

This review summarizes current knowledge and challenges in the diagnosis and management of low- and intermediate-grade lung neuroendocrine tumors. Treatment guidelines recommend consideration of systemic therapies for patients with unresectable, advanced bronchopulmonary disease. Additional data from randomized trials in lung neuroendocrine tumors are needed to support establishment of standard treatment regimens.
Keywords: Neuroendocrine tumors, Lung neoplasms, Carcinoid tumor, Disease management
Abstract
Neuroendocrine tumors (NET) of the lung represent approximately 25% of all primary lung tumors and can be classified as low grade (typical carcinoids), intermediate grade (atypical carcinoids), or high grade (large cell neuroendocrine carcinoma or small cell lung carcinoma). Low- and intermediate-grade lung NET are increasingly recognized as biologically distinct from high-grade lung NET based on clinical behavior and underlying molecular abnormalities. This review summarizes current knowledge and challenges in the diagnosis and management of low- and intermediate-grade lung NET. Accurate histopathologic classification of lung NET is critical to determining appropriate treatment options but can be challenging even for experts. For low- and intermediate-grade lung NET, surgery remains the mainstay of treatment for localized disease. Although no standard systemic therapy has been established for the treatment of advanced, unresectable disease, a number of promising treatment options are emerging, including somatostatin analogs, temozolomide-based chemotherapy, targeted therapy with mammalian target of rapamycin or vascular endothelial growth factor inhibitors, and peptide receptor radionuclide therapy. Given the difficulty in accurately diagnosing these tumors, and the paucity of data supporting establishment of standard systemic therapy options, management of patients within the setting of a multidisciplinary team, including specialists with expertise in NET, is recommended. Ongoing and future clinical trials hopefully will provide stronger evidence to support treatment recommendations for low- and intermediate-grade lung NET.
Implications for Practice:
Treatment of neuroendocrine tumors (NET), particularly those of lung origin, continues to evolve. This review seeks to educate oncologists on the most up-to-date options and supporting data regarding management of two rare lung neoplasms, typical and atypical carcinoid tumors. Although surgical resection has been the mainstay of treatment, several systemic options have been studied in the treatment of NET of various origins that may potentially play a role in treating typical carcinoid tumors and atypical carcinoid tumors.
Introduction
Neuroendocrine tumors (NET) arise from neuroendocrine cells in various anatomic sites, most commonly the gastrointestinal and pulmonary systems [1]. Bronchopulmonary or lung NET represent approximately 25% of primary lung neoplasms and 20%–25% of primary NET [1–3].
World Health Organization (WHO) 2004 criteria classify lung NET into 4 histologic variants: typical carcinoid (TC), atypical carcinoid (AC), large cell neuroendocrine carcinoma (LCNEC), and small cell lung carcinoma (SCLC) [4]. Lung NET may also be classified as grade 1 (TC), grade 2 (AC), or grade 3 (LCNEC or SCLC). TC and AC are rare (incidence is 1%–2%) [5–7], with increases seen in the U.S. (0.30 to 1.35 per 100,000 persons for the years 1973–2004) [2], partially attributable to improvements in imaging [8]. This review focuses on diagnosis and management of low- and intermediate-grade lung NET; as such, SCLC and LCNEC are not further discussed.
Successful treatment of lung NET relies on accurate and timely diagnosis, which may be challenging because patients remain asymptomatic for long periods or have nonspecific symptoms [5, 9]. Furthermore, distinguishing between histological variants requires expert pathology, as differences can be subtle [5].
Aside from use of somatostatin analogs (SSA) as standard of care for treating symptoms associated with functional carcinoids [10], currently, no established systemic treatments exist for patients with unresectable or metastatic TC or AC; most treatments are investigational [9].
Materials and Methods
PubMed was searched (years 2010–2015) for articles regarding the diagnosis and management of lung NET, including identification and differentiation, prognosis, treatment options, impact of multidisciplinary care, and future perspectives. Key search terms included “neuroendocrine tumor,” “lung NET,” “small cell lung carcinoma,” “large cell neuroendocrine carcinoma,” “typical carcinoid,” and “atypical carcinoid.” Additionally, ClinicalTrials.gov, and cancer-focused congresses such as those of the American Society of Clinical Oncology were searched (years 2012–2014) to identify relevant studies evaluating therapies in lung NET. Reference citations within relevant articles revealed further sources of value.
Pathophysiology
TC and AC are thought to originate from pulmonary neuroendocrine cells (PNECs), specialized epithelial cells occurring as solitary cells or clusters of 4–10 cells (neuroepithelial bodies) in the airways and lungs [3, 8]. In adults, neuroepithelial bodies function as chemoreceptors in the airway, secreting serotonin in response to hypoxia, inducing vasoconstriction in poorly ventilated areas of the lung and redirecting blood flow toward better ventilated areas [11, 12]. Diffuse idiopathic neuroendocrine cell hyperplasia (DIPNECH), a rare, preinvasive lesion that can give rise to TC and AC, is defined as hyperplasia of PNECs confined to respiratory epithelium without penetration through the basement membrane [13]. DIPNECH is an exaggerated adaptive response to hypoxia and chronic lung injury or infection. However, most cases, even those that progress to carcinoid, arise in otherwise normal lung tissue [3, 13]. DIPNECH can cause chronic nonproductive cough and interstitial lung disease, which can progress to obliterative bronchiolitis [13].
DIPNECH lesions extending beyond the basement membrane are known as tumorlets when nodular aggregates of neuroendocrine cells measure <0.5 cm (greatest diameter) and as lung NET when measuring ≥0.5 cm [3, 12]. Lung carcinoids are thought to arise from DIPNECH or tumorlets based on co-occurrence with established carcinoids [12].
Although lung NET are traditionally thought to represent a continuum of disease, TC and AC may represent distinct biological entities based on clinical behavior and underlying molecular abnormalities [12]. TCs and ACs commonly occur in nonsmokers with no discernible relationship to smoking [3]. They are also both associated with multiple endocrine neoplasia type 1 in 5%–10% of cases [9].
Diagnosis
Clinical Presentation
Lung NET are heterogeneous and can present with nonspecific symptoms that mimic more common conditions. Furthermore, up to half of patients are asymptomatic at diagnosis [6].
Nonfunctional (nonsecretory) lung NET, if symptomatic, present with symptoms relating to tumor mass, including cough, hemoptysis, recurrent pulmonary infection, dyspnea, and chest pain [5, 8, 9]. Approximately 70%–75% of TCs and ACs are located centrally in the major bronchi, with the remainder found in peripheral lung [3, 8]. Up to 90% of patients with central lung NET present with obstructive symptoms; peripheral tumors tend to be asymptomatic unless they are so extensive they cause dyspnea. Peripheral tumors are often discovered incidentally on investigation for other conditions [6, 8]. Obstructive symptoms by a carcinoid can mimic more prevalent respiratory conditions, including asthma, chronic obstructive pulmonary disease, and pneumonia. Often, a tumor is only suspected after initial treatment fails to improve symptoms, delaying diagnosis by months or years [5, 9].
Functional (secretory) lung NET typically present with symptoms including cutaneous flushing, diarrhea, and wheezing caused by excessive secretion of hormones and peptides by tumor cells [5]. An unusual carcinoid syndrome resulting from histamine release with associated prolonged generalized erythema can also be associated with lung NET [9]. However, classical carcinoid syndrome is less common in lung NET than in gastrointestinal NET (<2% vs. ∼10%) [3, 14].
Pathology
Histopathologic tumor classification has the greatest impact on prognosis and treatment in lung NET; accurate classification, therefore, is crucial [15]. WHO 2004 classifications for lung NET (Table 1) account for cell size and morphology, architectural growth patterns, mitotic index, and presence of necrosis [4].
Table 1.
Classification of lung neuroendocrine tumors based on World Health Organization 2004 criteria [3, 5, 6]
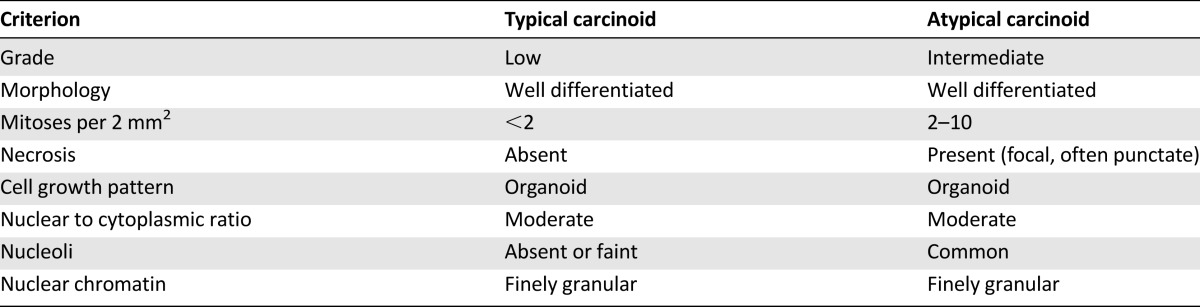
TC and AC are defined as tumors ≥0.5 cm with carcinoid morphology, and are differentiated by mitotic rate and presence or absence of necrosis [5, 6]. Differences between TC and AC can be subtle [3], and considerable interobserver variability in histopathologic classification among pathologists has been reported for TC and AC [16].
The Ki-67 index is not yet included for grading lung carcinoids. Inclusion of the Ki-67 index in WHO classification criteria is prognostically effective [17], and has been suggested for lung NET. TC and AC often have a low Ki-67 labeling index [18]. A study by Rindi et al. suggested discrimination of lung NET grade by using Ki-67 cutoff values of <4% for grade 1, 4%–24% for grade 2, and ≥25% for grade 3, and proposed a grading system based on the combination of Ki-67, mitotic count, and necrosis [19]. Other possible Ki-67 cutoffs have been suggested to distinguish TC from AC [20, 21], but none have been widely accepted as clinically useful.
Tumor Markers
Establishment of a NET diagnosis can be aided by immunohistochemical detection of general neuroendocrine markers, including chromogranin A (CgA), CD56, synaptophysin, and neuron-specific enolase (NSE) [15]. Most TCs and ACs are positive for CgA and/or synaptophysin by immunohistochemistry [15]. Synaptophysin is regarded as among the most specific markers of neuroendocrine differentiation, with higher sensitivity than CgA or NSE. Nevertheless, in a small proportion of TCs or ACs (particularly AC), not all neuroendocrine markers are expressed and a panel approach is recommended [3, 9].
Additionally, serum CgA may be used to monitor recurrence or response to treatment, as it frequently correlates with tumor volume. Care should be taken, as levels may be elevated with proton pump inhibitors, liver or renal failure, or chronic gastritis [9].
5-Hydroxyindoleacetic acid is a metabolite of serotonin. Urinary levels can be useful markers in patients with well-differentiated midgut NET [15], but are less useful in lung NET, as levels are frequently normal [9].
Imaging
If lung NET is suspected, imaging is performed to determine size and location of the primary tumor, and for staging. Computed tomography (CT) is recommended for imaging lung carcinoid tumors [8]. Magnetic resonance imaging (MRI) may also be useful for imaging mediastinal or abdominal metastasis [9]. For follow-up, European Society of Medical Oncology (ESMO) guidelines recommend performing CT imaging or MRI once yearly in patients who have undergone primary surgery for TC or AC, or every 3 months in those with metastatic disease receiving treatment with cytotoxic or biologic agents [15].
Given the shared structural similarities between TC and AC, radiologic findings alone cannot distinguish between them, necessitating the use of functional imaging with nuclear medicine [22]. Somatostatin (SST) receptors are expressed in approximately 80% of lung NET, predominantly SST2 receptors [5, 23]. SST receptor scintigraphy (SRS) with radiolabeled SSA 111In-labeled octreotide, in conjunction with single-photon emission CT, may also be used in perioperative staging for detection of nodal involvement and distant metastases, and is effective for visualizing TC and AC [24]. The North American Neuroendocrine Tumor Society (NANETS) guidelines recommend SRS to test for presence of SST-receptor expression, to evaluate appropriateness of peptide receptor radionuclide therapy (PRRT), or as part of follow-up imaging [9]. Positron emission tomographic (PET) scans with gallium-68 SSAs, including gallium 68-tetraazacyclododecane tetraacetic acid-octreotate (68Ga-DOTATATE) and 1,4,7,10-tetraazacyclododecane-NI,NII,NIII,NIIII-tetraacetic acid (D)-Phe1-thy3-octreotide (68Ga-DOTATOC), have shown higher sensitivity and specificity for SST-receptor imaging than octreoscan, and are likely to replace it for staging NET, including lung carcinoids [22, 25, 26]. 18F-fluorodeoxyglucose PET (FDG PET)/CT might have a role in diagnostic imaging of TCs and ACs [27], and has been suggested for tumors with Ki-67 index >10% [15]. Thus, there is an emerging role for FDG PET and gallium-68 PET for postoperative staging and evaluation for PRRT.
Staging Criteria
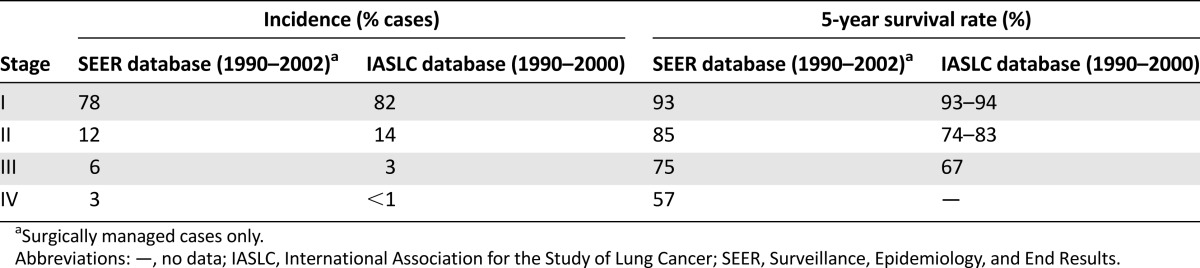
Tumor-node-metastasis staging of TC and AC is prognostic for survival using data from the Surveillance, Epidemiology, and End Results and International Association for the Study of Lung Cancer databases (Table 2) [28]. ACs are notably more aggressive than TCs, with a higher frequency of regional lymph node involvement (50%–60% vs. 10%–15%) and distant metastasis (∼20% vs. 2%–5%) [7, 29], and worse survival outcomes at 5 years (56%–79% vs. 87%–96%) and 10 years (35%–56% vs. 82%–87%) [7, 15, 29].
Table 2.
Stage distribution and 5-year survival rates of low- or intermediate-grade lung neuroendocrine tumors in the SEER and IASLC databases [28]
Treatment
The mainstay of treatment is surgery for localized TCs and ACs and for highly selected cases of advanced disease. TC and AC are relatively insensitive to chemotherapy and radiotherapy and no standard systemic therapy has been established. Targeted therapies may be beneficial, although these remain investigational for lung NET.
Localized Disease
Surgery
Complete surgical resection, with preservation of as much normal lung tissue as possible, is the standard of care for patients with localized TC or AC [5, 9]. For TC, surgical resection provides excellent outcomes, with 5- and 10-year survival rates of ∼90% and ∼80%, respectively, and very low recurrence rates (3%–5%) [30–32]. For AC, 5- and 10-year survival rates following surgery are lower, around 70% and 50%, respectively, owing to a much higher recurrence rate (∼25%) [30–32].
AC may require more aggressive surgery than TC [5, 33]. Although lung-sparing limited/sublobar resection provides similarly low recurrence rates to lobectomy or pneumonectomy in patients with TC, the risk of locoregional recurrence is increased in patients with AC [32–34]. The benefits of lymph node dissection are poorly defined in TC, although systemic lymph node sampling is recommended to define pathologic stage. Since AC has a much higher rate of lymph node involvement, lymph node dissection is often recommended, even in patients with clinical N0 disease [5].
Since AC has a much higher rate of lymph node involvement, lymph node dissection is often recommended, even in patients with clinical N0 disease.
Adjuvant Therapy
Given the high rate of recurrence, the potential role for adjuvant therapy is of interest. Use of adjuvant therapy is not well studied in lung NET and discordance exists among treatment recommendations. Whereas National Comprehensive Cancer Network (NCCN) guidelines recommend adjuvant chemotherapy with or without radiation therapy for patients with stage II/III AC [35], guidelines from the European Neuroendocrine Tumor Society (ENETS) recommend this only for AC patients with positive lymph nodes, particularly those with high proliferative index [26], and NANETS guidelines do not recommend this modality because of lack of supportive data [9]. Retrospective analyses suggest some patients might benefit from adjuvant chemotherapy [14, 36], whereas adjuvant radiotherapy has been shown to improve local control in some studies [37] but not others [14]. In one retrospective study in patients with pulmonary NET (including 36 patients with TC and 3 with AC), no local recurrences were observed after adjuvant radiation therapy in node-positive patients [37].
Treatment of Carcinoid Syndrome
Approximately 2% of lung NET demonstrate carcinoid syndrome [3]. SSAs are the mainstay of treatment for carcinoid syndrome, controlling symptoms by reducing excessive hormone production [9, 38]. Octreotide is the only SSA approved in the U.S. for treatment of carcinoid syndrome [39]; both lanreotide and octreotide are approved for this indication in Europe [40, 41]. Octreotide long-acting repeatable (LAR) provides good symptom control in 40%–90% of patients, although occasional rescue with short-acting octreotide is required [42–44]. Lanreotide has shown similar efficacy to octreotide in controlling symptoms of carcinoid syndrome [45, 46]. Interferon-α (2a or 2b) may also be used to treat carcinoid syndrome, either alone or with an SSA. As monotherapy, interferon-α provides similar symptom control but with a delayed onset of action and more side effects than SSAs [38, 47]. In patients with insufficient response to SSA alone, good symptom control can be achieved in combination with interferon-α [48].
Advanced Disease
Although SSAs are the standard of care for treating symptoms associated with functional carcinoids [10], there is currently no established systemic treatment for unresectable or metastatic TC or AC. Although several treatment approaches are proposed for advanced lung NET, none have been validated in large phase II/III trials [8]. Most clinical evidence comes from subgroup analyses of studies in patients with low- or intermediate-grade NET from various sites of origin or is extrapolated from studies evaluating NET from other primary sites. Most systemic treatments remain investigational for TC and AC [9].
Surgery
Surgery remains the only curative treatment for TC and AC, even for metastatic disease [8, 9]. Surgery is often recommended with curative intent in patients with limited hepatic metastases, with ∼20% achieving a cure [9]. Even without a cure, patients may benefit from symptom alleviation and prolonged disease-free survival [9]. If complete resection is not possible, palliative debulking surgery may be performed to relieve symptoms or prevent complications. However, in the presence of unresectable metastases, resection of a small asymptomatic primary tumor is not indicated [9, 49].
Somatostatin Analogs
Although octreotide is primarily used to treat carcinoid syndrome symptoms, it may also have antiproliferative activity [50]. The phase IIIB PROMID study evaluated octreotide LAR in patients with well-differentiated midgut NET, with or without functionally active tumors [50]. Compared with placebo, octreotide LAR prolonged progression-free survival (PFS) (14.3 vs. 6.0 months; hazard ratio [HR]: 0.34; p < .001) and improved stable disease rates (67% vs. 37% of patients), with similar responses in functional and nonfunctional tumors.
Similarly, lanreotide autogel improved PFS compared with placebo (HR: 0.47; 95% confidence interval [CI]: 0.30–0.73; p < .001) in nonfunctioning enteropancreatic NET in the phase III CLARINET study [51]. PFS was improved with lanreotide in midgut or pancreatic NET (pNET) versus placebo, whereas the small population of patients with hindgut NET (n = 14) had improved PFS with placebo. Based on these results, lanreotide was approved in the U.S. for treatment of gastroenteropancreatic NET [52], and marketing authorization application submission has been accepted for filing in Europe [53].
Although neither trial included lung NET, results are intriguing and support antitumor activity of SSAs in NET. Recent treatment guidelines for lung NET from NANETS, ENETS (Fig. 1), ESMO, and NCCN recommend octreotide and, now, lanreotide be considered for tumor control in bulky and/or symptomatic disease that cannot be completely resected [8, 9, 26, 49], particularly when SRS or PET imaging is positive.
Figure 1.

European Neuroendocrine Tumor Society (ENETS) Expert Consensus and Recommendations for Best Practice for Typical and Atypical Pulmonary Carcinoid [26]. (A): ENETS recommendations for the control of hormone-related symptoms. (B): ENETS recommendations for the control of hormone-related symptoms and tumor growth. aProgression is defined according to Response Evaluation Criteria in Solid Tumors. Reprinted with permission from: Caplin ME, Baudin E, Ferolla P et al. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol 2015;26:1604–1620 (published by Oxford University Press on behalf of the European Society for Medical Oncology).
Abbreviations: AC, atypical carcinoid; PRRT, peptide receptor radionuclide therapy; TC, typical carcinoid.
Chemotherapy
High-grade lung NET demonstrate sensitivity to platinum-based chemotherapy, with the standard regimen being cisplatin plus etoposide [54]. Platinum-based chemotherapy has demonstrated only minor activity in low- or intermediate-grade lung NET, with no standard regimen defined [9].
Recent data suggest temozolomide-based regimens have promise in low- or intermediate-grade NET, including lung NET [9, 15]. In a phase II study involving 13 patients with lung NET, temozolomide treatment resulted in partial response (PR) in 31% and stable disease in a further 31% [55]. A retrospective analysis of 31 patients with metastatic TC or AC treated with temozolomide showed a PR rate of 14% and stable disease in 52% [56]. Combination of temozolomide with capecitabine has shown promising results in a retrospective study involving 29 patients with low- to intermediate-grade NET (8 with lung NET). Tumor control (PR or stable disease) was achieved in 72% and median time to progression (TTP) was 9 months [57].
Whereas low- or intermediate-grade NET from other sites appear to respond to streptozocin-based regimens [58, 59], results in lung NET have been poor, suggesting TC and AC may be resistant to streptozocin [15, 60].
Current treatment guidelines recommend reserving cytotoxic chemotherapy for patients with progressive metastatic disease for whom no other feasible treatment options are available [9, 26, 49].
Targeted Therapy
Targeted therapies may provide an alternative for tumor control in TC or AC, particularly agents targeting the mammalian target of rapamycin (mTOR) pathway or angiogenesis. The mTOR pathway is functionally activated in a subset of low- and intermediate-grade lung NET, demonstrating greater expression of phosphorylated forms of mTOR and ribosomal S6 kinase versus high-grade lung NET [61]. TC and AC are highly vascular tumors known to express vascular endothelial growth factor (VEGF) [62, 63]. Moreover, VEGF expression is associated with poor prognosis [64]. Therefore, VEGF receptor (VEGFR)-targeted therapies may be useful. Subsets of TC have also been found to express c-Kit, platelet-derived growth factor receptor (PDGFR) α and β, and epidermal growth factor receptor [65].
Currently, two targeted therapies, everolimus and sunitinib, are approved for treatment of low- or intermediate-grade advanced pNET, but are not approved for lung NET [9, 49].
mTOR Pathway Inhibition
Everolimus is an mTOR inhibitor currently approved in the U.S. and Europe for treatment of unresectable, metastatic, well-differentiated pNET. Data suggest that everolimus is beneficial in well-differentiated lung NET. In primary TC and AC cell lines, everolimus was shown to reduce VEGF secretion and cell viability [66].
Data suggest that everolimus is beneficial in well-differentiated lung NET. In primary TC and AC cell lines, everolimus was shown to reduce VEGF secretion and cell viability.
In the RADIANT-2 trial, patients with well- to moderately differentiated NET of varying origin, including lung and carcinoid syndrome, were randomized to receive octreotide combined with everolimus or placebo. Per adjudicated central review (on which the primary endpoint was based), everolimus showed a trend toward improved PFS versus placebo (median: 16.4 vs. 11.3 months; p = .026), although the statistical boundary for significance was not met (p = .0246). However, per local investigator review (performed as a supportive analysis), PFS in the octreotide plus everolimus arm was significantly longer versus placebo (12.0 vs. 8.6 months; p = .018). Statistical analysis of the primary endpoint is thought to have been confounded by informative censoring [67]. In an exploratory analysis among 44 lung NET patients, everolimus appeared to improve PFS versus placebo (median: 13.6 vs. 5.6 months), although this was not statistically significant [68]. Tumor shrinkage was observed in 67% of everolimus recipients versus 27% of placebo recipients.
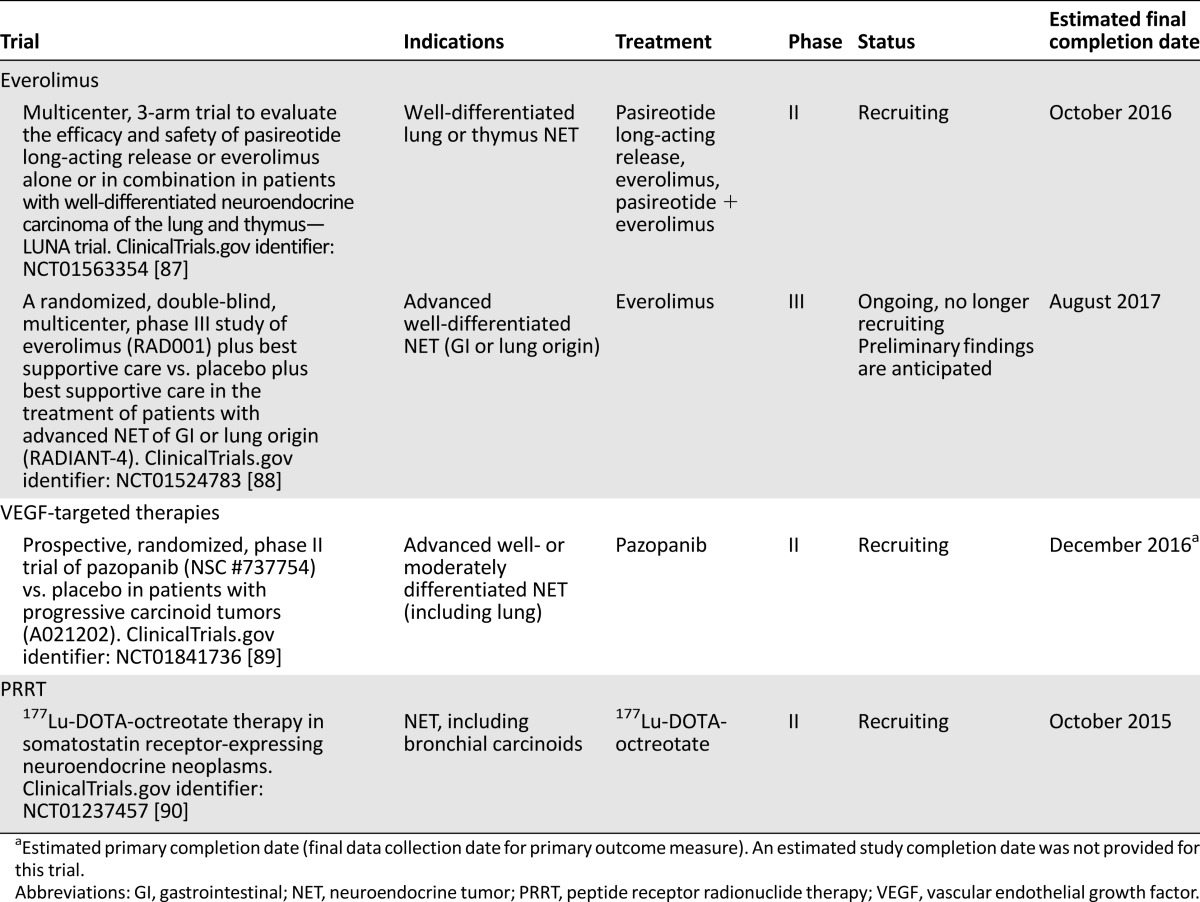
The phase II RAMSETE study extended these findings to nonfunctional, nonpNET (including lung NET), demonstrating a high rate of disease stabilization (55%) and a favorable median PFS (185 days) [69]. RADIANT-4, a large randomized, placebo-controlled, double-blind, phase III study (NCT01524783), assessed the efficacy and tolerability of everolimus in patients with well-differentiated, advanced, nonfunctional NET of lung or gastrointestinal origin, and is expected to definitively establish whether there is a role for everolimus in the therapy of lung carcinoid. Findings are expected to be reported in 2015. Ongoing studies of everolimus in the treatment of patients with lung NET are listed in Table 3.
Table 3.
Ongoing phase II/III trials including patients with low- or intermediate-grade lung NET
Antiangiogenic Agents
Sunitinib is an inhibitor of receptor tyrosine kinases for multiple tumor-cell growth factors including VEGFR-1, -2, and -3; PDGFR-α and -β; and c-kit, and is currently approved in the U.S. and Europe for treatment of unresectable, metastatic, well-differentiated pNET. However, no data are available for sunitinib specifically in lung NET. In a phase II study in patients with metastatic NET, sunitinib demonstrated antitumor activity in pNET (overall response rate [ORR]: 16.7% based on Response Evaluation Criteria in Solid Tumors, version 1.0 [RECIST v1.0]; median TTP: 7.7 months), whereas activity in other NET (including lung NET) could not be definitively determined [70].
Per preliminary findings from a phase II, single-arm study with the VEGFR inhibitor axitinib in 30 patients with unresectable or metastatic carcinoid tumors (3 of whom had lung NET), median time to treatment failure (TTF) was 8.99 months (SD ±7.18). Best radiographic response was PR in 3.3% and stable disease in 70% of patients [71]. In a phase II, single-arm study with the VEGFR inhibitor pazopanib, tumor response was noted in patients with pNET but not in those with carcinoid tumors (including lung NET) [72].
Bevacizumab, a monoclonal antibody targeting VEGF, is approved in the U.S. and elsewhere for the treatment of non-small cell lung cancer, metastatic colorectal cancer, glioblastoma, and metastatic renal cell carcinoma. As with sunitinib, no data are available specifically for lung NET.
A phase II trial compared bevacizumab (n = 22) with interferon-α-2b (n = 22) in metastatic low/intermediate-grade NET, including 4 lung NET, receiving octreotide [73]. Among bevacizumab recipients, 18% had PR, 77% had stable disease, and 5% had disease progression. Among interferon recipients, 68% had stable disease and 27% had disease progression. After 18 weeks, PFS rates were 95% for bevacizumab and 68% for interferon (p = .02). Bevacizumab, but not interferon, significantly reduced tumor blood flow. A follow-up study in 39 patients with low-grade NET confirmed single-agent bevacizumab reduced tumor blood flow. Bevacizumab plus everolimus reduced tumor blood flow even further, although everolimus alone had no effect [74]. As a follow-up to this small phase II trial, a phase III study compared octreotide plus interferon-α versus octreotide plus bevacizumab in patients with advanced, poor-prognosis NET (including atypical carcinoids). No significant differences in PFS were found between the interferon and the bevacizumab arms (16.6 vs. 15.4 months, respectively; p = .55), although the bevacizumab arm was associated with longer TTF and higher radiologic response [75]. Encouraging activity has been observed with bevacizumab plus temsirolimus in pNET [76], as well as bevacizumab plus temozolomide [77] or fluorouracil plus folinic acid plus oxaliplatin [78] in advanced NET. Ongoing studies with angiogenesis inhibitors in the treatment of patients with lung NET are listed in Table 3.
Peptide Receptor Radionuclide Therapy
In NET expressing SST receptors, PRRT may present an alternative treatment, with encouraging results observed, although most data come from single-center, retrospective studies. PRRT uses radiolabeled SSAs, where a high-energy β-emitting radionuclide such as 90Y or 177Lu is bound to an SSA to selectively deliver radiotherapy to SST receptor-positive tumor cells [38]. Various radiolabeled SSAs have been investigated, including 90Y-DOTATOC or 177Lu-DOTATATE [38]. The only data in lung NET come from a review of patients with foregut carcinoids treated with 177Lu-DOTATATE. The ORR was 50%; among 9 patients with lung NET, 5 had PR, 1 had minor response, and 2 had stable disease [79]. PRRT is still considered investigational in Europe and the U.S. due to a lack of data from prospective randomized studies [8, 9]. Nevertheless, recent ENETS consensus recommendations suggest PRRT may be an option to treat metastases from TC and AC demonstrating strong expression of SST receptors [26].
Multidisciplinary Team Approach
Owing to the nonspecificity of symptomatology, variable natural history, and limited high-level evidence from large randomized trials, diagnosis and management of lung NET are complex. Consultation with multiple specialists in clinical oncology, endocrinology, radiology/nuclear medicine, pathology, and surgery is required to manage aspects of disease [84]. In traditional care models, this can result in an overwhelming number of referrals, tests, evaluations/opinions, lack of continuity and uniformity in care, and treatment delays [81]. Furthermore, physicians may lack experience or education because of the rarity of lung NET, resulting in missed or delayed diagnoses [80].
A multidisciplinary team approach comprises coordinated care delivery by a team of healthcare professionals from various specialties, facilitating multiple consultations during a single patient visit [80]. This approach has demonstrated clear benefits in other malignancies, including improved diagnosis and staging accuracy, consistent use of recommended diagnostic tests, reduction in diagnosis and treatment delays, and improved compliance with evidence-based therapy [82–85]. Importantly, some centers using this approach demonstrated improved survival compared with traditional care [83, 85].
In Europe, ENETS has established certification criteria for Centers of Excellence for management of NET. Since program inception (2007/2008), 26 centers have been certified (http://www.enets.org/excellence.html). Although there is no equivalent program elsewhere, some institutions have independently established multidisciplinary resource centers [80].
Conclusion
Few treatment options are available for patients with lung NET whose tumors cannot be completely resected. Several promising systemic therapy options have emerged for treatment of NET, including everolimus, SSAs, temozolomide-based chemotherapy, and PRRT. Although limited evidence currently supports their efficacy in lung NET and none are approved for this specific population, treatment guidelines recommend consideration of systemic therapies (octreotide, lanreotide, everolimus, or chemotherapy) for patients with unresectable, advanced bronchopulmonary disease. Additional data from randomized trials in lung NET are needed to support establishment of standard treatment regimens.
Separate trials, when feasible, or at least stratification for primary tumor site, have been recommended in large trials [86]. To date, few prospective trials (and no randomized phase III trials) have been conducted exclusively in patients with lung NET. Several trials are ongoing to further assess the potential utility of promising systemic therapies, although most of these include lung NET patients as subsets of larger patient populations (Table 3). The largest clinical trial to date in patients with tumors of lung origin, the phase III RADIANT-4 study, is ongoing, with primary data anticipated in 2015. It is hoped that this and future trials will provide stronger evidence to support treatment recommendations for patients with lung NET.
Acknowledgments
I thank Sophia Shumyatsky, Pharm.D., and Harleigh E. Willmott, Ph.D., C.M.P.P., from ApotheCom, Yardley, Pennsylvania, for editorial assistance. Financial support for editorial assistance was provided by Novartis Pharmaceuticals Corporation, in compliance with international guidelines on Good Publication Practice.
Disclosures
Edward M. Wolin: Novartis, Ipsen, Celgene, Advanced Accelerator Applications (C/A).
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934–959. doi: 10.1002/cncr.11105. [DOI] [PubMed] [Google Scholar]
- 2.Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063–3072. doi: 10.1200/JCO.2007.15.4377. [DOI] [PubMed] [Google Scholar]
- 3.Rekhtman N. Neuroendocrine tumors of the lung: An update. Arch Pathol Lab Med. 2010;134:1628–1638. doi: 10.5858/2009-0583-RAR.1. [DOI] [PubMed] [Google Scholar]
- 4.Travis W, Brambilla E, Muller-Hermelink H, et al. World Health Organization classification of tumors. Pathology and genetics of tumors of the lung, pleura, thymus and heart. Lyon, France: IARC Press; 2004. [Google Scholar]
- 5.Gustafsson BI, Kidd M, Chan A, et al. Bronchopulmonary neuroendocrine tumors. Cancer. 2008;113:5–21. doi: 10.1002/cncr.23542. [DOI] [PubMed] [Google Scholar]
- 6.Travis WD. Advances in neuroendocrine lung tumors. Ann Oncol. 2010;21(suppl 7):vii65–vii71. doi: 10.1093/annonc/mdq380. [DOI] [PubMed] [Google Scholar]
- 7.Naalsund A, Rostad H, Strøm EH, et al. Carcinoid lung tumors--incidence, treatment and outcomes: a population-based study. Eur J Cardiothorac Surg. 2011;39:565–569. doi: 10.1016/j.ejcts.2010.08.036. [DOI] [PubMed] [Google Scholar]
- 8.Öberg K, Hellman P, Ferolla P, et al. Neuroendocrine bronchial and thymic tumors: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(suppl 7):vii120–vii123. doi: 10.1093/annonc/mds267. [DOI] [PubMed] [Google Scholar]
- 9.Phan AT, Oberg K, Choi J, et al. NANETS consensus guideline for the diagnosis and management of neuroendocrine tumors: well-differentiated neuroendocrine tumors of the thorax (includes lung and thymus) Pancreas. 2010;39:784–798. doi: 10.1097/MPA.0b013e3181ec1380. [DOI] [PubMed] [Google Scholar]
- 10.Toumpanakis C, Caplin ME. Update on the role of somatostatin analogs for the treatment of patients with gastroenteropancreatic neuroendocrine tumors. Semin Oncol. 2013;40:56–68. doi: 10.1053/j.seminoncol.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 11.Cutz E, Jackson A. Neuroepithelial bodies as airway oxygen sensors. Respir Physiol. 1999;115:201–214. doi: 10.1016/s0034-5687(99)00018-3. [DOI] [PubMed] [Google Scholar]
- 12.Swarts DR, Ramaekers FC, Speel EJ. Molecular and cellular biology of neuroendocrine lung tumors: evidence for separate biological entities. Biochim Biophys Acta. 2012;1826:255–271. doi: 10.1016/j.bbcan.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Davies SJ, Gosney JR, Hansell DM, et al. Diffuse idiopathic pulmonary neuroendocrine cell hyperplasia: An under-recognised spectrum of disease. Thorax. 2007;62:248–252. doi: 10.1136/thx.2006.063065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daddi N, Schiavon M, Filosso PL, et al. Prognostic factors in a multicentre study of 247 atypical pulmonary carcinoids. Eur J Cardiothorac Surg. 2014;45:677–686. doi: 10.1093/ejcts/ezt470. [DOI] [PubMed] [Google Scholar]
- 15.Hörsch D, Schmid KW, Anlauf M, et al. Neuroendocrine tumors of the bronchopulmonary system (typical and atypical carcinoid tumors): Current strategies in diagnosis and treatment. Conclusions of an expert meeting February 2011 in Weimar, Germany. Oncol Res Treat. 2014;37:266–276. doi: 10.1159/000362430. [DOI] [PubMed] [Google Scholar]
- 16.Swarts DR, van Suylen RJ, den Bakker MA, et al. Interobserver variability for the WHO classification of pulmonary carcinoids. Am J Surg Pathol. 2014;38:1429–1436. doi: 10.1097/PAS.0000000000000300. [DOI] [PubMed] [Google Scholar]
- 17.Rindi G, Klöppel G, Couvelard A, et al. TNM staging of midgut and hindgut (neuro) endocrine tumors: A consensus proposal including a grading system. Virchows Arch. 2007;451:757–762. doi: 10.1007/s00428-007-0452-1. [DOI] [PubMed] [Google Scholar]
- 18.Skov BG, Holm B, Erreboe A, et al. ERCC1 and Ki67 in small cell lung carcinoma and other neuroendocrine tumors of the lung: Distribution and impact on survival. J Thorac Oncol. 2010;5:453–459. doi: 10.1097/JTO.0b013e3181ca063b. [DOI] [PubMed] [Google Scholar]
- 19.Rindi G, Klersy C, Inzani F, et al. Grading the neuroendocrine tumors of the lung: An evidence-based proposal. Endocr Relat Cancer. 2013;21:1–16. doi: 10.1530/ERC-13-0246. [DOI] [PubMed] [Google Scholar]
- 20.Pelosi G, Rindi G, Travis WD, et al. Ki-67 antigen in lung neuroendocrine tumors: unraveling a role in clinical practice. J Thorac Oncol. 2014;9:273–284. doi: 10.1097/JTO.0000000000000092. [DOI] [PubMed] [Google Scholar]
- 21.Walts AE, Ines D, Marchevsky AM. Limited role of Ki-67 proliferative index in predicting overall short-term survival in patients with typical and atypical pulmonary carcinoid tumors. Mod Pathol. 2012;25:1258–1264. doi: 10.1038/modpathol.2012.81. [DOI] [PubMed] [Google Scholar]
- 22.Lococo F, Cesario A, Paci M, et al. PET/CT assessment of neuroendocrine tumors of the lung with special emphasis on bronchial carcinoids. Tumour Biol. 2014;35:8369–8377. doi: 10.1007/s13277-014-2102-y. [DOI] [PubMed] [Google Scholar]
- 23.Granberg D, Sundin A, Janson ET, et al. Octreoscan in patients with bronchial carcinoid tumours. Clin Endocrinol (Oxf) 2003;59:793–799. doi: 10.1046/j.1365-2265.2003.01931.x. [DOI] [PubMed] [Google Scholar]
- 24.Yellin A, Zwas ST, Rozenman J, et al. Experience with somatostatin receptor scintigraphy in the management of pulmonary carcinoid tumors. Isr Med Assoc J. 2005;7:712–716. [PubMed] [Google Scholar]
- 25.Krausz Y, Freedman N, Rubinstein R, et al. 68Ga-DOTA-NOC PET/CT imaging of neuroendocrine tumors: comparison with ¹¹¹In-DTPA-octreotide (OctreoScan®) Mol Imaging Biol. 2011;13:583–593. doi: 10.1007/s11307-010-0374-1. [DOI] [PubMed] [Google Scholar]
- 26.Caplin ME, Baudin E, Ferolla P, et al. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol. 2015;26:1604–1620. doi: 10.1093/annonc/mdv041. [DOI] [PubMed] [Google Scholar]
- 27.Moore W, Freiberg E, Bishawi M, et al. FDG-PET imaging in patients with pulmonary carcinoid tumor. Clin Nucl Med. 2013;38:501–505. doi: 10.1097/RLU.0b013e318279f0f5. [DOI] [PubMed] [Google Scholar]
- 28.Travis WD, Giroux DJ, Chansky K, et al. The IASLC Lung Cancer Staging Project: Proposals for the inclusion of broncho-pulmonary carcinoid tumors in the forthcoming (seventh) edition of the TNM Classification for Lung Cancer. J Thorac Oncol. 2008;3:1213–1223. doi: 10.1097/JTO.0b013e31818b06e3. [DOI] [PubMed] [Google Scholar]
- 29.Fink G, Krelbaum T, Yellin A, et al. Pulmonary carcinoid: presentation, diagnosis, and outcome in 142 cases in Israel and review of 640 cases from the literature. Chest. 2001;119:1647–1651. doi: 10.1378/chest.119.6.1647. [DOI] [PubMed] [Google Scholar]
- 30.Lou F, Sarkaria I, Pietanza C, et al. Recurrence of pulmonary carcinoid tumors after resection: implications for postoperative surveillance. Ann Thorac Surg. 2013;96:1156–1162. doi: 10.1016/j.athoracsur.2013.05.047. [DOI] [PubMed] [Google Scholar]
- 31.Aydin E, Yazici U, Gulgosteren M, et al. Long-term outcomes and prognostic factors of patients with surgically treated pulmonary carcinoid: our institutional experience with 104 patients. Eur J Cardiothorac Surg. 2011;39:549–554. doi: 10.1016/j.ejcts.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 32.Cañizares MA, Matilla JM, Cueto A, et al. Atypical carcinoid tumours of the lung: Prognostic factors and patterns of recurrence. Thorax. 2014;69:648–653. doi: 10.1136/thoraxjnl-2013-204102. [DOI] [PubMed] [Google Scholar]
- 33.Okoye CC, Jablons DM, Jahan TM, et al. Divergent management strategies for typical versus atypical carcinoid tumors of the thoracic cavity. Am J Clin Oncol. 2014;37:350–355. doi: 10.1097/COC.0b013e31827a7f6d. [DOI] [PubMed] [Google Scholar]
- 34.Fox M, Van Berkel V, Bousamra M, 2nd, et al. Surgical management of pulmonary carcinoid tumors: Sublobar resection versus lobectomy. Am J Surg. 2013;205:200–208. doi: 10.1016/j.amjsurg.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 35.National Comprehensive Cancer Network, Inc. NCCN Clinical Practice Guidelines in Oncology: Small cell lung cancer version 1.2016. Available at: http://www.nccn.org/professionals/physician_gls/pdf/sclc.pdf Accessed July 9, 2015.
- 36.Chong CR, Wirth LJ, Nishino M, et al. Chemotherapy for locally advanced and metastatic pulmonary carcinoid tumors. Lung Cancer. 2014;86:241–246. doi: 10.1016/j.lungcan.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carretta A, Ceresoli GL, Arrigoni G, et al. Diagnostic and therapeutic management of neuroendocrine lung tumors: A clinical study of 44 cases. Lung Cancer. 2000;29:217–225. doi: 10.1016/s0169-5002(00)00119-7. [DOI] [PubMed] [Google Scholar]
- 38.Pavel M, Kidd M, Modlin I. Systemic therapeutic options for carcinoid. Semin Oncol. 2013;40:84–99. doi: 10.1053/j.seminoncol.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 39.Novartis Pharmaceuticals Corp. Sandostatin LAR Depot (octreotide acetate for injectable suspension) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corp. Revised July 2014.
- 40.Novartis Pharmaceuticals UK Ltd. Sandostatin LAR 10 mg, 20 mg or 30 mg powder and solvent for suspension for injection. Surrey, UK: Novartis Pharmaceuticals UK Ltd. Updated January 2015.
- 41.Ipsen Ltd. Somatuline LA 30 mg, powder for suspension for injection. Slough, Berkshire, UK: Ipsen Limited. Updated November 2014.
- 42.Ruszniewski P, Ish-Shalom S, Wymenga M, et al. Rapid and sustained relief from the symptoms of carcinoid syndrome: Results from an open 6-month study of the 28-day prolonged-release formulation of lanreotide. Neuroendocrinology. 2004;80:244–251. doi: 10.1159/000082875. [DOI] [PubMed] [Google Scholar]
- 43.Rubin J, Ajani J, Schirmer W, et al. Octreotide acetate long-acting formulation versus open-label subcutaneous octreotide acetate in malignant carcinoid syndrome. J Clin Oncol. 1999;17:600–606. doi: 10.1200/JCO.1999.17.2.600. [DOI] [PubMed] [Google Scholar]
- 44.Oberg K, Norheim I, Theodorsson E. Treatment of malignant midgut carcinoid tumours with a long-acting somatostatin analogue octreotide. Acta Oncol. 1991;30:503–507. doi: 10.3109/02841869109092409. [DOI] [PubMed] [Google Scholar]
- 45.O’Toole D, Ducreux M, Bommelaer G, et al. Treatment of carcinoid syndrome: A prospective crossover evaluation of lanreotide versus octreotide in terms of efficacy, patient acceptability, and tolerance. Cancer. 2000;88:770–776. doi: 10.1002/(sici)1097-0142(20000215)88:4<770::aid-cncr6>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 46.Vinik A, Wolin EM, Audry H, et al. ELECT: A phase 3 study of efficacy and safety of lanreotide autogel/depot (LAN) treatment for carcinoid syndrome in patients with neuroendocrine tumors (NETs) J Clin Oncol. 2014;32(suppl 3):268a. [Google Scholar]
- 47.Dong M, Phan AT, Yao JC. New strategies for advanced neuroendocrine tumors in the era of targeted therapy. Clin Cancer Res. 2012;18:1830–1836. doi: 10.1158/1078-0432.CCR-11-2105. [DOI] [PubMed] [Google Scholar]
- 48.Janson ET, Oberg K. Long-term management of the carcinoid syndrome. Treatment with octreotide alone and in combination with alpha-interferon. Acta Oncol. 1993;32:225–229. doi: 10.3109/02841869309083916. [DOI] [PubMed] [Google Scholar]
- 49.National Comprehensive Cancer Network, Inc. NCCN Clinical Practice Guidelines in Oncology Neuroendocrine Tumors. Version 1. 2015; 2015:1–108. Available at: http://www.nccn.org/professionals/physician_gls/pdf/neuroendocrine.pdf. Accessed July 9, 2015.
- 50.Rinke A, Müller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID Study Group. J Clin Oncol. 2009;27:4656–4663. doi: 10.1200/JCO.2009.22.8510. [DOI] [PubMed] [Google Scholar]
- 51.Caplin ME, Pavel M, Ćwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371:224–233. doi: 10.1056/NEJMoa1316158. [DOI] [PubMed] [Google Scholar]
- 52.Ipsen Biopharmaceuticals, Inc. Somatuline depot (lanreotide) injection. Signes, France: Ipsen Pharma Biotech. Revised December 2014.
- 53.Ipsen Biopharmaceuticals, Inc. Ipsen announces acceptance of filings for somatuline in the treatment of GEP-NETs in the US with priority review and in Europe [press release]. Available at: http://www.businesswire.com/news/home/20140831005040/en/Ipsen-Announces-Acceptance-Filings-Somatuline%C2%AE-Treatment-GEP-NET1s Accessed August 10, 2015.
- 54.Fjällskog ML, Granberg DP, Welin SL, et al. Treatment with cisplatin and etoposide in patients with neuroendocrine tumors. Cancer. 2001;92:1101–1107. doi: 10.1002/1097-0142(20010901)92:5<1101::aid-cncr1426>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 55.Ekeblad S, Sundin A, Janson ET, et al. Temozolomide as monotherapy is effective in treatment of advanced malignant neuroendocrine tumors. Clin Cancer Res. 2007;13:2986–2991. doi: 10.1158/1078-0432.CCR-06-2053. [DOI] [PubMed] [Google Scholar]
- 56.Crona J, Fanola I, Lindholm DP, et al. Effect of temozolomide in patients with metastatic bronchial carcinoids. Neuroendocrinology. 2013;98:151–155. doi: 10.1159/000354760. [DOI] [PubMed] [Google Scholar]
- 57.Spada F, Fumagalli C, Antonuzzo L, et al. Capecitabine plus temozolomide (CAP-TEM) in patients with advanced neuroendocrine neoplasms (NEN): An Italian multicenter retrospective analysis. J Clin Oncol. 2014;32(suppl):281a. [Google Scholar]
- 58.Sun W, Lipsitz S, Catalano P, et al. Phase II/III study of doxorubicin with fluorouracil compared with streptozocin with fluorouracil or dacarbazine in the treatment of advanced carcinoid tumors: Eastern Cooperative Oncology Group Study E1281. J Clin Oncol. 2005;23:4897–4904. doi: 10.1200/JCO.2005.03.616. [DOI] [PubMed] [Google Scholar]
- 59.Moertel CG, Lefkopoulo M, Lipsitz S, et al. Streptozocin-doxorubicin, streptozocin-fluorouracil or chlorozotocin in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1992;326:519–523. doi: 10.1056/NEJM199202203260804. [DOI] [PubMed] [Google Scholar]
- 60.Granberg D, Eriksson B, Wilander E, et al. Experience in treatment of metastatic pulmonary carcinoid tumors. Ann Oncol. 2001;12:1383–1391. doi: 10.1023/a:1012569909313. [DOI] [PubMed] [Google Scholar]
- 61.Righi L, Volante M, Rapa I, et al. Mammalian target of rapamycin signaling activation patterns in neuroendocrine tumors of the lung. Endocr Relat Cancer. 2010;17:977–987. doi: 10.1677/ERC-10-0157. [DOI] [PubMed] [Google Scholar]
- 62.Terris B, Scoazec JY, Rubbia L, et al. Expression of vascular endothelial growth factor in digestive neuroendocrine tumours. Histopathology. 1998;32:133–138. doi: 10.1046/j.1365-2559.1998.00321.x. [DOI] [PubMed] [Google Scholar]
- 63.Ambs S, Bennett WP, Merriam WG, et al. Vascular endothelial growth factor and nitric oxide synthase expression in human lung cancer and the relation to p53. Br J Cancer. 1998;78:233–239. doi: 10.1038/bjc.1998.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Phan AT, Wang L, Xie K, et al. Association of VEGF expression with poor prognosis among patients with low-grade neuroendocrine carcinoma. J Clin Oncol. 2006;24(suppl):4091a. [Google Scholar]
- 65.Granberg D, Wilander E, Oberg K. Expression of tyrosine kinase receptors in lung carcinoids. Tumour Biol. 2006;27:153–157. doi: 10.1159/000092718. [DOI] [PubMed] [Google Scholar]
- 66.Zatelli MC, Minoia M, Martini C, et al. Everolimus as a new potential antiproliferative agent in aggressive human bronchial carcinoids. Endocr Relat Cancer. 2010;17:719–729. doi: 10.1677/ERC-10-0097. [DOI] [PubMed] [Google Scholar]
- 67.Pavel ME, Hainsworth JD, Baudin E, et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomised, placebo-controlled, phase 3 study. Lancet. 2011;378:2005–2012. doi: 10.1016/S0140-6736(11)61742-X. [DOI] [PubMed] [Google Scholar]
- 68.Fazio N, Granberg D, Grossman A, et al. Everolimus plus octreotide long-acting repeatable in patients with advanced lung neuroendocrine tumors: analysis of the phase 3, randomized, placebo-controlled RADIANT-2 study. Chest. 2013;143:955–962. doi: 10.1378/chest.12-1108. [DOI] [PubMed] [Google Scholar]
- 69.Pavel ME, Wiedenmann B, Capdevila J, et al. RAMSETE: A single arm, multicenter, single-stage phase II trial of RAD001 (everolimus) in advanced and metastatic silent neuro-endocrine tumours in Europe. J Clin Oncol. 2012;30(suppl):4122a. [Google Scholar]
- 70.Kulke MH, Lenz HJ, Meropol NJ, et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol. 2008;26:3403–3410. doi: 10.1200/JCO.2007.15.9020. [DOI] [PubMed] [Google Scholar]
- 71.Cives M, Strosbeg JR, Campos T, et al. A phase II study of axitinib in advanced carcinoid tumors: Preliminary results. J Clin Oncol. 2015;33(suppl):4100a. [Google Scholar]
- 72.Phan AT, Halperin DM, Chan JA, et al. Pazopanib and depot octreotide in advanced, well-differentiated neuroendocrine tumours: A multicentre, single-group, phase 2 study. Lancet Oncol. 2015;16:695–703. doi: 10.1016/S1470-2045(15)70136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yao JC, Phan A, Hoff PM, et al. Targeting vascular endothelial growth factor in advanced carcinoid tumor: A random assignment phase II study of depot octreotide with bevacizumab and pegylated interferon alpha-2b. J Clin Oncol. 2008;26:1316–1323. doi: 10.1200/JCO.2007.13.6374. [DOI] [PubMed] [Google Scholar]
- 74.Yao JC, Phan AT, Fogelman D, et al. Randomized run-in study of bevacizumab (B) and everolimus (E) in low- to intermediate-grade neuroendocrine tumors (LGNETs) using perfusion CT as functional biomarker. J Clin Oncol. 2010;28(suppl):4002a. [Google Scholar]
- 75.Yao JC, Guthrie K, Moran C, et al. SWOG S0518: Phase III prospective randomized comparison of depot octreotide plus interferon alpha-2b versus depot octreotide plus bevacizumab (NSC #704865) in advanced, poor prognosis carcinoid patients ( NCT00569127) J Clin Oncol. 2015;33(suppl):4004a. doi: 10.1200/JCO.2016.70.4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hobday TJ, Qin R, Reidy DL, et al. Multicenter phase II trial of temsirolimus (TEM) and bevacizumab (BEV) in pancreatic neuroendocrine tumor (PNET) J Clin Oncol. 2012;30(suppl):260a. doi: 10.1200/JCO.2014.56.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kulke MH, Stuart K, Earle CC, et al. A phase II study of temozolomide and bevacizumab in patients with advanced neuroendocrine tumors. J Clin Oncol. 2006;24(suppl):4044a. [Google Scholar]
- 78.Venook AP, Ko AH, Tempero MA, et al. Phase II trial of FOLFOX plus bevacizumab in advanced, progressive neuroendocrine tumors. J Clin Oncol. 2008;26(suppl):15545a. [Google Scholar]
- 79.van Essen M, Krenning EP, Bakker WH, et al. Peptide receptor radionuclide therapy with 177Lu-octreotate in patients with foregut carcinoid tumours of bronchial, gastric and thymic origin. Eur J Nucl Med Mol Imaging. 2007;34:1219–1227. doi: 10.1007/s00259-006-0355-4. [DOI] [PubMed] [Google Scholar]
- 80.Singh S, Law C. Multidisciplinary reference centers: The care of neuroendocrine tumors. J Oncol Pract. 2010;6:e11–e16. doi: 10.1200/JOP.2010.000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Metz DC, Choi J, Strosberg J, et al. A rationale for multidisciplinary care in treating neuroendocrine tumours. Curr Opin Endocrinol Diabetes Obes. 2012;19:306–313. doi: 10.1097/MED.0b013e32835570f1. [DOI] [PubMed] [Google Scholar]
- 82.Tamagno G, McGowan L, King R, et al. Initial impact of a systematic multidisciplinary approach on the management of patients with gastroenteropancreatic neuroendocrine tumor. Pancreas. 2010;39:281. doi: 10.1007/s12020-013-9910-5. [DOI] [PubMed] [Google Scholar]
- 83.Forrest LM, McMillan DC, McArdle CS, et al. An evaluation of the impact of a multidisciplinary team, in a single centre, on treatment and survival in patients with inoperable non-small-cell lung cancer. Br J Cancer. 2005;93:977–978. doi: 10.1038/sj.bjc.6602825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gabel M, Hilton NE, Nathanson SD. Multidisciplinary breast cancer clinics. Do they work? Cancer. 1997;79:2380–2384. [PubMed] [Google Scholar]
- 85.Stephens MR, Lewis WG, Brewster AE, et al. Multidisciplinary team management is associated with improved outcomes after surgery for esophageal cancer. Dis Esophagus. 2006;19:164–171. doi: 10.1111/j.1442-2050.2006.00559.x. [DOI] [PubMed] [Google Scholar]
- 86.Kulke MH, Siu LL, Tepper JE, et al. Future directions in the treatment of neuroendocrine tumors: Consensus report of the National Cancer Institute Neuroendocrine Tumor clinical trials planning meeting. J Clin Oncol. 2011;29:934–943. doi: 10.1200/JCO.2010.33.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.US National Institutes of Health. 3-arm trial to evaluate pasireotide LAR/everolimus alone/in combination in patients with lung/thymus NET - LUNA trial. Available at https://clinicaltrials.gov/ct2/show/NCT01563354. Accessed April 8, 2013.
- 88.US National Institutes of Health. Everolimus plus best supportive care vs placebo plus best supportive care in the treatment of patients with advanced neuroendocrine tumors (GI or lung origin) (RADIANT-4). Available at http://clinicaltrials.gov/ct2/show/NCT01524783. Accessed April 8, 2013.
- 89.US National Institutes of Health. Pazopanib hydrochloride in treating patients with progressive carcinoid tumors Available at https://clinicaltrials.gov/ct2/show/NCT01841736. Accessed October 6, 2014.
- 90.US National Institutes of Health. 177Lutetium-DOTA-octreotate therapy in somatostatin receptor-expressing neuroendocrine neoplasms. Available at http://clinicaltrials.gov/ct2/show/NCT01237457. Accessed October 6, 2014.