

Abstract
Antimicrobial peptides (AMPs) are at the front-line of host defense during infection and play critical roles both in reducing the microbial load early during infection and in linking innate to adaptive immunity. However, successful pathogens have developed mechanisms to resist AMPs. Although considerable progress has been made in elucidating AMP-resistance mechanisms of pathogenic bacteria in vitro, less is known regarding the in vivo significance of such resistance. Nevertheless, progress has been made in this area, largely by using murine models and, in two instances, human models of infection. Herein, we review progress on the use of in vivo infection models in AMP research and discuss the AMP resistance mechanisms that have been established by in vivo studies to contribute to microbial infection. We posit that in vivo infection models are essential tools for investigators to understand the significance to pathogenesis of genetic changes that impact levels of bacterial susceptibility to AMPs.
Keywords: Antimicrobial peptides, Bacteria, Cell envelope modifications, Innate host defense, In vivo models, Pathogenesis, Resistance mechanisms, Transporters
1. Introduction
Antimicrobial peptides (AMPs) are small, amphipathic peptides that attack microbial invaders of eukaryotic hosts (reviewed in [1, 2]). AMPs are considered “host antibiotics” as they are critical components of the innate host response to infection. Moreover, because they can exert direct and/or indirect antimicrobial action, they have also been termed host-defense peptides [3] due to their capacity to link innate immunity to adaptive immune systems. The efficacy of AMP action in vivo coupled with the ability of microbes to resist their action can influence the microbial population level before adaptive immune responses become available. The over-arching goal of this review is to highlight seminal reports that provide important insights into the significance of bacterial AMP-resistance mechanisms during infection, as assessed by vertebrate and other model systems.
Most AMPs have a net positive charge; by this cationic nature, AMPs are preferentially attracted to microbial cell surfaces, which are more negatively charged than eukaryotic cell surfaces. Once associated with the microbial surface, their amphipathic nature allows AMPs to insert into a microbe’s cell membrane, disrupting the integrity of the membrane and leading to osmotic lysis of the microbial cell. This generalized pathway offers microbes numerous opportunities to develop decreased susceptibility to AMPs, and bacteria frequently accomplish this by altering—sometimes subtly—cell envelope structures that impede AMP binding events.
AMPs are ubiquitous in eukaryotic organisms, from plants to animals and invertebrates to mammals. Mammalian AMPs include defensins, which form β-sheets, and the cathelicidins, which exhibit greater structural heterogeneity. The defensins contain six Cys residues and form three intramolecular disulfide bonds; defensins are subdivided into α-and β-defensins by the positions and bonding patterns of their cysteines. Humans produce six α-defensins, over 30 β-defensins, and one α-helical cathelicidin, termed LL-37. These cationic AMPs are expressed primarily in neutrophils, macrophages, epithelial cells, and specialized secretory cells, such as the Paneth cells of the gut. In phagocytes, AMPs reside in granules and contribute to phagolysosomal killing of engulfed microbes; AMPs are also released by phagocyte degranulation to kill extracellular pathogens. AMPs secreted by epithelial cells and Paneth cells contribute to the innate barrier defenses against infection.
In order to survive in eukaryotic hosts, bacterial pathogens have evolved mechanisms to overcome the antimicrobial activity of AMPs. They often use systems similar to mechanisms identified for resistance to classical antibiotics. In vitro studies utilizing a variety of AMP susceptibility assays and bacterial genetic tools have elucidated a number of mechanisms bacteria use to thwart AMP activity [4]; examples of bacterial AMP-resistance systems include degradation, target modification, decreased import, and energy-dependent transport.
A full understanding of how bacteria overcome AMP-mediated attack during infection requires a combination of in vitro and in vivo studies. In vitro work is critical to understanding the genes, proteins, and mechanisms involved in AMP resistance; however, the contribution of these in vitro-established mechanisms to bacterial disease can only be elucidated in vivo. Here, we review the literature on in vivo studies that examine the role of AMP resistance mechanisms in bacterial pathogenesis. We first describe the major in vivo models used in these studies; we then discuss the collective findings of in vitro and in vivo research that established AMP resistance mechanisms which contribute significantly to bacterial disease.
For the purposes of this review, we largely confined our focus to cationic AMPs and to in vivo studies that directly compared parent strains in vivo with isogenic derivatives in which genes involved in AMP resistance were inactivated. A number of in vivo studies have demonstrated the importance to pathogenesis of two-component signal transduction systems that modulate AMP resistance mechanisms, such as the extensively-studied PhoPQ system of Salmonella enterica serovar Typhimurium and other Gram-negative pathogens (recently reviewed in [5]). However, the regulatory networks governed by these two-component signal transduction systems include genes not involved in AMP resistance; thus, the specific contribution of AMP resistance in vivo is difficult to discern from mutants affecting the entire regulon. We have therefore focused this review primarily on mutants carrying loss-of-function mutations in structural genes involved in AMP resistance mechanisms.
It is first, however, necessary to define what we mean by AMP-resistance in the context of this review. Accordingly, since minimal inhibitory concentration (MIC) “breakpoints” typically used for classical antibiotics to separate sensitive from resistant strains are not easily determined for AMPs, we will refer to resistance mechanisms as those that endow bacteria with increased fitness or survivability during infection. In this regard, it is important to emphasize that some mutations or gene acquisitions may only have slight influences (2–4 fold changes) in bacterial susceptibility to AMPs under laboratory conditions; yet their influence in vivo is substantially greater when assessed in an infection model.
2. In vivo Models of AMP Resistance Mechanisms in Pathogenesis
2.1 Human models of infection
To study human pathogens, an ideal in vivo model would be a human experimental infection model. Of course, concerns such as safety to the human subjects and transmissibility to the public preclude the ability to perform human infection experiments with most bacterial pathogens. However, within limitations imposed for medical and ethical reasons, a few human experimental infection models are currently in use. These models provide the ability to accurately recapitulate the kinetics of natural, human disease. Importantly, human models of infection allow the study of a human pathogen in the context of the specific, host-derived pressures with which the pathogen evolved. One such host-derived pressure is attack by AMPs; as AMP resistance mechanisms are often specific for certain AMPs, it is beneficial to examine AMP resistance against the AMPs that pathogen encounters during infection. Another advantage of human models is that they can be adapted for testing new therapeutics for treatment of infections or vaccine candidates for disease prevention.
As with any model, human infection models have limitations. For subject safety reasons, these models are generally limited to local infections and to the early stages of disease, with treatment at the onset of symptoms or discomfort; long-term infections, systemic infections, and sequelae cannot be safely examined in human volunteers. To ensure control of the infection, human models are typically restricted to one or two well-characterized wild-type bacterial strains, and isogenic derivatives thereof, that are readily treatable and do not harbor plasmids or phages that could transmit genetic material to the host’s microbial flora. Many other aspects of human infection models, such as route of inoculation and dosage, are far less flexible than in animal models. Working within these limitations, however, human experimental infection models accurately reproduce naturally acquired disease and provide important information about the host-pathogen relationship that is directly relevant to humans. Two such models have been used to examine the role of AMP resistance mechanisms in human infectious disease [6, 7].
Haemophilus ducreyi is a human-specific pathogen that causes the sexually transmitted, genital ulcer disease chancroid as well as a non-sexually transmitted chronic limb ulceration syndrome [8–10]. Lacking nonhuman animal models that accurately mimicked human disease, Spinola et al. developed a human model of H. ducreyi infection in which healthy, adult volunteers are inoculated in the upper arm and followed through the papular and pustular stages of disease; for subject safety reasons, infections are terminated before lesions ulcerate [6, 11, 12]. Importantly, this model likely reflects early host defense events during development of both chancroid and chronic limb ulceration syndrome and allows for testing the importance of presumed virulence factors. For parent-mutant strain comparisons, each subject is inoculated with the parent strain on one arm and with the mutant strain on the other arm; thus, each subject serves as his or her own control. This human infection model was used to define the contribution to virulence of the Sap transporter, which protects H. ducreyi from AMPs in vitro (discussed in section 3.3.2; see also Fig. 1) [13]. This study was the first to establish a role for AMP resistance mechanisms in human disease.
Fig. 1. The Sap transporter contributes to virulence of H. ducreyi in human volunteers.
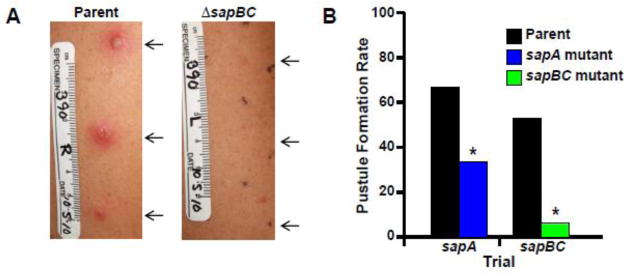
(A) Arms of volunteer # 390 at Day 6 post-inoculation with parent strain 35000HP (left panel) or mutant strain 35000HPsapBC (right panel) [79]. Arrows indicate sites of inoculation (outlined by black dots in right panel). Pustules formed at all parent-inoculated sites (left) but no mutant-inoculated sites (right) in this volunteer. Stickers on volunteer arms in photos indicate scale in cm, volunteer # (390), arm depicted (R, right arm; L, left arm), and date of photograph. (B) Pustule formation rates in human volunteers inoculated with parent strain 35000HP and either 35000HPsapA [13] or 35000HPsapBC [79]. For the mutants, the degree of attenuation in vivo correlated with the level of LL-37 resistance in vitro [79]. Asterisks indicate significant differences from parent strain in each trial (P < 0.05). Photographs courtesy of Stanley M. Spinola.
Neisseria gonorrhoeae, which causes the sexually transmitted infection gonorrhea, is another human-specific pathogen for which reliable nonhuman animal models were historically difficult to establish. Gonorrhea causes urethritis in males and cervicitis in females, with ascending female infections leading to salpingitis, pelvic inflammatory disease, and sterility. Cohen et al. developed a human infection model in which healthy male volunteers are inoculated intraurethrally with N. gonorrhoeae and followed until symptoms begin or urinalysis indicates colonization [7]. It is important to stress that complications associated with ascending infection preclude the use of women in experimental gonococcal infection. Thus, within the limitations of differences in the structure, physiology and host defenses in the male vs. female reproductive tract and how gonococcal virulence factors might function in these two environments, this human infection model has over the past twenty years provided important insights regarding gonococcal pathogenesis during early stages of infection (reviewed in [14]). In the infection model, parent-mutant comparisons are performed by co-infection with the two strains at a 1:1 ratio and determining the ratio (reported as a competitive index) of parent to mutant colonies recovered from the host; such co-infections demonstrate the relative fitness of isogenic mutants compared with wild-type strains in vivo [15]. With respect to AMP-resistance systems, Hobbs et al. recently used the human gonococcal infection model to determine the contribution of the lipid A phosphoethanolamine (PEA) transferase, LptA (discussed in section 3.1.1), to virulence during human infection; this was the first study to establish electrostatic repulsion of AMPs as a significant contributor to human disease [15].
2.2 Nonhuman in vivo models of infection
For most pathogens, no option exists for human experimentation; researchers rely instead on various nonhuman vertebrate and invertebrate models to study pathogenic mechanisms, including the importance of AMP-resistance systems, in vivo. These models, which we will refer to collectively as animal models, offer several advantages, including the generally lower cost of non-primate animal studies compared with human studies, the ability to study later stages of infection, and the ability to choose and even manipulate the genetic background of the host. Disadvantages of animal infection models include the inability to study host-restricted aspects of the disease or host response, and the difficulty in establishing the relevance to humans of results obtained in animals. Within these limitations, however, most bacterial pathogenesis studies have been performed in animals and have provided a wealth of information on host-pathogen interactions.
2.2.1 Mouse models of infection
The most widespread animal used to study AMP resistance mechanisms in vivo has been the mouse (Table 1). One advantage of using mice is the ability to use different routes of inoculation to examine different stages of disease. The most extensively used in vivo system for studying bacterial AMP-resistance mechanisms has been murine infections with S. Typhimurium, which causes gastroenteritis in humans and a typhoid fever-like disease in mice. This pathogen may be used in mice to model typhoid fever, in which S. Typhimurium is ingested and colonizes the small intestine, including Peyer’s Patches, from which the organism crosses the gut epithelial barrier, is taken up by macrophages, and disseminates to the reticuloendothelial system (RES); in the RES, the bacteria reside and multiply in mesenteric lymph nodes and the liver and spleen. By comparing the outcome of orally administered infections with those administered intraperitoneally, in which bacteria are directly taken up by macrophages, investigators can determine the contribution of specific virulence factors to early or late stages of disease. This approach showed that Mig-14, a regulator of AMP resistance, is dispensable for initial gut colonization but is required for survival in the RES [16]; in contrast, electrostatic repulsion by 4-aminoarabinose (Ara4N) (discussed in section 3.1.1) was found to be important for initial infection of the gut but not for later stages of disease [17, 18].
Table 1.
AMP resistance mechanisms examined in vivo
| Mechanism of AMP Resistance | Organism | Virulence Factor(s) | In vivo Model(s), route of inoculationa | Reference(s) |
|---|---|---|---|---|
| Electrostatic Repulsion | ||||
| PEA modification of lipid A | N. gonorrhoeae | LptA | Human, male, intraurethral | [15] |
| Mouse, female, intravaginal | [15, 30] | |||
| S. Typhimurium | PmrC | Mouse, oral and i.p. | [35] | |
| PEA modification of LOS core | S. Typhimurium | CptA | Mouse, oral and i.p. | [35] |
| PEA modification of LPS, flagella, glycans | C. jejuni | EptC | Mouse, oral Chick, oral |
[34] [34] |
| Aminoarabinose modification of LPS | S. Typhimurium | PmrHFIJKLM | Mouse, oral and i.p. | [17, 18] |
| Mouse, CRAMP−/−, oral | [18] | |||
| Mouse, MMP7−/−, oral | [18] | |||
| D-Ala modification of lipoteichoic acid | Group B Streptococcus | DltA | Rat, i.p. Mouse, i.n. and i.v. |
[42] [42] |
| L. monocytogenes | DltA | Mouse, i.v. | [45] | |
| S. aureus | DltA | Mouse, i.v. | [41] | |
| Rabbit, i.v. | [44] | |||
| L-Lys modification of cell membrane phosphatidylglycerol | S. aureus | MprF | Rabbit, i.v. | [44] |
| Mouse, i.v. | [43] | |||
| L. monocytogenes | MprF | Mouse, i.v. | [46] | |
| Galactosamine modification of lipid A | F. novicida | AlmEFG | Mouse, subcutaneous | [37] |
| Membrane fluidity/permeability | ||||
| Lipid A modified by acylation | K. pneumoniae | LpxM | Mouse, i.n. and i.v. | [48] |
| L. pneumophila | Rcp | Mouse, i.t. | [47] | |
| LPS truncation | ||||
| LPS core synthesis | P. multocida | HptA | Chicken, i.m. | [50] |
| H. influenzae, nontypeable | LpsA, OpsX, LgtF | Mouse, i.n. | [51] | |
| B. cenocepacia | HldA, HldD | Rat, i.t. | [49] | |
| B. bronchiseptica | Pgm | Mouse, i.n. | [52] | |
| Surface modification | ||||
| LPS biosynthesis | S. Typhimurium | Pgm | Mouse, i.v. | [53] |
| modification of unidentified surface structures | Y. pestis | PgmA | Mouse, i.n. and i.v. | [25] |
| Phosphate modification of LPS core | S. Typhimurium | WaaP | Mouse, oral, i.p., and i.v. | [54] |
| Phosphocholine modification of LPS core | P. multocida | PcgC | Chicken, i.m. and i.t. | [55] |
| Transport | ||||
| Efflux Pump | N. gonorrhoeae | MTR | Mouse, female, intravaginal | [62, 64] |
| V. cholerae | VexAB | Mouse, infant, oral | [65] | |
| Uptake transport | E. chrysanthemi | SapABCDF | Potato tuber, injection | [77] |
| Chicory leaves, injection | [82] | |||
| H. ducreyi | SapA | Human, cutaneous | [13] | |
| SapBC | Human, cutaneous | [79] | ||
| H. influenzae, nontypeable | SapA | Chinchilla otitis | [72, 75] | |
| SapD | Chinchilla otitis | [78] | ||
| S. Typhimurium | SapCDF, SapDF | Mouse, oral and i.p. | [73] | |
| YejABDF | Mouse, oral and i.p. | [90] | ||
| AMP binding proteins | ||||
| Bind AMP, prevent AMP from reaching cell membrane | Group B Streptococcus | PilB | Mouse, i.p. and i.v. | [89] |
| Group A Streptococcus | SIC | Mouse, i.p. | [91] | |
| Unclear | ||||
| Ferrous iron-binding protein | S. Typhimurium | Omb | Mouse, oral and i.p. | [92] |
Routes of inoculation: i.m., intramuscular; i.n., intranasal; i.p., intraperitoneal; i.t., intratracheal; i.v., intravenous
Another advantage of mouse models is the ability to use a genetically defined host. As with routes of inoculation, the host genetics can be varied to examine different stages of disease. For example, many mouse strains used in models of S. Typhimurium infection carry a mutation in Nramp1 that renders the mice highly susceptible to infection; thus, these mice typically succumb to infection before adaptive immune responses can develop. In contrast, mice with a wild-type Nramp1 gene product can become persistently infected with S. Typhimurium, but with little to no disease, for up to 1 year. Thus, Nramp1+/+ mice can be used to study long-term persistence of S. Typhimurium infection [19]; this model was used to demonstrate the importance of Mig-14-mediated AMP resistance in establishing the persistent carrier state of S. Typhimurium infection [20].
The availability of knockout mice lacking specific genes is an invaluable tool for understanding the host side of the host-pathogen relationship. Two knockout mouse strains are available that affect the repertoire of murine AMPs. Mice express the cathelicidin-related AMP (CRAMP), a homolog of LL-37; CRAMP−/− mice are hyper-susceptible to several bacterial infections, including a subcutaneous anthrax infection model [21] and Group A streptococcal skin and soft tissue infections [22]. Mouse intestinal crypts contain α-defensins called cryptdins, which are activated upon cleavage by matrix metalloprotease 7 (MMP7, Matrilysin). MMP7−/− mice cannot activate intracellular cryptdins; however, cryptdins secreted into the intestinal lumen are activated by other lumenal proteases. MMP7−/− mice are colonized to a greater extent by E. coli and are more susceptible to the lethal effects of S. Typhimurium infection [23]. Interestingly, when CRAMP−/− mice were challenged with Ara4N-deficient S. Typhimurium, no greater attenuation was observed than in wild-type mice [18]; these studies showed that CRAMP alone is not a major mediator of clearance of Ara4N-deficient S. Typhimurium.
Another tool available for in vivo studies in mice is transgenic animals expressing human genes. For example, transgenic mice expressing the human α-defensin HD-5 have helped to understand the importance of this AMP in combating S. Typhimurium infection and the contributions of resistance mechanisms to disease progression [24]. To be sure, additional mouse strains will be developed in the future for testing host and bacterial factors that modulate the efficacy of the innate immunity arm of host defense, which includes AMPs.
2.2.2 Other in vivo models
In addition to humans and mice, a variety of less common in vivo models have been used to examine the role of AMP resistance mechanisms in bacterial disease (Table 1). Several of these involve natural hosts or long-established models of specific diseases. Among the natural host models used in understanding AMP resistance mechanisms is the flea model of Yersinia pestis infection. Y. pestis, the causative agent of plague, is a vector-borne disease that primarily infects rodents and is transmitted by fleas. As insects produce AMPs, Felek et al. examined the effects of the AMP resistance gene pgmA (discussed in section 3.2) on the survival of Y. pestis in fleas and the bacterium’s transmission from fleas to mice [25]. Another natural host model is the chicken, which has been used to examine AMP resistance mechanisms of zoonotic pathogens, such as Campylobacter jejuni, a common colonizer of poultry and cause of food-borne gastroenteritis in people, and Pasteurella multocida, which causes fowl cholera in many avian species and can cause severe animal bite-related wound infections and opportunistic infections in humans. Other in vivo models that have been used to study AMP resistance mechanisms in a pathogen’s natural host include oysters, as a model for the marine pathogen Vibrio splendidus, and potatoes and witloof chicory leaves, which have been used to study the plant pathogen Erwinia chrysanthemi (recently renamed Dickeya dadantii). Although the extensive array of tools used with mice is not available in these models, they do have the advantage of being the natural setting for the pathogen of interest.
Although not a natural host, the chinchilla has long been used as a model for studying otitis media. The chinchilla model is well established and allows infection with several of the most common causes of inner ear infections, including Haemophilus influenzae. As will be discussed in section 3, all of these models have been used to examine the effects of AMP resistance mechanisms on virulence.
3. Antimicrobial Peptide Resistance Mechanisms that Contribute to Bacterial Disease
What have we learned from these in vivo infection models about the importance of AMP resistance mechanisms with respect to bacterial pathogenesis? A combination of in vitro and in vivo studies have defined an array of AMP resistance mechanisms; most of these fall mechanistically into a few categories. The best defined AMP resistance mechanisms directly shown to contribute to virulence in vivo are modification of cell surface structures with positively charged moieties for electrostatic repulsion of cationic AMPs and the use of molecular transporters to pump AMPs away from the cell membrane. In addition to these mechanisms, several virulence factors have been defined that affect AMP resistance and virulence by less well-understood mechanisms that primarily involve modifications to surface structures.
3.1 Electrostatic Repulsion
The bacterial cell surface is typically more negatively charged than eukaryotic cell surfaces; cationic AMPs are thus more highly attracted to bacterial cells than to the cells of the host. By adding positively charged molecules to the bacterial surface, a wide variety of bacterial pathogens render their cell surfaces less negatively charged and thus less attractive to cationic AMPs. In Gram-negative bacteria, electrostatic repulsion can be elicited by modifying surface structures with PEA or Ara4N. In Gram-positive bacteria, a similar result is achieved by adding D-Ala residues to cell wall polymers or lysine residues to membrane phospholipids. The contribution to virulence has been demonstrated in vivo for each of these mechanisms of electrostatic repulsion.
3.1.1 Gram-negative electrostatic repulsion mechanisms
One of the most thorough studies of the contribution of electrostatic repulsion to virulence has been performed with the human-specific pathogen N. gonorrhoeae. The lipooligosaccharide (LOS) of N. gonorrhoeae is decorated with PEA on its lipid A and core sugars [26]. While the core sugar PEA modifications have only minor effects on AMP resistance, loss of the lipid A PEA transferase, LptA, significantly impairs the organism’s ability to survive attack by AMPs [26]. The contribution of LptA to pathogenesis was recently demonstrated using two in vivo models of disease [15], namely the human male and female mouse experimental systems. In the human challenge model of N. gonorrhoeae infection (described in section 2.1), the lptA mutant was outcompeted ~ 100-fold by its isogenic wild-type parent strain, demonstrating the importance of lipid A-PEA modification to N. gonorrhoeae infection in humans [15].
The human model of N. gonorrhoeae infection is limited to male volunteers, because of the risks associated with ascending gonorrhea infections in women. However, the organism’s pathogenesis differs between the male urethra and the female reproductive tract [27]. To study female gonococcal disease, a mouse model was developed by A. Jerse and colleagues in which 17β-estradiol treatment, combined with streptomycin to reduce the level of competing commensal bacteria in the reproductive tract, renders female mice susceptible to vaginal infection [28, 29]. This model is a highly useful tool that allows investigators to test the significance of presumed gonococcal virulence factors during infection and draw inferences about pathogenesis, despite differences between mice and humans, which may not be fully appreciated by the sole use of common laboratory procedures involving test tubes or tissue culture systems. In brief, the model allows colonization of the female mouse genital tract, with recovery of bacteria from the vagina and cervix; ascending infection is seen in 17–20% of infected mice, mimicking the ascending reproductive tract infections in women. A purulent neutrophil response develops, similar to that in symptomatic women.
With this estradiol-treated mouse model, Hobbs et al. and Packiam et al. examined the role of LptA in female reproductive tract disease [15, 30]. Although single strain infection experiments showed little difference between the parent and isogenic lptA mutant strains for colonization, co-infection experiments demonstrated that the lptA mutant was significantly less fit than the parent strain in the female mouse reproductive tract [15, 30]. Interestingly, however, relative to the parent strain, the lptA mutant also induced a much weaker host inflammatory response, suggesting an immunostimulatory role for LptA in addition to its protective function [30]. A similar study is now underway (M. Hobbs, personal communication) in the human male infection model system. Nevertheless, based on the similar results obtained in the co-infection models, we posit that (at the very least for gonococci), the mouse model developed by A. Jerse has been validated for use in studies dealing with the in vivo significance of bacterial AMP-resistance mechanisms.
Unlike LptA in N. gonorrhoeae, whose only known substrate is lipid A, the LptA homolog in C. jejuni, EptC, modifies several diverse substrates, including lipid A, LOS core heptose I, the flagellar subunit protein FlgG, and N-linked glycans that decorate periplasmic proteins [31–34]. Loss of eptC increases bacterial sensitivity to polymyxin B and several human and avian AMPs, decreases motility, and lessens TLR4-MD2 complex-mediated responses [34]. In vivo, EptC is needed to colonize the gastrointestinal tracts of chickens and mice, suggesting an important role for PEA modification in both avian commensalism and mammalian disease [34].
In addition to PEA, a number of Gram-negative pathogens modify their lipopolysaccharide (LPS) with Ara4N. In vitro studies have established that Ara4N modification contributes significantly to AMP resistance. The best studied example of the in vivo contribution of Ara4N to virulence is in S. Typhimurium, in which Ara4N is generated and added to LPS by the products of pmrE and the pmrHFIJKLM operon. Using a pmrF mutant that was polar on the rest of the operon, Gunn et al. showed that the pmrF-M genes are not required for S. Typhimurium survival in mice when administered intraperitoneally but are required when the organism is administered orally [17, 18]; thus, the Ara4N modification likely protects S. Typhimurium from host defenses encountered in the intestine but not in macrophages that engulf the bacteria in the peritoneum. Interestingly, despite the in vitro connection between Ara4N modification and resistance to polymyxin B, virulence studies in knockout mice showed that the attenuation of Ara4N-deficient bacteria was not due to CRAMP [18]. However, the Ara4N-deficient bacteria induced an altered innate immune response compared with wild-type S. Typhimurium [18].
S. Typhimurium LPS is also modified with PEA moieties on the lipid A and core. These modifications are mediated by PEA transferases PmrC, which modifies lipid A, and CptA, which modifies the core Heptose I sugar. In mice inoculated either orally or intraperitoneally, neither the pmrC nor cptA mutant was attenuated for infection in single strain inoculation studies, nor was either mutant outcompeted by the parent strain in vivo [35]. However, a pmrC cptA double mutant used in co-infection with the isogenic parent strain showed a modest but statistically significant impairment [35]. Thus, in S. Typhimurium, PmrC and CptA provide only minor contributions to virulence; rather, Ara4N modification of LPS appears to be the dominant mechanism of electrostatic repulsion in this pathogen. Side-by-side comparisons of polymyxin B sensitivities confirmed that Ara4N modification contributes more than PEA modification to AMP resistance in S. Typhimurium [36]. In contrast, PEA modifications contribute more to virulence of N. gonorrhoeae and C. jejuni, which do not modify their LOS with Ara4N.
In addition to PEA and Ara4N, Gram-negative pathogens can use other amine-containing surface modifications to induce electrostatic repulsion. Francisella noivicida has an unusual outer membrane, in which 70% of the lipid A lacks KDO, core sugars, and O antigen; these “free” lipid A molecules are modified with galactosamine at the 1-phosphate position [37]. The deacetylase NaxD is required for this galactosamine modification; a naxD mutant is more negatively charged than its isogenic parent, more sensitive to polymyxin B, and less able to replicate in macrophages [37]. In vivo, the naxD mutant was less fit than the parent strain and significantly attenuated for lethality in mice [37]. Similarly, Vibrio cholerae, which causes the diarrheagenic disease cholera, adds Gly and diglycine moieties to its lipid A; although not examined in vivo, this modification significantly enhanced the organism’s resistance to polymyxin B in vitro [38]. Interestingly, the V. cholerae genes involved in Gly addition to lipid A, almEFG, bear structural and functional homology to those involved in addition of D-Ala to Gram-positive cell wall components (discussed in section 3.1.2), thus mechanistically bridging the divide between Gram-negative and Gram-positive cell wall modifications that exert electrostatic repulsion [38, 39].
3.1.2. Gram-positive electrostatic repulsion mechanisms
Addition of amino acids to the Gram-positive cell wall increases the surface charge and confers protection from AMPs in several important pathogens. In Staphylococcus aureus (and other Gram-positive pathogens including Clostridium difficile and Listeria monocytogenes), products of the dltABCD operon mediate addition of D-Ala esters to cell wall lipoteichoic acid and confer protection from neutrophil α-defensins HNP1-3 and from nonoxidative killing by human neutrophils [40, 41]. In a mouse model of S. aureus sepsis, a dltABCD mutant was impaired, relative to its isogenic parent, in disease progression, with significantly reduced rates of septic arthritis and mortality and reduced bacterial load in the kidneys [41]. Similarly, a dltA mutant in Streptococcus agalactiae, or Group B Streptococcus (GBS), which causes invasive infections, shows increased sensitivity to human α-defensins and to killing by human neutrophils and macrophages [42]. In vivo, the GBS dltA mutant exhibited a significantly higher LD50 in neonatal rats [42]. In mouse models of GBS disease, intranasal inoculation with wild-type bacteria led to pneumonia, and intravenous inoculation induced bacteremia, colonized the brain, and caused meningitis; the GBS dltA mutant was cleared from lungs too quickly to cause pneumonia and was unable to survive in the bloodstream or colonize the brain [42]. Together, these studies demonstrate the importance of D-Ala esterification of lipoteichoic acid to invasive disease by Gram-positive cocci.
Another mechanism of electrostatic repulsion in Gram-positive bacteria is addition of positively charged L-Lys to cell membrane phosphatidylglycerol, which is mediated by the multiple peptide resistance factor MprF. Deletion of mprF renders S. aureus more sensitive to cationic AMPs and to nonoxidative killing by human neutrophils [43]; in a mouse model of sepsis, the mprF mutant was similar to the dltABCD mutant in being attenuated for lethality, induction of septic arthritis, and colonization of kidneys, thus confirming a role for electrostatic repulsion in invasive S. aureus disease [43]. The in vivo contributions of these mechanisms to S. aureus virulence were directly compared in a rabbit model of infective endocarditis. In this model, sterile vegetations are artificially induced on the aortic valve, followed by intravenous inoculation with S. aureus strains [44]. Relative to their isogenic parent strain, both dltA and mprF mutants were more rapidly cleared from the bloodstream and impaired in their level of colonization of cardiac vegetations; however, only the dltA mutant was impaired for subsequent spread to the kidneys and spleen; the mprF mutant and parent strain colonized these organs to similar extents [44]. Thus, while both mechanisms contribute to invasive disease, D-Ala esterification of lipoteichoic acid appears to play a greater role in systemic disease caused by S. aureus.
Electrostatic repulsion of cationic AMPs also contributes to virulence of the intracellular pathogen L. monocytogenes. Both dltABCD-mediated D-Ala modification of lipoteichoic acid and mprF-mediated L-Lys addition to phosphatidylglycerol contribute to cationic AMP resistance in vitro [45, 46]. In separate studies utilizing a mouse model of invasive listeriosis, a dltA mutant was severely attenuated for survival in the bloodstream, liver and spleen and had a 10,000x increase in LD50 [45], and an mprF mutant was similarly attenuated for survival in the liver and spleen [46]. Thus, both intracellular and extracellular pathogens enhance their virulence by electrostatic repulsion.
3.2 Other Surface Modifications
In addition to the mechanisms of electrostatic repulsion described in section 3.1, numerous surface modifications, primarily of the Gram-negative cell wall, have been found to affect AMP resistance, though the exact mechanisms of action are not always clear. Resistance to cationic AMPs in several pathogens is associated with specific acylation events on lipid A. The lipid A palmitoyltransferase PagP adds palmitate to lipid A and enhances cationic AMP resistance, presumably by increasing the hydrophobicity of the outer membrane to prevent AMPs from translocating across the membrane into the periplasm. The PagP homolog Rcp is associated with virulence in Legionella pneumophila, in which co-infection of wild-type and isogenic rcp mutant revealed a fitness defect in the rcp mutant for colonizing the lungs [47]. Similarly, an lpxM mutant of Klebsiella pneumoniae, which expresses a pentaacylated lipid A, had increased sensitivity to α-helical cationic AMPs and was attenuated in mice for colonization of the lungs and spleen [48]. Although no in vitro growth defect was identified in these acylation mutants, the increased permeability of the outer membrane could make the mutants more vulnerable to complement or osmotic pressures in the host environment independent of AMP activity. Similarly, genes affecting synthesis of the core oligosaccharide of LPS or LOS are correlated with AMP resistance and virulence in several pathogens (Table 1) [49–51]. However, truncation of the core and loss of O-Ag likely impair bacterial defenses beyond those required for AMP resistance. Thus, the specific contribution of AMPs to the attenuation of core-truncated mutants can be difficult to ascertain.
Phosphoglucomutase (Pgm) is correlated with AMP resistance and virulence in several pathogens. PGM interconverts glucose-1-phosphate and glucose-6-phosphate and plays a role in production of nucleotide sugars used in biosynthesis of the LPS core. A pgm mutant in Bordetella bronchiseptica lacks O-Ag and expresses a truncated core oligosaccharide lacking hexoses [52]. This mutant showed increased sensitivity to oxidative stress and an insect AMP; in a mouse model of respiratory tract infection, the pgm mutant was cleared from lungs in 4 days while the isogenic parent caused disease that persisted for 7 weeks [52]. An S. Typhimurium pgm mutant expressed full LPS core and a truncated O-Ag; in vivo, the pgm mutant was attenuated for colonization of liver and spleen [53]. Interestingly, a Y. pestis mutant lacking Pgm expressed wild-type LPS yet lost AMP resistance, indicating that Y. pestis Pgm may affect a surface structure other than LPS; unlike pgm mutants in other bacteria, the Pgm-deficient Y. pestis strain was fully virulent in mouse and flea models of infection [25]. Thus, the contribution of Pgm to surface structures and to virulence varies substantially among pathogens.
Additional LPS modifications that enhance AMP resistance and virulence include modifying core sugars with phosphate or phosphocholine. Even though phosphorylation enhances the negativity of the cell surface, phosphorylation of the LPS core is associated with polymyxin B resistance in S. Typhimurium. A S. Typhimurium mutant lacking the sugar kinase WaaP generates full length LPS but lacks one heptose, phosphates on the remaining heptoses, and PEA on heptose I [54]. This waaP mutant had increased sensitivity to polymyxin B in vitro; in mouse models of Salmonella infection, the waaP mutant was attenuated for lethality and rapidly cleared from the liver and spleen, regardless of the route of inoculation [54]. P. multocida causes veterinary diseases, including fowl cholera in poultry. P. multocida modifies its LPS with phosphocholine, which increases its resistance to the chicken AMP fowlicidin-1 [55]. A phosphocholine-deficient mutant was unable to compete with the isogenic parent strain in a chicken model of fowl cholera; inoculation with the mutant alone caused disease but required a much longer time frame before symptoms appeared, indicating that phosphocholine enhances but is not required for virulence in chickens [55].
3.3 Transport-mediated AMP Resistance
3.3.1 Efflux Pumps
Efflux pumps protect bacteria by transporting harmful host-derived or other exogenous substances out of the cell (reviewed in [56]). In medically relevant pathogens, efflux pumps represent a major mechanism of antibiotic resistance. Additionally, efflux pumps of several pathogens are able to remove host-derived cationic AMPs. The best-studied example of an efflux pump mediating AMP resistance is the multiple transferable resistance (MTR) transporter of N. gonorrhoeae that confers protection against structurally hydrophobic antimicrobial agents [57]. The MTR transporter is a member of the resistance-nodulation-division (RND) family of Gram-negative efflux pumps and comprises three proteins: MtrD is an inner membrane transporter energized by the proton motive force across the cell membrane; MtrE forms an outer membrane channel; and MtrC is a periplasmic membrane fusion protein that stabilizes the MtrD-MtrE complex to form a transporter that crosses the entire cell wall [58–60]. Studies with the E. coli RND transporter AcrAB-TolC indicated that these transporters can remove substrates from the cytoplasm, periplasm, and inner membrane [61]. In N. gonorrhoeae, the MTR transporter is required for LL-37 resistance in vitro [57]; the pump also helps gonococci resist the antimicrobial action of progesterone and bile salts.
The role of MTR in pathogenesis was examined using the estradiol-treated mouse model of N. gonorrhoeae infection (discussed in section 3.1.1) [62]. In this model, a mutant lacking MtrD and MtrE was able to colonize mice, but the amount of recoverable bacteria was much lower than the parent strain [62]. These data were the first to demonstrate a role for an RND efflux pump in a genital tract infection. Subsequent in vivo studies with mutants lacking transcriptionally regulatory proteins or harboring cis-acting mutations that control expression of the mtrCDE operon revealed that mutations impacting promoter use or loss of the repressor MtrR could increase gonococcal fitness during infection [63]. In contrast, loss of a transcriptional activator (MtrA) decreased in vivo fitness of gonococci [64]. Given the similar results of lptA-positive and –negative gonococci in the human male and female mouse models of infection [15], it is likely that the presence and levels of the MTR efflux pump are important during human infection, as the pump would promote bacterial survival by exporting LL-37 (and potentially other host antimicrobials).
V. cholerae encodes six RND efflux transporters; Bina et al. sought to identify the role of these transporters in V. cholerae AMP resistance and virulence. They found that only one RND transporter, VexAB-TolC, conferred resistance to the cationic AMP polymyxin B. Like the N. gonorrhoeae MTR system, the VexAB system also conferred protection against detergents and other hydrophobic agents in V. cholerae [65]. In an infant mouse model of cholera, a peroral coinfection of wild-type and vexB mutant V. cholerae strains showed that the vexB mutant was outcompeted by its isogenic parent strain for survival in the small intestine [65]. Together, the N. gonorrhoeae and V. cholerae studies demonstrate that RND transporters enhance the in vivo fitness of pathogenic bacteria in multiple organ systems and correlate that fitness advantage with resistance to AMPs.
In vitro studies showed that an MTR-like RND efflux transporter in Neisseria meningitidis conferred resistance to polymyxin B, and an RND efflux transporter in H. ducreyi conferred resistance to LL-37, HBD-2, and HBD-3 [66, 67]. Although no in vivo studies were performed with isogenic mutants in these transporters, the data suggest that RND pumps may represent a widespread Gram-negative mechanism of AMP resistance.
Gram-positive pathogens also produce efflux pumps, although the correlation between active efflux and cationic AMP resistance is less well established. One study examining S. aureus isolates correlated the presence of the qacA gene, which encodes an efflux pump of the major facilitator superfamily, with resistance to a cationic, platelet-derived AMP; however, subsequent studies by the same group demonstrated that QacA-mediated AMP resistance was independent of QacA’s efflux activity [68, 69]. Thus, a definitive role in pathogenesis for Gram-positive bacteria by efflux of AMPs during infection has yet to be established.
3.3.2 Uptake Transporters
In addition to efflux pumps, uptake transporters can confer resistance to cationic AMPs. The sensitive to antimicrobial peptides (Sap) transporter is a peptide uptake transporter closely related to dipeptide permease (Dpp) and oligopeptide permease (Opp) uptake transporters that bring small peptides into the cytoplasm for degradation or recycling [70, 71]. The Sap transporter is expressed in many pathogens of the Gammaproteobacteria class, including the Enterobacteriaceae and Pasteurellaceae. The pentameric transporter consists of a periplasmic solute binding protein, SapA, which binds certain cationic AMPs, two inner membrane permease proteins, SapB and SapC, which form a channel for transport, and two cytoplasmic ATP-binding cassette proteins, SapD and SapF, that energize the transporter [71, 72].
The Sap transporter was first identified in S. Typhimurium and shown to confer protection against the model AMP protamine and crude extracts from human neutrophils, which contain LL-37 and the α-defensins human neutrophil peptide (HNP)-1, HNP-2, and HNP-3 [71, 73]. Transposon mutants affecting the sapCDF genes had a significantly higher LD50 than the parent strain, whether delivered orally or by the intraperitoneal route, in a mouse model of typhoid fever [73]. Subsequently, Sap transporters have been characterized in several human pathogens, including E. coli, H. ducreyi, H. influenzae, and Proteus mirabilis, and the plant pathogen E. chrysanthemi [13, 74–77]. In vitro studies in all these pathogens demonstrate that the Sap transporter contributes to AMP resistance; however, the specificity of AMPs transported by Sap varies among pathogens. For example, the Sap transporter of nontypeable H. influenzae (NTHI) is highly effective against the human cathelicidin LL-37 as well as human and chinchilla β-defensins, while the Sap transporter of H. ducreyi protects against LL-37 but has no effect on human α- or β-defensins, and the S. Typhimurium Sap transporter is ineffective against a rabbit defensin [13, 73, 75, 78, 79]. Similarly, the S. Typhimurium Sap transporter conferred protection against protamine, but the V. fischeri and E. chrysanthemi Sap transporters had no effect on protamine [73, 77, 80].
The importance of the Sap transporter in vivo has been established in several pathogens. In H. ducreyi, two studies examined the contributions to AMP resistance and virulence of the periplasmic solute binding protein SapA and the inner membrane SapBC channel. A nonpolar sapA mutant showed increased sensitivity to LL-37 compared with the isogenic parent strain [13], and a nonpolar sapBC mutant was significantly more sensitive to LL-37 than either the isogenic parent strain or the sapA mutant [79]. These data suggest that the periplasmic component, SapA, is not absolutely required for transport activity across the SapBC channel. In vivo studies with the human model of H. ducreyi infection similarly showed that the sapA mutant was partially attenuated for virulence, as measured by the rate of pustule formation at inoculated sites; by the same criteria, the sapBC mutant was fully attenuated for virulence (Fig. 1). Thus, the levels of attenuation in vivo directly correlated with the level of LL-37 sensitivity in vitro [79]. These were the first studies to establish that a bacterial AMP resistance mechanism contributes to human infectious disease.
Mason and colleagues examined the contribution to virulence of Sap transporter components in NTHI during middle ear infections in the chinchilla model of otitis media. Mutations in sapA or sapD rendered NTHI significantly impaired for colonization of the nasopharynx and middle ear [75, 78]. In co-infection studies between either the sapA or sapD mutant and the isogenic parent strain, neither mutant was able to compete with the wild-type strain, although the sapD mutant was cleared more rapidly than the sapA mutant in competition with the parent strain [75, 78]. In vitro studies show that, in addition to transporting AMPs, the NTHI Sap transporter imports heme into the cell [81]. The in vivo importance of the AMP transport activity of NTHI Sap was investigated by pretreating chinchillas with neutralizing antibody against chinchilla β-defensin-1 (cBD-1), followed by co-infection with wild-type and sapA mutant NTHI strains. With cBD-1 neutralized, the sapA mutant no longer showed a fitness defect relative to the wild-type strain; these data confirmed that the Sap transporter protects against AMPs in vivo [72].
The contribution of the Sap system to bacterial virulence extends beyond mammalian pathogens, as evidenced by studies in E. chrysanthemi, which causes soft rot diseases in many agricultural crop plants. In vivo studies with potato tubers and witloof chicory leaves demonstrated that a sapABCDF mutant caused significantly less rot than its isogenic parent; in competitive infections, the sapABCDF mutant was less fit than the parent strain [77, 82]. In vitro, the sapABCDF mutant was hypersensitive to the plant-derived AMPs snakin-1 and α-thionin but were unaffected by a potato-derived defensin or protamine [77].
Interestingly, in vivo studies of a Sap transporter in the symbiotic, bioluminescent bacterium Vibrio fischeri demonstrated that the transporter plays a significant role in colonizing the light organ of the bacterium’s host, the Hawaiian bobtail squid (Euprymna scolopes); however, the transporter has no apparent role in protecting V. fischeri from cationic AMPs [80]. These results suggest that the Sap transporter may have evolved a different function for this host-symbiont relationship than for the host-pathogen relationship. However, of the AMPs tested in this study, none was from the host squid, and only salmon-derived protamine was of marine origin [80]; the possibility remains that the V. fischeri Sap transporter is specific for AMPs derived from Euprymna or other marine animals.
3.4 Other Mechanisms of AMP Resistance
Proteolysis of AMPs is a fairly common mechanism of AMP resistance [83–86]; yet, few studies have shown a direct correlation between AMP resistance and the in vivo contribution of a protease to virulence. The outer membrane protein OmpT is the prototypical member of the so-called omptin family of cell surface proteases, which are expressed by many members of the Enterobacteriaceae. OmpT in E. coli and several other pathogens has been shown to cleave LL-37 and other AMPs; however, most omptin proteases have multiple host substrates. For example, the omptin Pla of Y. pestis cleaves plasminogen, and other factors involved in coagulation and fibrinolysis, as well as Fas ligand and LL-37 [87, 88]. Pla has long been recognized as an important virulence determinant in the spread of Y. pestis during infection; however, its multiple substrates makes determining the specific role of LL-37 degradation in virulence difficult.
Streptococcus pyogenes, or Group A Streptococcus (GAS), and GBS express surface or secreted proteins that bind cationic AMPs, preventing their interaction with the cell membrane. GBS expresses a pilus whose major subunit, PilB, protects the organism from cationic AMPs including LL-37, murine CRAMP, and polymyxin B; ectopic expression of pilB confers a similar AMP resistance profile to the nonpathogenic, AMP-sensitive Lactococcus lactis [89]. The pilB-expressing L. lactis strain demonstrated significantly increased binding to LL-37, suggesting that PilB mediates AMP resistance by binding AMPs and preventing their activity against the bacterial cell [89]. In a mouse model of bloodstream infection, the pilB mutant was attenuated for survival in the bloodstream and was significantly outcompeted by its isogenic parent strain in co-infection experiments; the pilB mutant was also significantly less lethal to mice inoculated by the intraperitoneal route [89]. pilB expression in L. lactis caused this nonpathogen to induce mortality in mice with an LD50 similar to the parental GBS strain [89].
4. Conclusions
In vivo model systems are powerful tools for establishing the importance of AMP resistance mechanisms to bacterial disease processes. As exemplified by the murine model of S. Typhimurium, in vivo infection models can also elucidate specific organs or stages of disease in which an AMP resistance mechanism is critical. One limitation of in vivo studies, however, is that the exact cause of decreased fitness of a loss-of-function mutant can be difficult to establish in vivo. Transporters and proteases often have additional substrates, and surface modifications may affect interactions between the bacterial surface and host components other than AMPs. Studies directly confirming that loss-of-fitness is due to the activity of an AMP, such as by use of a neutralizing anti-AMP antibody, are very few. Nevertheless, as discussed in this review, strong correlations have been established in many model systems between AMP sensitivity of a loss-of-function mutant in vitro and reduced fitness or virulence in vivo. Moreover, such in vivo studies unequivocally establish the important role of AMPs in the host innate immune barrier to bacterial infection.
As AMPs represent an ancient component of host resistance to infection, and microbes have likely co-evolved with human AMP producers, it is not surprising that successful pathogens have developed mechanisms to resist their action during infection. While numerous mechanisms of AMP-resistance have been identified and can be studied with great rigor under laboratory conditions, formal testing of the significance of resistance during human infections can only be done using model systems of infection. While organ and cell culture systems can provide valuable insights, they cannot fully replicate the environment in which microbe-AMP interactions occur; as such, whole models are essential to draw meaningful conclusions. Recognizing that non-human models of infection have limitations, we nevertheless conclude, as supported by recent results from human infection models, that AMP-resistance systems can profoundly impact the fate of bacteria during infection. As the overall AMP research field moves forward it is, however, fair to ask: why are whole animal systems, including humans, needed for AMP-resistance studies? Apart from providing important information regarding bacterial pathogenesis and host defense, the models also allow for testing new therapeutics that might be influenced by AMP-resistance systems. Moreover, attempts to cripple AMP-resistance systems might be a new strategy to augment host defense. The efficacy of new therapeutic approaches that must by-pass or target AMP-resistance will require an intact living system such as those described herein.
Research Highlights.
Antimicrobial peptides (AMP) protect the host from infection by pathogenic bacteria.
Bacterial pathogens have evolved mechanisms to repel, degrade, or expel AMPs.
In vitro studies have elucidated many bacterial mechanisms of AMP resistance.
Human and other in vivo models prove the importance of AMP resistance to disease.
Acknowledgments
We thank our numerous colleagues for their many contributions to AMP research and apologize to those whose work was not cited in this review. We are also grateful to our many collaborators and members of our research groups who have contributed so significantly to our work and have been instrumental in advancing the field of AMP research. W.M.S. is especially grateful to Drs. Marcia Hobbs and Ann Jerse for their important studies on N. gonorrhoeae that have been instrumental in advancing AMP-resistance research. M.E.B would particularly like to thank Dr. Stanley M. Spinola and his research team for their ongoing collaborative studies on the effects of AMP resistance mechanisms in human H. ducreyi infection.
Work performed in M.E.B’s laboratory has been supported by NIH grants R21 AI075008 and R21 AI096056, while work in W.M.S.’s laboratory has been supported by NIH grants R37 AI21150-29 and U19 AI031496-21 and a VA Merit Award from the Medical Research Service of the Department of Veterans Affairs. W.M.S. is the recipient of a Senior Research Career Scientist Award from the Medical Research Service of the Department of Veterans Affairs.
Abbreviations
- AMP(s)
antimicrobial peptide(s)
- Ara4N
4-aminoarabinose
- cBD
chinchilla β-defensin
- CRAMP
cathelicidin-related antimicrobial peptide
- Dpp
dipeptide permease
- GAS
Gropu A Streptococcus
- GBS
Group B Streptococcus
- HBD
human β-defensin
- HNP
human neutrophil peptide
- LOS
lipooligosaccharide
- LPS
lipopolysaccharide
- MIC
minimal inhibitory concentration
- MMP7
matrix metalloprotease 7
- MTR
multiple transferrable resistance
- NTHI
nontypeable Haemophilus influenzae
- Opp
oligopeptide permease
- PEA
phosphoethanolamine
- Pgm
phosphoglucomutase
- RES
reticuloendothelial system
- RND
resistance-nodulation-cell division
- Sap
sensitive to antimicrobial peptides
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Margaret E. Bauer, Email: mebauer@iupui.edu.
William M. Shafer, Email: wshafer@emory.edu.
References
- 1.Jenssen H, Hamill P, Hancock RE. Peptide antimicrobial agents. Clin Microbiol Rev. 2006;19:491–511. doi: 10.1128/CMR.00056-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pinheiro da Silva F, Machado MC. Antimicrobial peptides: clinical relevance and therapeutic implications. Peptides. 2012;36:308–314. doi: 10.1016/j.peptides.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 3.Brown KL, Hancock RE. Cationic host defense (antimicrobial) peptides. Curr Opin Immunol. 2006;18:24–30. doi: 10.1016/j.coi.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 4.Goytia M, Kandler JL, Shafer WM. Mechanisms and signficance of bacterial resistance to human cationic antimicrobial peptides. In: Hiemstra P, Zaat S, editors. Antimicrobial peptides and human disease. Springer Press; 2013. pp. 219–254. [Google Scholar]
- 5.Dalebroux ZD, Miller SI. Salmonellae PhoPQ regulation of the outer membrane to resist innate immunity. Curr Opin Microbiol. 2014;17:106–113. doi: 10.1016/j.mib.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spinola SM, Wild LM, Apicella MA, Gaspari AA, Campagnari AA. Experimental human infection with Haemophilus ducreyi. J Infect Dis. 1994;169:1146–1150. doi: 10.1093/infdis/169.5.1146. [DOI] [PubMed] [Google Scholar]
- 7.Cohen MS, Cannon JG, Jerse A, Charniga L, Isbey S, Whicker L. Human experimentation with Neisseria gonorrhoeae: rationale, methods, and implications for the biology of infection and vaccine development. J Infect Dis. 1994;169:532–537. doi: 10.1093/infdis/169.3.532. [DOI] [PubMed] [Google Scholar]
- 8.Lewis DA, Ison CA. Chancroid. Sex Transm Infect. 2006;82(Suppl IV):iv19–iv20. doi: 10.1136/sti.2006.023127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marks M, Chi KH, Vahi V, Pillay A, Sokana O, Pavluck A, Mabey DC, Chen CY, Solomon AW. Haemophilus ducreyi associated with skin ulcers among children, Solomon Islands. Emerg Infect Dis. 2014;20:1705–1707. doi: 10.3201/eid2010.140573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mitja O, Lukehart SA, Pokowas G, Moses P, Kapa A, Godornes C, Robson J, Cherian S, Houinei W, Kazadi W, Siba P, de Lazzari E, Bassat Q. Haemophilus ducreyi as a cause of skin ulcers in children from a yaws-endemic area of Papua New Guinea: a prospective cohort study. Lancet Glob Health. 2014;2:E235–E241. doi: 10.1016/S2214-109X(14)70019-1. [DOI] [PubMed] [Google Scholar]
- 11.Al-Tawfiq JA, Thornton AC, Katz BP, Fortney KR, Todd KD, Hood AF, Spinola SM. Standardization of the experimental model of Haemophilus ducreyi infection in human subjects. J Infect Dis. 1998;178:1684–1687. doi: 10.1086/314483. [DOI] [PubMed] [Google Scholar]
- 12.Janowicz DM, Ofner S, Katz BP, Spinola SM. Experimental infection of human volunteers with Haemophilus ducreyi: fifteen years of clinical data and experience. J Infect Dis. 2009;199:1671–1679. doi: 10.1086/598966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mount KL, Townsend CA, Rinker SD, Gu X, Fortney KR, Zwickl BW, Janowicz DM, Spinola SM, Katz BP, Bauer ME. Haemophilus ducreyi SapA contributes to cathelicidin resistance and virulence in humans. Infect Immun. 2010;78:1176–1184. doi: 10.1128/IAI.01014-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hobbs MM, Sparling PF, Cohen MS, Shafer WM, Deal CD, Jerse AE. Experimental gonococcal infection in male volunteers: cumulative experience with Neisseria gonorrhoeae strains FA1090 and MS11mkC. Front Microbiol. 2011;2:123. doi: 10.3389/fmicb.2011.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hobbs MM, Anderson JE, Balthazar JT, Kandler JL, Carlson RW, Ganguly J, Begum AA, Duncan JA, Lin JT, Sparling PF, Jerse AE, Shafer WM. Lipid A’s structure mediates Neisseria gonorrhoeae fitness during experimental infection of mice and men. mBio. 2013;4:e00892–00813. doi: 10.1128/mBio.00892-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valdivia RH, Cirillo DM, Lee AK, Bouley DM, Falkow S. mig-14 Is a horizontally acquired, host-induced gene required for Salmonella enterica lethal infection in the murine model of typhoid fever. Infect Immun. 2000;68:7126–7131. doi: 10.1128/iai.68.12.7126-7131.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gunn JS, Ryan SS, van Velkinburgh JC, Ernst RK, Miller SI. Genetic and functional analysis of a PmrA-PmrB-regulated locus necessary for lipopolysaccharide modification, antimicrobial peptide resistance, and oral virulence of Salmonella enterica serovar Typhimurium. Infect Immun. 2000;68:6139–6146. doi: 10.1128/iai.68.11.6139-6146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strandberg KL, Richards SM, Tamayo R, Reeves LT, Gunn JS. An altered immune response, but not individual cationic antimicrobial peptides, is associated with the oral attenuation of Ara4N-deficient Salmonella enterica serovar Typhimurium in mice. PLoS ONE. 2012;7:e49588. doi: 10.1371/journal.pone.0049588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monack DM, Bouley DM, Falkow S. Salmonella typhimurium persists within macrophages in the mesenteric lymph nodes of chronically infected Nramp1+/+ mice and can be reactivated by IFNγ neutralization. The J Exper Med. 2004;199:231–241. doi: 10.1084/jem.20031319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brodsky IE, Ghori N, Falkow S, Monack D. Mig-14 is an inner membrane-associated protein that promotes Salmonella typhimurium resistance to CRAMP, survival within activated macrophages and persistent infection. Mol Microbiol. 2005;55:954–972. doi: 10.1111/j.1365-2958.2004.04444.x. [DOI] [PubMed] [Google Scholar]
- 21.McGillivray SM, Ebrahimi CM, Fisher N, Sabet M, Zhang DX, Chen Y, Haste NM, Aroian RV, Gallo RL, Guiney DG, Friedlander AM, Koehler TM, Nizet V. ClpX contributes to innate defense peptide resistance and virulence phenotypes of Bacillus anthracis. J Innate Immun. 2009;1:494–506. doi: 10.1159/000225955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, Pestonjamasp V, Piraino J, Huttner K, Gallo RL. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature. 2001;414:454–457. doi: 10.1038/35106587. [DOI] [PubMed] [Google Scholar]
- 23.Wilson CL, Ouellette AJ, Satchell DP, Ayabe T, Lopez-Boado YS, Stratman JL, Hultgren SJ, Matrisian LM, Parks WC. Regulation of intestinal a-defensin activation by the metalloproteinase matrilysin in innate host defense. Science. 1999;286:113–117. doi: 10.1126/science.286.5437.113. [DOI] [PubMed] [Google Scholar]
- 24.Salzman NH, Ghosh D, Huttner KM, Paterson Y, Bevins CL. Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature. 2003;422:522–526. doi: 10.1038/nature01520. [DOI] [PubMed] [Google Scholar]
- 25.Felek S, Muszynski A, Carlson RW, Tsang TM, Hinnebusch BJ, Krukonis ES. Phosphoglucomutase of Yersinia pestis is required for autoaggregation and polymyxin B resistance. Infect Immun. 2010;78:1163–1175. doi: 10.1128/IAI.00997-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis LA, Choudhury B, Balthazar JT, Martin LE, Ram S, Rice PA, Stephens DS, Carlson R, Shafer WM. Phosphoethanolamine substitution of lipid A and resistance of Neisseria gonorrhoeae to cationic antimicrobial peptides and complement-mediated killing by normal human serum. Infect Immun. 2009;77:1112–1120. doi: 10.1128/IAI.01280-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edwards JL, Apicella MA. The molecular mechanisms used by Neisseria gonorrhoeae to initiate infection differ between men and women. Clin Microbiol Rev. 2004;17:965–981. doi: 10.1128/CMR.17.4.965-981.2004. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jerse AE. Experimental gonococcal genital tract infection and opacity protein expression in estradiol-treated mice. Infect Immun. 1999;67:5699–5708. doi: 10.1128/iai.67.11.5699-5708.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jerse AE, Wu H, Packiam M, Vonck RA, Begum AA, Garvin LE. Estradiol-treated Female mice as surrogate hosts for Neisseria gonorrhoeae genital tract infections. Front Microbiol. 2011;2:107. doi: 10.3389/fmicb.2011.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Packiam M, Yedery RD, Begum AA, Carlson RW, Ganguly J, Sempowski GD, Ventevogel MS, Shafer WM, Jerse AE. Phosphoethanolamine decoration of Neisseria gonorrhoeae lipid A plays a dual immunostimulatory and protective role during experimental genital tract infection. Infect Immun. 2014;82:2170–2179. doi: 10.1128/IAI.01504-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cullen TW, Trent MS. A link between the assembly of flagella and lipooligosaccharide of the Gram-negative bacterium Campylobacter jejuni. Proc Natl Acad Sci U S A. 2010;107:5160–5165. doi: 10.1073/pnas.0913451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cullen TW, Madsen JA, Ivanov PL, Brodbelt JS, Trent MS. Characterization of unique modification of flagellar rod protein FlgG by Campylobacter jejuni lipid A phosphoethanolamine transferase, linking bacterial locomotion and antimicrobial peptide resistance. J Biol Chem. 2012;287:3326–3336. doi: 10.1074/jbc.M111.321737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott NE, Nothaft H, Edwards AV, Labbate M, Djordjevic SP, Larsen MR, Szymanski CM, Cordwell SJ. Modification of the Campylobacter jejuni N-linked glycan by EptC protein-mediated addition of phosphoethanolamine. J Biol Chem. 2012;287:29384–29396. doi: 10.1074/jbc.M112.380212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cullen TW, O’Brien JP, Hendriksen DR, Giles DK, Hobb RI, Thompson SA, Brodbelt JS, Trent MS. EptC of Campylobacter jejuni mediates phenotypes involved in host interactions and virulence. Infect Immun. 2013;81:430–440. doi: 10.1128/IAI.01046-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamayo R, Choudhury B, Septer A, Merighi M, Carlson R, Gunn JS. Identification of cptA, a PmrA-regulated locus required for phosphoethanolamine modification of the Salmonella enterica serovar Typhimurium lipopolysaccharide core. J Bacteriol. 2005;187:3391–3399. doi: 10.1128/JB.187.10.3391-3399.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee H, Hsu FF, Turk J, Groisman EA. The PmrA-regulated pmrC gene mediates phosphoethanolamine modification of lipid A and polymyxin resistance in Salmonella enterica. J Bacteriol. 2004;186:4124–4133. doi: 10.1128/JB.186.13.4124-4133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Llewellyn AC, Zhao J, Song F, Parvathareddy J, Xu Q, Napier BA, Laroui H, Merlin D, Bina JE, Cotter PA, Miller MA, Raetz CR, Weiss DS. NaxD is a deacetylase required for lipid A modification and Francisella pathogenesis. Mol Microbiol. 2012;86:611–627. doi: 10.1111/mmi.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hankins JV, Madsen JA, Giles DK, Brodbelt JS, Trent MS. Amino acid addition to Vibrio cholerae LPS establishes a link between surface remodeling in Gram-positive and Gram-negative bacteria. Proc Natl Acad Sci US A. 2012;109:8722–8727. doi: 10.1073/pnas.1201313109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henderson JC, Fage CD, Cannon JR, Brodbelt JS, Keatinge-Clay AT, Trent MS. Antimicrobial peptide resistance of Vibrio cholerae results from an LPS modification pathway related to nonribosomal peptide synthetases. ACS Chem Biol. 2014;9:2382–2392. doi: 10.1021/cb500438x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peschel A, Otto M, Jack RW, Halbacher H, Jung G, Götz F. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem. 1999;274:8405–8410. doi: 10.1074/jbc.274.13.8405. [DOI] [PubMed] [Google Scholar]
- 41.Collins LV, Kristian SA, Weidenmaier C, Faigle M, Van Kessel KP, Van Strijp JA, Gotz F, Neumeister B, Peschel A. Staphylococcus aureus strains lacking D-alanine modifications of teichoic acids are highly susceptible to human neutrophil killing and are virulence attenuated in mice. J Infect Dis. 2002;186:214–219. doi: 10.1086/341454. [DOI] [PubMed] [Google Scholar]
- 42.Poyart C, Pellegrini E, Marceau M, Baptista M, Jaubert F, Lamy MC, Trieu-Cuot P. Attenuated virulence of Streptococcus agalactiae deficient in D-alanyl-lipoteichoic acid is due to an increased susceptibility to defensins and phagocytic cells. Mol Microbiol. 2003;49:1615–1625. doi: 10.1046/j.1365-2958.2003.03655.x. [DOI] [PubMed] [Google Scholar]
- 43.Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, Kalbacher H, Nieuwenhuizen WF, Jung G, Tarkowski A, van Kessel KPM, van Strijp JAG. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with L-lysine. J Exp Med. 2001;193:1067–1076. doi: 10.1084/jem.193.9.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weidenmaier C, Peschel A, Kempf VA, Lucindo N, Yeaman MR, Bayer AS. DltABCD- and MprF-mediated cell envelope modifications of Staphylococcus aureus confer resistance to platelet microbicidal proteins and contribute to virulence in a rabbit endocarditis model. Infect Immun. 2005;73:8033–8038. doi: 10.1128/IAI.73.12.8033-8038.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abachin E, Poyart C, Pellegrini E, Milohanic E, Fiedler F, Berche P, Trieu-Cuot P. Formation of D-alanyl-lipoteichoic acid is required for adhesion and virulence of Listeria monocytogenes. Mol Microbiol. 2002;43:1–14. doi: 10.1046/j.1365-2958.2002.02723.x. [DOI] [PubMed] [Google Scholar]
- 46.Thedieck K, Hain T, Mohamed W, Tindall BJ, Nimtz M, Chakraborty T, Wehland J, Jansch L. The MprF protein is required for lysinylation of phospholipids in listerial membranes and confers resistance to cationic antimicrobial peptides (CAMPs) on Listeria monocytogenes. Mol Microbiol. 2006;62:1325–1339. doi: 10.1111/j.1365-2958.2006.05452.x. [DOI] [PubMed] [Google Scholar]
- 47.Robey M, O’Connell W, Cianciotto NP. Identification of Legionella pneumophila rcp, a pagP-like gene that confers resistance to cationic antimicrobial peptides and promotes intracellular infection. Infect Immun. 2001;69:4276–4286. doi: 10.1128/IAI.69.7.4276-4286.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clements A, Tull D, Jenney AW, Farn JL, Kim SH, Bishop RE, McPhee JB, Hancock RE, Hartland EL, Pearse MJ, Wijburg OL, Jackson DC, McConville MJ, Strugnell RA. Secondary acylation of Klebsiella pneumoniae lipopolysaccharide contributes to sensitivity to antibacterial peptides. J Biol Chem. 2007;282:15569–15577. doi: 10.1074/jbc.M701454200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loutet SA, Flannagan RS, Kooi C, Sokol PA, Valvano MA. A complete lipopolysaccharide inner core oligosaccharide is required for resistance of Burkholderia cenocepacia to antimicrobial peptides and bacterial survival in vivo. J Bacteriol. 2006;188:2073–2080. doi: 10.1128/JB.188.6.2073-2080.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harper M, Boyce JD, Cox AD, St Michael F, Wilkie IW, Blackall PJ, Adler B. Pasteurella multocida expresses two lipopolysaccharide glycoforms simultaneously, but only a single form is required for virulence: identification of two acceptor-specific heptosyl I transferases. Infect Immun. 2007;75:3885–3893. doi: 10.1128/IAI.00212-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morey P, Viadas C, Euba B, Hood DW, Barberan M, Gil C, Grillo MJ, Bengoechea JA, Garmendia J. Relative contributions of lipooligosaccharide inner and outer core modifications to nontypeable Haemophilus influenzae pathogenesis. Infect Immun. 2013;81:4100–4111. doi: 10.1128/IAI.00492-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.West NP, Jungnitz H, Fitter JT, McArthur JD, Guzman CA, Walker MJ. Role of phosphoglucomutase of Bordetella bronchiseptica in lipopolysaccharide biosynthesis and virulence. Infect Immun. 2000;68:4673–4680. doi: 10.1128/iai.68.8.4673-4680.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paterson GK, Cone DB, Peters SE, Maskell DJ. The enzyme phosphoglucomutase (Pgm) is required by Salmonella enterica serovar Typhimurium for O-antigen production, resistance to antimicrobial peptides and in vivo fitness. Microbiol. 2009;155:3403–3410. doi: 10.1099/mic.0.029553-0. [DOI] [PubMed] [Google Scholar]
- 54.Yethon JA, Gunn JS, Ernst RK, Miller SI, Laroche L, Malo D, Whitfield C. Salmonella enterica serovar Typhimurium waaP mutants show increased susceptibility to polymyxin and loss of virulence in vivo. Infect Immun. 2000;68:4485–4491. doi: 10.1128/iai.68.8.4485-4491.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harper M, Cox A, St Michael F, Parnas H, Wilkie I, Blackall PJ, Adler B, Boyce JD. Decoration of Pasteurella multocida lipopolysaccharide with phosphocholine is important for virulence. J Bacteriol. 2007;189:7384–7391. doi: 10.1128/JB.00948-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Piddock LJ. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin Microbiol Rev. 2006;19:382–402. doi: 10.1128/CMR.19.2.382-402.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shafer WM, Qu X-D, Waring AJ, Lehrer RI. Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc Natl Acad Sci USA. 1998;95:1829–1833. doi: 10.1073/pnas.95.4.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hagman KE, Pan W, Spratt BG, Balthazar JT, Judd RC, Shafer WM. Resistance of Neisseria gonorrhoeae to antimicrobial hydrophobic agents is modulated by the mtrRCDE efflux system. Microbiol. 1995;141:611–622. doi: 10.1099/13500872-141-3-611. [DOI] [PubMed] [Google Scholar]
- 59.Hagman KE, Lucas CE, Balthazar JT, Snyder L, Nilles M, Judd RC, Shafer WM. The MtrD protein of Neisseria gonorrhoeae is a member of the resistance/nodulation/division protein family constituting part of an efflux system. Microbiol. 1997;143:2117–2125. doi: 10.1099/00221287-143-7-2117. [DOI] [PubMed] [Google Scholar]
- 60.Delahay RM, Robertson BD, Balthazar JT, Shafer WM, Ison CA. Involvement of the gonococcal MtrE protein in the resistance of Neisseria gonorrhoeae to toxic hydrophobic agents. Microbiol. 1997;143:2127–2133. doi: 10.1099/00221287-143-7-2127. [DOI] [PubMed] [Google Scholar]
- 61.Murakami S, Nakashima R, Yamaguchi A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature. 2002;419:587–593. doi: 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- 62.Jerse AE, Sharma ND, Simms AN, Crow ET, Snyder LA, Shafer WM. A gonococcal efflux pump system enhances bacterial survival in a female mouse model of genital tract infection. Infect Immun. 2003;71:5576–5582. doi: 10.1128/IAI.71.10.5576-5582.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zalucki YM, Dhulipala V, Shafer WM. Dueling regulatory properties of a transcriptional activator (MtrA) and repressor (MtrR) that control efflux pump gene expression in Neisseria gonorrhoeae. mBio. 2012;3:e00446–00412. doi: 10.1128/mBio.00446-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Warner DM, Folster JP, Shafer WM, Jerse AE. Regulation of the MtrC-MtrD-MtrE efflux-pump system modulates the in vivo fitness of Neisseria gonorrhoeae. J Infect Dis. 2007;196:1804–1812. doi: 10.1086/522964. [DOI] [PubMed] [Google Scholar]
- 65.Bina XR, Provenzano D, Nguyen N, Bina JE. Vibrio cholerae RND family efflux systems are required for antimicrobial resistance, optimal virulence factor production, and colonization of the infant mouse small intestine. Infect Immun. 2008;76:3595–3605. doi: 10.1128/IAI.01620-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tzeng Y-L, Ambrose KD, Zughaier S, Zhou X, Miller YK, Shafer WM, Stephens DS. Cationic antimicrobial peptide resistance in Neisseria meningitidis. J Bacteriol. 2005;187:5387–5396. doi: 10.1128/JB.187.15.5387-5396.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rinker SD, Trombley MP, Gu X, Fortney KR, Bauer ME. Deletion of mtrC in Haemophilus ducreyi increases sensitivity to human antimicrobial peptides and activates the CpxRA regulon. Infect Immun. 2011;79:2324–2334. doi: 10.1128/IAI.01316-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kupferwasser LI, Skurray RA, Brown MH, Firth N, Yeaman MR, Bayer AS. Plasmid-mediated resistance to thrombin-induced platelet microbicidal protein in Staphylococci: role of the qacA locus. Antimicrob Agents Chemother. 1999;43:2395–2399. doi: 10.1128/aac.43.10.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bayer AS, Kupferwasser LI, Brown MH, Skurray RA, Grkovic S, Jones T, Mukhopadhay K, Yeaman MR. Low-level resistance of Staphylococcus aureus to thrombin-induced platelet microbicidal protein 1 in vitro associated with qacA gene carriage is independent of multidrug efflux pump activity. Antimicrob Agents Chemother. 2006;50:2448–2454. doi: 10.1128/AAC.00028-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oldham ML, Davidson AL, Chen J. Structural insights into ABC transporter mechanism. Current Opin Struct Biol. 2008;18:726–733. doi: 10.1016/j.sbi.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parra-Lopez C, Baer MT, Groisman EA. Molecular genetic analysis of a locus required for resistance to antimicrobial peptides in Salmonella typhimurium. E M B O Journal. 1993;12:4053–4062. doi: 10.1002/j.1460-2075.1993.tb06089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shelton CL, Raffel FK, Beatty WL, Johnson SM, Mason KM. Sap transporter mediated import and subsequent degradation of antimicrobial peptides in Haemophilus. PLoS Pathog. 2011;7:e1002360. doi: 10.1371/journal.ppat.1002360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Groisman EA, Parra-Lopez C, Salcedo M, Lipps CJ, Heffron F. Resistance to host antimicrobial peptides is necessary for Salmonella virulence. Proc Natl Acad Sci U S A. 1992;89:11939–11943. doi: 10.1073/pnas.89.24.11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stumpe S, Bakker EP. Requirement of a large K+-uptake capacity and of extracytoplasmic protease activity for protamine resistance of Escherichia coli. Arch Microbiol. 1997;167:126–136. [PubMed] [Google Scholar]
- 75.Mason KM, Munson RS, Jr, Bakaletz LO. A mutation in the sap operon attenuates survival of nontypeable Haemophilus influenzae in a chinchilla model of otitis media. Infect Immun. 2005;73:599–608. doi: 10.1128/IAI.73.1.599-608.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McCoy AJ, Liu H, Falla TJ, Gunn JS. Identification of Proteus mirabilis mutants with increased sensitivity to antimicrobial peptides. Antimicrob Agents Chemother. 2001;45:2030–2037. doi: 10.1128/AAC.45.7.2030-2037.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.López-Solanilla E, García-Olmedo F, Rodríguez-Palenzuela P. Inactivation of the sapA to sapF locus of Erwinia chrysanthemi reveals common features in plant and animal bacterial pathogenesis. Plant Cell. 1998;10:917–924. [PMC free article] [PubMed] [Google Scholar]
- 78.Mason KM, Bruggeman ME, Munson RS, Bakaletz LO. The non-typeable Haemophilus influenzae Sap transporter provides a mechanism of antimicrobial peptide resistance and SapD-dependent potassium acquisition. Mol Microbiol. 2006;62:1357–1372. doi: 10.1111/j.1365-2958.2006.05460.x. [DOI] [PubMed] [Google Scholar]
- 79.Rinker SD, Gu X, Fortney KR, Zwickl BW, Katz BP, Janowicz DM, Spinola SM, Bauer ME. Permeases of the Sap transporter are required for cathelicidin resistance and virulence of Haemophilus ducreyi in humans. J Infect Dis. 2012;206:1407–1414. doi: 10.1093/infdis/jis525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lupp C, Hancock REW, Ruby EG. The Vibrio fisheri sapABCDF locus is required for normal growth, both in culture and in symbiosis. Arch Microbiol. 2002;179:57–65. doi: 10.1007/s00203-002-0502-7. [DOI] [PubMed] [Google Scholar]
- 81.Mason KM, Raffel FK, Ray WC, Bakaletz LO. Heme utilization by nontypeable Haemophilus influenzae is essential and dependent on Sap transporter function. J Bacteriol. 2011;193:2527–2535. doi: 10.1128/JB.01313-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.López-Solanilla E, Llama-Palacios A, Collmer A, García-Olmedo F, Rodríguez-Palenzuela P. Relative effects on virulence of mutations in the sap, pel, and hrp loci of Erwinia chrysanthemi. Mol Plant Microbe Interact. 2001;14:386–393. doi: 10.1094/MPMI.2001.14.3.386. [DOI] [PubMed] [Google Scholar]
- 83.Sieprawska-Lupa M, Mydel P, Krawczyk K, Wojcik K, Puklo M, Lupa B, Suder P, Silberring J, Reed M, Pohl J, Shafer W, McAleese F, Foster T, Travis J, Potempa J. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother. 2004;48:4673–4679. doi: 10.1128/AAC.48.12.4673-4679.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Belas R, Manos J, Suvanasuthi R. Proteus mirabilis ZapA metalloprotease degrades a broad spectrum of substrates, including antimicrobial peptides. Infect Immun. 2004;72:5159–5167. doi: 10.1128/IAI.72.9.5159-5167.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schmidtchen A, Frick I-M, Björck L. Dermatan sulphate is released by proteinases of common pathogenic bacteria and inactivate antibacterial α-defensin. Mol Microbiol. 2001;39:708–713. doi: 10.1046/j.1365-2958.2001.02251.x. [DOI] [PubMed] [Google Scholar]
- 86.Schmidtchen A, Frick I-M, Andersson E, Tapper H, Björck L. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol Microbiol. 2002;46:157–168. doi: 10.1046/j.1365-2958.2002.03146.x. [DOI] [PubMed] [Google Scholar]
- 87.Caulfield AJ, Walker ME, Gielda LM, Lathem WW. The Pla protease of Yersinia pestis degrades fas ligand to manipulate host cell death and inflammation. Cell Host Microbe. 2014;15:424–434. doi: 10.1016/j.chom.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Galvan EM, Lasaro MA, Schifferli DM. Capsular antigen fraction 1 and Pla modulate the susceptibility of Yersinia pestis to pulmonary antimicrobial peptides such as cathelicidin. Infect Immun. 2008;76:1456–1464. doi: 10.1128/IAI.01197-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Maisey HC, Quach D, Hensler ME, Liu GY, Gallo RL, Nizet V, Doran KS. A group B Streptococcal pilus protein promotes phagocyte resistance and systemic virulence. FASEB J. 2008;22:1715–1724. doi: 10.1096/fj.07-093963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Eswarappa SM, Panguluri KK, Hensel M, Chakravortty D. The yejABEF operon of Salmonella confers resistance to antimicrobial peptides and contributes to its virulence. Microbiol. 2008;154:666–678. doi: 10.1099/mic.0.2007/011114-0. [DOI] [PubMed] [Google Scholar]
- 91.Pence MA, Rooijakkers SH, Cogen AL, Cole JN, Hollands A, Gallo RL, Nizet V. Streptococcal inhibitor of complement promotes innate immune resistance phenotypes of invasive M1T1 group A Streptococcus. J Innate Immun. 2010;2:587–595. doi: 10.1159/000317672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Su JH, Chung YC, Lee HC, Tseng IC, Chang MC. Ferrous iron-binding protein Omb of Salmonella enterica serovar Choleraesuis promotes resistance to hydrophobic antibiotics and contributes to its virulence. Microbiol. 2009;155:2365–2374. doi: 10.1099/mic.0.026880-0. [DOI] [PubMed] [Google Scholar]