

Abstract
Background
The Caldanaerobacter subterraneus species includes thermophilic fermentative bacteria able to grow on carbohydrates substrates with acetate and L-alanine as the main products. In this study, comprehensive analysis of three genomes of C. subterraneus subspecies was carried in order to identify genes encoding key metabolic enzymes and to document the genomic basis for the evolution of these organisms.
Methods
Average nucleotide identity and in silico DNA relatedness were estimated for the studied C. subterraneus genomes. Genome synteny was evaluated using R2CAT software. Protein conservation was analyzed using mGenome Subtractor. Horizontal gene transfer was predicted through the GOHTAM pipeline (using tetranucleotide composition) and phylogenetic analyses (by maximum likelihood). Hydrolases were identified through the MEROPS and CAZy platforms.
Results
The three genomes of C. subterraneus showed high similarity, although there are substantial differences in their gene composition and organization. Each subspecies possesses a gene cluster encoding a carbon monoxide dehydrogenase (CODH) and an energy converting hydrogenase (ECH). The CODH gene is associated with an operon that resembles the Escherichia coli hydrogenase hyc/hyf operons, a novel genetic context distinct from that found in archetypical hydrogenogenic carboxydotrophs. Apart from the CODH-associated hydrogenase, these bacteria also contain other hydrogenases, encoded by ech and hyd genes. An Mbx ferredoxin:NADP oxidoreductase homolog similar to that originally described in the archaeon Pyrococcus furiosus was uniquely encoded in the C. subterraneus subsp. yonseiensis genome. Compositional analysis demonstrated that some genes of the CODH-ECH and mbx operons present distinct sequence patterns in relation to the majority of the other genes of each genome. Phylogenetic reconstructions of the genes from these operons and those from the ech operon are incongruent to the species tree. Notably, the cooS gene of C. subterraneus subsp. pacificus and its homologs in C. subterraneus subsp. tengcongensis and C. subterraneus subsp. yonseiensis form distinct clades. The strains have diverse hydrolytic enzymes and they appear to be proteolytic and glycolytic. Divergent glycosidases from 14 families, among them amylases, chitinases, alpha-glucosidases, beta-glucosidases, and cellulases, were identified. Each of the three genomes also contains around 100 proteases from 50 subfamilies, as well about ten different esterases.
Conclusions
Genomic information suggests that multiple horizontal gene transfers conferred the adaptation of C. subterraneus subspecies to extreme niches throughout the carbon monoxide utilization and hydrogen production. The variety of hydrolases found in their genomes indicate the versatility of the species in obtaining energy and carbon from diverse substrates, therefore these organisms constitute a remarkable resource of enzymes with biotechnological potential.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1955-9) contains supplementary material, which is available to authorized users.
Keywords: Caldanaerobacter subterraneus, Genome, Horizontal gene transfer, Hydrogenase, Carbon monoxide dehydrogenase, Glycosidase, Protease, Esterase, Phylogeny, Thermophile
Background
Thermophilic bacteria possess diverse adaptations in order to thrive under high temperatures [1, 2]. Therefore, these organisms are sources of potentially useful thermostable proteins, which is promising because of the increasing biotechnological interest in highly thermostable enzymes [3]. Besides, the genomic study of these organisms can provide insights on interesting metabolic features characteristic of these bacteria, like the ability to generate hydrogen gas as metabolic product, a promising renewable fuel. With the advent of high throughput technologies of DNA sequencing, many genomes of thermophilic bacteria are being unraveled (e.g. [4–7], and the in silico analysis of the large amount of generated data is a fundamental initial approach to understand the full potential of these organisms.
Caldanaerobacter subterraneus includes fermentative thermophilic bacteria with relatively low genomic GC content (under 40 %) able to grow on carbohydrate substrates with acetate L-alanine, H2, and CO2 as the main products that have been isolated from a variety of hot environments [8–11]. C. subterraneus subsp. pacificus (formerly known as Carboxydobrachium pacificum) is known to grow on CO hydrogenogenically [8]; C. subterraneus subsp. tengcongensis–formerly Thermoanaerobacter tengcongensis–and C. subterraneus subsp. yonseiensis (but not C. subterraneus subsp. subterraneus) have been reported to oxidize CO [11]; however there is no mention if they produce hydrogen from CO. In 2002, the genome of C. subterraneus subsp. tengcongensis was sequenced. A CODH gene cooS was found in the genome and ascribed to the acetogenic Wood-Ljungdahl pathway [12]. However, after this report it was noted that the genome lacks the acetyl-CoA synthase gene, indispensable for this pathway, and the CODH gene is clustered with ECH genes, suggesting that C. subteraneus subsp. tencongensis has the capacity for hydrogenogenic carboxydotrophy [13]. Recently, the genome of C. subterraneus subsp. yonseiensis has also been published [14], which can contribute to the understanding of the evolution of the metabolic features in this species relative to its sibling strains. Moreover, these genomes constitute helpful resources for cloning and expression of novel enzymes of biotechnological importance (e.g. [1, 2, 15]).
In this study, the genome of C. subterraneus subsp. pacificus was sequenced. This bacterium grows from 50 to 80 °C, and was isolated from a submarine thermal vent in Japan, unlike the other subspecies (C. subterraneus subsp. tengcongensis and C. subterraneus subsp. yonseiensis were isolated from terrestrial high temperature environments, and C. subterraneus subsp. subterraneus strains are oilfield isolates). C. subterraneus subsp. pacificus is known to be able to grow chemolithotrophically on CO, producing H2 and CO2 during growth [8].
The main objective of this study was to explore the differences among the three genomes by comparative analysis. The analyses were focused on inferring the physiological and evolutionary aspects of these organisms. The role of horizontal gene transfer (HGT) in shaping these three genomes was also evaluated and key metabolic genes and proteins with potential biotechnological application, such as carbon monoxide dehydrogenase, hydrogenases, proteases, glycosidases and esterases were identified.
Results and discussion
Phylogeny of the species
A phylogenetic tree was constructed using 16S rRNA gene sequences. Thermoanaerobacterales and other bacterial species were included to demonstrate the evolutionary context of C. subterraneus subspecies, and to use them as reference for comparative purposes against other gene dendrograms. The tree included available copies of 16S rRNA genes of Caldanaerobacter species and subspecies (Fig. 1). The resulting 16S rRNA tree is in agreement with previous information: Sokolova et al. [8] and Subbotina et al. [16] have also shown that the species later reassigned to the genus Caldanaerobacter [11] are very close to each other and form a clade adjacent to but distinct from the clade of Thermoanaerobacter species.
Fig. 1.
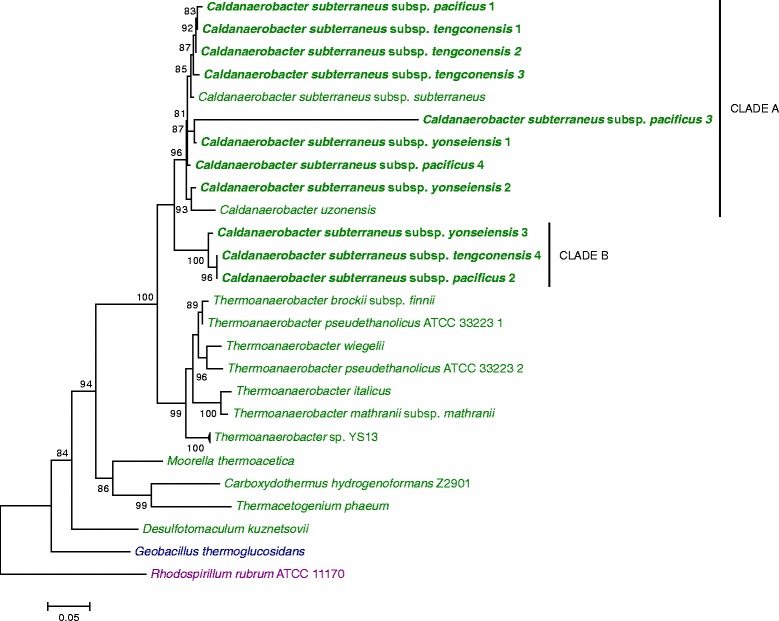
Evolutive history of Caldanaerobacter subterraneus subspecies. The 16S rRNA tree was constructed using the maximum-likelihood method. aLRT values greater than 70 % are shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. Caldanaerobacter subterraneus subspecies are in bold. Other Clostridiales species are in green, Bacillales species in blue. R. rubrum was used as outgroup (in purple)
C. subterraneus subsp. tengcongensis is known to exhibit an exceptionally high level of sequence divergence among its intragenomic 16S rRNA gene copies (6.7 %) [17]. As demonstrated in Fig. 1, the C. subterraneus subspecies with available genomes exhibit multiple 16S rRNA gene copies that are separated in two main clades (Clade A and Clade B, Fig. 1). This clade separation most probably represents the most ancient gene duplication that occurred before the diversification of subspecies. Considering Clade B, C. subterraneus subsp. pacificus is closer to C. subterraneus subsp. tengcongensis than to C. subterraneus subsp. yonseiensis. This pattern is not evident in Clade A due to the presence of intra-subspecies multiple 16S rRNA genes that interfere with interpretation of the true phylogenetic relationships among these subspecies.
Genomes overview and horizontal gene transfer detection
As expected, the average nucleotide identity (ANI) and in silico prediction of in vitro DNA-DNA hybridization (DDH) values for the genomes of C. subterraneus subspecies confirmed the conclusion of Fardeau et al. [11] about the affiliation of the C. subterraneus subspecies within the same species and once more showed the closest proximity of C. subterraneus subsp. pacificus to C. subterraneus subsp. tengcongensis (ANI value was 98.8 % and DDH value 85 %) than to C. subterraneus subsp. yonseiensis (ANI value was 98.0 % and DDH value was 80 %).
The genomes of C. subterraneus subsp. pacificus and C. subterraneus subsp. yonseiensis present a similar pattern of high colinearity with the C. subterraneus subsp. tengcongensis genome (Fig. 2a). These data are reinforced by the results of homology score (H-value) distribution of the CDSs, which shows a high number of common CDSs between C. subterraneus subsp. tengcongensis and the other two subspecies (Fig. 2b).
Fig. 2.
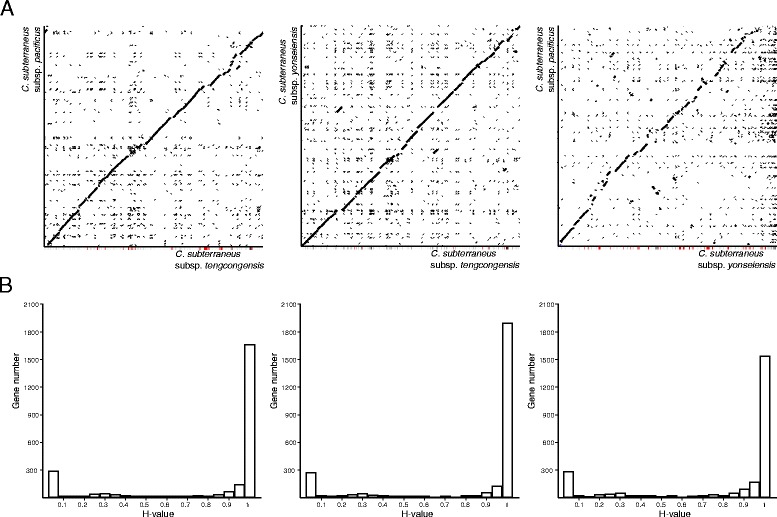
Comparison of C. subterraneus genomes. a Synteny plots demonstrating the high collinearity of the C. subterraneus genomes. C. subterraneus subsp. pacificus vs. C. subterraneus subsp. tengcongensis (left), C. subterraneus subsp. yonseiensis vs. C. subterraneus subsp. tengcongensis (middle), and C. subterraneus subsp. pacificus vs. C. subterraneus subsp. yonseiensis (right). b Histograms of H-values (a homology measure, see Methods) for all predicted proteins of C. subterraneus subspecies. C. subterraneus subsp. pacificus vs. C. subterraneus subsp. tengcongensis (left), C. subterraneus subsp. yonseiensis vs. C. subterraneus subsp. tengcongensis (middle), and C. subterraneus subsp. pacificus vs. C. subterraneus subsp. yonseiensis (right)
Table 1 shows a comparison of the general features of the three C. subterraneus genomes. Although these genomes all present low overall GC content (~37.7 %), their rRNAs and tRNAs have higher GC content (higher than 59.0 %), which corroborates the recognized positive correlation between the GC content of the rRNA and tRNA and optimal growth temperatures of prokaryotes [17].
Table 1.
Overview of C. subterraneus genomes
| C. subterraneus subsp. pacificus | C. subterraneus subsp. tengcongensis | C. subterraneus subsp. yonseiensis | |
|---|---|---|---|
| Genome size (Mb) | 2.39 | 2.69 | 2.7 |
| Genome GC content (%) | 37.7 | 37.8 | 37.7 |
| Number of contigs | 135 | 1 (complete chromosome) | 102 |
| CDS | 2511 | 2588 | 2711 |
| Operon | 871 | 1291 | 880 |
| Hypothetical proteinsa | 962 (38.31 %) | 855 (33.04 %) | 836 (30.84 %) |
| Average gene length | 819 | 905 | 834 |
| rRNA | 11 | 12 | 18 |
| rRNA average GC content (%) | 59.8 | 59.81 | 59.3 |
| tRNA | 49 | 56 | 59 |
| tRNA average GC content (%) | 60.26 | 60.12 | 59.98 |
| Number of horizontally transferred CDSsb | 173 (6.88 %) | 121 (4.67 %) | 127 (4.68 %) |
| Origin | Pacific Ocean hot vents | Terrestrial hot spring | Geothermal hot stream |
| Reference | This study | [12] | [14] |
aPercentage of hypothetical proteins of all genome proteins is in parentheses
bDetected by GOHTAM. In parentheses is the percentage of horizontally transferred CDSs of all CDSs present in the genome
Genes that putatively could have been acquired via horizontal transfer were identified in all three genomes. In C. subterraneus subsp. pacificus and C. subterraneus subsp. yonseiensis, most of the putative horizontally transferred genes correspond to hypothetical proteins, 99 of 173 (57.2 %), and 75 of 127 (59.1 %), respectively. C. subterraneus subsp. tengcongensis presents a lower proportion of hypothetical genes that could have been horizontally transferred, 55 of 121 CDSs (45.5 %). Also, some of the xenologous CDSs are transposases (9 in C. subterraneus subsp. tengcongensis, 8 in C. subterraneus subsp. pacificus, and 5 in C. subterraneus subsp. yonseiensis).
CODH dehydrogenase and Hyf/Hyc hydrogenase
Carbon monoxide dehydrogenases (CODH) are enzymes that catalyze the interconversion of CO and CO2, and they vary in their functional roles in the cell [18, 19]. All three C. subterraneus subspecies examined in this study possess a cooS gene encoding a CODH that is upstream of a hydrogenase gene cluster, with an invariant gene order identical to that found in Geobacillus thermoglucosidans strains [20].
In Fig. 3, the cooS genetic contexts of C. subterraneus subspecies and G. thermoglucosidans strains are contrasted to those from model organisms for studying carboxydotrophy, such as Carboxydothermus hydrogenoformans, Moorella thermoacetica, Rhodospirillum rubrum (Bacteria), and Thermococcus onnurineus (Archaea). The species C. hydrogenoformans, for example, possesses five cooS paralogs distributed along the genome, and their genetic contexts provide clues on the physiological roles of the CODHs in this organism [19]. As in C. subterraneus, in R. rubrum and C. hydrogenoformans, hydrogenase genes are also clustered with a cooS gene, and are identified by the prefix coo. In these organisms, it was suggested that the CODH and the coo hydrogenase gene cluster includes genes encoding proteins required for proton translocation, fundamental for energy conservation [21, 22]. Although the hydrogenase genes of C. subterraneus have homologous counterparts in the R. rubrum and C. hydrogenoformans coo hydrogenase genes, the former ones are more similar to the hyf/hyc genes from Escherichia coli, encoding the hydrogenase module of formate hydrogen lyase complexes [23]. A homologous hyf/hyc operon with identical genetic organization to that from C. subterraneus subspecies is also present in M. thermoacetica (Fig. 3), where it also includes formate dehydrogenase genes and it is thought to encode a formate hydrogen lyase complex [4]. In the archaeon T. onnurineus, the cooS gene is associated to hyf-hyc homologs (Fig. 3), which are fundamental for carboxydotrophic hydrogenogenesis [24, 25]. Interestingly, the organization of these hydrogenase genes is identical to that found in the hyc operon of E. coli [26] where the hyfDEF homologs are absent (in Fig. 3, the hydrogenase genes were named as hyf in order to permit a clear identification of the homologous genes among the considered species). The hyc and hyf operons encode for paralogous energy-converting Ni-Fe hydrogenases Hyd-3 and Hyd-4 of E. coli, which have significant similarity to the components of NADH:quinone oxidoreductase (complex I), suggesting their implication in energy metabolism [23]. Therefore, although distinct from the coo hydrogenase, it is likely that in C. subterraneus the Hyc/Hyf proteins and CODH form a complex responsible to extract energy by CO oxidation. This metabolism is often stated to be ancient, R. Hedderich [27], for example, suggested that energy-converting hydrogenases may have been originally associated with CODH.
Fig. 3.
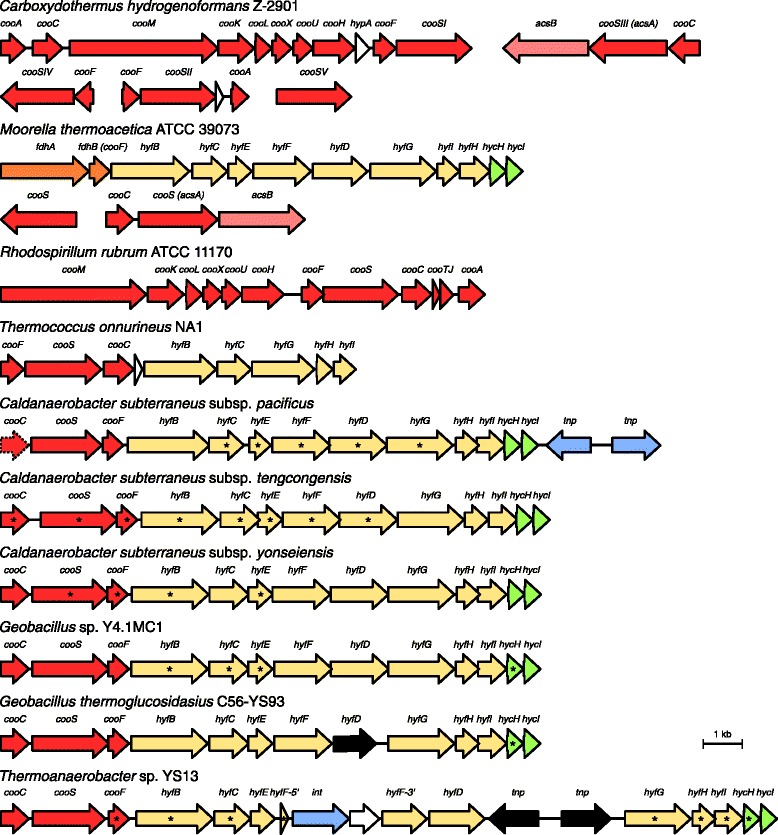
Comparison of the CODH-hydrogenase gene cluster organization between C. subterraneus and other species. Arrows represent genes and their respective direction of transcription. Asterisks indicate significant prediction of HGT in C. subterraneus and Geobacillus thermoglucosidans strains
Although Bao et al. [12] suggested that the CODH could also be utilized to fix carbon throughout the Wood-Ljungdahl pathway in C. subterraneus subsp. tengcongensis, it is improbable, because the genome of this strain does not present a gene putatively encoding for the key enzyme acetyl coenzyme A synthase (acsB) [4]. As well, this gene was not found in the other two subspecies of C. subterraneus suggesting that the same argument can be applied to all these three subspecies, which indicates a limitation for the use of carbon monoxide or carbon dioxide as a carbon source. This contrasts to the capabilities of other thermophilic, CO-utilizing, hydrogenogenic or acetogenic Firmicutes (e.g., C. hydrogenoformans [28] and M. thermoacetica [4]) (Fig. 3).
As demonstrated in Additional file 1: Figure S1, the CODHs of all three C. subterraneus subspecies contain the conserved amino acid residues important for the activity of this enzyme when compared to the archetypical CODHs deposited in PDB database. However, the CODH of C. subterraneus subsp. pacificus has important distinctive characteristics in relation to those of the other two subspecies of C. subterraneus, as the absence of the regions 450–454 and 537–544 (Additional file 1: Figure S1). In fact, this CODH has 66 % identity with its counterpart from Methanosarcina acetivorans (NP_618172.1), while in relation to those from C. subterraneus subsp. tengcongensis and C. subterraneus subsp. yonseiensis has 50 and 49 % identity, respectively. This observation, Blast searches and their alignments indicate that these proteins are not true orthologs but rather pseudoorthologs (xenologs). To investigate this finding, the phylogeny of these CODHs was analyzed. The resulting phylogenetic tree suggests a recent inter-phylum transfer of CODH gene from C. subterraneus subsp. tengcongensis to Thermodesulfobacterium thermophilum and possible more ancient CODH gene transference between Bacteria and Archaea (Fig. 4). The tree also confirmed that the CODHs of Caldanaerobacter subspecies can be classified in different lineages (Fig. 4).
Fig. 4.
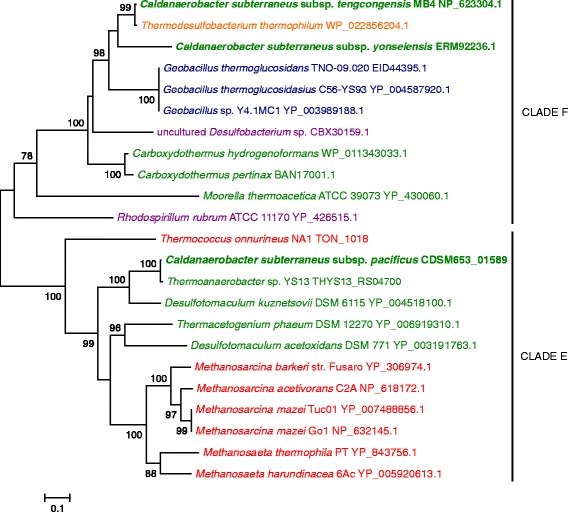
Evolutive history of CODH (CooS) from C. subterraneus subspecies. Details are as shown in Fig. 1, unless specified otherwise. Thermodesulfobacterium is in orange, and Archaea are in red. Accession number or locus tag are adjacent the species name. This tree is mid-point rooted. Classification of clades as in Techtmann et al. [29]
Considering the species 16S rRNA tree as reference (Fig. 1), this result was unexpected. The CODH of C. subterraneus subsp. pacificus clusters with the homologous counterpart of Thermoanaerobacter sp. YS13 (having 98.8 % identity with this protein), and both are relatively distant from the CODHs from the other C. subterraneus subspecies. Following the classification described in Techtmann et al. [29], the CODH of C. subterraneus subsp. pacificus belongs to “Clade E”. On the other hand, the CODHs from C. subterraneus subsp. tengcongensis and C. subterraneus subsp. yonseiensis are immersed in “Clade F” (Fig. 4) and are rather closely related to the homologous proteins from Geobacillus thermoglucosidans strains (Order Bacillales), which are relatively distant from Caldanaerobacter (Order Clostridiales) (Fig. 1). However, despite their affiliation with the same clade, the CODHs of C. subterraneus subsp. tengcongensis and C. subterraneus subsp. yonseiensis are distant enough from each other to exclude their vertical inheritance from the LCA (last common ancestor) of these subspecies. While most of C. subterraneus subsp. yonseiensis proteins have H-values higher than 0.95 in relation to the C. subterraneus subsp. tengcongensis proteins (Fig. 2 and Additional file 2: Table S1), the CODH (O163_06470) has a H-value of 0.78 (Additional file 2: Table S1). These results suggest that the CODH evolutionary history of C. subterraneus includes several recent HGT events.
Taking these observations into account, all genes of the CODH-hydrogenase gene cluster were investigated for HGT by means of detection of phylogenetic discrepancies and parametric methods. Becq et al. [30] showed tetranucleotide composition and codon usage analyses had mean specificity of 87.8 and 89.2 %, and mean sensitivity of 77.2 and 91.5 %, respectively, when these methods were tested with artificial genomes. These values vary depending on intrinsic genome characteristics of the recipient organism, and on the origin of the HGT (e.g. a DNA sequence from a phylogenetically close related donor can be poorly detected by these methods). The greatest advantage of parametric methods is they do not rely on sequence data banks as phylogenetic approaches do [31]. However, phylogenetic reconstruction is necessary to infer historical events from sequences [32].
Parametric analysis of nucleotide composition revealed that some CODH-hydrogenase genes from G. thermoglucosidans and prominently those from C. subterraneus subspecies have differential sequence patterns in relation to the “standard” gene sequence pattern of each genome (Fig. 3 and Additional file 2: Table S1), which suggests that these genes could have been acquired by horizontal transfer. The presence of a transposase gene downstream the CODH-hydrogenase gene cluster in C. subterraneus subsp. pacificus represents an additional evidence supporting this hypothesis (Fig. 3).
The close relationship of the hyf/hyc hydrogenase gene cluster between C. subterraneus and G. thermoglucosidans strains was confirmed by phylogenetic analyses. Apparently, these hyc and hyf genes shared common evolutionary histories and were acquired together as a cluster, not individually (Fig. 3 and Additional file 2: Table S1). Considering the phylogenetic distance between Geobacillus and C. subterraneus, the high identity levels between their CODH-hydrogenase proteins (~70–80 %) can hardly be interpreted as a result of vertical inheritance from the LCA but are rather a result of acquisition of the cluster by the C. subterraneus lineage via HGT or its acquisition by both C. subterraneus and G. thermoglucosidans lineages from the same source or sources related to each other.
In general, in most of the phylogenetic trees of the CODH-hydrogenase gene cluster, the three subspecies of C. subterraneus form monophyletic clades with Thermoanaerobacter sp. YS13 (Additional file 3: Figure S2 and Additional file 4: Figure S3), with the exception of cooF and cooS genes (Fig. 4). In fact, the hyc and hyf genes of Thermoanaerobacter sp. YS13 have identity values higher than 96 % at the nucleotide level when compared to the C. subterraneus subsp. tengcongensis orthologous genes. These observations and the presence of genes implicated with transposition within its CODH-hydrogenase gene cluster (Fig. 4) support the idea that this strain inherited the CODH-hydrogenase gene cluster from C. subterraneus. Since C. subterraneus subsp. pacificus is closer to C. subterraneus subsp. tengcongensis than any other organism in most phylogenetic reconstructions (Fig. 1 and Additional file 4: Figure S3) and gene comparisons (hyfCDEGHI and hycH are identical at nucleotide sequence level), it is likely that the gene transfer event occurred before the diversification of these subspecies. This observation implies that most probably their LCA harbored a “Clade E” CODH (as C. subterraneus subsp. pacificus). In spite of our contention that the hyf/hyc hydrogenase gene cluster was acquired as a cluster, not as individual genes, there is evidence that the cooF and cooS genes from C. subterraneus have distinct evolutionary histories with respect to the other genes of the cluster. An important point is that the GC contents of cooF and cooS genes from C. subterraneus subsp. tengcongensis (ca. 57 and 61 % respectively) and C. subterraneus subsp. yonseiensis (ca. 51 and 49 % respectively) are much higher than the mean gene GC content of these genomes (ca. 38 %) (Additional file 2: Table S1). Together with the above-mentioned considerable differences in the amino acid and nucleotide sequence patterns, the GC content data suggests that the CODHs of C. subterraneus subsp. tengcongensis and C. subterraneus subsp. yonseiensis were acquired recently via independent HGT events from prokaryotes having a higher genomic GC content after the diversification of the subspecies.
An important point to consider regarding the CODH tree is that its composition is very diverse taxonomically and does not reflect properly species relationships, indicating that HGT played an important role in the current distribution of carboxydotrophy among prokaryotes. Independent studies already pointed out that HGT of the cooS gene likely took place in several thermophilic species [29, 33, 34]. Despite the fact that the donor and acceptor organisms in these instances may be phylogenetically remote, they are usually able to grow in anaerobic environments at similar ranges of temperature and pH [33, 35]. The acquisition of new physiological characteristics would putatively allow the recipient organisms to be recruited to new thermophilic consortia, and consequently, the horizontal transference of important genes for adaptation to specialized niches would be facilitated.
Our analysis revealed at least four recent independent HGT events in the evolutionary history of the CODH-hydrogenase gene cluster: (1) cooS and cooF replacement in C. subterraneus subsp. tengcongensis; (2) cooS and cooF replacement in and C. subterraneus subsp. yonseiensis; (3) transfer of the cluster as whole to a recent ancestor of Thermoanaerobacter sp. YS13 from a common ancestor of C. subterraneus subsp. pacificus and C. subterraneus subsp. tengcongensis; (4) cooS. cooF, and cooC transfer from C. subterraneus subsp. tengcongensis to Thermodesulfobacterium thermophilum.
Other hydrogenases
Besides the CODH-associated hydrogenase, C. subterraneus sbsp. tengcongensis harbors additional hydrogenases, a NiFe hydrogenase (encoded by ech genes) and a NADH-dependent Fe-only hydrogenase (encoded by hyd genes), which putatively catalyze the production of H2 from excess of reducing equivalents formed during the fermentation of saccharides at low p(H2) [21]. The genes encoding these enzymes have been identified in C. subterraneus subsp. pacificus (although some ech are incomplete in the current genome assembly) and C. subterraneus subsp. yonseiensis. As pointed out by Calteau et al. [36] and Soboh et al. [21], both hydrogenase genes were wrongly assigned as NADH:ubiquinone oxidoreductase genes in C. subterraneus subsp. tengcongensis genome because of automatic annotation process. This error was also introduced to the C. subterraneus subsp. yonseiensis genome, and in the previous version of the genome of C. subterraneus subsp. pacificus. However, in the latest annotation of the genome of C. subterraneus subsp. pacificus, the proper description of these genes has been included.
Homologs of E. coli hyp genes are located upstream the ech genes in C. subterraneus. In E. coli, hyp genes are essential for the maturation of the hydrogenases [23]. The synteny of hyp genes adjacent to ech genes could indicate their role in the maturation of Ech hydrogenase. However, it is noteworthy that possibly Hyp proteins could act on this as well as other hydrogenases, notably the CODH associated hydrogenase, which resembles the E. coli Hyc hydrogenase (Hyd-3), target of HypA and HypC proteins [37]. In M. thermoacetica, the hypABFCDE operon, and in the Geobacillus thermoglucosidans strains investigated in this study, the hypAB genes are located downstream the hyf/hyc operon, which represents additional evidence supporting the probable interaction of their gene products.
Calteau et al. [36] had suggested that ech genes from an archaeon related to Methanosarcina were transferred horizontally to a C. subterraneus subsp. tengcongensis ancestor, however our analyses throughout parametric methods did not detect divergent sequence patterns in these genes (Additional file 2: Table S1). On the other hand, our phylogenetic analyses for most ech genes (with the exception of echB) demonstrated that C. subterraneus are immersed in the Thermoanaerobacter clade (Additional file 5: Figure S4). Consequently, the C. subterraneus subspecies last common ancestor would not have acquired the ech genes directly from an archaeon, but more likely indirectly through a Thermoanaerobacter species. Therefore, the alternative hypothesis by Calteau et al. [36] would be in agreement with our observations suggesting an initial transfer of these genes from an archaeon to a bacterial lineage followed by a second bacterium to bacterium transference. The hyp and hyd genes showed characteristics expected for this species in accordance to sequence composition (Additional file 2: Table S1) and phylogeny (Additional file 6: Figure S5 and Additional file 7: Figure S6).
E. coli has multiple hydrogenases that act differently depending on carbon source availability and on pH [38]. Similarly, hydrogenases from C. subterraneus are expected to be active under different environmental conditions which would increase fitness in a variety of extreme environmental situations and carbon sources that C. subterraneus subspecies encounter in their natural niches [8].
The Mbx ferredoxin:NADP oxidoreductase
From all C. subterraneus subspecies investigated in this study, C. subterraneus subsp. yonseiensis uniquely encodes an mbx gene cluster (genes O163_11500 to O163_11560). Its products are highly similar to Pyrococcus furiosus Mbx proteins (identities ranging from 30 to 60 %), which were automatically misannotated as NADH-ubiquinone oxidoreductase subunits and were initially described as encoding a putative fourth hydrogenase in P. furiosus [39]. However, according to the currently prevailing views, substantiated by the Adams lab [40, 41], Mbx is not a hydrogenase but a ferredoxin:NADP oxidoreductase, one of the differentiating features being the lack of the two CxxC Ni-binding motifs characteristic of [NiFe]-hydrogenases in the MbxL (HyfG) subunit (including O163_11555 in C. subterraneus subsp. yonseiensis). The genes O163_11495 and O163_11565 that flank the mbx operon in C. subterraneus subsp. yonseiensis were also found in the other C. subterraneus genomes (Fig. 5). The gene O163_11495 encodes a putative G-D-S-L family lipolytic protein (Additional file 8: Table S3), therefore it does not seem to be functionally related to Mbx hydrogenase, and the gene O163_11565 encodes for a putative cation transporter. Preliminary blast searches using the nucleotide region spanning from the gene O163_11495 to the gene O163_11565 revealed 97 % identity to a genomic region from Thermoanaerobacter wiegelii. Although these organisms are rather closely related (Fig. 1), the high identity between these genome fragments is unexpected (the ANI between their genomes is 82 %) and represents a strong indication of HGT. In fact, as demonstrated in Fig. 5 and Additional file 2: Table S1, some of the mbx genes show differential tetranucleotide composition in C. subterraneus subsp. yonseiensis. Phylogenetic analyses of each deduced Mbx protein corroborated the hypothesis that most mbx genes in C. subterraneus subsp. yonseiensis grouped with Thermoanaerobacter species (Additional file 9: Figure S7). Furthermore, in the phylogenetic reconstruction of the gene O163_11495, C. subterraneus subsp. yonseiensis is closer to Thermoanaerobacter than to other C. subterraneus subspecies, and in the phylogeny of the gene O163_11565, homologs from all C. subterraneus subspecies are immersed in a Thermoanaerobacter species clade (Additional file 9: Figure S7).
Fig. 5.
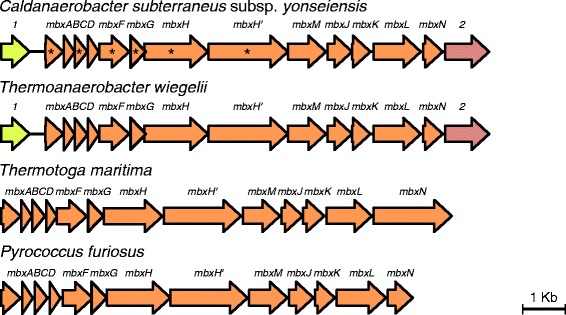
Comparison of the Mbx gene cluster organization across different species. Arrows represent genes and their respective direction of transcription. Asterisks indicate significant prediction of HGT in C. subterraneus subsp. yonseiensis. 1–putative G-D-S-L family lipolytic protein gene; 2–putative cation transporter gene
Evidence suggests that the mbx operon could have been transferred from a Thermoanaerobacter species to C. subterraneus subsp. yonseiensis. Calteau et al. [36] suggested that the mbx genes could have been originally transferred from Archaea to Bacteria. As the case of ech genes, this evolutionary event would have preceded the bacteria-to-bacteria HGTs.
Glycosidases
Glycosidases from thermophiles have many industrial and biotechnological applications [42], thus the wealth of glycosidases in these species motivates detailed study. C. subterraneus subsp. tengcongensis presents 25 glycosidases distributed in 13 families, while C. subterraneus subsp. yonseiensis harbors 17 glycosidases from 8 families, and most of which have homologous counterparts in C. subterraneus subsp. tengcongensis. C. subterraneus subsp. pacificus have 21 glycosidases from 12 families, and two of them are specific to this subspecies (Additional file 2: Table S1 and Additional file 10: Table S2).
At this time three glycoside hydrolases deduced from C. subterraneus subsp. tengcongensis genome have been biochemically characterized. Two of them are starch-hydrolyzing enzymes, a glucoamylase (TTE1813) [43] and an alphaglucosidase (TTE0006) [15], and both have homologs in the other two C. subterrraneus subspecies (Additional file 10: Table S2). Exoglucohydrolases similar to these ones are extensively utilized for the hydrolysis of starch to glucose in industrial processes for food and ethanol production [15, 43]. Additional alphaglucosidases from the GH31 family remaining to be investigated were found in the genomes of C. subterraneus subsp. tengcongensis (TTE1934) and C. subterraneus subsp. pacificus (CDSM653_01802) (Additional file 10: Table S2). These orthologs have 32 % identity with the protein MalA of the archaeon Sulfolobus solfataricus, which has a substrate preference for maltose and maltooligosaccharides [44]. Their neighbor genes are sugar permease genes in both C. subterraneus genomes, and they present different tetranucleotide composition, suggesting a likely horizontal inheritance for these genes (Additional file 2: Table S1).
The third type of identified glycosidase is a cellulase (endoglucanase) (TTE0359) [45], which was also found in C. subterraneus subsp. yonseiensis (Additional file 10: Table S2). This enzyme is able to break the internal bonds of cellulose, generating glucans of different lengths that are substrate for other enzymes to glucose production. One of these enzymes is the betaglucosidase, which hydrolyzes cellobiose disaccharides to glucose. Putative betaglucosidases from families GH1 and GH3 were found in the genomes of C. subterraneus subsp. tengcongensis and C. subterraneus subsp. pacificus, but the genome of C. subterraneus subsp. yonseiensis only hosts one belonging to the GH3 family (Additional file 10: Table S2). Currently, intensive studies of such enzymes are being carried out, due to their implication in the saccharification of lignocellulosic materials such as sugarcane bagasse for production of biofuel [46]. Also, it is worth noting that four putative enzymes originally annotated as hypothetical in C. subterraneus subsp. tengcongensis genome belong by similarity to the GH18 family of glycosidases, and they were found in the other two C. subterraneus genomes (Additional file 10: Table S2). This family is known by containing chitinases, enzymes that hydrolyze chitin, one of the most common biopolymers in nature [47]. Bacterial chitinases can be utilized as biological control of fungi and insects, but are also suitable for protoplast generation and the treatment of shellfish waste [48].
Esterases
Esterases are widely utilized in industry for production of pharmaceuticals, detergents, biodiesel and other compounds [49, 50]. At least five esterases of C. subterraneus subsp. tengcongensis have been biochemically characterized [1, 2, 51–54]. These enzymes share high thermal stability at temperatures above 60 °C, and they use different substrates, as mentioned in Additional file 8: Table S3. Besides these esterases, Levisson et al. [55] detected through in silico approaches four additional esterases in C. subterraneus subsp. tengcongensis genome (Additional file 8: Table S3). In our study, it was verified that C. subterraneus subsp. yonseiensis possesses homologs of each one of the esterases referenced above. Two of them were not located in C. subterraneus subsp. pacificus genome, however using the LIPABASE proteins as reference, a specific lipase was found (CDSM653_00572). It matched a lipase from Acinetobacter baumannii [EMBL:A3M3C1], but because the coverage was 36 % and identity 31 %, more detailed studies are necessary to evaluate its catalytic properties.
Proteases
Proteases are ubiquitous to all life forms, with in vivo functions ranging from protein turnover to growth substrate hydrolysis and amino acid acquisition. They have a highly diverse range of applications, such as tenderization of meat, composing detergent formulations, leather processing, molecular biology applications and peptide synthesis [56].
Around 100 proteases from 50 distinct subfamilies were found in each C. subterraneus genome (Additional file 11: Table S4). Among these proteins, metallo- and serineproteases were the most common. We note that the M42 subfamily proteases were originally annotated as cellulase-like proteins in C. subterraneus subsp. tengcongensis (Additional file 11: Table S4), but it is likely another case of misannotation. Dutoit et al. [57] verified experimentally that two proteins annotated as cellulases in the Thermotoga maritima and Clostridium thermocellum genomes were actually M42 aminopeptidases.
Although C. subterraneus possesses many proteases, only one peptidase has been already characterized, a serine protease named as tengconlysin (TTE0824) [58]. Therefore the potential of C. subterraneus as a source for proteases is underexploited.
Conclusions
The study of C. subterraneus genomes is important to understand the adaptations allowing them to thrive in extreme habitats, as well as to analyze enzymes with biotechnological potential showing functionality under high temperatures. H2 is an important compound to the chemical industry, and a future clean biofuel [59]. Genomic data indicate that C. subterraneus is able to produce H2 throughout different hydrogenase systems, markedly one associated with a CODH that permits obtaining energy from carbon monoxide, widely available in syngas and other industrial fuel gases. Horizontal gene transfer seems to be an important evolutionary driving force in carboxydotrophy and hydrogenogenesis in this species, abilities that permitted it to survive in niches where multiple inorganic and organic substrates may be available at low concentrations. In this sense, it is also worth noting that these bacteria encode a wide repertoire of hydrolase genes, such as glycosidases, esterases and proteases that act on a wide variety of substrates to provide them with carbon and energy. Therefore, the metabolic versatility of this species makes it a good source to target for novel enzymes with biotechnological potential.
Methods
Bacterial strain, genome sequencing, and operon prediction
C. subterraneus subsp. pacificus was isolated from a submarine hot vent in Okinawa Trough [8]. Genome DNA was mainly sequenced and assembled at the J. Craig Venter Institute. Contigs of C. subterraneus subp. pacificus genome were automatically annotated with the xBase platform [60] using as reference the C. subterraneus subsp. tengcongensis genome. Genes of interest were inspected carefully and had their annotation refined manually. Operons were predicted using DOOR software [61]. This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession ABXP00000000. The version described in this paper is version ABXP02000000.
16S rRNA gene phylogeny
Most of the 16S rRNA gene sequences were retrieved from the SILVA rRNA Database (Additional file 12: Table S5) [62]. Sequences were aligned using SINA software [63], and gap positions were removed. Phylogenetic reconstructions were performed using the Phylogeny.fr platform [64] with the maximum likelihood method implemented in the PhyML program (v3.0 aLRT) [65]. For each phylogeny, the GTR (Generalized Time Reversible) substitution model was selected assuming an estimated proportion of invariant sites and 4 gamma-distributed rate categories to account for rate heterogeneity across sites. The gamma shape parameter was estimated directly from the data. Reliability for internal branching was assessed using the aLRT (approximate Likelihood Ratio Test) [66].
Comparison of C. subterraneus genomes
In addition to the genome sequenced in this study, two genomes of C. subterraneus are publicly available from the following subspecies: C. subterraneus subsp. yonseiensis (AXDC00000000.1) and C. subterraneus subsp. tengcongensis (NC_003869.1). Only the C. subterraneus subsp. tengcongensis genome is complete. Therefore, the following comparative analyses were made using this genome as reference.
The average nucleotide identity (ANI) values (species boundary is 95 %) for the genomes of Caldanaerobacter subterraneus subspecies were determined [67]. The in silico prediction of in vitro DNA-DNA hybridization (DDH) values (species boundary is 70 %) were calculated using GGDC 2.0 BLAST+ and recommended formula 2 [68].
Synteny plots were generated with the R2CAT software [69], aligning and ordering the contigs of C. subterraneus subsp. pacificus and C. subterraneus subsp. yonseiensis against C. subterraneus subsp. tengcongensis genome.
mGenome Subtractor [70] was utilized to compare the conservation of proteins of C. subterraneus subsp. pacificus and C. subterraneus subsp. yonseiensis genomes in relation to those from C. subterraneus subsp. tengcongensis.
The homology score (H-value) between two proteins is the product of the identity level (expressed as a value between 0 and 1) and of the ratio of the match length to query length [70]. Conserved proteins were defined by having a homology score H-value above 0.64.
Horizontal gene transfer analysis
The genomes of C. subterraneus and the genomes from related species of interest were utilized for HGT detection throughout the GOHTAM platform [71], which detects horizontal gene transfers based on tetranucleotide composition and/or codon usage. GC content of the CDSs was computed using EMBOSS package [72, 73]. Genes containing a GC content higher or lower than two standard deviations from the average CDS GC content for all CDSs in a genome were highlighted.
Other phylogenetic analyses
Amino acid sequences of Caldanaerobacter subterraneus subspecies were utilized as query in blastp searches against the Genbank NR database. The most similar sequences were retrieved. Also, for some proteins (e.g. CooS), homologous counterparts from PDB database were also retrieved. Subsequently, amino acid sequences were aligned using MUSCLE software embedded in MEGA [74]. Sites from the alignment containing gaps were removed. The phylogeny was constructed on the Phylogeny.fr platform basically as described previously above, but this time the WAG substitution model [75] was utilized.
Genome mining for hydrolases
Predicted proteins of the three subspecies of C. subterraneus were used as queries in blastp searches for glycohydrolase, esterase, peptidase and lipase databases. For protease identification the batch blast tool from MEROPS database [76] was utilized. For each protein, the hit with the lowest e-value (<10−10) was considered. Glycoside hydrolases were identified using CAT (Cazymes Analysis Tool) [77] with the following parameters: complete genome and e-value threshold of 10−10. Only glycosidases with domain and length consistency were considered. Esterases homologous to those already identified in C. subterraneus subsp. tengcongesis [1, 2, 51–55] were identified in the other subspecies genomes using as criterion the H-value >0.64. Lipases were searched among the C. subterraneus proteins using the blastp tool embedded in Bioedit [78] against the lipase database LIPABASE [79], with an e-value cutoff set to 10−10.
Availability of supporting data
Supporting data are included as Additional files. Phylogenetic data have been deposited at TreeBASE under the accession URL http://purl.org/phylo/treebase/phylows/study/TB2:S18113.
Acknowledgements
FHS received scholarship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil), process number 2403-13-7. The work of AVL and TGS was supported by the Russian Scientific Fund grant no. 14-24-00165. JMG acknowledges funding through projects CSD2009-00006, CGL2014-58762-P and GEN2006-26423-E from the Spanish Ministry of Economy and Competitiveness and RNM2529 and BIO288 from the Andalusian Government; Feder funds cofinanced these projects. C. subterraneus pacificus genome sequencing was carried out by the J. Craig Venter Institute through the Microbial Genome Sequencing Project sponsored by The Gordon and Betty Moore Foundation’s Marine Microbiology Initiative. Funding from the mobility programme 003-ABEL-CM-2013 (NILS Science and Sustainability programme, EEA grants) is also acknowledged. FTR acknowledges the support of a US National Science Foundation Grant. FTR acknowledges grant support from the Center for Dark Energy Biosphere Investigations (C-DEBI), an NSF Research Center at the University of Southern California. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Additional files
Alignment of CODHs (CooS) from C. subterraneus with archetypical CODHs from other prokaryotes. Cp C. subterraneus subsp. pacificus, Ct C. subterraneus subsp. tengcongensis (NP_623304.1), Cy C. subterraneus subsp. yonseiensis (ERM92236.1), Ch Carboxydothermus hydrogenoformans (WP_011343033.1), Mt Moorella thermoacetica ATCC 39073 (YP_430060.1), Rr Rhodospirillum rubrum ATCC_11170 (YP_426515.1). Black boxes represent 100 % identity. Purple letters–Cluster C, red letters–Cluster B and green letters–Cluster D, as defined by Dobbek et al. [80]. (PDF 33 kb)
Genome analyses of C. subterraneus subspecies and other bacteria. (XLSX 965 kb)
Evolutive history of CooC and CooF proteins from C. subterraneus subspecies. The tree was constructed using the maximum-likelihood method. aLRT values greater than 70 % are shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. Caldanaerobacter subterraneus subspecies are in bold. Other Clostridiales species are in green, Bacillales in blue, Proteobacteria in purple, Thermodesulfobacteria in orange, Archaea in red, and other bacteria in black. Accession number or locus tag are adjacent the species name. The tree is mid-point rooted. (PDF 57 kb)
Evolutive history of Hyc and Hyf proteins from Caldanaerobacter subterraneus subspecies. The subtrees were extracted from trees constructed using the maximum-likelihood method. Details are as shown in Additional file 3: Figure S2, unless specified otherwise. (PDF 59 kb)
Evolutive history of Ech hydrogenase from C. subterraneus subspecies. The subtrees were extracted from trees constructed using the maximum-likelihood method. Details are as shown in Additional file 3: Figure S2, unless specified otherwise. (PDF 55 kb)
Evolutive history of Hyp proteins from C. subterraneus subspecies. The subtrees were extracted from trees constructed using the maximum-likelihood method. Details are as shown in Additional file 3: Figure S2, unless specified otherwise. (PDF 58 kb)
Evolutive history of Hyd proteins from C. subterraneus subspecies. The subtrees were extracted from trees constructed using the maximum-likelihood method. Details are as shown in Additional file 3: Figure S2, unless specified otherwise. (PDF 56 kb)
Esterases found in C. subterraneus genomes. (XLSX 7 kb)
Evolutive history of Mbx from C. subterraneus subsp. yonseiensis. Details are as shown in Additional file 3: Figure S2, unless specified otherwise. (PDF 70 kb)
Glycohydrolases found in C. subterraneus genomes. (XLSX 7 kb)
Proteases found in C. subterraneus genomes. (XLSX 11 kb)
16S rRNA sequences utilized in this study. (XLSX 11 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
FHS designed the study, carried out most of the analyses, and wrote the manuscript. AVL carried out some of the analyses, contributed to the discussion of the CODH-hydrogenase gene cluster evolution, and helped to draft the manuscript. TGS and FTR discussed the HGT analysis and helped to draft the manuscript. JMG coordinated the study, and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
FH Sant’Anna, Email: fhsantanna@yahoo.com.br.
AV Lebedinsky, Email: a.lebedinsky@mail.ru.
TG Sokolova, Email: tatso2204@gmail.com.
FT Robb, Email: frobb@som.umaryland.edu.
JM Gonzalez, Email: jmgrau@irnase.csic.es.
References
- 1.Rao L, Xue Y, Zhou C, Tao J, Li G, Lu JR, et al. A thermostable esterase from Thermoanaerobacter tengcongensis opening up a new family of bacterial lipolytic enzymes. Biochim Biophys Acta. 2011;1814:1695–702. doi: 10.1016/j.bbapap.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 2.Moriyoshi K, Koma D, Yamanaka H, Sakai K, Ohmoto T. Expression and characterization of a thermostable acetylxylan esterase from Caldanaerobacter subterraneus subsp. tengcongensis involved in the degradation of insoluble cellulose acetate. Biosci Biotechnol Biochem. 2013;77:2495–8. doi: 10.1271/bbb.130568. [DOI] [PubMed] [Google Scholar]
- 3.De Champdoré M, Staiano M, Rossi M, D’Auria S. Proteins from extremophiles as stable tools for advanced biotechnological applications of high social interest. J R Soc Interface. 2007;4:183–91. doi: 10.1098/rsif.2006.0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pierce E, Xie G, Barabote RD, Saunders E, Han CS, Detter JC, et al. The complete genome sequence of Moorella thermoacetica (f. Clostridium thermoaceticum) Environ Microbiol. 2008;10:2550–73. doi: 10.1111/j.1462-2920.2008.01679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oehler D, Poehlein A, Leimbach A, Müller N, Daniel R, Gottschalk G, et al. Genome-guided analysis of physiological and morphological traits of the fermentative acetate oxidizer Thermacetogenium phaeum. BMC Genomics. 2012;13:723. doi: 10.1186/1471-2164-13-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao Y, Caspers MP, Abee T, Siezen RJ, Kort R. Complete genome sequence of Geobacillus thermoglucosidans TNO-09.020, a thermophilic sporeformer associated with a dairy-processing environment. J Bacteriol. 2012;194:4118. doi: 10.1128/JB.00318-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Visser M, Worm P, Muyzer G, Pereira IAC, Schaap PJ, Plugge CM, et al. Genome analysis of Desulfotomaculum kuznetsovii strain 17(T) reveals a physiological similarity with Pelotomaculum thermopropionicum strain SI(T) Stand Genomic Sci. 2013;8:69–87. doi: 10.4056/sigs.3627141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sokolova T, Gonzalez J, Kostrikina N, Chernyh N, Tourova T, Kato C, et al. Carboxydobrachium pacificum gen. nov., sp. nov., a new anaerobic, thermophilic, CO-utilizing marine bacterium from Okinawa Trough. Int J Syst Evol Microbiol. 2001;51:141–9. doi: 10.1099/00207713-51-1-141. [DOI] [PubMed] [Google Scholar]
- 9.Xue Y, Xu Y, Liu Y, Ma Y, Zhou P. Thermoanaerobacter tengcongensis sp. nov., a novel anaerobic, saccharolytic, thermophilic bacterium isolated from a hot spring in Tengcong, China. Int J Syst Evol Microbiol. 2001;51:1335–41. doi: 10.1099/00207713-51-4-1335. [DOI] [PubMed] [Google Scholar]
- 10.Kim B, Grote R, Lee D, Antranikian G, Pyun Y. Thermoanaerobacter yonseiensis sp. nov., a novel extremely thermophilic, xylose-utilizing bacterium that grows at up to 85 degrees C. Int J Syst Evol Microbiol. 2001;51:1539–48. doi: 10.1099/00207713-51-4-1539. [DOI] [PubMed] [Google Scholar]
- 11.Fardeau M-L, Bonilla Salinas M, L’Haridon S, Jeanthon C, Verhé F, Cayol J-L, et al. Isolation from oil reservoirs of novel thermophilic anaerobes phylogenetically related to Thermoanaerobacter subterraneus: reassignment of T. subterraneus, Thermoanaerobacter yonseiensis, Thermoanaerobacter tengcongensis and Carboxydibrachium pacificum to. Int J Syst Evol Microbiol. 2004;54(Pt 2):467–74. doi: 10.1099/ijs.0.02711-0. [DOI] [PubMed] [Google Scholar]
- 12.Bao Q, Tian Y, Li W, Xu Z, Xuan Z, Hu S, et al. A complete sequence of the T. tengcongensis genome. Genome Res. 2002;12:689–700. doi: 10.1101/gr.219302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sokolova TG, Henstra A-M, Sipma J, Parshina SN, Stams AJM, Lebedinsky AV. Diversity and ecophysiological features of thermophilic carboxydotrophic anaerobes. FEMS Microbiol Ecol. 2009;68:131–41. doi: 10.1111/j.1574-6941.2009.00663.x. [DOI] [PubMed] [Google Scholar]
- 14.Lee S-J, Lee Y-J, Park G-S, Kim B-C, Lee SJ, Shin J-H, et al. Draft genome sequence of an anaerobic and extremophilic bacterium, Caldanaerobacter yonseiensis, isolated from a geothermal hot stream. Genome Announc. 2013;1:e00923-13. doi: 10.1128/genomeA.00923-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou C, Xue Y, Ma Y. Enhancing the thermostability of alpha-glucosidase from Thermoanaerobacter tengcongensis MB4 by single proline substitution. J Biosci Bioeng. 2010;110:12–7. doi: 10.1016/j.jbiosc.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 16.Subbotina IV, Chernyh NA, Sokolova TG, Kublanov IV, Bonch-Osmolovskaya EA, Lebedinsky AV. Oligonucleotide probes for the detection of representatives of the genus Thermoanaerobacter. Microbiology. 2003;72:331–9. doi: 10.1023/A:1024256217575. [DOI] [Google Scholar]
- 17.Galtier N, Lobry JR. Relationships between genomic G + C content, RNA secondary structures, and optimal growth temperature in prokaryotes. J Mol Evol. 1997;44:632–6. doi: 10.1007/PL00006186. [DOI] [PubMed] [Google Scholar]
- 18.Ferry JG. CO dehydrogenase. Annu Rev Microbiol. 1995;49:305–33. doi: 10.1146/annurev.mi.49.100195.001513. [DOI] [PubMed] [Google Scholar]
- 19.Wu M, Ren Q, Durkin AS, Daugherty SC, Brinkac LM, Dodson RJ, et al. Life in hot carbon monoxide: the complete genome sequence of Carboxydothermus hydrogenoformans Z-2901. PLoS Genet. 2005;1:e65. doi: 10.1371/journal.pgen.0010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sokolova T, Lebedinsky A. CO-oxidizing anaerobic thermophilic prokaryotes. In: Satyanarayana T, Littlechild J, Kawarabayasi Y, editors. Thermophilic microbes in environmental and industrial biotechnology: biotechnology of thermophiles. 2. Netherlands: Springer; 2013. pp. 203–31. [Google Scholar]
- 21.Soboh B, Linder D, Hedderich R. Purification and catalytic properties of a CO-oxidizing:H2-evolving enzyme complex from Carboxydothermus hydrogenoformans. Eur J Biochem. 2002;269:5712–21. doi: 10.1046/j.1432-1033.2002.03282.x. [DOI] [PubMed] [Google Scholar]
- 22.Fox JD, Kerby RL, Roberts GP, Ludden PW. Characterization of the CO-induced, CO-tolerant hydrogenase from Rhodospirillum rubrum and the gene encoding the large subunit of the enzyme. J Bacteriol. 1996;178:1515–24. doi: 10.1128/jb.178.6.1515-1524.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forzi L, Sawers RG. Maturation of [NiFe]-hydrogenases in Escherichia coli. Biometals. 2007;20:565–78. doi: 10.1007/s10534-006-9048-5. [DOI] [PubMed] [Google Scholar]
- 24.Lim JK, Kang SG, Lebedinsky AV, Lee J-H, Lee HS. Identification of a novel class of membrane-bound [NiFe]-hydrogenases in Thermococcus onnurineus NA1 by in silico analysis. Appl Environ Microbiol. 2010;76:6286–9. doi: 10.1128/AEM.00123-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim M-S, Bae SS, Kim YJ, Kim TW, Lim JK, Lee SH, et al. CO-dependent H2 production by genetically engineered Thermococcus onnurineus NA1. Appl Environ Microbiol. 2013;79:2048–53. doi: 10.1128/AEM.03298-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Böhm R, Sauter M, Böck A. Nucleotide sequence and expression of an operon in Escherichia coli coding for formate hydrogenlyase components. Mol Microbiol. 1990;4:231–43. doi: 10.1111/j.1365-2958.1990.tb00590.x. [DOI] [PubMed] [Google Scholar]
- 27.Hedderich R. Energy-converting [NiFe] hydrogenases from archaea and extremophiles: ancestors of complex I. J Bioenerg Biomembr. 2004;36:65–75. doi: 10.1023/B:JOBB.0000019599.43969.33. [DOI] [PubMed] [Google Scholar]
- 28.Henstra AM, Stams AJ. Deep conversion of carbon monoxide to hydrogen and formation of acetate by the anaerobic thermophile Carboxydothermus hydrogenoformans. Int J Microbiol. 2011; 2011: 641582. doi: 10.1155/2011/641582 [DOI] [PMC free article] [PubMed]
- 29.Techtmann SM, Lebedinsky AV, Colman AS, Sokolova TG, Woyke T, Goodwin L, et al. Evidence for horizontal gene transfer of anaerobic carbon monoxide dehydrogenases. Front Microbiol. 2012;3:132. doi: 10.3389/fmicb.2012.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Becq J, Churlaud C, Deschavanne P. A benchmark of parametric methods for horizontal transfers detection. PLoS One. 2010;5:e9989. doi: 10.1371/journal.pone.0009989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lawrence JG, Ochman H. Reconciling the many faces of lateral gene transfer. Trends Microbiol. 2002;10:1–4. doi: 10.1016/S0966-842X(01)02282-X. [DOI] [PubMed] [Google Scholar]
- 32.Eisen JA. Horizontal gene transfer among microbial genomes: new insights from complete genome analysis. Curr Opin Genet Dev. 2000;10:606–11. doi: 10.1016/S0959-437X(00)00143-X. [DOI] [PubMed] [Google Scholar]
- 33.Techtmann SM, Colman AS, Robb FT. “That which does not kill us only makes us stronger”: the role of carbon monoxide in thermophilic microbial consortia. Environ Microbiol. 2009;11:1027–37. doi: 10.1111/j.1462-2920.2009.01865.x. [DOI] [PubMed] [Google Scholar]
- 34.Gonzalez JM, Robb FT. Genetic analysis of Carboxydothermus hydrogenoformans carbon monoxide dehydrogenase genes cooF and cooS. FEMS Microbiol Lett. 2000;191:243–7. doi: 10.1111/j.1574-6968.2000.tb09346.x. [DOI] [PubMed] [Google Scholar]
- 35.Wagner ID, Wiegel J. Diversity of thermophilic anaerobes. Ann N Y Acad Sci. 2008;1125:1–43. doi: 10.1196/annals.1419.029. [DOI] [PubMed] [Google Scholar]
- 36.Calteau A, Gouy M, Perrière G. Horizontal transfer of two operons coding for hydrogenases between bacteria and archaea. J Mol Evol. 2005;60:557–65. doi: 10.1007/s00239-004-0094-8. [DOI] [PubMed] [Google Scholar]
- 37.Jacobi A, Rossmann R, Böck A. The hyp operon gene products are required for the maturation of catalytically active hydrogenase isoenzymes in Escherichia coli. Arch Microbiol. 1992;158:444–51. doi: 10.1007/BF00276307. [DOI] [PubMed] [Google Scholar]
- 38.Trchounian K. Transcriptional control of hydrogen production during mixed carbon fermentation by hydrogenases 4 (hyf) and 3 (hyc) in Escherichia coli. Gene. 2012;506:156–60. doi: 10.1016/j.gene.2012.06.084. [DOI] [PubMed] [Google Scholar]
- 39.Silva PJ, van den Ban EC, Wassink H, Haaker H, de Castro B, Robb FT, et al. Enzymes of hydrogen metabolism in Pyrococcus furiosus. Eur J Biochem. 2000;267:6541–51. doi: 10.1046/j.1432-1327.2000.01745.x. [DOI] [PubMed] [Google Scholar]
- 40.Schut GJ, Bridger SL, Adams MWW. Insights into the metabolism of elemental sulfur by the hyperthermophilic archaeon Pyrococcus furiosus: characterization of a coenzyme A- dependent NAD(P)H sulfur oxidoreductase. J Bacteriol. 2007;189:4431–41. doi: 10.1128/JB.00031-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schut GJ, Boyd ES, Peters JW, Adams MWW. The modular respiratory complexes involved in hydrogen and sulfur metabolism by heterotrophic hyperthermophilic archaea and their evolutionary implications. FEMS Microbiol Rev. 2013;37:182–203. doi: 10.1111/j.1574-6976.2012.00346.x. [DOI] [PubMed] [Google Scholar]
- 42.Vieille C, Zeikus GJ. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol Mol Biol Rev. 2001;65:1–43. doi: 10.1128/MMBR.65.1.1-43.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng Y, Xue Y, Zhang Y, Zhou C, Schwaneberg U, Ma Y. Cloning, expression, and characterization of a thermostable glucoamylase from Thermoanaerobacter tengcongensis MB4. Appl Microbiol Biotechnol. 2010;87:225–33. doi: 10.1007/s00253-010-2439-0. [DOI] [PubMed] [Google Scholar]
- 44.Rolfsmeier M, Blum P. Purification and characterization of a maltase from the extremely thermophilic crenarchaeote Sulfolobus solfataricus. J Bacteriol. 1995;177:482–5. doi: 10.1128/jb.177.2.482-485.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liang C, Xue Y, Fioroni M, Rodríguez-Ropero F, Zhou C, Schwaneberg U, et al. Cloning and characterization of a thermostable and halo-tolerant endoglucanase from Thermoanaerobacter tengcongensis MB4. Appl Microbiol Biotechnol. 2011;89:315–26. doi: 10.1007/s00253-010-2842-6. [DOI] [PubMed] [Google Scholar]
- 46.Kuhad RC, Gupta R, Singh A. Microbial cellulases and their industrial applications. Enzyme Res. 2011;2011:280696. doi: 10.4061/2011/280696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dahiya N, Tewari R, Hoondal GS. Biotechnological aspects of chitinolytic enzymes: a review. Appl Microbiol Biotechnol. 2006;71:773–82. doi: 10.1007/s00253-005-0183-7. [DOI] [PubMed] [Google Scholar]
- 48.Bhattacharya D, Nagpure A, Gupta RK. Bacterial chitinases: properties and potential. Crit Rev Biotechnol. 2008;27:21–8. doi: 10.1080/07388550601168223. [DOI] [PubMed] [Google Scholar]
- 49.Hasan F, Shah AA, Hameed A. Industrial applications of microbial lipases. Enzyme Microb Technol. 2006;39:235–51. doi: 10.1016/j.enzmictec.2005.10.016. [DOI] [Google Scholar]
- 50.Panda T, Gowrishankar BS. Production and applications of esterases. Appl Microbiol Biotechnol. 2005;67:160–9. doi: 10.1007/s00253-004-1840-y. [DOI] [PubMed] [Google Scholar]
- 51.Zhang J, Liu J, Zhou J, Ren Y, Dai X, Xiang H. Thermostable esterase from Thermoanaerobacter tengcongensis: high-level expression, purification and characterization. Biotechnol Lett. 2003;25:1463–7. doi: 10.1023/A:1025076121700. [DOI] [PubMed] [Google Scholar]
- 52.Grosse S, Bergeron H, Imura A, Boyd J, Wang S, Kubota K, et al. Nature versus nurture in two highly enantioselective esterases from Bacillus cereus and Thermoanaerobacter tengcongensis. Microb Biotechnol. 2010;3:65–73. doi: 10.1111/j.1751-7915.2009.00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Royter M, Schmidt M, Elend C, Höbenreich H, Schäfer T, Bornscheuer UT, et al. Thermostable lipases from the extreme thermophilic anaerobic bacteria Thermoanaerobacter thermohydrosulfuricus SOL1 and Caldanaerobacter subterraneus subsp. tengcongensis. Extremophiles. 2009;13:769–83. doi: 10.1007/s00792-009-0265-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abokitse K, Wu M, Bergeron H, Grosse S, Lau PCK. Thermostable feruloyl esterase for the bioproduction of ferulic acid from triticale bran. Appl Microbiol Biotechnol. 2010;87:195–203. doi: 10.1007/s00253-010-2441-6. [DOI] [PubMed] [Google Scholar]
- 55.Levisson M, van der Oost J, Kengen SWM. Carboxylic ester hydrolases from hyperthermophiles. Extremophiles. 2009;13:567–81. doi: 10.1007/s00792-009-0260-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sinha R, Khare SK. Thermostable proteases. In: Satyanarayana T, Littlechild J, Kawarabayasi Y, editors. Thermophilic microbes in environmental and industrial biotechnology: biotechnology of thermophiles. 2. Netherlands: Springer; 2013. pp. 859–80. [Google Scholar]
- 57.Dutoit R, Brandt N, Legrain C, Bauvois C. Functional characterization of two M42 aminopeptidases erroneously annotated as cellulases. PLoS One. 2012;7:e50639. doi: 10.1371/journal.pone.0050639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koma D, Yamanaka H, Moriyoshi K, Ohmoto T, Sakai K. Overexpression and characterization of thermostable serine protease in Escherichia coli encoded by the ORF TTE0824 from Thermoanaerobacter tengcongensis. Extremophiles. 2007;11:769–79. doi: 10.1007/s00792-007-0103-0. [DOI] [PubMed] [Google Scholar]
- 59.Lee H-S, Vermaas WFJ, Rittmann BE. Biological hydrogen production: prospects and challenges. Trends Biotechnol. 2010;28:262–71. doi: 10.1016/j.tibtech.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 60.Chaudhuri RR, Loman NJ, Snyder LAS, Bailey CM, Stekel DJ, Pallen MJ. xBASE2: a comprehensive resource for comparative bacterial genomics. Nucleic Acids Res. 2008;36(Database issue):D543–6. doi: 10.1093/nar/gkm928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mao X, Ma Q, Zhou C, Chen X, Zhang H, Yang J, et al. DOOR 2.0: presenting operons and their functions through dynamic and integrated views. Nucleic Acids Res. 2014;42(Database issue):D654–9. doi: 10.1093/nar/gkt1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yarza P, Richter M, Peplies J, Euzeby J, Amann R, Schleifer K-H, et al. The All-Species Living Tree project: a 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst Appl Microbiol. 2008;31:241–50. doi: 10.1016/j.syapm.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 63.Pruesse E, Peplies J, Glöckner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28:1823–9. doi: 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008;36(Web Server issue):W465–9. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 66.Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol. 2006;55:539–52. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- 67.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57(Pt 1):81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 68.Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 2013;14:60. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Husemann P, Stoye J. r2cat: synteny plots and comparative assembly. Bioinformatics. 2010;26:570–1. doi: 10.1093/bioinformatics/btp690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shao Y, He X, Harrison EM, Tai C, Ou H-Y, Rajakumar K, et al. mGenomeSubtractor: a web-based tool for parallel in silico subtractive hybridization analysis of multiple bacterial genomes. Nucleic Acids Res. 2010;38(Web Server issue):W194–200. doi: 10.1093/nar/gkq326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ménigaud S, Mallet L, Picord G, Churlaud C, Borrel A, Deschavanne P. GOHTAM: a website for “Genomic Origin of Horizontal Transfers, Alignment and Metagenomics”. Bioinformatics. 2012;28:1270–1. doi: 10.1093/bioinformatics/bts118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–7. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 73.Carver T, Bleasby A. The design of Jemboss: a graphical user interface to EMBOSS. Bioinformatics. 2003;19:1837–43. doi: 10.1093/bioinformatics/btg251. [DOI] [PubMed] [Google Scholar]
- 74.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Whelan S, Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol. 2001;18:691–9. doi: 10.1093/oxfordjournals.molbev.a003851. [DOI] [PubMed] [Google Scholar]
- 76.Rawlings ND, Waller M, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014;42(Database issue):D503–9. doi: 10.1093/nar/gkt953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Park BH, Karpinets TV, Syed MH, Leuze MR, Uberbacher EC. CAZymes Analysis Toolkit (CAT): web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology. 2010;20:1574–84. doi: 10.1093/glycob/cwq106. [DOI] [PubMed] [Google Scholar]
- 78.Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8. [Google Scholar]
- 79.Messaoudi A, Belguith H, Ghram I, Ben Hamida J. LIPABASE: a database for “true” lipase family enzymes. Int J Bioinform Res Appl. 2011;7:390–401. doi: 10.1504/IJBRA.2011.043770. [DOI] [PubMed] [Google Scholar]
- 80.Dobbek H, Svetlitchnyi V, Gremer L, Huber R, Meyer O. Crystal structure of a carbon monoxide dehydrogenase reveals a [Ni-4Fe-5S] cluster. Science. 2001;293:1281–5. doi: 10.1126/science.1061500. [DOI] [PubMed] [Google Scholar]