

Abstract
Obstructions to replication fork progression, referred to collectively as DNA replication stress, challenge genome stability. In Saccharomyces cerevisiae, cells lacking RTT107 or SLX4 show genome instability and sensitivity to DNA replication stress and are defective in the completion of DNA replication during recovery from replication stress. We demonstrate that Slx4 is recruited to chromatin behind stressed replication forks, in a region that is spatially distinct from that occupied by the replication machinery. Slx4 complex formation is nucleated by Mec1 phosphorylation of histone H2A, which is recognized by the constitutive Slx4 binding partner Rtt107. Slx4 is essential for recruiting the Mec1 activator Dpb11 behind stressed replication forks, and Slx4 complexes are important for full activity of Mec1. We propose that Slx4 complexes promote robust checkpoint signaling by Mec1 by stably recruiting Dpb11 within a discrete domain behind the replication fork, during DNA replication stress.
Keywords: checkpoint kinase, replication stress, DNA damage response, Dpb11, Slx4
Introduction
DNA replication errors are a major source of genome instability and a driving force of cancer (Hanahan & Weinberg, 2000, 2011), aging (Lopez-Otin et al, 2013), and other human diseases. A variety of obstructions to DNA replication fork progression, collectively known as DNA replication stress, challenge the accuracy and timely completion of DNA replication. These challenges include DNA secondary structures and repetitive DNA, strong protein–DNA interactions, transcription complexes, DNA lesions, inhibition of DNA polymerases, and depletion of dNTPs (Zeman & Cimprich, 2014). DNA replication stress exposes stretches of single-stranded DNA (ssDNA) at stalled replication forks and this is the primary signal for activation of the S phase checkpoint (Costanzo et al, 2003; Zou & Elledge, 2003; Byun et al, 2005), an evolutionarily conserved signal transduction cascade that facilitates the completion of DNA replication and delays cell cycle progression.
Checkpoint activation is initiated by the localization of several proteins to stalled replication forks. In Saccharomyces cerevisiae, the central checkpoint kinase Mec1 (human ATR) is recruited by physical interaction of the Mec1 binding partner Ddc2 with replication protein A (RPA)-coated ssDNA (Zou & Elledge, 2003). RPA-coated ssDNA also recruits the Rad24 clamp loader to 5′ ssDNA-dsDNA junctions, where it loads the 9-1-1 clamp (composed of Rad17-Mec3-Ddc1 in budding yeast) onto DNA (Zou et al, 2003; Majka et al, 2006a). The Ddc1 subunit of 9-1-1 promotes Mec1 activity both directly and by binding to another Mec1 activator, Dpb11 (human TopBP1), following Ddc1 phosphorylation by Mec1 (Wang & Elledge, 2002; Majka et al, 2006b; Mordes et al, 2008; Navadgi-Patil & Burgers, 2008, 2009b; Puddu et al, 2008). In the presence of its activators, Mec1 orchestrates a cellular response that regulates replisome function to prevent defective replication and fork collapse (Tercero & Diffley, 2001; Cobb et al, 2003, 2005; Lou et al, 2008; De Piccoli et al, 2012) and regulates replication origin firing, DNA repair, fork restart, and cell cycle progression [reviewed in Cimprich & Cortez (2008) and Friedel et al (2009)].
Early during the checkpoint response, Mec1 phosphorylates numerous substrates at stalled replication forks including checkpoint sensors like RPA (Brush et al, 1996; Brush & Kelly, 2000) and Ddc1 (Majka et al, 2006b; Navadgi-Patil & Burgers, 2009b), DNA replication proteins such as Psf1 (De Piccoli et al, 2012), and Ser129 of histone H2A (Downs et al, 2000; Redon et al, 2003; Cobb et al, 2005). Phosphorylation of Ddc1 recruits the Mec1 activator Dpb11 (Furuya et al, 2004; Puddu et al, 2008), serving to amplify the Mec1 signal (Mordes et al, 2008), and Mec1 phosphorylation of histone H2A promotes recruitment of the checkpoint mediator protein Rad9 (Nakamura et al, 2004; Toh et al, 2006; Hammet et al, 2007). Thus, the role of Mec1 is the local detection and signaling of replication stress and DNA damage. Downstream of Mec1, the major effector kinase Rad53 (human CHK2) is recruited by Rad9, which functions redundantly with Mrc1 to promote Rad53 phosphorylation and activation (Navas et al, 1996; Emili, 1998; Sun et al, 1998; Vialard et al, 1998; Alcasabas et al, 2001; Gilbert et al, 2001; Osborn & Elledge, 2003; Sweeney et al, 2005). Activated Rad53 functions globally in late origin inhibition, DNA damage-dependent gene transcription, and cell cycle signaling [reviewed in Branzei & Foiani (2006) and Segurado & Tercero (2009)].
The roles of other Mec1 targets are not as well understood. The multi-BRCT (BRCA-1 C-Terminal homology) domain protein Rtt107 and its binding partner Slx4 are both Mec1 targets during replication stress (Rouse, 2004; Flott & Rouse, 2005; Roberts et al, 2006). Phosphorylated Slx4 competes with Rad9 for binding to the amino-terminal BRCT pair of Dpb11, resulting in decreased Rad53 signaling (Ohouo et al, 2013; Cussiol et al, 2015). Here, we define the steps in assembly of protein complexes containing Rtt107 and Slx4 on chromatin during DNA replication stress and demonstrate that Slx4 complexes accumulate behind the replisome. We find that Slx4 is recruited with Rtt107, which itself is recruited via a BRCT-dependent interaction with Mec1-phosphorylated histone H2A. Rtt107 binding is essential for Slx4 function in promoting the completion of DNA synthesis during replication stress. Slx4, in concert with the checkpoint clamp subunit Ddc1, is critical for stable recruitment of Dpb11, which in turn promotes full activation of Mec1 and phosphorylation of Mec1 targets behind the stressed replication forks. Together, our data indicate that formation of Slx4 complexes during replication stress promotes checkpoint signaling by Mec1 and does so in a discrete domain distal to the stressed replication forks.
Results
Slx4 accumulates on chromatin distal to stressed DNA replication forks
Slx4 is required for normal DNA synthesis after DNA replication stress caused by the alkylating agent methyl methanesulfonate (MMS) (Roberts et al, 2006; Flott et al, 2007), and Slx4 forms nuclear foci typical of DNA damage response proteins during replication stress (Tkach et al, 2012). To better characterize Slx4 focus formation, we imaged cells expressing Slx4-GFP from the SLX4 locus in mid-logarithmic phase (Fig1A and B). Slx4 foci were absent in most unbudded (G1 phase) cells, but were abundant in cells with a small bud (a morphology that is typical of S phase). Slx4 foci decreased in large budded cells, and decreased further following anaphase, suggesting that Slx4 is recruited to foci during S phase and that the signals for Slx4 recruitment are reduced in G2/M. When cells were released synchronously into S phase, formation of Slx4 foci was evident in 67% of cells, and rapidly decreased as cells progressed into G2 (Fig1C and D), consistent with Slx4 foci forming during S phase. When mid-logarithmic phase cultures were treated with MMS, the fraction of small-budded cells with one or more Slx4 focus increased from 88% to 98%, and the fraction of small-budded cells with three or more Slx4 foci increased from 25% to 59% (Fig1B). Together, these data suggest that Slx4 is recruited to nuclear foci during S phase and that Slx4 focus formation is stimulated by the presence of replication stress induced by MMS. These data further suggest that Slx4 might function directly at replication forks during DNA replication stress, either endogenous or induced by MMS.
Figure 1.

Slx4 is recruited behind replication forks during MMS-induced DNA replication stress
- Intracellular localization of Slx4-GFP in logarithmic phase cells, either untreated or treated for 90 min with 0.035% MMS. Examples of cells with the indicated morphologies are shown. The nuclear periphery is marked with Nup49-RFP, the outline of the cells is indicated by the dashed line, and the scale bar indicates 5 μm.
- Slx4-GFP foci were quantified in logarithmic phase cells, either untreated or treated for 90 min with 0.035% MMS. Cells with each of the indicated morphologies were assessed, and the fraction of cells with each morphology that had 1, 2, or ≥ 3 foci is plotted. 477 (-MMS) and 619 (+MMS) cells were evaluated in the experiment shown, which is a representative of two replicates.
- Cells were arrested in G1 and released synchronously into the cell cycle. The fraction of cells with 1, 2, or ≥ 3 Slx4-GFP foci was quantified at the indicated times, and the average of three replicates is plotted.
- The DNA contents of cells from the samples in (C) were measured by flow cytometry and are plotted as histograms. The positions of 1C and 2C DNA contents are indicated.
- ChIP-seq analysis was performed following synchronous release of SLX4-FLAG cells into S phase in the presence of 0.035% MMS for 60 min. Slx4 ChIP enrichment scores on chromosome 10 are shown. Early origins are indicated by green bars and late origins by red bars.
- The replication profile compares the relative copy number of DNA sequences in the input sample from the SLX4-FLAG cells released into S phase in the presence of 0.035% MMS for 60 min to a DNA sample prepared from G1-arrested wild-type cells. Copy number along chromosome 10 is plotted.
- The median (± standard error) Slx4 ChIP enrichment score and replication profile (Rep) across n = 108 early-firing origins, in wild-type cells, are plotted.
- The distributions of Slx4 ChIP enrichment scores at early- and late-firing origins genome-wide are shown as a boxplot. The median is indicated by the horizontal bar, the box spans the first through third quartiles, the whiskers extend to the last data points within 1.5 times the interquartile range, and outliers are plotted as circles. The distributions were compared using the Wilcoxon rank-sum test.
- ChIP-seq analysis was performed on DPB3-FLAG and SLX4-FLAG cells at 30, 60, and 90 min following synchronous release into S phase in the presence of 0.035% MMS. The median (± standard error) Dpb3 (left) and Slx4 (right) ChIP enrichment score across n = 108 early-firing origins are plotted.
We used chromatin immunoprecipitation coupled to deep sequencing (ChIP-seq) to assess Slx4 binding genome-wide during synchronous progression through S phase in the presence of MMS (Fig1E). The presence of MMS slows replication fork progression, facilitating detection of fork-associated proteins. Slx4 binds DNA sequences that are proximal to early-firing replication origins (Fig1E shows enrichment in the Slx4 chromatin immunoprecipitate along the length of chromosome 10). We extracted the enrichment values for 50 kb on either side of each of the 108 yeast replication origins that are known to fire in early S phase and plotted the median enrichment scores to produce a genome-wide view of enrichment at early origins in the Slx4 ChIP (Fig1G). The distributions of enrichments across early- and late-firing origins are shown in Fig1H, and indicate that Slx4 binds preferentially to early origin proximal sequences, relative to late origin proximal sequences. The median enrichment of each early origin shows a modest negative correlation (r = −0.26) with the time of origin activation (Yabuki et al, 2002), indicating that origins that fire earlier in S phase are more likely to show stronger enrichment in the Slx4 ChIP (Supplementary Fig S1A). We also mapped the coordinates of replicated DNA by measuring the increase in DNA copy number (Fig1F and G) and noted that Slx4 binding sites corresponded precisely to the positions of newly replicated DNA surrounding early origins (Fig1G).
To determine whether Slx4 binds at DNA replication forks during replication stress, we compared the chromosomal location of the DNA polymerase ε subunit Dpb3 to that of Slx4 during synchronous progression through S phase in the presence of MMS (Fig1I and Supplementary Fig S1B–D). At 30 min, Dpb3 formed a tight peak centered on early origins. By 60 min, Dpb3 binding had become distinctly bi-modal, consistent with bidirectional replication fork movement away from the early origins. After 90 min, Dpb3 peaks were further from the origins and little Dpb3 was detected directly at the early replication origins. We confirmed that the position of Dpb3 corresponds to the edges of the copy number peaks at each of the time points (Supplementary Fig S1C), indicating that Dpb3 is located at sites of DNA synthesis, as expected. By contrast to the Dpb3 pattern, Slx4 binding at early-firing origins was barely detected at 30 min despite DNA polymerase clearly being engaged near replication origins, and so association of Slx4 with chromatin is temporally distinct from association of polymerase. After 60 and 90 min of DNA replication stress, there was a dramatic increase in Slx4 binding, yet Slx4 remained centered on the replication origins and did not assume a bi-modal pattern. Therefore, Slx4 accumulates on chromatin in a region that is distinct from that occupied by the replication machinery, suggesting that Slx4 binds chromatin behind the DNA replication forks during MMS-induced replication stress.
Binding to Rtt107 is sufficient for Slx4 recruitment
Slx4 binds constitutively and directly to Rtt107 (Roberts et al, 2006), and several lines of evidence suggest that Slx4 and Rtt107 function in concert during DNA replication stress (Roberts et al, 2006; Ohouo et al, 2010, 2013). Slx4 also binds directly to Dpb11, in a phosphorylation-dependent manner (Ohouo et al, 2010, 2013; Gritenaite et al, 2014). We tested if Dpb11 or Rtt107 mediate Slx4 recruitment behind replication forks during replication stress (Fig2). We performed ChIP-seq analysis with Slx4-7MUT and Slx4-486A, both of which interact with Rtt107 but are compromised for Dpb11 interaction (Fig2A and B; Ohouo et al, 2010, 2013; Gritenaite et al, 2014). Both Slx4-7MUT and Slx4-486A give ChIP patterns that are indistinguishable from wild-type Slx4 (compare Fig2C and D to Fig1B). We conclude that the interaction between Slx4 and Dpb11 is not important for recruiting Slx4 behind stressed replication forks.
Figure 2.

Slx4 recruitment behind DNA replication forks requires physical interaction with Rtt107
- A, B Analysis of Slx4, Slx4-bd, Slx4-7MUT, and Slx4-486A binding to Rtt107 (A) and Dpb11 (B) by co-immunoprecipitation. The input extract and the anti-Flag immunoprecipitates are shown. Immunoblots were probed with anti-HA or anti-Flag antibodies, as indicated. Binding to Dpb11 was tested with and without treatment of asynchronous cells with 0.035% MMS for 2 h.
- C–F ChIP-seq analysis of Slx4 was performed following synchronous release of (C) slx4-7MUT-FLAG, (D) slx4-486A-FLAG, (E) SLX4-FLAG rtt107Δ, and (F) slx4-bd-FLAG cells into S phase in the presence of 0.035% MMS for 60 min. Slx4 ChIP enrichment scores on chromosome 10 are shown.
- G The distributions of Slx4 ChIP enrichment scores at early- and late-firing origins genome-wide are shown as a boxplot for slx4-7MUT-FLAG, slx4-486A-FLAG, SLX4-FLAG rtt107Δ, and slx4-bd-FLAG cells. The median is indicated by the horizontal bar, the box spans the first through third quartiles, the whiskers extend to the last data points within 1.5 times the interquartile range, and outliers are plotted as circles. The distributions at early origins were compared using the Wilcoxon rank-sum test. The P-values for Slx4-7MUT vs Slx4-bd and Slx4-486A vs Slx4-bd are shown. The P-values for Slx4-7MUT and Slx4-486A vs Slx4 in rtt107Δ were similar, < 10−33.
- H, I The median (± standard error) copy number across n = 108 early-firing origins is plotted for wild-type and rtt107Δ cells (H) and for slx4-7MUT, slx4-486A, and slx4-bd cells (I) following synchronous release into S phase in the presence of 0.035% MMS for 60 min.
Source data are available online for this figure.
We next performed ChIP-seq of Slx4 in an rtt107Δ strain (Fig2E) and found that deletion of RTT107 abolished recruitment of Slx4. Importantly, the absence of Slx4 binding at chromosome coordinates flanking early origins in rtt107Δ was not due to an absence of DNA replication forks in these regions, as the DNA replication profile of rtt107Δ was highly similar to wild-type (Fig2H). To test whether binding of Slx4 to Rtt107 was the key determinant of Slx4 recruitment, we mapped the region of Slx4 that binds to Rtt107, using yeast two-hybrid assays, to two regions spanning amino acids 286–598 (Supplementary Fig S2A and B). Deletion of this region of Slx4 eliminates binding to Rtt107 in co-IP assays when the slx4-bd (binding defective) allele is expressed from its native locus in yeast cells (Fig2A). Slx4-bd also lacks the region that binds to Dpb11 (Fig2B). Importantly, slx4-bd is not synthetic-lethal with sgs1Δ and so is not a null allele like slx4Δ (Supplementary Fig S2C). Slx4-bd is expressed at the same level as Slx4 (Supplementary Fig S2D), and Slx4-bd retains its ability to bind the Slx1 nuclease (Supplementary Fig S2E), indicating that Slx4-bd retains most of the known Slx4 functions. Slx4-bd failed to associate with regions behind stressed replication forks (Fig2F and G) without affecting replication kinetics (Fig2I), indicating that interaction with Rtt107 mediates the recruitment of Slx4 behind stressed DNA replication forks.
Mec1 phosphorylation of H2A recruits Rtt107 distal to stressed DNA replication forks
Rtt107 binds directly to H2A phosphorylated on serine 129 (H2A-S129-P) in vitro (Li et al, 2012) and in vivo (Ohouo et al, 2013), and this interaction is conserved in fission yeast (Williams et al, 2010) and humans (Yan et al, 2011). As phosphorylated H2A serves as a binding platform for multiple DNA damage response proteins with BRCT motifs (Kobayashi et al, 2002; Stewart et al, 2003; Ward et al, 2003; van Attikum et al, 2004; Morrison et al, 2004; Nakamura et al, 2004; Hammet et al, 2007), and BRCT motif interaction with H2A-S129-P is required for Brc1 (fission yeast Rtt107) to assemble into DNA damage response foci (Williams et al, 2010), phosphorylated H2A could function directly in Rtt107 recruitment to chromatin during MMS-induced replication stress.
Rtt107 binds to origin proximal regions following DNA replication stress induced by hydroxyurea (Roberts et al, 2008). We first tested if Rtt107 was similarly recruited during MMS-induced replication stress. The ChIP-seq profile of Rtt107 was highly similar to that of Slx4 (Fig3A and B), indicating that Rtt107 and Slx4 accumulate at the same chromatin locations behind stressed forks. To test the role of H2A serine 129 phosphorylation, we performed ChIP-seq with an h2a-s129a strain that lacks the Mec1 phosphoacceptor amino acid in H2A and found that Rtt107 binding to early origin proximal regions was greatly diminished (Fig3C, D, and F), relative to wild-type. The median view across 108 early origins shows that Rtt107 binding is reduced across the entire interval and lacks the specificity for the origin proximal regions observed when S129 is intact (Fig3D). These data point to the existence of a minor S129-independent mode of Rtt107 recruitment to chromatin, a mode that lacks the normal degree of spatial specificity. Again, replicated DNA peaks in h2a-s129a cells (Fig3E) were highly similar to those in wild-type, suggesting that diminished recruitment of Rtt107 was not caused indirectly by altered replication kinetics. We conclude that Rtt107 accumulates behind MMS-stressed replication forks by binding to histone H2A that has been phosphorylated by Mec1. Reduced Rtt107 recruitment in h2a-s129a cells correlated with a reduction in Rtt107 phosphorylation by Mec1 (Roberts et al, 2006) (Fig3G), indicating that Rtt107 is a Mec1 target when recruited to chromatin, where Mec1 is active.
Figure 3.

Phosphorylated histone H2A recruits Rtt107 behind replication forks during MMS-induced DNA replication stress
- ChIP-seq analysis was performed following synchronous release of RTT107-FLAG cells into S phase in the presence of 0.035% MMS for 60 min. Rtt107 ChIP enrichment scores on chromosome 10 are shown. Early origins are indicated by green bars and late origins by red bars.
- The median (± standard error) Rtt107 ChIP enrichment scores are plotted with the median (± standard error) Slx4 ChIP enrichment scores across n = 108 early-firing origins, in wild-type cells.
- ChIP-seq analysis was performed following synchronous release of RTT107-FLAG h2a-s129a cells into S phase in the presence of 0.035% MMS for 60 min. Rtt107 ChIP enrichment scores on chromosome 10 are shown.
- The median (± standard error) Rtt107 ChIP enrichment scores are plotted across n = 108 early-firing origins, for wild-type and h2a-s129a cells.
- The median (± standard error) replication profile across n = 108 early-firing origins is plotted for wild-type and h2a-s129a cells.
- The distributions of Rtt107 ChIP enrichment scores at early- and late-firing origins genome-wide are shown as boxplots. The median is indicated by the horizontal bar, the box spans the first through third quartiles, the whiskers extend to the last data points within 1.5 times the interquartile range, and outliers are plotted as circles. The distributions were compared using the Wilcoxon rank-sum test.
- Immunoblot analysis of Rtt107-FLAG in wild-type and h2a-s129a cells before and after treatment of asynchronous cultures with 0.035% MMS for the indicated times.
Source data are available online for this figure.
Slx4 promotes stable Dpb11 recruitment distal to stressed DNA replication forks
Since our data indicate that the Mec1 activator Dpb11 (Mordes et al, 2008; Navadgi-Patil & Burgers, 2008) does not play a role in recruiting Slx4 to chromatin during replication stress, we asked if Slx4 is important for recruitment of Dpb11 (Fig4). We first demonstrated that Dpb11 is recruited to chromatin during MMS-induced replication stress (Fig4A). The pattern of enrichment in Dpb11 ChIP-seq has two properties. First, Dpb11 localizes in a broad peak, centered on early-firing origins (Fig4A and C), much like Rtt107 and Slx4, suggesting that Dpb11 is recruited behind MMS-stressed replication forks. The second property of the Dpb11 enrichment pattern is a sharp narrow peak, corresponding to just a few kilobases spanning early origins (Fig4A). Deletion of either SLX4 or RTT107 eliminates the broad Dpb11 peak behind stressed forks (Fig4A and C) with little effect on the narrow origin proximal peak. Replication profiles were highly similar in the three strains (Fig4B). The remaining narrow Dpb11 peak could represent Dpb11 that is recruited by Rad9 or Ddc1 (Puddu et al, 2008; Navadgi-Patil & Burgers, 2009b; Pfander & Diffley, 2011), or that is functioning in initiation reactions (Tanaka & Araki, 2013). We repeated the Dpb11 ChIP-seq in rad9Δ and ddc1Δ (Fig4D). Deletion of RAD9 had no effect on Dpb11 recruitment, whereas deletion of DDC1 resulted in a pattern of Dpb11 binding that was highly similar to that seen in slx4Δ. Replication profiles are slightly more advanced in rad9Δ and ddc1Δ compared to wild-type (Fig4E), consistent with Rad53 deactivation promoting replication fork restart (Szyjka et al, 2008). None of the gene deletions had a detectable effect on Dpb11 expression (Fig4F). We conclude that Slx4 and Ddc1 are both required, and neither is sufficient, for stable association of Dpb11 distal to MMS-stressed forks.
Figure 4.
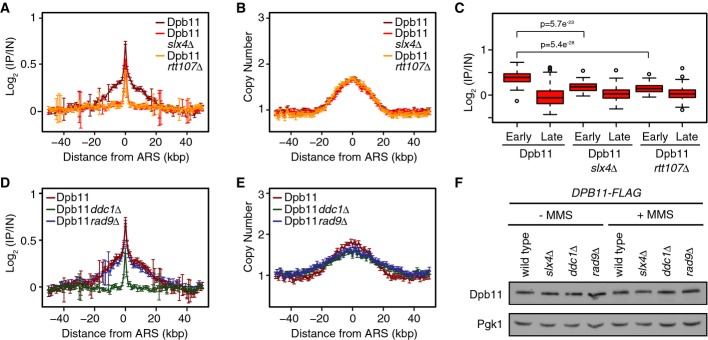
Slx4 and Ddc1 recruit the Mec1 activator Dpb11 behind stressed replication forks
- A, B The median (± standard error) Dpb11 ChIP enrichment scores (A) and DNA copy number (B) across n = 108 early-firing origins in wild-type, slx4Δ, and rtt107Δ cells are plotted.
- C The distributions of Dpb11 ChIP enrichment scores at early- and late-firing origins genome-wide are shown as a boxplot. The median is indicated by the horizontal bar, the box spans the first through third quartiles, the whiskers extend to the last data points within 1.5 times the interquartile range, and outliers are plotted as circles. The distributions at early origins were compared using the Wilcoxon rank-sum test.
- D, E The median (± standard error) Dpb11 ChIP enrichment scores (D) and replication profile (E) across n = 108 early-firing origins in wild-type, ddc1Δ, and rad9Δ cells are plotted.
- F The level of Dpb11 was assessed by immunoblot analysis of the indicated strains, in the presence and absence of 0.035% MMS. Dbp11-FLAG was detected with an anti-FLAG antibody. Pgk1 levels were assessed as a loading control.
Source data are available online for this figure.
Slx4 promotes Mec1 signaling during DNA replication stress
In response to DNA double strand breaks (DSBs), Dpb11 promotes Rad9 phosphorylation by simultaneously binding to Mec1 and Rad9 (Pfander & Diffley, 2011). During MMS treatment, Slx4 competes with Rad9 for Dpb11 binding (Ohouo et al, 2013) and is required for phosphorylation of Rtt107 by Mec1 (Roberts et al, 2006), raising the possibility that recruitment of Dpb11 by Slx4 interaction promotes Mec1 signaling. We asked if Slx4 has a general effect on Mec1 activity in vivo, using mass spectrometry to compare phosphorylation of S/T-Q Mec1 target sites in wild-type and slx4Δ after treatment with MMS (Fig5A and Supplementary Table S1). As previously reported, the abundance of Rad53 target phosphopeptides decreased in wild-type cells compared to slx4Δ (Ohouo et al, 2013). By contrast, the abundance of 15 phosphopeptides containing Mec1 phosphorylation sites (of 38 total detected) increased by at least 20% in wild-type cells when compared to slx4Δ, indicating that Slx4 promotes phosphorylation at those sites by Mec1.
Figure 5.
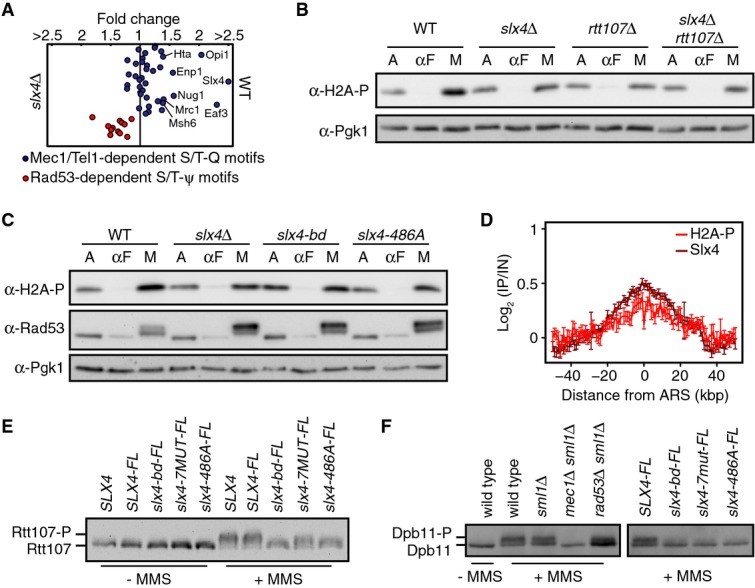
Slx4 regulates local Mec1 signaling during DNA replication stress
- Quantitative phosphoproteome analysis of the relative abundance of known Mec1 targets in wild-type and slx4Δ cells following growth for 1 h in 0.01% MMS. Mec1-/Tel1-dependent Rad53-independent (blue circles; S/T-Q motifs) and Rad53-dependent (red circles; S/T-ψ motifs) phospho events are plotted as the wild-type: slx4Δ abundance ratio, on a linear scale.
- Immunoblot analysis of H2A-S129 phosphorylation (H2A-S129-P) in the indicated strains. Cultures were sampled during asynchronous growth (A), after G1 arrest with α-factor (αF), and after release into S phase in 0.035% MMS for 60 min (M). Immunoblots were probed with anti-H2A-S129-P antibodies and with anti-Pgk1 antibodies (as a loading control).
- Immunoblot analysis of H2A-S129 phosphorylation and Rad53 phosphorylation in the indicated strains. Cultures were treated as in (B), with anti-Pgk1 staining of the immunoblot used as the loading control.
- The median (± standard error) H2A-S129-P and Slx4 ChIP enrichment score across n = 108 early-firing origins in wild-type cells is plotted.
- Immunoblot analysis of Rtt107 phosphorylation in strains expressing the indicated Slx4 proteins. Cells were treated with or without 0.035% MMS for 2 h. The epitope-tagged Rtt107 was detected with anti-HA antibodies.
- Immunoblot analysis of Dpb11 phosphorylation in the indicated strains. Cells were treated with or without 0.035% MMS for 2 h. The immunoblot was probed with anti-HA antibodies to detect the HA epitope-tagged Dpb11 present in all strains.
Source data are available online for this figure.
Of particular interest, the abundance of phosphorylated Ser129 of histone H2A increased by 1.4-fold in wild-type in the phosphoproteome analysis. We assessed phosphorylation of histone H2A Ser129 directly, using immunoblots and a phospho-specific antibody. When G1-arrested cells were released into S phase in the presence of MMS, H2A phosphorylation was consistently decreased in slx4Δ relative to wild-type, by approximately 1.5-fold (Fig5B; mean of three replicates = 0.66 ± 0.04). Similar decreases were observed in rtt107Δ and slx4Δ rtt107Δ (Fig5B), and in slx4-bd and slx4-486A (Fig5C). We mapped the location of H2A-Ser129-P during replication stress, and compared it to the location of Slx4, by chromatin-IP of each protein from the same sample (Fig5D). Phosphorylated H2A was enriched at the same chromosome coordinates as Slx4 (Pearson’s r = 0.91 for median enrichments across 50 kb flanking the 108 early origins, Supplementary Fig S3), although the peak was smaller and noisier. Nonetheless, when considered in light of the properties of Slx4 localization, these data indicate that the peak of Mec1-phosphorylated H2A coincides with regions that contain newly replicated DNA, near early-firing replication origins, behind the replication forks.
Although neither was detected in the phosphoproteome analysis, Rtt107 and Dpb11 are Mec1 targets during replication stress (Rouse, 2004; Roberts et al, 2006; Ohouo et al, 2010), and our data indicate that both proteins localize to the same chromatin regions as Slx4. We tested whether Slx4 complexes also promoted Mec1 phosphorylation of Rtt107 and Dpb11, as was the case with H2A phosphorylation. We detected Mec1 phosphorylation of Rtt107 by the distinctive mobility shift of Rtt107 (Rouse, 2004; Roberts et al, 2006) and analyzed Rtt107 from SLX4, slx4-bd, slx4-7MUT, and slx4-486A cells, in the presence and absence of MMS-induced replication stress (Fig5E). Rtt107 phosphorylation was reduced in slx4-7MUT compared to wild-type, and severely depleted in slx4-bd and slx4-486A, consistent with Slx4-486A being more defective in Dpb11 binding than Slx4-7MUT (Ohouo et al, 2013). These data indicate that Rtt107-Slx4-Dpb11 interactions facilitate Rtt107 phosphorylation by Mec1. We also analyzed Dpb11 phosphorylation. As is the case in UV damage (Puddu et al, 2008) and in vitro (Mordes et al, 2008), Dpb11 is phosphorylated in a Mec1-dependent (and partially Rad53 independent) manner in MMS (Ohouo et al (2010) and Fig5F). Dpb11 phosphorylation is compromised when Slx4 cannot bind to Rtt107 and Dpb11 (Fig5F). We conclude that efficient phosphorylation of Dpb11 by Mec1 requires stable association of Dpb11 with Slx4 complexes. Together, our data are consistent with Slx4 complex formation promoting Mec1 signaling behind the stressed fork by recruiting or retaining the Mec1 activator Dpb11, and resulting in enhanced phosphorylation of Rtt107, Dpb11, and H2A.
Slx4 complex assembly promotes recovery from MMS-induced DNA replication stress
Cells lacking SLX4 are sensitive to MMS (Chang et al, 2002; Fricke & Brill, 2003), show delayed S phase progression during recovery from MMS treatment (Roberts et al, 2006; Flott et al, 2007), and have increased spontaneous DNA damage (Roberts et al, 2006). We compared the in vivo function of Slx4-7MUT and Slx4-486A, which are recruited behind stalled forks, to that of Slx4-bd, which is not. We found that slx4-bd was sensitive to MMS (Fig6A) and progressed through S phase more slowly than SLX4 during recovery from MMS (Fig6B). In both assays, slx4-bd displayed the same phenotype as slx4Δ, indicating that the functions of Slx4 in MMS resistance and MMS recovery require interaction with Rtt107. By contrast, slx4-7MUT and slx4-486A were less sensitive to MMS, and slx4-7MUT recovered from MMS more rapidly than slx4-bd, slx4-486A, and slx4Δ (Fig6A and B). These data are consistent with Slx4-Rtt107 and Slx4-Dpb11 interactions both contributing to replication stress resistance and indicate a critical role for Rtt107-Slx4-Dpb11 complex assembly behind stalled forks for Slx4 function in vivo.
Figure 6.

Slx4 recruitment by Rtt107 promotes recovery from DNA replication stress
- A The indicated strains were serially diluted ten-fold, spotted on YPD or YPD + MMS plates, and grown at 30°C for 3 days.
- B Cells were arrested in G1 with α-factor, released synchronously into S phase in the presence of 0.035% MMS for 1 h, and allowed to recover in fresh media for 3 h. Samples were taken for analysis by flow cytometry at the indicated times.
- C–E Wild-type, slx4Δ, and slx4-bd cells expressing thymidine kinase were arrested in G1 and released into S phase in the presence of 400 μg/ml BrdU and 0.035% MMS. (C) Samples were collected at 15 and 30 min for BrdU track length analysis. The distributions of BrdU track lengths are displayed as boxplots. The median is indicated by the horizontal bar, the box spans the first through third quartiles, the whiskers extend to the last data points within 1.5 times the interquartile range, and outliers are plotted as circles. Median BrdU track length is shown, and P-values were determined using a Mann–Whitney U-test. (D) Following 60 min of treatment, MMS was washed out and cells were allowed to recover in fresh media containing BrdU for 90 min before samples were taken for BrdU gap analysis. Mean values of 3 independent experiments and standard error are shown for the percentage of fibers with gaps following recovery from MMS. At least 190 fibers were analyzed for each replicate, and the P-values were determined using a t-test. (E) Representative DNA fibers extracted from different micrographs and assembled using Photoshop are shown. Green is BrdU-labeled nascent DNA, and red is unlabeled parental DNA. The scale bar is 50 kbp.
We assessed the effect of disrupting Rtt107-Slx4-Dpb11 complex assembly behind stalled forks by measuring replication fork rate during replication stress and by counting the number of unreplicated gaps during recovery from replication stress. Wild-type, slx4Δ, and slx4-bd cells were pulse labeled with BrdU during progression through S phase in the presence of MMS and during recovery from MMS. Following molecular combing of the labeled DNA, we measured the lengths of replication tracks on individual DNA fibers isolated during MMS treatment (Fig6C) and determined the fraction of fibers isolated during recovery that contained unreplicated regions (Fig6D and E). The slx4Δ and slx4-bd cells displayed a mild but statistically insignificant slowing of DNA replication forks during replication stress, suggesting that Slx4 function has little effect on replication fork movement when replication stress is present. By contrast, slx4Δ [as previously reported (Flott et al, 2007)] and slx4-bd showed a marked increase in the number of unreplicated regions during recovery from MMS, indicating that assembly of Slx4 complexes behind stalled replication forks is critical for the timely completion of DNA replication following replication stress.
Discussion
We propose that Slx4 protein complexes assemble in response to MMS-induced DNA replication stress and, in concert with the checkpoint clamp subunit Ddc1, promote the recruitment or retention of Dpb11, which in turn increases Mec1 activity. Slx4 foci form during S phase, and increase in abundance when replication stress is present, as do Rtt107 and Dpb11 foci (Chin et al, 2006; Germann et al, 2011; Tkach et al, 2012). Similarly, Slx4 ChIP signal increases over time during MMS treatment, suggesting Slx4 complexes are forming on chromatin in response to replication stress. H2A-Rtt107, Slx4-Dpb11, and Ddc1-Dpb11 interactions are promoted by checkpoint kinase phosphorylations that are greatly increased during replication stress. Slx4 foci form in the majority of S phase cells even in the absence of MMS, so we expect that H2A-Rtt107-Slx4-Dpb11 complexes also form during an unperturbed S phase, at individual stressed forks, but to a lesser extent than we observe during MMS-induced replication stress. Finally, because ChIP-seq presents a population view, we cannot exclude the possibility that the Rtt107-Slx4-Dpb11 signal is contributed by a minor species of replication forks that lag behind the majority of forks, which we localize by Dpb3 ChIP and replication profile analysis. In such a scenario, we still expect that Slx4 complexes would be spatially resolved from the replication machinery, as none of the key components of the Slx4 complexes (H2A-S129P, Dpb11, and the 9-1-1 clamp) are components of the replisome.
When replication stress is present, Slx4 localizes to sites distal to replication forks by physical interaction with Rtt107, and recruitment by Rtt107 is essential for Slx4 function during replication stress. Rtt107 is recruited by binding to histone H2A phosphorylated on Ser129 by Mec1. Once recruited, Slx4 is phosphorylated by Mec1 and bound by the Mec1 activator Dpb11, likely generating a H2A-Ser129-P/Rtt107/Slx4/Dpb11 multiprotein complex that promotes Mec1 phosphorylation of at least three targets that reside in the same region behind stressed replication forks: Rtt107, Dpb11, and H2A. Assembly of this complex bears striking similarities to assembly of the H2A-Ser129-P/Rad9/Dpb11 complex, with Rtt107-Slx4 replacing Rad9. In both cases, a pair of BRCT motifs mediates H2A-Ser129-P recognition (Hammet et al, 2007; Li et al, 2012; Ohouo et al, 2013) and binding to Dpb11 is regulated by CDK phosphorylation (Pfander & Diffley, 2011; Ohouo et al, 2013; Gritenaite et al, 2014). Efficient recruitment of both complexes to lesions or stressed forks likely depends on multiple BRCT-mediated interactions. Binding of Ddc1 to BRCT motifs 3 and 4 of Dpb11 is presumed to be important for Dpb11 recruitment because the interaction is required for robust checkpoint activation via Rad9/Dpb11 complexes (Furuya et al, 2004; Puddu et al, 2008; Pfander & Diffley, 2011), and we find that Ddc1 is required for chromatin binding of Dpb11. Likewise, binding of Slx4 to BRCT motifs 1 and 2 of Dpb11 is important for maximum Mec1 activity and for MMS resistance (Ohouo et al, 2010). Consistent with a two-site model of coordinated BRCT–phosphoprotein interactions (Cussiol et al, 2015), we propose that Slx4 functions in concert with Ddc1 to recruit or stably retain the Mec1 activator Dpb11 on chromatin, distal to the replication machinery, during the DNA replication stress response (Fig7A and B).
Figure 7.
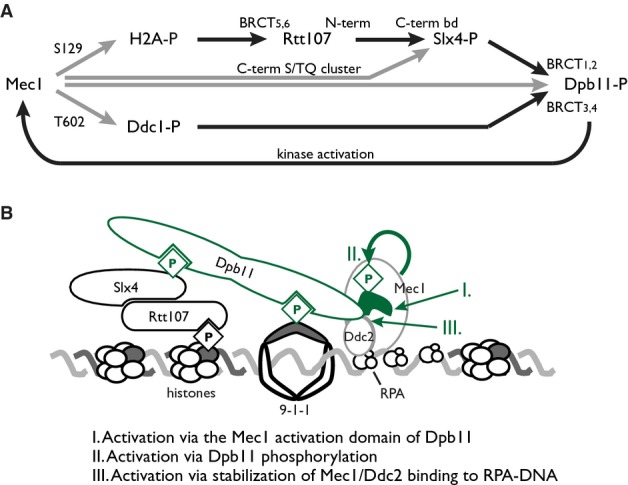
Model of Mec1 activation promoted by Slx4 complex assembly
- Mec1 phosphorylation of H2A serine 129 nucleates assembly of Slx4 complexes by providing a binding site for the C-terminal BRCT pair of Rtt107. Slx4 binds Rtt107 constitutively via its C-terminal Rtt107 binding domain. Phosphorylation of Slx4, by Cdk and Mec1, creates binding sites for the N-terminal BRCT pair of Dpb11, whereas the C-terminal BRCT pair binds Ddc1 phosphorylated on threonine 602 by Mec1. Engagement of both Dpb11 BRCT pairs results in stable complex formation distal to the MMS-stressed replication fork, and promotes Mec1 kinase activity, resulting in a positive feedback loop that amplifies Mec1 signaling. Phosphorylations are indicated by gray lines, and protein–protein interactions are indicated by black lines.
- A model of Slx4 complex assembly, with three possible modes of Mec1 activation indicated.
Signaling and repair behind the stressed replication fork
We find that during MMS-induced replication stress, Slx4 is localized in a pattern that is spatially distinct from that of the DNA polymerase epsilon subunit Dpb3. As long as 90 min after release from G1 into MMS, Slx4 localizes on chromatin in a gradient with low amounts co-localizing with Dpb3 and the leading edge of DNA synthesis marked by increasing copy number, and the maximum amount co-localizing with the most fork-distal regions. Similar localization profiles are seen for the Slx4 binding partners Rtt107, Dpb11, and H2A-S129P. We infer that it is unlikely that Slx4 complexes function at the replication fork, or in concert with the replication machinery, although it is formally possible that a small amount of Slx4 complexes are present at, or extremely proximal to, the replication fork. We propose that the pool of Slx4 that we detect distal to replication forks is functional, as it co-localizes with Dpb11 and H2A-S129P, and Slx4 mutants that fail to assemble complexes distal to the forks display MMS sensitivity and replication defects. At this point, we are unable to distinguish between Slx4 complexes being assembled once the fork has passed and Slx4 being deposited by the passing fork.
Slx4 complexes compete with Rad9 complexes to down-regulate Rad53 signaling during replication stress (Ohouo et al, 2013; Cussiol et al, 2015). Recent co-immunoprecipitation analysis suggests that Rad9-Dpb11-Ddc1 complexes form early during MMS-induced replication stress and transition to Slx4-Dpb11-Ddc1 complexes at later times (Cussiol et al, 2015). While we have not directly assessed Rad9 recruitment here, our evidence that Slx4 complexes are distal to the stressed forks is consistent with models in which Slx4 competes with Rad9 to assemble signaling complexes during fork stress (Ohouo et al, 2010, 2013; Cussiol et al, 2015). There are at least two protein–protein contact sites where such a competition could occur, H2A-S129P and BRCT 1 and 2 of Dpb11. Our data suggest that in the chromatin context, binding of Slx4 is largely independent of Dpb11. Slx4 mutants that do not interact with Dpb11 bind chromatin in patterns that are highly similar to wild-type Slx4, via interaction with Rtt107, and binding of Dpb11 itself requires Slx4. Thus, the major species of Slx4 is not likely to be recruited by pre-existing Dpb11 complexes, suggesting that H2A-S129P could be the principal site of competition between Rad9 and Rtt107-Slx4. A more detailed understanding of the transition mechanism awaits kinetic and affinity data for the relevant protein–protein interactions.
Why are Rtt107-Slx4-Dpb11 complexes assembled (or retained) behind the replication fork? One possibility is that there is little necessity for repair of MMS damage to occur at the fork itself. Indeed, repair at the replication fork could in many cases be sub-optimal as it would increase the time required to complete S phase and unnecessarily restrain proliferation. Repair processes that are uncoupled from DNA replication, as has been demonstrated for some post-replication repair events (Daigaku et al, 2010; Karras & Jentsch, 2010), would allow completion of DNA replication independently of repair. Slx4-Dpb11 complexes were recently shown to contribute to the resolution of DNA repair intermediates by binding the Mus81-Mms4 structure-specific nuclease in G2/M (Gritenaite et al, 2014), and we find that failure to assemble Slx4 complexes results in persistence of unreplicated regions and an elongated S phase. One interesting possibility is that Slx4-Dpb11 complexes serve a signaling function during S phase to promote the timely completion of DNA replication and then transition to a repair function during G2.
The role of Slx4 in recruiting Dpb11 to stressed replication forks
Dpb11 functions both in the initiation of DNA replication and in checkpoint activation, roles that are conserved in fission yeast (Cut5/Rad4), Xenopus, and human (TopBP1). Following initiation, Dpb11 does not appear to move with the elongating replication forks (Masumoto et al, 2000), suggesting that Dpb11 must be recruited again to DNA lesions or sites of replication stress (Navadgi-Patil & Burgers, 2009a). Consistent with this idea, Dpb11 forms nuclear foci in response to DNA breaks and replication stress, and these foci are distinct from sites of replication initiation marked by Sld3 (Germann et al, 2011). Two pathways for Dpb11 recruitment have been proposed, involving the checkpoint clamp subunit Ddc1 and DNA polymerase epsilon, both of which interact with Dpb11 (Araki et al, 1995; Wang & Elledge, 2002; Furuya et al, 2004; Puddu et al, 2008). We define the chromatin sites of Dpb11 recruitment during replication stress, and find that Dpb11 binds to regions near early-firing origins, in a pattern that is similar to Rtt107 and Slx4. The Dpb11 pattern is spatially distinct from that of the DNA polymerase epsilon subunit Dpb3, suggesting that Dpb11 recruitment is not via interaction with polymerase. Unexpectedly, recruitment of Dpb11 to the broad region behind replication forks required Slx4, which binds directly to Dpb11 via a BRCT-phospho-Slx4 interaction (Ohouo et al, 2010; Gritenaite et al, 2014). Dpb11 recruitment also requires Ddc1, but interaction between Dpb11 and Rad9 does not appear to be important. Since both Slx4 and Ddc1 are required for Dpb11 recruitment, but neither is sufficient, we propose that stable recruitment of Dpb11 behind stressed replication forks relies on engagement of both BRCT pairs of Dpb11, the N-terminal pair to Slx4 (Ohouo et al, 2013; Cussiol et al, 2015) and the C-terminal pair to Ddc1 (Wang & Elledge, 2002). One particularly interesting aspect of Dpb11 biology is the plasticity of its protein–protein interactions. As such, we have no expectation that the mode of recruitment that we find in MMS should predominate during recruitment of Dpb11 to other types of lesions. Indeed, even in MMS we observed a peak of Dpb11 very near to replication origins that did not show the same genetic dependencies as the bulk of Dpb11.
Amplification of Mec1 signaling
Mec1 phosphorylation of histone H2A is an early event during the response to replication stress (Cobb et al, 2005). Phosphorylation of H2A by Mec1 is promoted by Ddc1, Dpb11, and Ddc2 (Majka et al, 2006b; Mordes et al, 2008; Navadgi-Patil & Burgers, 2008, 2009b; Puddu et al, 2011; Bandhu et al, 2014), but recruitment of Mec1 to lesions independent of Ddc1 and Dpb11 is sufficient for basal kinase activity and H2A phosphorylation (Bandhu et al, 2014). Mec1 kinase activity is stimulated by interaction with Ddc2 (Bandhu et al, 2014) and by kinase activating motifs on both Ddc1 and Dpb11 (Mordes et al, 2008; Navadgi-Patil & Burgers, 2009b; Navadgi-Patil et al, 2011). We find that Slx4 also promotes Mec1-mediated H2A-Ser129-P formation by binding to Rtt107 and Dpb11, as MMS-induced H2A-Ser129-P levels are decreased in slx4Δ, slx4-bd, slx4-486A, and rtt107Δ cells. Of particular interest, Mec1 is active at the same chromosomal sites where Slx4 is recruited, since Slx4 and H2A-Ser129-P co-localize extensively.
Likely due to the multiple modes of Mec1 activation that can be employed, the reduction in Mec1 activity in the absence of Slx4 is modest. Nonetheless, Slx4 is required for full activation of Mec1 during MMS-induced replication stress. Consistent with previous evidence that Dpb11 is important for full activation of Mec1 (Puddu et al, 2008), we suggest that Slx4 activation of Mec1 occurs via Slx4-dependent recruitment of Dpb11 during MMS-induced replication stress. We propose three possible mechanisms by which Slx4 could stimulate Mec1 activity (Fig7B). In the first, Slx4 promotes the binding of Dpb11 behind stressed replication forks, allowing the unstructured C-terminal domain of Dpb11 to interact with independently recruited Mec1 (Mordes et al, 2008; Navadgi-Patil & Burgers, 2008). Consistent with this model, the ATR activation domain of the fission yeast Dpb11 homolog is particularly important for checkpoint signal amplification in S phase (Lin et al, 2012). In the second model, Slx4 promotes phosphorylation of Dpb11 by Mec1, resulting in enhanced activity of Mec1 as demonstrated in vitro (Mordes et al, 2008). In the third, assembly of Slx4-Dpb11 complexes stabilizes the association of Mec1-Ddc2 with chromatin, possibly via Dpb11-Ddc2 contacts (Mordes et al, 2008), promoting stable occupancy by Mec1 and enhancing the access of Mec1 to its chromatin-bound substrates. These possibilities are not mutually exclusive: Enhanced recruitment or retention of Dpb11 behind the fork could promote Mec1 phosphorylation of Dpb11, promoting Mec1 activation and establishing a positive feedback loop, for example. The increase in H2A phosphorylation that is promoted by Slx4 could further increase Rtt107 recruitment, which we find depends on H2A phosphorylation, also resulting in positive feedback (Fig7A) and signal amplification. Amplification of the Mec1 signal could have the interesting effect of promoting the abundance and stability of Slx4-Dpb11 complexes distal to the stressed fork, facilitating a transition from Rad9-Dpb11-Ddc1 complexes to Slx4-Dpb11-Ddc1 complexes (Cussiol et al, 2015). The central role of H2A phosphorylation in formation of Rtt107-Slx4-Dpb11 complexes during DNA replication stress is entirely consistent with the role of H2A phosphorylation by Mec1 in promoting genome stability, particularly when replication stress is present (Szilard et al, 2010).
Diverse Slx4 functions during DNA replication stress
Slx4 appears to have at least three functions during DNA replication stress. Slx4 dampens checkpoint signaling to Rad53 by competing with Rad9 for Dpb11 binding (Ohouo et al, 2013), Slx4 promotes resolution of DNA repair intermediates in G2/M by recruiting the Mus81-Mms4 nuclease via interaction with Dpb11 (Gritenaite et al, 2014), and Slx4 promotes full Mec1 activation by recruiting Dpb11 behind stressed replication forks. The relative contribution of these three functions to slx4Δ phenotypes, including MMS resistance, slow S phase progression, and failure to complete DNA replication, is difficult to resolve because a separation of function mutant in SLX4 does not currently exist, and the nexus of all three Slx4 functions is Dpb11. Extensive use has been made of slx4-486A, but it is clearly defective in all three functions, presumably because it is severely compromised in Dpb11 binding. Epistasis experiments with mus81Δ slx4-486A suggest that joint molecule resolution is particularly important for MMS resistance (Gritenaite et al, 2014), but Mus81/Mms4 likely plays little role in the slow S phase progression that is evident in slx4 mutants, as mms4Δ mutants do not display this phenotype during MMS recovery (AB and GWB, unpublished). Determining the relative contributions of the Slx4 functions to the replication stress response and to genome stability awaits more precise molecular dissection of the Slx4 pathways.
Materials and Methods
Yeast strains and media
All yeast strains used in this study are derivatives of BY4741 (Brachmann et al, 1998) or W303, and are listed in Supplementary Table S2. Strains were constructed using genetic crosses and standard PCR-based gene disruption techniques. Standard yeast media and growth conditions were used.
Chromatin immunoprecipitation and deep sequencing
Chromatin immunoprecipitation was performed using Flag epitope-tagged versions of each indicated protein, as previously described (Roberts et al, 2008), with modifications. Logarithmically growing cells at 23°C were arrested in G1 with 1.2 μM α-factor for 2.5 h, released into S phase with 100 μg/ml pronase (Sigma P5147) in the presence of 0.035% MMS (v/v) for 60 min (unless stated otherwise), and cross-linked with formaldehyde. Cells were harvested and washed twice with cold TBS (20 mM Tris–HCl pH 7.5, 150 mM NaCl), resuspended in FA-lysis buffer (50 mM HEPES pH 7.5, 2 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 150 mM NaCl) containing 0.05% SDS, lysed, and sonicated. Immunoprecipitates were washed sequentially with 1 ml of FA-lysis buffer, high salt wash buffer (FA-lysis buffer containing 1 M NaCl), wash buffer 2 (FA-lysis buffer containing 0.5 M NaCl), wash buffer 3 (50 mM HEPES pH 7.5, 0.25 M LiCl, 2 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 1% NP-40, 10 mM Tris–HCl pH 8.0), and TE (10 mM Tris–HCl pH 8.0, 1 mM EDTA). Protein–DNA complexes were eluted, cross-links were reversed, protein and RNA was digested, and DNA was isolated by phenol/chloroform extraction and ethanol precipitation. Sequencing libraries were generated using the Nextera™ XT DNA Sample Preparation Kit (Illumina) with custom index primers for the PCR amplification step. Libraries were quantified using a 2100 Bioanalyzer (Agilent) and the KAPA SYBR FAST Universal qPCR Kit (KAPA Biosystems), and sequenced using the HiSeq 2500 (Illumina) by multiplexing 12–18 samples per lane, or using the MiSeq (Illumina) by multiplexing 8 samples, to generate at least 10 million reads per sample. Each chromatin immunoprecipitation was performed at least twice, with a representative sample shown.
Sequencing data analysis
Input and IP samples from each experiment were sequenced on an Illumina HiSeq 2500 (50 nt single-end read, or 100 nt paired-end reads for SLX4-FLAG, SLX4-FLAG rtt107Δ, slx4-7mut-FLAG, and slx4-bd-FLAG). All sequencing data are deposited in the Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra; Study accession SRP048361). The number of reads for each sample ranges from 5.5 M to 24.2 M. The quality of sequencing reads was first assessed using FastQC. (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc). All samples have a median PHRED score of 30 or higher for all positions. Sequenced reads were mapped to the S. cerevisiae reference genome version WS220 [downloaded from the Saccharomyces Genome Database (Cherry et al, 2012; Engel et al, 2014)] using Bowtie2 (version 2.0.0) (Langmead & Salzberg, 2012) with default settings, except for forcing end-to-end alignment. Greater than 90% mapping rates were achieved for all samples, yielding a minimum 22× coverage for all samples (Supplementary Table S3). In order to reduce any bias from DNA sequencing, the data were normalized by the ratio of coverage for each IP and input pair prior to each comparison. We used a 100-bp sliding window with a step size of 50 bp to calculate enrichment scores as a log2 ratio of normalized read counts for each IP:input pair.
The chromosome coordinates of confirmed and likely (but not dubious) replication origins were extracted from OriDB (http://cerevisiae.oridb.org), and compared to our list of origins with annotated ARS consensus sequence (ACS) (Berbenetz et al, 2010) to arrive at a set of 412 replication origins (Supplementary Table S4). Origins were grouped as either early firing or late firing based on annotations in SGD (http://www.yeastgenome.org) and OriDB, and filtered to include only those origins that bind Dpb3 in early S phase (unpublished data) as early firing, yielding 108 early-firing origins. The chromosome coordinate of each replication origin was then defined either as the location of ACS (Berbenetz et al, 2010) or as the centre of the OriDB ARS coordinates for origins without a defined ACS.
To quantify protein enrichment at all annotated replication origins, we collected the enrichment scores for regions 5,000 bp upstream and downstream of each replication origin and calculated the mean of enrichments. The distributions of enrichments for the 108 early origins and the 304 late origins were plotted as boxplots and compared using the Wilcoxon rank-sum test using R.
To visualize protein binding across all early- and late-firing regions, we extracted enrichment values for 1-kb bins across 50 kb upstream and downstream of each replication origin coordinate (centered at the replication origins). Each point represents the median of enrichment scores within the bin, and each error bar represents standard error.
The increase in copy number was used to detect replicated regions in each input sample (Yabuki et al, 2002; Koren et al, 2014). We used VarScan 2 (version 2.3.5) to compare sequencing read counts from the input sample with read counts from a reference sample from a G1-arrested strain (BY4741), using the default settings (Koboldt et al, 2012) to detect replicated regions. Replication profiles are plotted either as histograms across Chromosome X or as averages of all early or late origins.
To compare enrichment in ChIP of Slx4 to the timing of replication origin activation, we calculated the median enrichment value across 50 kb upstream and downstream of each replication origin coordinate (centered at the replication origins), for the 108 early origins as defined above. Enrichment was plotted against Trep from Yabuki et al (2002), as curated in OriDB (http://cerevisiae.oridb.org). The Pearson’s correlation was calculated in R using the ‘rcorr’ package. To compare enrichment in ChIP of Slx4 to Rtt107, H2A-S129-P, Dpb3, and an Slx4 replicate, we calculated the median enrichment across 108 early origins for each 1-kb bin spanning 50 kb upstream and downstream of each replication origin coordinate (centered at the replication origin) and made the pairwise comparisons. Pearson’s correlations were calculated in R using the ‘rcorr’ package.
Yeast two-hybrid
Diploids harboring plasmids expressing the Gal4 DNA-binding domain (DBD) alone or fused to full-length Rtt107 and plasmids expressing the Gal4 activation domain (AD) alone or fused to full-length Slx4 or Slx4 fragments (Supplementary Table S5) were grown overnight in SD-leu-trp, serially diluted, and spotted onto SD-leu-trp-his plates. Plates were incubated at 30°C for 3–4 days.
Whole cell extracts, immunoblotting and immunoprecipitation
Logarithmically growing cells at 30°C were treated with or without 0.035% MMS for 2 h before cells were collected and fixed with 10% trichloroacetic acid, and whole cell extracts were prepared (Pellicioli et al, 1999). Alternatively, cells were harvested and treated with 0.1 M NaOH, and whole cell extracts were prepared (Kushnirov, 2000). Proteins were resolved by SDS–PAGE and subjected to immunoblotting with α-FLAG M2, α-HA (Roche 11867423001; or 12CA5), α-PGK (Novex 459250), α-VSV-G (Roche 11667351001), α-H2A-S129-P (abcam ab15083), or α-Rad53 (abcam ab104232) antibodies. Native extracts for immunoprecipitation were prepared from 5 × 108 cells as previously described (Shimomura et al, 1998), with some modifications. Cell pellets were resuspended in FA-lysis buffer containing 1 mM DTT, 2 mM sodium fluoride, 1 mM sodium ortho-vanadate, and protease inhibitors (1× Complete Mini, EDTA-free protease inhibitor cocktail (Roche 11836170001), 2.5 μg/ml aprotinin, 10 mM β-glycerophosphate, 5 μg/ml leupeptin, 2 μg/ml pepstatin A, 1 mM PMSF, and 5 μg/ml TLCK) and then lysed with glass beads. Cleared extracts were immunoprecipitated with α-FLAG M2 antibody. Beads were washed twice with 0.5 ml FA-lysis buffer as above and eluted in 5× SDS loading buffer.
DNA damage sensitivity
Yeast strains were grown overnight in YPD, serially diluted, and spotted onto YPD plates containing the indicated concentrations of MMS (Sigma-Aldrich). Plates were incubated at 30°C for 2–3 days.
Cell synchronization, flow cytometry and microscopy
Logarithmically growing cells at 23°C or 30°C were arrested in G1 with 1.2 μM alpha factor for 2 h, released into S phase with 100 μg/ml pronase, and treated immediately with 0.035% MMS (v/v) for 60 min. Where indicated, cells were then harvested, washed with YPD, and released into fresh YPD to recover for 3 h. Cultures were sampled at the indicated times and processed for flow cytometry as described (Bellay et al, 2011). DNA contents were measured using a FACSCalibur flow cytometer, and data were plotted as histograms using FlowJo Flow Cytometry Analysis Software, version 9.7.5. For analysis of Slx4-GFP nuclear foci, yeast strain AYY174 was grown to mid-log phase in YPD, diluted into fresh YPD, and cultured overnight to OD600 = 0.3. For analysis of asynchronous cells, cells were treated for 90 minutes with 0.035% MMS, or cultured without MMS, harvested, and washed once in low-fluorescence medium with or without MMS before imaging. For analysis of cells during synchronous cell cycle progression, cells were arrested in G1 with 1.2 μM α-factor for 2 h at 30°C and released into the cell cycle with 100 μg/ml pronase. Cells were harvested and washed once in low-fluorescence medium before imaging. Eleven z slices with a 0.4-μm step size images were acquired using Volocity imaging software (PerkinElmer) controlling a Leica DMI6000 confocal fluorescence microscope with fluorescein isothiocyanate, Texas Red, and differential interference contrast filter sets (Quorum Technologies). Images were scored by visual inspection for Slx4-GFP foci.
Molecular combing
SLX4 alleles were introduced into the E1670 background [MATa ade2-1 trp1-1 can1-100 his3-11,15 leu2-3,112 RAD5 + GAL psi+ ura3::URA3/GPD-TK7x (Lengronne et al, 2001)]. Cultures were grown in YPAD to OD600 = 0.25 and arrested in G1 by addition of 2.5 μM α-factor for 75 min at 30°C, followed by an additional aliquot of 1 μM α-factor for 75 min. Cultures were released from G1 by addition of 100 μg/ml pronase with 0.035% MMS. Samples were collected after 15 and 30 min for BrdU track length analysis, fixed with 0.1% (w/v) sodium azide, and incubated for 10 min on ice. Parallel samples were fixed in ethanol and analyzed by flow cytometry. The cultures were allowed to proceed for a total of 60 min in MMS before being washed out into fresh media containing BrdU and allowed to recover for an additional 90 min when samples were taken for BrdU gap analysis. DNA combing and detection with anti-BrdU and anti-DNA antibodies was performed as described (Cheung-Ong et al, 2012). DNA fibers were imaged using an Axio Imager microscope with a 63× objective. Individual coverslips were blinded before image acquisition to avoid bias in the analysis. Images were processed to maximize signal intensity, and fluorescent tracks were measured in ImageJ (http://rsb.info.nih.gov/ij). Track lengths were converted from pixels to kilobase pairs using a conversion factor based on combing λ-DNA.
Phosphoproteome analysis
SILAC labeling of yeast, IMAC, and quantitative phosphoproteome analysis were carried out as described (Ohouo et al, 2013). All calculations and plot generation were performed using a custom-designed web tool, which analyzed data files generated by the SORCERER software (Sage-N Research). Normalized protein/peptide abundances and appropriate abundance ratios were calculated. MATLAB (Mathworks) was used to plot the data. Results of the phosphoproteome analysis were filtered using a list of kinase checkpoint-dependent phosphopeptides previously identified (Smolka et al, 2007; Chen et al, 2010). Phosphopeptides plotted were manually inspected for phospho site assignment and quantitation.
Acknowledgments
We thank Heather Griffiths for the yeast two-hybrid mapping of the Rtt107 binding region of Slx4, Tania Roberts and Johnny Tkach for strain construction, Charles Boone for plasmids, Christophe Redon for strains, Joe Mellor and Fritz Roth for custom index primers used in sequencing library preparation, and Achille Pellicioli for sharing data prior to publication. Thanks also to George Brush, Antony Carr, Dan Durocher, and Jason Hendry for comments on the manuscript. This work was supported by Canadian Cancer Society Research Institute grant 702310 and Canadian Institutes of Health Research grant MOP-79368 (to GWB), a Natural Science and Engineering Research Council of Canada Post-Graduate Scholarship (to DG), Natural Sciences and Engineering Research Council of Canada Discovery Grant number 327612 (to ZZ), National Institutes of Health grant R01-GM097272 (to MBS), and by a MEXT Grant-in-Aid for Scientific Research on Innovative Areas (to KS).
Author contributions
AB designed and carried out the experiments, wrote the paper, and edited the paper. TK analyzed ChIP-seq and DNA copy number data and edited the paper. DG designed, carried out, and analyzed DNA combing experiments and edited the paper. JC designed and carried out ChIP-seq analysis of H2A-Ser129-P and edited the paper. FBO designed, carried out the phosphoproteome analysis, and analyzed the phosphoproteome data and edited the paper. AY designed, carried out, and analyzed fluorescence microscopy experiments. JO performed analysis of Rad53 activation and constructed strains. RN analyzed ChIP-seq data. AG performed initial analysis of Dpb11 phosphorylation. KS analyzed ChIP-seq data and edited the paper. MS designed the H2A-Ser129-P ChIP-seq and phosphoproteome analysis and edited the paper. ZZ analyzed ChIP-seq and DNA copy number data and edited the paper. GB designed the experiments, wrote the paper, and edited the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figures
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Source Data for Supplementary Figure S2
Review Process File
Source Data for Figure 2
Source Data for Figure 3G
Source Data for Figure 4F
Source Data for Figure 5
References
- Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJ, Bousset K, Furuya K, Diffley JF, Carr AM, Elledge SJ. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol. 2001;3:958–965. doi: 10.1038/ncb1101-958. [DOI] [PubMed] [Google Scholar]
- Araki H, Leem SH, Phongdara A, Sugino A. Dpb11, which interacts with DNA polymerase II(epsilon) in Saccharomyces cerevisiae, has a dual role in S-phase progression and at a cell cycle checkpoint. Proc Natl Acad Sci USA. 1995;92:11791–11795. doi: 10.1073/pnas.92.25.11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Attikum H, Fritsch O, Hohn B, Gasser SM. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell. 2004;119:777–788. doi: 10.1016/j.cell.2004.11.033. [DOI] [PubMed] [Google Scholar]
- Bandhu A, Kang J, Fukunaga K, Goto G, Sugimoto K. Ddc2 mediates Mec1 activation through a Ddc1- or Dpb11-independent mechanism. PLoS Genet. 2014;10:e1004136. doi: 10.1371/journal.pgen.1004136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellay J, Atluri G, Sing TL, Toufighi K, Costanzo M, Ribeiro PS, Pandey G, Baller J, VanderSluis B, Michaut M, Han S, Kim P, Brown GW, Andrews BJ, Boone C, Kumar V, Myers CL. Putting genetic interactions in context through a global modular decomposition. Genome Res. 2011;21:1375–1387. doi: 10.1101/gr.117176.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbenetz NM, Nislow C, Brown GW. Diversity of eukaryotic DNA replication origins revealed by genome-wide analysis of chromatin structure. PLoS Genet. 2010;6:e1001092. doi: 10.1371/journal.pgen.1001092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M. The Rad53 signal transduction pathway: replication fork stabilization, DNA repair, and adaptation. Exp Cell Res. 2006;312:2654–2659. doi: 10.1016/j.yexcr.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Brush GS, Morrow DM, Hieter P, Kelly TJ. The ATM homologue MEC1 is required for phosphorylation of replication protein A in yeast. Proc Natl Acad Sci USA. 1996;93:15075–15080. doi: 10.1073/pnas.93.26.15075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brush GS, Kelly TJ. Phosphorylation of the replication protein A large subunit in the Saccharomyces cerevisiae checkpoint response. Nucleic Acids Res. 2000;28:3725–3732. doi: 10.1093/nar/28.19.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, Bellaoui M, Boone C, Brown GW. A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc Natl Acad Sci USA. 2002;99:16934–16939. doi: 10.1073/pnas.262669299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Albuquerque CP, Liang J, Suhandynata RT, Zhou H. A proteome-wide analysis of kinase-substrate network in the DNA damage response. J Biol Chem. 2010;285:12803–12812. doi: 10.1074/jbc.M110.106989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, Christie KR, Costanzo MC, Dwight SS, Engel SR, Fisk DG, Hirschman JE, Hitz BC, Karra K, Krieger CJ, Miyasato SR, Nash RS, Park J, Skrzypek MS, Simison M, et al. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 2012;40:D700–D705. doi: 10.1093/nar/gkr1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung-Ong K, Song KT, Ma Z, Shabtai D, Lee AY, Gallo D, Heisler LE, Brown GW, Bierbach U, Giaever G, Nislow C. Comparative chemogenomics to examine the mechanism of action of DNA-targeted platinum-acridine anticancer agents. ACS Chem Biol. 2012;7:1892–1901. doi: 10.1021/cb300320d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin JK, Bashkirov VI, Heyer WD, Romesberg FE. Esc4/Rtt107 and the control of recombination during replication. DNA Repair (Amst) 2006;5:618–628. doi: 10.1016/j.dnarep.2006.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb JA, Bjergbaek L, Shimada K, Frei C, Gasser SM. DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J. 2003;22:4325–4336. doi: 10.1093/emboj/cdg391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb JA, Schleker T, Rojas V, Bjergbaek L, Tercero JA, Gasser SM. Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev. 2005;19:3055–3069. doi: 10.1101/gad.361805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell. 2003;11:203–213. doi: 10.1016/s1097-2765(02)00799-2. [DOI] [PubMed] [Google Scholar]
- Cussiol JR, Jablonowski CM, Yimit A, Brown GW, Smolka MB. Dampening checkpoint signalling via coordinated BRCT-domain interactions. EMBO J. 2015;34:1704–1717. doi: 10.15252/embj.201490834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigaku Y, Davies AA, Ulrich HD. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature. 2010;465:951–955. doi: 10.1038/nature09097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Piccoli G, Katou Y, Itoh T, Nakato R, Shirahige K, Labib K. Replisome stability at defective DNA replication forks is independent of S phase checkpoint kinases. Mol Cell. 2012;45:696–704. doi: 10.1016/j.molcel.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Downs JA, Lowndes NF, Jackson SP. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature. 2000;408:1001–1004. doi: 10.1038/35050000. [DOI] [PubMed] [Google Scholar]
- Emili A. MEC1-dependent phosphorylation of Rad9p in response to DNA damage. Mol Cell. 1998;2:183–189. doi: 10.1016/s1097-2765(00)80128-8. [DOI] [PubMed] [Google Scholar]
- Engel SR, Dietrich FS, Fisk DG, Binkley G, Balakrishnan R, Costanzo MC, Dwight SS, Hitz BC, Karra K, Nash RS, Weng S, Wong ED, Lloyd P, Skrzypek MS, Miyasato SR, Simison M, Cherry JM. The reference genome sequence of Saccharomyces cerevisiae: then and now. G3: Genes - Genomes - Genetics. 2014;4:389–398. doi: 10.1534/g3.113.008995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flott S, Rouse J. Slx4 becomes phosphorylated after DNA damage in a Mec1/Tel1-dependent manner and is required for repair of DNA alkylation damage. Biochem J. 2005;391:325–333. doi: 10.1042/BJ20050768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flott S, Alabert C, Toh GW, Toth R, Sugawara N, Campbell DG, Haber JE, Pasero P, Rouse J. Phosphorylation of Slx4 by Mec1 and Tel1 regulates the single-strand annealing mode of DNA repair in budding yeast. Mol Cell Biol. 2007;27:6433–6445. doi: 10.1128/MCB.00135-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke WM, Brill SJ. Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Genes Dev. 2003;17:1768–1778. doi: 10.1101/gad.1105203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedel AM, Pike BL, Gasser SM. ATR/Mec1: coordinating fork stability and repair. Curr Opin Cell Biol. 2009;21:237–244. doi: 10.1016/j.ceb.2009.01.017. [DOI] [PubMed] [Google Scholar]
- Furuya K, Poitelea M, Guo L, Caspari T, Carr AM. Chk1 activation requires Rad9 S/TQ-site phosphorylation to promote association with C-terminal BRCT domains of Rad4TOPBP1. Genes Dev. 2004;18:1154–1164. doi: 10.1101/gad.291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germann SM, Oestergaard VH, Haas C, Salis P, Motegi A, Lisby M. Dpb11/TopBP1 plays distinct roles in DNA replication, checkpoint response and homologous recombination. DNA Repair (Amst) 2011;10:210–224. doi: 10.1016/j.dnarep.2010.11.001. [DOI] [PubMed] [Google Scholar]
- Gilbert CS, Green CM, Lowndes NF. Budding yeast Rad9 is an ATP-dependent Rad53 activating machine. Mol Cell. 2001;8:129–136. doi: 10.1016/s1097-2765(01)00267-2. [DOI] [PubMed] [Google Scholar]
- Gritenaite D, Princz LN, Szakal B, Bantele SC, Wendeler L, Schilbach S, Habermann BH, Matos J, Lisby M, Branzei D, Pfander B. A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev. 2014;28:1604–1619. doi: 10.1101/gad.240515.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammet A, Magill C, Heierhorst J, Jackson SP. Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep. 2007;8:851–857. doi: 10.1038/sj.embor.7401036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Karras GI, Jentsch S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell. 2010;141:255–267. doi: 10.1016/j.cell.2010.02.028. [DOI] [PubMed] [Google Scholar]
- Kobayashi J, Tauchi H, Sakamoto S, Nakamura A, Morishima K, Matsuura S, Kobayashi T, Tamai K, Tanimoto K, Komatsu K. NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr Biol. 2002;12:1846–1851. doi: 10.1016/s0960-9822(02)01259-9. [DOI] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Handsaker RE, Kamitaki N, Karlic R, Ghosh S, Polak P, Eggan K, McCarroll SA. Genetic variation in human DNA replication timing. Cell. 2014;159:1015–1026. doi: 10.1016/j.cell.2014.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnirov VV. Rapid and reliable protein extraction from yeast. Yeast. 2000;16:857–860. doi: 10.1002/1097-0061(20000630)16:9<857::AID-YEA561>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengronne A, Pasero P, Bensimon A, Schwob E. Monitoring S phase progression globally and locally using BrdU incorporation in TK(+) yeast strains. Nucleic Acids Res. 2001;29:1433–1442. doi: 10.1093/nar/29.7.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Liu K, Li F, Wang J, Huang H, Wu J, Shi Y. Structure of C-terminal tandem BRCT repeats of Rtt107 protein reveals critical role in interaction with phosphorylated histone H2A during DNA damage repair. J Biol Chem. 2012;287:9137–9146. doi: 10.1074/jbc.M111.311860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SJ, Wardlaw CP, Morishita T, Miyabe I, Chahwan C, Caspari T, Schmidt U, Carr AM, Garcia V. The Rad4(TopBP1) ATR-activation domain functions in G1/S phase in a chromatin-dependent manner. PLoS Genet. 2012;8:e1002801. doi: 10.1371/journal.pgen.1002801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou H, Komata M, Katou Y, Guan Z, Reis CC, Budd M, Shirahige K, Campbell JL. Mrc1 and DNA polymerase epsilon function together in linking DNA replication and the S phase checkpoint. Mol Cell. 2008;32:106–117. doi: 10.1016/j.molcel.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majka J, Binz SK, Wold MS, Burgers PM. Replication protein A directs loading of the DNA damage checkpoint clamp to 5’-DNA junctions. J Biol Chem. 2006a;281:27855–27861. doi: 10.1074/jbc.M605176200. [DOI] [PubMed] [Google Scholar]
- Majka J, Niedziela-Majka A, Burgers PM. The checkpoint clamp activates Mec1 kinase during initiation of the DNA damage checkpoint. Mol Cell. 2006b;24:891–901. doi: 10.1016/j.molcel.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masumoto H, Sugino A, Araki H. Dpb11 controls the association between DNA polymerases alpha and epsilon and the autonomously replicating sequence region of budding yeast. Mol Cell Biol. 2000;20:2809–2817. doi: 10.1128/mcb.20.8.2809-2817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordes DA, Nam EA, Cortez D. Dpb11 activates the Mec1-Ddc2 complex. Proc Natl Acad Sci USA. 2008;105:18730–18734. doi: 10.1073/pnas.0806621105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison AJ, Highland J, Krogan NJ, Arbel-Eden A, Greenblatt JF, Haber JE, Shen X. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell. 2004;119:767–775. doi: 10.1016/j.cell.2004.11.037. [DOI] [PubMed] [Google Scholar]
- Nakamura TM, Du LL, Redon C, Russell P. Histone H2A phosphorylation controls Crb2 recruitment at DNA breaks, maintains checkpoint arrest, and influences DNA repair in fission yeast. Mol Cell Biol. 2004;24:6215–6230. doi: 10.1128/MCB.24.14.6215-6230.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navadgi-Patil VM, Burgers PM. Yeast DNA replication protein Dpb11 activates the Mec1/ATR checkpoint kinase. J Biol Chem. 2008;283:35853–35859. doi: 10.1074/jbc.M807435200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navadgi-Patil VM, Burgers PM. A tale of two tails: activation of DNA damage checkpoint kinase Mec1/ATR by the 9-1-1 clamp and by Dpb11/TopBP1. DNA Repair (Amst) 2009a;8:996–1003. doi: 10.1016/j.dnarep.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navadgi-Patil VM, Burgers PM. The unstructured C-terminal tail of the 9-1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol Cell. 2009b;36:743–753. doi: 10.1016/j.molcel.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navadgi-Patil VM, Kumar S, Burgers PM. The unstructured C-terminal tail of yeast Dpb11 (human TopBP1) protein is dispensable for DNA replication and the S phase checkpoint but required for the G2/M checkpoint. J Biol Chem. 2011;286:40999–41007. doi: 10.1074/jbc.M111.283994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas TA, Sanchez Y, Elledge SJ. RAD9 and DNA polymerase epsilon form parallel sensory branches for transducing the DNA damage checkpoint signal in Saccharomyces cerevisiae. Genes Dev. 1996;10:2632–2643. doi: 10.1101/gad.10.20.2632. [DOI] [PubMed] [Google Scholar]
- Ohouo PY, Bastos de Oliveira FM, Almeida BS, Smolka MB. DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol Cell. 2010;39:300–306. doi: 10.1016/j.molcel.2010.06.019. [DOI] [PubMed] [Google Scholar]
- Ohouo PY, Bastos de Oliveira FM, Liu Y, Ma CJ, Smolka MB. DNA-repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature. 2013;493:120–124. doi: 10.1038/nature11658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn AJ, Elledge SJ. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 2003;17:1755–1767. doi: 10.1101/gad.1098303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicioli A, Lucca C, Liberi G, Marini F, Lopes M, Plevani P, Romano A, Di Fiore PP, Foiani M. Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J. 1999;18:6561–6572. doi: 10.1093/emboj/18.22.6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfander B, Diffley JF. Dpb11 coordinates Mec1 kinase activation with cell cycle-regulated Rad9 recruitment. EMBO J. 2011;30:4897–4907. doi: 10.1038/emboj.2011.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puddu F, Granata M, Di Nola L, Balestrini A, Piergiovanni G, Lazzaro F, Giannattasio M, Plevani P, Muzi-Falconi M. Phosphorylation of the budding yeast 9-1-1 complex is required for Dpb11 function in the full activation of the UV-induced DNA damage checkpoint. Mol Cell Biol. 2008;28:4782–4793. doi: 10.1128/MCB.00330-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puddu F, Piergiovanni G, Plevani P, Muzi-Falconi M. Sensing of replication stress and Mec1 activation act through two independent pathways involving the 9-1-1 complex and DNA polymerase epsilon. PLoS Genet. 2011;7:e1002022. doi: 10.1371/journal.pgen.1002022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon C, Pilch DR, Rogakou EP, Orr AH, Lowndes NF, Bonner WM. Yeast histone 2A serine 129 is essential for the efficient repair of checkpoint-blind DNA damage. EMBO Rep. 2003;4:678–684. doi: 10.1038/sj.embor.embor871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts TM, Kobor MS, Bastin-Shanower SA, Ii M, Horte SA, Gin JW, Emili A, Rine J, Brill SJ, Brown GW. Slx4 Regulates DNA Damage Checkpoint-dependent Phosphorylation of the BRCT Domain Protein Rtt107/Esc4. Mol Biol Cell. 2006;17:539–548. doi: 10.1091/mbc.E05-08-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts TM, Zaidi IW, Vaisica JA, Peter M, Brown GW. Regulation of rtt107 recruitment to stalled DNA replication forks by the cullin rtt101 and the rtt109 acetyltransferase. Mol Biol Cell. 2008;19:171–180. doi: 10.1091/mbc.E07-09-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse J. Esc4p, a new target of Mec1p (ATR), promotes resumption of DNA synthesis after DNA damage. EMBO J. 2004;23:1188–1197. doi: 10.1038/sj.emboj.7600129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segurado M, Tercero JA. The S-phase checkpoint: targeting the replication fork. Biol Cell. 2009;101:617–627. doi: 10.1042/BC20090053. [DOI] [PubMed] [Google Scholar]
- Shimomura T, Ando S, Matsumoto K, Sugimoto K. Functional and physical interaction between Rad24 and Rfc5 in the yeast checkpoint pathways. Mol Cell Biol. 1998;18:5485–5491. doi: 10.1128/mcb.18.9.5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolka MB, Albuquerque CP, Chen SH, Zhou H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci USA. 2007;104:10364–10369. doi: 10.1073/pnas.0701622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- Sun Z, Hsiao J, Fay DS, Stern DF. Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science. 1998;281:272–274. doi: 10.1126/science.281.5374.272. [DOI] [PubMed] [Google Scholar]
- Sweeney FD, Yang F, Chi A, Shabanowitz J, Hunt DF, Durocher D. Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr Biol. 2005;15:1364–1375. doi: 10.1016/j.cub.2005.06.063. [DOI] [PubMed] [Google Scholar]
- Szilard RK, Jacques PE, Laramee L, Cheng B, Galicia S, Bataille AR, Yeung M, Mendez M, Bergeron M, Robert F, Durocher D. Systematic identification of fragile sites via genome-wide location analysis of gamma-H2AX. Nat Struct Mol Biol. 2010;17:299–305. doi: 10.1038/nsmb.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szyjka SJ, Aparicio JG, Viggiani CJ, Knott S, Xu W, Tavare S, Aparicio OM. Rad53 regulates replication fork restart after DNA damage in Saccharomyces cerevisiae. Genes Dev. 2008;22:1906–1920. doi: 10.1101/gad.1660408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Araki H. Helicase activation and establishment of replication forks at chromosomal origins of replication. Cold Spring Harb Perspect Biol. 2013;5:a010371. doi: 10.1101/cshperspect.a010371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tercero JA, Diffley JF. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature. 2001;412:553–557. doi: 10.1038/35087607. [DOI] [PubMed] [Google Scholar]
- Tkach JM, Yimit A, Lee AY, Riffle M, Costanzo M, Jaschob D, Hendry JA, Ou J, Moffat J, Boone C, Davis TN, Nislow C, Brown GW. Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat Cell Biol. 2012;14:966–976. doi: 10.1038/ncb2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh GW, O’Shaughnessy AM, Jimeno S, Dobbie IM, Grenon M, Maffini S, O’Rorke A, Lowndes NF. Histone H2A phosphorylation and H3 methylation are required for a novel Rad9 DSB repair function following checkpoint activation. DNA Repair (Amst) 2006;5:693–703. doi: 10.1016/j.dnarep.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Vialard JE, Gilbert CS, Green CM, Lowndes NF. The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J. 1998;17:5679–5688. doi: 10.1093/emboj/17.19.5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Elledge SJ. Genetic and physical interactions between DPB11 and DDC1 in the yeast DNA damage response pathway. Genetics. 2002;160:1295–1304. doi: 10.1093/genetics/160.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, Minn K, Jorda KG, Chen J. Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J Biol Chem. 2003;278:19579–19582. doi: 10.1074/jbc.C300117200. [DOI] [PubMed] [Google Scholar]
- Williams JS, Williams RS, Dovey CL, Guenther G, Tainer JA, Russell P. gammaH2A binds Brc1 to maintain genome integrity during S-phase. EMBO J. 2010;29:1136–1148. doi: 10.1038/emboj.2009.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabuki N, Terashima H, Kitada K. Mapping of early firing origins on a replication profile of budding yeast. Genes Cells. 2002;7:781–789. doi: 10.1046/j.1365-2443.2002.00559.x. [DOI] [PubMed] [Google Scholar]
- Yan W, Shao Z, Li F, Niu L, Shi Y, Teng M, Li X. Structural basis of gammaH2AX recognition by human PTIP BRCT5-BRCT6 domains in the DNA damage response pathway. FEBS Lett. 2011;585:3874–3879. doi: 10.1016/j.febslet.2011.10.045. [DOI] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- Zou L, Liu D, Elledge SJ. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci USA. 2003;100:13827–13832. doi: 10.1073/pnas.2336100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Source Data for Supplementary Figure S2
Review Process File
Source Data for Figure 2
Source Data for Figure 3G
Source Data for Figure 4F
Source Data for Figure 5