

ABSTRACT
The Kaposi's sarcoma-associated herpesvirus (KSHV) LANA protein is essential for the replication and maintenance of virus genomes in latently KSHV-infected cells. LANA also drives dysregulated cell growth through a multiplicity of mechanisms that include altering the activity of the cellular kinases extracellular signal-regulated kinase (ERK) and glycogen synthase kinase 3 (GSK-3). To investigate the potential impact of these changes in enzyme activity, we used protein microarrays to identify cell proteins that were phosphorylated by the combination of ERK and GSK-3. The assays identified 58 potential ERK-primed GSK-3 substrates, of which 23 had evidence for in vivo phosphorylation in mass spectrometry databases. Two of these, SMAD4 and iASPP, were selected for further analysis and were confirmed as ERK-primed GSK-3 substrates. Cotransfection experiments revealed that iASPP, but not SMAD4, was targeted for degradation in the presence of GSK-3. iASPP interferes with apoptosis induced by p53 family members. To determine the importance of iASPP to KSHV-infected-cell growth, primary effusion lymphoma (PEL) cells were treated with an iASPP inhibitor in the presence or absence of the MDM2 inhibitor Nutlin-3. Drug inhibition of iASPP activity induced apoptosis in BC3 and BCBL1 PEL cells but did not induce poly(ADP-ribose) polymerase (PARP) cleavage in virus-negative BJAB cells. The effect of iASPP inhibition was additive with that of Nutlin-3. Interfering with iASPP function is therefore another mechanism that can sensitize KSHV-positive PEL cells to cell death.
IMPORTANCE KSHV is associated with several malignancies, including primary effusion lymphoma (PEL). The KSHV-encoded LANA protein is multifunctional and promotes both cell growth and resistance to cell death. LANA is known to activate ERK and limit the activity of another kinase, GSK-3. To discover ways in which LANA manipulation of these two kinases might impact PEL cell survival, we screened a human protein microarray for ERK-primed GSK-3 substrates. One of the proteins identified, iASPP, showed reduced levels in the presence of GSK-3. Further, blocking iASPP activity increased cell death, particularly in p53 wild-type BC3 PEL cells.
INTRODUCTION
The Kaposi's sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen (LANA) is expressed in all KSHV-infected cells, including the tumor cells of the KSHV-associated malignancies Kaposi's sarcoma, primary effusion lymphoma (PEL), and muticentric Castleman disease. LANA functions in the replication and maintenance of latent, episomal KSHV genomes (1, 2) by binding to the KSHV origin of replication in the terminal repeats (3, 4), recruiting cellular replication proteins (5–11), and tethering the KSHV episomal genomes to host chromosomes (12–18). Sequences in the KSHV terminal repeats are bound by the C terminus of LANA, and the X-ray crystal structures of the human and murine gammaherpesvirus 68 (MHV68) LANA DNA-binding domains have been solved (19–21).
KSHV infection leads to a global reprogramming of cell gene expression (22–26) and modification of cell signaling pathways (27–31). In addition to its replication functions, LANA also participates in this reprogramming. LANA mediates transcriptional upregulation and downregulation (32). Chromatin immunoprecipitation (ChIP) analyses have found LANA associated with the same sequences in the host genome as were characterized in the viral terminal repeats, in association with a separate characterized binding motif and binding indirectly through protein-protein interactions (33–35). LANA participates in epigenetic silencing (36–39), increased transcription factor activity (40–46), and modulation of interferon signaling (33, 47), the cell cycle (42, 48, 49) and kinase activity (50–54).
Glycogen synthase kinase 3 (GSK-3) alpha and beta are ubiquitous and constitutively active serine/threonine kinases that regulate multiple cellular pathways, including the Wnt pathway (55). LANA-mediated inactivation of GSK-3 (42, 56) has the known consequences of blocking differentiation (57) and stabilizing cMyc (44, 58). GSK-3 phosphorylation of substrates generally requires a priming phosphorylation by a second kinase at the +4 position in the recognition sequence S/T-X-X-X-S/T. LANA also leads to activation of the mitogen-activated protein kinase (MAPK) extracellular signal-regulated kinase 1 (ERK1) (44), a known priming kinase for GSK-3. The inactivation of GSK-3 by LANA has the potential to contribute to other pathways that are altered after KSHV infection. GSK-3 phosphorylation often results in tagging of the substrate for proteasomal degradation. To identify novel cell substrates whose GSK-3 phosphorylation and degradation could potentially be modified in LANA-expressing cells, we performed GSK-3 phosphorylation assays using ERK1 as the priming kinase and a protein array displaying 4,191 unique human proteins. Bioinformatic algorithms were incorporated into the analysis of the phosphorylation data obtained from the kinase assays. A particularly interesting ERK-primed, GSK-3 phosphorylated protein identified in this screen was the inhibitor of p53-mediated apoptosis, iASPP.
MATERIALS AND METHODS
Human ORF cloning and microarray production.
Using the Gateway recombinant cloning system (Invitrogen), 4,191 nonredundant human open reading frames (ORFs) consisting of a collection of genes encoding 1,371 human transcription factors, 238 transcriptional coregulators, 689 RNA-binding proteins, 257 DNA-binding proteins, 146 DNA repair proteins, 286 chromosome-organizing proteins, 332 protein kinases, and 652 mitochondrial proteins as well as a panel of 589 proteins involved in various other cellular processes were shuttled from the selected entry clones of the Ultimate Human ORF Collection (Invitrogen) or from the entry clones generated in our own laboratories into a yeast high-copy expression vector (pEGH-A) that produces glutathione S-transferase (GST)-His6 fusion proteins under the control of the galactose-inducible GAL1 promoter. Plasmids were rescued into Escherichia coli and verified by restriction endonuclease digestion. Plasmids with inserts of the correct size were transformed into yeast. Proteins were purified as GST-His6 fusion proteins from yeast using a high-throughput protein purification protocol (59). Purified human proteins were arrayed in a 384-well format and printed on FullMoon glass slides (FullMoon Biosystems) in duplicate. The printing quality and quantity of the immobilized proteins on a representative batch of chips were monitored using anti-GST antibody followed by Cy5-labeled secondary antibody (60).
Protein array phosphorylation assays.
Phosphorylation assays were performed using a protocol similar to that previously described (59). Purified ERK1 and GSK-3β (Cell Signaling) were added to 250 μl of kinase buffer (50 mM Tris-HCl and 25 mM HEPES-KOH at pH 7.5) containing 100 mM NaCl, 10 mM MgCl2, 1 mM MnCl2, 1 mM dithiothreitol (DTT), 1 mM EGTA, 2 mM NaVO4, 2 mM NaF, 0.01% cold ATP, and [γ-32P]ATP [33.3 nM final concentration]). Each reaction mixture was then incubated on microarrays and coverslipped before being placed in a humidity chamber at 30°C for 30 min. Following the termination of the reaction, the slides were subjected to three 10-min washes in Tris-buffered saline (TBS)–0.1% Tween 20 (pH 7.5), three 10-min washes in 0.5% SDS, and one quick rinse with distilled water before being spun dry and exposed to X-ray film (Kodak). Control slides were incubated with kinase buffer without added kinase and processed in parallel. Exposures were taken for each microarray assayed at 10, 17, and 24 h. The slides were scanned, and the phosphorylation signals were acquired using the GenePix software.
Data analysis.
Data analysis included four steps: local background correction, normalization, identification of phosphorylation events, and removal of signals from control incubations. The signal intensity of each protein on the array was quantified by dividing the median foreground intensity by the median intensity of its local background. To identify positive signals on the microarrays, the standard deviation from the signal intensity distribution was estimated as described previously (60). The cutoff was two standard deviations above the mean. Each protein was printed in duplicate, and a positive was scored only if both spots showed a signal intensity higher than the cutoff value. To identify signals arising from autophosphorylation events, control assays were performed with kinase buffer lacking exogenous added kinase. Those proteins that exhibited autophosphorylation activity were excluded from the substrate list.
Plasmids.
The open reading frames for SMAD4 and iASPP from the Ultimate Human ORF Collection (Invitrogen) were subcloned into pDEST-SG5-Flag (pJHA411) using the Gateway system (Invitrogen). Mutations in SMAD4 were introduced using the QuikChange II site-directed mutagenesis kit. GST-SMAD4 and GST-iASPP(401–828) proteins were expressed in bacteria from a modified pGEX2T vector (pGH413). SG5-HA-GSK-3β was obtained from F. McCormick (University of California, San Francisco).
Liquid kinase assays.
Purified GST-SMAD4, GST-SMAD4 mutants, and GST-iASPP(401–828) on beads were washed twice with ERK buffer (25 mM Tris-HCl [pH 7.5], 10 mM MgCl2, 1 mM DTT, 1 mM EGTA, 20 μM cold ATP). Samples were incubated in kinase buffer with 10 μCi of [γ-32P]ATP and 25 mU of activated ERK1 (Upstate) or 10 μCi of [γ-32P]ATP and 0.1 U of GSK-3β (Cell Signaling) for 30 min at 30°C. For primed reactions, samples were incubated in 20 μM cold ATP and 25 mU of activated ERK1 for 30 min at 30°C, washed 6 times with lysis buffer (50 mM Tris-HCl [pH 7.8], 1 mM DTT, 0.5 mM EDTA, 50 mM NaCl, 0.2% Nonidet P-40, and 5% glycerol), and incubated in kinase buffer containing 10 μCi of [γ-32P]ATP and 0.1 U of GSK-3β for 30 min at 30°C. In the wash control reaction, the GSK-3β was omitted. Finally, reaction mixtures were washed twice with ice-cold washing buffer (50 mM Tris-HCl [pH 7.9], 10 mM MgCl2, 2 mM DTT, 100 mM NaCl) and separated by gel electrophoresis. Radiolabeled polypeptides were detected by autoradiography.
Western blotting.
HA-GSK-3β and Flag-SMAD4 or Flag-iASPP were transfected individually or together (0.6 μg each) with 4 μl of Lipofectamine 2000 (Invitrogen) into HeLa cells grown in six-well plates. Two days later, the cells were rinsed with 1 ml of ice-cold phosphate-buffered saline and lysed in 1 ml radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.4], 1% NP-40, 0.25% Na deoxycholate, 150 mM NaCl, and 1 mM EDTA) containing protease inhibitor cocktail (1 mM phenylmethylsulfonyl fluoride [PMSF], 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 μg/ml pepstatin) and phosphatase inhibitor cocktail (Sigma). After sonication (Branson Sonifier 450) on ice for 1 min, the cell lysates were centrifuged at 10,000 × g for 10 min. Samples were resuspended in 60 μl of 2× Laemmli sampling buffer (Bio-Rad) and heated at 95°C for 5 min. The samples were resolved on a 4 to 20% SDS-polyacrylamide gel (Invitrogen). The proteins on the gel were transferred to a nitrocellulose membrane and subjected to immunoblot analysis with anti-Flag or anti-actin antibodies (Sigma). Endogenous iASPP proteins in B cell lines was detected using anti-iASPP antibody (Sigma). Poly(ADP-ribose) polymerase (PARP) cleavage was measured using antibody to cleaved PARP (Cell Signaling).
Cell cultures and cell growth.
PEL and B cell lines were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (Corning). TREx-BCBL1 cells were obtained from J. Jung (University of Southern California). The CellTiter-Glo luminescence assay kit (Promega) was used according to the manufacturer's instructions to determine cellular ATP levels as a measure of cell growth. Nutlin-3 (N6287) and JNJ-7706621 (S1249) were obtained from Sigma and Selleckchem, respectively.
RESULTS
Establishment of the screening assay.
We previously used protein microarrays to identify substrates of human kinases, monitor changes in the human phospho-proteome, identify kinases that phosphorylate viral proteins, and identify cellular and viral substrates of viral kinases (61–65). We have now adapted this approach to identify proteins that are potentially ERK-primed GSK-3 substrates. To confirm that the requirement for priming phosphorylation was retained when GSK-3β was assayed using the protein chip format and to establish conditions for the assays, human protein arrays were incubated in the presence of kinase buffer containing [γ-32P]ATP to measure autophosphorylation of the proteins imprinted on the array, ERK1 in kinase buffer containing [γ-32P]ATP to identify ERK1 substrates, or GSK-3β in kinase buffer containing [γ-32P]ATP to evaluate the requirement for priming for GSK-3β phosphorylation (Fig. 1). The signals seen on the control array (Fig. 1A) represent autophosphorylating kinases. Comparison with ERK1 result shows that added ERK1 is able to phosphorylate a large number of additional proteins on the microarray (Fig. 1B). However, when GSK-3β was incubated with the array, the results were very similar to those with the control. The results confirm that in the array assays, GSK-3β activity is limited in the absence of priming phosphorylation (Fig. 1C). The small number of GSK-3 signals over the control may represent proteins that do not require priming, proteins that require priming in vivo but can be inefficiently phosphorylated in vitro in the absence of priming, or proteins that purify from the yeast as phosphorylated proteins and are consequently preprimed.
FIG 1.

Protein array kinase assays. Examples of autoradiographic exposures of the human protein arrays after incubation with kinase buffer containing [γ-32P]ATP (A), [γ-32P]ATP plus ERK1 (B), [γ-32P]ATP plus GSK-3β (C), or ERK1 plus cold ATP, a wash step, and then [γ-32P]ATP plus GSK-3β (D) are shown.
For ERK-primed GSK-3β phosphorylation, a protocol was developed in which the microarray was first treated with ERK1 in kinase buffer containing cold ATP at 30°C for 30 min and then washed extensively with a nondenaturing detergent to remove the ERK1 protein. Next, the array was incubated with GSK-3β in the presence of kinase buffer containing [γ-32P]ATP. After an additional 30 min of incubation at 30°C, the array was washed under denaturing conditions, dried, and exposed to X-ray film (Fig. 1D).
Identification of GSK-3β substrates that require ERK1 priming.
Phosphorylated proteins on each array were identified using an algorithm that measures the relative signal intensity of each spot at a cutoff value of two standard deviations (SD) above background. Proteins identified on the autophosphorylation control arrays and the arrays monitoring residual ERK activity and GSK-3 alone were removed from those appearing on the ERK1-primed GSK-3β list. This resulted in a data set of 148 proteins that were potential ERK1-primed GSK-3β substrates (see Table S1 in the supplemental material). Identifying substrates primed with ERK1 is complicated by the fact that there are no ERK inhibitors that are active in vitro and that can be used to abolish ERK activity after the priming phosphorylation. Visual inspection of results of phosphorylation assays in which the ERK-primed array was washed and then incubated with the radiolabeled ATP in kinase buffer revealed that residual ERK1 activity was detectable and that in many cases this resulted in proteins that had an intense signal on the ERK-primed GSK-3β array also showing a signal, albeit weaker, on the ERK control. This outcome precluded a definitive determination of primed GSK-3β substrates using the simple approach of subtracting the ERK1 control phosphorylation data from the ERK1-primed GSK-3β phosphorylation data. We consequently applied bioinformatic analysis to the data set.
To further evaluate the prospective ERK1-primed GSK-3β data set, we applied an algorithm, termed M3 (motif discovery based on microarray and MS/MS), to identify the dominant motif present in the 148 potential substrates. M3 predicts consensus phosphorylation motifs by combining kinase substrate interaction data with in vivo phosphorylation sites, determined mainly by tandem mass spectrometry (MS/MS) analysis, and uses an iterative approach to identify associated pS/T sites within each data set (62). The short amino acid sequences (15-mers) containing these sites were then binned into groups according to their identified kinase substrate interaction relationships. The dominant LOGO identified from the initial ERK1-primed GSK-3β data set was S/T-X-X-X-S (see Fig. S1A in the supplemental material). Fifty-five of the 148 substrates identified had this motif, which conforms to the known GSK-3β consensus phosphorylation motif. ERK1 can also phosphorylate threonine to prime for GSK-3β. The Phospho.elm website was used to screen the amino acid sequences of the remaining 93 proteins from the potential ERK1-primed GSK-3β data set for proteins that had an S/T-X-X-X-T phosphorylation motif. Only three proteins were identified as having phosphorylation events occurring on these amino acids. The combined analyses resulted in a list of 58 proteins that were putative ERK1-primed GSK-3 substrates (Table 1).
TABLE 1.
Putative ERK1-primed GSK-3 substrates identified
| Protein | Protein (continued) | Protein (continued) |
|---|---|---|
| ABI2 | EP400 | NUP35 |
| AFF4 | ERF | PCTK3 |
| ANXA2 | FLJ20105 | PER1 |
| BCOR | FMNL2 | PPP1R13L |
| BTBD12 | FOSL1 | RAD23B |
| C19orf21 | FOSL2 | RBM12 |
| C20orf19 | FOXP4 | RPA2 |
| CARHSP1 | GDI2 | SMAD4 |
| CC2D1A | HCLS1 | SOX9 |
| CENTG1 | HIRIP3 | STUB1 |
| CHAF1B | HSF1a | SUHW4 |
| CNOT2 | KLF4 | TBC1D22A |
| CRK | LIG1 | TCEAL6 |
| CSTF2T | LIMD1 | TRIP10 |
| CTTN | MAGEB6 | TSC22D3 |
| DAZAP1 | MEF2C | ZHX1 |
| DCP1A | NFATC1 | ZNF503 |
| EIF3S4 | NFATC3a | ZYX |
| EIF4B | NFATC4a | |
| ELK1 | NUP133 |
Known GSK-3β substrate.
Reanalysis of the remaining 90 proteins from the initial list (see Table S1 in the supplemental material) generated a different LOGO consistent with ERK1 signal bleedthrough (see Fig. S1B in the supplemental material). A direct examination of the microarray data confirmed that 74% of these proteins were visualized on the ERK phosphorylation and/or the ERK priming data sets.
The strongest candidates among the 58 proteins with the S/T-X-X-X-S/T motif would be those for which data existed for in vivo phosphorylation at the motif serine/threonine residues. The MS/MS in vivo phosphorylation database was interrogated, and 23 of the 58 proteins had evidence for in vivo phosphorylation at both the priming and the GSK-3 phosphorylation sites or at one or the other of these sites (see Table S2 in the supplemental material).
Validation of SMAD4 and iASPP as ERK-primed GSK-3β substrates.
We selected for follow-up two proteins from Table S2 in the supplemental material that had not previously been shown to be GSK-3 substrates experimentally, namely, SMAD4 and iASPP (also known as PPP1R13L). SMADs mediate transcriptional responses to transforming growth factor β (TGF-β) and bone morphogenic protein (BMP) and are divided into 3 classes: the receptor-regulated SMADs that transmit TGF-β signaling (SMAD2 and -3) and bone morphogenetic protein signaling (SMAD1, -5, and -8), the inhibitory SMADs (SMAD6 and -7), and a common SMAD, SMAD4, that partners with the other SMADs to form the transcriptionally active trimeric complex (66). Inactivation of SMAD4 is common in pancreatic and colorectal cancers but less so in other human cancers (67, 68). GSK-3 phosphorylation of SMAD4 has not been investigated. There are 3 sites in the SMAD4 amino acid sequence that fit the consensus for ERK primed GSK-3 phosphorylation. These are the amino acid pairings S5/T9P, T273/T277P, and S517/T521P.
To validate SMAD4 phosphorylation, in vitro kinase assays were performed using purified GST-SMAD4 and variants that carried mutations in the potential individual GSK-3 phosphorylation sites (S5A, T273A, and S517A) (Fig. 2A and C). ERK1 strongly phosphorylated the GST-SMAD4 protein (Fig. 2B, lane E), while GSK-3 was not active in the absence of priming phosphorylation (Fig. 2B, lane G) but did phosphorylate GST-SMAD4 after ERK1 priming (Fig. 2B, lane Ec+G). The control phosphorylation in which ERK priming was followed by washing steps and then addition of radiolabeled ATP in kinase buffer showed that little residual ERK activity was retained after the washing steps (Fig. 2B, lane Ec). GST-SMAD4 mutated for all three potential GSK-3 sites (S5A, T273A, and S517A) showed no GSK-3 phosphorylation above the level seen with the control priming ERK1 reaction, indicating that one or more of these sites are phosphorylated by GSK-3 (Fig. 2B, lane Ec versus lane Ec+G). GSK-3 phosphorylation was reduced but detectable upon mutation of the potential GSK-3 site at T273, suggesting that T273 is one, but possibly not the only, GSK-3 phosphorylation site. Phosphorylation of the combined GSK-3 sites at T273A and S517A was similar to that seen with mutation of T273A alone (Fig. 2B, lane Ec versus lane Ec+G). These results are compatible with GSK-3 phosphorylation at T273 with a less preferred site potentially at S5 of SMAD4.
FIG 2.

SMAD4 is an ERK-primed GSK-3β substrate. (A) Locations of consensus paired ERK1/2 (E) and GSK-3 (G) phosphorylation sites in SMAD4. (B) Autoradiographs of GST-SMAD4 and GST-SMAD4 mutants after incubation with [γ-32P]ATP (A), [γ-32P]ATP plus ERK1 (E), [γ-32P]ATP plus GSK-3β (G), or ERK1 plus cold ATP, a wash step, and then [γ-32P]ATP (Ec) or plus GSK-3β and [γ-32P]ATP (Ec+G). (C) Coomassie blue-stained gel of the GST-SMAD4 and GST-SMAD4 mutants.
The tumor suppressor p53 mediates cell cycle arrest or apoptosis in response to DNA damage-inducing stimuli. p53 transcriptional activity is regulated through a variety of posttranslational modifications and through protein-protein interactions (69). ASPPs (ankyrin repeat-, SH3 domain-, and proline-rich region-containing proteins) specifically affect p53-mediated apoptotic responses (70). ASPP1 and ASPP2 enhance the proapoptotic function of p53 by promoting the binding of p53 to proapoptotic gene targets, while iASSP (PPP1R13L) prevents the transcriptional activity of p53 bound to these promoters (71, 72). The 828-amino-acid iASPP protein localizes to both the cytoplasm and the nucleus, while the 407-amino-acid iASPP splice variant that has common C-terminal sequences with the 828-amino-acid protein localizes to the nucleus (73, 74). We examined iASPP expression in a variety of B cell lines and detected expression of both the long and short forms of iASPP in all the cell lines, including the KSHV-positive PEL cell lines JSC-1, BCBL1, and BC3 (Fig. 3). Expression was abundant in the tumor-derived cell lines and lower in the lymphoblastoid cell line (LCL) that was established by in vitro infection with Epstein-Barr virus (EBV). This is consistent with reports of upregulated expression of iASPP being associated with malignant transformation (75, 76).
FIG 3.
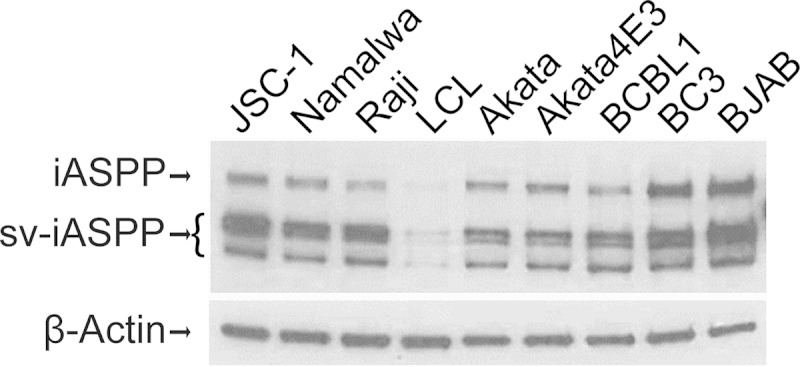
iASPP protein expression in lymphoma cell lines. Western blotting using anti-iASPP antibody is shown. JSC-1, BCBL1, and BC3 are KSHV-positive PEL cell lines. Namalwa, Raji, LCL, and Akata are EBV-positive cell lines. Akata4E3 and BJAB are virus-negative lymphoma cell lines. β-Actin served as the loading control. sv-iASPP, splice-variant iASPP.
To confirm that iASPP was an ERK-primed GSK-3 substrate, an in vitro phosphorylation assay was performed on a GST-iASPP protein containing carboxy-terminal iASPP sequences from amino acid 401 to 828. This region includes all of the sequences in common between the long and short iASPP proteins. GST-iASPP(401–828) was strongly phosphorylated by ERK (Fig. 4A, lane 1). The majority of the ERK signal was removed by washing (Fig. 4A, lane 2), and GSK-3β showed minimal activity in the absence of ERK priming (Fig. 4A, lane 3). GSK-3β was capable of phosphorylating GST-iASPP after ERK priming (Fig. 4A, lane 4). Incubation with radioactive ATP alone did not produce any signal (Fig. 4A, lane 5).
FIG 4.
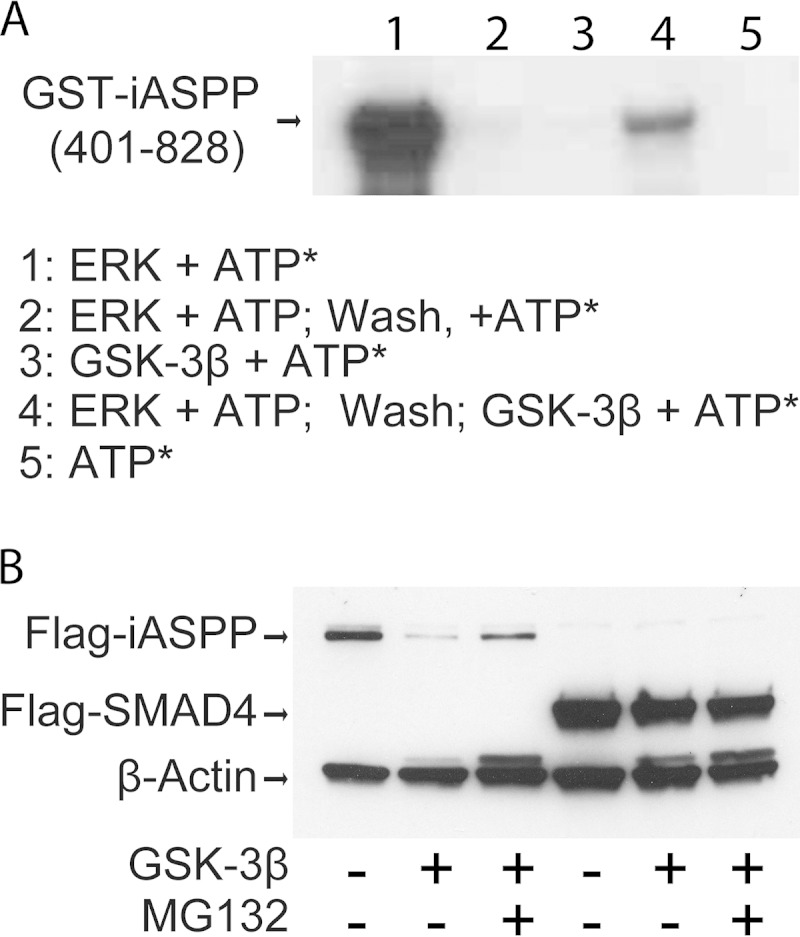
GSK-3β phosphorylation decreases iASPP protein levels. (A) Autoradiograph showing in vitro phosphorylation of GST-iASPP by ERK1 and ERK1-primed GSK-3β. ATP*, [γ-32P]ATP. (B) Western blot examining protein levels of Flag-iASPP and Flag-SMAD4 in the presence or absence of cotransfected GSK-3β and the proteasome inhibitor MG132.
GSK-3β phosphorylation decreases the stability of iASPP.
The effect of GSK-3β expression on the protein levels of iASPP and SMAD4 was examined in transfected HeLa cells. Coexpression of GSK-3β reduced the level of the 100-kDa Flag-iASPP protein, and the protein level was partially restored upon treatment with the proteasome inhibitor MG132 (Fig. 4B). In contrast, coexpression of GSK-3β with Flag-SMAD4 did not significantly affect the level of Flag-SMAD4 (Fig. 4B). β-Actin served as a loading control.
Targeting iASPP impacts PEL cell growth.
Treatment with the MDM2 inhibitor Nutlin-3 leads to increased apoptosis and PARP-1 cleavage in p53 wild-type and p53 mutant PEL cell lines (77–79). The effect of Nutlin-3 on p53 mutant PEL cell lines is partially mediated through its ability to also disrupt the interaction between MDM2 and p73 (77). Binding of iASPP to p53 family members is dependent upon phosphorylation of iASPP, and the kinase inhibitor JNJ-7706621 was recently shown to abolish this phosphorylation when used at 2 and 5 μM concentrations (80). We tested the impact on PEL cell growth of activating p53 family members through inhibition of iASPP using JNJ-7706621 alone or in combination with the MDM2 inhibitor Nutlin-3 (Fig. 5). The growth of KSHV-negative p53 mutant BJAB cells was unaffected by 10 μM Nutlin-3. JNJ-7706621 (5 μM) alone or in combination with 10 μM Nutlin-3 reduced cell growth by 26% after 3 days of treatment (Fig. 5A). BCBL1 p53 mutant PEL cells are known to be relatively resistant to Nutlin-3 compared to other PEL cell lines (77–79). BCBL1 exhibited a similar sensitivity to 5 μM JNJ-7706621 as to 10 μM Nutlin-3 (35% versus 26% reduction). The combination of Nutlin-3 and JNJ-7706621 was additive, with a 69% reduction in growth (Fig. 5B). BC3 p53 wild-type PEL cells were highly sensitive to treatment with 5 μM JNJ-7706621, which resulted in a 93% reduction in cell growth at 3 days. The combination of Nutlin-3 and JNJ-7706621 was completely cytotoxic (Fig. 5C). In light of the sensitivity of BC3 cells to JNJ-7706621, the effect of treatment with a lower dose of 2 μM JNJ-7706621 was also examined. After 3 days of treatment, the reduction in growth of BC3 cells treated with 2 μM JNJ-7706621 was similar to that of BC3 cells treated with Nutlin-3 (35% versus 48% growth reduction). The combination of the two drugs was again completely cytotoxic (Fig. 5D).
FIG 5.

Targeting iASPP increases apoptosis of BCBL1 and BC3 cells. (A to C) Growth of BJAB (A), BCBL1 (B), and BC3 (C) cells with or without treatment with Nutlin-3 (10 μM), JNJ-7706621 (5 μM), or the combination of Nutlin-3 and JNJ-7706621. The data points are averages of two measurements. (D) Relative metabolic activity of BC3 cells untreated or treated for 2 days with 10 μM Nutln-3, 2 μM JNJ-7706621, or the combination of Nutlin-3 and JNJ-7706621. ***, P < 0.001. (E) Western blot examining cleavage of endogenous PARP in BCBL1, BC3, and BJAB cells. Cells were harvested after treatment for 2 days with Nutlin-3, JNJ-7706621, or the drug combination. The anti-PARP antibody used is specific for cleaved PARP (PARP*). β-Actin served as the loading control.
The effect of JNJ-7706621 on cell growth curves could be mediated through either decreased cell proliferation or increased cell death. We examined PARP cleavage in cells treated for 2 days with Nutlin-3, JNJ-7706621, or a combination of the two drugs as a measure of caspase induction and apoptosis (Fig. 5E). The slowing in growth of BJAB cells treated with 5 μM JNJ-7706621 or the drug combination was not associated with any increase in PARP cleavage and therefore is likely to reflect the effect of JNJ-7706621 on the cell cycle. BCBL1 cells showed an increase in PARP cleavage at 5 μM JNJ-7706621 that was greater than that seen with 10 μM Nutlin-3, and, allowing for the decrease in actin signal, PARP cleavage was slightly increased by the combination treatment with Nutlin-3 and JNJ-7706621. Treatment of BC3 cells with either 5 μM JNJ-7706621 or the combination of Nutlin-3 plus 2 or 5 μM JNJ-7706621 led to a dramatic increase in PARP cleavage, indicating that JNJ-7706621 was an efficient inducer of BC3 PEL cell death.
Drug treatment did not induce the KSHV lytic cycle.
It has been reported that inhibition of CDK1 with Purvalanol A or short hairpin RNA (shRNA) knockdown leads to reactivation of the KSHV lytic cycle (81). JNJ-7706621 inhibits CDK1/CycB with a 50% inhibitory concentration (IC50) of 9 μM (82). To ensure that lytic reactivation was not contributing to the inhibition of PEL cell growth in cells treated with JNJ-7706621, we examined the protein level of the immediate early lytic cycle regulator RTA. RTA expression did not increase in TREx-BCBL1 or BC3 cells treated with either 2 μM or 5 μM JNJ-7706621 for 3 days (Fig. 6). Therefore, at the concentrations of JNJ-7706621 used in our experiments, lytic cycle reactivation was not a factor in the treatment-induced PEL cell growth inhibition.
FIG 6.
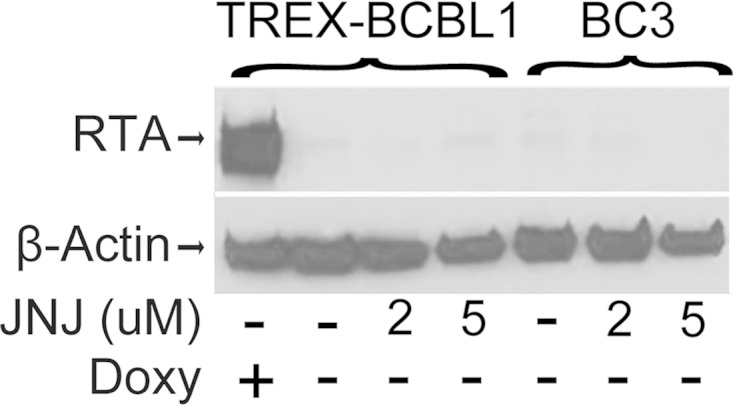
Lack of lytic induction after JNJ-7706621 treatment. The Western blot shows RTA expression in TREX-BCBL1 and BC3 PEL cells treated with the indicated concentrations of JNJ-7706621 (JNJ) for 3 days. RTA expression in doxycycline (Doxy)-induced TREx-BCBL1 cells served as a positive control. β-Actin served as the loading control.
DISCUSSION
Proteomic arrays have been used for global analysis of protein modification by phosphorylation, ubiquitination, sumoylation, acetylation, and S-nitrosylation (83). Here, we sought to expand the use of these arrays for kinase substrate identification by undertaking an analysis of phosphorylation by a kinase, GSK-3, that requires prior priming of its substrates by a second kinase. ERK is a common priming kinase for GSK-3. The KSHV LANA protein affects the activity of both ERK and GSK-3, increasing ERK activity and impairing nuclear GSK-3 activity. Applying the analysis to the combination of ERK-primed GSK-3 phosphorylation on the human protein arrays therefore had the potential to identify novel cell substrates that might be relevant to KSHV biology.
The kinase assays performed on the protein arrays with GSK-3β alone demonstrated that GSK-3β retained the requirement for priming phosphorylation in this setting, while the combination of ERK prephosphorylation followed by incubation with GSK-3β resulted in extensive substrate phosphorylation. Bioinformatic analysis of the substrates phosphorylated by GSK-3 after ERK priming revealed that all 148 substrates contained a serine or threonine residue followed by a proline and were consequently valid candidates for ERK phosphorylation. However, only 58 contained the ERK phosphorylation site at the +4 position of a consensus GSK-3 motif. This outcome is consistent with residual ERK protein being retained on the arrays after the washing step and contributing to the phosphorylation signal generated upon the subsequent addition of isotopically labeled ATP and GSK-3β. Identification of ERK-primed GSK-3 substrates therefore required the protein array phosphorylation data be combined with bioinformatic analysis. Direct identification of substrates for GSK-3 should be possible in cases where inhibitors of the chosen priming kinase could be incorporated into the assay. Unfortunately, there are currently no ERK inhibitors that are active in vitro.
The David Gene Functional Classification tool (david.abcc.ncifcrf.gov) identified transcriptional regulation as the dominant (P value, 2.2E−9) functional classification for the proteins in Table 1 that scored as ERK-primed GSK-3β substrates. We followed up on two proteins in this category, SMAD4 and iASPP. GSK-3 is known to phosphorylate SMAD3 at Ser-204 in the SMAD3 linker region, an event that decreases the transcriptional activity of SMAD3 (84), and Thr-66 outside the linker region, which leads to SMAD3 degradation (85). However, TGF-β signaling is reduced in PEL cells by LANA-mediated epigenetic silencing of the TGF-β type II receptor (36), reducing the likelihood that GSK-3 would impact TGF-β downstream signaling. GSK-3 sites in the SMAD1/5/8 proteins can be phosphorylated by GSK3 after priming by MAPK. Phosphorylation of the SMAD1 linker by ERK and GSK3 induces proteasomal degradation of SMAD1, leads to increased cytoplasmic retention of SMAD1, and regulates the duration of SMAD1-mediated signaling (86, 87). LANA binds activated SMAD1 and enhances SMAD1 upregulation of Id1 transcription, and inhibition of BMP-mediated SMAD1 phosphorylation with Dorsomorphin inhibited the growth of KSHV-transformed mesenchymal precursor in soft agar and inhibited tumor growth in nude mice (88). SMAD4 partners with the receptor-regulated SMADs to form the transcriptionally active complex, and a change in SMAD4 stability could therefore impact SMAD1 induced signaling. However, we saw no reproducible difference in SMAD4 protein stability after GSK-3 phosphorylation. It remains possible that GSK-3 phosphorylation of SMAD4 may have some other regulatory consequence.
iASPP binds p53 and the p53 family members p63 and p73 and prevents their activation of apoptotic genes while not affecting cell cycle regulatory responses (70, 89–91). p53 is functionally inactivated in PEL cells by LANA, but this inactivation can be overcome by targeting MDM2 degradation of p53. The MDM2 inhibitor Nutlin-3 induces PEL cell death, with p53 wild-type cells being more sensitive than PEL cells harboring p53 mutations. Reactivation of p53 function is an attractive anticancer strategy, and several drugs targeting the p53-MDM2 interaction are under development (92). Nutlin-3 treatment produces tumor regression in a mouse xenograft model of PEL, but in advanced tumors the effect was limited by induction of the KSHV lytic cycle, which led to an impairment in p53-mediated apoptotic responses (79). We saw increased PEL cell death when Nutlin-3 was combined with an anti-iASPP drug treatment. iASPP specifically blocks p53 apoptotic responses, and thus the identification of an additional mechanism that could reinforce MDM2 disruption and p53 activation in PEL cells may have therapeutic relevance for treatment of KSHV-associated disease.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jun Seop Jeong for creating the microarray printing program and Jae Jung for TREx-BCBL1 cells.
This work was supported in part by Public Health Services grants R21CA171814 to S.D.H. and R01GM076102 to H.Z. A UNCF-Merck Postdoctoral Fellowship, the Burroughs Wellcome Fund, and NREF supported C.R.G.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01072-15.
REFERENCES
- 1.Ye FC, Zhou FC, Yoo SM, Xie JP, Browning PJ, Gao SJ. 2004. Disruption of Kaposi's sarcoma-associated herpesvirus latent nuclear antigen leads to abortive episome persistence. J Virol 78:11121–11129. doi: 10.1128/JVI.78.20.11121-11129.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ballestas ME, Kaye KM. 2011. The latency-associated nuclear antigen, a multifunctional protein central to Kaposi's sarcoma-associated herpesvirus latency. Future Microbiol 6:1399–1413. doi: 10.2217/fmb.11.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu J, Garber AC, Renne R. 2002. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J Virol 76:11677–11687. doi: 10.1128/JVI.76.22.11677-11687.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ballestas ME, Kaye KM. 2001. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J Virol 75:3250–3258. doi: 10.1128/JVI.75.7.3250-3258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dheekollu J, Chen HS, Kaye KM, Lieberman PM. 2013. Timeless-dependent DNA replication-coupled recombination promotes Kaposi's sarcoma-associated herpesvirus episome maintenance and terminal repeat stability. J Virol 87:3699–3709. doi: 10.1128/JVI.02211-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun Q, Tsurimoto T, Juillard F, Li L, Li S, De Leon Vazquez E, Chen S, Kaye K. 2014. Kaposi's sarcoma-associated herpesvirus LANA recruits the DNA polymerase clamp loader to mediate efficient replication and virus persistence. Proc Natl Acad Sci U S A 111:11816–11821. doi: 10.1073/pnas.1404219111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Purushothaman P, McDowell ME, McGuinness J, Salas R, Rumjahn SM, Verma SC. 2012. Kaposi's sarcoma-associated herpesvirus-encoded LANA recruits topoisomerase IIbeta for latent DNA replication of the terminal repeats. J Virol 86:9983–9994. doi: 10.1128/JVI.00839-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shamay M, Liu J, Li R, Liao G, Shen L, Greenway M, Hu S, Zhu J, Xie Z, Ambinder RF, Qian J, Zhu H, Hayward SD. 2012. A protein array screen for Kaposi's sarcoma-associated herpesvirus LANA interactors links LANA to TIP60, PP2A activity, and telomere shortening. J Virol 86:5179–5191. doi: 10.1128/JVI.00169-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu J, Liu E, Renne R. 2009. Involvement of SSRP1 in latent replication of Kaposi's sarcoma-associated herpesvirus. J Virol 83:11051–11063. doi: 10.1128/JVI.00907-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stedman W, Deng Z, Lu F, Lieberman PM. 2004. ORC, MCM, and histone hyperacetylation at the Kaposi's sarcoma-associated herpesvirus latent replication origin. J Virol 78:12566–12575. doi: 10.1128/JVI.78.22.12566-12575.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verma SC, Choudhuri T, Kaul R, Robertson ES. 2006. Latency-associated nuclear antigen (LANA) of Kaposi's sarcoma-associated herpesvirus interacts with origin recognition complexes at the LANA binding sequence within the terminal repeats. J Virol 80:2243–2256. doi: 10.1128/JVI.80.5.2243-2256.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piolot T, Tramier M, Coppey M, Nicolas JC, Marechal V. 2001. Close but distinct regions of human herpesvirus 8 latency-associated nuclear antigen 1 are responsible for nuclear targeting and binding to human mitotic chromosomes. J Virol 75:3948–3959. doi: 10.1128/JVI.75.8.3948-3959.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cotter MA II, Robertson ES. 1999. The latency-associated nuclear antigen tethers the Kaposi's sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 264:254–264. doi: 10.1006/viro.1999.9999. [DOI] [PubMed] [Google Scholar]
- 14.Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, Kaye KM. 2006. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311:856–861. doi: 10.1126/science.1120541. [DOI] [PubMed] [Google Scholar]
- 15.Kelley-Clarke B, De Leon-Vazquez E, Slain K, Barbera AJ, Kaye KM. 2009. Role of Kaposi's sarcoma-associated herpesvirus C-terminal LANA chromosome binding in episome persistence. J Virol 83:4326–4337. doi: 10.1128/JVI.02395-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.You J, Srinivasan V, Denis GV, Harrington WJ Jr, Ballestas ME, Kaye KM, Howley PM. 2006. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen interacts with bromodomain protein Brd4 on host mitotic chromosomes. J Virol 80:8909–8919. doi: 10.1128/JVI.00502-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krithivas A, Fujimuro M, Weidner M, Young DB, Hayward SD. 2002. Protein interactions targeting the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus to cell chromosomes. J Virol 76:11596–11604. doi: 10.1128/JVI.76.22.11596-11604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsumura S, Persson LM, Wong L, Wilson AC. 2010. The latency-associated nuclear antigen interacts with MeCP2 and nucleosomes through separate domains. J Virol 84:2318–2330. doi: 10.1128/JVI.01097-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hellert J, Weidner-Glunde M, Krausze J, Richter U, Adler H, Fedorov R, Pietrek M, Ruckert J, Ritter C, Schulz TF, Luhrs T. 2013. A structural basis for BRD2/4-mediated host chromatin interaction and oligomer assembly of Kaposi sarcoma-associated herpesvirus and murine gammaherpesvirus LANA proteins. PLoS Pathog 9:e1003640. doi: 10.1371/journal.ppat.1003640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Domsic JF, Chen HS, Lu F, Marmorstein R, Lieberman PM. 2013. Molecular basis for oligomeric-DNA binding and episome maintenance by KSHV LANA. PLoS Pathog 9:e1003672. doi: 10.1371/journal.ppat.1003672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Correia B, Cerqueira SA, Beauchemin C, Pires de Miranda M, Li S, Ponnusamy R, Rodrigues L, Schneider TR, Carrondo MA, Kaye KM, Simas JP, McVey CE. 2013. Crystal structure of the gamma-2 herpesvirus LANA DNA binding domain identifies charged surface residues which impact viral latency. PLoS Pathog 9:e1003673. doi: 10.1371/journal.ppat.1003673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong YK, Foreman K, Shin JW, Hirakawa S, Curry CL, Sage DR, Libermann T, Dezube BJ, Fingeroth JD, Detmar M. 2004. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat Genet 36:683–685. doi: 10.1038/ng1383. [DOI] [PubMed] [Google Scholar]
- 23.Moses AV, Jarvis MA, Raggo C, Bell YC, Ruhl R, Luukkonen BG, Griffith DJ, Wait CL, Druker BJ, Heinrich MC, Nelson JA, Fruh K. 2002. Kaposi's sarcoma-associated herpesvirus-induced upregulation of the c-kit proto-oncogene, as identified by gene expression profiling, is essential for the transformation of endothelial cells. J Virol 76:8383–8399. doi: 10.1128/JVI.76.16.8383-8399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang HW, Trotter MW, Lagos D, Bourboulia D, Henderson S, Makinen T, Elliman S, Flanagan AM, Alitalo K, Boshoff C. 2004. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat Genet 36:687–693. doi: 10.1038/ng1384. [DOI] [PubMed] [Google Scholar]
- 25.Poole LJ, Yu Y, Kim PS, Zheng QZ, Pevsner J, Hayward GS. 2002. Altered patterns of cellular gene expression in dermal microvascular endothelial cells infected with Kaposi's sarcoma-associated herpesvirus. J Virol 76:3395–3420. doi: 10.1128/JVI.76.7.3395-3420.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoo SM, Zhou FC, Ye FC, Pan HY, Gao SJ. 2005. Early and sustained expression of latent and host modulating genes in coordinated transcriptional program of KSHV productive primary infection of human primary endothelial cells. Virology 343:47–64. doi: 10.1016/j.virol.2005.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma-Walia N, Naranatt PP, Krishnan HH, Zeng L, Chandran B. 2004. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-phosphatidylinositol 3-kinase-rho GTPase signal pathways and cytoskeletal rearrangements. J Virol 78:4207–4223. doi: 10.1128/JVI.78.8.4207-4223.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma-Walia N, Krishnan HH, Naranatt PP, Zeng L, Smith MS, Chandran B. 2005. ERK1/2 and MEK1/2 induced by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J Virol 79:10308–10329. doi: 10.1128/JVI.79.16.10308-10329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh VV, Dutta D, Ansari MA, Dutta S, Chandran B. 2014. Kaposi's sarcoma-associated herpesvirus induces the ATM and H2AX DNA damage response early during de novo infection of primary endothelial cells, which play roles in latency establishment. J Virol 88:2821–2834. doi: 10.1128/JVI.03126-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naranatt PP, Krishnan HH, Svojanovsky SR, Bloomer C, Mathur S, Chandran B. 2004. Host gene induction and transcriptional reprogramming in Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: insights into modulation events early during infection. Cancer Res 64:72–84. doi: 10.1158/0008-5472.CAN-03-2767. [DOI] [PubMed] [Google Scholar]
- 31.George Paul A, Sharma-Walia N, Kerur N, White C, Chandran B. 2010. Piracy of prostaglandin E2/EP receptor-mediated signaling by Kaposi's sarcoma-associated herpes virus (HHV-8) for latency gene expression: strategy of a successful pathogen. Cancer Res 70:3697–3708. doi: 10.1158/0008-5472.CAN-09-3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.An FQ, Compitello N, Horwitz E, Sramkoski M, Knudsen ES, Renne R. 2005. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus modulates cellular gene expression and protects lymphoid cells from p16 INK4A-induced cell cycle arrest. J Biol Chem 280:3862–3874. doi: 10.1074/jbc.M407435200. [DOI] [PubMed] [Google Scholar]
- 33.Lu F, Tsai K, Chen HS, Wikramasinghe P, Davuluri RV, Showe L, Domsic J, Marmorstein R, Lieberman PM. 2012. Identification of host-chromosome binding sites and candidate gene targets for Kaposi's sarcoma-associated herpesvirus LANA. J Virol 86:5752–5762. doi: 10.1128/JVI.07216-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu J, Yang Y, Turner PC, Jain V, McIntyre LM, Renne R. 2014. LANA binds to multiple active viral and cellular promoters and associates with the H3K4methyltransferase hSET1 complex. PLoS Pathog 10:e1004240. doi: 10.1371/journal.ppat.1004240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mercier A, Arias C, Madrid AS, Holdorf MM, Ganem D. 2014. Site-specific association with host and viral chromatin by Kaposi's sarcoma-associated herpesvirus LANA and its reversal during lytic reactivation. J Virol 88:6762–6777. doi: 10.1128/JVI.00268-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Bartolo DL, Cannon M, Liu YF, Renne R, Chadburn A, Boshoff C, Cesarman E. 2008. KSHV LANA inhibits TGF-beta signaling through epigenetic silencing of the TGF-beta type II receptor. Blood 111:4731–4740. doi: 10.1182/blood-2007-09-110544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shamay M, Krithivas A, Zhang J, Hayward SD. 2006. Recruitment of the de novo DNA methyltransferase Dnmt3a by Kaposi's sarcoma-associated herpesvirus LANA. Proc Natl Acad Sci U S A 103:14554–14559. doi: 10.1073/pnas.0604469103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim KY, Huerta SB, Izumiya C, Wang DH, Martinez A, Shevchenko B, Kung HJ, Campbell M, Izumiya Y. 2013. Kaposi's sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen regulates the KSHV epigenome by association with the histone demethylase KDM3A. J Virol 87:6782–6793. doi: 10.1128/JVI.00011-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gunther T, Schreiner S, Dobner T, Tessmer U, Grundhoff A. 2014. Influence of ND10 components on epigenetic determinants of early KSHV latency establishment. PLoS Pathog 10:e1004274. doi: 10.1371/journal.ppat.1004274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.An J, Lichtenstein AK, Brent G, Rettig MB. 2002. The Kaposi sarcoma-associated herpesvirus (KSHV) induces cellular interleukin 6 expression: role of the KSHV latency-associated nuclear antigen and the AP1 response element. Blood 99:649–654. doi: 10.1182/blood.V99.2.649. [DOI] [PubMed] [Google Scholar]
- 41.An J, Sun Y, Rettig MB. 2004. Transcriptional coactivation of c-Jun by the KSHV-encoded LANA. Blood 103:222–228. doi: 10.1182/blood-2003-05-1538. [DOI] [PubMed] [Google Scholar]
- 42.Fujimuro M, Wu FY, ApRhys C, Kajumbula H, Young DB, Hayward GS, Hayward SD. 2003. A novel viral mechanism for dysregulation of beta-catenin in Kaposi's sarcoma-associated herpesvirus latency. Nat Med 9:300–306. doi: 10.1038/nm829. [DOI] [PubMed] [Google Scholar]
- 43.Lim C, Gwack Y, Hwang S, Kim S, Choe J. 2001. The transcriptional activity of cAMP response element-binding protein-binding protein is modulated by the latency associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J Biol Chem 276:31016–31022. doi: 10.1074/jbc.M102431200. [DOI] [PubMed] [Google Scholar]
- 44.Liu J, Martin HJ, Liao G, Hayward SD. 2007. The Kaposi's sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J Virol 81:10451–10459. doi: 10.1128/JVI.00804-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murakami Y, Yamagoe S, Noguchi K, Takebe Y, Takahashi N, Uehara Y, Fukazawa H. 2006. Ets-1-dependent expression of vascular endothelial growth factor receptors is activated by latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus through interaction with Daxx. J Biol Chem 281:28113–28121. doi: 10.1074/jbc.M602026200. [DOI] [PubMed] [Google Scholar]
- 46.Verma SC, Borah S, Robertson ES. 2004. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J Virol 78:10348–10359. doi: 10.1128/JVI.78.19.10348-10359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cloutier N, Flamand L. 2010. Kaposi sarcoma-associated herpesvirus latency-associated nuclear antigen inhibits interferon (IFN) beta expression by competing with IFN regulatory factor-3 for binding to IFNB promoter. J Biol Chem 285:7208–7221. doi: 10.1074/jbc.M109.018838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ottinger M, Christalla T, Nathan K, Brinkmann MM, Viejo-Borbolla A, Schulz TF. 2006. Kaposi's sarcoma-associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J Virol 80:10772–10786. doi: 10.1128/JVI.00804-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar A, Sahu SK, Mohanty S, Chakrabarti S, Maji S, Reddy RR, Jha AK, Goswami C, Kundu CN, Rajasubramaniam S, Verma SC, Choudhuri T. 2014. Kaposi sarcoma herpes virus latency associated nuclear antigen protein release the G2/M cell cycle blocks by modulating ATM/ATR mediated checkpoint pathway. PLoS One 9:e100228. doi: 10.1371/journal.pone.0100228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bajaj BG, Verma SC, Lan K, Cotter MA, Woodman ZL, Robertson ES. 2006. KSHV encoded LANA upregulates Pim-1 and is a substrate for its kinase activity. Virology 351:18–28. doi: 10.1016/j.virol.2006.03.037. [DOI] [PubMed] [Google Scholar]
- 51.Fujimuro M, Hayward SD. 2004. Manipulation of glycogen-synthase kinase-3 activity in KSHV-associated cancers. J Mol Med 82:223–231. doi: 10.1007/s00109-003-0519-7. [DOI] [PubMed] [Google Scholar]
- 52.Sun Z, Xiao B, Jha HC, Lu J, Banerjee S, Robertson ES. 2014. Kaposi's sarcoma-associated herpesvirus-encoded LANA can induce chromosomal instability through targeted degradation of the mitotic checkpoint kinase Bub1. J Virol 88:7367–7378. doi: 10.1128/JVI.00554-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cai Q, Xiao B, Si H, Cervini A, Gao J, Lu J, Upadhyay SK, Verma SC, Robertson ES. 2012. Kaposi's sarcoma herpesvirus upregulates Aurora A expression to promote p53 phosphorylation and ubiquitylation. PLoS Pathog 8:e1002566. doi: 10.1371/journal.ppat.1002566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roupelieva M, Griffiths SJ, Kremmer E, Meisterernst M, Viejo-Borbolla A, Schulz T, Haas J. 2010. Kaposi's sarcoma-associated herpesvirus Lana-1 is a major activator of the serum response element and mitogen-activated protein kinase pathways via interactions with the Mediator complex. J Gen Virol 91:1138–1149. doi: 10.1099/vir.0.017715-0. [DOI] [PubMed] [Google Scholar]
- 55.McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL, Montalto G, D'Assoro AB, Libra M, Nicoletti F, Maestro R, Basecke J, Rakus D, Gizak A, Demidenko ZN, Cocco L, Martelli AM, Cervello M. 2014. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 5:2881–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fujimuro M, Hayward SD. 2003. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus manipulates the activity of glycogen synthase kinase-3beta. J Virol 77:8019–8030. doi: 10.1128/JVI.77.14.8019-8030.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu J, Martin H, Shamay M, Woodard C, Tang QQ, Hayward SD. 2007. Kaposi's sarcoma-associated herpesvirus LANA protein downregulates nuclear glycogen synthase kinase 3 activity and consequently blocks differentiation. J Virol 81:4722–4731. doi: 10.1128/JVI.02548-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bubman D, Guasparri I, Cesarman E. 2007. Deregulation of c-Myc in primary effusion lymphoma by Kaposi's sarcoma herpesvirus latency-associated nuclear antigen. Oncogene 26:4979–4986. doi: 10.1038/sj.onc.1210299. [DOI] [PubMed] [Google Scholar]
- 59.Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, Lan N, Jansen R, Bidlingmaier S, Houfek T, Mitchell T, Miller P, Dean RA, Gerstein M, Snyder M. 2001. Global analysis of protein activities using proteome chips. Science 293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- 60.Hu S, Xie Z, Onishi A, Yu X, Jiang L, Lin J, Rho HS, Woodard C, Wang H, Jeong JS, Long S, He X, Wade H, Blackshaw S, Qian J, Zhu H. 2009. Profiling the human protein-DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell 139:610–622. doi: 10.1016/j.cell.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woodard CL, Goodwin CR, Wan J, Xia S, Newman R, Hu J, Zhang J, Hayward SD, Qian J, Laterra J, Zhu H. 2013. Profiling the dynamics of a human phosphorylome reveals new components in HGF/c-Met signaling. PLoS One 8:e72671. doi: 10.1371/journal.pone.0072671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Newman RH, Hu J, Rho HS, Xie Z, Woodard C, Neiswinger J, Cooper C, Shirley M, Clark HM, Hu S, Hwang W, Jeong JS, Wu G, Lin J, Gao X, Ni Q, Goel R, Xia S, Ji H, Dalby KN, Birnbaum MJ, Cole PA, Knapp S, Ryazanov AG, Zack DJ, Blackshaw S, Pawson T, Gingras AC, Desiderio S, Pandey A, Turk BE, Zhang J, Zhu H, Qian J. 2013. Construction of human activity-based phosphorylation networks. Mol Syst Biol 9:655. doi: 10.1038/msb.2013.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Woodard C, Shamay M, Liao G, Zhu J, Ng AN, Li R, Newman R, Rho HS, Hu J, Wan J, Qian J, Zhu H, Hayward SD. 2012. Phosphorylation of the chromatin binding domain of KSHV LANA. PLoS Pathog 8:e1002972. doi: 10.1371/journal.ppat.1002972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li R, Zhu J, Xie Z, Liao G, Liu J, Chen MR, Hu S, Woodard C, Lin J, Taverna SD, Desai P, Ambinder RF, Hayward GS, Qian J, Zhu H, Hayward SD. 2011. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe 10:390–400. doi: 10.1016/j.chom.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu J, Liao G, Shan L, Zhang J, Chen MR, Hayward GS, Hayward SD, Desai P, Zhu H. 2009. Protein array identification of substrates of the Epstein-Barr virus protein kinase BGLF4. J Virol 83:5219–5231. doi: 10.1128/JVI.02378-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gaarenstroom T, Hill CS. 2014. TGF-beta signaling to chromatin: how Smads regulate transcription during self-renewal and differentiation. Semin Cell Dev Biol 32:107–118. doi: 10.1016/j.semcdb.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 67.Maitra A, Hruban RH. 2008. Pancreatic cancer. Annu Rev Pathol 3:157–188. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reid MD, Saka B, Balci S, Goldblum AS, Adsay NV. 2014. Molecular genetics of pancreatic neoplasms and their morphologic correlates: an update on recent advances and potential diagnostic applications. Am J Clin Pathol 141:168–180. doi: 10.1309/AJCP0FKDP7ENVKEV. [DOI] [PubMed] [Google Scholar]
- 69.Kruse JP, Gu W. 2009. Modes of p53 regulation. Cell 137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Trigiante G, Lu X. 2006. ASPP [corrected] and cancer. Nat Rev Cancer 6:217–226. doi: 10.1038/nrc1818. [DOI] [PubMed] [Google Scholar]
- 71.Bergamaschi D, Samuels Y, O'Neil NJ, Trigiante G, Crook T, Hsieh JK, O'Connor DJ, Zhong S, Campargue I, Tomlinson ML, Kuwabara PE, Lu X. 2003. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet 33:162–167. doi: 10.1038/ng1070. [DOI] [PubMed] [Google Scholar]
- 72.Samuels-Lev Y, O'Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, Campargue I, Naumovski L, Crook T, Lu X. 2001. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell 8:781–794. doi: 10.1016/S1097-2765(01)00367-7. [DOI] [PubMed] [Google Scholar]
- 73.Zhang X, Diao S, Rao Q, Xing H, Liu H, Liao X, Wang M, Wang J. 2007. Identification of a novel isoform of iASPP and its interaction with p53. J Mol Biol 368:1162–1171. doi: 10.1016/j.jmb.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 74.Slee EA, Gillotin S, Bergamaschi D, Royer C, Llanos S, Ali S, Jin B, Trigiante G, Lu X. 2004. The N-terminus of a novel isoform of human iASPP is required for its cytoplasmic localization. Oncogene 23:9007–9016. doi: 10.1038/sj.onc.1208088. [DOI] [PubMed] [Google Scholar]
- 75.Liu Z, Zhang X, Huang D, Liu Y, Zhang X, Liu L, Li G, Dai Y, Tan H, Xiao J, Tian Y. 2012. Elevated expression of iASPP in head and neck squamous cell carcinoma and its clinical significance. Med Oncol 29:3381–3388. doi: 10.1007/s12032-012-0306-9. [DOI] [PubMed] [Google Scholar]
- 76.Zhang X, Wang M, Zhou C, Chen S, Wang J. 2005. The expression of iASPP in acute leukemias. Leuk Res 29:179–183. doi: 10.1016/j.leukres.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 77.Santag S, Jager W, Karsten CB, Kati S, Pietrek M, Steinemann D, Sarek G, Ojala PM, Schulz TF. 2013. Recruitment of the tumour suppressor protein p73 by Kaposi's sarcoma herpesvirus latent nuclear antigen contributes to the survival of primary effusion lymphoma cells. Oncogene 32:3676–3685. doi: 10.1038/onc.2012.385. [DOI] [PubMed] [Google Scholar]
- 78.Petre CE, Sin SH, Dittmer DP. 2007. Functional p53 signaling in Kaposi's sarcoma-associated herpesvirus lymphomas: implications for therapy. J Virol 81:1912–1922. doi: 10.1128/JVI.01757-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sarek G, Kurki S, Enback J, Iotzova G, Haas J, Laakkonen P, Laiho M, Ojala PM. 2007. Reactivation of the p53 pathway as a treatment modality for KSHV-induced lymphomas. J Clin Invest 117:1019–1028. doi: 10.1172/JCI30945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lu M, Breyssens H, Salter V, Zhong S, Hu Y, Baer C, Ratnayaka I, Sullivan A, Brown NR, Endicott J, Knapp S, Kessler BM, Middleton MR, Siebold C, Jones EY, Sviderskaya EV, Cebon J, John T, Caballero OL, Goding CR, Lu X. 2013. Restoring p53 function in human melanoma cells by inhibiting MDM2 and cyclin B1/CDK1-phosphorylated nuclear iASPP. Cancer Cell 23:618–633. doi: 10.1016/j.ccr.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 81.Li X, Chen S, Sun R. 2012. Cdk1 inhibition induces mutually inhibitory apoptosis and reactivation of Kaposi's sarcoma-associated herpesvirus. J Virol 86:6668–6676. doi: 10.1128/JVI.06240-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Emanuel S, Rugg CA, Gruninger RH, Lin R, Fuentes-Pesquera A, Connolly PJ, Wetter SK, Hollister B, Kruger WW, Napier C, Jolliffe L, Middleton SA. 2005. The in vitro and in vivo effects of JNJ-7706621: a dual inhibitor of cyclin-dependent kinases and aurora kinases. Cancer Res 65:9038–9046. doi: 10.1158/0008-5472.CAN-05-0882. [DOI] [PubMed] [Google Scholar]
- 83.Sutandy FX, Qian J, Chen CS, Zhu H. 2013. Overview of protein microarrays. Curr Protoc Protein Sci Chapter 27:Unit 27 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Millet C, Yamashita M, Heller M, Yu LR, Veenstra TD, Zhang YE. 2009. A negative feedback control of transforming growth factor-beta signaling by glycogen synthase kinase 3-mediated Smad3 linker phosphorylation at Ser-204. J Biol Chem 284:19808–19816. doi: 10.1074/jbc.M109.016667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guo X, Ramirez A, Waddell DS, Li Z, Liu X, Wang XF. 2008. Axin and GSK3-β control Smad3 protein stability and modulate TGF-β signaling. Genes Dev 22:106–120. doi: 10.1101/gad.1590908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fuentealba LC, Eivers E, Ikeda A, Hurtado C, Kuroda H, Pera EM, De Robertis EM. 2007. Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 131:980–993. doi: 10.1016/j.cell.2007.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sapkota G, Alarcon C, Spagnoli FM, Brivanlou AH, Massague J. 2007. Balancing BMP signaling through integrated inputs into the Smad1 linker. Mol Cell 25:441–454. doi: 10.1016/j.molcel.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 88.Liang D, Hu H, Li S, Dong J, Wang X, Wang Y, He L, He Z, Gao Y, Gao SJ, Lan K. 2014. Oncogenic herpesvirus KSHV hijacks BMP-Smad1-Id signaling to promote tumorigenesis. PLoS Pathog 10:e1004253. doi: 10.1371/journal.ppat.1004253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bell HS, Ryan KM. 2008. iASPP inhibition: increased options in targeting the p53 family for cancer therapy. Cancer Res 68:4959–4962. doi: 10.1158/0008-5472.CAN-08-0182. [DOI] [PubMed] [Google Scholar]
- 90.Notari M, Hu Y, Koch S, Lu M, Ratnayaka I, Zhong S, Baer C, Pagotto A, Goldin R, Salter V, Candi E, Melino G, Lu X. 2011. Inhibitor of apoptosis-stimulating protein of p53 (iASPP) prevents senescence and is required for epithelial stratification. Proc Natl Acad Sci U S A 108:16645–16650. doi: 10.1073/pnas.1102292108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cai Y, Qiu S, Gao X, Gu SZ, Liu ZJ. 2012. iASPP inhibits p53-independent apoptosis by inhibiting transcriptional activity of p63/p73 on promoters of proapoptotic genes. Apoptosis 17:777–783. doi: 10.1007/s10495-012-0728-z. [DOI] [PubMed] [Google Scholar]
- 92.Zawacka-Pankau J, Selivanova G. 2015. Pharmacological reactivation of p53 as a strategy to treat cancer. J Intern Med 277:248–259. doi: 10.1111/joim.12336. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.