

Background: ERp57 is a disulfide isomerase up-regulated in prion related-disorders, but its impact on PrP biology is unknown.
Results: ERp57 gain- and loss-of-function can increase or reduce, respectively, PrP levels in neurons, both in cell culture and animal models.
Conclusion: ERp57 regulates steady-state prion protein levels.
Significance: ERp57 is a cellular factor involved in the synthesis and folding of PrP, representing a novel therapeutic target in prion-related diseases.
Keywords: endoplasmic reticulum (ER), endoplasmic reticulum stress (ER stress), prion disease, protein-disulfide isomerase, transgenic mice, calnexin/calreticulin cycle, ERp57 protein, prion protein
Abstract
Although the accumulation of a misfolded and protease-resistant form of the prion protein (PrP) is a key event in prion pathogenesis, the cellular factors involved in its folding and quality control are poorly understood. PrP is a glycosylated and disulfide-bonded protein synthesized at the endoplasmic reticulum (ER). The ER foldase ERp57 (also known as Grp58) is highly expressed in the brain of sporadic and infectious forms of prion-related disorders. ERp57 is a disulfide isomerase involved in the folding of a subset of glycoproteins in the ER as part of the calnexin/calreticulin cycle. Here, we show that levels of ERp57 increase mainly in neurons of Creutzfeldt-Jacob patients. Using gain- and loss-of-function approaches in cell culture, we demonstrate that ERp57 expression controls the maturation and total levels of wild-type PrP and mutant forms associated with human disease. In addition, we found that PrP physically interacts with ERp57, and also with the closest family member PDIA1, but not ERp72. Furthermore, we generated a conditional knock-out mouse for ERp57 in the nervous system and detected a reduction in the steady-state levels of the mono- and nonglycosylated forms of PrP in the brain. In contrast, ERp57 transgenic mice showed increased levels of endogenous PrP. Unexpectedly, ERp57 expression did not affect the susceptibility of cells to ER stress in vitro and in vivo. This study identifies ERp57 as a new modulator of PrP levels and may help with understanding the consequences of ERp57 up-regulation observed in human disease.
Introduction
Prion-related disorders (PrDs)5 are fatal and rare neurodegenerative disorders characterized by spongiform degeneration of the brain accompanied by the accumulation of a misfolded form of the prion protein (PrP) (1). PrDs can be classified as sporadic, familiar, and infectious forms, affecting both humans and other mammals, where Creutzfeldt-Jacob disease (CJD) is the most frequent form in humans. The “protein-only” hypothesis postulates that the pathogenesis of infectious PrD forms involves a conformational change of wild-type PrP (here referred to as PrPC) to a protease-resistant form, initiated by a direct interaction between them (2). In infectious forms of the disease, PrP misfolding occurs mainly in the plasma membrane and in the endocytic pathway, whereas in familial variants the pathological changes in the conformation of PrP are proposed to occur during its synthesis at the endoplasmic reticulum (ER) (3). Human PrPC biosynthesis involves a series of post-translational modifications, including the addition of N-linked glycosylations at Asn181 and Asn197, the formation of a disulfide bridge between Cys179 and Cys214, and the addition of a GPI anchor at Ser230, among other post-translational modifications (1, 4). About 10% of PrPC is not properly folded at the ER and is removed by the proteasome through the ER-associated degradation (ERAD) pathway (5, 6). Once folded, PrPC is transported to the plasma membrane where it locates mainly to lipid raft microdomains via its GPI anchor (4, 7). Although PrP misfolding is the triggering step initiating PrDs, the cellular factors involved in the folding/misfolding of PrP are unknown.
Different reports have shown that the accumulation of misfolded PrP induces ER stress in infectious PrD forms (8–16), although in familial forms of PrDs the involvement of protein folding stress responses is less clear (17–19). ER stress triggers an adaptive reaction known as the unfolded protein response (UPR), which controls the expression of a diverse group of target genes involved in protein folding, quality control mechanisms, and ERAD (20, 21). When these pro-survival mechanisms are unable to recover ER proteostasis, the UPR triggers apoptosis (22). ER stress has been proposed to have two main consequences on PrD progression as follows: (i) it may contribute to neurological impairment due to the repression of the synthesis of a cluster of synaptic proteins (11, 12), and (ii) it may operate as a signal to trigger neuronal loss (9). In vitro experiments have shown that a vicious cycle may operate in prion pathogenesis where prion misfolding predisposes cells to ER stress, which then may facilitate partial misfolding of PrPC and, thus, prion replication (15, 23–26). Importantly, ER stress is also emerging as a driver of most common neurodegenerative diseases, including Alzheimer disease, Parkinson disease, and amyotrophic lateral sclerosis (27).
A proteomic study of sporadic CJD brain tissue revealed that the major protein up-regulated in this pathology is the ER foldase ERp57 (also known as Grp58 or PDIA3) (28). This observation was then confirmed in sporadic cases and also new variant CJD cases, in addition to animal models of infectious PrD (8–10, 13, 14, 18, 23). ERp57 is a member of the protein-disulfide isomerase (PDI) family, a group of ∼21 proteins that catalyze the formation and isomerization of disulfide bonds thereby facilitating protein folding (29). Accumulating evidence supports a functional role of PDIs in a variety of protein misfolding disorders affecting the nervous system (30). ERp57 is a central component of the calnexin (CNX) and calreticulin (CRT) cycle, involved in the folding and quality control of a subgroup of glycoproteins in the ER (31, 32). Although genetic ablation of ERp57 expression in mice is embryonically lethal (33, 34), Erp57-deficient cells do not develop drastic alterations in the folding of glycosylated proteins, and only a small subgroup of putative substrates are affected (35, 36). Beside its role in the CNX and CRT cycle, ERp57 is required as a scaffold protein for the assembly of the heavy chain of the MHC class I peptide loading complex, a function independent of its enzymatic activity involving covalent bonding with tapasin (37–39). Additional functions for ERp57 are reported, including the modulation of ER calcium homeostasis and STAT3 signaling (34, 40, 41). Although PDIs have been proposed to have neuroprotective activities (30), a drug screening identified a pro-apoptotic role of ERp57 and PDIA1 in models of neurodegeneration (42).
Only a few studies have attempted to address the impact of ERp57 in PrDs using cell culture models, and we showed that ERp57 expression protects cells against the toxicity of infectious PrP forms (10). An interactome analysis indicated that PrPC physically associates with ERp57, and pharmacological inhibition of PDI activity increases the levels of prion replication in vitro (10, 43). PrPC also binds to CNX and CRT (44). Expression of PDIA1, the closest family member to ERp57, is also induced in PrD rodent models. PDIA1 expression has protective effects against mutant PrP associated with human disease, reducing ER stress levels (18). In vitro studies also suggested that disulfide bonds may contribute to PrP misfolding and aggregation (45–48). Based on this evidence, here we investigate the possible impact of ERp57 in the expression of PrP using gain- and loss-of-function approaches both in cell culture models and genetically modified mice. Our results support an active involvement of ERp57 in the fine-tuning of PrP protein levels.
Experimental Procedures
Human Samples
The study was conducted according to the provisions of the Helsinki Declaration and was designed in accordance with the relevant Chilean legislation and carried out with the approval of the Ethics Committee of the El Salvador Hospital, Santiago, Chile. Autopsies and human sample use were approved by the Ethics Committee of the Faculty of Medicine, University of Chile, and by the FONDECYT funding agency (protocol number CBA 0323 FMUCH).
Histological Analysis
For histological analysis of human tissue, 10-μm-thick sections were obtained from formalin-fixed, paraffin-embedded blocks of the brains of CJD and control subjects (49). The paraffin-embedded sections were deparaffinized in xylene, followed by rehydration in a decreasing concentration of ethanol solutions. For routine pathological examination, deparaffinized sections from all blocks were stained with hematoxylin and eosin. Sections for immunohistochemistry (IHC) were incubated in 10 mm sodium citrate buffer, pH 6.0, and heated three times in a microwave oven for 5 min for antigen recovery, washed in TBS IHC wash buffer, treated with formic acid for 5 min, and washed again. Sections were then pretreated with 0.3% H2O2 in methanol for 30 min at room temperature to inhibit endogenous peroxidase activity. After washing twice with TBS IHC wash buffer for 5 min each, sections were blocked with 3% normal horse serum for 30 min at room temperature, followed by incubation with anti-ERp57 (1:100, Santa Cruz Biotechnology), anti-PDIA1 (1:100, Abcam), anti-ERp72 (1:100, StressGen), anti-KDEL (1:100, StressGen), and anti-PrP 6D11 (1:500, SIGNET) in a humidified chamber at 4 °C overnight. Negative control sections were incubated with a negative control reagent (Dako) instead of primary antibodies. After washing twice with TBS IHC wash buffer for a total time of 5 min, the sections were incubated with the respective biotinylated secondary antibody for 30 min at room temperature, rinsed twice with TBS IHC wash buffer for a total of 5 min, followed by incubation with the avidin-biotin ABC kit (Vector Laboratories) for 30 min at room temperature. After rinsing with TBS IHC wash buffer, peroxidase labeling was visualized with 3,3′-diaminobenzidine (Impact DAB, Vector Laboratories) for 3 min at room temperature. Sections were then rinsed in tap water, dehydrated for 10 min, cleared, and mounted.
For histological analysis of mouse tissue, animals were anesthetized using ketamine/xylazine and perfused intracardially with ice-cold saline followed by 4% paraformaldehyde in PBS, pH 7.4. After 24 h post-fixation in 4% paraformaldehyde, brains were cryoprotected in 30% sucrose in PBS. Twenty five-micrometer-thick coronal brain sections were obtained using a Leica cryostat (Leica, Nussloch, Germany). Sections were pretreated with 3% H2O2 in methanol for 30 min at room temperature followed by incubation in citrate buffer, pH 6.0, for 15 min at 95 °C for antigen recovery. After two washes in PBS for 5 min each, sections were blocked with 5% BSA, 0.3% Triton X-100 in PBS for 1 h at room temperature followed by incubation with anti-NeuN (1:300, Millipore) and anti-GFAP (1:250, Dako) or anti-ERp57 (1:100, Santa Cruz Biotechnology) in a humidified chamber at 4 °C overnight. After washing three times in PBS, sections were incubated with goat anti-mouse IgG Alexa Fluor 594 and goat anti-rabbit IgG Alexa Fluor 488 (1:1000, Invitrogen) or HRP-conjugated goat anti-rabbit IgG (1:1000, Invitrogen) secondary antibodies for 1 h at room temperature. Immunohistochemistry was performed using 3,3′-diaminobenzidine HRP substrate kit (Vector Laboratories) using the manufacturer's instructions.
Cell Culture, Plasmids, and Cell Transfections
Murine embryonic fibroblasts (MEFs), HEK293T, Neuro2a, and NSC34 cells were cultured in DMEM supplemented with 5% fetal bovine serum and antibiotics (10,000 units/ml penicillin, 10 μg/ml streptomycin), at 37 °C and 5% CO2. Transfections were performed using Effectene (Qiagen) according to the manufacturer's instructions. Expression vectors of 3F4-tagged PrP mutants (PrPCTM, PrPPG14, PrPD177N, and PrPE199K) and GFP fusion proteins were provided by David Harris (Washington University) (50). The generation of PrPC-3F4 and PrPC-GFP constructs was described previously (26). The construct encoding GFP-tagged amyloid precursor protein (APP-GFP) was a gift from Patricia Burgos (Universidad Austral de Chile, Chile) (51). Plasmids to express PDIA1, ERp57, and ERp72 tagged with the V5 epitope were provided by Dr. Neil Bulleid (University of Glasgow, Scotland, UK) (52). The KDEL-DsRED was obtained from Clontech. Lentiviral expression vector pLKO.1 carrying shRNA against ERp57 (target sequence 5′-GACCAGTTTATGTTTGTGGTT-3′) or luciferase (for Mock control) were from The Broad Institute (Boston, MA). Lentiviral particles were generated by standard methods and biosafety rules using HEK293T cells (53, 54). Stable knockdown of ERp57 (and Mock control) in Neuro2a cells was performed by lentiviral transduction of constructs encoding shRNA for ERp57 (or luciferase) followed by selection with puromycin. NSC34 cells stably expressing ERp57 were generated by transfection using Effectene (Qiagen) following the manufacturer's instructions. After 48 h of transfection, cells were selected using G418 (1.3 mg/ml).
SDS-PAGE and Western Blot Analysis
Cell culture pellets or brain tissue was homogenized on ice in RIPA buffer (20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 0.5% Triton X-100) containing a protease inhibitor mixture (Roche Applied Science, Basel, Switzerland). Protein concentrations were determined by micro-BCA assay (Pierce). The equivalent of 30–50 μg of total protein was generally loaded onto 10% SDS-polyacrylamide gels and analyzed by Western blot. The following antibodies and dilutions were used: anti-PrP (6D11) (1:5000, SIGNET); anti-PrP (3F4) (1:5000, Abcam); anti-ERp57 (1:2000, Santa Cruz Biotechnology); anti-PDIA1 (1:3000, Abcam); anti-ERO1Lα (1:2000, Novus Biologicals); anti-calnexin (1:2000, StressGen); anti-BiP (1:3000, Abcam); anti-β-actin (1:2000, Cell Signaling); anti-Hsp90 (1:3000, Santa Cruz Biotechnology); and anti-V5 (1:5000, Invitrogen). After the incubation with the primary antibody, membranes were incubated for 1 h at room temperature with HRP-conjugated secondary antibodies (all from Invitrogen). After washing, detection was performed by enhanced chemiluminescence assay (Amersham Biosciences, Cardiff, UK).
RNA Extraction and Quantitative Real Time PCR
Total RNA from tissues was isolated using TRIzol as recommended by the supplier (Life Technologies, Inc., 15596-018). The cDNA was synthesized with SuperScript III reverse transcriptase (Life Technologies, Inc., 11754250) using random primers p(dN)6 (Roche Applied Science). Quantitative PCRs were performed using standard protocols (55). Actin mRNA was monitored as a housekeeping control. The following primers were used: Erp57, forward 5′-GAGGCTTGCCCCTGAGTATG-3′ and reverse 5′-GTTGGCAGTGCAATCCACC-3′; Xbp1s, forward 5′-TGCTGAGTCCGCAGCAGGTG-3′ and reverse 5′-GACTAGCAGACTCTGGGGAAG-3′; Prp, forward 5′-TCATCCCACGATCAGGAAGAT-3′ and reverse 5′-TGCGTCACCCAGTACCAGAA-3′; Edem, forward 5′-AAGCCCTCTGGAACTTGCG-3′ and reverse 5′-AACCCAATGGCCTGTCTGG-3′; Bip, forward 5′-TCATCGGACGCACTTGGAA-3′ and reverse 5′-CAACCACCTTGAATGGCAAGA-3′; Pdia1, forward 5′-AGTTCGCCCCAACCAGTACTT-3′ and reverse 5′-CAAGATCAAGCCCCACCTGAT-3′; Chop, forward 5′-GTCCCTAGCTTGGCTGACAGA-3′ and reverse 5′-TGGAGAGCGAGGGCTTTG-3′; and actin, forward 5′-CTCAGGAGGAGCAATGATCTTGAT-3′ and reverse 5′-TACCACCATGTACCCAGGCA-3′. Splicing of XBP1 mRNA was also evaluated by conventional PCR using primer: Xbp1, forward 5′-ACACGCTTGGGAATGGACAC-3′ and reverse 5′-CCATGGGAAGATGTTCTGGG-3′, or by conventional PCR using primers Xbp1, forward 5′-AAACAGAGTAGCAGCGCAGACTGC-3′ and reverse 5′-GGATCTCTAAAACTAGAGGCTTGGTG-3′, followed by digestion with the restriction enzyme PstI as described previously (56).
Proteinase K (PK), Filter Trap, and PNGase F Experiments
PK assays were performed using a protocol described previously (26). Twenty micrograms of total protein in 1% Nonidet P-40 buffer were treated for 30 min at 37 °C with different concentrations of PK (2, 4, and 6 μg/ml). Proteolysis was stopped by adding phenylmethylsulfonyl fluoride followed by SDS-PAGE sample buffer and then heating the samples for 5 min at 95 °C. For filter trap, 25 μg of total protein in 1% Nonidet P-40 buffer was diluted in PBS containing 1% SDS to a final concentration of 0.25 μg/μl and filtered through cellulose acetate membrane with a pore size of 0.22 μm in a dot-blot apparatus (Bio-Rad). For loading control, 25 μg of total protein was analyzed by SDS-PAGE and Western blot or, in addition, diluted in PBS and loaded onto PVDF membrane in a dot-blot apparatus followed by Ponceau S staining. For deglycosylation assays, samples were treated with PNGase F (New England Biolabs) following the manufacturer's recommendations. Briefly, samples were cooled to 25 °C, and then the reaction buffer and 10 units of PNGase F were added. After 1 h at 37 °C, SDS-PAGE sample buffer was added, and samples were heated for 5 min at 95 °C followed by electrophoresis. Finally, samples were analyzed by Western blot using the anti-PrP (6D11) or (3F4) antibodies.
Pulse and Chase Experiments
Eighteen hours after transfection with expression vectors for 3F4-tagged PrP, MEFs were starved for 15 min in Met/Cys-free medium, pulsed for 10 min with 50 μCi of [35S]Met/Cys in 1 ml of starvation medium/dish, and chased for the indicated times with DMEM supplemented with 5 mm “cold” Met/Cys. Post-nuclear supernatants were prepared by solubilization of cells in 800 μl/dish ice-cold 2% CHAPS in HEPES-buffered saline, pH 6.8, containing 20 mm N-ethylmaleimide and protease inhibitors. CHAPS-insoluble material was separated by a 10-min centrifugation at 10,000 × g. Immunoprecipitations were performed by adding protein A beads (Sigma; 1:10, w/v swollen in HEPES-buffered saline) and the anti-PrP (3F4) antibody to the cleared extract followed by incubation for 2 h at 4 °C. Immunoprecipitates were extensively washed three times with 0.5% CHAPS in HEPES-buffered saline and resuspended in sample buffer for SDS-PAGE. Relevant bands were quantified using the ImageQuant software (GE Healthcare). Gels were also exposed to BioMax films (Eastman-Kodak Co.) and scanned with an AGFA scanner (Mortsel, Belgium).
Cycloheximide-chase experiments were performed in MEFs transfected with PrP-GFP or APP-GFP constructs. Briefly, after 24 h of transfection, cells were re-plated in a 48-well format and then 24 h later treated with 50 μg/ml cycloheximide for different time points. After the treatment, cells were detached by trypsinization and analyzed by flow cytometry.
Immunoprecipitations
HEK293T cells were transfected using Effectene transfection reagent. After 48 h, cells were collected and washed once in 1 ml of PBS pH 7.4. Subsequently, cells were resuspended in 500 μl of Nonidet P-40 buffer (0.2% Nonidet P-40, 50 mm Tris-HCl, pH 7.5, 150 mm NaCl) plus a protease inhibitor mix (Roche Applied Science) and incubated overnight at 4 °C. Cell lysate was centrifuged at 10,000 × g for 5 min at 4 °C. Subsequently, the supernatant was incubated with V5 antibody-coated agarose beads (V5 protein purification kit, MBL International) under constant agitation for 4 h at 4 °C. Then, the beads were washed three times with Nonidet P-40 buffer, and the antibody-bound complexes were released by incubation with V5 peptide (MBL International) for 30 min at room temperature. Finally, the supernatant was obtained by centrifugation and analyzed by Western blot.
Generation of Conditional ERp57 Knock-out Mice
The ERp57 floxed mice were described previously where exons 2 and 3 were flanked with two loxP sites (33), and mice were kindly provided by Dr. Günther Hämmerling (German Cancer Research Center, Heidelberg, Germany). Mice expressing Cre recombinase under the control of the nestin promoter were obtained from The Jackson Laboratory (B6.Cg-Tg(Nes-cre)1Kln/J, 003771). To generate mice deficient in ERp57 in the central nervous system (CNS), ERp57 floxed animals were crossed with Nestin-Cre transgenic mice as we described before (8). Mice genotypes were designated as follows: ERp57WT (wild-type), ERp57HET (heterozygous, carrying one knock-out allele), and ERp57cKO (conditional knock-out). All experiments and animal care follow the Institutional Review Board's Animal Care of the University of Chile (CBA number 0305 FMUCH). The following primers were used for genotyping of mice: Erp57 floxed allele, forward 5′-CGCCAGCCTCTCCATTTAG-3′ and reverse 5′-CAGAGATCCTGCCTCTG-3′; Cre forward 5′-GCGGTCTGGCAGTAAAAACTATC-3′ and reverse 5′-GTGAAACAGCATTGCTGTCACTT-3′.
Generation of ERp57 Transgenic Mouse Model
The transgenic mouse line overexpressing ERp57 was recently described (57) using the expression plasmid MoPrP.XhoI (58). In brief, the human ERp57 cDNA (Gene ID 2923) was introduced into the plasmid using the XhoI restriction site. The resulting plasmid expressing the ERp57 sequence under the control of the PrP promoter was used to generate transgenic mice at the “Centro de Estudios Científicos,” Valdivia, Chile. The plasmid was purified and linearized for microinjection in mice with an FVB background. The primers used for genotyping were forward 5′-AATTCCTGGATGCTGGGCACAAAC-3′ and reverse 5′-TCTGCTTGTCATCGTCGTCCTTGT-3′. Selected transgenic mouse lines were backcrossed into a C57BL/6 background for more than 10 generations. All experiments and animal care followed the Institutional Review Board's Animal Care, University of Chile (CBA number 0305 FMUCH).
Pharmacological Induction of ER Stress
For induction of ER stress in vitro, the ER stressors tunicamycin, thapsigargin, or brefeldin A were added to the cell culture medium followed by measurement of ER stress markers by Western blot or quantitative real time PCR analysis as we reported previously (56, 59). The MTT method was employed to measure cell viability according to the manufacturer's instructions (Promega). For induction of ER stress in vivo, mice received a single intraperitoneal injection of tunicamycin (5 mg/kg) diluted in sterile 150 mm glucose solution as described before (60). Control mice received an intraperitoneal injection of vehicle (5% DMSO in 150 mm glucose solution). Mice were euthanized, and tissue was collected 24 h after injection.
Statistical Analysis
For statistical analysis, GraphPad Prism software version 5.01 and SigmaPlot were used. All graphs show means with S.E. Significance was calculated using Student's t test or one- or two-way ANOVA with Bonferroni post hoc test.
Results
ERp57 Levels Are Increased in Neurons of CJD Patients
Although it is reported that ERp57 levels are augmented in post-mortem brain tissue derived from CJD patients (9, 28), the specific cell types responding have not been defined. To analyze the expression pattern of ERp57 in the brain, we performed immunohistochemistry studies. We confirmed the diagnosis of CJD patients by H&E staining and immunohistochemistry of PrP. The three subjects analyzed presented signs of spongiosis, neuronal loss, and gliosis (Fig. 1, A and B). We then evaluated the expression pattern of ERp57 in the cerebellum using immunohistochemistry and found higher ERp57 levels in Purkinje cells of CJD patients compared with control subjects (Fig. 1C). We also monitored the expression levels of other ER chaperones in these patient samples. Analysis of PDIA1 distribution, the closest homologue to ERp57, also revealed to different extents an up-regulation in CJD cases, whereas ERp72 did not show clear changes compared with control subjects (Fig. 1D). KDEL staining, which mostly recognizes BiP/Grp78 and Grp94, depicted a slight increase in some of the CJD cases analyzed with variable results. These results confirmed previous findings supporting the up-regulation of ERp57 levels in post-mortem brain tissue derived from CJD cases.
FIGURE 1.

Detection of ERp57 in brain tissue of patients with Creutzfeldt-Jakob disease. A–D, brain tissue from three CJD patients and three control subjects was fixed in formalin. After a month of fixation, tissue was embedded in paraffin and cut into 10-μm-thick slices. Cerebral tissues were then stained with hematoxylin and eosin (H&E) (A), processed for immunohistochemistry using anti-PrP (3F4) antibody in cortex (B), or anti-ERp57 (C), anti-PDIA1, anti-ERp72, and anti-KDEL (D) antibodies in cerebellum. Black arrowheads indicate positive reaction to ERp57 in Purkinje cells. Scale bars, 50 μm.
ERp57 Deficiency Reduces PrP Levels
Because ERp57 selectively catalyzes the folding and disulfide bond formation of a subset of glycosylated proteins, we decided to analyze the expression levels of endogenous PrPC in MEFs that are deficient for ERp57 (ERp57KO) (36). We observed a reduction in the steady-state levels of PrPC in ERp57KO cells when compared with wild-type control cells by Western blot analysis (Fig. 2A). Quantification of relative PrPC levels in ERp57KO MEFs indicated a reduction near 25% (Fig. 2A, right panel). To confirm these results in a different cell culture system, we knocked down ERp57 in Neuro2a cells after stable delivery of an shRNA construct using lentiviral vectors. A control shRNA against luciferase mRNA was employed (Mock). Targeting ERp57 resulted in significant reduction of PrPC steady-state levels (Fig. 2B).
FIGURE 2.

PrP levels in ERp57-deficient cells. A, endogenous levels of PrP in ERp57-deficient (ERp57KO) and wild-type control (ERp57WT) MEFs were evaluated by Western blot (left panel) using the anti-PrP (6D11) antibody. Quantification of three independent experiments (right panel) was performed. Hsp90 levels were monitored as a loading control. B, detection of endogenous levels of PrP in Neuro2a cells stably knocked down for ERp57. Neuro2a cells stably expressing shRNA against luciferase were used as control (Mock). β-Actin was monitored as a loading control. M, Mock; E, ERp57. C, ERp57-deficient and wild-type control MEFs were transfected with 3F4-tagged PrPC or PrPD177N constructs. After 48 h, PrP levels were analyzed by Western blot using the anti-PrP (3F4) antibody. Hsp90 was used as a loading control. D, ERp57-deficient and wild-type control MEFs were transfected with PrPC-GFP or PrPD177N-GFP constructs. After 48 h, GFP fluorescence was analyzed by flow cytometry. Three independent experiments were performed. E, ERp57-deficient and wild-type MEFs were transfected with a construct encoding APP-GFP. After 48 h, cells were analyzed by flow cytometry (left panel) or fluorescence microscopy (right panel). F, Neuro2a cells were transfected with constructs encoding 3F4-tagged PrPC or its PrD-related mutants. Transfection with the empty vector was used as control (Mock). After 48 h, protein (upper panel) and mRNA (lower panel) levels of endogenous ERp57 were measured by Western blot and real time PCR, respectively. G, levels of spliced XBP1 mRNA were monitored in Neuro2a cells overexpressing PrPC or its PrD-related mutants described in F using a PstI- and RT-PCR-based assay (upper panel) or real time PCR to specifically amplify the spliced variant (lower panel). H, Neuro2a cells were transfected with constructs encoding 3F4-tagged PrPC or PrPD177N and 48 h later treated with different concentrations of thapsigargin. After 16 h of treatment, levels of Erp57 (upper panel) and Xbp1s (lower panel) mRNA were measured by real time PCR. Bars in F–H indicate average and standard deviation of three determinations. When indicated, statistical analysis was performed using Student's t test in A and one-way ANOVA with Bonferroni post hoc test in D. Mean ± S.E. with p values: n.s., p > 0.05; *, p ≤ 0.05; ***, p ≤ 0.001. Densitometric analyses were performed using Image J.
We then tested the effects of ERp57 deficiency on the levels of a mutant PrP form linked to CJD. We transiently transfected ERp57KO and control MEFs with expression vectors for PrPC or the murine PrPD177N variant, equivalent to human mutation D178N linked to CJD, in addition to empty vector (Mock). All proteins contained the 3F4 tag to differentiate them from the endogenous protein. We then determined PrP levels using the anti-PrP 3F4 antibody and observed that ERp57 deficiency resulted in lower levels of both PrP forms (Fig. 2C). To corroborate these findings, ERp57KO MEFs were transiently transfected with PrP-GFP fusion constructs (PrPC-GFP and PrPD177N-GFP), and the percentage of cells expressing GFP was quantified by flow cytometry analysis (Fig. 2D). Transfection of a GFP plasmid alone showed similar expression in both ERp57 WT and KO cells (Fig. 2D). Again, ERp57 deficiency significantly reduced the levels of wild-type and mutant PrP fused to GFP.
We then performed control experiments to determine whether changes in PrP observed in ERp57KO cells are due to a generalized deficit in protein folding in the ER. To this aim, we overexpressed amyloid precursor protein (APP) fused to GFP (APP-GFP), another protein that traffics through the secretory pathway that undergoes glycosylation. We monitored the levels and distribution of APP using flow cytometry and fluorescent microscopy analysis and did not observe changes in APP-GFP levels or its expression pattern when ERp57WT and ERp57KO MEFs were compared (Fig. 2E). These results suggest that ERp57 deficiency may affect client proteins, such as PrP, in a more specific manner.
Although mutant PrP variants associated with familial PrDs are retained at the ER, data linking their expression with the induction of an ER stress response is controversial (17–19). To study the impact of mutant PrP on ER physiology, we expressed in Neuro2a cells several PrP mutants including the murine PrPD177N variant, PrPPG14 (nine-octapeptide insertion) linked to fatal familial insomnia, PrPE199K (equivalent to human PrPE200K) associated with familial CJD, and the point mutant L9R/3AV that generates an abnormal pathogenic form called PrPCTM (carboxyl-terminal mutant) (61–63). All constructs contained the human PrP-3F4 epitope for detection (Fig. 2F) (50). These mutants were selected because of previous studies showing their partial retention at the ER and Golgi compartments (50, 64, 65). Unexpectedly, expression of all these mutant PrP forms did not trigger the up-regulation of ERp57 at the protein (Fig. 2F, upper panel) or mRNA level (Fig. 2F, lower panel). Similarly, analysis of XBP1 mRNA splicing, a classical marker of ER stress (22), did not reveal any changes upon mutant PrP expression (Fig. 2G). We then tested whether mutant PrP overexpression could enhance the induction of ERp57 after treatment with the pharmacological ER stress agent thapsigargin. Again, the up-regulation of Erp57 or spliced Xbp1 mRNAs were not altered in Neuro2a cells expressing wild-type or mutant PrP (Fig. 2H). In summary, our results suggest that PrD-linked mutant PrP does not trigger the up-regulation of ERp57 but under resting conditions it controls PrP steady-state levels.
ERp57 Overexpression Enhances PrP Expression
We then performed gain-of-function studies by stably overexpressing a V5-tagged version of ERp57 (35) in the NSC34 neuronal cell line. First, we analyzed the expression levels of endogenous murine PrPC by Western blot. Consistent with our previous results, augmented steady-state levels of PrPC were observed in these cells when compared with control Mock cells (Fig. 3A). We then assessed the effects of ERp57-V5 overexpression on the levels of a set of PrP mutants linked to PrDs. We confirmed the altered localization of these PrP mutants using GFP fusion constructs together with the ER marker KDEL-dsRED, observing reduced expression at the plasma membrane and increased intracellular accumulation (Fig. 3B). We then transiently transfected PrP-3F4 constructs in NSC34 cells stably overexpressing ERp57-V5 and determined the relative levels of PrP after 48 h using Western blot analysis. In agreement with our loss-of-function experiments, overexpression of ERp57 increased the levels of PrPC and the mutants D177N, PG14, and E199K (Fig. 3C). ERp57 had only a slight effect on PrPCTM, which had low expression levels. We then evaluated the misfolding of PrP in these experiments by treating protein extracts with different concentrations of PK. Consistent with increased levels of PrP upon ERp57-V5 overexpression, the relative amount of PK-resistant forms of PrP was also augmented under these conditions (Fig. 3D). These observations indicate that ERp57 expression modulates steady-state levels of PrP wild-type and mutant forms in different cell culture models.
FIGURE 3.
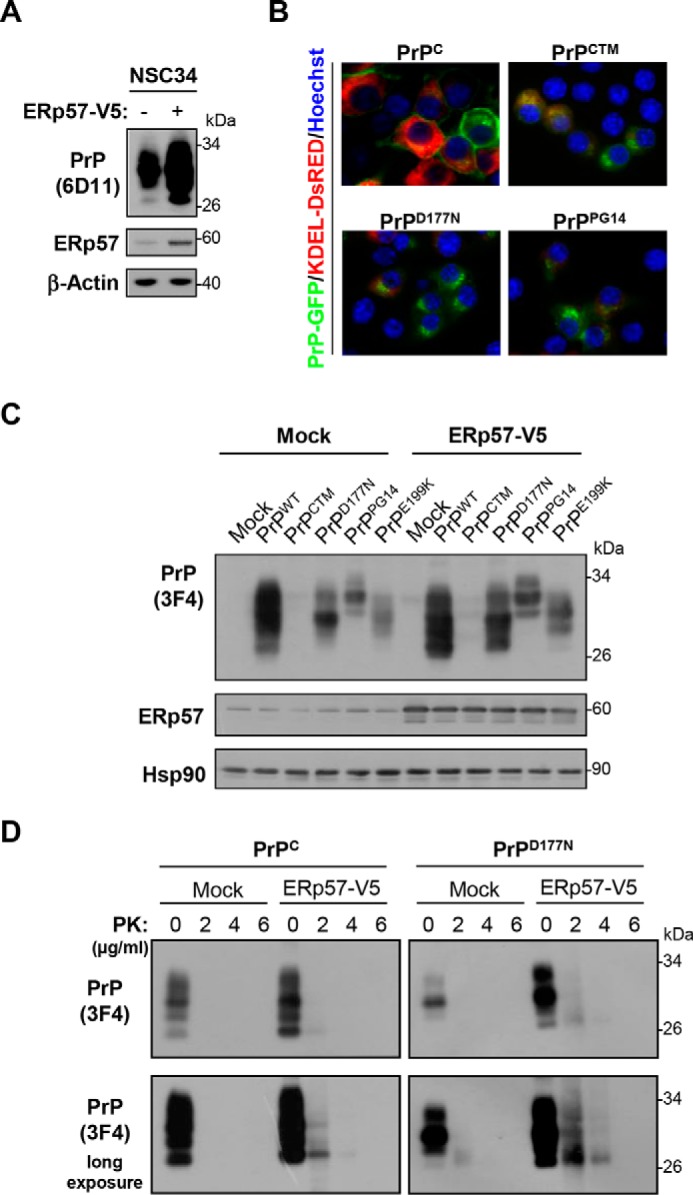
PrP levels in NSC34 cells overexpressing ERp57. A, NSC34 cells were transfected with empty vector (pcDNA3, Mock) or an ERp57 expression vector. After 24 h of transfection, cells were selected with the antibiotic G418 for 2 weeks. Then, ERp57 and endogenous PrP levels were analyzed by Western blot. β-Actin was used as a loading control. B, NSC34 cells were transiently co-transfected with different PrP-GFP constructs (PrPC-GFP, PrPCTM-GFP, PrPD177N-GFP, or PrPPG14-GFP) together with vector encoding for the ER marker KDEL-dsRED (red). Localization of PrP (green) was analyzed by confocal microscopy. The nucleus was stained with Hoechst (blue). C, NSC34 cells stably overexpressing ERp57 were transiently transfected with constructs encoding 3F4-tagged PrPc and its PrDs-related mutants (PrPCTM, PrPD177N, PrPPG14, and PrPE199K). After 48 h, the steady-state levels of PrP were analyzed by Western blot using the anti-PrP (3F4) antibody. Hsp90 was monitored as a loading control. D, PK resistance of PrPC and PrPD177N was assessed in protein extracts from NSC34 cells transiently co-expressing ERp57 for 48 h by incubation with the indicated amounts of the protease for 30 min at 37 °C followed by Western blot analysis.
ERp57 Deficiency Alters PrP Maturation
Based on the observation that ERp57 controls the expression of PrP at basal levels, we decided to monitor in detail its synthesis and maturation in ERp57KO cells. We performed pulse-chase experiments in ERp57KO and wild-type MEFs transiently transfected with 3F4-tagged PrPC. In control ERp57WT cells, the classical pattern of PrP maturation was observed over time, where nonglycosylated, mono-glycosylated, and di-glycosylated PrP bands were visualized, in addition to PrP species with higher molecular weight showing more complex glycosylations (Fig. 4A, left panel). The nature of these bands was confirmed after the treatment of protein extracts with PNGase to deglycosylate PrP (Fig. 4B). We then studied the synthesis and maturation of PrPC in ERp57-deficient cells between 10 and 180 min after radioactive pulse (Fig. 4A, right panel). Under these conditions, we found a nearly 50% reduction in the average half-life of total overexpressed PrPC in ERp57-deficient cells (Fig. 4A). Importantly, no differences in the rate of PrP synthesis were found between ERp57KO and control cells as measured by the PrP radioactive signal detected when we started the chase (t = 0–10 min) (Fig. 4A). Surprisingly, the band pattern of PrP in SDS-PAGE analysis performed without the reducing agent DTT in the sample buffer remained the same, suggesting the absence of an altered oligomerization into S–S-linked assemblies in ERp57-deficient cells (Fig. 4A, lower panels), which is in contrast to the results reported for some ERp57 substrates (36).
FIGURE 4.

Effects of ERp57 on PrP maturation. A, ERp57-deficient (ERp57KO) and wild-type control (ERp57WT) MEFs were transfected with a construct for PrPC-3F4. After 16 h, a radioactive pulse was conducted with 0.10 mCi of 35S-Promix per plate. The chase of PrP protein was performed from 10 to 180 min. Then cells were lysed, and PrP was purified by immunoprecipitation using anti-PrP (3F4) antibody. Samples were analyzed in 12% SDS-polyacrylamide gels under thiol-reducing (+DTT, upper panel) and nonreducing (−DTT, lower panel) conditions and revealed by autoradiography. White, gray, and black arrowheads indicate di-, mono-, and nonglycosylated forms of PrP, respectively. The asterisk indicates higher molecular weight forms of PrP containing more complex glycosylations. B, deglycosylation analysis of PrPC in MEFs after treatment of protein extracts with PNGase F. The different glycosylation states of PrP are identified as in A. C, filter-tap analysis of aggregates of endogenous PrPC in ERp57-deficient and control MEFs under basal conditions. Dot-blots on PVDF membrane followed by Ponceau S staining or Western blot analysis of Hsp90 and PrP were performed as loading controls. Bands were cropped from their original position for the clarity of presentation. All samples were run in the same electrophoresis and detected in the same Western blot. D, thiol reduction assay using dithiothreitol (DTT) was performed to determine the dependence of PrPC aggregates on disulfide cross-links using filter-trap analysis. Dot-blot on PVDF membrane followed by Ponceau S staining was used as the loading control. E, ERp57-deficient and wild-type MEFs were transfected with PrPC-GFP or APP-GFP constructs. After 48 h, cells were treated with 50 μg/ml cycloheximide (CHX) for indicated time points, and fluorescence intensity was measured by flow cytometry. The initial fluorescence was normalized to 1 unit for control cells to monitor relative protein decay over time. Data represent the average and standard error of three independent experiments. Statistical analysis was performed using two-way ANOVA with Bonferroni post hoc test. Mean ± S.E. shown; **, p ≤ 0.01.
To further assess whether PrP forms protein aggregates undetectable by Western blot, we carried out filter trap analysis to evaluate the presence of large species that are retained in 0.22-μm pores of cellulose acetate membranes. Remarkably, we observed that ERp57-deficiency in MEFs promotes the aggregation of endogenous PrP at basal levels (Fig. 4C). Moreover, these large aggregates were susceptible to DTT treatment, suggesting disulfide-dependent interactions (Fig. 4D). These results suggest that a reduced folding efficiency of PrP in ERp57KO cells leads to aberrant intermolecular disulfide bonds in the protein.
We then complemented these experiments by monitoring the decay of PrPC-GFP by flow cytometry analysis in cells treated with cycloheximide to inhibit protein synthesis. Again, a lower stability of PrPC was confirmed in ERp57KO MEFs when compared with control cells (Fig. 4E, upper panel). In contrast, APP-GFP showed similar stability in both cell lines under the same experimental conditions (Fig. 4E, lower panel). Taken together, these results suggest that ERp57 may participate in the folding or the maturation of PrPC.
ERp57 Forms a Protein Complex with PrP
Based on our results showing that ERp57 modulates PrPC levels and affects its maturation, we then investigated the possible physical interaction between PrPC and ERp57. We co-transfected a PrPC-3F4 construct together with a V5-tagged version of ERp57 in HEK cells. After 48 h, we immunoprecipitated ERp57-V5 and analyzed the possible association with PrPC using Western blot analysis. We observed an interaction between ERp57-V5 and PrPC-3F4, where the mono- and di-glycosylated forms of PrP were preferentially co-immunoprecipitated (Fig. 5A). As control, we then analyzed the possible association of PrP with V5-tagged PDIA1 or ERp72, two structurally related proteins to ERp57 (29). Immunoprecipitation of PrPC-3F4 revealed only an interaction with PDIA1, but not with ERp72 (Fig. 5A). We then performed similar experiments with the PrPD177N mutant. We detected an association between PrPD177N and ERp57-V5 with a similar efficiency of co-immunoprecipitation as PrPC (Fig. 5B). However, the di-glycosylated form of PrPD177N was enriched in these experiments (Fig. 5B). These results suggest that ERp57 and PDIA1 form protein complexes with PrP.
FIGURE 5.

Immunoprecipitation of PrP with ERp57 and PDIA1. A, HEK293T cells were co-transfected with expression vectors for V5-tagged PDIA1, ERp57, or ERp72 together with 3F4-tagged PrPC. After 48 h, V5-tagged proteins were immunoprecipitated (IP) and eluted with V5 peptide. The possible interaction of PDIs with PrP was analyzed by Western blot (WB) using anti-PrP (3F4) and anti-V5 antibodies. Hsp90 was monitored as a loading control. The inputs and elutions (IP: V5) are shown. B, HEK293T cells were co-transfected with expression vectors for V5-tagged ERp57 together with 3F4-tagged PrPC or PrPD177N. After 48 h, V5-tagged proteins were immunoprecipitated and eluted with V5 peptide. The possible interaction of ERp57 with PrP was analyzed by Western blot using anti-PrP (3F4) and anti-V5 antibodies. The inputs and elutions (IP: V5) are shown.
ERp57 Expression Does Not Influence the Susceptibility of Cells to ER Stress
Our results together with previous reports suggest that ERp57 may have a dual activity on PrDs, modulating PrP synthesis and the protection of cells from the ER stress reaction generated in the disease. Thus, we decided to monitor the impact of ERp57 expression on the susceptibility of cells to experimental ER stress. ERp57KO and control MEFs were treated with the ER stress agent tunicamycin for 16 h, and the expression levels of different ER folding components were monitored, including ERp57, PDIA1, CNX, ERO1Lα, and BiP. No changes were observed between genotypes at basal levels or in cells undergoing ER stress (Fig. 6A), suggesting the lack of clear compensatory changes upon Erp57 deletion. In agreement with these results, analysis of cell viability in cells treated with three different ER stress agents indicated no differential vulnerability of ERp57-deficient cells (Fig. 6B).
FIGURE 6.

Effect of ERp57 expression on ER stress responses. A, ERp57WT and ERp57KO MEFs were treated with the ER stressor tunicamycin (Tm) for 16 h, and the levels of indicated ER chaperones and cofactors were measured by Western blot analysis. Hsp90 was employed as a loading control. B, ERp57WT and ERp57KO MEFs were treated or not treated (NT) with Tm, thapsigargin (Thg), or brefeldin A (Bref A), and cell viability was assessed 16 h later by the MTT assay. Data represent mean and S.D. of three measurements. C, NSC34 cells transiently overexpressing V5-tagged ERp57 or Mock were treated with Tm for the indicated time points. ER stress responses were assessed by monitoring the levels of XBP1 spliced (Xbp1s) and unspliced (Xbp1u) mRNAs, in addition to Chop mRNA levels by semi-quantitative RT-PCR. Actin was used as housekeeping control. D, NSC34 cells overexpressing V5-tagged ERp57 and Mock control cells were treated with the ER stressor Tm for 16 h, and the levels of indicated ER folding components were measured by Western blot analysis. Hsp90 was employed as a loading control. E, NSC34 cells overexpressing V5-tagged ERp57 and Mock control cells were treated with Tm or Thg, and cell viability was assessed 16 h later by the MTT assay. Data represents mean and S.D. of three measurements.
We then performed similar experiments in NSC34 cells overexpressing ERp57-V5. Overexpression of ERp57 did not reduce the activation of Xbp1 mRNA splicing or the up-regulation of the ER stress pro-apoptotic factor Chop in cells treated with tunicamycin (Fig. 6C). Consistent with these results, no changes were observed in the levels of a panel of ER folding factors in ERp57-V5 overexpressing cells at basal levels or after treatment with tunicamycin (Fig. 6D). Finally, these cells did not shown any protection against ER stress when cell viability was monitored with the MTT assay (Fig. 6E). In summary, our results indicate that ERp57 expression does not influence the global response to ER stress, but it has a specific effect on controlling the levels of PrP.
PrP Expression Pattern Is Altered in the Brain of a Conditional ERp57 Knock-out Mouse
Although ERp57 deficiency in mice is embryonic lethal, a viable conditional B cell-specific knock-out mouse was described before (33). We generated a CNS-specific ERp57 knock-out mouse by crossing that floxed animal with a Cre transgenic mouse under the control of the Nestin promoter. Targeting Erp57 in the nervous system bypassed embryonic lethality because we were able to generate ERp57 heterozygous (ERp57HET) and conditional knock-out mice (ERp57cKO). We confirmed the reduction of Erp57 expression in both genotypes at the mRNA level using real time PCR with a near 50% reduction in ERp57HET animals and a complete loss of ERp57 mRNA expression in ERp57cKO mice (Fig. 7A). These results were then validated in brain cortex, hippocampus, and cerebellum using Western blot analysis (Fig. 7B).
FIGURE 7.

Endogenous PrP levels on a CNS-specific conditional knock-out mouse for ERp57. A, Erp57 floxed animals were crossed with Nestin-Cre transgenic mice to generate CNS-specific knock-out and heterozygous animals. The expression levels of ERp57 were evaluated by quantitative real time PCR in brain cortex tissue of control (ERp57WT), heterozygous (ERp57HET), and homozygous (ERp57cKO) animals. B, ERp57 levels were analyzed in mice of all three genotypes by Western blot of brain cortex, hippocampus, and cerebellum. As a loading control, β-actin levels were determined. C, level of PrP mRNA was determined by real time PCR in brain cortex of all three genotypes. D, PrP levels were analyzed by Western blot using the anti-PrP (6D11) antibody in cortex tissue of ERp57WT (n = 4), ERp57HET (n = 4), and ERp57cKO (n = 4) animals. ERp57 levels were also determined. β-Actin levels were measured as loading control. E, PrP bands were quantified by densitometric analysis using the ImageJ program. Di-glycosylated (left panel), mono-glycosylated (middle panel), and nonglycosylated (right panel) forms of endogenous PrP were quantified and normalized to the loading control. F, expression levels of Bip, Edem1, Pdia1, and Chop mRNA were determined in the cortex of ERp57WT, ERp57HET, and ERp57cKO animals by real time PCR. All values were normalized to actin mRNA. G, levels of indicated ER folding components were determined in the cerebellum of ERp57WT, ERp57HET, and ERp57cKO animals by Western blot. β-Actin was employed as a loading control. H, immunofluorescence against NeuN (red) and GFAP (green) to visualize the content of total neurons and astrocytes, respectively, in spinal cord ventral horn (n = 5). Statistical analyses were performed using one-way ANOVA and Bonferroni multiple comparison test. Mean ± S.E. is shown; p values are as follows: n.s., p > 0.05; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
Next, we evaluated mRNA levels of PrP in brain cortex of ERp57-deficient mice (Fig. 7C). Despite the slight induction observed in ERp57HET animals, deficiency of ERp57 in the nervous system had no major impact on transcription of PrP (Fig. 7C). Then, the protein pattern of endogenous PrP was evaluated in the brain of ERp57-deficient mice (Fig. 7D). Quantification of different PrP species revealed a significant reduction of the mono-glycosylated PrP form in the cortex of ERp57cKO mice (Fig. 7E, middle panel). We also found a small change in the levels of the nonglycosylated PrP form (Fig. 7E, right panel), although no differences in the levels of the di-glycosylated form were detected (Fig. 7E, left panel).
To assess whether targeting ERp57 in the brain alters ER proteostasis, we monitored the mRNA levels of a few ER stress-responsive genes, including Bip, Edem1, Pdi, and Chop. Overall, no changes where observed when the three genotypes were compared using cerebellar extracts (Fig. 7F). Similarly, analysis of the expression levels of CNX, ERO1Lα, and BiP did not reveal any up-regulation in ERp57-deficient animals (Fig. 7G). Qualitative immunofluorescence analysis indicated that ERp57cKO mice do not show evident signs of neuronal loss or astrogliosis in CNS tissue (Fig. 7H). In summary, these data indicate that ERp57 deficiency in the nervous system impacts the steady-state levels of PrP expression in vivo.
Overexpression of ERp57 in Neurons Increases PrP Expression in Vivo
We recently generated a transgenic mouse that overexpresses a FLAG-tagged version of human ERp57 under the control of the PrP promoter (ERp57-Tg) (57). The overexpression of ERp57-FLAG in transgenic mice was confirmed in the cerebellum using Western blot analysis, observing a near 1.5-fold increase in ERp57 levels compared with nontransgenic littermates (non-Tg) (Fig. 8A). The overexpression of ERp57 was also confirmed by immunohistochemistry (Fig. 8B). We then monitored PrP levels in these brain extracts. Remarkably, a significant increase in the total levels of PrP was observed in the cerebellum of 2-month-old ERp57-FLAG transgenic mice (Fig. 8C), whereas the mRNA levels of PrP were not altered (Fig. 8D).
FIGURE 8.
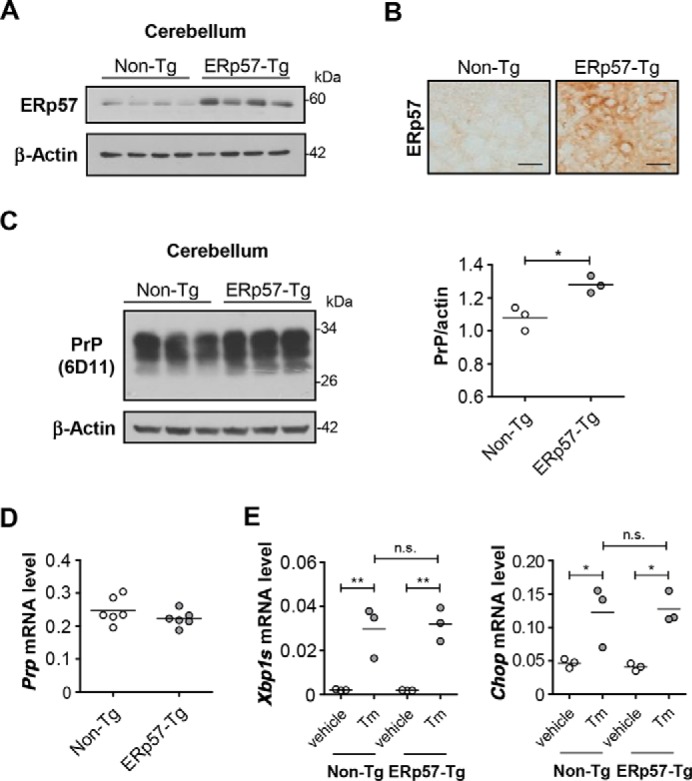
Endogenous PrP levels in animals overexpressing ERp57 in the CNS. A neuron-specific Erp57 transgenic mouse was generated using the PrP promoter to drive ERp57-FLAG expression. A, ERp57 protein levels were evaluated in the cerebellum of ERp57 transgenic mice (n = 4) compared with nontransgenic littermates (n = 4) by Western blot analysis. β-Actin was used as loading control. B, immunohistochemistry to ERp57 was also employed to confirm overexpression of the human protein in brain cortex of transgenic mice compared with nontransgenic littermates. Scale bar, 20 μm. C, endogenous PrP levels were assessed in the cerebellum of ERp57 transgenic animals (n = 3) compared with nontransgenic littermates (n = 3). Anti-PrP (6D11) antibody was used for Western blot detection. β-Actin was used as loading control. Densitometric analysis of PrP levels is shown in the right panel. D, level of PrP mRNA in cerebellum of ERp57 transgenic animals was determined by real time PCR. E, ERp57 transgenic mice and nontransgenic littermates were injected intraperitoneally with 1 μg of the ER stressor tunicamycin (Tm)/g of body weight or vehicle (150 mm glucose). After 24 h, levels of Xbp1s and Chop mRNA were determined by real time PCR in brain cortex. All values were normalized to actin mRNA (n = 3). Statistical analyses were performed using Student's t test in (C) and one-way ANOVA with Bonferroni multiple comparison test in E. Mean ± S.E. shown; p values are as follows: n.s., p > 0.05; *, p ≤ 0.05; **, p ≤ 0.01.
We then monitored the susceptibility of ERp57-FLAG overexpressing mice to ER stress. We intraperitoneally injected these animals with the ER stress agent tunicamycin and then measured the levels of the ER stress markers Chop and spliced XBP1 mRNA in the brain using real time PCR. Consistent with our in vitro experiments, ERp57 overexpression did not alter the susceptibility of brain cells to undergo ER stress (Fig. 8E). Taken together, these results suggest that enforced expression of ERp57 in the brain augments PrP levels.
Discussion
Increased levels of ERp57 is widely reported in diseases such as Alzheimer disease, amyotrophic lateral sclerosis, Parkinson disease, Huntington disease, and PrD, among other pathologies (30), although its biological significance has remained elusive. The induction of ERp57 has been associated with a UPR due to the accumulation of misfolded proteins inside the ER, contributing to the folding and quality control of glycoproteins as part of the CNX/CRT cycle. Here, we investigated the impact of ERp57 on PrP biogenesis. First, we confirmed the upregulation of ERp57 in patients diagnosed with sporadic CJD and identified a specific response in neurons. Because PrP is a disulfide bond-containing glycoprotein, we hypothesized that ERp57 could directly participate in the biosynthesis of PrP. Our results show that ERp57 deficiency alters the maturation and decreases the levels of both endogenous and exogenous PrPC, whereas ERp57 overexpression has the opposite effect. In addition, similar results were obtained when a panel of disease-related PrP mutants was expressed, suggesting that PrPC and PrP mutant forms are possible clients of the ERp57-folding pathway. In agreement with this idea, we also established that both PrPC and PrPD177N physically interact with ERp57. Furthermore, we confirmed that PrPC also associates with PDIA1 but not with ERp72. Alternatively, it may be feasible that ERp57 and PDIA1 act on different conformational pools of unfolded/misfolded PrP, because PDIA1 is also part of the ERAD pathway (66) and may therefore target unfolded/misfolded PrP toward degradation. Of note, a recent report suggested that calnexin mediates a novel PrP clearance pathway under ER stress termed “rapid ER stress-induced export” (67). This pathway involves the export of misfolded GPI proteins to the plasma membrane for subsequent degradation by the lysosome. It may be interesting to explore in the future whether ERp57 also modulates the degradation of PrP by rapid ER stress-induced export.
To investigate the significance of ERp57 on the biology of PrP in vivo, we first generated a CNS-specific ERp57 knock-out mouse model. Homozygous mice showed a near full loss of ERp57 in the CNS, whereas heterozygous animals showed a 50% reduction in ERp57 mRNA and protein levels. ERp57-deficient animals did not show major changes in the levels of the di-glycosylated PrP at steady state; however, we confirmed the reduction of the mono- and nonglycosylated form of endogenous PrP in ERp57 knock-out animals. This may be explained by compensatory mechanisms involving CNX and CRT. Another possibility is the functional replacement of ERp57 by other PDIs in neurons in vivo, including PDIA1. Furthermore, we have examined PrP levels in transgenic mice overexpressing ERp57 under the control of the prion promoter. We have recently reported a functional characterization of this transgenic line showing that overexpression of ERp57 improves recovery in peripheral nerve regeneration after mechanical injury to sciatic nerve (57), indicating that the overexpressed chaperone is functional in the nervous system. Interestingly, PrP expression is induced in models of peripheral nerve degeneration (68), and its deficiency triggers myelin defects (69). Because mice overexpressing ERp57 displayed augmented levels of PrP in the nervous system, it remains to be determined whether part of the beneficial effects of overexpressing ERp57 are due to enhancement of PrP expression.
Although ERp57 and PDIA1 have been suggested to protect cells against ER stress (70) due to its crucial role in assisting the folding of glycosylated proteins, we did not find evidence supporting a major role of ERp57 in the susceptibility of cells to ER stress and cell death in vitro and in vivo using multiple experimental systems. These observations may be explained by the fact that ERp57 possibly catalyze the folding of a small subset of glycosylated proteins in the secretory pathway (34–36). The expression of other PDI family members and other compensatory systems may maintain the global homeostasis of disulfide bond formation at the ER in neurons lacking ERp57 expression. Unexpectedly, we did not observe drastic compensatory changes in other PDIs or ER foldases when ERp57 expression was targeted, which again supports the concept that ERp57 may assist the folding of a small subset of ER client proteins, and its deficiency does not cause a general perturbation of redox folding in the ER.
In future experiments, we plan to investigate whether ERp57 modulates the progression of protein misfolding disorders. Overall, this study identifies a new component regulating PrP levels that may have relevance for understanding the contribution of the cellular context during prion pathogenesis.
Author Contributions
M. T. designed the study and experiments, performed experiments, analyzed data, and wrote the paper. D. B. M. designed experiments, performed experiments, analyzed data, and wrote the paper. J. M. M. designed experiments, performed experiments, and analyzed data. U. W. designed experiments, performed experiments, analyzed data, and wrote the paper. V. H. C. performed experiments and analyzed data. T. S. performed experiments and analyzed data. C. A. performed experiments and analyzed data. P. R. performed experiments and analyzed data. S. M. performed experiments and analyzed data. N. M. performed experiments and analyzed data. C. V. performed experiments and analyzed data. L. C. designed experiments and analyzed the data. C. S. designed experiments and analyzed the data. M. M. designed experiments and analyzed the data. C. H. designed the study and experiments, analyzed the data, and wrote the paper. All authors have read and approved the final version of the manuscript.
This work was supported in part by FONDECYT Grants 1140549 and 1100176, Millennium Institute Grant P09-015-F, Ring Initiative Grant ACT1109, FONDEF Grant D11I1007, CONICYT Grant USA2013-0003, ECOS-CONICYTC13S02, The Michael J. Fox Foundation for Parkinson Research, The Frick Foundation, Amyotrophic Lateral Sclerosis Therapy Alliance, Muscular Dystrophy Association, Foundation Grant COPEC-UC (to C. H.), FONDECYT Grant 11121524 (to S. M.), FONDECYT Postdoctoral Fellowships 3110067 (to U. W.) and 3130351 (to D. B. M.), CONICYT Ph.D. fellowship (to M. T. and V. H. C.), and Master fellowship (to P. R.). The authors declare that they have no conflicts of interest with the contents of this article.
- PrD
- prion-related disorder
- PrP
- prion protein
- PrPC
- wild-type PrP
- PDI
- protein-disulfide isomerase
- CJD
- Creutzfeldt-Jacob disease
- CNX
- calnexin
- CRT
- calreticulin
- MEF
- murine embryonic fibroblast
- ER
- endoplasmic reticulum
- UPR
- unfolded protein response
- ERAD
- ER-associated degradation
- APP
- amyloid precursor protein
- Tm
- tunicamycin
- Bref A
- brefeldin A
- IHC
- immunohistochemistry
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PNGase F
- N-glycosidase F
- ANOVA
- analysis of variance.
References
- 1. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Soto C. (2011) Prion hypothesis: the end of the controversy? Trends Biochem. Sci. 36, 151–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hetz C. A., Soto C. (2006) Stressing out the ER: a role of the unfolded protein response in prion-related disorders. Curr. Mol. Med. 6, 37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Puig B., Altmeppen H., Glatzel M. (2014) The GPI-anchoring of PrP: implications in sorting and pathogenesis. Prion 8, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yedidia Y., Horonchik L., Tzaban S., Yanai A., Taraboulos A. (2001) Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. EMBO J. 20, 5383–5391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma J., Lindquist S. (2001) Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc. Natl. Acad. Sci. U.S.A. 98, 14955–14960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vey M., Pilkuhn S., Wille H., Nixon R., DeArmond S. J., Smart E. J., Anderson R. G., Taraboulos A., Prusiner S. B. (1996) Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc. Natl. Acad. Sci. U.S.A. 93, 14945–14949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hetz C., Lee A.-H., Gonzalez-Romero D., Thielen P., Castilla J., Soto C., Glimcher L. H. (2008) Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 757–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hetz C., Russelakis-Carneiro M., Maundrell K., Castilla J., Soto C. (2003) Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 22, 5435–5445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hetz C., Russelakis-Carneiro M., Wälchli S., Carboni S., Vial-Knecht E., Maundrell K., Castilla J., Soto C. (2005) The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J. Neurosci. 25, 2793–2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moreno J. A., Halliday M., Molloy C., Radford H., Verity N., Axten J. M., Ortori C. A., Willis A. E., Fischer P. M., Barrett D. A., Mallucci G. R. (2013) Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 5, 206ra138. [DOI] [PubMed] [Google Scholar]
- 12. Moreno J. A., Radford H., Peretti D., Steinert J. R., Verity N., Martin M. G., Halliday M., Morgan J., Dinsdale D., Ortori C. A., Barrett D. A., Tsaytler P., Bertolotti A., Willis A. E., Bushell M., Mallucci G. R. (2012) Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 485, 507–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rane N. S., Kang S. W., Chakrabarti O., Feigenbaum L., Hegde R. S. (2008) Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration. Dev. Cell. 15, 359–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Steele A. D., Hetz C., Yi C. H., Jackson W. S., Borkowski A. W., Yuan J., Wollmann R. H., Lindquist S. (2007) Prion pathogenesis is independent of caspase-12. Prion 1, 243–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Torres M., Castillo K., Armisén R., Stutzin A., Soto C., Hetz C. (2010) Prion protein misfolding affects calcium homeostasis and sensitizes cells to endoplasmic reticulum stress. PLoS ONE 5, e15658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xu K., Zhu X.-P. (2012) Endoplasmic reticulum stress and prion diseases. Rev. Neurosci. 23, 79–84 [DOI] [PubMed] [Google Scholar]
- 17. Quaglio E., Restelli E., Garofoli A., Dossena S., De Luigi A., Tagliavacca L., Imperiale D., Migheli A., Salmona M., Sitia R., Forloni G., Chiesa R. (2011) Expression of mutant or cytosolic PrP in transgenic mice and cells is not associated with endoplasmic reticulum stress or proteasome dysfunction. PLoS ONE 6, e19339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang S. B, Shi Q., Xu Y., Xie W. L., Zhang J., Tian C., Guo Y., Wang K., Zhang B. Y., Chen C., Gao C., Dong X. P. (2012) Protein-disulfide isomerase regulates endoplasmic reticulum stress and the apoptotic process during prion infection and PrP mutant-induced cytotoxicity. PLoS ONE 7, e38221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang X., Shi Q., Xu K., Gao C., Chen C., Li X. L., Wang G. R., Tian C., Han J., Dong X. P. (2011) Familial CJD associated PrP mutants within transmembrane region induced CTM-PrP retention in ER and triggered apoptosis by ER stress in SH-SY5Y cells. PLoS ONE 6, e14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hetz C. (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102 [DOI] [PubMed] [Google Scholar]
- 21. Walter P., Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 22. Hetz C., Chevet E., Oakes S. A. (2015) Proteostasis control by the unfolded protein response. Nat. Cell Biol. 17, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kang S. W., Rane N. S., Kim S. J., Garrison J. L., Taunton J., Hegde R. S. (2006) Substrate-specific translocational attenuation during ER stress defines a pre-emptive quality control pathway. Cell 127, 999–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Orsi A., Fioriti L., Chiesa R., Sitia R. (2006) Conditions of endoplasmic reticulum stress favor the accumulation of cytosolic prion protein. J. Biol. Chem. 281, 30431–30438 [DOI] [PubMed] [Google Scholar]
- 25. Nunziante M., Ackermann K., Dietrich K., Wolf H., Gädtke L., Gilch S., Vorberg I., Groschup M., Schätzl H. M. (2011) Proteasomal dysfunction and endoplasmic reticulum stress enhance trafficking of prion protein aggregates through the secretory pathway and increase accumulation of pathologic prion protein. J. Biol. Chem. 286, 33942–33953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hetz C., Castilla J., Soto C. (2007) Perturbation of endoplasmic reticulum homeostasis facilitates prion replication. J. Biol. Chem. 282, 12725–12733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hetz C., Mollereau B. (2014) Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249 [DOI] [PubMed] [Google Scholar]
- 28. Yoo B. C., Krapfenbauer K., Cairns N., Belay G., Bajo M., Lubec G. (2002) Overexpressed protein-disulfide isomerase in brains of patients with sporadic Creutzfeldt-Jakob disease. Neurosci. Lett. 334, 196–200 [DOI] [PubMed] [Google Scholar]
- 29. Feige M. J., Hendershot L. M. (2011) Disulfide bonds in ER protein folding and homeostasis. Curr. Opin. Cell Biol. 23, 167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Andreu C. I., Woehlbier U., Torres M., Hetz C. (2012) Protein-disulfide isomerases in neurodegeneration: from disease mechanisms to biomedical applications. FEBS Lett. 586, 2826–2834 [DOI] [PubMed] [Google Scholar]
- 31. Jessop C. E., Tavender T. J., Watkins R. H., Chambers J. E., Bulleid N. J. (2009) Substrate specificity of the oxidoreductase ERp57 is determined primarily by its interaction with calnexin and calreticulin. J. Biol. Chem. 284, 2194–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Coe H., Michalak M. (2010) ERp57, a multifunctional endoplasmic reticulum resident oxidoreductase. Int. J. Biochem. Cell Biol. 42, 796–799 [DOI] [PubMed] [Google Scholar]
- 33. Garbi N., Tanaka S., Momburg F., Hämmerling G. J. (2006) Impaired assembly of the major histocompatibility complex class I peptide-loading complex in mice deficient in the oxidoreductase ERp57. Nat. Immunol. 7, 93–102 [DOI] [PubMed] [Google Scholar]
- 34. Coe H., Jung J., Groenendyk J., Prins D., Michalak M. (2010) ERp57 modulates STAT3 signaling from the lumen of the endoplasmic reticulum. J. Biol. Chem. 285, 6725–6738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jessop C. E., Chakravarthi S., Garbi N., Hämmerling G. J., Lovell S., Bulleid N. J. (2007) ERp57 is essential for efficient folding of glycoproteins sharing common structural domains. EMBO J. 26, 28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Soldà T., Garbi N., Hämmerling G. J., Molinari M. (2006) Consequences of ERp57 deletion on oxidative folding of obligate and facultative clients of the calnexin cycle. J. Biol. Chem. 281, 6219–6226 [DOI] [PubMed] [Google Scholar]
- 37. Garbi N., Hämmerling G., Tanaka S. (2007) Interaction of ERp57 and tapasin in the generation of MHC class I-peptide complexes. Curr. Opin. Immunol. 19, 99–105 [DOI] [PubMed] [Google Scholar]
- 38. Wearsch P. A., Cresswell P. (2007) Selective loading of high-affinity peptides onto major histocompatibility complex class I molecules by the tapasin-ERp57 heterodimer. Nat. Immunol. 8, 873–881 [DOI] [PubMed] [Google Scholar]
- 39. Peaper D. R., Cresswell P. (2008) The redox activity of ERp57 is not essential for its functions in MHC class I peptide loading. Proc. Natl. Acad. Sci. U.S.A. 105, 10477–10482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Turano C., Gaucci E., Grillo C., Chichiarelli S. (2011) ERp57/GRP58: a protein with multiple functions. Cell. Mol. Biol. Lett. 16, 539–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Y., Camacho P. (2004) Ca2+-dependent redox modulation of SERCA 2b by ERp57. J. Cell Biol. 164, 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hoffstrom B. G., Kaplan A., Letso R., Schmid R. S., Turmel G. J., Lo D. C., Stockwell B. R. (2010) Inhibitors of protein-disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat. Chem. Biol. 6, 900–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Watts J. C., Huo H., Bai Y., Ehsani S., Jeon A. H., Won A. H., Shi T., Daude N., Lau A., Young R., Xu L., Carlson G. A., Williams D., Westaway D., Schmitt-Ulms G. (2009) Interactome analyses identify ties of PrP and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog. 5, e1000608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Capellari S., Zaidi S. I., Urig C. B., Perry G., Smith M. A., Petersen R. B. (1999) Prion protein glycosylation is sensitive to redox change. J. Biol. Chem. 274, 34846–34850 [DOI] [PubMed] [Google Scholar]
- 45. Lucassen R., Nishina K., Supattapone S. (2003) In vitro amplification of protease-resistant prion protein requires free sulfhydryl groups. Biochemistry 42, 4127–4135 [DOI] [PubMed] [Google Scholar]
- 46. Tompa P., Tusnády G. E., Friedrich P., Simon I. (2002) The role of dimerization in prion replication. Biophys. J. 82, 1711–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Welker E., Wedemeyer W. J., Scheraga H. A. (2001) A role for intermolecular disulfide bonds in prion diseases? Proc. Natl. Acad. Sci. U.S.A. 98, 4334–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yanai A., Meiner Z., Gahali I., Gabizon R., Taraboulos A. (1999) Subcellular trafficking abnormalities of a prion protein with a disrupted disulfide loop. FEBS Lett. 460, 11–16 [DOI] [PubMed] [Google Scholar]
- 49. Torres M., Cartier L., Matamala J., Hernandez N., Woehvier U., Hetz C. (2012) Altered prion protein expression pattern in CSF as a biomarker for Creutzfeldt-Jakob Disease. PLoS One 7, e36159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ivanova L., Barmada S., Kummer T., Harris D. A. (2001) Mutant prion proteins are partially retained in the endoplasmic reticulum. J. Biol. Chem. 276, 42409–42421 [DOI] [PubMed] [Google Scholar]
- 51. Burgos P. V., Mardones G. A., Rojas A. L., daSilva L. L., Prabhu Y., Hurley J. H., Bonifacino J. S. (2010) Sorting of the Alzheimer's disease amyloid precursor protein mediated by the AP-4 complex. Dev. Cell 18, 425–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jessop C. E., Watkins R. H., Simmons J. J., Tasab M., Bulleid N. J. (2009) Protein disulphide isomerase family members show distinct substrate specificity: P5 is targeted to BiP client proteins. J. Cell Sci. 122, 4287–4295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moffat J., Grueneberg D. A., Yang X., Kim S. Y., Kloepfer A. M., Hinkle G., Piqani B., Eisenhaure T. M., Luo B., Grenier J. K., Carpenter A. E., Foo S. Y., Stewart S. A., Stockwell B. R., Hacohen N., et al. (2006) A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298 [DOI] [PubMed] [Google Scholar]
- 54. Hetz C., Thielen P., Matus S., Nassif M., Court F., Kiffin R., Martinez G., Cuervo A. M., Brown R. H., Glimcher L. H. (2009) XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 23, 2294–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lisbona F., Rojas-Rivera D., Thielen P., Zamorano S., Todd D., Martinon F., Glavic A., Kress C., Lin J. H., Walter P., Reed J. C., Glimcher L. H., Hetz C. (2009) BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1α. Mol. Cell 33, 679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rodriguez D. A., Zamorano S., Lisbona F., Rojas-Rivera D., Urra H., Cubillos-Ruiz J. R., Armisen R., Henriquez D. R., Cheng E. H., Letek M., Vaisar T., Irrazabal T., Gonzalez-Billault C., Letai A., Pimentel-Muiños F. X., et al. (2012) BH3-only proteins are part of a regulatory network that control the sustained signalling of the unfolded protein response sensor IRE1α. EMBO J. 31, 2322–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Castillo V., Oñate M., Woehlbier U., Rozas P., Andreu C., Medinas D., Valdes P., Osorio F., Mercado G., Vidal R. L., Kerr B., Court F. A., Hetz C. (2015) Functional role of the disulfide isomerase ERp57 in axonal regeneration. PLoS ONE 10, e0136620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Borchelt D. R., Davis J., Fischer M., Lee M. K., Slunt H. H., Ratovitsky T., Regard J., Copeland N. G., Jenkins N. A., Sisodia S. S., Price D. L. (1996) A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet. Anal. 13, 159–163 [DOI] [PubMed] [Google Scholar]
- 59. Rojas-Rivera D., Armisén R., Colombo A., Martínez G., Eguiguren A. L., Díaz A., Kiviluoto S., Rodríguez D., Patron M., Rizzuto R., Bultynck G., Concha M. L., Sierralta J., Stutzin A., Hetz C. (2012) TMBIM3/GRINA is a novel unfolded protein response (UPR) target gene that controls apoptosis through the modulation of ER calcium homeostasis. Cell Death Differ. 19, 1013–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hetz C., Bernasconi P., Fisher J., Lee A.-H., Bassik M. C., Antonsson B., Brandt G. S., Iwakoshi N. N., Schinzel A., Glimcher L. H., Korsmeyer S. J. (2006) Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1α. Science 312, 572–576 [DOI] [PubMed] [Google Scholar]
- 61. Stewart R. S., Piccardo P., Ghetti B., Harris D. A. (2005) Neurodegenerative illness in transgenic mice expressing a transmembrane form of the prion protein. J. Neurosci. 25, 3469–3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chiesa R., Piccardo P., Ghetti B., Harris D. A. (1998) Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 21, 1339–1351 [DOI] [PubMed] [Google Scholar]
- 63. Dossena S., Imeri L., Mangieri M., Garofoli A., Ferrari L., Senatore A., Restelli E., Balducci C., Fiordaliso F., Salio M., Bianchi S., Fioriti L., Morbin M., Pincherle A., Marcon G., et al. (2008) Mutant prion protein expression causes motor and memory deficits and abnormal sleep patterns in a transgenic Mouse model. Neuron 60, 598–609 [DOI] [PubMed] [Google Scholar]
- 64. Drisaldi B., Stewart R. S., Adles C., Stewart L. R., Quaglio E., Biasini E., Fioriti L., Chiesa R., Harris D. A. (2003) Mutant PrP is delayed in its exit from the endoplasmic reticulum, but neither wild-type nor mutant PrP undergoes retrotranslocation prior to proteasomal degradation. J. Biol. Chem. 278, 21732–21743 [DOI] [PubMed] [Google Scholar]
- 65. Stewart R. S., Drisaldi B., Harris D. A. (2001) A transmembrane form of the prion protein contains an uncleaved signal peptide and is retained in the endoplasmic reticulum. Mol. Biol. Cell 12, 881–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ashok A., Hegde R. S. (2008) Retrotranslocation of prion proteins from the endoplasmic reticulum by preventing GPI signal transamidation. Mol. Biol. Cell 19, 3463–3476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Satpute-Krishnan P., Ajinkya M., Bhat S., Itakura E., Hegde R. S., Lippincott-Schwartz J. (2014) ER stress-induced clearance of misfolded GPI-anchored proteins via the secretory pathway. Cell 158, 522–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Moya K. L., Hässig R., Breen K. C., Volland H., Di Giamberardino L. (2005) Axonal transport of the cellular prion protein is increased during axon regeneration. J. Neurochem. 92, 1044–1053 [DOI] [PubMed] [Google Scholar]
- 69. Bremer J., Baumann F., Tiberi C., Wessig C., Fischer H., Schwarz P., Steele A. D., Toyka K. V., Nave K.-A., Weis J., Aguzzi A. (2010) Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 13, 310–318 [DOI] [PubMed] [Google Scholar]
- 70. Uehara T., Nakamura T., Yao D., Shi Z.-Q., Gu Z., Ma Y., Masliah E., Nomura Y., Lipton S. A. (2006) S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441, 513–517 [DOI] [PubMed] [Google Scholar]