunoprecipitation and genome-wide transcript analysis. J Bacteriol. 2003;185:1911–1922. doi: 10.1128/JB.185.6.1911-1922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dineen SS, McBride SM, Sonenshein AL. Integration of metabolism and virulence by Clostridium difficile CodY. J Bacteriol. 2010;192:5350–5362. doi: 10.1128/JB.00341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Majerczyk CD, Dunman PM, Luong TT, Lee CY, Sadykov MR, Somerville GA, Bodi K, Sonenshein AL. Direct targets of CodY in Staphylococcus aureus. J Bacteriol. 2010;192:2861–2877. doi: 10.1128/JB.00220-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pohl K, Francois P, Stenz L, Schlink F, Geiger T, Herbert S, Goerke C, Schrenzel J, Wolz C. CodY in Staphylococcus aureus: a regulatory link between metabolism and virulence gene expression. J Bacteriol. 2009;191:2953–2963. doi: 10.1128/JB.01492-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Malke H, Ferretti JJ. CodY-affected transcriptional gene expression of Streptococcus pyogenes during growth in human blood. J Med Microbiol. 2007;56:707–714. doi: 10.1099/jmm.0.46984-0. [DOI] [PubMed] [Google Scholar]
- 132.Kreth J, Chen Z, Ferretti J, Malke H. Counteractive balancing of transcriptome expression involving CodY and CovRS in Streptococcus pyogenes. J Bacteriol. 2011;193:4153–4165. doi: 10.1128/JB.00061-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Guedon E, Serror P, Ehrlich SD, Renault P, Delorme C. Pleiotropic transcriptional repressor CodY senses the intracellular pool of branched-chain amino acids in Lactococcus lactis. Mol Microbiol. 2001;40:1227–1239. doi: 10.1046/j.1365-2958.2001.02470.x. [DOI] [PubMed] [Google Scholar]
- 134.Bennett HJ, Pearce DM, Glenn S, Taylor CM, Kuhn M, Sonenshein AL, Andrew PW, Roberts IS. Characterization of relA and codY mutants of Listeria monocytogenes: identification of the CodY regulon and its role in virulence. Mol Microbiol. 2007;63:1453–1467. doi: 10.1111/j.1365-2958.2007.05597.x. [DOI] [PubMed] [Google Scholar]
- 135.Brinsmade SR, Kleijn RJ, Sauer U, Sonenshein AL. Regulation of CodY activity through modulation of intracellular branched-chain amino acid pools. J Bacteriol. 2010;192:6357–6368. doi: 10.1128/JB.00937-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ratnayake-Lecamwasam M, Serror P, Wong KW, Sonenshein AL. Bacillus subtilis CodY represses early-stationary-phase genes by sensing GTP levels. Genes Dev. 2001;15:1093–1103. doi: 10.1101/gad.874201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Handke LD, Shivers RP, Sonenshein AL. Interaction of Bacillus subtilis CodY with GTP. J Bacteriol. 2008;190:798–806. doi: 10.1128/JB.01115-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Brinsmade SR, Sonenshein AL. Dissecting complex metabolic integration provides direct genetic evidence for CodY activation by guanine nucleotides. J Bacteriol. 2011;193:5637–5648. doi: 10.1128/JB.05510-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hendriksen WT, Bootsma HJ, Estevao S, Hoogenboezem T, de Jong A, de Groot R, Kuipers OP, Hermans PW. CodY of Streptococcus pneumoniae: link between nutritional gene regulation and colonization. J Bacteriol. 2008;190:590–601. doi: 10.1128/JB.00917-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Petranovic D, Guedon E, Sperandio B, Delorme C, Ehrlich D, Renault P. Intracellular effectors regulating the activity of the Lactococcus lactis CodY pleiotropic transcription regulator. Mol Microbiol. 2004;53:613–621. doi: 10.1111/j.1365-2958.2004.04136.x. [DOI] [PubMed] [Google Scholar]
- 141.Levdikov VM, Blagova E, Colledge VL, Lebedev AA, Williamson DC, Sonenshein AL, Wilkinson AJ. Structural rearrangement accompanying ligand binding in the GAF domain of CodY from Bacillus subtilis. J Mol Biol. 2009;390:1007–1018. doi: 10.1016/j.jmb.2009.05.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kriel A, Bittner AN, Kim SH, Liu K, Tehranchi AK, Zou WY, Rendon S, Chen R, Tu BP, Wang JD. Direct regulation of GTP homeostasis by (p)ppGpp: a critical component of viability and stress resistance. Mol Cell. 2012;48:231–241. doi: 10.1016/j.molcel.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kriel A, Brinsmade SR, Tse JL, Tehranchi A, Bittner A, Sonenshein AL, Wang JD. GTP dysregulation in Bacillus subtilis cells lacking (p)ppGpp results in phenotypic amino acid auxotrophy and failure to adapt to nutrient downshift and regulate biosynthesis genes. J Bacteriol. 2014;196:189–201. doi: 10.1128/JB.00918-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Geiger T, Goerke C, Fritz M, Schafer T, Ohlsen K, Liebeke M, Lalk M, Wolz C. Role of the (p)ppGpp synthase RSH, a RelA/SpoT homolog, in stringent response and virulence of Staphylococcus aureus. Infect Immun. 2010;78:1873–1883. doi: 10.1128/IAI.01439-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Guedon E, Sperandio B, Pons N, Ehrlich SD, Renault P. Overall control of nitrogen metabolism in Lactococcus lactis by CodY, and possible models for CodY regulation in Firmicutes. Microbiology. 2005;151:3895–3909. doi: 10.1099/mic.0.28186-0. [DOI] [PubMed] [Google Scholar]
- 146.Belitsky BR, Sonenshein AL. Genetic and biochemical analysis of CodY-binding sites in Bacillus subtilis. J Bacteriol. 2008;190:1224–1236. doi: 10.1128/JB.01780-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Belitsky BR, Sonenshein AL. Contributions of multiple binding sites and effector-independent binding to CodY-mediated regulation in Bacillus subtilis. J Bacteriol. 2011;193:473–484. doi: 10.1128/JB.01151-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wray LV, Jr, Fisher SH. Bacillus subtilis CodY operators contain overlapping CodY binding sites. J Bacteriol. 2011;193:4841–4848. doi: 10.1128/JB.05258-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Belitsky BR, Sonenshein AL. Genome-wide identification of Bacillus subtilis CodY-binding sites at single-nucleotide resolution. Proc Natl Acad Sci U S A. 2013;110:7026–7031. doi: 10.1073/pnas.1300428110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shivers RP, Dineen SS, Sonenshein AL. Positive regulation of Bacillus subtilis ackA by CodY and CcpA: establishing a potential hierarchy in carbon flow. Mol Microbiol. 2006;62:811–822. doi: 10.1111/j.1365-2958.2006.05410.x. [DOI] [PubMed] [Google Scholar]
- 151.Belitsky BR, Sonenshein AL. Roadblock repression of transcription by Bacillus subtilis CodY. J Mol Biol. 2011;411:729–743. doi: 10.1016/j.jmb.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Belitsky BR. Indirect repression by Bacillus subtilis CodY via displacement of the activator of the proline utilization operon. J Mol Biol. 2011;413:321–336. doi: 10.1016/j.jmb.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Karlsson S, Lindberg A, Norin E, Burman LG, Akerlund T. Toxins, butyric acid, and other short-chain fatty acids are coordinately expressed and down-regulated by cysteine in Clostridium difficile. Infect Immun. 2000;68:5881–5888. doi: 10.1128/iai.68.10.5881-5888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Dineen SS, Villapakkam AC, Nordman JT, Sonenshein AL. Repression of Clostridium difficile toxin gene expression by CodY. Mol Microbiol. 2007;66:206–219. doi: 10.1111/j.1365-2958.2007.05906.x. [DOI] [PubMed] [Google Scholar]
- 155.Li J, Ma M, Sarker MR, McClane BA. CodY is a global regulator of virulence-associated properties for Clostridium perfringens type D strain CN3718. mBio. 2013;4:e00770–00713. doi: 10.1128/mBio.00770-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Montgomery CP, Boyle-Vavra S, Roux A, Ebine K, Sonenshein AL, Daum RS. CodY deletion enhances in vivo virulence of community-associated methicillin-resistant Staphylococcus aureus clone USA300. Infect Immun. 2012;80:2382–2389. doi: 10.1128/IAI.06172-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rivera FE, Miller HK, Kolar SL, Stevens SM, Jr, Shaw LN. The impact of CodY on virulence determinant production in community-associated methicillin-resistant Staphylococcus aureus. Proteomics. 2012;12:263–268. doi: 10.1002/pmic.201100298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Majerczyk CD, Sadykov MR, Luong TT, Lee C, Somerville GA, Sonenshein AL. Staphylococcus aureus CodY negatively regulates virulence gene expression. J Bacteriol. 2008;190:2257–2265. doi: 10.1128/JB.01545-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Novick RP. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol Microbiol. 2003;48:1429–1449. doi: 10.1046/j.1365-2958.2003.03526.x. [DOI] [PubMed] [Google Scholar]
- 160.Batzilla CF, Rachid S, Engelmann S, Hecker M, Hacker J, Ziebuhr W. Impact of the accessory gene regulatory system (Agr) on extracellular proteins, codY expression and amino acid metabolism in Staphylococcus epidermidis. Proteomics. 2006;6:3602–3613. doi: 10.1002/pmic.200500732. [DOI] [PubMed] [Google Scholar]
- 161.Reiss S, Pane-Farre J, Fuchs S, Francois P, Liebeke M, Schrenzel J, Lindequist U, Lalk M, Wolz C, Hecker M, Engelmann S. Global analysis of the Staphylococcus aureus response to mupirocin. Antimicrob Agents Chemother. 2012;56:787–804. doi: 10.1128/AAC.05363-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tu Quoc PH, Genevaux P, Pajunen M, Savilahti H, Georgopoulos C, Schrenzel J, Kelley WL. Isolation and characterization of biofilm formation-defective mutants of Staphylococcus aureus. Infect Immun. 2007;75:1079–1088. doi: 10.1128/IAI.01143-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.van Schaik W, Chateau A, Dillies MA, Coppee JY, Sonenshein AL, Fouet A. The global regulator CodY regulates toxin gene expression in Bacillus anthracis and is required for full virulence. Infect Immun. 2009;77:4437–4445. doi: 10.1128/IAI.00716-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chateau A, van Schaik W, Six A, Aucher W, Fouet A. CodY regulation is required for full virulence and heme iron acquisition in Bacillus anthracis. FASEB J. 2011;25:4445–4456. doi: 10.1096/fj.11-188912. [DOI] [PubMed] [Google Scholar]
- 165.Frenzel E, Doll V, Pauthner M, Lucking G, Scherer S, Ehling-Schulz M. CodY orchestrates the expression of virulence determinants in emetic Bacillus cereus by impacting key regulatory circuits. Mol Microbiol. 2012;85:67–88. doi: 10.1111/j.1365-2958.2012.08090.x. [DOI] [PubMed] [Google Scholar]
- 166.Lindback T, Mols M, Basset C, Granum PE, Kuipers OP, Kovacs AT. CodY, a pleiotropic regulator, influences multicellular behaviour and efficient production of virulence factors in Bacillus cereus. Environ Microbiol. 2012;14:2233–2246. doi: 10.1111/j.1462-2920.2012.02766.x. [DOI] [PubMed] [Google Scholar]
- 167.Hsueh YH, Somers EB, Wong AC. Characterization of the codY gene and its influence on biofilm formation in Bacillus cereus. Arch Microbiol. 2008;189:557–568. doi: 10.1007/s00203-008-0348-8. [DOI] [PubMed] [Google Scholar]
- 168.Lobel L, Sigal N, Borovok I, Ruppin E, Herskovits AA. Integrative genomic analysis identifies isoleucine and CodY as regulators of Listeria monocytogenes virulence. PLoS Genet. 2012;8:e1002887. doi: 10.1371/journal.pgen.1002887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Malke H, Steiner K, McShan WM, Ferretti JJ. Linking the nutritional status of Streptococcus pyogenes to alteration of transcriptional gene expression: the action of CodY and RelA. Int J Med Microbiol. 2006;296:259–275. doi: 10.1016/j.ijmm.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 170.Caymaris S, Bootsma HJ, Martin B, Hermans PW, Prudhomme M, Claverys JP. The global nutritional regulator CodY is an essential protein in the human pathogen Streptococcus pneumoniae. Mol Microbiol. 2010;78:344–360. doi: 10.1111/j.1365-2958.2010.07339.x. [DOI] [PubMed] [Google Scholar]
- 171.Bouillaut L, Self WT, Sonenshein AL. Proline-dependent regulation of Clostridium difficile Stickland metabolism. J Bacteriol. 2013;195:844–854. doi: 10.1128/JB.01492-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Stickland LH. Studies in the metabolism of the strict anaerobes (Genus Clostridium): The reduction of proline by Cl. sporogenes. Biochem J. 1935;29:288–290. doi: 10.1042/bj0290288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Jackson S, Calos M, Myers A, Self WT. Analysis of proline reduction in the nosocomial pathogen Clostridium difficile. J Bacteriol. 2006;188:8487–8495. doi: 10.1128/JB.01370-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Brekasis D, Paget MS. A novel sensor of NADH/NAD+ redox poise in Streptomyces coelicolor A3(2) EMBO J. 2003;22:4856–4865. doi: 10.1093/emboj/cdg453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Schau M, Chen Y, Hulett FM. Bacillus subtilis YdiH is a direct negative regulator of the cydABCD operon. J Bacteriol. 2004;186:4585–4595. doi: 10.1128/JB.186.14.4585-4595.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Larsson JT, Rogstam A, von Wachenfeldt C. Coordinated patterns of cytochrome bd and lactate dehydrogenase expression in Bacillus subtilis. Microbiology. 2005;151:3323–3335. doi: 10.1099/mic.0.28124-0. [DOI] [PubMed] [Google Scholar]
- 177.Gyan S, Shiohira Y, Sato I, Takeuchi M, Sato T. Regulatory loop between redox sensing of the NADH/NAD(+) ratio by Rex (YdiH) and oxidation of NADH by NADH dehydrogenase Ndh in Bacillus subtilis. J Bacteriol. 2006;188:7062–7071. doi: 10.1128/JB.00601-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Pagels M, Fuchs S, Pane-Farre J, Kohler C, Menschner L, Hecker M, McNamarra PJ, Bauer MC, von Wachenfeldt C, Liebeke M, Lalk M, Sander G, von Eiff C, Proctor RA, Engelmann S. Redox sensing by a Rex-family repressor is involved in the regulation of anaerobic gene expression in Staphylococcus aureus. Mol Microbiol. 2010;76:1142–1161. doi: 10.1111/j.1365-2958.2010.07105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Mehmeti I, Jonsson M, Fergestad EM, Mathiesen G, Nes IF, Holo H. Transcriptome, proteome, and metabolite analyses of a lactate dehydrogenase-negative mutant of Enterococcus faecalis V583. Appl Environ Microbiol. 2011;77:2406–2413. doi: 10.1128/AEM.02485-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wietzke M, Bahl H. The redox-sensing protein Rex, a transcriptional regulator of solventogenesis in Clostridium acetobutylicum. Appl Microbiol Biotechnol. 2012;96:749–761. doi: 10.1007/s00253-012-4112-2. [DOI] [PubMed] [Google Scholar]
- 181.Sickmier EA, Brekasis D, Paranawithana S, Bonanno JB, Paget MS, Burley SK, Kielkopf CL. X-ray structure of a Rex-family repressor/ NADH complex insights into the mechanism of redox sensing. Structure. 2005;13:43–54. doi: 10.1016/j.str.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 182.Richardson AR, Libby SJ, Fang FC. A nitric oxide-inducible lactate dehydrogenase enables Staphylococcus aureus to resist innate immunity. Science. 2008;319:1672–1676. doi: 10.1126/science.1155207. [DOI] [PubMed] [Google Scholar]
- 183.Aboulnaga el H, Pinkenburg O, Schiffels J, El-Refai A, Buckel W, Selmer T. Effect of an oxygen-tolerant bifurcating butyryl coenzyme A dehydrogenase/electron-transferring flavoprotein complex from Clostridium difficile on butyrate production in Escherichia coli. J Bacteriol. 2013;195:3704–3713. doi: 10.1128/JB.00321-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sohling B, Gottschalk G. Molecular analysis of the anaerobic succinate degradation pathway in Clostridium kluyveri. J Bacteriol. 1996;178:871–880. doi: 10.1128/jb.178.3.871-880.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sorensen KI, Hove-Jensen B. Ribose catabolism of Escherichia coli: characterization of the rpiB gene encoding ribose phosphate isomerase B and of the rpiR gene, which is involved in regulation of rpiB expression. J Bacteriol. 1996;178:1003–1011. doi: 10.1128/jb.178.4.1003-1011.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Daddaoua A, Krell T, Ramos JL. Regulation of glucose metabolism in Pseudomonas: the phosphorylative branch and Entner-Doudoroff enzymes are regulated by a repressor containing a sugar isomerase domain. J Biol Chem. 2009;284:21360–21368. doi: 10.1074/jbc.M109.014555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kohler PR, Choong EL, Rossbach S. The RpiR-like repressor IolR regulates inositol catabolism in Sinorhizobium meliloti. J Bacteriol. 2011;193:5155–5163. doi: 10.1128/JB.05371-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yamamoto H, Serizawa M, Thompson J, Sekiguchi J. Regulation of the glv operon in Bacillus subtilis: YfiA (GlvR) is a positive regulator of the operon that is repressed through CcpA and cre. J Bacteriol. 2001;183:5110–5121. doi: 10.1128/JB.183.17.5110-5121.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhu Y, Nandakumar R, Sadykov MR, Madayiputhiya N, Luong TT, Gaupp R, Lee CY, Somerville GA. RpiR homologues may link Staphylococcus aureus RNAIII synthesis and pentose phosphate pathway regulation. J Bacteriol. 2011;193:6187–6196. doi: 10.1128/JB.05930-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Tojo S, Satomura T, Morisaki K, Yoshida K, Hirooka K, Fujita Y. Negative transcriptional regulation of the ilv-leu operon for biosynthesis of branched-chain amino acids through the Bacillus subtilis global regulator TnrA. J Bacteriol. 2004;186:7971–7979. doi: 10.1128/JB.186.23.7971-7979.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Shivers RP, Sonenshein AL. Bacillus subtilis ilvB operon: an intersection of global regulons. Mol Microbiol. 2005;56:1549–1559. doi: 10.1111/j.1365-2958.2005.04634.x. [DOI] [PubMed] [Google Scholar]
- 192.Tojo S, Satomura T, Morisaki K, Deutscher J, Hirooka K, Fujita Y. Elaborate transcription regulation of the Bacillus subtilis ilv-leu operon involved in the biosynthesis of branched-chain amino acids through global regulators of CcpA, CodY and TnrA. Mol Microbiol. 2005;56:1560–1573. doi: 10.1111/j.1365-2958.2005.04635.x. [DOI] [PubMed] [Google Scholar]
- 193.Grandoni JA, Fulmer SB, Brizzio V, Zahler SA, Calvo JM. Regions of the Bacillus subtilis ilv-leu operon involved in regulation by leucine. J Bacteriol. 1993;175:7581–7593. doi: 10.1128/jb.175.23.7581-7593.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Tojo S, Kumamoto K, Hirooka K, Fujita Y. Heavy involvement of stringent transcription control depending on the adenine or guanine species of the transcription initiation site in glucose and pyruvate metabolism in Bacillus subtilis. J Bacteriol. 2010;192:1573–1585. doi: 10.1128/JB.01394-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Jourlin-Castelli C, Mani N, Nakano MM, Sonenshein AL. CcpC, a novel regulator of the LysR family required for glucose repression of the citB gene in Bacillus subtilis. J Mol Biol. 2000;295:865–878. doi: 10.1006/jmbi.1999.3420. [DOI] [PubMed] [Google Scholar]
- 196.Mittal M, Pechter KB, Picossi S, Kim HJ, Kerstein KO, Sonenshein AL. Dual role of CcpC protein in regulation of aconitase gene expression in Listeria monocytogenes and Bacillus subtilis. Microbiology. 2013;159:68–76. doi: 10.1099/mic.0.063388-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hartmann T, Zhang B, Baronian G, Schulthess B, Homerova D, Grubmuller S, Kutzner E, Gaupp R, Bertram R, Powers R, Eisenreich W, Kormanec J, Herrmann M, Molle V, Somerville GA, Bischoff M. Catabolite control protein E (CcpE) is a LysR-type transcriptional regulator of TCA cycle activity in Staphylococcus aureus. J Biol Chem. 2013;288:36116–361128. doi: 10.1074/jbc.M113.516302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Crooke AK, Fuller JR, Obrist MW, Tomkovich SE, Vitko NP, Richardson AR. CcpA-independent glucose regulation of lactate dehydrogenase 1 in Staphylococcus aureus. PloS One. 2013;8:e54293. doi: 10.1371/journal.pone.0054293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ravcheev DA, Li X, Latif H, Zengler K, Leyn SA, Korostelev YD, Kazakov AE, Novichkov PS, Osterman AL, Rodionov DA. Transcriptional regulation of central carbon and energy metabolism in bacteria by redox-responsive repressor Rex. J Bacteriol. 2012;194:1145–1157. doi: 10.1128/JB.06412-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Larson TJ, Ehrmann M, Boos W. Periplasmic glycerophosphodiester phosphodiesterase of Escherichia coli, a new enzyme of the glp regulon. J Biol Chem. 1983;258:5428–5432. [PubMed] [Google Scholar]
- 201.Pitts AC, Tuck LR, Faulds-Pain A, Lewis RJ, Marles-Wright J. Structural insight into the Clostridium difficile ethanolamine utilisation microcompartment. PloS One. 2012;7:e48360. doi: 10.1371/journal.pone.0048360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Garsin DA. Ethanolamine utilization in bacterial pathogens: roles and regulation. Nat Rev Microbiol. 2010;8:290–295. doi: 10.1038/nrmicro2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Joseph B, Przybilla K, Stuhler C, Schauer K, Slaghuis J, Fuchs TM, Goebel W. Identification of Listeria monocytogenes genes contributing to intracellular replication by expression profiling and mutant screening. J Bacteriol. 2006;188:556–568. doi: 10.1128/JB.188.2.556-568.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Maadani A, Fox KA, Mylonakis E, Garsin DA. Enterococcus faecalis mutations affecting virulence in the Caenorhabditis elegans model host. Infect Immun. 2007;75:2634–2637. doi: 10.1128/IAI.01372-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Newsholme P, Curi R, Gordon S, Newsholme EA. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem J. 1986;239:121–125. doi: 10.1042/bj2390121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 207.Tugal D, Liao X, Jain MK. Transcriptional control of macrophage polarization. Arterioscler Thromb Vasc Biol. 2013;33:1135–1144. doi: 10.1161/ATVBAHA.113.301453. [DOI] [PubMed] [Google Scholar]
- 208.Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229:176–185. doi: 10.1002/path.4133. [DOI] [PubMed] [Google Scholar]
- 209.Mahdavian Delavary B, van der Veer WM, van Egmond M, Niessen FB, Beelen RH. Macrophages in skin injury and repair. Immunobiology. 2011;216:753–762. doi: 10.1016/j.imbio.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 210.Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. 2009;9:609–617. doi: 10.1038/nri2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Palsson-McDermott EM, O’Neill LA. The Warburg effect then and now: from cancer to inflammatory diseases. Bioessays. 2013;35:965–973. doi: 10.1002/bies.201300084. [DOI] [PubMed] [Google Scholar]
- 212.Newsholme P, Gordon S, Newsholme EA. Rates of utilization and fates of glucose, glutamine, pyruvate, fatty acids and ketone bodies by mouse macrophages. Biochem J. 1987;242:631–636. doi: 10.1042/bj2420631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hothersall JS, Gordge M, Noronha-Dutra AA. Inhibition of NADPH supply by 6-aminonicotinamide: effect on glutathione, nitric oxide and superoxide in J774 cells. FEBS Lett. 1998;434:97–100. doi: 10.1016/s0014-5793(98)00959-4. [DOI] [PubMed] [Google Scholar]
- 215.Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4:13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wu G, Bazer FW, Davis TA, Kim SW, Li P, Marc Rhoads J, Carey Satterfield M, Smith SB, Spencer TE, Yin Y. Arginine metabolism and nutrition in growth, health and disease. Amino Acids. 2009;37:153–168. doi: 10.1007/s00726-008-0210-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.De Groote MA, Fang FC. NO inhibitions: antimicrobial properties of nitric oxide. Clin Infect Dis. 1995;21(Suppl 2):S162–165. doi: 10.1093/clinids/21.supplement_2.s162. [DOI] [PubMed] [Google Scholar]
- 218.Shay JE, Celeste Simon M. Hypoxia-inducible factors: crosstalk between inflammation and metabolism. Semin Cell Dev Biol. 2012;23:389–394. doi: 10.1016/j.semcdb.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 219.Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, Chawla A. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Lehrke M, Lazar MA. The many faces of PPARgamma. Cell. 2005;123:993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 222.Kiss M, Czimmerer Z, Nagy L. The role of lipid-activated nuclear receptors in shaping macrophage and dendritic cell function: From physiology to pathology. J Allergy Clin Immunol. 2013;132:264–286. doi: 10.1016/j.jaci.2013.05.044. [DOI] [PubMed] [Google Scholar]
- 223.Mandard S, Patsouris D. Nuclear control of the inflammatory response in mammals by peroxisome proliferator-activated receptors. PPAR Res. 2013;2013:613864. doi: 10.1155/2013/613864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Bloch H, Segal W. Biochemical differentiation of Mycobacterium tuberculosis grown in vivo and in vitro. J Bacteriol. 1956;72:132–141. doi: 10.1128/jb.72.2.132-141.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, Schoolnik GK. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J Exp Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.de Carvalho LP, Fischer SM, Marrero J, Nathan C, Ehrt S, Rhee KY. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem Biol. 2010;17:1122–1131. doi: 10.1016/j.chembiol.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 227.Marrero J, Rhee KY, Schnappinger D, Pethe K, Ehrt S. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc Natl Acad Sci U S A. 2010;107:9819–9824. doi: 10.1073/pnas.1000715107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Munoz-Elias EJ, McKinney JD. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med. 2005;11:638–644. doi: 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Gould TA, van de Langemheen H, Munoz-Elias EJ, McKinney JD, Sacchettini JC. Dual role of isocitrate lyase 1 in the glyoxylate and methylcitrate cycles in Mycobacterium tuberculosis. Mol Microbiol. 2006;61:940–947. doi: 10.1111/j.1365-2958.2006.05297.x. [DOI] [PubMed] [Google Scholar]
- 230.Brock M, Buckel W. On the mechanism of action of the antifungal agent propionate. Eur J Biochem. 2004;271:3227–3241. doi: 10.1111/j.1432-1033.2004.04255.x. [DOI] [PubMed] [Google Scholar]
- 231.Rocco CJ, Escalante-Semerena JC. In Salmonella enterica, 2-methylcitrate blocks gluconeogenesis. J Bacteriol. 2010;192:771–778. doi: 10.1128/JB.01301-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Lee W, VanderVen BC, Fahey RJ, Russell DG. Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. J Biol Chem. 2013;288:6788–6800. doi: 10.1074/jbc.M112.445056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Savvi S, Warner DF, Kana BD, McKinney JD, Mizrahi V, Dawes SS. Functional characterization of a vitamin B12-dependent methyl-malonyl pathway in Mycobacterium tuberculosis: implications for propionate metabolism during growth on fatty acids. J Bacteriol. 2008;190:3886–3895. doi: 10.1128/JB.01767-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Rajaram MV, Brooks MN, Morris JD, Torrelles JB, Azad AK, Schlesinger LS. Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-activated receptor gamma linking mannose receptor recognition to regulation of immune responses. J Immunol. 2010;185:929–942. doi: 10.4049/jimmunol.1000866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Mahajan S, Dkhar HK, Chandra V, Dave S, Nanduri R, Janmeja AK, Agrewala JN, Gupta P. Mycobacterium tuberculosis modulates macrophage lipid-sensing nuclear receptors PPARgamma and TR4 for survival. J Immunol. 2012;188:5593–5603. doi: 10.4049/jimmunol.1103038. [DOI] [PubMed] [Google Scholar]
- 236.Almeida PE, Carneiro AB, Silva AR, Bozza PT. PPARgamma expression and function in mycobacterial infection: roles in lipid metabolism, immunity, and bacterial killing. PPAR Res. 2012;2012:383829. doi: 10.1155/2012/383829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Chatterjee SS, Hossain H, Otten S, Kuenne C, Kuchmina K, Machata S, Domann E, Chakraborty T, Hain T. Intracellular gene expression profile of Listeria monocytogenes. Infect Immun. 2006;74:1323– 1338. doi: 10.1128/IAI.74.2.1323-1338.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Joseph B, Przybilla K, Stuhler C, Schauer K, Slaghuis J, Fuchs TM, Goebel W. Identification of Listeria monocytogenes genes contributing to intracellular replication by expression profiling and mutant screening. J Bacteriol. 2006;188:556–568. doi: 10.1128/JB.188.2.556-568.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Chico-Calero I, Suarez M, Gonzalez-Zorn B, Scortti M, Slaghuis J, Goebel W, Vazquez-Boland JA. Hpt, a bacterial homolog of the microsomal glucose- 6-phosphate translocase, mediates rapid intracellular proliferation in Listeria. Proc Natl Acad Sci U S A. 2002;99:431–436. doi: 10.1073/pnas.012363899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Stoll R, Goebel W. The major PEP-phosphotransferase systems (PTSs) for glucose, mannose and cellobiose of Listeria monocytogenes, and their significance for extra- and intracellular growth. Microbiology. 2010;156:1069–1083. doi: 10.1099/mic.0.034934-0. [DOI] [PubMed] [Google Scholar]
- 241.Gillmaier N, Gotz A, Schulz A, Eisenreich W, Goebel W. Metabolic responses of primary and transformed cells to intracellular Listeria monocytogenes. PloS One. 2012;7:e52378. doi: 10.1371/journal.pone.0052378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Purves J, Cockayne A, Moody PC, Morrissey JA. Comparison of the regulation, metabolic functions, and roles in virulence of the glyceraldehyde-3-phosphate dehydrogenase homologues gapA and gapB in Staphylococcus aureus. Infect Immun. 2010;78:5223–5232. doi: 10.1128/IAI.00762-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Hochgrafe F, Wolf C, Fuchs S, Liebeke M, Lalk M, Engelmann S, Hecker M. Nitric oxide stress induces different responses but mediates comparable protein thiol protection in Bacillus subtilis and Staphylococcus aureus. J Bacteriol. 2008;190:4997–5008. doi: 10.1128/JB.01846-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Richardson AR, Payne EC, Younger N, Karlinsey JE, Thomas VC, Becker LA, Navarre WW, Castor ME, Libby SJ, Fang FC. Multiple targets of nitric oxide in the tricarboxylic acid cycle of Salmonella enterica serovar typhimurium. Cell Host Microbe. 2011;10:33–43. doi: 10.1016/j.chom.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Richardson AR, Dunman PM, Fang FC. The nitrosative stress response of Staphylococcus aureus is required for resistance to innate immunity. Mol Microbiol. 2006;61:927–939. doi: 10.1111/j.1365-2958.2006.05290.x. [DOI] [PubMed] [Google Scholar]
- 246.Fuller JR, Vitko NP, Perkowski EF, Scott E, Khatri D, Spontak JS, Thurlow LR, Richardson AR. Identification of a lactate-quinone oxidoreductase in Staphylococcus aureus that is essential for virulence. Front Cell Infect Microbiol. 2011;1:19. doi: 10.3389/fcimb.2011.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Weiner MA, Read TD, Hanna PC. Identification and characterization of the gerH operon of Bacillus anthracis endospores: a differential role for purine nucleosides in germination. J Bacteriol. 2003;185:1462–1464. doi: 10.1128/JB.185.4.1462-1464.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Sorg JA, Sonenshein AL. Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol. 2008;190:2505–2512. doi: 10.1128/JB.01765-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Janoir C, Deneve C, Bouttier S, Barbut F, Hoys S, Caleechum L, Chapeton-Montes D, Pereira FC, Henriques AO, Collignon A, Monot M, Dupuy B. Adaptive strategies and pathogenesis of Clostridium difficile from in vivo transcriptomics. Infect Immun. 2013;81:3757–3769. doi: 10.1128/IAI.00515-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Antunes LC, Han J, Ferreira RB, Lolic P, Borchers CH, Finlay BB. Effect of antibiotic treatment on the intestinal metabolome. Antimicrob Agents Chemother. 2011;55:1494–1503. doi: 10.1128/AAC.01664-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, Sonnenburg JL. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature. 2013;502:96–99. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Roux A, Todd DA, Velázquez JV, Cech NB, Sonenshein AL. CodY-mediated regulation of the Staphylococcus aureus Agr system integrates nutritional and population density signals. J Bacteriol. 2014;196:1184–1196. doi: 10.1128/JB.00128-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Vitko NP, Spahich NA, Richardson AR. Glycolytic dependency of high-level nitric oxide resistance and virulence in Staphylococcus aureus. MBio. 2015;6(2):0045–15. doi: 10.1128/mBio.00045-15. [DOI] [PMC free article] [PubMed] [Google Scholar]