

Abstract
Background and Purpose
Oxidative stress and subsequent activation of inflammatory responses is a widely accepted consequence of exposure to environmental toxins. TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin), a well-known environmental toxin, exerts its toxicity through many signalling mechanisms, with liver being the principal organ affected. However, an effective antidote to TCDD-induced toxicity is unknown. The present study evaluated the effect of eicosapentaenoic acid (EPA), an n3 fatty acid, on TCDD-induced toxicity.
Experimental Approach
In cultures of HepG2 cells, the EPA/AA ratio was determined using gas chromatography, oxidative stress and inflammatory responses through reactive oxygen species (ROS) levels, antioxidant status, [Ca2+]i, nuclear migration of two redox-sensitive transcription factors, NF-κB p65 and Nrf-2, expression of MAP kinase (p-Erk, p-p38), NF-κB p65, COX-2 and Nrf-2. Cellular changes in ΔΨm, acidic vesicular organelle formation, cell cycle analysis and scanning electron microscopy analysis were performed.
Key Results
EPA offered significant cytoprotection by increasing EPA/AA ratios in cell membranes, inhibiting ROS generation, enhancing antioxidant status and modulating nuclear translocation of redox-sensitive transcription factors (NF-κB p65 and Nrf-2) and expression of NF-κB p65, COX-2 and Nrf-2. Furthermore, TCDD-induced upstream events of MAPK phosphorylation, the increase in [Ca2+]i levels and cell surface changes in microvilli were significantly inhibited by EPA. EPA treatment maintained ΔΨm and prevented formation of acidic vesicular organelles.
Conclusion and Implications
The present study demonstrates for the first time some underlying molecular mechanisms of cytoprotection exerted by EPA against TCDD-induced oxidative stress and inflammatory responses.
Table of Links
| TARGETS |
|---|
| Enzymes |
| COX-2 |
| CYP1A1 |
| ERK2 |
| p-38 kinase |
| LIGANDS |
|---|
| AA, arachidonic acid |
| EPA, eicosapentaenoic acid |
| U0126 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
It is well accepted that free radicals exert their effect on cellular processes through cell signalling pathways leading to toxicity. TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin), a potent liver carcinogen, is known to induce reactive oxygen species (ROS), which plays a central role in TCDD toxicity. The mechanism involves activation of the cytochrome P450 CYP1A1, as well as arachidonic acid (AA) and its metabolites (Nebert et al., 2000; Bui et al., 2012). TCDD elicits its toxicity through multiple signalling mechanisms involving activation of the aryl hydrocarbon receptor (AhR) and AA release from membrane phospholipids (Tan et al., 2002; Giudetti and Cagnazzo, 2012). Thus, direct downstream events of CYP1A1 activation through AhR and formation of AA metabolites lead to sustained oxidative stress and subsequent inflammatory responses (Kraemer et al., 1996; Vogel et al., 1997; Dong and Matsumura, 2008; Sciullo et al., 2009). In addition, activated macrophages in liver cells show sustained NF-κB activation, which contributes to chronic inflammatory responses (Sunami et al., 2012). As TCDD induces toxic responses by several routes, intense efforts have been made to identify its mechanism of action. However, approaches taken to alleviate TCDD-induced toxicity are few and dietary intervention by natural compounds could offer significant protection. Thus, an effective treatment that could target both AA and ROS generation and subsequent downstream events would be an effective strategy to prevent TCDD-induced toxicity.
Polyunsaturated fatty acids (PUFAs) in cell membranes are responsible for membrane fluidity, cell–cell interaction and cellular signalling (Benatti et al., 2004). The ratio of n3 and n6 PUFAs in the cell membrane determines various signalling events to external stimuli. A high n3/n6 ratio is important for redox homeostasis in the body, which is primarily influenced by diet (Simopoulos, 2002; Mullen et al., 2010). Several epidemiological studies have reported that consumption of n3 fatty acids decreased the incidence of inflammatory diseases by modulating the n3/n6 ratio (Kremer et al., 1987; Kremer, 2000; Calder, 2006). Eicosapentaenoic acid (EPA), an n3 PUFA, is a potent antioxidant and anti-inflammatory agent and regulates the expression of various cytoprotective antioxidant enzymes (Yin et al., 2007; Brooks et al., 2008). In addition, EPA and AA compete for membrane incorporation leading to changes in membrane n3/n6 ratios, thereby modulating various signalling pathways. EPA is known to be protective against valproate-induced liver oxidative stress (El-Mowafy et al., 2011), neurotoxicity (Kawashima et al., 2008), rheumatoid arthritis (Kremer et al., 1990), diabetes (Sarbolouki et al., 2013) and cardiotoxicity (von Schacky and Harris, 2007). The protection offered by EPA against TCDD-induced toxicity was published earlier by Turkez et al. (2012) through basic cytotoxicity and oxidative stress assays, without the exact molecular mechanisms of EPA's actions being delineated.
The present study focused on the cytoprotective effect of EPA on TCDD-induced toxicity in HepG2 cells, with specific reference to oxidative stress, inflammatory responses and redox signalling through two important transcription factors: NF-κB and Nrf-2. The study demonstrates for the first time sequence of some molecular mechanisms underlying the cytoprotective effect of EPA against TCDD-induced cytotoxicity.
Methods
Cell culture conditions and treatments
The HepG2 cell line was obtained from the National Centre for Cell Science, Pune, India. The cells were grown in 25 cm2/75 cm2 flasks supplemented with DMEM with 5% FBS (v/v) containing 100 units·mL−1 penicillin and 30 μg·mL−1 streptomycin in a CO2 incubator with humidified atmosphere of 95% air and 5% CO2. Cells at 80% confluence were used for all treatments.
Fatty acid preparation
EPA stock was prepared in 100% ethanol and stored at −20°C in the dark. Before EPA treatment, the fatty acid was conjugated with fatty acid free-BSA at a molar ratio of 3:1 in 5% FBS containing DMEM media for 1 h at 37°C. The final concentrations of ethanol during treatment did not exceed 0.01%.
Cell viability by MTT assay
Cell viability was determined by MTT assay (Mossman, 1983). HepG2 cells were used at a density of 1 × 104 cells per well. In order to determine optimum dosages and exposure times, preliminary cytotoxicity and cytoprotective studies were carried out. Dose and time responsive study was performed with different concentrations of TCDD (0.1, 1, 10, 50 and 100 nM) for different time points (24, 48 and 72 h). From the above fixed dose and time point (10 nM TCDD for 48 h), cytoprotection offered by EPA was determined by pre-treating, co-treating and post-treating cells with EPA at different concentrations (10, 20, 30, 40 μM) for different time points (24 and 48 h) against TCDD-induced cytotoxicity. TCDD was dissolved in DMSO (final solvent concentration <0.01%). Combined solvent toxicity of EPA and TCDD (<0.02%) did not show any cytotoxicity. After the respective treatment periods, the cells were incubated for 5 h with MTT (5 mg·mL−1), and purple formazan crystals were read spectrophotometrically at 570 nm (Bio-Tek Instruments, Winooski, VT, USA). Treatment schedule was followed unless otherwise defined: Cells were treated with TCDD for 48 h, with and without EPA pre-treatment for 48 h. For EPA control, the cells were treated with 40 μM EPA alone for 48 h.
Membrane incorporation of EPA by gas chromatography
Membrane incorporation of EPA was analysed as described previously (Morrison and Smith, 1964). HepG2 cells (5 × 106 per group) were treated with TCDD for 48 h in the presence or absence of EPA for 48 h. Lipids were extracted (Folch et al., 1957) and saponified by methylation to their corresponding fatty acid methyl esters (FAMEs) using BF3-methanol. The FAMEs were separated by gas liquid chromatography (Varian Inc. Corporate Headquarters, Palo Alto, CA, USA) equipped with a capillary column (Elite 225, 30 m long and 0.25 mm diameter) and a flame ionization detector in the presence of hydrogen and air, using nitrogen as a carrier gas. The data were analysed using chromcard software (Thermo Fisher Scientific, San Jose, CA, USA), and the values of AA and EPA were calculated from the mean percentage of the total fatty acids.
Estimation of [Ca2+]i level
Intracellular calcium levels were determined using Fura-2 AM (Sul et al., 2009). Time course study on TCDD-induced intracellular calcium level showed the maximum increase at 45 min (preliminary data not shown). Cells were treated with EPA in the presence or absence of TCDD for 45 min. For pre-treatment, cells were treated with EPA for 48 h, washed with PBS and incubated with Fura-2 AM for 1 h, followed by TCDD treatment for 45 min. [Ca2+]i levels were measured at an excitation wavelength (510 nm) and emission wavelength cycling between 340 and 380 nm. The ratio of intensities (340/380) is proportional to the changes in the [Ca2+]i. The values were expressed as relative percentage of Fura-2 AM fluorescence compared with the control.
Measurement of CYP1A1 activity
The CYP1A1 activity was determined using the reaction mixture containing 5 mM MgCl2, 5 μM 7-ethoxy resorufin and 10 μM dicoumarol. Conversion of 7-ethoxy resorufin to resorufin was measured spectrofluorimetrically (excitation 530 nm; emission 590 nm) (Peters et al., 2004). The CYP1A1 activity was expressed as picomol resorufin formed· min−1 (mg protein)−1.
Estimation of ROS
ROS were determined by using DCF-DA (Royall and Ischiropoulos, 1993). TCDD-induced ROS generation peaked at 6 h (preliminary data not shown). Cells were treated with or without EPA pre-treatment (48 h) followed by addition of DCF-DA for 30 min and TCDD for 6 h. The samples were re-suspended in PBS and read in a Hitachi spectrofluorometer (Hitachi Spectrofluorometer, Tokyo, Japan; excitation 480 nm, emission 520 nm). The values were expressed as relative percentage of DCF fluorescence compared with the control.
Estimation of GSH and antioxidant enzyme activities
The GSH levels were measured spectrofluorimetrically using ortho-phthalaldehyde (OPT) (Hissin and Hilf, 1976). The samples that were precipitated with orthophosphoric acid were treated with OPT (5 mg·mL−1) and incubated in the dark for 10 min, and analysed in a spectrofluorometer (excitation wavelength, 350 nm; emission wavelength, 420 nm). The cell extract was prepared with 50 mM Tris, 5 mM EDTA, 10 μg·mL−1 PMSF, pH 7.6. The sonicated samples were centrifuged at 1700× g for 5 min at 4°C. The supernatant was aliquoted and stored at −80°C. The protein content was determined as described previously (Lowry et al., 1951).
NAD(P)H:quinone oxidoreductase 1 (NQO1) activity
The resorufin consumption was measured for 3 min at an interval of 1 min (excitation 530 nm; emission wavelength 585 nm) (Nims et al., 1984).
Superoxide dismutase (SOD) activity
Auto-oxidation of pyrogallol was measured by an increase in absorbance at 420 nm at 30 s intervals for 3 min (Marklund and Marklund, 1974).
Catalase (CAT) activity
The reaction was initiated by the addition of H2O2 to a final concentration of 10 mM and the change in absorbance was measured at 240 nm at 30 s intervals for 3 min. The specific activity of catalase was calculated based on the extinction coefficient of hydrogen peroxide, E240 = 43.6 M−1·cm−1 (Aebi, 1974).
Glutathione-S-transferase (GST) activity
The change in absorbance was measured at 340 nm for 3 min at 30 s intervals. The specific activity was calculated based on the extinction coefficient of GS-CDNB, E340 = 0.0096 μM−1·cm−1 (Habig et al., 1974).
Glutathione peroxidase (GPx) activity
GPx activity was determined as described previously (Rotruck et al., 1973). Glutathione consumed was calculated from a standard graph using linear regression analysis.
Nuclear localization study – immunofluorescence
Time course study on TCDD-induced NF-κB p65 nuclear localization was carried out at different time points. From the shortlisted time point (7 h), the effect of EPA was studied with TCDD treatment for 7 h, with or without EPA pre-treatment for 48 h. To determine the time point at which EPA mediated Nrf-2 localization, cells were pre-treated with EPA followed by TCDD treatment at different time points (Figure 4A). Further, the effect of EPA and TCDD on the level of Nrf-2 localization was determined from the appropriate time point. Briefly, cells (1 × 104) were allowed to attach on a polylysine-coated coverslip. Following treatment, cells were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for 10 min and then exposed to primary antibodies (NF-κB p65 or Nrf-2) at 1:100 dilutions overnight at 4°C and treated with FITC-conjugated secondary antibody (1:2000) for 1 h at room temperature. Nuclei were stained with DAPI for 2 min and viewed under Olympus FV1000 confocal laser scanning microscope (Na et al., 2006).
Figure 4.

EPA mediates nuclear migration and up-regulation of Nrf-2 against TCDD-induced oxidative stress. (A) EPA mediates Nrf-2 migration in response to TCDD-induced oxidative stress: The cells were pre-treated with EPA followed by TCDD treatment at different time points [48 h (EPA) + 3 h (TCDD); 48 h (EPA) + 5 h (TCDD); 48 h (EPA) + 7 h (TCDD); 48 h (EPA) + 9 h (TCDD)]. Nrf-2 nuclear localization was visualized using mouse monoclonal Nrf-2 primary antibody followed by addition of FITC-conjugated secondary antibody. The nuclei were visualized using DAPI. Control and cells treated with 48 h (EPA) + 3 h (TCDD); 48 h (EPA) + 5 h (TCDD) show cytoplasmic sequestration of Nrf-2. Cells treated with 48 h (EPA) + 7 h (TCDD); 48 h (EPA) + 9 h (TCDD) show nuclear localization of Nrf-2, visualized as turquoise blue colour. CL, cytoplasmic localization; NL, nuclear localization. (B) Effect of EPA and TCDD on Nrf-2 nuclear migration: The cells were treated with TCDD for 7 h in the presence and absence of EPA for 48 h. Nrf-2 nuclear localization was visualized using mouse monoclonal Nrf-2 primary antibody followed by addition of FITC-conjugated secondary antibody. The nuclei were visualized using DAPI. Control and EPA-alone-treated cells showed retention of Nrf-2 in the cytoplasm. Cells exposed to TCDD for 7 h show partial Nrf-2 nuclear localization. Pre-treatment with EPA followed by TCDD treatment shows nuclear localization of Nrf-2 which was visualized as turquoise blue colour. CL, cytoplasmic localization; NL, nuclear localization. (C) EPA up-regulates Nrf-2 expression: Cells were treated with TCDD for 48 h in the presence/absence of EPA for 48 h. (D) Densitometric analysis of Nrf-2 expression: The results shown are means ± SE of three independent experiments. Significant up-regulation (*P < 0.05) of Nrf-2 expression was observed in TCDD treatment compared with the control cells. Pre-treatment with EPA followed by TCDD showed a further increase (+++P < 0.001) compared with the TCDD-treated cells. NS, non-significantly different from control; Student's t-test.
Preparation of whole cell lysates and nuclear fractionation for Western blotting
Cell samples (approx 5 × 106 cells) were used for determining protein expression. Preliminary studies on TCDD-induced time course expressions showed maximum expression of MAP kinases (1 h), NF-κB p65 (12 h), COX-2 (15 h) and Nrf-2 (48 h) (data not shown). The effects of EPA on these proteins were monitored at the above-mentioned time points with or without EPA pre-treatment for 48 h. All controls received solvents, and for the EPA control group, the cells were treated with EPA for 48 h.
Whole cell lysate
Cells were lysed with whole cell lysis buffer (RIPA buffer) and stored at −80°C. The samples were exposed to one cycle of freezing and thawing and the protein concentration of the supernatant was measured (Lowry et al. (1951). For preparation of nuclear extracts, cells were lysed with ice-cold cytosolic extraction buffer {10 mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM EDTA (pH 8.0), 0.1 mM EGTA (pH 7.0), 1 mM DTT, 10% NP-40, 1 mM PMSF and 0.5 μL protease inhibitor cocktail [AEBSF [4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride] (104 mM), aprotinin (80 μM), bestatin (4 mM), E-64 (1.4 mM), leupeptin (2 mM), pepstatin A (1.5 mM)]} and incubated in ice for 20 min. The cells were then centrifuged at 10 000× g for 5 min. The supernatant was removed and ice-cold nuclear extraction buffer {10 mM HEPES (pH 7.9), 400 mM NaCl, 1 mM EDTA (pH 8.0), 1 mM EGTA (pH 7.0), 1 mM DTT, 0.5 mM PMSF and 0.1 μL protease inhibitor cocktail [AEBSF (104 mM), aprotinin (80 μM), bestatin (4 mM), E-64 (1.4 mM), leupeptin (2 mM), pepstatin A (1.5 mM)]} was added to the pellet and incubated in ice for 30 min. The extract was centrifuged at 10 000× g for 15 min, and the supernatant containing nuclear extract was transferred to a pre-chilled tube, protein concentration was determined and stored at −80°C until further analysis.
Western blotting: Erk (p44/p42), p38, NF-κB p65, COX-2, Nrf-2 expression
Western blot analysis was carried out as described previously Towbin et al. (1979). About 50 μg of protein samples was loaded on an equal protein basis, separated on a 10% SDS-PAGE and electroblotted onto nitrocellulose membranes. The membrane was probed with specific monoclonal primary antibodies at 1:200 dilution in 5% (w/v) non-fat milk overnight, followed by incubation with secondary antibody (HRP-conjugated rabbit anti-mouse antibody) at (1:5000) for 1 h. The bands were developed using the DAB/H2O2 colour development system and densitometric analysis was carried out using ImageJ software (Wayne Rasband, Bethesda, MD, USA).
Determination of mitochondrial membrane potential (Δψm)
Mitochondrial membrane potential was determined as described previously (Marchetti et al., 1996). DiOC6 (3) is a cell permeable marker that specifically accumulates in the mitochondria depending upon ΔΨm. After treatment, the cells were washed with PBS, treated with 50 nM DiOC6 (3) and incubated at 37°C in a CO2 incubator for 30 min. Simultaneously, a positive control of the mitochondrial uncoupler CCCP was added 15 min prior to the addition of DiOC6 (3). Following which, cells were suspended in 1.0 mL of PBS and read in a spectrofluorometer (excitation: 488 nm and emission: 500 nm). The values are expressed as relative percentage of mitochondrial membrane potential compared with the control.
Scanning electron microscopy
Cells were fixed in 2.0% (w/v) paraformaldehyde and 2.5% (v/v) glutaraldehyde in 0.1 M Na cacodylate buffer, pH 7.4. Secondary fixation was performed using 1.0% aqueous osmium tetroxide (w/v) in 0.1 M Na cacodylate buffer, pH 7.4. The samples were then serially dehydrated in ethanol, critical point dried and analysed in a JEOL6390 LA scanning elctron microscope (JOEL Ltd., Tokyo, Japan).
Cell cycle measurements
Cells were fixed in 80% (v/v) ice-cold methanol at 4°C overnight. Following which, cells were washed with PBS and treated with 1 mL of propidium iodide (50 μg·mL−1 in sodium citrate buffer containing 0.1% Triton X-100 and 20 μg·mL−1 RNase) and analysed in a BD 290 flow cytometer (J. Trotter, San Diego, CA, USA) using Win MDI flow cytometric software (BD Biosciences, San Jose, CA, USA).
Acridine orange (AO)/ethidium bromide (ETBR) staining
Cell death induced by TCDD was determined as described previously (Meiyanto et al., 2007). After treatment, cells were treated with AO/ETBR for 30 min, mounted on a slide and images captured with an Olympus fluorescent microscope in green filter (excitation wavelength: 536 nm; emission wavelength: 617 nm; 200 cells were scored for each treatment).
Data analysis
Data were analysed by one-way anova followed by Tukey's analysis for all assays except densitometric analysis, which was assessed using a Student's t- test.
Materials
TCDD was a kind gift from Dr. Howard G. Shertzer, Professor, University of Cincinnati. EPA (99% purity), Fura-2 AM (Fura-2-acetoxymethyl ester), 7-ethoxy resorufin, dicumarol, resorufin, DAPI and electron microscopy grade chemicals were obtained from Sigma Aldrich Private Ltd, Bangalore, India. DMEM, FBS and all other chemicals were purchased from Hi-Media Laboratories, Mumbai, India. DCF-DA (2′,7′-dichlorodihydrofluoresceindiacetate), OPT (ortho-phthalaldehyde), DiOC6 (3,3′-dihexyloxacarbocyanine iodide) and CCCP [carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone] were obtained from Calbiochem, San Diego, CA, USA. RIPA lysis and extraction buffer were from Life Technologies, Inc., Carlsbad, CA, USA. Primary monoclonal antibodies for NF-κB p65, COX-2, Nrf-2, Lamin and β-actin were obtained from Santa Cruz Biotechnology, Santa Cruz, CA, USA. Phospho-MAPK family antibody sampler kit consisting of monoclonal antibodies for phospho-p44/42 MAPK (Thr202/Tyr204), phospho-p38 MAPK (Thr180/Tyr182), anti-rabbit IgG, HRP-linked antibody and U0126 (MEK 1/2) was purchased from Cell Signaling Technology, Danvers, MA, USA, and Upstate, Lake Placid, NY, USA. Western blot membranes were obtained from Whatman, Clifton, NJ, USA. Diaminobenzidine (DAB)/H2O2 colour development kit for Western blots and fluorescein isothiocyanate (FITC)-conjugated secondary antibody for immunofluorescence were obtained from Bangalore Genei, Bangalore, India.
Results
EPA protected HepG2 cells from TCDD-induced cytotoxicity by maintaining cell viability
The results from MTT assays following exposure of HepG2 cels to TCCD showed a dose-dependent decrease in cell viability up to 10 nM for 48 h (P < 0.001). Above 10nM TCCD did not show dose-dependent cytotoxicity. Similar cytotoxic effects of TCDD were observed after 72 h exposure. So, minimum dose and exposure time (i.e. 10 nM for 48 h) with maximum cytotoxic effect were used for further studies (Figure 1A). Cytoprotection offered by EPA against TCDD-induced toxicity was determined by pre-treatment, co-treatment and post-treatment with different concentrations of EPA. Figure 1B shows reduction in cell viability to 76% by TCDD treatment at 10 nM for 48 h (P < 0.001). Pre-, co- and post-treatment of EPA at different concentrations and time points showed that 24 h EPA treatment did not offer significant cytoprotection against TCDD-induced toxicity (data not shown), while 48 h EPA treatment in different treatment schedules offered a dose-dependent cytoprotection. Pre-treatment with EPA offered better cytoprotection against TCDD-induced toxicity, exhibiting a dose-dependent increase in cell viability, with 40 μM EPA pre-treatment yielding a cell viability of 95% (P < 0.001). As pre-treatment with EPA showed better protection against TCDD-induced cytotoxicity, further study was carried out with pre-treatment of cells with 40 μM EPA for 48 h followed by TCDD (10 nM) treatment for 48 h (Figure 1B).
Figure 1.

Effect of EPA on TCDD-induced cytotoxicity in HepG2 cells. (A) TCDD-induced cytotoxicity: HepG2 cells were treated with TCDD (0.1, 1, 10, 50 and 100 nM) for different exposure time (24, 48 and 72 h). Cell viability was determined by MTT assay and the results are expressed as percentage of cell viability. Results shown are mean ± SE (n = 9), which are three independent experiment performed in triplicate. ***P < 0.001; **P < 0.01; *P < 0.05, significantly different from control. NS, non-significantly different from control. (B) EPA protects against TCDD-induced toxicity: Cells were treated with 10 nM TCDD and different doses of EPA (10, 20, 30, 40 μM). Pre-treatment (EPA 48 h followed by TCDD 48 h), co-treatment (EPA + TCDD – 48 h) and post-treatment (TCDD 48 h followed by EPA 48 h) of EPA was performed to analyse the cytoprotective effect of EPA against TCDD-induced cytotoxicity. Cell viability was determined by MTT assay and the results are expressed as percentage of cell viability. Results shown are means ± SE (n = 9), which are three independent experiment performed in triplicate. ***P < 0.001, significantly different from control. NS, non-significantly different from control. +++P < 0.001; ++P < 0.01; +P < 0.05, significantly different from TCDD treated group. (C) EPA modulates the EPA/AA ratio: The ratio of EPA/AA during EPA and TCDD treatment was calculated and the results represented are means ± SE (n = 3;, three independent experiments). ***P < 0.001, significantly different from control. NS, non-significantly different from control. +P < 0.05, significantly different from the TCDD-treated group (one-way anova followed by Tukey's multiple comparison test).
Membrane incorporation of EPA and modulation of EPA/AA ratio
Membrane incorporation of EPA and modulation of the EPA/AA ratio is a key event through which EPA mediates its cytoprotective effect. EPA treatment resulted in increased EPA levels in membrane phospholipids when compared to the control group. Pre-treatment with EPA followed by TCDD treatment resulted in a significant increase in the EPA/AA ratio of cell membranes, compared with cells treated with TCDD alone (Figure 1C).
Effect of EPA on TCDD-induced cellular oxidative stress, Ca2+ deregulation and MAPK activation
EPA inhibited TCDD-induced CYP1A1 and ROS generation
TCDD increased CYP1A1 activity (P < 0.001), compared with the control cells (Figure 2A). Cells treated with EPA alone showed no difference from control cells. Pre-treatment with EPA followed by TCDD treatment showed a decline in CYP1A1 activity (P < 0.01). We then measured ROS levels in the presence or absence of EPA, followed by TCDD treatment (Figure 2B). TCDD treatment increased ROS levels with a relative fluorescence of 202% (P < 0.001), compared with the control group. Pre-treatment with EPA reduced the levels of ROS (P < 0.001), compared with those in the group treated with TCDD alone. Figure 2C shows the images of ROS fluorescence in cells treated in the presence of EPA and TCDD.
Figure 2.
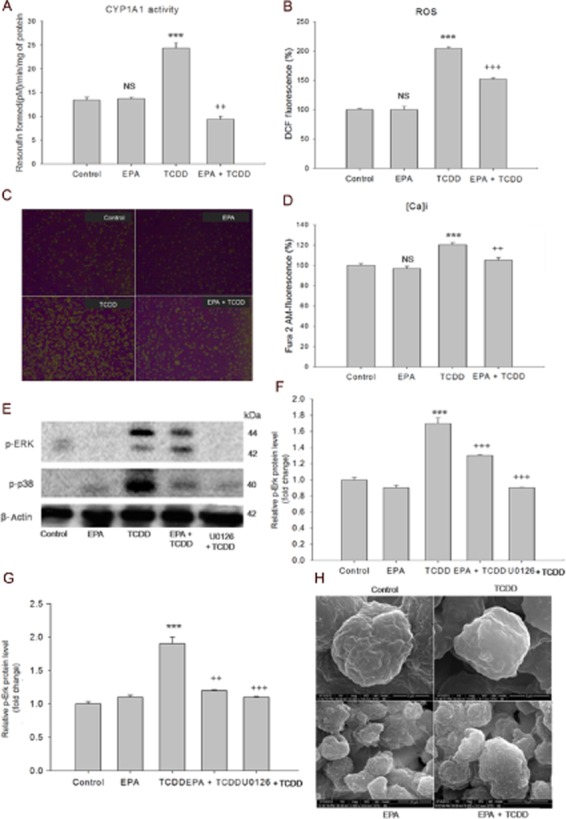
EPA inhibits TCDD-induced oxidative stress, regulates [Ca2+]i levels and prevents MAPK activation. (A) EPA inhibits CYP1A1 activity: HepG2 cells were treated with TCDD for 48 h in the presence/absence of EPA for 48 h. Conversion of 7-ethoxy resorufin to resorufin by CYP1A1 was measured fluorimetrically. The results are expressed as picomol resorufin formed min-1 (mg protein)-1. (B) EPA inhibits TCDD-induced ROS generation: HepG2 cells were treated with TCDD treatment for 6 in the presence/absence of 40 μM EPA for 48 h. The fluorescence intensity is expressed as percentage of relative DCF fluorescence. The results shown are means ± SE (n = 9; three independent experiments performed in triplicate). ***P < 0.001, significantly different from control. NS, non-significantly different from control. ++P < 0.01; +++P < 0.001, significantly different from the TCDD-treated group (one-way anova followed by Tukey's multiple comparison test). (C) Effect of EPA and TCDD on DCF fluorescence: Control cells and cells treated with EPA show low DCF fluorescence. Cells with TCDD show high fluorescence compared with control cells. Pre-treatment with EPA followed by TCDD shows low DCF fluorescence. Magnification: 100×. (D) EPA maintains TCDD-induced intracellular [Ca2+]i levels. HepG2 cells were treated with TCDD for 45 min in the presence/absence of EPA for 48 h. The results were expressed as percentage of relative fluorescence of Fura 2-AM. The results shown are means ± SE (n = 9; three independent experiments performed in triplicate). ***P < 0.001, significantly different from control. NS, non-significantly different from control. ++P < 0.01, significantly different from the TCDD-treated group. (E) EPA prevents TCDD-induced MAP kinases. HepG2 cells were treated with TCDD for 1 h in the presence and absence of EPA treatment for 48 h. For positive control, cells were treated with MAPK inhibitor U0126 for 1 h prior to TCDD treatment. After the treatment period, proteins were extracted and subjected to Western blot analysis. Densitometric analysis was performed using ImageJ software. (F) and (G) show densitometric analysis on the effect of EPA on TCDD-induced MAP kinases. The results shown are means ± SE of three independent experiments. ***P < 0.001, significantly different from control. NS, non-significantly different from control. +++P < 0.001; ++P < 0.01, significantly different from the TCDD-treated group; Student's t-test. (H) EPA prevents TCDD-induced loss of microvilli. SEM analysis: HepG2 cells were treated with TCDD for 48 h in the presence or absence of EPA for 48 h. Control and EPA treatment shows normal cell morphology with microvilli (15 000×) and (2000×) respectively. TCDD – Cells show complete loss of microvilli with the appearance of dome or sac-like structures (10 000×). EPA pre-treatment followed by TCDD treatment restored the normal cell surface morphology with the appearance of microvilli (5000×).
EPA maintained [Ca2+]i levels and prevented MAPK phosphorylation (Erk (p44/p42) and p38)
Treatment with TCDD increased [Ca2+]i levels (120%) (P < 0.001) compared with the control and pre-treatment with EPA blocked this increase (104%; P < 0.001) (Figure 2D). Increased levels of p-Erk (p44/p42) and p-p38 (P < 0.001) were observed during TCDD treatment, compared with the control group. Pre-treatment with EPA decreased MAPK phosphorylation (P < 0.001), compared with cells treated with TCDD alone. Treatment with the MAP kinase inhibitor (10 μM – U1026) for 1 h before TCDD treatment down-regulated p-Erk (p44/p42) and p-p38 (P < 0.001), compared with TCDD treatment, clearly indicating that TCDD-induced phosphorylation of these MAPKs was significantly inhibited by EPA treatment (Figure 2E, F, G).
EPA prevented TCDD-induced changes in cell surface microvilli
Cell surface changes induced by TCDD and EPA were determined by scanning electron microscopy. Control cells and the cells treated with EPA alone showed normal cell morphology with the presence of microvilli. TCDD treatment resulted in complete loss of microvilli, with ballooning of microvilli. In contrast, cells pre-treated with EPA maintained characteristic normal cell morphology after exposure to TCCD, with the appearance of microvilli (Figure 2H).
Effect of EPA on TCDD-induced redox signalling
EPA prevented TCDD-induced nuclear migration of NF-κB p65
NF-κB p65 is a pleiotropic transcription factor, which acts as a central mediator of oxidative stress and inflammation. In unstressed conditions, NF-κB p65 is sequestered in the cytoplasm bound to inhibitor IκB. In response to oxidative stress, IκB is degraded, which results in subsequent nuclear localization of NF-κB p65 and initiation of inflammatory responses. A time course revealed that TCDD-mediated NF-κB p65 nuclear migration after 7 h (Figure 3A). Pre-treatment with EPA followed by TCDD treatment for 7 h showed cytoplasmic sequestration of NF-κB, while control cells and cells treated with EPA alone did not alter NF-κB p65 nuclear migration (Figure 3B).
Figure 3.
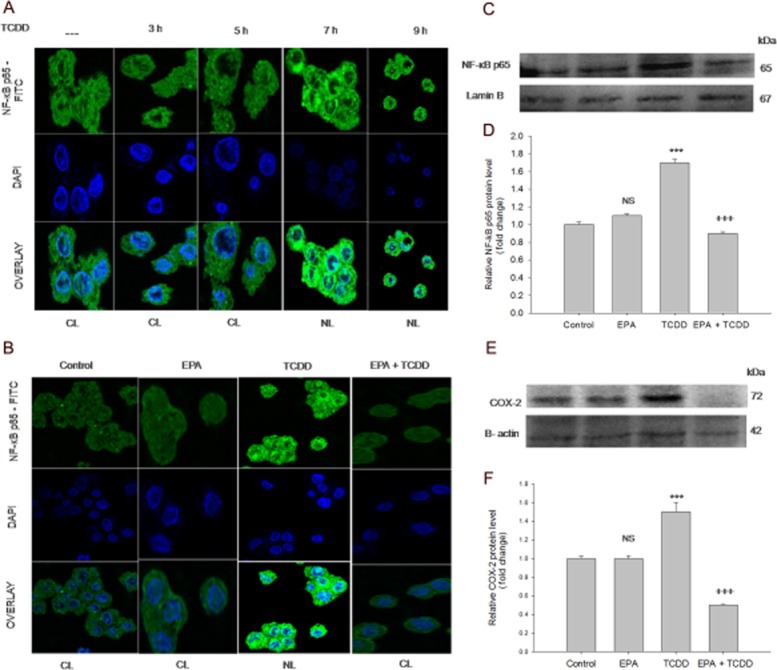
EPA mediates anti-inflammatory effects: prevents expression and nuclear localization of NF-κB p65, and COX-2 expression. (A) TCDD induces NF-κB p65 nuclear localization: The cells were treated with TCDD for different time points (10 nM TCDD for 3, 5, 7 and 9 h). NF-κB p65 nuclear localization was visualized using mouse monoclonal NF-κB p65 primary antibody followed by addition of FITC-conjugated secondary antibody. The nuclei were visualized using DAPI. Control, TCDD 3 h, TCDD 5 h show NF-κB p65 sequestration in the cytoplasm. TCDD 7 h and TCDD 9 h show NF-κB p65 nuclear localization (visualized as turquoise blue colour). CL, cytoplasmic localization; NL, nuclear localization. (B) EPA prevents TCDD-induced NF-κB p65 nuclear localization: The cells were treated with TCDD for 7 h in the presence/absence of EPA for 48 h. Control and EPA treated cells showed NF-κB p65 sequestration in the cytoplasm (indicate unstressed conditions). Cells exposed to TCDD for 7 h shows NF-κB p65 nuclear localization, visualized as turquoise blue colour. Pre-treatment with EPA followed by TCDD treatment shows cytoplasmic retention of NF-κB p65. CL, cytoplasmic localization; NL, nuclear localization. (C) EPA down regulates NF-κB p65 expression: Cells were treated with TCDD for 12 h in the presence/absence of EPA for 48 h. (D) Densitometric analysis of NF-κB p65 expression: The results shown are means ± SE of three independent experiments. ***P < 0.001, significantly different from control. NS, non-significantly different from control. +++P < 0.001, significantly different from the TCDD-treated group; Student's t-test. (E). EPA down-regulates COX-2 expression: Cells were treated with TCDD for 15 h in the presence/absence of EPA for 48 h. (F) Densitometric analysis of COX-2 expression: The results shown are means ± SE of three independent experiments. ***P < 0.001, significantly different from control. NS, non-significantly different from control. +++P < 0.001, significantly different from the TCDD-treated group; Student's t-test.
EPA inhibited TCDD-induced NF-κB p65 and COX-2 expressions
In order to determine the effect of EPA on TCDD-induced pro-inflammatory response, NF-κB p65 and COX-2 expressions were studied by Western blot analysis. EPA pre-treatment resulted in significant down-regulation of NF-κB p65 and COX-2 expressions. Densitometric analysis showed increased NF-κB p65 and COX-2 (P < 0.001) expression in TCDD-treated cells, compared with the control cells. Pre-treatment with EPA down-regulated NF-κB p65 and COX-2 expression (P < 0.001), compared with cells treated with TCDD alone (Figure 3C, D).
EPA augments Nrf-2 nuclear localization and expression
Nrf-2, a redox-sensitive transcription factor, migrates into the nucleus and offers cytoprotection by inducing antioxidant enzymes. The results from the time course study revealed that EPA induced Nrf-2 nuclear localization after 7 h in response to TCDD-induced oxidative stress (Figure 4A). Furthermore, Figure 4B shows the effect of EPA and TCDD on Nrf-2 nuclear migration. Control cells and the cells treated with EPA alone show sequestration of Nrf-2 in the cytoplasm, revealing the unstressed state inside the cells. Cells treated with TCDD show partial nuclear localization of Nrf-2. Extensive Nrf-2 nuclear localization was observed in EPA pre-treatment followed by TCDD treatment. Figure 4C and D show the effect of EPA and TCDD on Nrf-2 expression: increased Nrf-2 expression (P < 0.05) was observed in TCDD-treated cells, compared with the control cells. EPA treatment alone did not result in any significant change in the protein expression, compared with the control cells. Pre-treatment with EPA up-regulated Nrf-2 expression (P < 0.001), compared with cells treated with TCDD alone.
EPA offers cytoprotection by modulating antioxidant status
The cellular antioxidant status is an indicator of redox homeostasis. In the present study, the effect of EPA and TCDD on various non-enzymic and enzymic antioxidants was determined. The level of GSH was decreased by TCDD treatment (P < 0.01) and Pre-treatment with EPA offered a significant increase in GSH levels (P < 0.001). TCDD treatment significantly reduced the activities of antioxidant enzymes (SOD, CAT, GST, GPx and NQO1) when compared to the control. Cells pre-treated with EPA showed increased levels of the antioxidant status, compared with TCDD-treated cells, restoring values to near control or higher (Figure 5A, B, C, D, F).
Figure 5.

EPA enhances antioxidant status. HepG2 cells were treated with TCDD for 48 h in the presence/absence of EPA treatment for 48 h. NQO1 activity was determined using fluorimetric measurement of resorufin consumption. NQO1 activity 1 U = pmol of resorufin consumption min-1 (mg protein)-1. SOD 1 U = the amount of enzyme required to give 50% inhibition of pyrogallol auto-oxidation. Catalase 1 U = the amount of enzyme that consumes 1 nmol H2O2 per minute. GST 1 U = the amount of enzyme that conjugates 1 μmol CDNB per minute. GPx 1 U = the amount of enzyme that converts 1 μmol GSH to GSSG in the presence of H2O2 per minute. GSH is expressed as μmol GSH (mg protein)-1. The results were expressed relative percentage of antioxidant enzyme activity compared with the control. The results shown are mean of ± SD (n = 9; the results of three independent experiments carried out in triplicate). ***P < 0.001, **P < 0.05, significantly different from control. NS, non-significantly different from control. +++P < 0.001; ++P < 0.01, is significantly different from the TCDD-treated group; one-way anova followed by Tukey's multiple comparison test.
EPA prevented TCDD-induced loss of ΔΨm and acidic vesicular organelles (AVOs)
Loss of mitochondrial potential is an indicator of the functional status of the mitochondria. Cells treated with TCDD for 48 h showed loss of mitochondrial membrane potential (P < 0.01) when compared to the control. Pre-treatment with EPA resulted in a significant increase in mitochondrial membrane potential (P < 0.01), compared with TCDD-treated cells (Figure 6A). In order to determine the nature of cell death induced by TCDD, AO/ETBR staining was carried out (Figure 6B, C). The differential scoring of AO and ETBR staining revealed significant (P < 0.001) formation of AVOs (48%) and no significant level of apoptosis in TCDD-treated cells, compared with control, while cells pretreated with EPA showed decreased AVO formation (3%); (P < 0.001), compared with the TCDD treatment. Flow cytometric analysis was carried out to validate our microscopic observations. Accumulation of cells in the subG1 phase of the cell cycle is characteristic of apoptosis. Flow cytometric analysis showed that TCDD treatment did not increase accumulation in subG1 phase (3.53%), compared with the control cells (1.45%). Similarly, treatment with EPA alone (1.74%) or EPA pre-treatment followed by TCDD (1.51%) treatment did not change subG1 accumulation, indicating that the mechanism of TCDD-induced toxicity did not involve apoptosis (Supporting Information Table S1).
Figure 6.
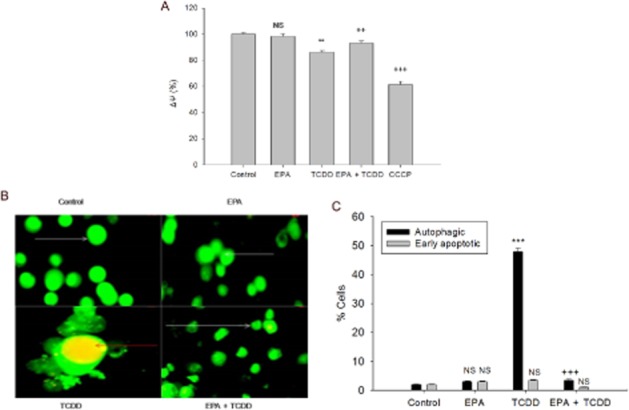
EPA attenuates TCDD-induced cellular damage. (A) Mitochondrial membrane potential (ΔψM): The cells were treated with TCDD for 48 h in the presence or absence of EPA for 48 h. The cells that were treated with 50 μM CCCP for 15 min were used as positive control. The results were expressed as percentage of relative of DiOC6 (3) fluorescence. ***P < 0.001; **P < 0.01, significantly different from control. NS, non-significantly different from control. ++P < 0.01, significantly different from the TCDD-treated group. (B) EPA prevents TCDD-induced AVO formation: Cells were treated with TCDD for 48 h in the presence or absence of EPA pre-treatment for 48 h. Control cells and cells treated with EPA show normal cell morphology with green florescence (white arrows). Cells treated with TCDD with orange-red spots indicate AVOs (red arrows). Cells pre-treated with EPA followed by TCDD treatment with normal cellular morphology with green florescence. Magnification: 100×. (C) Percentage of cells with apoptosis and autophagy. For each treatment group, 200 cells were scored for differential uptake of the stain and the results were calculated in percentage and represented as means ± SE, from three independent experiments. ***P < 0.001, significantly different from control. NS, non-significantly different from control. +++P < 0.001, significantly different from the TCDD-treated group; one-way anova followed by Tukey's multiple comparison test.
Discussion
TCDD is an environmental toxin that has extensive effects on cellular mechanisms. TCDD-induced toxic effects are mediated by redox imbalance and AA release (Rifkind, 2006; Wan et al., 2014). TCDD-induced AA release from membrane phospholipids occurs through alterations in Ca2+ homeostasis and oxidative stress conditions. The released AA acts as a substrate for COX-2, lipoxygenase and cytochrome P450s and results in the formation of eicosanoids and thereby mediates liver toxicity through its pro-inflammatory mechanisms (Lin et al., 2011; Bui et al., 2012). As AA and ROS are known to play a major role in TCDD-induced toxic events, one of the hypothesized mechanisms for the protective action of EPA was through its membrane incorporation. The ratio of EPA and AA in membrane phospholipids determines the activation and/or suppression of various redox and inflammatory pathways. The present study showed significant incorporation of EPA in membrane phospholipids with a rise in the EPA/AA ratio. Ca2+ is an important second messenger responsible for the induction of various signal transduction pathways and identified as an important mechanism through which TCDD mediates its toxic effects (Kim et al., 2007; Piaggi et al., 2007). TCDD-induced MAPK activation and subsequent redox imbalance through CYP1A1 expression has been demonstrated earlier (Tan et al., 2002). The present study showed that EPA treatment protected against TCDD-induced toxicity by regulating Ca2+ homeostasis, down-regulating p-Erk (44/42) and p-p38 kinases, decreasing CYP1A1 activity and generating ROS. An interesting observation during TCDD treatment showed ballooning of the microvilli on the cell surface. Generally, actin-based cytoskeletal core structure of the microvilli acts as a diffusion barrier and plays a prime role in maintaining low Ca2+ levels inside the cytoplasm (Lange and Gartzke, 2001). The maintenance of microvillar shape is dependent upon Ca2+ homeostasis, as dysregulation of Ca2+ homeostasis leads to microfilament depolymerization through activation of kinase pathways (Lassing and Lindberg, 1985; Lange et al., 1998). Pre-treatment with EPA preserved the normal cell morphology with microvillus structures revealing the protective role of EPA against TCDD-induced calcium dysregulation. earlier studies had shown that membrane incorporation of EPA and regulation of Ca2+ levels play an important role in cytoprotection by EPA (Hallaq et al., 1990; Schmocker et al., 2007; Ishii et al., 2009).
Oxidative stress is closely related to inflammation and TCDD-induced oxidative stress, and its inflammatory role was monitored with two important redox-sensitive transcription factors: NF-κB and Nrf-2. These two transcription factors exert opposing effects, as NF-κB p65 influences oxidative stress and pro-inflammation, while Nrf-2 enhances antioxidant status and suppresses inflammation (Shen et al., 2005). Data from the time courses revealed that TCDD-induced NF-κB p65 nuclear localization was a downstream event to ROS generation. A recent study by Vogel et al. (2014) demonstrated the critical role of NF-κB p65 in the regulation of AhR-dependent gene expression and mediation of inflammatory responses. Further, EPA showed significant anti-inflammatory action by prevention of TCDD-induced nuclear migration of NF-κB p65 and subsequent down-regulation of NF-κB p65 and COX-2 expression. Inhibition of NF-κB P65 activity by EPA has been reported earlier against LPS-induced inflammation in RAW cells (Lo et al., 1999), THP-1 monocytes (Zhao et al., 2004) and human kidney cells (Li et al., 2005). The transcription factor Nrf-2 has gained prominence as it is activated by natural chemopreventive agents and plays a pivotal role by transcribing genes in the antioxidant response element, thus offering protection against oxidative stress (Kwak et al., 2002; Balogun et al., 2003). It is therefore reasonable that activation of Nrf-2 occurs as a cellular adaptation to oxidative stress caused by various stimuli (McDonald et al., 2010; Speciale et al., 2011), as we observed in the present study through mild activation and expression during TCDD exposure. TCDD-induced Nrf-2 expression has been reported earlier by Miao et al. (2005). It is apparent from the present results that this level of Nrf-2 induction remained ineffective against TCDD-induced sustained oxidative stress and inflammatory states, suggesting that it is more of an adaptive activation rather than one that could lead to cytoprotection. Kensler et al. (2007) reported that Nrf-2 signalling played a role in cell survival as an adaptive response against environmental stress. As Nrf-2 is modulated by antioxidants and chemopreventive agents, the effects of EPA on Nrf-2 nuclear localization was tested in response to TCDD-induced stress. The results show that EPA mediated Nrf-2 nuclear localization at 7 h after TCDD exposure. The increased expression of Nrf-2 during EPA pre-treatment corroborates with the increase in the antioxidant status of the cell, confirming a cytoprotective effect of EPA, against TCDD-induced stress. Wang et al. (2010) showed that the EPA suppressed LPS-induced inflammation through the Nrf-2 pathway. In addition, the role of oxidized EPA and EPA derivatives (17-EFOX-D6, 17-EFOX-D5) in cytoprotection was mediated by Nrf-2 and Nrf-2-dependent antioxidant gene expressions (Gao et al., 2007; Groeger et al., 2010). The results of the present study clearly demonstrated that EPA exerted its cytoprotective activity by induction of Nrf-2 and suppression of NF-κB, which explained the antioxidant and anti-inflammatory properties of EPA (Van Beelen et al., 2006; Zuniga et al., 2011). A growing body of evidence suggests that there could be considerable crosstalk between these redox-regulated transcription factors (Bellezza et al., 2010; Tusi et al., 2010), which needs to be explored further. Reports show that NF-κB negatively regulates Nrf-2 expression, thereby enhancing inflammatory responses (Liu et al., 2008). An interesting observation in the present study was that EPA alone did not affect Nrf-2 nuclear migration or its expression in the absence of TCCD, but only during TCDD-induced oxidative stress, clearly demonstrating its potential role under unfavourable conditions.
Loss of mitochondrial membrane potential, decrease in antioxidant enzymes, resistance to apoptosis and formation of AVOs lead us to believe that prolonged and sustained oxidative stress might result in an autophagic response. Autophagy is normally induced under unfavourable stressful conditions by the formation of autophagolysosomes (Kanzawa et al., 2005). Although this is a new type of TCDD-induced stress response in HepG2 cells, the mechanism of autophagy has been previously described in kidney cells by Fiorito et al. (2011). Autophagy-induced stress responses have recently gained importance and exploring its role would help in better understanding of the mechanisms involved in TCDD-induced liver toxicity. Earlier reports showed the importance of ROS and loss of mitochondrial membrane potential in the regulation of autophagy (Azad et al., 2009; Huang et al., 2011). Conversely, mild activation of Nrf-2 has also been linked to anti-apoptotic effects (Niture and Jaiswal, 2013), which is consistent with the present study. Nrf-2 has also been implicated in drug resistance and cell survival (Stępkowski and Kruszewski, 2011). Thus, many of the actions of Nrf-2 in cytoprotection, anti-apoptotic effect and drug resistance are all dependent upon the strength and duration of the oxidative stress and the chemical nature of the compound in action. However, further studies must be carried out to explore the diverse mechanisms of action of TCDD.
In conclusion, combating the detrimental effects of environmental toxins through dietary intervention is an area that is gaining importance. This study has found new data to add to the many mechanisms through which TCDD exerts its toxic effects through the temporal distribution of NF-κB and Nrf-2 transcription factors, disappearance of cell surface microvilli and autophagic induction. Further, we have demonstrated for the first time that dietary treatment with EPA modulated the EPA/AA ratio in membrane phospholipids and offered cytoprotection by maintaining Ca2+ homeostasis, thereby preventing the toxic downstream events of TCDD-induced oxidative stress and inflammatory responses. All these effects of EPA are important attributes of this fatty acid as summarised in the scheme in Figure 7.
Figure 7.
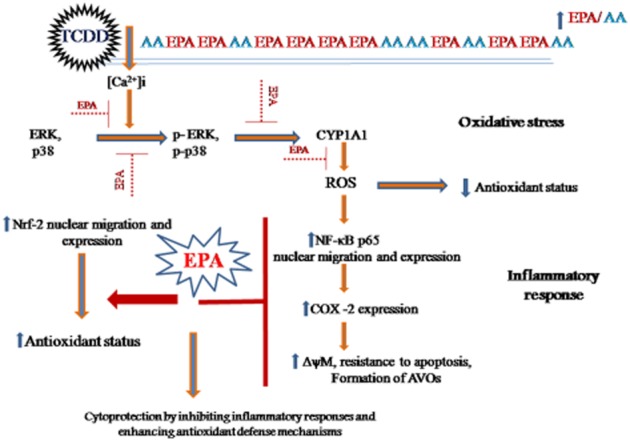
Schematic representation of EPA-mediated cytoprotection against TCDD-induced toxicity.
Glossary
- AA
arachidonic acid
- AEBSF
4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride
- AhR
aryl hydrocarbon receptor
- AVO
acidic vesicular organelles
- CCCP
carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone
- DAB
diaminobenzidine
- DCF-DA
2′,7′-dichlorodihydrofluoresceindiacetate
- DiOC6
3,3′-dihexyloxacarbocyanine iodide
- EPA
eicosapentaenoic acid
- FITC
fluorescein isothiocyanate
- Fura-2 AM
Fura-2-acetoxymethyl ester
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NQO1
NAD(P)H dehydrogenase quinone1
- Nrf-2
nuclear factor erythroid 2-related factor 2
- OPT
ortho-phthalaldehyde
- ROS
reactive oxygen species
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
Author contributions
K. S. P. contributed to the concept/design, data acquisition, data analysis, interpretation, drafting of the manuscript and critical revision of the manuscript. K. R. contributed to data interpretation and critical revision of the manuscript. P. P. contributed to data acquisition. C.-Y. H. and V. V. prepared and edited the manuscript. V. V. P. contributed to the concept/design, data interpretation and critical revision of the manuscript.
Conflicts of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.13247
Table S1 Cell cycle analysis.
References
- Aebi H. Catalase. In: Bergmeyer H, editor. Methods of Enzymatic Analysis. New York: Academic Press; 1974. pp. 673–684. [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal. 2009;11:777–790. doi: 10.1089/ars.2008.2270. [DOI] [PubMed] [Google Scholar]
- Balogun E, Hoque M, Gong P, Killeen E, Green CJ, Foresti JA, et al. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf-2 and the antioxidant response element. Biochem J. 2003;371:887–895. doi: 10.1042/BJ20021619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellezza I, Mierla AL, Minelli A. Nrf-2 and NF-κB and their concerted modulation in cancer pathogenesis and progression. Cancers. 2010;2:483–497. doi: 10.3390/cancers2020483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benatti P, Peluso G, Nicolai R, Calvani M. Polyunsaturated fatty acids: biochemical nutritional and epigenetic properties. J Am Coll Nutr. 2004;23:281–302. doi: 10.1080/07315724.2004.10719371. [DOI] [PubMed] [Google Scholar]
- Brooks JD, Milne GL, Yin H, Sanchez SC, Porter NA, Morrow JD. Formation of highly reactive cyclopentenone isoprostane compounds (A3/J3-isoprostanes) in vivo from eicosapentaenoic acid. J Biol Chem. 2008;283:12043–12055. doi: 10.1074/jbc.M800122200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui P, Solaimani P, Wu X, Hankinson O. 2,3,7,8-Tetrachlorodibenzo-p-dioxin treatment alters eicosanoid levels in several organs of the mouse in an aryl hydrocarbon receptor-dependent fashion. Toxicol Appl Pharmacol. 2012;259:143–151. doi: 10.1016/j.taap.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calder PC. n-3 polyunsaturated fatty acids inflammation and inflammatory diseases. Am J Clin Nutr. 2006;83:1505S–1519S. doi: 10.1093/ajcn/83.6.1505S. [DOI] [PubMed] [Google Scholar]
- Dong B, Matsumura F. Roles of cytosolic phospholipase A2 and Src kinase in the early action of 2,3,7,8-tetrachlorodibenzo-p-dioxin through a non-genomic pathway in MCF10A cells. Mol Pharmacol. 2008;74:255–263. doi: 10.1124/mol.107.044669. [DOI] [PubMed] [Google Scholar]
- El-Mowafy AM, Abdel-Dayem MA, Abdel-Aziz A, El-Azab MF, Said SA. Eicosapentaenoic acid ablates valproate-induced liver oxidative stress and cellular derangement without altering its clearance rate: dynamic synergy and therapeutic utility. Biochim Biophys Acta. 2011;1811:460–467. doi: 10.1016/j.bbalip.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Fiorito F, Ciarcia R, Granato GE, Marfe G, Iovane V, Florio S, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induced autophagy in a bovine kidney cell line. Toxicology. 2011;290:258–270. doi: 10.1016/j.tox.2011.10.004. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane-Stanely GH. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem. 1957;226:497–507. [PubMed] [Google Scholar]
- Gao L, Wang J, Sekhar KR, Yin H, Yared NF, Schneider SN, et al. Novel n-3 fatty acid oxidation products activate Nrf-2 by destabilizing the association between keap1 and cullin3. J Biol Chem. 2007;282:2529–2537. doi: 10.1074/jbc.M607622200. [DOI] [PubMed] [Google Scholar]
- Giudetti AM, Cagnazzo R. Beneficial effects of n-3 PUFA on chronic airway inflammatory diseases. Prostaglandins Other Lipid Mediat. 2012;99:57–67. doi: 10.1016/j.prostaglandins.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Groeger AL, Cipollina C, Cole MP, Woodcock SR, Bonacci G, Rudolph TK, et al. Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids. Nat Chem Biol. 2010;6:433–441. doi: 10.1038/nchembio.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habig WH, Pabst MJ, Jakoby WB. Glutathione S-transferases: the first enzymatic step in mercapturic acid formation. J Biol Chem. 1974;249:7130–7139. [PubMed] [Google Scholar]
- Hallaq H, Sellmayer A, Smith TW, Leaf A. Protective effect of eicosapentaenoic acid on ouabain toxicity in neonatal rat cardiac myocytes. Proc Natl Acad Sci U S A. 1990;87:7834–7838. doi: 10.1073/pnas.87.20.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hissin PJ, Hilf R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal Biochem. 1976;74:214–226. doi: 10.1016/0003-2697(76)90326-2. [DOI] [PubMed] [Google Scholar]
- Huang J, Lam GY, Brumell JH. Autophagy signaling through reactive oxygen species. Antioxid Redox Signal. 2011;14:2215–2231. doi: 10.1089/ars.2010.3554. [DOI] [PubMed] [Google Scholar]
- Ishii H, Horie Y, Ohshima S, Anezaki Y, Kinoshita N, Dohmen T, et al. Eicosapentaenoic acid ameliorates steatohepatitis and hepatocellular carcinoma in hepatocyte-specific PTEN-deficient mice. J Hepatol. 2009;50:562–571. doi: 10.1016/j.jhep.2008.10.031. [DOI] [PubMed] [Google Scholar]
- Kanzawa T, Zhang L, Xiao L, Germano IM, Kondo Y, Kondo S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene. 2005;24:980–991. doi: 10.1038/sj.onc.1208095. [DOI] [PubMed] [Google Scholar]
- Kawashima A, Harada T, Imada K, Yano T, Mizuguchi K. Eicosapentaenoic acid inhibits interleukin-6 production in interleukin-1beta-stimulated C6 glioma cells through peroxisome proliferator-activated receptor gamma. Prostaglandins Leukot Essent Fatty Acids. 2008;79:59–65. doi: 10.1016/j.plefa.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Kim SY, Lee HG, Choi EJ, Park KY, Yang JH. TCDD alters PKC signaling pathways in developing neuronal cells in culture. Chemosphere. 2007;67:S421–S427. doi: 10.1016/j.chemosphere.2006.05.138. [DOI] [PubMed] [Google Scholar]
- Kraemer SA, Arthur KA, Denison MS, Smith WL, DeWitt DL. Regulation of prostaglandin endoperoxide H synthase-2 expression by 2,3,7,8,-tetrachlorodibenzo-p-dioxin. Arch Biochem Biophys. 1996;330:319–328. doi: 10.1006/abbi.1996.0259. [DOI] [PubMed] [Google Scholar]
- Kremer JM. n-3 fatty acid supplements in rheumatoid arthritis. Am J Clin Nutr. 2000;71:349S–351S. doi: 10.1093/ajcn/71.1.349s. [DOI] [PubMed] [Google Scholar]
- Kremer JM, Jubiz W, Michalek A, Rynes RI, Bartholomew LE, Bigaouette J, et al. Fish-oil fatty acid supplementation in active rheumatoid arthritis. A double-blinded, controlled, crossover study. Ann Intern Med. 1987;106:497–503. doi: 10.7326/0003-4819-106-4-497. [DOI] [PubMed] [Google Scholar]
- Kremer JM, Lawrence DA, Jubiz W, DiGiacomo R, Rynes R, Bartholomew LE, et al. Dietary fish oil and olive oil supplementation in patients with rheumatoid arthritis. Clinical and immunologic effects. Arthritis Rheum. 1990;33:810–820. doi: 10.1002/art.1780330607. [DOI] [PubMed] [Google Scholar]
- Kwak MK, Itoh K, Yamamoto M, Kensler TW. Enhanced expression of the transcription factor Nrf-2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the Nrf-2 promoter. Mol Cell Biol. 2002;22:2883–2892. doi: 10.1128/MCB.22.9.2883-2892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange K, Gartzke J. Microvillar cell surface as a natural defense system against xenobiotics: a new interpretation of multidrug resistance. Am J Physiol Cell Physiol. 2001;281:C369–C385. doi: 10.1152/ajpcell.2001.281.2.C369. [DOI] [PubMed] [Google Scholar]
- Lange K, Brandt U, Gartzke J, Bergmann J. Action of insulin on the surface morphology of hepatocytes: role of phosphatidylinositol 3-kinase in insulin-induced shape change of microvilli. Exp Cell Res. 1998;239:139–151. doi: 10.1006/excr.1997.3894. [DOI] [PubMed] [Google Scholar]
- Lassing I, Lindberg U. Specific interaction between phosphatidylinositol 4 5-bisphosphate and profilactin. Nature. 1985;314:472–474. doi: 10.1038/314472a0. [DOI] [PubMed] [Google Scholar]
- Li H, Ruan XZ, Powis SH, Fernando R, Mon WY, Wheeler DC, et al. EPA and DHA reduce LPS-induced inflammation responses in HK-2 cells: evidence for a PPAR-gamma-dependent mechanism. Kidney Int. 2005;67:867–874. doi: 10.1111/j.1523-1755.2005.00151.x. [DOI] [PubMed] [Google Scholar]
- Lin S, Yang Z, Liu H, Cai Z. Metabolomic analysis of liver and skeletal muscle tissues in C57BL/6J and DBA/2J mice exposed to 2,3,7,8-tetrachlorodibenzo-pdioxin. Mol Biosyst. 2011;7:1956–1965. doi: 10.1039/c1mb05057e. [DOI] [PubMed] [Google Scholar]
- Liu GH, Qu J, Shen X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim Biophys Acta. 2008;1783:713–727. doi: 10.1016/j.bbamcr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Lo CJ, Chiu KC, Fu M, Lo R, Helton S. Fish oil decreases macrophage tumor necrosis factor gene transcription by altering the NF kappa B activity. J Surg Res. 1999;82:216–221. doi: 10.1006/jsre.1998.5524. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Marchetti P, Hirsch T, Zamzami N, Castedo M, Decaudin D, Susin SA, et al. Mitochondrial permeability transition triggers lymphocyte apoptosis. J Immunol. 1996;157:4830–4836. [PubMed] [Google Scholar]
- Marklund S, Marklund G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem. 1974;47:469–474. doi: 10.1111/j.1432-1033.1974.tb03714.x. [DOI] [PubMed] [Google Scholar]
- McDonald JT, Kim K, Norris AJ, Vlashi E, Phillips TM, Lagadec C, et al. Ionizing radiation activates the Nrf-2 antioxidant response. Cancer Res. 2010;70:8886–8895. doi: 10.1158/0008-5472.CAN-10-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiyanto E, Agustina D, Supardjan AM, Da'I M. PVG-O induces apoptosis on T47D breast cancer cell line through caspase-3 activation. J Kedokteran Yarsi. 2007;15:75–79. [Google Scholar]
- Miao W, Hu L, Scrivens PJ, Batist G. Transcriptional regulation of NF-E2 p45-related factor (Nrf-2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: direct cross-talk between phase I and II drug-metabolizing enzymes. J Biol Chem. 2005;280:20340–20348. doi: 10.1074/jbc.M412081200. [DOI] [PubMed] [Google Scholar]
- Morrison WR, Smith LM. Preparation of fatty acid methyl esters and dimethyl acetals from lipids with boron fluoride-methanol. J Lipid Res. 1964;5:600–608. [PubMed] [Google Scholar]
- Mossman T. Rapid colorimetric assay for cellular growth and survival to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Mullen A, Loscher CE, Roche HM. Anti-inflammatory effects of EPA and DHA are dependent upon time and dose-response elements associated with LPS stimulation in THP-1-derived macrophages. J Nutr Biochem. 2010;21:444–450. doi: 10.1016/j.jnutbio.2009.02.008. [DOI] [PubMed] [Google Scholar]
- Na HJ, Lee G, Oh HY, Jeon KS, Kwon HJ, Ha KS, et al. 4-O-Methylgallic acid suppresses inflammation-associated gene expression by inhibition of redox-based NF-kappaB activation. Int Immunopharmacol. 2006;6:1597–1608. doi: 10.1016/j.intimp.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response cell cycle control and apoptosis. Biochem Pharmacol. 2000;59:65–85. doi: 10.1016/s0006-2952(99)00310-x. [DOI] [PubMed] [Google Scholar]
- Nims RW, Prough RA, Lubet RA. Cytosol-mediated reduction of resorufin: a method for measuring quinone oxidoreductase. Arch Biochem Biophys. 1984;229:459–465. doi: 10.1016/0003-9861(84)90176-0. [DOI] [PubMed] [Google Scholar]
- Niture SK, Jaiswal AK. Nrf2-induced antiapoptotic Bcl-xL protein enhances cell survival and drug resistance. Free Radic Biol Med. 2013;57:119–131. doi: 10.1016/j.freeradbiomed.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters AK, van Londen K, Bergman A, Bohonowych J, Denison MS, van den Berg M, et al. Effects of polybrominated diphenyl ethers on basal and TCDD-induced ethoxyresorufin activity and cytochrome P450-1A1 expression in MCF-7 HepG2 and H4IIE cells. Toxicol Sci. 2004;82:488–496. doi: 10.1093/toxsci/kfh284. [DOI] [PubMed] [Google Scholar]
- Piaggi S, Novelli M, Martino L, Masini M, Raggi C, Orciuolo E, et al. Cell death and impairment of glucose-stimulated insulin secretion induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in beta-cell line INS-1E. Toxicol Appl Pharmacol. 2007;220:333–340. doi: 10.1016/j.taap.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Rifkind AB. CYP1A in TCDD toxicity and in physiology-with particular reference to CYP dependent arachidonic acid metabolism and other endogenous substrates. Drug Metab Rev. 2006;38:291–335. doi: 10.1080/03602530600570107. [DOI] [PubMed] [Google Scholar]
- Rotruck JT, Pope AL, Ganther HE, Swanson AB, Hafeman DG, Hoekstra WG. Selenium: biochemical role as a component of glutathione peroxidase. Science. 1973;179:588–590. doi: 10.1126/science.179.4073.588. [DOI] [PubMed] [Google Scholar]
- Royall JA, Ischiropoulos H. Evaluation of 2 7-dichlorofluorescein and dihydrorhodamine 123 as fluorescent probes for intracellular H2O2 in cultured endothelial cells. Arch Biochem Biophys. 1993;302:348–355. doi: 10.1006/abbi.1993.1222. [DOI] [PubMed] [Google Scholar]
- Sarbolouki S, Javanbakht MH, Derakhshanian H, Hosseinzadeh P, Zareei M, Hashemi SB, et al. Eicosapentaenoic acid improves insulin sensitivity and blood sugar in overweight type 2 diabetes mellitus patients: a double-blind randomised clinical trial. Singapore Med J. 2013;54:387–390. doi: 10.11622/smedj.2013139. [DOI] [PubMed] [Google Scholar]
- von Schacky C, Harris WS. Cardiovascular benefits of omega-3 fatty acids. Cardiovasc Res. 2007;73:310–315. doi: 10.1016/j.cardiores.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Schmocker C, Weylandt KH, Kahlke L, Wang J, Lobeck H, Tiegs G, et al. Omega-3 fatty acids alleviate chemically induced acute hepatitis by suppression of cytokines. Hepatology. 2007;45:864–869. doi: 10.1002/hep.21626. [DOI] [PubMed] [Google Scholar]
- Sciullo EM, Dong B, Vogel CF, Matsumura F. Characterization of the pattern of the non-genomic signaling pathway through which TCDD-induces early inflammatory responses in U937 human macrophages. Chemosphere. 2009;74:1531–1537. doi: 10.1016/j.chemosphere.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen G, Jeong WS, Hu R, Kong AN. Regulation of Nrf2 NF-kappaB and AP-1 signaling pathways by chemopreventive agents. Antioxid Redox Signal. 2005;7:1648–1663. doi: 10.1089/ars.2005.7.1648. [DOI] [PubMed] [Google Scholar]
- Simopoulos AP. Omega-3 fatty acids in inflammation and autoimmune diseases. J Am Coll Nutr. 2002;21:495–505. doi: 10.1080/07315724.2002.10719248. [DOI] [PubMed] [Google Scholar]
- Speciale A, Anwar S, Ricciardi E, Chirafisi J, Saija A, Cimino F. Cellular adaptive response to glutathione depletion modulates endothelial dysfunction triggered by TNF-α. Toxicol Lett. 2011;207:291–297. doi: 10.1016/j.toxlet.2011.09.017. [DOI] [PubMed] [Google Scholar]
- Stępkowski TM, Kruszewski MK. Molecular cross-talk between the NRF2/KEAP1 signaling pathway autophagy and apoptosis. Free Radic Biol Med. 2011;50:1186–1195. doi: 10.1016/j.freeradbiomed.2011.01.033. [DOI] [PubMed] [Google Scholar]
- Sul D, Kim HS, Choa EK, Lee M, Kim HS, Jung WW, et al. 2,3,7,8-TCDD neurotoxicity in neuroblastoma cells is caused by increased oxidative stress intracellular calcium levels and tau phosphorylation. Toxicology. 2009;255:65–71. doi: 10.1016/j.tox.2008.10.006. [DOI] [PubMed] [Google Scholar]
- Sunami Y, Leithäuser F, Gul S, Fiedler K, Güldiken N, Espenlaub S, et al. Hepatic activation of IKK/NFκB signaling induces liver fibrosis via macrophage-mediated chronic inflammation. Hepatology. 2012;56:1117–1128. doi: 10.1002/hep.25711. [DOI] [PubMed] [Google Scholar]
- Tan Z, Chang X, Puga A, Xia Y. Activation of mitogen-activated protein kinases (MAPKs) by aromatic hydrocarbons: role in the regulation of aryl hydrocarbon receptor (AHR) function. Biochem Pharmacol. 2002;64:771–780. doi: 10.1016/s0006-2952(02)01138-3. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Nat Acad Sci U S A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkez H, Geyikoglu F, Mokhtar YI, Togar B. Eicosapentaenoic acid protects against 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced hepatic toxicity in cultured rat hepatocytes. Cytotechnology. 2012;64:15–25. doi: 10.1007/s10616-011-9386-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusi SK, Ansari N, Amini M, Amirabad AD, Shafiee A, Khodagholi F. Attenuation of NF-kappaB and activation of Nrf-2 signaling by 124-triazine derivatives protects neuron-like PC12 cells against apoptosis. Apoptosis. 2010;15:738–751. doi: 10.1007/s10495-010-0496-6. [DOI] [PubMed] [Google Scholar]
- Van Beelen VA, Aarts JM, Reus A, Mooibroek H, Sijtsma L, Bosch D, et al. Differential induction of electrophile-responsive element-regulated genes by n-3 and n-6 polyunsaturated fatty acids. FEBS Let. 2006;580:4587–4590. doi: 10.1016/j.febslet.2006.07.028. [DOI] [PubMed] [Google Scholar]
- Vogel C, Donat S, Döhr O, Kremer J, Esser C, Roller M, et al. Effect of subchronic 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure on immune system and target gene responses in mice: calculation of benchmark doses for CYP1A1 and CYP1A2 related enzyme activities. Arch Toxicol. 1997;71:372–382. doi: 10.1007/s002040050401. [DOI] [PubMed] [Google Scholar]
- Vogel CF, Khan EM, Leung PS, Gershwin ME, Chang WL, Wu D, et al. Cross-talk between aryl hydrocarbon receptor and the inflammatory response: a role for nuclear factor-κB. J Biol Chem. 2014;289:1866–1875. doi: 10.1074/jbc.M113.505578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan C, Liu J, Nie X, Zhao J, Zhou S, Duan Z, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces premature senescence in human and rodent neuronal cells via ROS-dependent mechanisms. PLoS ONE. 2014;9:e89811. doi: 10.1371/journal.pone.0089811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Khor TO, Saw CL, Lin W, Wu T, Huang Y, et al. Role of Nrf2 in suppressing LPS-induced inflammation in mouse peritoneal macrophages by polyunsaturated fatty acids docosahexaenoic acid and eicosapentaenoic acid. Mol Pharm. 2010;7:2185–2193. doi: 10.1021/mp100199m. [DOI] [PubMed] [Google Scholar]
- Yin H, Brooks JD, Gao L, Porter NA, Morrow JD. Identification of novel autoxidation products of the ω-3 fatty acid eicosapentaenoic acid in vitro and in vivo. J Biol Chem. 2007;282:29890–29901. doi: 10.1074/jbc.M703108200. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Joshi-Barve S, Barve S, Chen LH. Eicosapentaenoic acid prevents LPS-induced TNF-alpha expression by preventing NF-kappa B activation. J Am Coll Nutr. 2004;23:71–78. doi: 10.1080/07315724.2004.10719345. [DOI] [PubMed] [Google Scholar]
- Zuniga J, Cancino M, Medina F, Varela P, Vargas R, Tapia G, et al. N-3 PUFA supplementation triggers PPAR-α activation and PPAR-α/NF-κB interaction: anti-inflammatory implications in liver ischemia-reperfusion injury. PLoS ONE. 2011;6:e28502. doi: 10.1371/journal.pone.0028502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Cell cycle analysis.